
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A microporous Cu2+ MOF based on a pyridyl isophthalic acid Schiff base ligand with high CO2 uptake†‡
Andreas
Kourtellaris
a,
Eleni E.
Moushi
a,
Ioannis
Spanopoulos
b,
Christos
Tampaxis
bc,
Georgia
Charalambopoulou
c,
Theodore A.
Steriotis
c,
Giannis S.
Papaefstathiou
d,
Pantelis N.
Trikalitis
b and
Anastasios J.
Tasiopoulos
 *a
*a
aDepartment of Chemistry, University of Cyprus, 1678 Nicosia, Cyprus. E-mail: atasio@ucy.ac.cy; Fax: +357 22895451; Tel: +357 22892765
bDepartment of Chemistry, University of Crete, Voutes 71003, Heraklion, Greece
cNational Center for Scientific Research Demokritos, 15341 Agia Paraskevi Attikis, Greece
dLaboratory of Inorganic Chemistry, Department of Chemistry, National and Kapodistrian University of Athens, Panepistimiopolis Zografou 157 71, Greece
First published on 20th September 2016
Abstract
A new Cu2+ complex that was isolated from the initial use of 5-((pyridin-4-ylmethylene)amino)isophthalic acid (PEIPH2) in 3d metal–organic framework (MOF) chemistry is reported. Complex {[Cu3(PEIP)2(5-NH2-mBDC)(DMF)]·7DMF}∞ denoted as Cu-PEIP·7DMF was isolated from the reaction of Cu(NO3)2·2.5H2O with PEIPH2 in N,N-dimethylformamide (DMF) at 100 °C and contains both the PEIP2− ligand and its 5-NH2-mBDC2− fragment. After the structure and properties of Cu-PEIP were known an analogous complex was prepared by a rational synthetic method that involved the reaction of Cu(NO3)2·2.5H2O, 5-((pyridin-4-ylmethyl)amino)isophthalic acid (PIPH2 – the reduced analogue of PEIPH2) and 5-NH2-mBDCH2 in DMF at 100 °C. Cu-PEIP comprises two paddle-wheel [Cu2(COO)4] units and exhibits a 3D-framework with a unique trinodal underlying network and point symbol (4.52)4(42·54·64·83·92)2(52·84). This network consists of pillared kgm-a layers containing a hexagonal shaped cavity with a relatively large diameter of ∼8–9 Å surrounded by six trigonal shaped ones with a smaller diameter of ∼4–5 Å and thus resembles the structure of HKUST-1. Gas sorption studies revealed that Cu-PEIP exhibits a 1785 m2 g−1 BET area as well as high CO2 sorption capacity (4.75 mmol g−1 at 273 K) and CO2/CH4 selectivity (8.5 at zero coverage and 273 K).
Introduction
Metal–organic frameworks (MOFs) have attracted tremendous interest in the last decade due to their unique structural features1 and potential applications in gas storage, separation,2,3 magnetism,4 catalysis,5 sensing6 and so on. The key factor for the discovery of MOFs with novel structural features and potentially interesting physical properties is the development of new synthetic strategies. Appropriate organic ligands are very important in this context, since even small changes in the flexibility, length, or symmetry of the ligands can result in a remarkable diversity of architectures and functions.7,8 Although several elements of rational design have been introduced in MOF chemistry,9 exploratory synthetic methods are still a fruitful source of novel materials with interesting properties.10An apparent strategy toward new MOFs with interesting properties consists of the use of N- and O-donor polytopic organic ligands.8a Specifically, pyridyl-carboxylates have been confirmed as excellent ligands to assemble multidimensional coordination polymers due to the ability of the N donor atoms to bind most of the metal ions and the high bridging capability of the carboxylate groups. As a result, the carboxylate ligands can bridge several metal ions to form stable oligonuclear or polynuclear secondary building units (SBUs) which are linked through carboxylate or pyridyl N groups to afford multidimensional coordination polymers.8a,11 One type of pyridyl polycarboxylate linkers that have attracted significant attention are the tritopic ones consisting of pyridyl and isophthalic acid moieties (for some examples see Scheme 1 in the ESI‡).12–14 When ligands possessing the isophthalic acid moiety are employed in Cu2+ chemistry, they often afford structures which are similar to that of HKUST-1 with a tbo topology.15 These structures either contain a layer with the kgm-a (augmented kagomé) or the sql-a (augmented square lattice) topology that are present in HKUST-1 or display an overall tbo topology,12d,e,16 since the latter (tbo) can be regarded either as a pillared kgm or pillared sql network. Thus, such ligands clearly have the potential to lead in materials resembling the structure and possibly the sorption properties of HKUST-1 and for this reason they have attracted intense interest.12–14,16 MOFs with the tbo topology are highly desirable for gas sorption applications since: (a) interpenetration is not allowed and therefore high surface areas are often obtained, (b) a relatively high density of open metal sites is created, leading to high gas uptake and (c) the structure contains additional strongly interacting sites (pockets and windows) that increase the overall gas uptake.16a
We are interested in the use of Schiff base ligands containing the isophthalic acid group for the construction of new functional MOFs. In fact we have reported a series of MOFs based on the CIPH3 ligand (CIPH3 = 5-(4-carboxybenzylideneamino)isophthalic acid) with interesting single-crystal-to-single-crystal (SCSC) transformation and sorption properties.9c,17 This ligand is more elongated than other commonly used tricarboxylic acids (such as trimesic acid) and possesses the semi-rigid C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N moiety that introduces into the isolated compounds some but not unlimited flexibility. For all these reasons CIPH3 was proven capable to afford MOFs with interesting properties. As an extension of this work we employed in MOF chemistry the ligand 5-((pyridin-4-ylmethylene)amino)isophthalic acid (PEIPH2) (Scheme 1 in the ESI‡) that is similar to CIPH3 but contains a pyridyl group in place of the benzoic acid moiety. This ligand carries all the advantages of the other pyridyl-isophthalic acid linkers mentioned above and displays unpredictable behavior when it is involved in reactions with metal ion sources. This arises from the fact that both PEIPH2 and its constituent moieties are present in the solution containing the ligand and thus it is possible for them to appear in the resulting compounds giving rise to a variety of new products. Surprisingly the coordination chemistry of PEIPH2 is actually unexplored with the only known compounds with this ligand being some organotin MOFs.14
N moiety that introduces into the isolated compounds some but not unlimited flexibility. For all these reasons CIPH3 was proven capable to afford MOFs with interesting properties. As an extension of this work we employed in MOF chemistry the ligand 5-((pyridin-4-ylmethylene)amino)isophthalic acid (PEIPH2) (Scheme 1 in the ESI‡) that is similar to CIPH3 but contains a pyridyl group in place of the benzoic acid moiety. This ligand carries all the advantages of the other pyridyl-isophthalic acid linkers mentioned above and displays unpredictable behavior when it is involved in reactions with metal ion sources. This arises from the fact that both PEIPH2 and its constituent moieties are present in the solution containing the ligand and thus it is possible for them to appear in the resulting compounds giving rise to a variety of new products. Surprisingly the coordination chemistry of PEIPH2 is actually unexplored with the only known compounds with this ligand being some organotin MOFs.14
We herein report a new Cu2+ MOF {[Cu3(PEIP)2(5-NH2-mBDC)(DMF)]·7DMF}∞ denoted as Cu-PEIP·7DMF which represents the initial 3d metal complex with the ligand PEIPH2. Although this compound was prepared by a serendipitous self-assembly synthetic procedure, after its structure was known the synthesis of an analogous complex was targeted with high priority and achieved following a rational synthetic method. Cu-PEIP exhibits a 3D-framework with a unique trinodal underlying network and point symbol (4·52)4(42·54·64·83·92)2(52·84). This network consists of pillared kgm-a layers and thus resembles the structure of HKUST-1. Cu-PEIP shows a significant BET area (1785 m2 g−1) as well as high CO2 sorption capacity (4.75 mmol g−1 at 273 K) and CO2/CH4 selectivity (8.5 at zero coverage and 273 K).
Experimental
Materials
Reagent grade chemicals were obtained from Aldrich and used without further purification. Water was distilled in-house. PEIPH2 and PIPH2 were synthesized according to the reported literature.13d,14Synthesis of Cu-PEIP
Method A: PEIPH2 (0.08 g, 0.296 mmol) was dissolved (after sonication for 3 minutes) in 5 mL N,N-dimethylformamide (DMF) in a 20 mL glass vial and solid Cu(NO3)2·2.5H2O (0.08 g, 0.344 mmol) was added to this solution. The mixture was sonicated for 3 min, sealed and then, heated without stirring at 100 °C for 24 h. During this period, green polyhedral-like crystals of Cu-PEIP were formed. They were isolated by filtration, washed several times with DMF and diethylether, and dried under vacuum. The yield was 70%. Elemental analysis: Anal. Calc. for C60H77O20N13Cu3 (Cu-PEIP·7DMF) C 48.34, H 5.21, N 12.21, Found: C 48.23, H 5.11, N 12.09.Method B: Method A was repeated, using 5-NH2-mBDCH2 (0.08 g, 0.442 mmol) and 4-pyridinecarboxaldehyde (43 μL, 0.456 mmol) instead of PEIPH2. The yield was ∼65%.
Synthesis of Cu-PIP
PIPH2 (0.08 g, 0.294 mmol) and 5-NH2-mBDCH2 (0.05 g, 0.276 mmol) were dissolved (after sonication for 3 minutes) in DMF (5 mL) in a 20 mL glass vial and solid Cu(NO3)2·2.5H2O (0.08 g, 0.344 mmol) was added to this solution. The mixture was sonicated for 3 min, sealed and then, heated without stirring at 100 °C for 24 h. During this period, green crystalline solid of Cu-PIP was formed. The microcrystalline solid was isolated by filtration, washed several times with DMF and diethylether, and dried under vacuum. The yield was 46%. Anal. Calc. for C60H81O20N13Cu3 (Cu-PIP·7DMF) C 48.20, H 5.46, N 12.18, Found: C 47.94, H 5.36, N 12.03.Physical measurements
Elemental analysis (C, H, N) was performed by the in-house facilities of the University of Cyprus, Chemistry Department. IR spectra were recorded on KBr pellets in the 4000–400 cm−1 range using a Shimadzu Prestige -21 spectrometer. PXRD patterns were recorded on a Shimazdu 6000 Series X-ray diffractometer (Cu Kα radiation, λ = 1.5418 Å). Thermal stability studies were performed with a Shimadzu TGA 50 thermogravimetric analyzer.Gas sorption measurements
Low-pressure argon, hydrogen, carbon dioxide and methane adsorption measurements were carried out on an Autosorb 1-MP instrument from Quantachrome equipped with multiple pressure transducers for highly accurate analyses and an oil-free vacuum system. High pressure excess adsorption measurements were conducted manometrically on a PCTPro-2000 instrument (Setaram) with the aid of a regular valve sealed stainless-steel measuring cell. The NIST database was implemented for estimating the compressibility of gases (H2, CH4, CO2) and helium was used for dead volume calibrations at 273–303 K. In order to avoid potential helium sorption errors, the dead volume at 77 K was calculated through a reference curve obtained by using different volumes of non-adsorbing materials (Pyrex glass). Ultra-high purity grade Ar (99.999%), He (99.999%), H2 (99.999%), CO2 (99.999%) and CH4 (99.9995%) were used for all adsorption measurements. Prior to analysis, as-made Cu-PEIP was soaked in CH2Cl2 at room temperature for three (3) days during which the supernatant solution was replaced six (6) times. The dichloromethane suspended samples were transferred inside the chamber of a supercritical CO2 dryer (Bal-Tec CPD 030) and CH2Cl2 was exchanged with liquid CO2 over a period of 5 hours at 8 °C. During this period, liquid CO2 was vented under positive pressure every 5 minutes. The rate of CO2 venting was always kept below the rate of filling so as to maintain full drying conditions inside the chamber. Following venting, the temperature was raised to 40 °C (above the critical temperature of CO2), kept there for 1 hour and then slowly vented over the period of 1 hour. The dried sample was transferred immediately inside a pre-weighted, argon filled 9 mm cell and closed using CellSeal™ provided by Quantachrome to prevent the intrusion of oxygen and atmospheric moisture during transfers and weighing. The cell was then transferred to the outgassing station where the sample was evacuated under dynamic vacuum at room temperature until the outgas rate was less than 2 mTorr min−1. After evacuation, the sample and cell were re-weighed to obtain the precise mass of the evacuated sample. Finally, the tube was transferred to the analysis port of the gas adsorption instrument. Likewise, for high pressure measurements, the dried sample was transferred sealed to an argon filled glove box (Labstar, MBraun, H2O and O2 <0.5 ppm), and a quantity of approx. 180 mg was inserted into a pre-weighted stainless-steel container. The sample mass was calculated and the container was sealed with a high pressure valve. The whole cell was removed from the glove box and was attached to the high pressure manometric apparatus. All-metal face seals were used. After suitable evacuation of the apparatus the sample was further outgassed overnight at room temperature under high vacuum (p < 10−6 mbar). The outgassing procedure was repeated after each high pressure measurement.Single crystal X-ray crystallography
Single crystal X-ray diffraction data were collected on an Oxford-Diffraction Supernova diffractometer, equipped with a CCD area detector utilizing Cu Kα (λ = 1.5418 Å) radiation. A suitable crystal was mounted on a Hampton cryoloop with Paratone-N oil and transferred to a goniostat where it was cooled for data collection. Empirical absorption corrections (multiscan based on symmetry-related measurements) were applied using CrysAlis RED software.18 The structure was solved by direct methods using SIR200419 and refined on F2 using full-matrix least-squares with SHELXL97.20 Software packages used were as follows: CrysAlis CCD for data collection,18 CrysAlis RED for cell refinement and data reduction,18 WINGX for geometric calculations,21 and DIAMOND22 and X-Seed23 for molecular graphics. The non-H atoms were treated anisotropically, whereas the aromatic H atoms were placed in calculated, ideal positions and refined as riding on their respective carbon atoms. Electron density contributions from disordered guest molecules were handled using the SQUEEZE procedure from the PLATON software suit.24 Selected crystal data for Cu-PEIP are summarized in Table 1. CCDC 1492970 contains the supplementary crystallographic data for this paper.| Complex | Cu-PEIP |
|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = {∑[w(|Fo|2 − |Fc|2)2]/∑[w(|Fo|4)]}1/2. | |
| Empirical formula | C39H25Cu3N6O13 |
| Formula weight | 976.30 |
| Temperature (K) | 100(2) |
| Radiation | Cu Kα (λ = 1.54180 Å) |
| Crystal system | Monoclinic |
| Space group | I2/m |
| a (Å) | 13.657(5) |
| b (Å) | 18.686(5) |
| c (Å) | 31.503(5) |
| β (°) | 101.532(5) |
| V (Å3) | 7877(4) |
| Z | 4 |
| D c (g cm−3) | 0.823 |
| μ (mm−1) | 1.266 |
| Refls coll. | 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 001 001 |
| Unique refls | 7255 |
| R int | 0.0367 |
|
R
1a [I > 2σ(I)] |
0.0749 |
| wR2b (all data) |
0.2441 |
| GOF | 1.157 |
| Δρmin/max (e Å3) | 1.279/−0.658 |
Results and discussion
Over the last few years, we have been systematically investigating the use of semi-rigid polytopic ligands, such as CIPH3 as a method for the synthesis of new MOFs.9c,17 An extension of these investigations included the use of PEIPH2, which is similar to CIPH3 but contains a pyridyl group in the place of the benzoic acid moiety, in MOF chemistry. Compound Cu-PEIP was initially prepared from the reaction of Cu(NO3)2·2.5H2O and PEIPH2 in DMF at 100 °C in 70% yield. A similar reaction was performed that contained instead of the pre-formed Schiff base ligand PEIPH2 its constituent moieties, i.e. 5-NH2-mBDCH2 and 4-pyridinecarboxaldehyde aiming at the in situ formation of the ligand and the isolation of the final product through a one-pot reaction (by omitting the ligand preparation step). This was realized from the reaction of Cu(NO3)2·2.5H2O, 5-NH2-mBDCH2 and 4-pyridinecarboxaldehyde in DMF at 100 °C in 65% yield. Interestingly, Cu-PEIP was isolated as excellent quality green polyhedral-like crystals in very high yields with both synthetic methods. When the structure of the compound Cu-PEIP was known the synthesis of an analogous complex was targeted and achieved by following a rational synthetic procedure. This procedure involved the use in the reaction mixture of the reduced analogue of PEIPH2, i.e. ligand PIPH2 that remains intact in solution, and 5-NH2-mBDCH2. Thus, compound Cu-PIP was prepared from the reaction of Cu(NO3)2·2.5H2O, PIPH2 and 5-NH2-mBDCH2 in DMF at 100 °C in 46% yield. The identity and purity of the bulk products of Cu-PEIP and Cu-PIP and their structural relation were confirmed by PXRD, elemental analysis and infrared spectroscopy (Fig. S1–S4 in the ESI‡).Compound Cu-PEIP crystallizes in the monoclinic space group I2/m. There are two crystallographically unique Cu2+ ions in the structure (Cu1 and Cu2), both adopting a square pyramidal coordination geometry (Fig. 1): Cu1 is coordinated with four carboxylic oxygen atoms 2 × (O1–O2) of four different PEIP2− ligands and one terminal DMF (O7) solvent molecule, whereas Cu2 is connected with two carboxylate oxygen atoms (O3–O4) from two different PEIP2− anions, two carboxylate oxygen atoms (O5–O6) from two different 5-NH2-mBDC2− ligands and one nitrogen atom from a third PEIP2− ligand (N1). This connectivity gives rise to a 3-D framework, which comprises two paddle-wheel [Cu2(COO)4] SBUs and exhibits large channels running mainly along the a-axis (Fig. 2a). The first type of dinuclear SBU, [Cu2]A, is formed by two symmetry-equivalent Cu1 metal ions, which are bridged by four syn,syn-μ2-COO− groups originating from four PEIP2− ligands, whereas the second paddle-wheel type SBU, [Cu2]B, consists of two symmetry-equivalent Cu2 ions, which are bridged by four syn,syn-μ2-COO− groups originating from two PEIP2− and two 5-NH2-mBDC2− ligands. The axial positions in [Cu2]A are occupied by two oxygen atoms from two DMF molecules, while those in [Cu2]B by two pyridyl nitrogen atoms from two PEIP2− ligands. The 5-NH2-mBDC2− ligands bridge two [Cu2]B SBUs with a [Cu2]B⋯[Cu2]B separation of ∼9.3 Å while the isophthalate moiety of the PEIP2− ligands bridges a [Cu2]A to a [Cu2]B SBU with the [Cu2]A⋯[Cu2]B separation being ∼9.2 Å. In this arrangement, the paddle-wheels and the isophthalate moieties of both ligands create an undulated (3·6·3·6) semi-regular plane net (kgm-a lattice) parallel to the bc plane (Fig. 2b). This layer contains two types of cavities; a hexagonal shaped with a relatively large diameter of ∼8–9 Å surrounded by six trigonal shaped ones with a smaller diameter of ∼4–5 Å as found by PLATON24 (taking into account the van der Waals radii of the atoms). The trigonal shaped cavities are composed of one [Cu2]A and two [Cu2]B SBUs and are filled with the terminal DMF molecules bound to [Cu2]A SBUs. The hexagonal shaped cavities are composed of two [Cu2]A separating two pairs of two [Cu2]B SBUs. The sides of the hexagonal cavities comprise the isophthalate moieties of two approximately parallel 5-NH2-mBDC2− ligands separating two adjacent isophthalate moieties of two pairs of PEIP2− ligands.
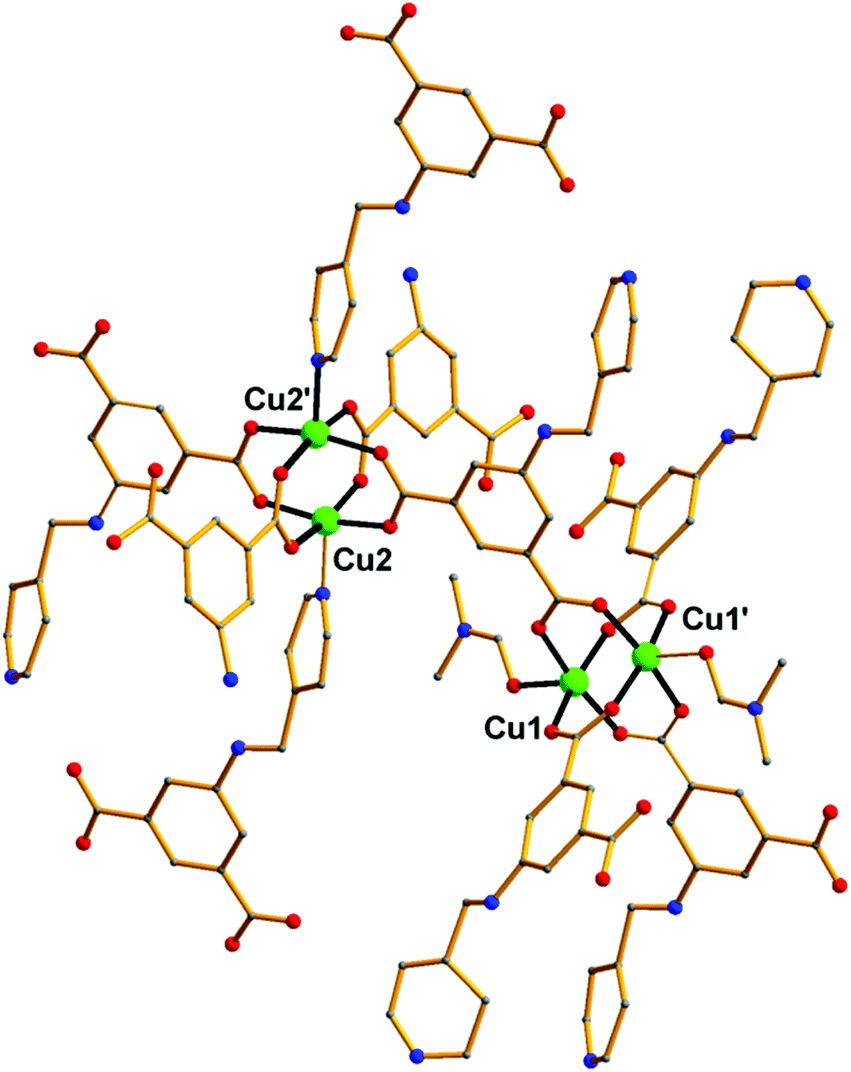 | ||
| Fig. 1 Representation of the connectivity of the [Cu2+]2 paddle wheel SBUs through the PEIP2− ligand in Cu-PEIP. Colour code: Cu green, O red, N blue, C gray. H atoms are omitted for clarity. | ||
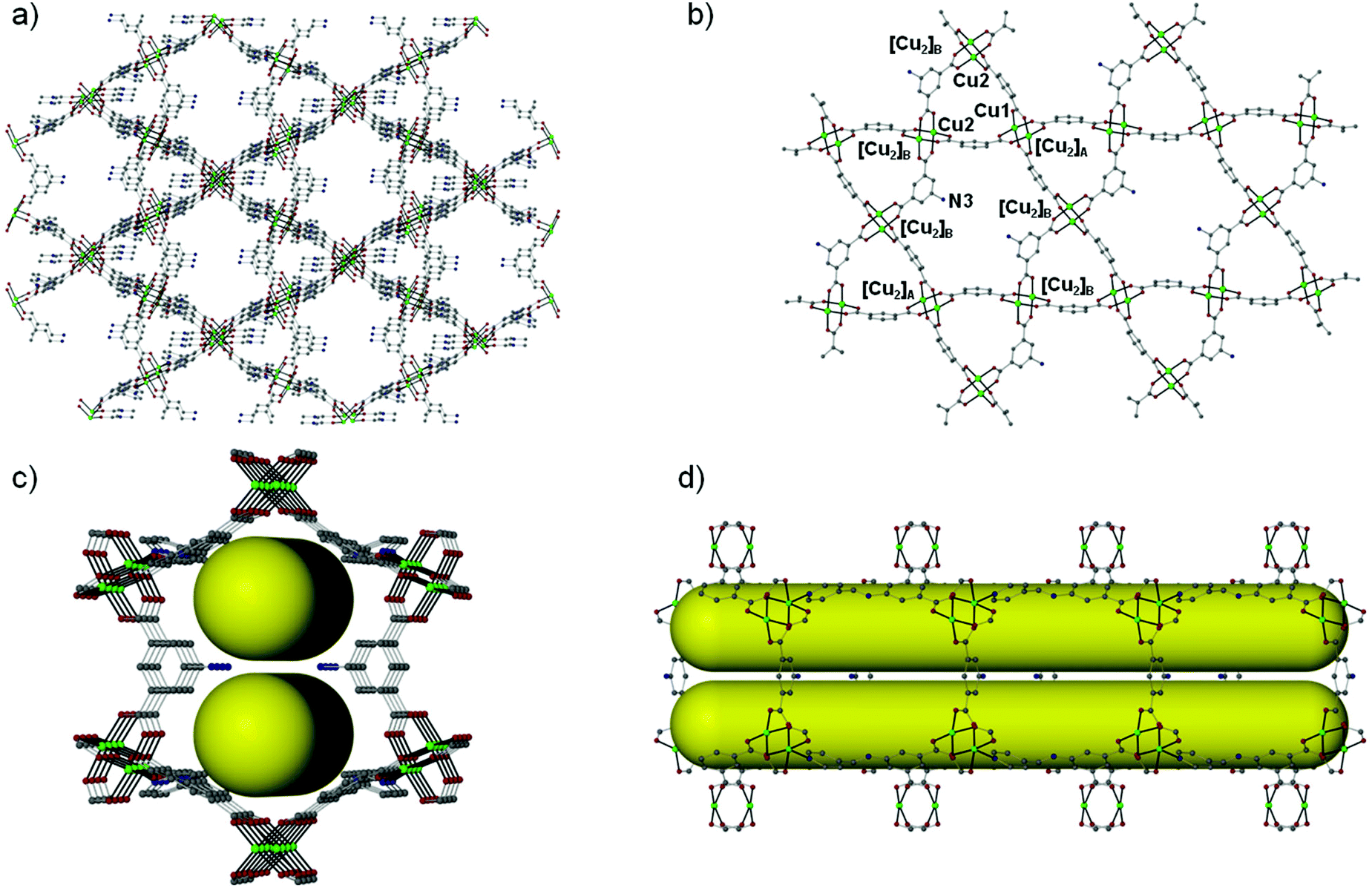 | ||
| Fig. 2 Representations of (a) the 3D framework along the a axis, (b) the (3·6·3·6) semi-regular net (augmented Kagomé lattice: kgm-a), (c) the compartmentalized hexagonal channels along the a axis and (d) the compartmentalized channel projected along the a axis for complex Cu-PEIP. Colour code: Cu green, O red, N blue, C gray. H atoms are omitted for clarity. | ||
The kgm-a layers are pillared through the pyridyl groups of the PEIP2− ligands giving rise to hexagonal channels running parallel to the a axis (Fig. 2c and d). Since there are only four PEIP2− ligands composing the hexagonal cavities, only four [Cu2]B SBUs act as connection points in the pillaring. The 5-NH2-mBDC2− ligands point their amino group toward the hexagonal cavities, thus compartmentalizing the cavities and the hexagonal channels as shown in Fig. 2c and d. Since part of the isophthalate ligands and part of the [Cu2] SBUs participate in the pillaring there is enough space left uncovered giving rise to pores through the other two directions of the material. The pores accommodate DMF molecules.
The solvent-accessible volume calculated by PLATON24 is 5241 Å3 and corresponds to 66.5% of the unit-cell volume (7877.0 Å3). A representation of the voids using the structure visualization program MERCURY25 reveals a continuous and complex 3D pore network, where the large compartmentalized hexagonal cavities communicate through relatively wide channels with a diameter ≥4 Å (Fig. 3).
 | ||
| Fig. 3 Representation of the pore network (shown in yellow) of Cu-PEIP (only pores and channels with diameter ≥4 Å are shown) along the b axis. | ||
From the topological point of view the [Cu2]A paddle-wheel SBUs possessing two DMF molecules on the Cu1 apical sites serve as 4-coordinated nodes, while the [Cu2]B SBUs possessing two pyridyl N atoms on the apical positions of the Cu2 ions act as 6-c nodes. The PEIP2− ligands bridge a [Cu2]A to a [Cu2]B through the isophthalate moiety and a [Cu2]B through the pyridyl N atom, thus serving as a 3-c node. The 5-NH2-mBDC2− ligand simply bridges two [Cu2]B SBUs and therefore is topologically silent (serves as a bridge). In this arrangement a 3,4,6-coordinated trinodal network with the stoichiometry (3-c)4(4-c)(6-c)2 and point symbol (4·52)4(42·54·64·83·92)2(52·84) forms which is, so far, unique (Fig. S5–S8 in the ESI‡). The network adopted by Cu-PEIP resembles the tbo net adopted by HKUST-1 and the eea net.12d,e,15,16 All three nets can be considered as pillared kgm-a layers. The augmented Kagomé lattice (kgm-a) is based on [Cu2(COO)4] SBUs and isophthalate moieties which alternate to create big hexagonal cavities comprising six paddle-wheels and six isophthalate moieties. In tbo the kgm-a layers are separated (pillared) by the third carboxylate of the trimesic acid while in the eea net the layers are separated by the pending pyridyl groups of the pyridyl-isophthalate ligands. In both tbo and eea nets, all six bridging ligands around each hexagonal cavity serve as pillars to both sides of the layer while in Cu-PEIP only four PEIP2− ligands serve as pillars since two out of the six bridges around the hexagonal cavities are 5-NH2-mBDC2− ligands which cannot connect neighboring kgm-a layers.
The thermal stability of Cu-PEIP and Cu-PIP was investigated by means of the thermogravimetric analysis (TGA) technique (Fig. S9 and S10 in the ESI‡). The TGA curves for the two complexes are essentially identical and thus only that of Cu-PEIP shall be discussed in detail. The TGA curve of Cu-PEIP indicates that this compound is decomposed through a multi-step process which is completed at a fairly low temperature (∼400 °C). The first two steps that are related to the removal of the lattice and bound DMF solvent molecules appear in the temperature range 30 °C to 260 °C and correspond to ∼40% of the material's total mass. This value is in agreement with the corresponding calculated value for Cu-PEIP·7DMF (∼39.2%). The next steps are associated with the decomposition of the two bridging ligands PEIP2− and 5-NH2-mBDC2− and are completed at ∼400 °C.
The relatively large solvent accessible volume present in Cu-PEIP prompted us to investigate its gas sorption properties. Several activation methods were employed, however the most efficient one involved the use of dichloromethane to remove the guest solvent molecules followed by treatment with supercritical CO2. Argon sorption measurements at 87 K revealed a type-I isotherm (Fig. 4), typical for a microporous solid, from which the apparent BET area was found to be 1785 m2 g−1 (Langmuir, 1814 m2 g−1), close to the geometric surface area (2059 m2 g−1) calculated from the single crystal structure using Poreblazer.26 The total pore volume is 0.64 cm3 g−1 at relative pressure, p/po, 0.99, which is slightly lower compared to the value of 0.75 cm3 g−1 calculated26 from the crystal structure. Given that the PXRD pattern of the activated sample suggests an intact framework, the difference in the pore volume could be explained by trapped organic molecules inside the pores of Cu-PEIP. The pore size distribution, calculated using Non-Local Density Functional Theory (NLDFT) after a successful fitting of the Ar adsorption isotherm data using a suitable NLDFT kernel (Fig. S11 in the ESI‡), shows two major peaks centered at 6 Å and 9 Å (inset in Fig. 4), in agreement with the crystallographic analysis.
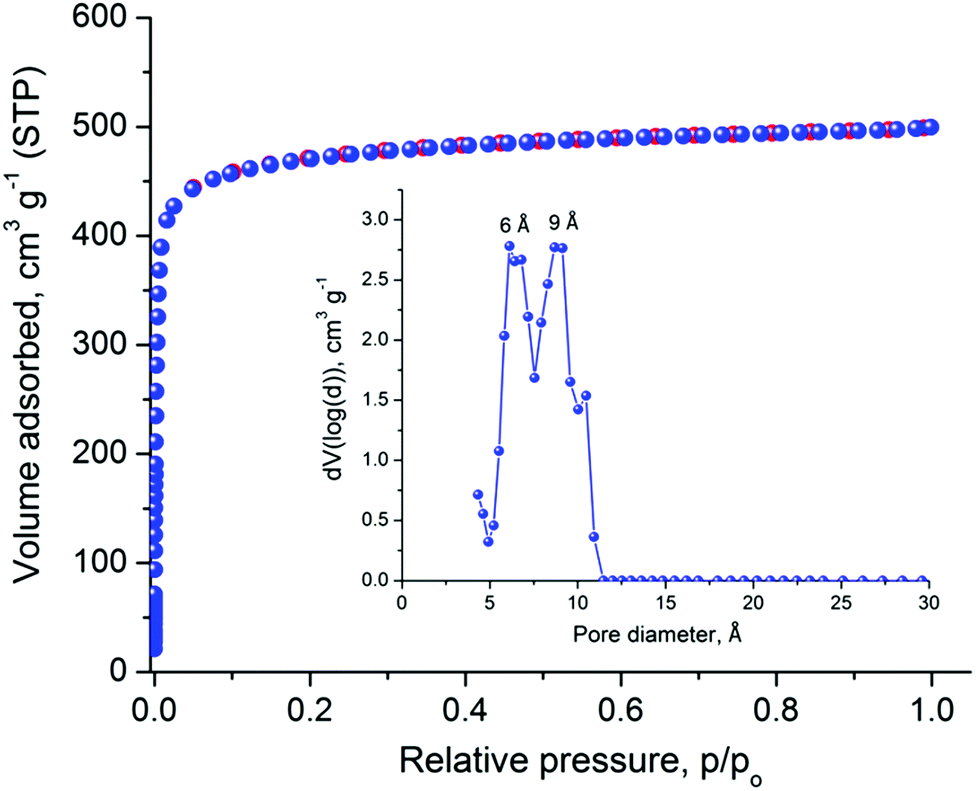 | ||
| Fig. 4 Argon sorption isotherm of Cu-PEIP recorded at 87 K and pore size distribution curve calculated by NLDFT (inset). | ||
The high porosity of Cu-PEIP in combination with the presence of unsaturated Cu(II) sites and –NH2 groups, prompted us to further investigate the gas sorption properties by recording low and high pressure isotherms of CO2, CH4 and H2 at different temperatures from which the total uptake, isosteric heat of adsorption (Qst) and CO2/CH4, selectivity were calculated. We note that amine functionalized MOFs are highly desirable, especially for CO2 capture, because high uptake and selectivity are expected due to favorable acid–base interactions.27
The CO2 uptake at 1 bar is 4.75 mmol g−1 (20.9 wt%) and 2.80 mmol g−1 (12.3 wt%) at 273 K and 298 K, respectively (Fig. 5). These values are within the same range of representative and high performance NH2-functionalized MOFs including NH2-MIL-101(Cr) (3.2 and 1.9 mmol g−1 at 273 K and 298 K),28 Zn(Atz)2 (4.35 mmol g−1 at 1.2 bar and 273 K),29 NH2-MIL-125(Ti) (5.9 mmol g−1 at 273 K)30 and NH2-UiO-66 (between 2.89 and 3.04 mmol g−1 at 298 K).31 The isosteric heat of adsorption, Qst, was calculated to be 34.2 kJ mol−1 at zero coverage (Fig. 6 and S12). Such a moderate value, which is lower compared to some NH2-functionalized MOFs, such as NH2-MIL-101(Cr) (52 kJ mol−1)28 and Zn(Atz)2 (40.8 kJ mol−1),29 is highly desirable because of the anticipated lower regeneration energy demand.27 At elevated pressures, very high gravimetric and volumetric CO2 uptake is observed, reaching 317 cm3 g−1 (14.2 mmol g−1) and 242 cm3 cm−3, respectively, at 298 K and 25 bar, while the corresponding saturation uptake at 50 bar is 347 cm3 g−1 (15.5 mmol g−1) and 264 cm3 g−1 (Fig. 5b). For comparison, the observed volumetric uptake at 25 bar and 298 K is slightly lower than MOF-177 (273 cm3 cm−3),32 HKUST-1 (276 cm3 cm−3)33 and Mg-MOF-74 (285 cm3 cm−3)34 but higher than MOF-5 (225 cm3 cm−3),32 gea-MOF-1 (224 cm3 cm−3),35 MOF-210 (127 cm3 cm−3)32 and MOF-200 (112 cm3 cm−3).32
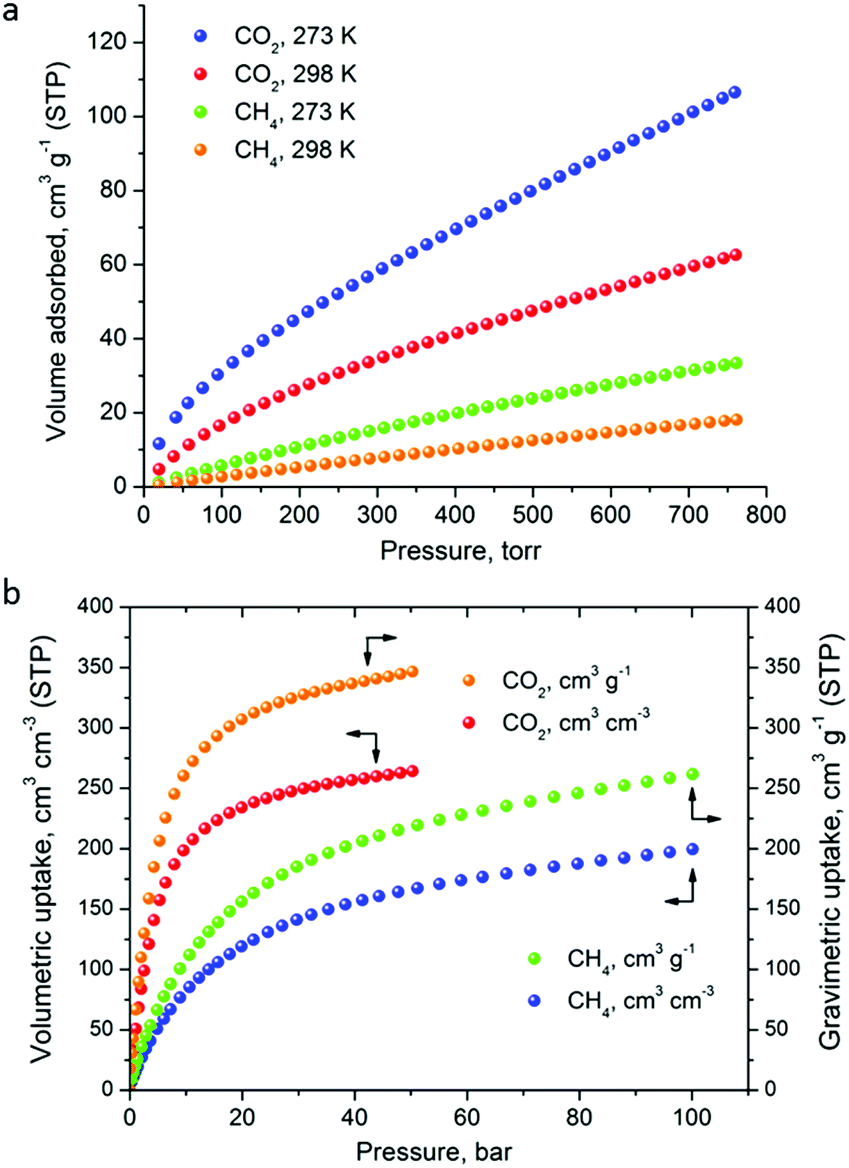 | ||
| Fig. 5 (a) Low pressure CO2 and CH4 sorption isotherms of Cu-PEIP at the indicated temperatures, up to 1 bar. (b) The corresponding high pressure isotherms recorded at 298 K. | ||
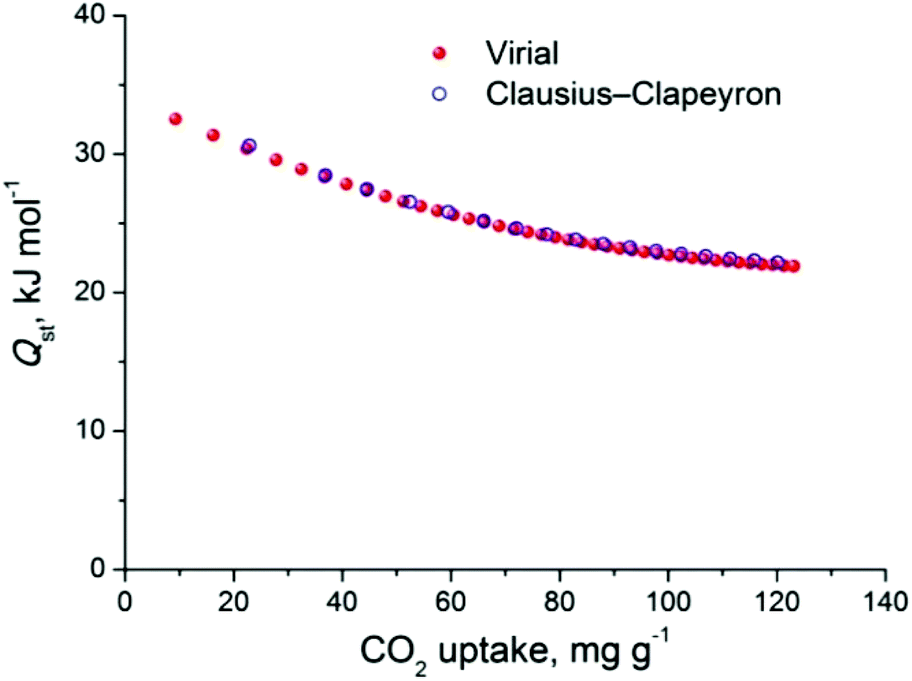 | ||
| Fig. 6 CO2 isosteric heat of adsorption (Qst) in Cu-PEIP as a function of surface coverage, calculated from a virial-type analysis. The corresponding Clausius–Clapeyron calculation is also shown, for comparison. | ||
The CH4 adsorption isotherms up to 1 bar, shown in Fig. 5a revealed an uptake of 33.4 cm3 g−1 (1.49 mmol g−1) and 18.1 cm3 g−1 (0.81 mmol g−1) at 273 K and 298 K, respectively. From these isotherms, the calculated isosteric heat of adsorption, Qst, at zero coverage using a virial-type equation, was estimated to be 21.2 kJ mol−1 and remained nearly constant as a function of the surface coverage (Fig. S13 and S14‡). Interestingly, this value is higher compared to HKUST-1 (17 kJ mol−1) and among the highest reported for MOFs (Table S1‡).36,37 For example, Ni-MOF-74 with a very high density of open metal sites showed a Qst value of 21.4 kJ mol−1.36 Very recently, a novel MOF without open metal sites, denoted as MAF-38, showed a Qst value of 21.6 kJ mol−1 at zero coverage.38 Despite the high Qst value for Cu-PEIP, a relatively high CO2/CH4 selectivity at low pressures, calculated using the IAST model, is observed reaching 8.5 at 273 K and 8.9 at 298 K.
High-pressure CH4 measurements revealed that the total gravimetric (cm3 g−1) and volumetric (cm3 cm−3) uptake at 298 K, is 197 and 150 at 35 bar, 233 and 176 at 65 bar and 246 and 187 at 80 bar, respectively. The resulting gravimetric CH4 working storage capacities in the pressure ranges 5–35 bar (US DOE standard),39 5–65 bar and 5–80 bar are estimated to be 131 cm3 g−1, 167 cm3 g−1 and 180 cm3 g−1, respectively. The corresponding volumetric working capacities are 99 cm3 cm−3, 125 cm3 cm−3 and 136 cm3 cm−3. These values are lower compared to the best performing MOFs (Tables S1 and S2 in the ESI‡) due to the combination of high Qst and a moderate surface area in Cu-PEIP. Compared to Ni-MOF-74, a MOF with a high Qst for CH4 and a slightly lower BET area (1350 m2 g−1), Cu-PEIP performs better in terms of the gravimetric working capacity at 5–65 bar (0.110 vs. 0.077 g g−1) and 5–80 bar (0.129 vs. 0.091 g g−1) (Tables S1 and S2 in the ESI‡).
Low pressure, H2 sorption isotherms recorded up to 1 bar at 77 K and 87 K revealed an uptake of 206 cm3 g−1 (2.06 wt%) and 143 cm3 g−1 (1.43 wt%), respectively, which is higher than the benchmark MOF-177 (142 and 82 cm3 g−1) (Fig. S15‡).40 The calculated Qst at zero coverage is 8.74 kJ mol−1 (Fig. S16 and S17‡), which is significantly higher than some well-known and high performance MOFs41 including MOF-5 and MOF-177 (4.4 kJ mol−1).40 High pressure H2 measurements, revealed an uptake of 614 cm3 g−1 (5.5 wt%) which is lower than the ultrahigh surface area MOFs such as MOF-210 and NU-100 (Fig. S18‡). However, at 20 bar, due to a high Qst, the uptake is 4.1 wt%, comparable to the best performing MOFs.
Conclusions
Summarizing, a new Cu2+ MOF is reported that was isolated from the initial use of ligand PEIPH2 in 3d MOF chemistry. Interestingly, Cu-PEIP displays novel structural features although pyridyl isophthalic acid tritopic ligands have been extensively employed in Cu MOF chemistry due to their ability to afford MOFs that display structural similarity with HKUST-1. The unique structural features of Cu-PEIP arose from the presence in its structure of both PEIPH2 and 5-NH2-mBDCH2 due to the capability of the tritopic Schiff base ligand to partially decompose in solution and appear in the reaction mixture together with its constituent moieties. After the structure of Cu-PEIP was known the synthesis of an analogous compound was targeted and achieved by employing a rational synthetic procedure. The crystal structure of Cu-PEIP exhibits a 3D-framework that contains large pores (∼9 Å) and solvent accessible volume (∼66.5%). In addition, it displays a unique trinodal underlying network consisting of pillared kgm-a layers and thus resembles the structure of HKUST-1. Gas sorption studies revealed that Cu-PEIP displays a significant BET area of 1785 m2 g−1 and high CO2 uptake, reaching 4.75 mmol g−1 at 273 K and 1 bar (2.80 mmol g−1 at 298 K) with good CO2/CH4 selectivity (8.5/8.9 at 273 K/298 K). Furthermore, high pressure gas sorption studies revealed that the volumetric CO2 uptake at 25 bar (242 cm3 cm−3 at 298 K) is higher compared to representative high surface area MOFs, including MOF-5, MOF-200 and MOF-210. This study emphasizes the usefulness of the elements of serendipity introduced from the employment of polytopic Schiff base ligands in MOF chemistry for the synthesis of microporous MOFs with unique structural features and interesting sorption properties. Further investigations are in progress focusing on the isolation of Cu-PEIP analogues containing various functionalized derivatives of isophthalic acid and/or more elongated tritopic pyridyl isophthalic acid ligands.Acknowledgements
This work was supported by the Cyprus Research Promotion Foundation Grant ΔIΔAKTΩP/0609/43 which is co-funded by the Republic of Cyprus and the European Regional Development Fund. EM and AJT thank the University of Cyprus for an internal postdoctoral fellowship to EM.Notes and references
- (a) M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed; (b) D. Bradshaw, J. B. Claridge, E. J. Cussen, T. J. Prior and M. J. Rosseinsky, Acc. Chem. Res., 2005, 38, 273–282 CrossRef CAS PubMed; (c) G. Ferey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- (a) N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed; (b) S. Zheng, T. Wu, J. Zhang, M. Chow, R. A. Nieto, P. Feng and X. H. Bu, Angew. Chem., Int. Ed., 2010, 49, 5362–5366 CrossRef CAS PubMed; (c) B. F. Abrahams, M. J. Grannas, T. A. Hudson and R. Robson, Angew. Chem., Int. Ed., 2010, 49, 1087–1089 CrossRef CAS PubMed.
- (a) H. Hayashi, A. P. Cote, H. Furukawa, M. O'Keeffe and O. M. Yaghi, Nat. Mater., 2007, 6, 501–506 CrossRef CAS PubMed; (b) R. E. Morris and P. S. Wheatley, Angew. Chem., Int. Ed., 2008, 47, 4966–4981 CrossRef CAS PubMed; (c) S. Noro, S. Kitagawa, M. Kondo and K. Seki, Angew. Chem., Int. Ed., 2000, 39, 2081–2084 CrossRef.
- (a) E. E. Moushi, T. C. Stamatatos, W. Wernsdorfer, V. Nastopoulos, G. Christou and A. J. Tasiopoulos, Angew. Chem., Int. Ed., 2006, 45, 7722–7725 CrossRef CAS PubMed; (b) P. Dechambenoit and J. R. Long, Chem. Soc. Rev., 2011, 40, 3249–3265 RSC.
- (a) L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838–846 CrossRef CAS PubMed; (b) J.-S. Qin, D.-Y. Du, W. Guan, X.-J. Bo, Y.-F. Li, L.-P. Guo, Z.-M. Su, Y.-Y. Wang, Y.-Q. Lan and H.-C. Zhou, J. Am. Chem. Soc., 2015, 137, 7169–7177 CrossRef CAS PubMed.
- (a) Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815–5840 RSC; (b) A. Douvali, A. C. Tsipis, S. V. Eliseeva, S. Petoud, G. S. Papaefstathiou, C. D. Malliakas, I. Papadas, G. S. Armatas, I. Margiolaki, M. G. Kanatzidis, T. Lazarides and M. J. Manos, Angew. Chem., Int. Ed., 2015, 54, 1651–1656 CrossRef CAS PubMed.
- (a) O. M. Yaghi, H. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474–484 CrossRef CAS; (b) M. Eddaoudi, J. Kim, D. Vodak, A. Sudik, J. Wachter, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4900–4904 CrossRef CAS PubMed.
- (a) X.-L. Zhao and W.-Y. Sun, CrystEngComm, 2014, 16, 3247–3258 RSC; (b) B. Manna, A. V. Desai and S. K. Ghosh, Dalton Trans., 2016, 45, 4060–4072 RSC.
- (a) S. M. Cohen, Chem. Rev., 2012, 112, 970–1000 CrossRef CAS PubMed; (b) O. Karagiaridi, W. Bury, J. E. Mondloch, J. T. Hupp and O. K. Farha, Angew. Chem., Int. Ed., 2014, 53, 4530–4540 CrossRef CAS PubMed; (c) E. J. Kyprianidou, T. Lazarides, S. Kaziannis, C. Kosmidis, G. Itskos, M. J. Manos and A. J. Tasiopoulos, J. Mater. Chem. A, 2014, 2, 5258–5266 RSC.
- (a) N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed; (b) Z.-J. Lin, J. Lü, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC.
- (a) C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 1466–1496 CrossRef CAS PubMed; (b) E. E. Moushi, A. Kourtellaris, I. Spanopoulos, M. J. Manos, G. S. Papaefstathiou, P. N. Trikalitis and A. J. Tasiopoulos, Cryst. Growth Des., 2015, 15, 185–193 CrossRef CAS.
- (a) M.-S. Chen, Z.-S. Bai, T. Okamura, Z. Su, S.-S. Chen, W.-Y. Sun and N. Ueyama, CrystEngComm, 2010, 12, 1935–1944 RSC; (b) X.-J. Deng, W. Gu, L. Wang, L.-F. Zeng and X. Z. Liu, Z. Anorg. Allg. Chem., 2011, 637, 708–712 CrossRef CAS; (c) M.-S. Chen, M. Chen, S. Takamizawa, T. Okamura, J. Fan and W.-Y. Sun, Chem. Commun., 2011, 47, 3787–3789 RSC; (d) Y. Xiong, Y.-Z. Fan, R. Yang, S. Chen, M. Pan, J.-J. Jiang and C.-Y. Su, Chem. Commun., 2014, 50, 14631–14634 RSC; (e) Z. Chen, K. Adil, L. J. Weselinski, Y. Belmabkhout and M. Eddaoudi, J. Mater. Chem. A, 2015, 3, 6276–6281 RSC.
- (a) X. Zhang, Y.-Y. Huang and Y.-G. Yao, Inorg. Chem. Commun., 2013, 28, 49–51 CrossRef; (b) L. Qin, J.-S. Hu, L.-F. Huang, Y.-Z. Li, Z.-J. Guo and H.-G. Zheng, Cryst. Growth Des., 2010, 10, 4176–4183 CrossRef CAS; (c) X. Zhang, J. Cheng, F. Chen, M. Sun and Y. Yao, Inorg. Chem. Commun., 2011, 14, 358–361 CrossRef CAS; (d) M. C. Das and P. K. Bharadwaj, J. Am. Chem. Soc., 2009, 131, 10942–10949 CrossRef CAS PubMed; (e) M. C. Das and P. K. Bharadwaj, Chem. – Eur. J., 2010, 16, 5070–5077 CrossRef CAS PubMed; (f) A. Karmakar, L. M. D. R. S. Martins, S. Hazra, M. F. C. G. Da Silva and A. J. L. Pombeiro, Cryst. Growth Des., 2016, 16, 1837–1849 CrossRef CAS; (g) M.-H. Xie, X.-L. Yang and C.-D. Wu, Chem. – Eur. J., 2011, 17, 11424–11427 CrossRef CAS PubMed.
- (a) V. Chandrasekhar, C. Mohapatra and R. Metre, Cryst. Growth Des., 2013, 13, 4607–4614 CrossRef CAS; (b) V. Chandrasekhar and C. Mohapatra, Cryst. Growth Des., 2013, 13, 4655–4658 CrossRef CAS.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- (a) V. Guillerm, D. Kim, J. F. Eubank, R. Luebke, X. Liu, K. Adil, M. S. Lah and M. Eddaoudi, Chem. Soc. Rev., 2014, 43, 6141–6172 RSC; (b) J. F. Eubank, H. Mouttaki, A. J. Cairns, Y. Belmabkhout, L. Wojtas, R. Luebke, M. Alkordi and M. Eddaoudi, J. Am. Chem. Soc., 2011, 133, 14204–14207 CrossRef CAS PubMed; (c) X. Liu, M. Oh and M. S. Lah, Inorg. Chem., 2011, 50, 5044–5053 CrossRef CAS PubMed; (d) X. Liu, M. Oh and M. S. Lah, Cryst. Growth Des., 2011, 11, 5064–5071 CrossRef CAS; (e) S. Xiang, J. Huang, L. Li, J. Zhang, L. Jiang, X. Kuang and C.-Y. Su, Inorg. Chem., 2011, 50, 1743–1748 CrossRef CAS PubMed; (f) B. Moulton, J. Lu, R. Hajndl, S. Hariharan and M. J. Zaworotko, Angew. Chem., Int. Ed., 2002, 41, 2821–2824 CrossRef CAS; (g) S. A. Bourne, J. Lu, A. Mondal, B. Moulton and M. J. Zaworotko, Angew. Chem., Int. Ed., 2001, 40, 2111–2113 CrossRef CAS.
- (a) E. J. Kyprianidou, G. S. Papaefstathiou, M. J. Manos and A. J. Tasiopoulos, CrystEngComm, 2012, 14, 8368–8373 RSC; (b) M. J. Manos, E. J. Kyprianidou, G. S. Papaefstathiou and A. J. Tasiopoulos, Inorg. Chem., 2012, 51, 6308–6314 CrossRef CAS PubMed; (c) C. G. Efthymiou, E. J. Kyprianidou, C. J. Milios, M. J. Manos and A. J. Tasiopoulos, J. Mater. Chem. A, 2013, 1, 5061–5069 RSC.
- Oxford Diffraction, CrysAlis CCD and CrysAlis RED, Oxford Diffraction Ltd., Abingdon, UK, 2008 Search PubMed.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 38, 381–388 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- K. Brandenburg, DIAMOND, Version 2003.2001d, Crystal Impact GbR: Bonn, Germany, 2006 Search PubMed.
- http://www.ccp14.ac.uk/ccp/web-mirrors/x-seed/ .
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. Van De Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- L. Sarkisov and A. Harrison, Mol. Simul., 2011, 37, 1248–1257 CrossRef CAS.
- Y. Lin, C. Kong and L. Chen, RSC Adv., 2016, 6, 32598–32614 RSC.
- Y. Lin, C. Kong and L. Chen, RSC Adv., 2012, 2, 6417–6419 RSC.
- R. Vaidhyanathan, S. S. Iremonger, K. W. Dawson and G. K. H. Shimizu, Chem. Commun., 2009, 5230–5232 RSC.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- J. Ethiraj, E. Albanese, B. Civalleri, J. G. Vitillo, F. Bonino, S. Chavan, G. C. Shearer, K. P. Lillerud and S. Bordiga, ChemSusChem, 2014, 7, 3382–3388 CrossRef CAS PubMed.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- J. Moellmer, A. Moeller, F. Dreisbach, R. Glaeser and R. Staudt, Microporous Mesoporous Mater., 2011, 138, 140–148 CrossRef CAS.
- J. M. Simmons, H. Wu, W. Zhou and T. Yildirim, Energy Environ. Sci., 2011, 4, 2177–2185 CAS.
- V. Guillerm, Ł. J. Weseliński, Y. Belmabkhout, A. J. Cairns, V. D'Elia, Ł. Wojtas, K. Adil and M. Eddaoudi, Nat. Chem., 2014, 6, 673–680 CAS.
- Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657–5678 RSC.
- I. Spanopoulos, C. Tsangarakis, E. Klontzas, E. Tylianakis, G. Froudakis, K. Adil, Y. Belmabkhout, M. Eddaoudi and P. N. Trikalitis, J. Am. Chem. Soc., 2016, 138, 1568–1574 CrossRef CAS PubMed.
- J.-M. Lin, C.-T. He, Y. Liu, P.-Q. Liao, D.-D. Zhou, J.-P. Zhang and X.-M. Chen, Angew. Chem., Int. Ed., 2016, 55, 4674–4678 CrossRef CAS PubMed.
- Y. Peng, V. Krungleviciute, I. Eryazici, J. T. Hupp, O. K. Farha and T. Yildirim, J. Am. Chem. Soc., 2013, 135, 11887–11894 CrossRef CAS PubMed.
- H. Furukawa, M. A. Miller and O. M. Yaghi, J. Mater. Chem., 2007, 17, 3197–3204 RSC.
- D. P. Broom, C. J. Webb, K. E. Hurst, P. A. Parilla, T. Gennett, C. M. Brown, R. Zacharia, E. Tylianakis, E. Klontzas, G. E. Froudakis, T. A. Steriotis, P. N. Trikalitis, D. L. Anton, B. Hardy, D. Tamburello, C. Corgnale, B. A. Van Hassel, D. Cossement, R. Chahine and M. Hirscher, Appl. Phys. A, 2016, 122, 151 CrossRef.
Footnotes |
| † Dedicated to Professor Mercouri G. Kanatzidis on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Structural figures, PXRD, IR, TGA and additional gas-sorption data. CCDC 1492970. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6qi00273k |
| This journal is © the Partner Organisations 2016 |