
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controllable synthesis of mesostructures from TiO2 hollow to porous nanospheres with superior rate performance for lithium ion batteries†
Hao
Ren
a,
Jiajia
Sun
a,
Ranbo
Yu
*a,
Mei
Yang
b,
Lin
Gu
c,
Porun
Liu
d,
Huijun
Zhao
d,
David
Kisailus
e and
Dan
Wang
bd
aDepartment of Physical Chemistry, School of Metallurgical and Ecological Engineering, University of Science & Technology Beijing, No. 30, Xueyuan Road, Haidian District, Beijing 100083, P. R. China. E-mail: ranboyu@ustb.edu.cn
bNational Key Laboratory of Biochemical Engineering, Institute of Process Engineering, Chinese Academy of Sciences, No. 1, Bei Er Tiao, Zhongguancun, Beijing 100190, P. R. China
cLaboratory for Advanced Materials & Electron Microscopy, Beijing National Laboratory for Condensed Matter Physics, Institute of Physics, Chinese Academy of Sciences, Beijing 100190, P. R. China
dCentre for Clean Environment and Energy, Gold Coast Campus, Griffith University, Southport, Queensland 4222, Australia
eDepartment of Chemical and Environmental Engineering, University of California, Riverside, CA 92521, USA
First published on 26th October 2015
Abstract
Uniform TiO2 nanospheres from hollow, core–shell and mesoporous structures have been synthesized using quasi-nano-sized carbonaceous spheres as templates. The TiO2 nanospheres formed after calcination at 400 °C are composed of ∼7 nm nanoparticles and the shells of the hollow TiO2 nanospheres are as thin as a single layer of nanoparticles. The ultrafine nanoparticles endow the hollow and mesoporous TiO2 nanospheres with short lithium ion diffusion paths leading to high discharge specific capacities of 211.9 and 196.0 mA h g−1 at a current rate of 1 C (167.5 mA g−1) after 100 cycles, and especially superior discharge specific capacities of 125.9 and 113.4 mA h g−1 at a high current rate of up to 20 C. The hollow and mesoporous TiO2 nanospheres also show superior cycling stability with long-term discharge capacities of 103.0 and 110.2 mA h g−1, respectively, even after 3000 cycles at a current rate of 20 C.
Introduction
Lithium-ion batteries (LIBs) are one of the most promising and widely used technologies because of their safety, stable cycle life and low cost.1 Due to the great demand for high capacity LIBs, potential candidates have been intensively studied for decades.2–5 Recently, transition metal oxides such as Fe2O3, Co3O4, Mn2O3, CuO and NiO have been introduced as anodes and show higher capacities.6–14 However, since these anode materials are used at low discharge voltages (below 1 V vs. Li+/Li), they suffer from the formation of an unstable solid electrolyte interface (SEI) film on the electrode surface, which affects their safety.15,16 Recently, with the blossom of electric vehicles, highly powered and safer anodes are necessary.17 In view of this, anode materials like Li4Ti5O12 and TiO2 with high discharge voltages under which the electrolyte is stable are of great interest.18–20TiO2 is widely applied in many fields including environmental remediation, sensing and energy applications.21–24 As an anode material for LIBs, it has a high discharge voltage (up to 1 V) which can prevent the formation of a SEI film. In addition, the volume expansion during lithiation and delithiation processes is ∼3%, which reduces strain and permits a stable morphology resulting in a long cycle life.25 Moreover, TiO2 is abundant, environmentally friendly and chemically stable. However, the limited specific capacity and poor cycling performance of TiO2 resulting from the low ionic and electrical conductivities have obstructed its practical use.26–28 An effective way to decrease the lithium ion diffusion path lengths is to use nanosized TiO2 particles. Thus, the mean diffusion time of the lithium ions in electrode materials could be strongly decreased in nanoparticles according to the following formula: Teq = L2/D (Teq: mean diffusion time, L: diffusion length, D: diffusion coefficient).29,30 Unfortunately, in practical applications, the aggregation of nanoparticles during the charge/discharge processes will lead to another challenge.31 In order to solve these problems, purposely designed hierarchical mesostructures such as hierarchical microspheres, hollow microspheres and core–shell hollow microspheres have been proposed and synthesized. These micro-sized particles can provide good structural stability while nanoparticles can enhance the interfacial contact area and shorten the transition path of the lithium ions and electrons.32–35 We have successfully designed and fabricated mesostructured multi-shelled Fe2O3 and TiO2 hollow spheres which can improve the specific capacity, and have demonstrated that thin shelled microspheres show a better performance compared to thick shelled spheres.7,36 To our knowledge, no reports focus on very thin shelled hollow spheres. Moreover, the nanoparticles of the multi-shelled TiO2 hollow microspheres are currently larger than 20 nm.36 We anticipate that smaller sized nanoparticles are more favorable for shorter lithium ion diffusion times, thus leading to a better rate performance.
Herein we have successfully prepared TiO2 hollow nanospheres (TiO2-HNSs) with much thinner shells composed of nanoparticles as small as about 7 nm using a quasi-nano-sized carbonaceous sphere template method. The shell of the TiO2-HNSs was found to be as thin as a single layer of nanoparticles. By tuning the precursor absorbance and calcination processes, TiO2 mesoporous nanospheres (TiO2-PNSs) have also been fabricated. These kinds of nanospheres show superior performance, especially at high charge/discharge current rates. This results from their hollow and porous structures, which can provide easy charge transfer routes during lithiation and delithiation processes.
Results and discussion
In order to decrease the nanoparticle size in the TiO2-HNSs and TiO2-PNSs, the calcination temperature could be decreased.37,38 However, the micro-sized carbonaceous microspheres with diameters of around 3 μm synthesized using sucrose as a precursor could not be easily decomposed at such low temperatures after adsorbing the Ti source (Fig. S1†). Thus, much smaller quasi-nano-sized carbonaceous spheres (CNSs) with an average diameter of approximately 260 nm were synthesized via a hydrothermal process with glucose instead of sucrose as the precursor,39,40 which easily decompose at 400 °C (Fig. S1 and S2†). Based on previous work,36,40 a 3 M TiCl4 aqueous solution was used as the precursor since smaller sized Ti-coordinated cations can be formed and they are more easily adsorbed by the negatively charged carbonaceous spheres. By systematically controlling the adsorption time of the Ti source and controlling the calcination process to regulate the combustion kinetics and diffusion dynamics (see Fig. 1 and Table S1†), we are able to tune the mesostructures of the resulting products. Essentially, after leaving the CNSs in a 3 M TiCl4 aqueous solution for 6 h, the as-soaked CNSs were calcined at 400 °C for 4 h at a heating rate of 10 °C min−1 to remove the CNS templates. During the CNS removal process, the Ti source radially diffuses from the outside to the inside of the CNSs until they coalesce to form a shell. In fact, controlling the time for the precursor adsorption onto and within the CNSs significantly affected the resulting structures. In the CNSs soaked for 6 h, TiO2-HNSs were formed (Fig. 1a and 2a), which is ascribed to the scarce but opportune adsorbance of the Ti precursor. However, at reduced times (i.e., 4 h) nearly-collapsed hollow nanospheres (Fig. S3a†) formed due to a reduced adsorbance, which was confirmed by thermogravimetric (TG) analysis (Fig. S1†). In contrast, increasing the soaking time to 12 h in a 3 M TiCl4 aqueous solution increased the adsorbance of TiCl4 onto and within the CNSs (Fig. S1†). Under these conditions, following calcination treatment at 400 °C for 4 h at a heating rate of 10 °C min−1, the excess Ti source remaining after forming a shell would promote the formation of a core inside the shell during the decomposition of the as-soaked CNSs, yielding TiO2 core–shell nanospheres (Fig. 1b and S3b†). If the calcination heating rate of the CNSs soaked for 12 h was decreased to 1 °C min−1, the decomposition rate of the CNSs was reduced and the Ti source would slowly shrink and aggregate continuously with the CNS templates until TiO2-PNSs were eventually formed (Fig. 1c and 2b). It's reasonable that the CNSs soaked for 6 h could lead to porous nanospheres with loosely aggregated nanoparticles (TiO2-PNS-LS) since the adsorbance of the Ti source was reduced (Fig. 1d, S1, S3c and S3d†). Moreover, TiO2 hollow nanospheres and porous nanospheres composed of about 15 nm sized nanoparticles (termed TiO2-HNS-500 and TiO2-PNS-500, respectively) were also prepared using the same conditions as those for the TiO2-HNSs and TiO2-PNSs, respectively, with the exception of modifying the calcination temperature to 500 °C (Fig. 1e, f, S4a and S4b†). The larger particle size is attributed to the higher calcination temperature yielding grain growth resulting in thicker shells in the TiO2-HNS-500 specimen compared to the TiO2-HNS samples.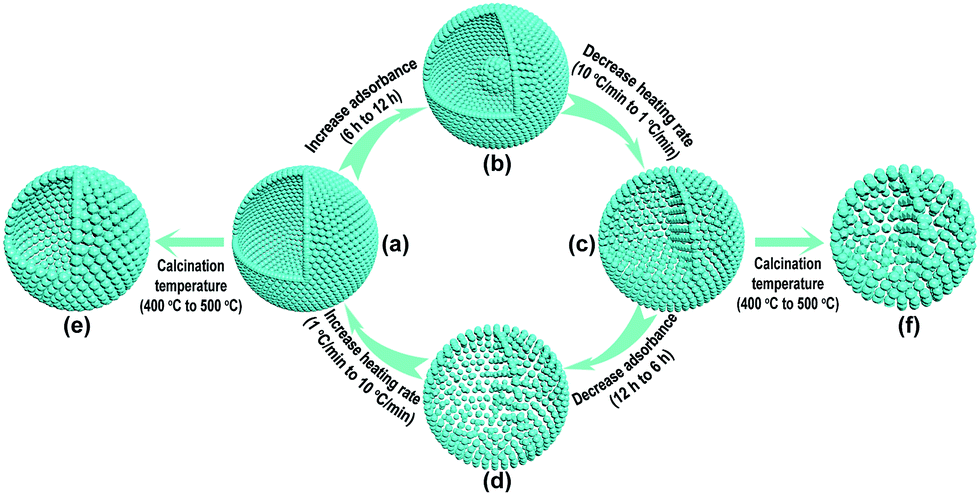 | ||
| Fig. 1 Schematic diagram of the controllable synthesis for each product. (a) TiO2-HNS; (b) TiO2-core–shell; (c) TiO2-PNS; (d) TiO2-PNS-LS; (e) TiO2-HNS-500 and (f) TiO2-PNS-500. | ||
The TiO2-HNS and TiO2-PNS structures are confirmed by TEM micrographs in Fig. 2a and b. Both these two kinds of nanospheres are uniform with a diameter of about 80 nm and are composed of about 7 nm sized nanoparticles (Fig. S5†). The shell of the TiO2-HNSs is only as thin as about 7 nm confirmed further using the spherical aberration corrected TEM images as shown in Fig. 2c, which means that the shell is composed of only a single layer of nanoparticles. The SEM micrographs in Fig. S6a and b† further show that both the TiO2-HNSs and the TiO2-PNSs are uniformly dispersed. The specific surface areas and pore size distributions are investigated using N2 adsorption/desorption analysis and shown in Fig. 3a and b. The TiO2-HNSs have a higher specific surface area of 65.8 m2 g−1 than that of 51.3 m2 g−1 for the TiO2-PNSs. Moreover, the size of the mesopores in the TiO2-PNSs is about 4 nm while the TiO2-HNSs have more much larger pores around 20 nm as well as a few pores around 4 nm. The effects of calcination heating rate and temperature on the mesoporous structures have also been investigated as shown in Fig. S7,† which demonstrates that a high heating rate results in large pores and a high calcination temperature would destroy the pores.
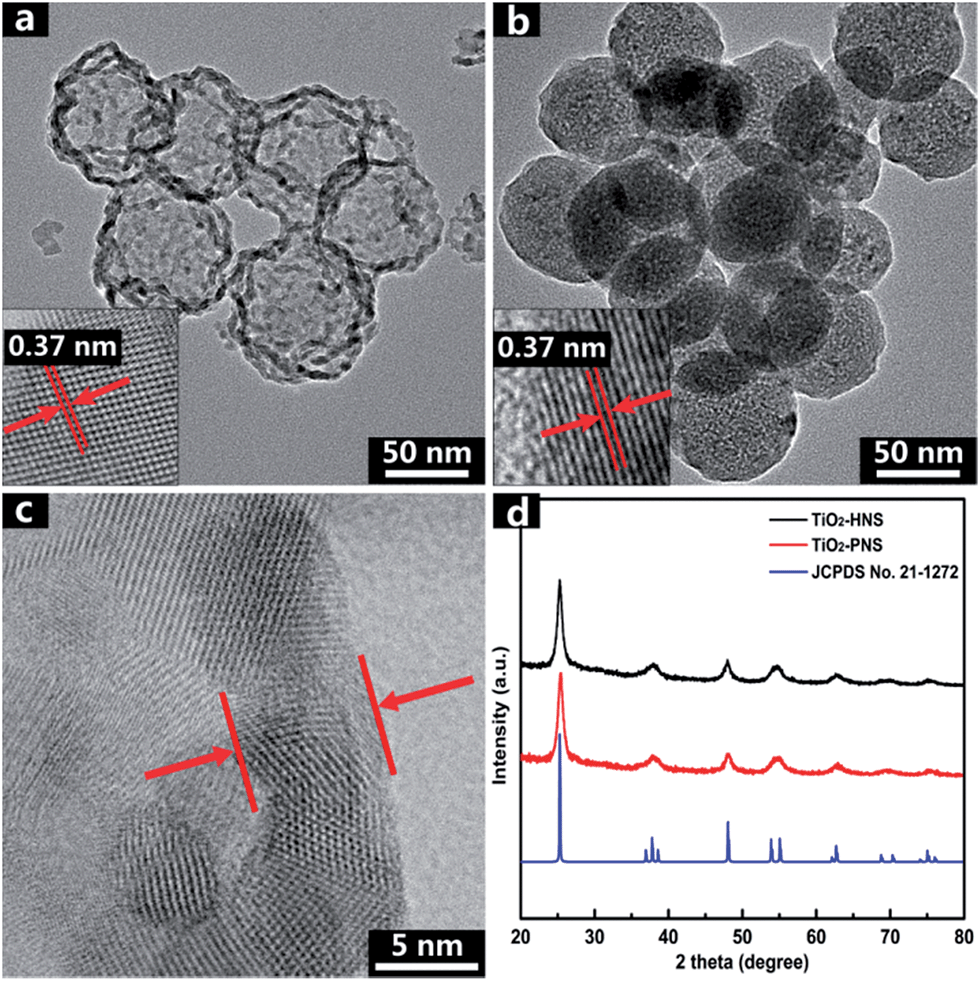 | ||
| Fig. 2 TEM micrographs of (a) TiO2-HNSs and (b) TiO2-PNSs. (c) Spherical aberration corrected TEM micrograph of the TiO2-HNS shell, and (d) XRD patterns of TiO2-HNSs and TiO2-PNSs. | ||
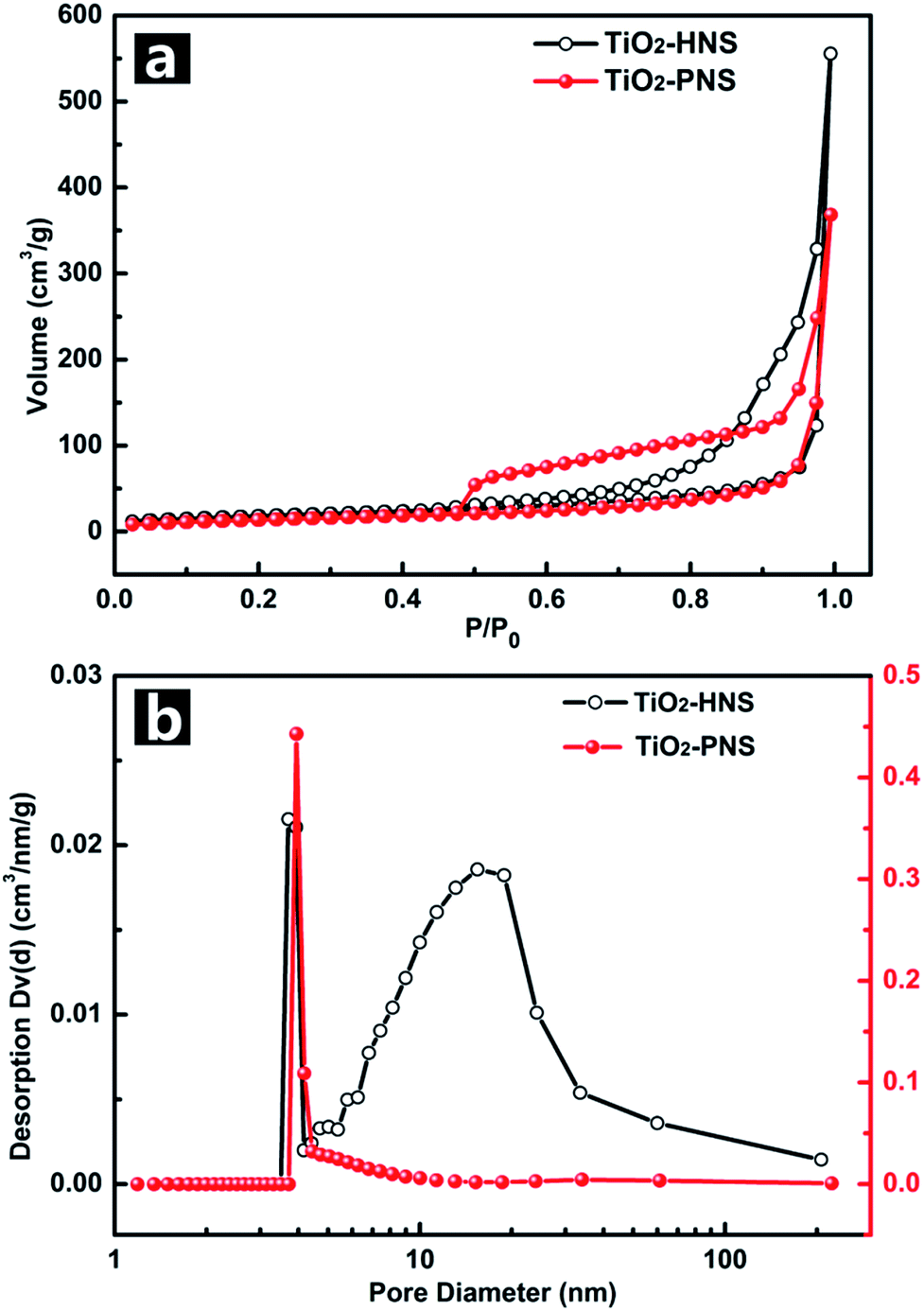 | ||
| Fig. 3 (a) Nitrogen adsorption/desorption isotherms and (b) Barret–Joyner–Halenda (BJH) pore-size distribution curves of the TiO2-HNSs and TiO2-PNSs. | ||
High resolution TEM (HRTEM) micrographs in the insets of Fig. 2a and b reveal the lattices of the TiO2-HNSs and TiO2-PNSs, which correspond to the {100} plane of anatase.41 The crystallinities of both the TiO2-HNSs and TiO2-PNSs were analyzed using X-ray powder diffraction (XRD), which confirmed the presence of crystallized anatase (JCPDS card no. 21-1272, space group: I41/amd, a = 3.7845 Å, c = 9.5143 Å) (Fig. 2d). The crystallite diameters of the TiO2-HNSs and TiO2-PNSs were calculated to be 7.3 nm and 7.1 nm, respectively, using the Scherrer formula.42 Raman spectra were used to further identify the composition of the TiO2-HNSs and TiO2-PNSs as shown in Fig. S8.† The Raman lines at around 144, 197 and 639 cm−1 can be assigned as the Eg modes of anatase TiO2. The Raman line at around 399 cm−1 is assigned to the B1g mode of anatase and the Raman line at around 519 cm−1 is assigned to the A1g or B1g mode of anatase.43,44 No other Raman lines are observed, which means that the samples are pure anatase. The XRD patterns of TiO2-HNS-500 and TiO2-PNS-500 samples demonstrate that trace rutile is formed besides the main anatase phase as shown in Fig. S9† and the sizes of nanoparticles are calculated to be 16.1 nm and 15.2 nm, respectively, which is attributed to the higher calcination temperature. The small nanoparticles in the TiO2-HNSs and TiO2-PNSs are believed to be effective in the high charge/discharge current rate performance in anodes for LIBs as previously described.
The electrochemical behaviors of the TiO2-HNS and TiO2-PNS electrodes were measured using cyclic voltammetry (CV) at a scan rate of 1 mV s−1 between 1.0 V and 3.0 V (Fig. 4a). Apparent anodic and cathodic peaks are observed in both electrodes at about 2.2 V and 1.7 V, which is in agreement with the previous literature.45 Slight peaks below 1.5 V are also found for both the samples, which can be attributed to an irreversible capacity loss.46 The TiO2-HNSs show a higher current density than that of the TiO2-PNSs manifested by a higher level charge separation and electrochemical conductivity.34 The Nyquist plots for the samples in Fig. 4b show a single semicircle in the high frequency region corresponding to the charge-transfer resistance (Rct) and a sloping straight line in the low frequency range corresponding to solid-state diffusion of lithium (Zw).47,48 The Rct of the TiO2-HNSs is smaller than that of the TiO2-PNSs, suggesting that the charge transfer for the former occurs prior to that of the latter. This may be ascribed to a higher specific surface area that enables greater contact with the electrolyte and larger pores around 20 nm (besides the 4 nm small pores), which may facilitate better electrolyte transport and strain release compared to that in the TiO2-PNSs (Fig. 3a, b and Table S2†). It is thus predicted that the performance of the TiO2-HNSs as anodes for LIBs would be enhanced.
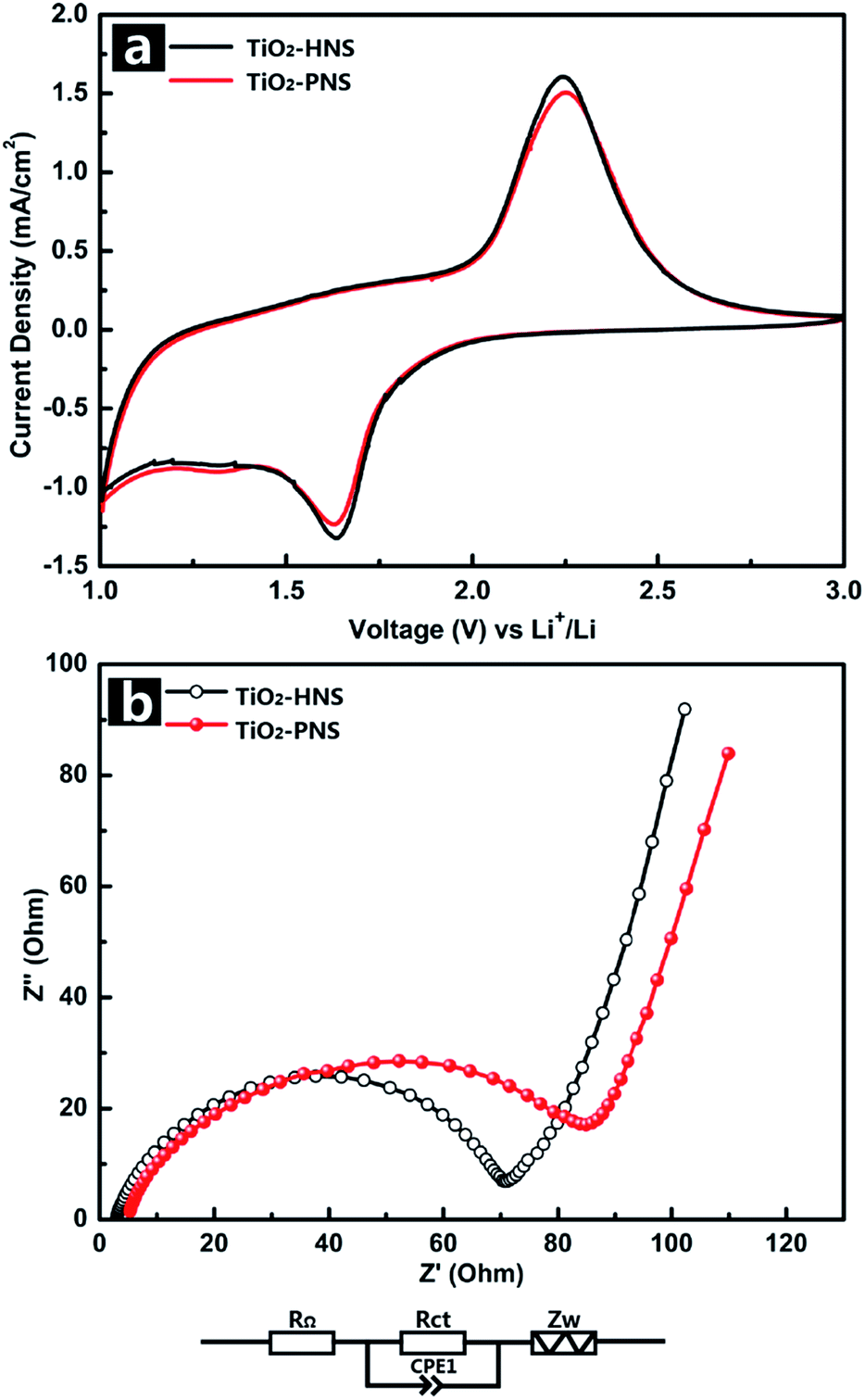 | ||
| Fig. 4 (a) Cyclic voltammetry profiles of the TiO2-HNSs and TiO2-PNSs at a scan rate of 1 mV s−1 between 1.0 V and 3.0 V for the first cycle, and (b) electrochemical impedance spectra (EIS) of the TiO2-HNSs and TiO2-PNSs (RΩ: external resistance, Rct: charge transfer resistance, CPE1: constant phase element, Zw: Warburg impedance). | ||
Standard TiO2/Li half-cells were used to measure the lithium storage properties of the TiO2-HNSs and TiO2-PNSs as anodes. Fig. 5a displays the first charge–discharge voltage profiles of the samples at a current rate of 1 C (167.5 mA g−1) between 1.0 V and 3.0 V. The potential falls to a plateau of 1.7 V quickly and then gradually declines to the cut-off potential of 1.0 V, consistent with previous literature.49,50 An initial discharge capacity of 295.2 mA h g−1 and charge capacity of 228.2 mA h g−1 are achieved for the TiO2-HNSs, resulting in a coulombic efficiency of 77.3%. The TiO2-PNSs exhibit an initial discharge capacity of 256.3 mA h g−1 and charge capacity of 195.9 mA h g−1, leading to a coulombic efficiency of 76.4%. The slightly lower coulombic efficiency is ascribed to the irreversible capacity loss, corroborating the CV measurement. Fig. 5b shows the cycling performance of the TiO2-HNSs and TiO2-PNSs at a current rate of 1 C between 1.0 V and 3.0 V for 100 cycles. After 100 cycles, the TiO2-HNSs show a higher capacity than the TiO2-PNSs (211.9 mA h g−1vs. 196.0 mA h g−1). The higher capacity of the TiO2-HNSs is ascribed to the higher specific surface area, which can lead to more surface lithium storage51 (Table S2†). Although demonstrating a slightly lower capacity, the TiO2-PNSs show a capacity retention of 92.8% with respect to the reversible specific capacities after 100 cycles, which is higher than that of the TiO2-HNSs (86.9%). This results from the very thin shell of the TiO2-HNSs, which is composed of only a single layer of nanoparticles and can fracture due to volume expansion during the charge and discharge processes (as seen in the TEM micrographs after 100 cycles, Fig. S10†). However, the TiO2-PNSs still maintain an unchanged stable structure leading to outstanding cycling performance, significantly better than that of commercial TiO2 Degussa P25.36 Moreover, the mass energy density of the TiO2-PNSs would be higher because of their more effective volumetric occupation compared to the TiO2-HNSs.
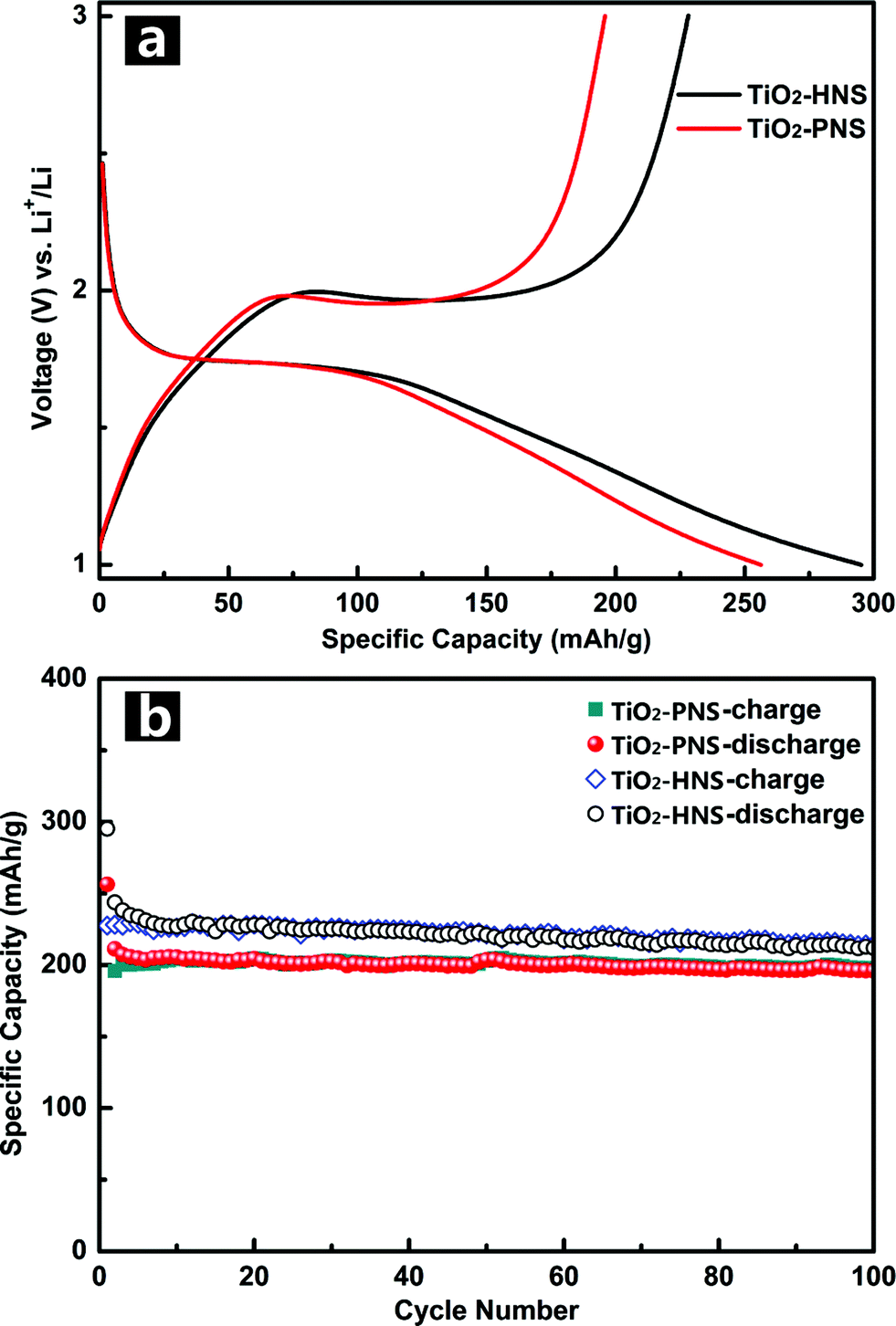 | ||
| Fig. 5 (a) Initial charge–discharge voltage profiles and (b) cycling performance of the TiO2-HNSs and TiO2-PNS at a current rate of 1 C between 1.0 and 3.0 V. | ||
Considering the importance of the potential for high rate applications, the cycling performance of the TiO2-HNSs and TiO2-PNSs at various charge and discharge current rates was measured. As shown in Fig. 6a, the capacity drops slowly as the current rate is increased up to 20 C (approximately 3 min for a charge or discharge process) from 1 C. A capacity of 125.9 mA h g−1 for the TiO2-HNSs and 113.4 mA h g−1 for the TiO2-PNSs can be achieved even at a high current rate of 20 C. A high discharge capacity of 205.4 mA h g−1 for the TiO2-HNSs and 190.1 mA h g−1 for the TiO2-PNSs can still be achieved when the current rate is reduced back to 1 C. The high discharge capacities at various current rates are summarized in Table S3† and are presented in Fig. 6b. This superior rate performance can be attributed to the special stable structures and fine particle sizes, which lead to very short lithium ion diffusion lengths and electron pathways during the lithium insertion and expulsion.30,31,52
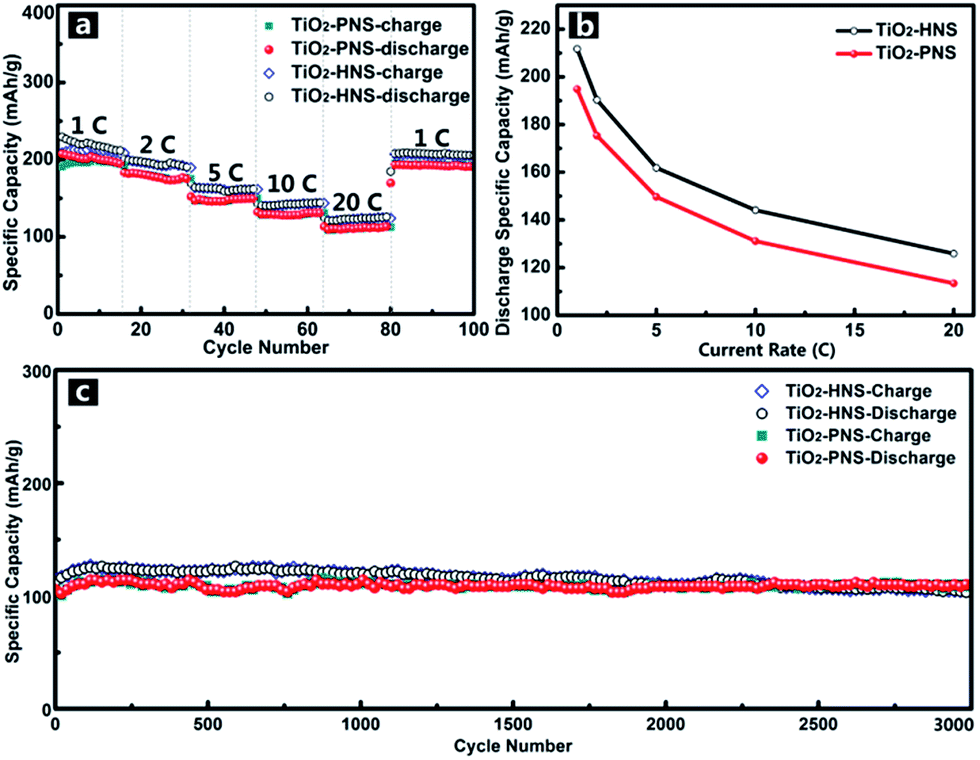 | ||
| Fig. 6 (a) Cycling performance at various charge–discharge current rates of the TiO2-HNSs and TiO2-PNSs between 1.0 and 3.0 V. (b) Statistics of the discharge specific capacities at various current rates according to the rate performance in (a). (Each discharge capacity summarized here is the last cycle of each current rate.) (c) Long-term cycling performance of the TiO2-HNSs and TiO2-PNSs at a high current rate of 20 C for 3000 cycles. | ||
In comparison, the rate performance of the TiO2-HNS-500 and TiO2-PNS-500 samples composed of larger nanoparticles was also measured as shown in Fig. S11† and the data are summarized in Table S3.† At a current rate of 1 C, the TiO2-HNS-500 sample shows a discharge capacity of 172.5 mA h g−1 while the TiO2-PNS-500 sample shows a discharge capacity of 145.7 mA h g−1. Discharge capacities of 65.4 and 54.7 mA h g−1 for the TiO2-HNS-500 and TiO2-PNS-500 samples, respectively, are achieved at a high current rate of 20 C. The capacities at 20 C are only 37.91% and 37.54% of the capacities at 1 C for the TiO2-HNS-500 and TiO2-PNS-500 samples, respectively, which are much lower than the results of 59.47% and 58.20% for the TiO2-HNSs and TiO2-PNSs, respectively. This results from the smaller nanoparticles in the TiO2-HNS and TiO2-PNS specimens, which could decrease the lithium ion diffusion lengths and shorten the electron pathways during the charge and discharge processes.52
The long-term cycling performance of the TiO2-HNSs and TiO2-PNSs at a high current rate of 20 C has also been demonstrated (Fig. 6c). Even after 3000 cycles, the TiO2-HNSs and TiO2-PNSs still maintain high discharge capacities of 103.0 mA h g−1 and 110.2 mA h g−1, respectively, with high retentions of 80.97% and 95.2% compared to the highest discharge capacities around the 116th cycle, respectively. The TiO2-PNSs show a significantly lower capacity loss because of their more stable structure than that of the TiO2-HNSs. The superior long-term cycling performance endows these nanospheres with great potential for application.
Conclusions
In summary, uniform TiO2 hollow and mesoporous nanospheres composed of ultrasmall nanoparticles were successfully fabricated using quasi-nano-sized carbonaceous spheres with a diameter of 260 nm as templates. The hollow and mesoporous structures could be controlled by controlling the heating rate of the calcination process and the adsorbance of TiCl4. When measured as anodes for LIBs, both TiO2 hollow and mesoporous nanospheres show good cycling performance, high specific capacity and high current rate performance. High specific capacities of 211.9 and 196.0 mA h g−1 are achieved for TiO2 hollow and mesoporous nanospheres at a current rate of 1 C after 100 cycles. Even at a high current rate of 20 C, they can still retain high specific capacities of 125.9 and 113.4 mA h g−1. TiO2 hollow and mesoporous nanospheres demonstrate long-term discharge capacities of 103.0 and 110.2 mA h g−1 after 3000 cycles at 20 C. The superior performance especially at high current rates is ascribed to the stable porous structures composed of ultrasmall nanoparticles, which enhance the interfacial contact area with the electrolyte and contribute to much shorter lithium ion diffusion lengths and electron transfer pathways. These TiO2 nanospheres have a great potential to be used for safe and high charge/discharge current rate devices.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (no. 21031005, 21271021, 51202248, 51472025, 51572261), and National Science Fund for Distinguished Young Scholars (no. 21325105).Notes and references
- M. Yoshio, R. J. Brodd and A. Kozawa, Lithium-Ion Batteries. Science and Technologies, Springer, New York, 2009 Search PubMed.
- Y. G. Wang, H. Q. Li, P. He, E. Hosono and H. S. Zhou, Nanoscale, 2010, 2, 1294–1305 RSC.
- H. Kim, M. Seo, M. Park and J. Cho, Angew. Chem., Int. Ed., 2010, 49, 2146–2149 CrossRef CAS PubMed.
- L. Bazin, S. Mitra, P. L. Taberna, P. Poizot, M. Gressier, M. J. Menu, A. Barnabé, P. Simon and J. M. Tarascon, J. Power Sources, 2009, 188, 578–582 CrossRef CAS.
- X. L. Huang, D. Xu, S. Yuan, D. L. Ma, S. Wang, H. Y. Zheng and X. B. Zhang, Adv. Mater., 2014, 26, 7264–7270 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- S. M. Xu, C. M. Hessel, H. Ren, R. B. Yu, Q. Jin, M. Yang and D. Wang, Energy Environ. Sci., 2014, 7, 632–637 CAS.
- K. Z. Cao, L. F. Jiao, H. Q. Liu, Y. C. Liu, Y. J. Wang, Z. P. Guo and H. T. Yuan, Adv. Energy Mater., 2014, 5, 201401421, DOI:10.1002/aenm.201401421.
- M. Du, C. H. Xu, J. Sun and L. Gao, J. Mater. Chem. A, 2013, 1, 7154–7158 CAS.
- X. D. Xu, R. G. Cao, S. Jeong and J. Cho, Nano Lett., 2012, 12, 4988–4991 CrossRef CAS PubMed.
- J. Y. Wang, N. L. Yang, H. J. Tang, Z. H. Dong, Q. Jin, M. Yang, D. Kisailus, H. J. Zhao, Z. Y. Tang and D. Wang, Angew. Chem., 2013, 125, 6545–6548 CrossRef.
- K. Z. Cao, L. F. Jiao, H. Xu, H. Q. Liu, H. Y. Kang, Y. Zhao, Y. C. Liu, Y. J. Wang and H. T. Yuan, Adv. Sci., 2015, 1500185, DOI:10.1002/advs.201500185.
- S. Yuan, X. L. Huang, D. L. Ma, H. G. Wang, F. Z. Meng and X. B. Zhang, Adv. Mater., 2014, 26, 2273–2279 CrossRef CAS PubMed.
- Z. C. Bai, Z. C. Ju, C. L. Guo, Y. T. Qian, B. Tang and S. L. Xiong, Nanoscale, 2014, 6, 3268–3273 RSC.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- M. Y. Nie, D. P. Abraham, Y. J. Chen, A. Bose and B. L. Lucht, J. Phys. Chem. C, 2013, 117, 13403–13412 CAS.
- S. Goriparti, E. Miele, F. D. Angelis, E. D. Fabrizio, R. P. Zaccaria and C. Capiglia, J. Power Sources, 2014, 257, 421–443 CrossRef CAS.
- Z. H. Chen, I. Belharouak, Y. K. Sun and K. Amine, Adv. Funct. Mater., 2013, 23, 959–969 CrossRef CAS.
- J. Liu, K. P. Song, P. A. van Aken, J. Maier and Y. Yu, Nano Lett., 2014, 14, 2597–2603 CrossRef CAS PubMed.
- H. G. Jung, S. W. Oh, J. Ce, N. Jayaprakash and Y. K. Sun, Electrochem. Commun., 2009, 11, 756–759 CrossRef CAS.
- J. Du, X. Y. Lai, N. L. Yang, J. Zhai, D. Kisailus, F. B. Su, D. Wang and L. Jiang, ACS Nano, 2011, 5, 590–596 CrossRef CAS PubMed.
- O. K. Varghese, D. W. Gong, M. Paulose, K. G. Ong, E. C. Dickey and C. A. Grimes, Adv. Mater., 2003, 15, 624–627 CrossRef CAS.
- N. L. Yang, Y. Y. Liu, H. Wen, Z. Y. Tang, H. J. Zhao, Y. L. Li and D. Wang, ACS Nano, 2013, 7, 1504–1512 CrossRef CAS PubMed.
- J. Du, J. Qi, D. Wang and Z. Y. Tang, Energy Environ. Sci., 2012, 5, 6914–6918 CAS.
- M. Wagemaker, G. J. Kearley, A. A. van Well, H. Mutka and F. M. Mulder, J. Am. Chem. Soc., 2003, 125, 840–848 CrossRef CAS PubMed.
- F. Gligor and S. W. de Leeuw, Solid State Ionics, 2006, 177, 2741–2746 CrossRef CAS.
- G. Q. Zhang, H. B. Wu, T. Song, U. Paik and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 12590–12593 CAS.
- J. P. Wang, Y. Bai, M. Y. Wu, J. Yin and W. F. Zhang, J. Power Sources, 2009, 191, 614–618 CrossRef CAS.
- Q. F. Zhang, E. Uchaker, S. L. Candelaria and G. Z. Cao, Chem. Soc. Rev., 2013, 42, 3127–3171 RSC.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS PubMed.
- Y. G. Guo, J. S. Hu and L. J. Wan, Adv. Mater., 2008, 20, 2878–2887 CrossRef CAS.
- T. B. Lan, Y. B. Liu, J. Dou, Z. S. Hong and M. D. Wei, J. Mater. Chem. A, 2014, 2, 1102–1106 CAS.
- Z. Y. Wang, L. Zhou and X. W. Lou, Adv. Mater., 2012, 24, 1903–1911 CrossRef CAS PubMed.
- S. Yoon and A. Manthiram, J. Phys. Chem. C, 2011, 115, 9410–9416 CAS.
- J. Qi, X. Y. Lai, J. Y. Wang, H. J. Tang, H. Ren, Y. Yang, Q. Jin, L. J. Zhang, R. B. Yu, G. H. Ma, Z. G. Su, H. J. Zhao and D. Wang, Chem. Soc. Rev., 2015, 44, 6749–6773 RSC.
- H. Ren, R. B. Yu, J. Y. Wang, Q. Jin, M. Yang, D. Mao and D. Wang, Nano Lett., 2014, 14, 6679–6684 CrossRef CAS PubMed.
- J. W. Kang, D. H. Kim, V. Mathew, J. S. Lim, J. H. Gim and J. Kim, J. Electrochem. Soc., 2011, 158, A59–A62 CrossRef CAS.
- X. M. Sun and Y. D. Li, Angew. Chem., Int. Ed., 2004, 43, 3827–3831 CrossRef CAS PubMed.
- X. Y. Lai, J. Li, B. A. Korgel, Z. H. Dong, Z. M. Li, F. B. Su, J. Du and D. Wang, Angew. Chem., 2011, 123, 2790–2793 CrossRef.
- V. D. Hildenbrand, H. Fuess, G. Pfaff and P. Reynders, Z. Physiol. Chem., 1996, 194, 139–150 CrossRef CAS.
- Q. J. Xiang, J. G. Yu, W. G. Wang and M. Jaroniec, Chem. Commun., 2011, 47, 6906–6908 RSC.
- N. M. Kinsinger, A. Dudchenko, A. Wong and D. Kisailus, ACS Appl. Mater. Interfaces, 2013, 5, 6247–6254 CAS.
- H. Q. Liu, K. Z. Cao, X. H. Xu, L. F. Jiao, Y. J. Wang and H. T. Yuan, ACS Appl. Mater. Interfaces, 2015, 7, 11239–11245 CAS.
- X. X. Xue, W. Ji, Z. Mao, H. J. Mao, Y. Wang, X. Wang, W. D. Ruan, B. Zhao and J. R. Lombardi, J. Phys. Chem. C, 2012, 116, 8792–8797 CAS.
- J. Wang, Y. K. Zhou, Y. Y. Hu, R. O’Hayre and Z. P. Shao, J. Phys. Chem. C, 2011, 115, 2529–2536 CAS.
- H. B. Wu, X. W. Lou and H. H. Hng, Chem.–Eur. J., 2012, 18, 3132–3135 CrossRef PubMed.
- H. Han, T. Song, E. K. Lee, A. Devadoss, Y. Jeon, J. Ha, Y. C. Chung, Y. M. Choi, Y. G. Jung and U. Paik, ACS Nano, 2012, 6, 8308–8315 CrossRef CAS PubMed.
- R. W. Mo, Z. Y. Lei, K. N. Sun and D. Rooney, Adv. Mater., 2014, 26, 2084–2088 CrossRef CAS PubMed.
- S. Ding, J. S. Chen, D. Luan, F. Y. C. Boey, S. Madhavi and X. W. Lou, Chem. Commun., 2011, 47, 5780–5782 RSC.
- Z. Q. Wang, L. Xiang, H. Xu, Y. Yang, Y. J. Cui, H. G. Pan, Z. Y. Wang, B. L. Chen and G. D. Qian, J. Mater. Chem. A, 2014, 2, 12571–12575 CAS.
- A. G. Dylla, G. Henkelman and K. J. Stevenson, Acc. Chem. Res., 2013, 46, 1104–1112 CrossRef CAS PubMed.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, thermogravimetric heating curves, SEM micrographs, additional TEM micrographs, Raman spectra, N2 adsorption–desorption results, EIS after 3 cycles at a current of 1 C and a summary of the cycling performance at various charge/discharge current densities are included. See DOI: 10.1039/c5sc03203b |
| This journal is © The Royal Society of Chemistry 2016 |