
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting polymorphism in second sphere coordination: thermal transformation, NLO properties and selective mechanochemical synthesis†
Hai-Bin
Yu
a,
Hai-Tao
Li
a,
Peng
Zhang
a,
Antonino
Famulari
b,
Fang
Guo
*a,
Ilaria
Bargigia
c and
Javier
Martí-Rujas
*c
aCollege of Chemistry, Liaoning University, Shenyang 110036, China. E-mail: fguo@lnu.edu.cn
bDipartimento di Chimica Materiali e Ingegneria Chimica “Giulio Natta”, Politecnico di Milano, Via L. Mancinelli 7, 20131 Milan, Italy
cCenter for Nano Science and Technology@Polimi, Istituto Italiano di Tecnologia, Via Pascoli 70/3, 20133 Milano, Italy. E-mail: javier.rujas@iit.it; Tel: +392399 989
First published on 9th March 2016
Abstract
The racemic organic building block L acting as a first sphere ligand yields a new second sphere adduct [LH]+·[FeCl4]− crystallizing as racemic polymorphs α and β. Solid-state DFT calculations show that polymorph β is the kinetic adduct as observed experimentally. The β-phase crystallizes in the polar space group Pna21 and displays NLO properties. Mechanochemical synthesis yields only the β-phase.
The combination of organic cations and metal anions to synthesize hybrid metal–organic structures via second sphere coordination was used in the early 80s.1 The importance of second sphere coordination has been demonstrated in supramolecular chemistry2 recognition phenomena in biological systems,3 metal-based anion receptors4–6 and catalysis.7 In the solid state, second sphere coordination has been studied much less, but recently, gas adsorption,8 metal-ion separation,9 structural rearrangements,10 and chiral systems11 have been reported.
As defined by Gibb's phase rule, polymorphism refers to the situation in which two or more crystalline phases have the same chemical composition but different structures. Polymorphism,12 usually confined to pharmaceutical organic materials,13 is of fundamental importance in academy and industry due to its effect on the solid-state properties of a solid material. Interestingly, polymorphic examples of hybrid metal–organic materials self-assembled via outer sphere coordination are rare despite the long history of such interactions. Self-assembling organic molecules and metals in a second sphere adduct in which the organic part is racemic can indeed increase the options of discovering a variety of structures such as racemic second sphere adducts, crystallization of pure R or S enantiomers (i.e., conglomerate), or even polymorphism in the second sphere adduct. However, to the best of our knowledge, racemic organic crystals as first sphere ligands have not been applied to explore polymorphism in second sphere adducts.
Herein, we report a racemic crystalline form of the organic N,N,N′,N′-tetrabenzyl-trans-1,2-diaminocyclohexane (L), and how racemic polymorphism is also observed in the second sphere adduct [LH]+·[FeCl4]−. Upon the self-assembly of L with FeCl3·6H2O and HCl, [LH]+·[FeCl4]− crystallizes as two polymorphs: α-phase (P21/c) and β-phase (Pna21). Both polymorphs contain R and S enantiomers in the structure, and so are racemic. Weak, attractive C–H⋯Cl and repulsive C–H⋯H–C supramolecular interactions play important roles in driving the molecules into non-polar and polar packing. Solid-state DFT calculations showed that the most stable structure is the thermodynamic α-phase. The structural transformation from the acentric and polar Pna21 to the centric P21/c structure is described upon thermal treatment in the solid state. The selective formation of the β-phase can be obtained by means of liquid-assisted grinding (LAG). Moreover, the single crystal second harmonic generation (SHG) behavior for the polar β-phase is reported, which shows significant efficiency compared to that of urea.
Ligand L was synthesized by reacting trans-1,2-diaminocyclohexane and benzyl bromide in the presence of LiOH/CH2Cl2 (Scheme 1).14 Crystals of L suitable for single crystal XRD (SC-XRD) were obtained upon evaporation from ethanol. In the crystalline state, L packs in the C2/c space group with half a molecule in the asymmetric unit (see the ESI†). The crystal structure of L is racemic14 while the acentric (R,R),N,N′-tetrabenzyl-1,2-diaminocyclohexane crystallizing in the monoclinic I2 space group is the enantiopure form.15 The bulk powder of the as synthesized L belongs to the phase crystallizing in the C2/c space group.14 In L, the bond angles around N1 were 112.9(1)°, 112.4(1)°, and 110.0(1)° for C1–N1–C2, C1–N1–C5 and C2–N1–C5, respectively. This geometric information indicates that the conformation around each nitrogen atom is near sp3 hybridised.
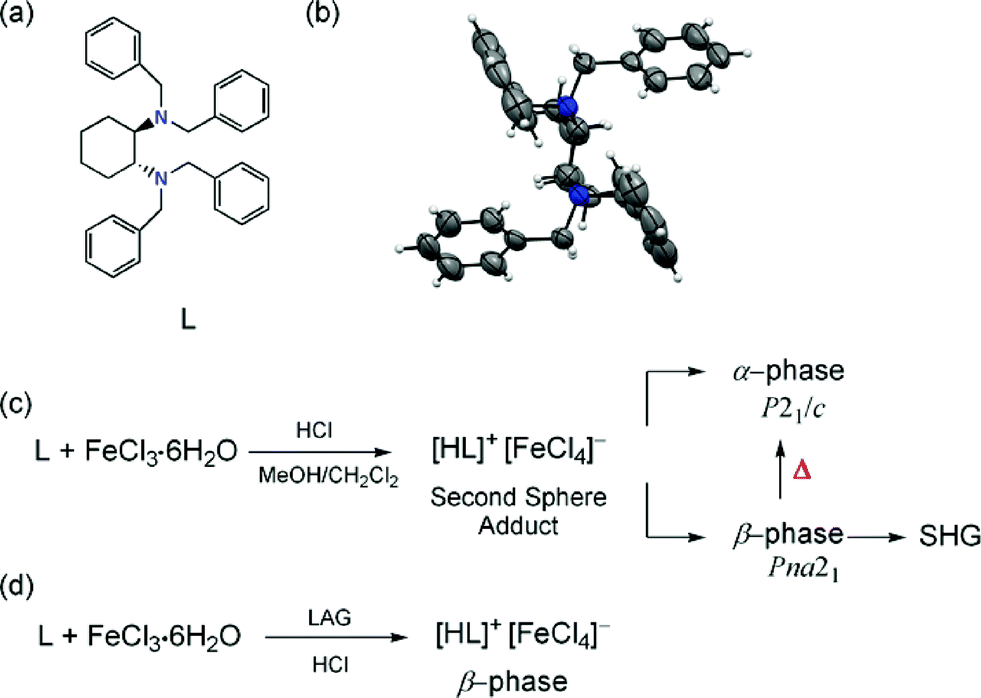 | ||
| Scheme 1 Chemical structure (a) and a view of the molecular structure. Displacement ellipsoids are drawn at the 50% probability level and H atoms are shown as small spheres of arbitrary radius (b). Synthetic route for the second sphere adduct [LH]+·[FeCl4]− giving rise to α- and β-phases in solution (c). LAG synthesis of the β-phase (d). | ||
The lone pairs of each nitrogen atom are oriented towards the diamine pocket formed by the two N,N′-benzyl substituents on each side.
The ability of L to form second sphere adducts was explored in the solid state. The second sphere supramolecular structure was prepared by mixing a CH2Cl2/MeOH solution of FeCl3·6H2O with L in the presence of HCl. After three days, crystals with two different habits were obtained: a large amount of yellow hexagonal crystals (α) and a small amount of yellow needle crystals (β).14
The structural characterization of α- and β-phases was carried out by SC-XRD. X-ray crystallography studies reveal that α- and β-phases are racemic polymorphs that crystallize in the monoclinic space group P21/c and orthorhombic space group Pna21, respectively. The different phases have been confirmed by X-ray powder diffraction (XRPD) experiments (Fig. 4). The β-phase belongs to a noncentrosymmetric and polar space group. It is worth noting that polar crystals are highly demanded in materials science because they can display piezoelectric, pyroelectric, ferroelectric, and SHG properties (vide ante).
Both second sphere adducts contain an independent [FeCl4]− anion and a [HL]+ cation in the asymmetric unit. Each [FeCl4]− anion in α- and β-phases has almost the same coordination environment and adopts a distorted tetrahedral geometry coordinated by four Cl atoms.14
It is worth mentioning that although the monocation is protonated and the N–H group in the diamine pocket is a potential hydrogen bonding donor, the structural analysis indicated that it is “locked” by the four extended benzyl rings. There are no charge-assisted hydrogen bonds among [HL]+ and the [FeCl4]− anion participating in the structural formation. Thus, no quasi-chelating motif is observed in the supramolecular adduct.19c Instead, weak interactions such as C–H⋯Cl contacts play a critical role in the self-assembly of the second sphere adduct.
For the α-phase, as seen in Fig. 2, each [FeCl4]− anion interacts with three neighbouring [HL]+ cations through five C–H⋯Cl interactions, with the methylene group (i. C11–H11A⋯Cl3: 3.798(6) Å, 149.1(4)°; ii. C11–H11B⋯Cl1: 3.486(6) Å, 128.8(4)°), and the other two are from [HL]+ at (x, y, z) with the benzene ring and the methylene group, respectively (iii. C15–H15⋯Cl2: 3.794(8) Å, 151.8(4)°; iv. C13–H13A⋯Cl2: 3.771(6) Å, 145.0(4)°). The structure contains both enantiomers. The 1D chains are further connected with each other through extensive C–H⋯Cl interaction (v. C24–H24A⋯Cl1: 3.83(1) Å, 160.6(7)°), forming a helical layer along the b-axis, as seen in Fig. S10.†
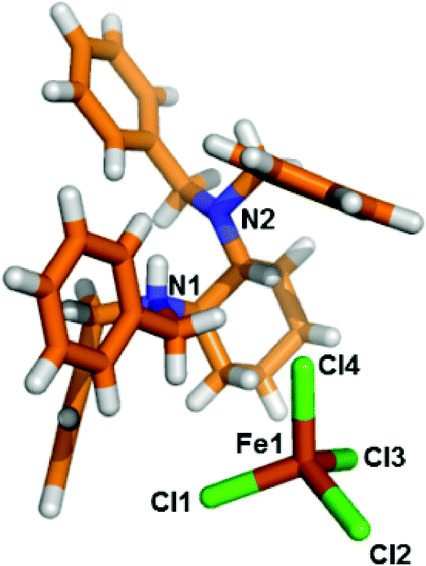 | ||
| Fig. 1 Stick model of the asymmetric unit of [LH]+·[FeCl4]− (α-phase). Color code: carbon: orange; nitrogen: blue; chloride: green; iron: brown; hydrogen: white. | ||
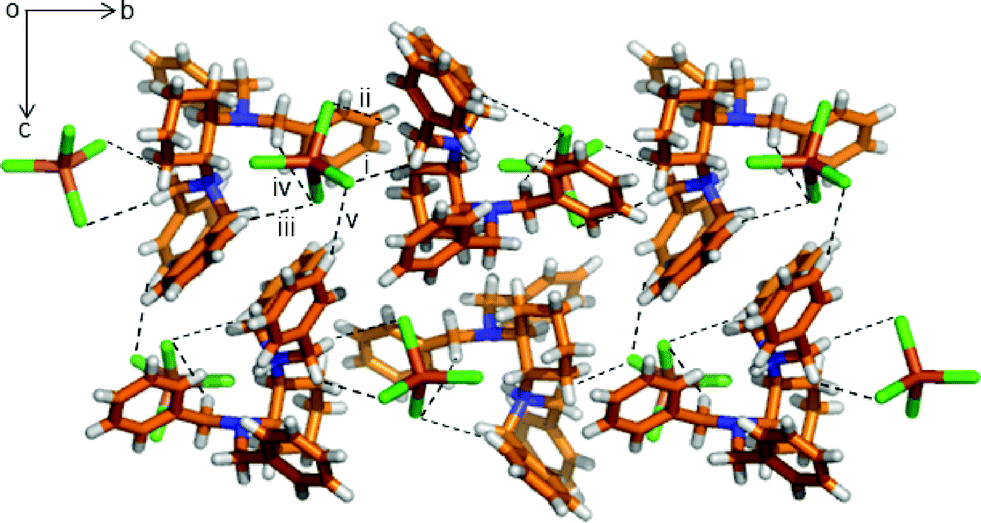 | ||
| Fig. 2 Crystal structure of the α-phase viewed along the a-axis showing the various C–H⋯Cl interactions among [HL]+ cations and [FeCl4]− anions. The hydrogen bonding interactions are shown with a dashed black line. Color code as in Fig. 1. | ||
A rather diverse set of interactions are observed in the β-phase. In this structure, the [FeCl4]− anion interacts with two [HL]+ cations (Fig. 3). The first anion–cation interaction is through one C–H⋯Cl contact with the methylene group (i. C14–H14B⋯Cl1: 3.78(2) Å, 150.4(9)°).
 | ||
| Fig. 3 Crystal structure of the β-phase viewed along a-axis formed by C–H⋯Cl interactions among [HL]+ cations and [FeCl4]− anions and repulsive C–H⋯H–C intermolecular interactions. The hydrogen bonding interactions are shown with a dashed black line (a). Packing of half a unit cell (a-axis) showing the non-centrosymmetric and polar orientation (black arrows) of layers of the enantiomers R (b) and S (c). Color code as in Fig. 1. | ||
Then the second ion pair interaction involves contacts ii. C21–H21B⋯Cl3: 3.62(2) Å, 129.1(9)° and iii. C19–H19⋯Cl1: 3.61(2) Å, 129(1)°. As in the α-phase, the structure is composed of both enantiomers of L but arranged in a non-centrosymmetric manner (Fig. S12†).14 There are also C–H⋯H–C repulsive interactions between the aryl and methylene groups (iv. C14–H14A⋯H33: 2.4881(3) Å). Further proof of the different crystal packings for both polymorphs is confirmed by IR experiments which show that the C–N and C–H stretching bands of –CH2 are different for polymorphs α and β (Fig. S14 and S15†).
Therefore, weak attractive C–H⋯Cl and repulsive C–H⋯H–C intermolecular interactions have played important roles in driving the molecules into different packing assemblies. Such non-attractive contacts are also relevant in maintaining the stability of the supramolecular adduct. The polar nature in the β-phase can be observed in the orientation of the [HL]+ cations “towards” the b-axis as shown by the black arrows in Fig. 3b and c. The overall crystal packing is non-centrosymmetric (Fig. S12†). Racemates tend to crystallize in centrosymmetric crystals; however, a racemate can crystallize in centro- and non-centrosymmetric space groups as polymorphic forms as in this case.
Given that the calculated densities for polymorphs α and β are very similar (1.300 and 1.334 g cm−3, respectively), it may be assumed that the free energy of these two polymorphic forms are quite similar.16 Thus, in order to investigate the stability of the two phases, solid-state Density Functional Theory (DFT) calculations were performed.14
The calculations were accomplished by fixing cell parameters and by optimising atomic coordinates within the experimental lattices. Explicit van der Waals corrections were included in the calculations17 as these terms can be important for subtle inter- and intra-particle interactions (i.e., attractive and repulsive).18 This approach has been proved to be adequate in a number of cases, such as supramolecular complexes,19 crystalline systems containing charged particles20 and crystalline phases of thiophene-based oligomers and polymers.21
The calculations showed that the α-phase (the lower density system) possesses stronger cation–anion interactions, within the crystalline phase, with respect to those corresponding to the β-phase (the higher density system) by about 16 or 14 kcal mol−1 (i.e., higher sublimation energy) obtained by DND (double-numeric quality basis set plus polarisation functions on heavy atoms, i.e. comparable to Gaussian 6-31G*) and DNP (double-numeric quality basis set plus polarisation functions on all atoms, comparable to Gaussian 6-31G**) sets, respectively. Therefore, according to the DFT results, the α-phase is the thermodynamic structure (lower energy) and the β-phase is the kinetic structure (higher energy). Thus, the β-phase could be transformed into the α-phase in the solid state, for instance by heating.
To further investigate the above mentioned phase transition of the β to the α form in the solid state, we have carried out ex situ XRPD experiments on heated samples of the β form (Fig. 4). The microcrystalline β-phase was heated from room temperature to 140 °C. When the β-phase reached 80 °C the XRPD pattern shows that there was only one phase (β), but reaching 100 °C a mixture of β and α-phases is observed (see the ESI†). However, when 110 °C was achieved a complete phase transformation occurs and only the α-phase is present (Fig. 4c). Further heating shows that the crystallite melts at ca. 140 °C. The phase transition and the melting of the second sphere adducts have been corroborated by DSC experiments.14 Clearly, the polymorphic phase transition is carried out in the solid state.
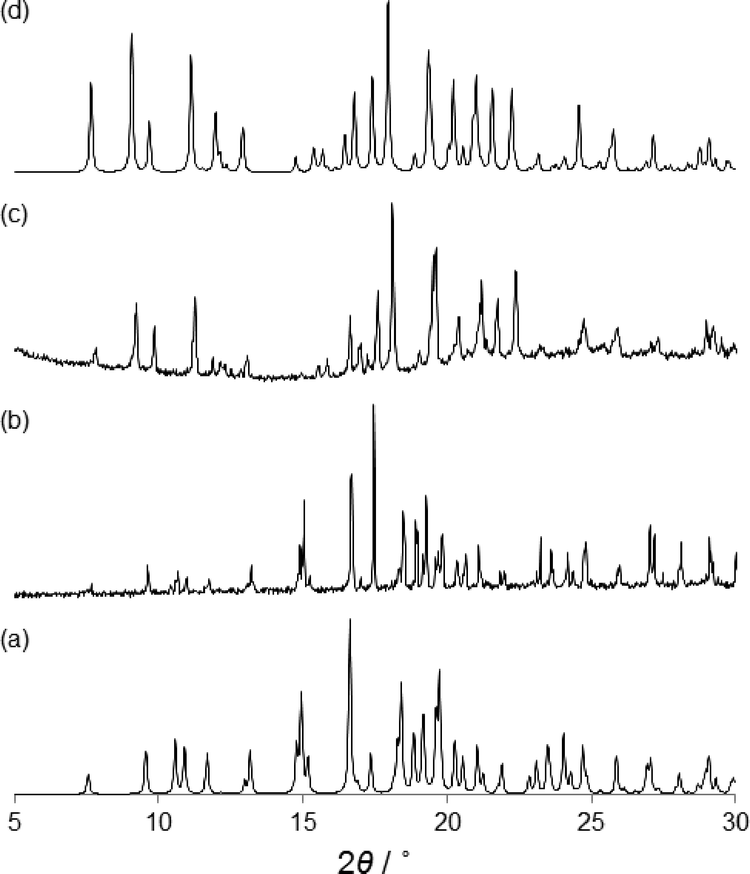 | ||
| Fig. 4 The simulated isotropic XRPD patterns of β (a) and α (d) phases calculated from single crystal structures. Experimental XRPD pattern corresponding to the β phase at room temperature (b) and that of the β phase at 110 °C measured ex situ at room temperature (c). | ||
Therefore, in light of the observed results, we can describe the α-phase as the thermodynamic phase and the β phase as the kinetic product. The formation of the β-phase can be described as the structure pertaining to a local minima in the energy landscape during the self-assembly of the metal anions and organic cations (i.e., small activation energy). The formation of the thermodynamic α-phase corresponds to the global minimum, which in this case is obtained by applying temperature to the system, the β-phase.
In general, controlling the formation of a desired polymorphic form is difficult. Here we have monitored various crystallization methods using different solvents such as methanol, ethanol, THF, propanol, acetone, acetonitrile, ethyl acetate, and cyclohexane, but the outcomes resulted in mixtures of the two phases, an indication of their comparable energies. As an alternative crystallization route, solid-state grinding has become an efficient method for co-crystal grinding,22 affording good opportunities for the selective preparation of a given polymorph.23 Herein we found that using LAG it is possible to control the formation of only the β polymorph. In the presence of various solvents, the grinding ligand L and FeCl3·6H2O with one drop of HCl gave the β-phase but not the α-phase (Fig. S16†).24 Therefore, the selective formation of the kinetic and noncentrosymmetric phase is possible via LAG but not from solution crystallization. Kinetic products have been obtained by mechanochemical methods in other systems.23c
Crucially, the properties of materials in the solid state depend not only on their building blocks, but also on how these subcomponents are arranged within the crystalline lattice. In the β-phase the organic cations are oriented in one particular way (Fig. 3), and for this reason the space group where crystallizes is non-centrosymmetric and polar (Pna21). This is relevant because the β-phase can show nonlinear optical properties such as SHG as well as ferroelectricity and piezoelectricity.25 To test this phenomenon, SHG experiments were carried out using single crystals of the β-phase.14,26
From the experimental results shown in Fig. 5, the β-phase displays a single crystal SHG efficiency 0.18 times that of urea.27 We note that the single crystal after the SHG experiment has the ability to withstand laser irradiation as it was not burned out and survived at least three measurements. This is important for potential applications in areas of photonics such as electro optics and frequency conversion. To the best of our knowledge, this is one of the few reports of single crystal SHG measured in a second sphere adduct on polar crystals.
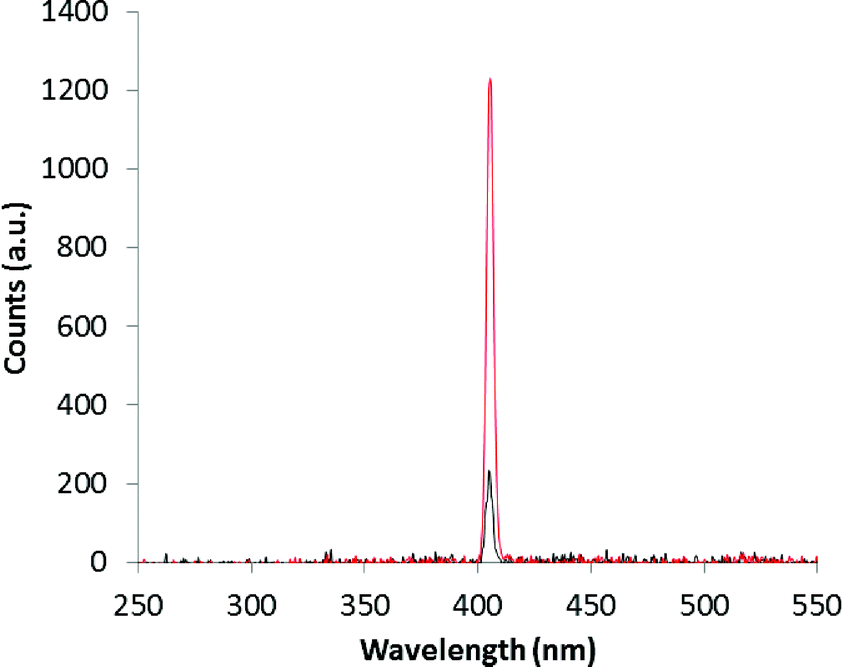 | ||
| Fig. 5 Single crystal SHG signals for urea (red) and the β-phase (black) of the super-complex [LH]+·[FeCl4]−. | ||
In summary, the potential of using racemic organic molecules as templates in the self-assembly with [FeCl4]− has led to the discovery of a new racemic polymorphic system in a second sphere adduct.28 X-ray crystallography reveals that in the β-phase the [HL]+ cations are oriented within the crystalline lattice giving its polar nature despite being a racemic crystal, with good SHG efficiency. Mechanochemical LAG has demonstrated that only the β-phase is obtained in the solid state. DFT solid phase calculations in combination with X-ray experiments showed that the thermodynamic product is the α-phase.
The stability and non-linear optical (NLO) behavior of the β-phase could render this material suitable for functional applications in areas such as electro-optics and frequency conversion. Ferroelectricity and piezoelectricity phenomena can also be exploited in the second sphere adduct reported due to the polar nature of the β-phase. Second sphere adducts self-assembled with racemic or enantiopure organic building blocks certainly are good candidates to synthesize new hybrid metal–organic structures with potential in many technological applications. We are currently designing new organic building blocks to be used as first sphere coordination in outer sphere adducts.
Acknowledgements
This research was supported by NSFC (no. 21571090, 20772054), the program for Liaoning Excellent Talents in University (LJQ 2011003), and Liaohe Scholar support from Liaoning University. A. F. acknowledges the support of CINECA and PRIN (project 2010XLLNM3_007).Notes and references
- (a) B. Dietrich, M. W. Hosseini, J. M. Lehn and R. B. Sessions, J. Am. Chem. Soc., 1981, 103, 1282 CrossRef CAS; (b) H. M. Coulquhoun, J. F. Stoddart, D. J. Williams, J. B. Wolstenholme and R. Zarzycki, Angew. Chem., Int. Ed. Engl., 1981, 20, 1051 CrossRef; (c) H. A. Bencini, A. Bianchi, P. Dapporto, E. Garcia-España, M. Micheloni, P. Paoletti and P. Paoli, J. Chem. Soc., Chem. Commun., 1990, 753 RSC.
- (a) M. C. T. Fyfe, P. T. Glink, S. Menzer, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed. Engl., 1997, 36, 2068 CrossRef CAS; (b) B. A. Blight, K. A. Van Noortwyk, J. A. Wisner and M. C. Jennings, Angew. Chem., Int. Ed., 2005, 44, 1499 CrossRef CAS PubMed; (c) B. A. Blight, J. A. Wisner and M. C. Jennings, Chem. Commun., 2006, 4593 RSC.
- S. E. Sherman and S. J. Lippard, Chem. Rev., 1987, 87, 1153 CrossRef CAS.
- S. Loeb, in Comprehensive Supramolecular Chemistry, ed., Atwood, J. L., Davies, J. E. D., MacNicol, D. D. and Vögtle, F., Elsevier Science: New York, 1996, vol. 1, p. 733 Search PubMed.
- D. A. Beauchamp and S. J. Loeb, Chem. – Eur. J., 2002, 8, 5084 CrossRef CAS.
- D. J. Mercer and S. J. Loeb, Chem. Soc. Rev., 2010, 39, 3612 RSC.
- G. M. Jacobsen, J. Y. Yang, B. Twamley, A. D. Wilson, R. M. Bullock, M. R. DuBois and D. L. DuBois, Energy Environ. Sci., 2008, 1, 167 CAS.
- S. A. Dalrymple and G. K. H. Shimizu, J. Am. Chem. Soc., 2007, 129, 12114 CrossRef CAS PubMed.
- (a) J. R. Turkington, V. Cocalia, K. Kendall, C. A. Morrison, P. Richardson, T. Sassi, P. A. Tasker, P. J. Bailey and K. C. Sole, Inorg. Chem., 2012, 51, 12805 CrossRef CAS PubMed; (b) Z. Liu, M. Frasconi, J. Lei, Z. J. Brown, Z. Zhu, D. Cao, J. Iehl, G. Liu, A. C. Fahrenbach, O. K. Farha, J. T. Hupp, C. A. Mirkin, Y. Y. Botros and J. F. Stoddart, Nat. Commun., 2013, 4, 1855 CrossRef PubMed.
- (a) P. Dechambenoit, S. Ferlay, N. Kyritsakas and M. W. Hosseini, J. Am. Chem. Soc., 2008, 130, 17106 CrossRef CAS PubMed; (b) J. Martí-Rujas and M. Cametti, New J. Chem., 2014, 38, 1385 RSC; (c) F. Guo, X. Wang, H.-Y. Guan, H.-B. Yu, L. Li, S.-S. Chen, A. Famulari and J. Martí-Rujas, Cryst. Growth Des., 2015, 15, 2842 CrossRef CAS; (d) L. Li, F. Maddalena, M. Oliveros, M. Caironi, F. Guo and J. Martí-Rujas, CrystEngComm, 2014, 16, 10888 RSC; (e) L. Li, J. Tong, F. Guo and J. Martí-Rujas, CrystEngComm, 2016 10.1039/C5CE02093J.
- (a) S. Ferlay, O. Félix, M. W. Hosseini, J. M. Planeix and N. Kyritsakas, Chem. Commun., 2002, 702 RSC; (b) H.-C. Yu, L. Li, J. Gao, J. Tong, W. Zheng, M. Cametti, A. Famulari, S. V. Meille, F. Guo and J. Martí-Rujas, Dalton. Trans., 2015, 44, 15960 RSC.
- A. J. Cruz-Cabeza and J. Bernstein, Chem. Rev., 2014, 114, 2170 CrossRef CAS PubMed.
- (a) S. Aitipamula, P. S. Chow and R. B. H. Tan, CrystEngComm, 2014, 16, 3451 RSC; (b) N. K. Duggirala, M. L. Perry, Ö. Almarsson and M. J. Zaworotko, Chem. Comm., 2016, 52, 640 RSC, see the ESI† for further details.
- See the ESI† for further details.
- E. Boyd, G. S. Coumbarides, J. Eames, R. V. H. Jones, M. Motevalli, R. A. Stenson and M. J. Suggate, J. Chem. Crystallogr., 2006, 36, 263 CrossRef CAS.
- A. Burger and R. Ramberger, Microchim. Acta, 1979, 2, 259 CrossRef CAS.
- S. Grimme, J. Chem. Phys., 2006, 124, 34108 CrossRef PubMed.
- (a) A. Baggioli, S. V. Meille, G. Raos, R. Po, M. Brinkmann and A. Famulari, Int. J. Quantum Chem., 2013, 113, 2154 CrossRef CAS; (b) A. Baggioli and A. Famulari, Phys. Chem. Chem. Phys., 2014, 16, 3983 RSC.
- (a) G. Yu, S. Yin, Y. Liu, Z. Shuai and D. Zhu, J. Am. Chem. Soc., 2003, 125, 14816 CrossRef CAS PubMed; (b) E. Maccaroni, L. Malpezzi, A. Famulari and N. Masciocchi, J. Pharm. Biomed. Anal., 2012, 60, 65 CrossRef CAS PubMed; (c) H. Y. Guan, Z. Wang, A. Famulari, X. Wang, F. Guo and J. Martí-Rujas, Inorg. Chem., 2014, 53, 7438 CrossRef CAS PubMed; (d) F. Guo, H. D. Shao, Q. Yang, A. Famulari and J. Martí-Rujas, CrystEngComm, 2014, 16, 969 RSC.
- (a) F. Guo, M.-Q. Zhang, A. Famulari and J. Martí-Rujas, CrystEngComm, 2013, 15, 6237 RSC; (b) H.-L. Liu, Y.-F. Xie, Z. G. Pan, A. Famulari, F. Guo, Z. Zhou and J. Martí-Rujas, Cryst. Growth Des., 2014, 14, 6528 CrossRef CAS.
- (a) A. Famulari, G. Raos, A. Baggioli, M. Casalegno, R. Po and S. V. Meille, J. Phys. Chem. B, 2012, 116, 14504 CrossRef CAS PubMed; (b) M. Casalegno, A. Baggioli, A. Famulari, S. V. Meille, T. Nicolini, R. Po and G. Raos, Materials for organic photovoltaics: insights from detailed structural models and molecular simulations, EPJ WEB OF CONFERENCES, 2012, p. 02002, DOI:10.1051/epjconf/20123302002; (c) T. Nicolini, A. Famulari, T. Gatti, J. Martí-Rujas, F. Villafiorita Monteleone, E. V. Canesi, C. Botta, E. Parisini, S. V. Meille and C. Bertarelli, J. Phys. Chem. Lett., 2014, 5, 2171 CrossRef CAS PubMed.
- A. V. Trask and W. Jones, Top. Curr. Chem., 2005, 254, 41 CAS.
- (a) A. V. Trask, W. D. S. Motherwell and W. Jones, Chem. Commun., 2004, 890 RSC; (b) D. Cinčić, I. Brekalo and B. Kaitner, Cryst. Growth Des., 2012, 15, 44 CrossRef; (c) M. Zbačnik, I. Nogalo, D. Cinčić and B. Kaitner, CrystEngComm, 2015, 17, 7870 RSC; (d) Y. Zhou, F. Guo, C. E. Hughes, D. L. Brown, T. R. Pesket and K. D. M. Harris, Cryst. Growth Des., 2015, 15, 2901 CrossRef CAS.
- A. V. Trask, W. D. S. Motherwell and W. Jones, Chem. Commun., 2004, 890 RSC.
- M. Cametti, I. Bargigia and J. Martí-Rujas, Dalton. Trans., 2016, 45, 1674 RSC.
- For examples of control of polymorphism for SHG in organic crystals: (a) J. Xu, S. Semin, D. Niedzialek, P. H. J. Kouwer, E. Fron, E. Coutino, M. Savoini, Y. Li, J. Hofkens, H. Uji-I, D. Beljonne, T. Rasing and A. E. Rowan, Adv. Mat., 2013, 25, 2084 CrossRef CAS PubMed; (b) J. Xu, S. Semin, J. Cremers, L. Wang, M. Savoini, E. Fron, E. Coutino, T. Chervy, C. Wang, Y. Li, H. Liu, Y. Li, P. Tinnemans, P. H. J. Kouwer, T. W. Ebbesen, J. Hofkens, D. Beljonne, A. E. Rowan and T. Rasing, Adv. Opt. Mater., 2015, 3, 948 CrossRef CAS.
- SHG efficiencies usually are compared with a standard material such as α-quartz, KDP (KH2PO4) or urea. KDP has an efficiency 16 times that of α-quartz, whereas urea has an efficiency 400 times that of α-quartz.
- An example of non-racemic polymoprhism was reported: P. Dechambenoit, S. Ferlay, M. W. Hosseini and N. Kyritsakas, Chem. Commun., 2007, 4626 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, crystal structure description, additional figures. CCDC 1452830–1452832 (α-phase, β-phase, L) contain the supplementary crystallographic data for this paper. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ce00366d |
| This journal is © The Royal Society of Chemistry 2016 |