
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe hydrogen dependent CO2 reductase: the first completely CO tolerant FeFe-hydrogenase†
Pierre
Ceccaldi
a,
Kai
Schuchmann‡
b,
Volker
Müller
b and
Sean J.
Elliott
*a
aDepartment of Chemistry, Boston University, 590 Commonwealth Avenue, Massachusetts 02215, USA. E-mail: elliott@bu.edu
bMolecular Microbiology and Bioenergetics, Institute of Molecular Biosciences, Johann Wolfgang Goethe University Frankfurt/Main, Max-von-Laue-Strasse 9, 60438 Frankfurt, Germany
First published on 24th October 2016
Abstract
The Hydrogen Dependent Carbon dioxide Reductase (HDCR) from Acetobacterium woodii presents a promising solution to the issue of H2 storage by reversibly coupling H2 oxidation to CO2 reduction. We here report on the electrocatalytic properties of the hydrogenase (Hase) module in the intact complex, including (an)aerobic oxidation, CO inhibition and the first systematic analysis of the catalytic bias (CB) of a Hase. CB depends on pH, regardless of the H2 concentration, despite a higher affinity for H2 than other FeFe-Hases. Remarkably, CO inhibition is fully reversible under all oxidation states of the active site, making HDCR the first “syngas-friendly” FeFe-Hase.
Broader contextMolecular hydrogen (H2) has been considered as a clean energy source for several decades, yet a number of critical obstacles must be overcome to allow hydrogen to be widely usable. In particular, the extreme explosive hazard of H2 prevents safe storage of large quantities, and one way chemists have sought to overcome this restriction is to transform H2 into another chemical that would serve as a storage unit, such as formate. Here, we report on the mechanistic traits of the Hydrogen Dependent Carbon dioxide Reductase from Acetobacterium woodii (HDCR), an enzymatic system recently reported by our co-authors, that is capable of the direct interface between a FeFe hydrogenase and a CO2 reductase. |
The consideration of dihydrogen (H2) as a potential new energy vector requires balancing the issue of gas storage and the possibility of an explosive hazard. Carbon dioxide (CO2) has been considered as an H2 storage unit through formic acid or formate:1 a concept used by acetogenic bacteria that can grow on H2 and CO2 only, thus using H2 and CO2 as energy and carbon source, respectively, through the Wood–Ljungdahl pathway (WLP) in Acetobacterium woodii (ref. 2, 3 and references therein). The methyl branch of WLP starts with the four-subunit Hydrogen Dependent Carbon dioxide Reductase (HDCR),2 the object of the present study (Fig. 1). Two subunits contain one catalytic site each: FdhF2 houses a Mo-bisPGD cofactor4 that reduces CO2 to formate, while the H2-oxidizing H-cluster characteristic of FeFe-Hases5 is bound by HydA2. These two active sites are connected to one another via eleven predicted FeS clusters distributed between all four subunits. This direct and reversible intramolecular coupling of H2 activation to CO2 reduction in a soluble enzyme is, to the better of our knowledge, unique in nature (see ref. 6 for a more complex example that a priori catalyses the reverse reaction only). HDCR catalysis is currently enigmatic, as it is unclear how the two enzymatic activities are coupled to one another, and how each catalytic component can serve as a standalone, monofunctional enzyme. In solution, HDCR turns over H2 102 times faster than CO2, thus accurate characterization of the HDCR Hase properties constitutes a required first-step to understand the electrochemical coupling between the two activities. Here, we use the lens of catalytic protein film electrochemistry (PFE) to examine HDCR for the first time. PFE has been extensively used for examining complex activity features of Hases, such as catalytic bias,7 inhibition by the small molecules CO8,9 and O2,10–12 as well as (in)activation processes.13–17 Here, we present the first electrochemical characterization of the intact HDCR complex, focusing on its Hase activity. We find that HDCR possesses the canonical properties of FeFe-Hases in terms of catalytic bias and oxidative inactivation, but reveals an unprecedented fully reversible inhibition by carbon monoxide.
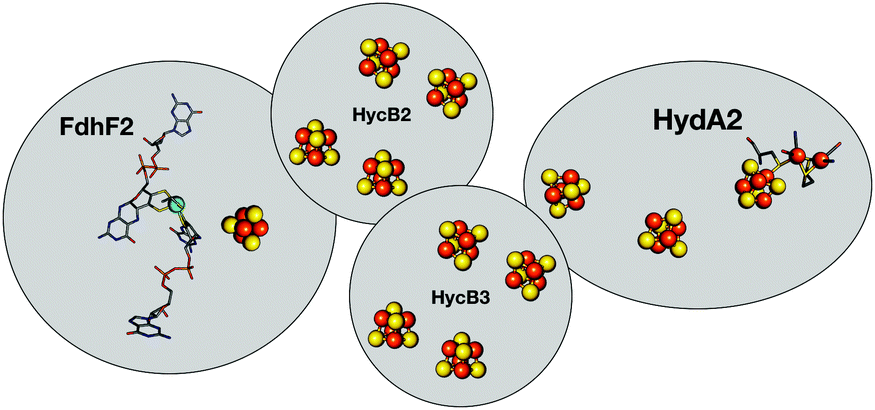 | ||
| Fig. 1 Schematic representation of HDCR.2 The formate dehydrogenase unit FdhF2 belongs to the bisPGD superfamily,4 and the HydA2 subunit is a typical FeFe-hydrogenase, with two FeS clusters besides the active site. Depicted cluster numbers and stoichiometries are predicted on the basis of amino acid sequence. | ||
Hases are reversible enzymes that catalyze H2 oxidation as well as proton reduction,18 yet FeFe-Hases are typically biased for proton reduction, which manifests itself by displaying low affinity for H2 (KM ∼ 0.6 atm and Ki = 5–30 atm)19 compared to NiFe-Hases that favor H2 oxidation.8,20 We initially examined the Hase activity of HDCR, as it was unclear if the complex may be a priori biased toward H2 evolution or oxidation. To do so, we defined the catalytic bias as the ratio of proton reduction and H2 oxidation activity under given conditions of pH and H2 pressure. Fig. 2 shows cyclic voltammograms (CVs) of a HDCR-coated rotating electrode, under 1 atm of H2 between pH 5 and 8. The electrode potential was swept at 0.1 V s−1 to prevent any redox (in)activation process15,16 and to limit film-loss during the experiment. All CVs cross the zero current axis at the value of the 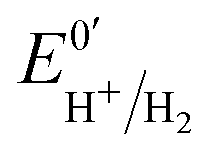 , as do all reversible hydrogenases. At pH 5 under 1 atm of H2 and an overpotential of 0.25 V, HDCR reduces protons 10 times faster than it oxidizes H2, thus revealing its bias towards proton reduction. That a FeFe-Hase favors proton reduction under acidic conditions is of no surprise (e.g. see Fig. 3 in ref. 8), but as pH increases, the ratio between reductive and oxidative activity decreases.
, as do all reversible hydrogenases. At pH 5 under 1 atm of H2 and an overpotential of 0.25 V, HDCR reduces protons 10 times faster than it oxidizes H2, thus revealing its bias towards proton reduction. That a FeFe-Hase favors proton reduction under acidic conditions is of no surprise (e.g. see Fig. 3 in ref. 8), but as pH increases, the ratio between reductive and oxidative activity decreases.
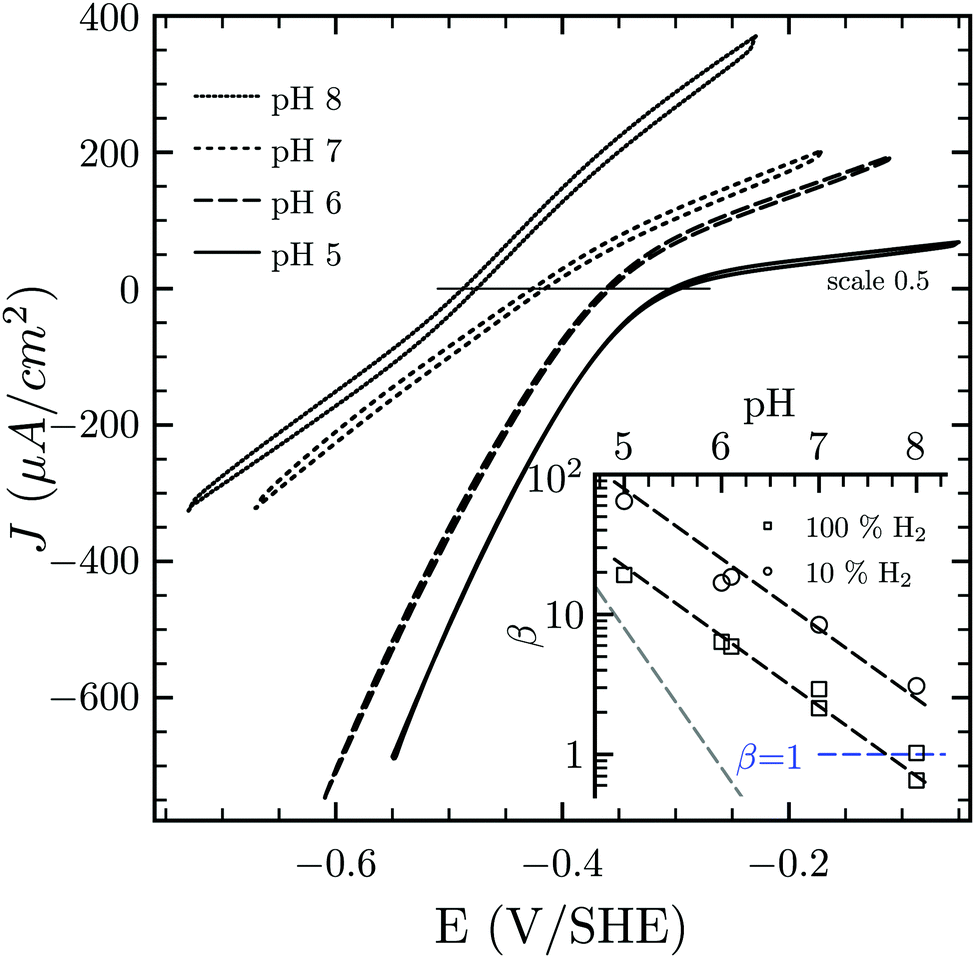 | ||
| Fig. 2 Catalytic bias of the hydrogenase activity of HDCR. Cyclic voltammogram of HDCR adsorbed on pyrolytic graphite disk electrode (≈3.1 mm2) at 30 °C, pH 5, 6, 7 and 8 under 1 atm of H2. Scan rate, 0.1 V s−1; ω = 3k rpm. The inset shows the pH dependence of β calculated as the ratio of the slopes of the voltammogram at high driving force21,22 (the dash lines follow the eqn (S1) (ESI†) with the slope [H+]0.5 in black and [H+]1 in grey for comparison). | ||
To analyse the pH dependence of the bias of HDCR-Hase, we built on the conclusions from Léger and coworkers (see eqn (9) and Fig. 5 in ref. 21) that showed the proportionality between the maximum turn-over rate k2 and the shape of the steady-state voltammogram.22 We thus consider the catalytic bias β as the ratio of the slopes for H2 formation or oxidation at high driving-force (Fig. 2 inset). Values of β distribute along the black dash lines, of which the slope is 0.5. This suggests that the catalytic bias is directly proportional to [H+]0.5, with no apparent effect of [H2] on this behaviour. Lowering H2 pressure at a given pH does increase β (i.e. the circles plot above the squares in the figure inset), but has no effect on its pH dependence (see ESI,† Section S2). We hope this first systematic study of catalytic bias under different thermodynamic conditions (pH and [H2]) will inspire other groups to run comparable studies to thus arrive at an universal “fingerprint” of the catalytic bias of Hases.
H2 either acts as a substrate or an inhibitor whether one considers the H2 oxidation or evolution. Consequently, lowering H2 pressure directly increases proton reduction and lowers H2 oxidation activity. Because the electrochemical setup allows for measurement of the hydrogenase activity while varying the H2 concentration in the cell, we measured the apparent affinity for H2 in the case of H2 oxidation (KM) or evolution (Ki), using multi-step chronoamperometry. This consists of monitoring the activity over time at a fixed electrode potential on each potential step (Fig. 3). The H2 pressure is varied by subsequently turning on and off its flow, thus inducing an exponential relaxation of [H2] in the electrochemical cell (panel A), which further results in a transient catalytic response (panel B, black trace). Under reductive conditions, the decrease of [H2] reversibly increases proton reduction current, and conversely decreases H2 oxidation current. The H2 oxidation current does not drop to zero when the H2 stream is stopped, due to the presence of some H2 remaining in the cell (black trace at t ≈ 1600 s in Fig. 3B). This residual H2 concentration can be measured independently through the monitoring of the open circuit potential, which shifts in accord with [H2] (Fig. S1 and Section S3, ESI†). To measure parameters Ki and KM, we analysed the data using QSoas, an open-source program that permits quantitative analysis of one-dimensional signals.23 We fitted the experimental data to the following equations:
iH2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) production =i([H2] = 0)/(1 + [H2]/Ki) production =i([H2] = 0)/(1 + [H2]/Ki) | (1) |
| iH2oxidation =i([H2] = ∞)/(1 + KM/[H2]) | (2) |
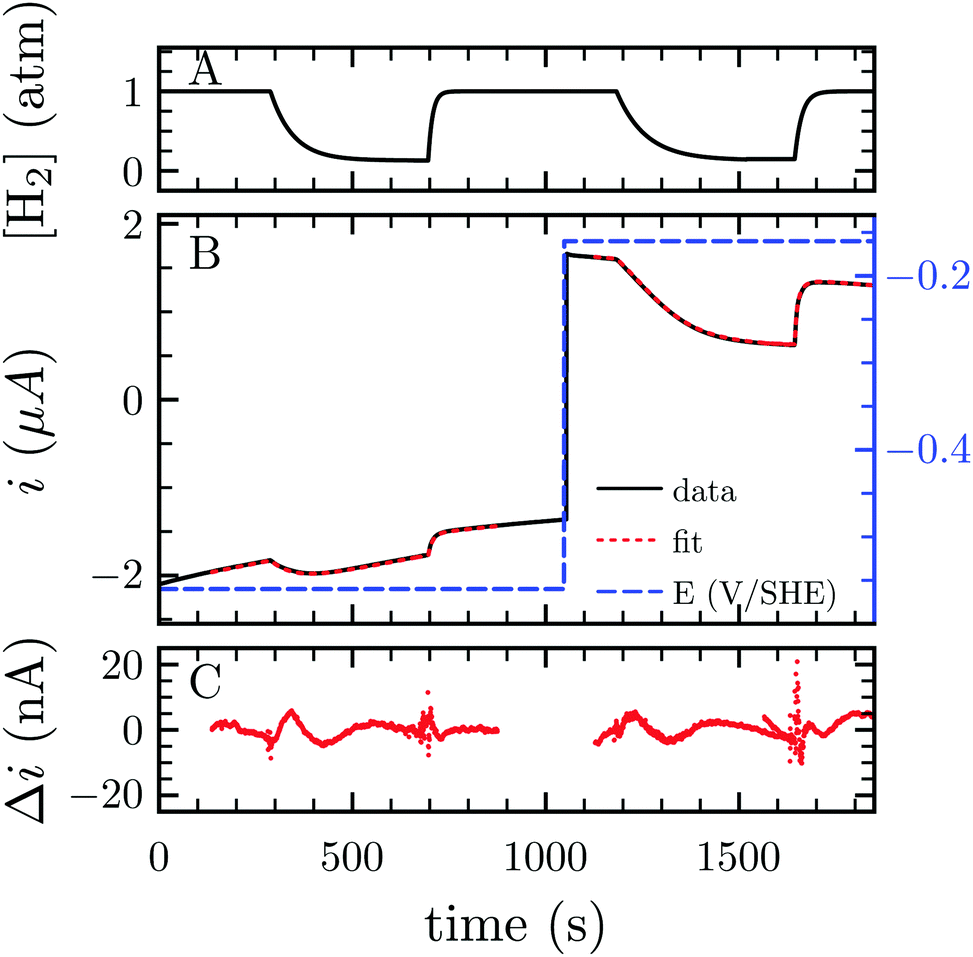 | ||
| Fig. 3 Measurement of Ki and KM for H2 at −560 and −160 mV per SHE, respectively. (A) H2 concentration against time. (B) Current corrected for non-faradic contribution (black, note the small but constant decay in current in absolute values, due to film-loss), best fit to eqn (1) and (2) (red) and electrode potential (blue) against time. (C) Difference between the fit and the data. Best fit parameters to the eqn (1) and (2) returned Ki = 6.1 atm (the average of 3 measurements returned Ki = 6.4 ± 1.3 atm) and KM = 0.26 atm (N = 3, KM = 0.24 ± 0.02 atm), respectively. Experimental conditions: 30 °C, pH 7, ω = 3k rpm. | ||
FeFe-Hases are inhibited by carbon monoxide (CO), and HDCR is no exception. Schuchmann and Müller reported that CO inhibits HDCR hydrogenase activity: 50% of inhibition is observed at 0.2 μM CO in the assay (see Figure S8 in ref. 2). Here, we examined the kinetics of CO inhibition of HDCR by monitoring its change in activity upon transient CO exposure (Fig. 4). The experiment consists in injecting a small volume of a CO-saturated buffer solution in the electrochemical cell, after which the inhibitor is flushed away by the constant H2 flow (panel A).
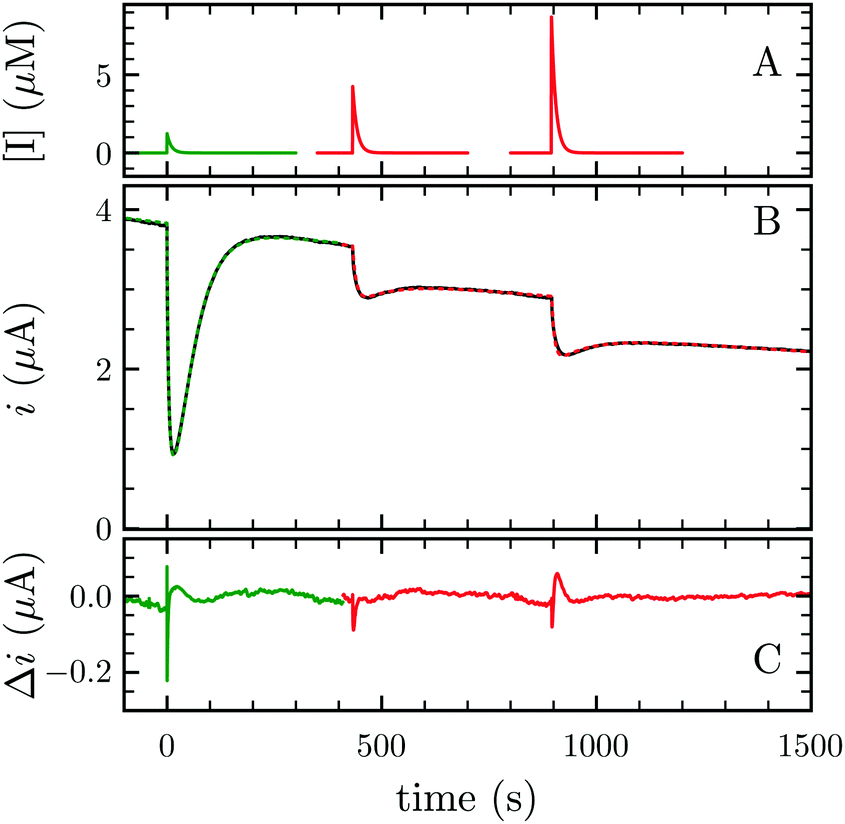 | ||
| Fig. 4 CO and O2 inhibition of H2 oxidation activity of A. woodii HDCR at +40 mV per SHE. (A) CO (green) and O2 (red) concentration against time. (B) Resulting current (black trace) and best fit of model 1 and 2 (green and red dashes, respectively). The best parameters of eqn (3) in ref. 27 to the CO inhibition (green trace) returned kin = 0.18 ± 0.01 s−1 μM−1 and kout = 0.02 ± 0.001 s−1; fitting the parameters of scheme to the aerobic inactivation returned kin = 4.8 ± 0.4 s−1 mM−1, kout = 5.5 ± 1 × 10−3 s−1 and k3 = 7 ± 1.4 × 10−3 s−1. (C) Difference between the fit and the data. Experimental conditions: pH 7, 1 atm H2, T = 30 °C ω = 3k rpm. | ||
Using QSoas23 and the procedure introduced by Leroux and coworkers and since developed by our first author and others,9,27–29 we fitted the transients of CO exposure with the parameters of Scheme 1, which considers reversible CO binding to HDCR in a single rate limiting step. Fitting parameters are the maximal current, the two rate constants of CO binding and release, the time-constant at which CO is flushed out of the cell and film-loss (see ESI† text). As noted previously, Ki for CO will depend upon [H2] (here 1 atm), and thus kin is an apparent rate constant. The best fit returned kin = 0.18 ± 0.01 s−1 μM−1 and kout = 0.02 ± 0.005 s−1, giving the inhibition constant KCOi = 0.11 ± 0.01 μM, thus three and ten times smaller than the reported values for the Cr and Ca FeFe-Hases9 obtained under the same conditions (pH 7, 1 atm H2, 30 °C). This larger inhibition by CO of HDCR might appear as a disadvantage for use of HDCR-based catalysts to process CO-containing syngas, nonetheless this is counterbalanced, and we think overcome by the full reversibility of the inhibition (see below).
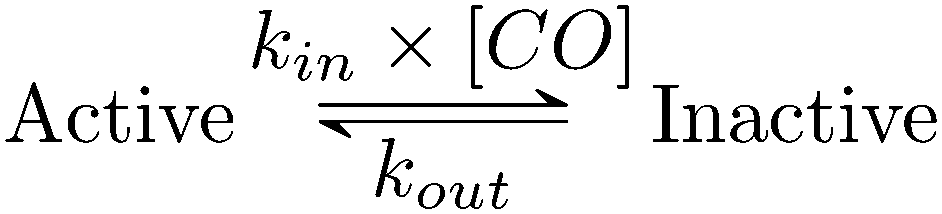 | ||
| Scheme 1 Kinetic model of CO inhibition of HDCR. CO binds with the bimolecular rate constant kin and unbinds with the unimolecular rate constant kout. Here, kin is not corrected for protection by H2, thus should be considered an apparent rate constant. | ||
Three distinct redox states of the H-cluster have been identified by FTIR and EPR spectroscopy: Hox and Hred differ by one electron and convert to one another at E0′ = −0.4 V per SHE at pH 8, as observed with Cr HydA1.30,31 Under turn-over conditions, CO reversibly binds to the Hox and Hred states, the latter leading to the disruption of the active site.9 The “super-reduced” state of the H-cluster (Hsred), one electron more reduced than Hred, appears at E0′ = −0.47 V per SHE30 and seems less sensitive to CO than Hred, as it is generated upon reduction of the Hred-CO state.31 Here we used CO as a probe of the oxidation state of the HDCR H-cluster, attempting to detect the three intermediate states through their differential sensitivity to CO. We repeated experiments similar to the one presented in Fig. 4 with different values of E across a ≈600 mV potential range, and with sequential injections of varying CO concentrations in order to extend CO exposure of the HDCR film and detect a possible slow irreversible step following CO binding on Hred state9,32 (Fig. S4, ESI†).
The kinetic parameters of CO inhibition as a function of electrode potential are plotted in Fig. 5 (note the log scale on the y axis). Starting from the high potential values, the inactivation slows down when approaching 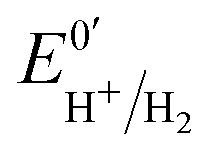 , then accelerates upon further reduction. This trend is similar to Ca and Cr HydA9 and appears as indirect evidence of redox changes within the H-cluster. Fitting the redox dependences of kCOin and kCOout returned
, then accelerates upon further reduction. This trend is similar to Ca and Cr HydA9 and appears as indirect evidence of redox changes within the H-cluster. Fitting the redox dependences of kCOin and kCOout returned 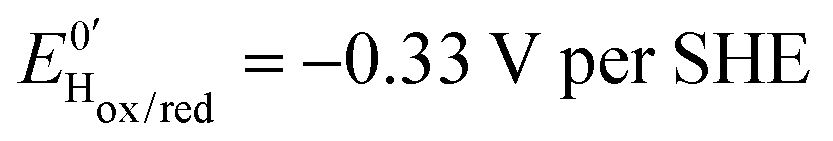 , and
, and 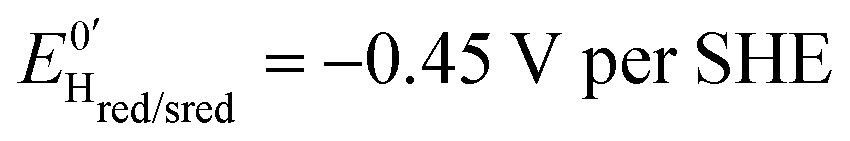 , similar to reported values.31
, similar to reported values.31
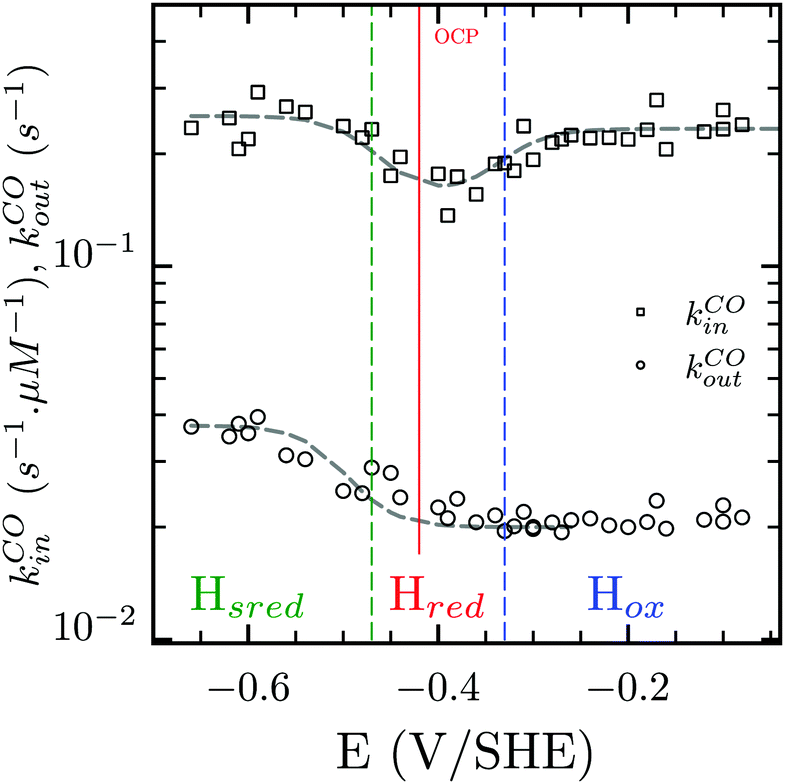 | ||
Fig. 5 Redox dependence of rate constants kin and kout from Scheme 1. The vertical lines mark the positions of the reduction potential of H+/H2 couple at pH 7 (red) and the redox transitions undergoing at the H-cluster according to ref. 31 (we assume 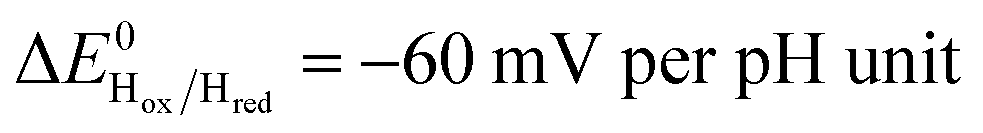 ). ). | ||
The most original feature of CO inhibition of HDCR is that the enzyme fully reactivates upon CO escape from the cell in all three oxidation states of the H-cluster. This observation is contrary to all PFE-based CO inhibition studies of FeFe-Hases to date, where CO binding to the H-cluster in the stability range of the Hred state provokes irreversible damage, assigned to the disruption of the bridge between the Fe2 subcluster and the adjacent cubane.8,9,32 In the case of HDCR, we took great care to expose the enzyme to high concentrations of inhibitor (up to 50 μM ≈500 × KCOi) so as to not miss an irreversible reaction of the CO-bound states (Fig. S4, ESI†). In the time scale of our experiments we only detected fully reversible inhibition, which could be simulated using the parameters of Scheme 1. In contrast, for Ca HydA1 the irreversible inhibition by CO is pronounced for concentrations much lower than the ones used here (see first transient in Fig. 1D and E in ref. 9), even though CO binds to the active site ≈75 times slower than in HDCR. Therefore if CO caused irreversible damage to HDCR, we would detect it.
Besides favoring proton reduction and being (up to the present work) sensitive to CO damage under certain conditions, FeFe-Hases also undergo oxidative (in)activation, with or without implication of O2. Fourmond and coworkers demonstrated that Ca and Cr FeFe-Hases produce three inactive species through anaerobic oxidation, two of which reactivate upon reduction.15 Such reactivatable oxidized states can be detected in CV experiments through the “Eswitch” inflection point on the reductive scan, which corresponds to the potential at which reactivation becomes faster than scan rate.12,14,17 The CVs presented in Fig. 6 display one or two Eswitch features depending on scan rate and temperature (Fig. S5, ESI†): HDCR anaerobic oxidation produces at least two species that reactivate upon reduction at different rates. The third, irreversibly inactivated species detected in previous studies15 could not be assigned here as this process combines with film-loss. Notably, these rates are approximately 100 times slower than in the case of previously reported enzymes.15
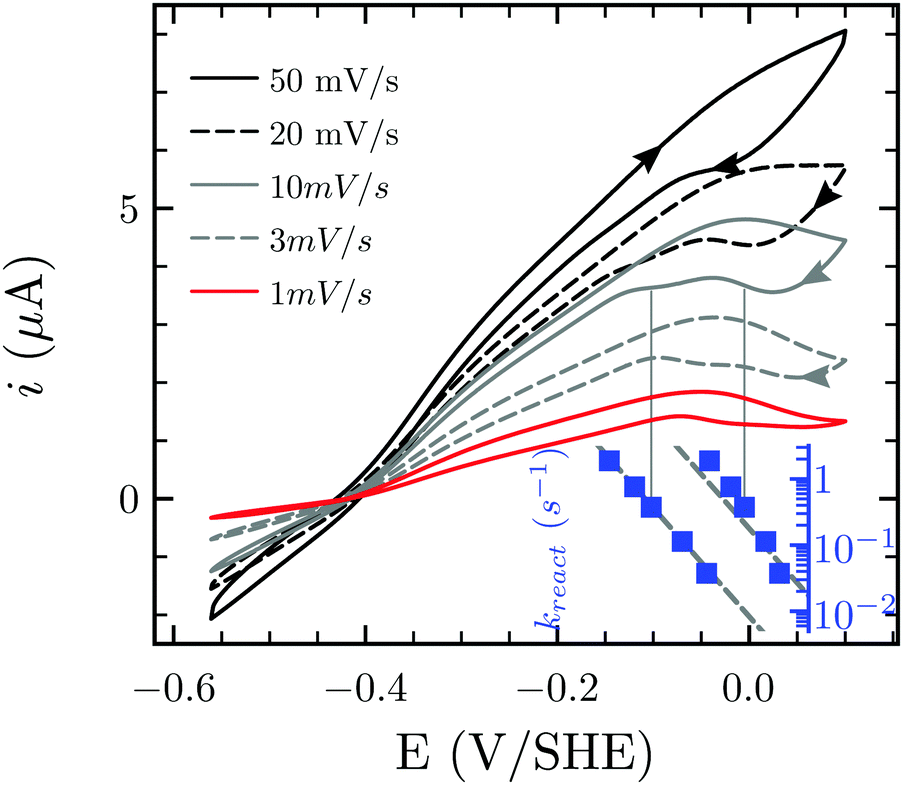 | ||
| Fig. 6 Anaerobic oxidative inactivation measured by cyclic voltammetry. The position of the reactivation waves at high potential shifts to more reductive values at increasing scan rate. The inset shows (on the same x axis) the reactivation rate constant at the position of Eswitch. Experimental conditions: T = 30 °C, pH 7.0, 1 atm H2, ω = 3k rpm. | ||
Finally Orain and coworkers demonstrated the partial reversibility of O2 attack on the H-cluster,12,33 a mechanism firstly suggested by two groups8,11 that we here test and validate for HDCR. We studied the aerobic inactivation of HDCR by measuring the H2 oxidation activity upon transient exposure to O2 (Fig. 4 red traces). We chose to perform this study at 30 °C rather than at 12 °C as in ref. 12, in order to best report on the determination of the rates of the underlying anaerobic processes. When exposed to O2, HDCR inactivates more slowly than with CO (note the inhibitor concentration plotted in the upper panel). Upon flushing O2 from the cell, activity first increases before stabilizing at a lower level than before O2 injection, suggesting that the aerobic inactivation occurs in two steps, only the second being irreversible. We fitted the data with the parameters of the kinetic model presented in Scheme 2,12 by using the reactivation rate constant determined by the position of Eswitch in CV (0.038 s−1, inset Fig. 6). Analogously to the works of Orain and coworkers, the partial reversibility of the O2 binding was necessary to satisfyingly reproduce the experimental traces, thus showing how HDCR reacts with O2.
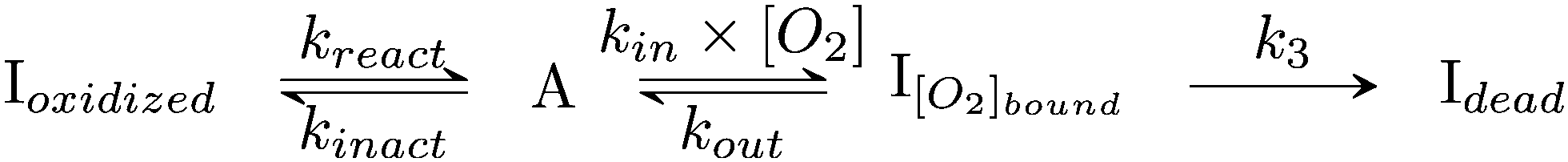 | ||
| Scheme 2 Kinetic model of aerobic inactivation of HDCR-Hase. The enzyme is in equilibrium between one active (A) and two inactive species, and Ioxidized is detected through the Eswitch in Fig. 6. The unimolecular reaction following O2 binding leads to the dead-end species Idead. | ||
The kinetic parameters of CO and O2 inhibition are summarized in Table 1. Even though we have no direct experimental evidence that O2 reacts with the active site, the likelihood with the works by Orain and coworkers strongly suggests that O2 targets the Fe2 subcluster. The reactivation step following oxygen binding (monomolecular rate kout) was very recently shown not to be the simple, non redox unbinding of O2 but rather its full reduction to water.26 That HDCR-Hase active site is eventually destroyed upon O2 exposure may prevent its applicability in industrial processes, but that is discarding recent developments in hydrogels as protective layers of oxygen sensitive catalysts.34–36
| I | k in (s−1 mM−1) | k out (×10−3 s−1) | k 3 (10−3 s−1) |
|---|---|---|---|
| CO | 20 ± 3 | 25 ± 5 | n.d. |
| O2 | 6.5 ± 0.1 | 5.4 ± 0.1 | 3.2 ± 0.12 |
At first glance, the entire HDCR complex displays a typical FeFe-Hase phenotype: it strongly favors proton reduction compared to NiFe-Hases, is inhibited by CO and O2, and produces two reversibly inactive species upon oxidation. Yet, CO inhibition is entirely reversible for HDCR (at least under the conditions we used here), which likely arises from specific structural properties that are yet to be deciphered. Thus, our results pave the way for a deeper investigation of the HDCR electrochemical behavior, and provide the essential underpinnings of future studies of HDCR reactivity towards CO2 and formate. Our findings suggest that structural studies of HDCR will likely unlock the mystery of how to design a FeFe-Hase that remains undamaged by CO. Such CO tolerance of a FeFe-Hase, detected here for the first time, is of great value if one wants to use enzyme-based catalysts to process CO-containing syngas.
References
- I. A. C. Pereira, Science, 2013, 342, 1329–1330 CrossRef CAS PubMed.
- K. Schuchmann and V. Müller, Science, 2013, 342, 1382–1385 CrossRef CAS PubMed.
- K. Schuchmann and V. Müller, Nat. Rev. Microbiol., 2014, 12, 809–821 CrossRef CAS PubMed.
- S. Grimaldi, B. Schoepp-Cothenet, P. Ceccaldi, B. Guigliarelli and A. Magalon, Biochim. Biophys. Acta, Bioenerg., 2013, 1827, 1048–1085 CrossRef CAS PubMed.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13–23 CrossRef CAS PubMed.
- J. S. McDowall, B. J. Murphy, M. Haumann, T. Palmer, F. A. Armstrong and F. Sargent, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E3948–E3956 CrossRef CAS PubMed.
- A. Abou Hamdan, S. Dementin, P.-P. Liebgott, O. Gutierrez-Sanz, P. Richaud, A. L. De Lacey, M. Rousset, P. Bertrand, L. Cournac and C. Léger, J. Am. Chem. Soc., 2012, 134, 8368–8371 CrossRef CAS PubMed.
- G. Goldet, C. Brandmayr, S. T. Stripp, T. Happe, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 14979–14989 CrossRef CAS PubMed.
- C. Baffert, L. Bertini, T. Lautier, C. Greco, K. Sybirna, P. Ezanno, E. Etienne, P. Soucaille, P. Bertrand, H. Bottin, I. Meynial-Salles, L. De Gioia and C. Léger, J. Am. Chem. Soc., 2011, 133, 2096–2099 CrossRef CAS PubMed.
- C. Léger, S. Dementin, P. Bertrand, M. Rousset and B. Guigliarelli, J. Am. Chem. Soc., 2004, 126, 12162–12172 CrossRef PubMed.
- C. Baffert, M. Demuez, L. Cournac, B. Burlat, B. Guigliarelli, P. Bertrand, L. Girbal and C. Léger, Angew. Chem., Int. Ed., 2008, 47, 2052–2054 CrossRef CAS PubMed.
- C. Orain, L. Saujet, C. Gauquelin, P. Soucaille, I. Meynial-Salles, C. Baffert, V. Fourmond, H. Bottin and C. Léger, J. Am. Chem. Soc., 2015, 137, 12580–12587 CrossRef CAS PubMed.
- V. Fourmond, P. Infossi, M.-T. Giudici-Orticoni, P. Bertrand and C. Léger, J. Am. Chem. Soc., 2010, 132, 4848–4857 CrossRef CAS PubMed.
- A. A. Hamdan, P.-P. Liebgott, V. Fourmond, O. Gutiérrez-Sanz, A. L. De Lacey, P. Infossi, M. Rousset, S. Dementin and C. Léger, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19916–19921 CrossRef PubMed.
- V. Fourmond, C. Greco, K. Sybirna, C. Baffert, P.-H. Wang, P. Ezanno, M. Montefiori, M. Bruschi, I. Meynial-Salles, P. Soucaille, J. Blumberger, H. Bottin, L. De Gioia and C. Léger, Nat. Chem., 2014, 6, 336–342 CrossRef CAS PubMed.
- V. Hajj, C. Baffert, K. Sybirna, I. Meynial-Salles, P. Soucaille, H. Bottin, V. Fourmond and C. Léger, Energy Environ. Sci., 2014, 7, 715–719 CAS.
- P. Ceccaldi, M. C. Marques, V. Fourmond, I. C. Pereira and C. Léger, Chem. Commun., 2015, 51, 14223–14226 RSC.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- V. Fourmond, C. Baffert, K. Sybirna, S. Dementin, A. Abou-Hamdan, I. Meynial-Salles, P. Soucaille, H. Bottin and C. Léger, Chem. Commun., 2013, 49, 6840–6842 RSC.
- C. Léger, S. Dementin, P. Bertrand, M. Rousset and B. Guigliarelli, J. Am. Chem. Soc., 2004, 126, 12162–12172 CrossRef PubMed.
- C. Léger, A. K. Jones, S. P. J. Albracht and F. A. Armstrong, J. Phys. Chem. B, 2002, 106, 13058–13063 CrossRef.
- C. Léger and P. Bertrand, Chem. Rev., 2008, 108, 2379–2438 CrossRef PubMed.
- V. Fourmond, Anal. Chem., 2016, 88, 5050–5052 CrossRef CAS PubMed.
- J. Cohen, K. Kim, P. King, M. Seibert and K. Schulten, Structure, 2005, 13, 1321–1329 CrossRef CAS PubMed.
- T. Lautier, P. Ezanno, C. Baffert, V. Fourmond, L. Cournac, J. C. Fontecilla-Camps, P. Soucaille, P. Bertrand, I. Meynial-Salles and C. Léger, Faraday Discuss., 2011, 148, 385–407 RSC.
- A. Kubas, C. Orain, D. De Sancho, L. Saujet, M. Sensi, C. Gauquelin, I. Meynial-Salles, P. Soucaille, H. A. Bottin, C. Baffert, V. Fourmond, R. B. Best, J. Blumberger and C. Léger, Nat. Chem., 2016 DOI:10.1038/nchem.2592.
- F. Leroux, S. Dementin, B. Burlat, L. Cournac, A. Volbeda, S. Champ, L. Martin, B. Guigliarelli, P. Bertrand, J. Fontecilla-Camps, M. Rousset and C. Léger, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11188–11193 CrossRef CAS PubMed.
- P.-P. Liebgott, F. Leroux, B. A. A. Burlat, S. A. Dementin, C. Baffert, T. Lautier, V. Fourmond, P. Ceccaldi, C. Cavazza, I. Meynial-Salles, P. Soucaille, J. C. Fontecilla-Camps, B. Guigliarelli, P. Bertrand, M. Rousset and C. Léger, Nat. Chem. Biol., 2009, 6, 63–70 CrossRef PubMed.
- P. Ceccaldi, E. Etienne, S. Dementin, B. Guigliarelli, C. Léger and B. Burlat, Biochim. Biophys. Acta, Bioenerg., 2016, 1857, 454–461 CrossRef CAS PubMed.
- A. Adamska, A. Silakov, C. Lambertz, O. Rüdiger, T. Happe, E. Reijerse and W. Lubitz, Angew. Chem., Int. Ed., 2012, 51, 11458–11462 CrossRef CAS PubMed.
- A. Adamska-Venkatesh, D. Krawietz, J. Siebel, K. Weber, T. Happe, E. Reijerse and W. Lubitz, J. Am. Chem. Soc., 2014, 136, 11339–11346 CrossRef CAS PubMed.
- C. E. Foster, T. Krämer, A. F. Wait, A. Parkin, D. P. Jennings, T. Happe, J. E. McGrady and F. A. Armstrong, J. Am. Chem. Soc., 2012, 134, 7553–7557 CrossRef CAS PubMed.
- K. D. Swanson, M. W. Ratzloff, D. W. Mulder, J. H. Artz, S. Ghose, A. Hoffman, S. White, O. A. Zadvornyy, J. B. Broderick, B. Bothner, P. W. King and J. W. Peters, J. Am. Chem. Soc., 2015, 137, 1809–1816 CrossRef CAS PubMed.
- N. Plumeré, O. Rüdiger, A. A. A. Oughli, R. Williams, J. Vivekananthan, S. Pöller, W. Schuhmann and W. Lubitz, Nat. Chem., 2014, 6, 822–827 CrossRef PubMed.
- A. A. Oughli, F. Conzuelo, M. Winkler, T. Happe, W. Lubitz, W. Schuhmann, O. Rüdiger and N. Plumeré, Angew. Chem., Int. Ed., 2015, 54, 12329–12333 CrossRef CAS PubMed.
- V. Fourmond, S. Stapf, H. Li, D. Buesen, J. Birrell, O. Rüdiger, W. Lubitz, W. Schuhmann, N. Plumeré and C. Léger, J. Am. Chem. Soc., 2015, 137, 5494–5505 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods. Comment on pH dependence of catalytic bias. Measure of H2 concentration by chronopotentiometry. Supplementary figures. See DOI: 10.1039/c6ee02494g |
| ‡ Present address: Center for Biomolecular Sciences, University of Nottingham, Nottingham, UK. |
| This journal is © The Royal Society of Chemistry 2017 |