
In the searching for zwitterionic intermediates on reaction paths of [3 + 2] cycloaddition reactions between 2,2,4,4-tetramethyl-3-thiocyclobutanone S-methylide and polymerizable olefins†
Radomir Jasiński
Cracow University of Technology, Institute of Organic Chemistry and Technology, Warszawska 24, 31-155 Cracow, Poland. E-mail: radomir@chemia.pk.edu.pl
First published on 18th November 2015
Abstract
DFT calculations show that – both in a weakly polar and in a strongly polar medium – the [3 + 2]-cycloaddition of 2,2,4,4-tetramethyl-3-thiocyclobutanone S-methylide with nitroethene takes place according to a polar, two-step mechanism with a zwitterionic intermediate. On the other hand, a similar reaction with less electrophilic t-butyl methacrylate is a single-step process. However, zwitterionic structures with “extended” conformation may be formed in both cases considered here, in a reaction path competitive to cycloaddition.
[3 + 2]-cycloaddition reactions comprise the most universal synthesis method of 5-membered heterocylic compounds.1–3 Practically any compound of this type may be synthesised using these reactions. At the same time, the latest reports clearly undermine the entrenched view of a single step mechanism of cycloaddition reactions as the only possible mechanism, regardless of structure of the reagents. Mechanistic aspects of the [3 + 2]-cycloaddition reaction draw ever increasing attention of organic chemists: it has been shown recently that a two-step, zwitterionic mechanism governs [3 + 2]-cycloaddition reactions between N-methylnitrone and fluorinated alkenes,4 azomethine ylides and dialkyl-2,3-dicyanobut-2-enedioates,5 di(tert-butyl)diazomethane and 1,2-bis(trifluoromethyl)ethene-1,2-dicarbonitrile6 and between nitrile N-oxides and electron-rich alkenes.7
Some time ago, Huisgen i Mlostoń8 advanced a hypothesis that reactions of 2,2,4,4-tetramethyl-3-thiocyclobutanone S-methylide (1) with polymerizable olefins, such as e.g. t-butyl methacrylate (2) and nitroethene (5) (Scheme 1) take place according to a similar mechanism. They based their hypothesis on the fact that they identified some amounts of polymers of the aforemented alkenes in post-reaction mixtures. They assumed that the polymerisation process is initiated by zwitterions 3 and 6. However, they were unable to obtain any proof of this assumption. At the same time, taking into account the clearly electrophilic nature of dipolarophiles and the unequal shielding of reaction centres in both reagents, the zwitterionic mechanism of both considered reactions may be considered as probable. Complex quantum-chemical studies were performed within the presented work in order to provide an answer to the aforementioned doubts.9
 | ||
| Scheme 1 Postulated, zwitterionic mechanisms for [3 + 2] cycloadditions between 2,2,4,4-tetramethyl-3-thiocyclobutanone S-methylide 1 and polymerizable olefins. | ||
Firstly, electronic properties of the reagents were characterized10 (see Table S1 in ESI†). It turned out that ylid 1 should be classified as a member of the group of strongly nucleophilic reagents (N > 4 eV). On the other hand, both analysed alkenes have a distinct electrophilic nature. Nitroethene is definitely the stronger electrophile of the two compounds (ω > 2.5 eV). During analysis of differences in global electrophilicity within individual reagent pairs it may be noted that the 1 + 2 reaction will be of weakly polar nature. On the other hand, the 1 + 5 cycloaddition should be interpreted as a clearly polar process. The aforementioned fact explains the correlation between the nature of local interactions and the observed regioselectivity of cycloaddition. In case of the weakly polar 1 + 2 cycloaddition, the nature of local nucleophile–electrophile interactions10 does not have a decisive influence on the course of reaction and its regioselectivity is decided by steric factors. As a consequence, the reaction leads to a relatively less sterically crowded 4,4-disubstituted thiolane 4. On the other hand, in case of the strongly polar 1 + 5 process, regioselectivity is decided by interactions between the strongly nucleophilic, terminal carbon atom of ylide 1 and the strongly electrophilic centre on the nitroethene β atom (see ESI†). This leads to 3-nitrothiolane 7 as the only reaction product.
Analysis of reactivity indices enables the nature of interactions in an elementary act of cycloadditions to be described. However, this does not allow one to diagnose the nature of critical structure along the path of reagents conversion into the adducts. Simulations of the energy profiles of the reaction had to be performed in order to obtain such a diagnosis.
It turned out that in a toluene medium, the [3 + 2]-cycloaddition process 1 + 2 takes place as a single step process (Fig. 1). It requires an activation barrier in excess of 25 kcal mol−1 to be overcome (see ESI†). During the transition state (TSA) of this reaction, both bonds σ required for formation of the heterocyclic ring are formed (Fig. 2). The degree of their progression is, however, weakly varied (r ≈ 2.55 Å). The weakly polar nature of the transition stage is additionally confirmed by the low value of the GEDT index11 (below 0.1e). This is understood taking into account the previously discussed electronic properties of reagents. However, a reaction in which a zwitterionic adduct with “extended” conformation (8) is formed may compete with the cycloaddition process. This compound is not, however, a stable configuration from the thermodynamic point of view and may easily decompose into individual reagents, which may then substantially irreversibly (−ΔG > 40 kcal mol−1) bind and form closed cycloadducts. However, it should be noted that even such a labile intermediate may initiate the polymerisation process mentioned by the Authors of work.8 In general, the mechanism of transformations taking place during the 1 + 2 reaction should be illustrated as presented in Scheme 2.
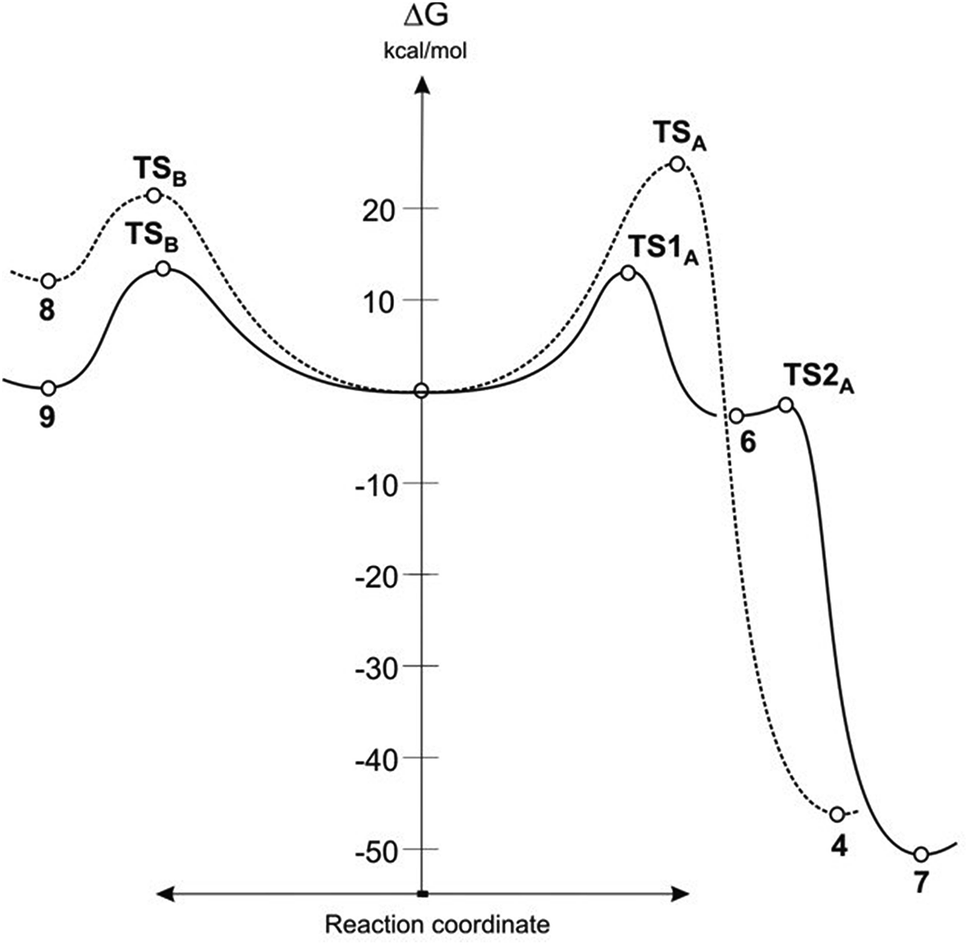 | ||
| Fig. 1 Gibbs free energy profiles for reaction between ylide 1 and alkenes 2 and 5 according to B3LYP/6-31G(d) calculations. | ||
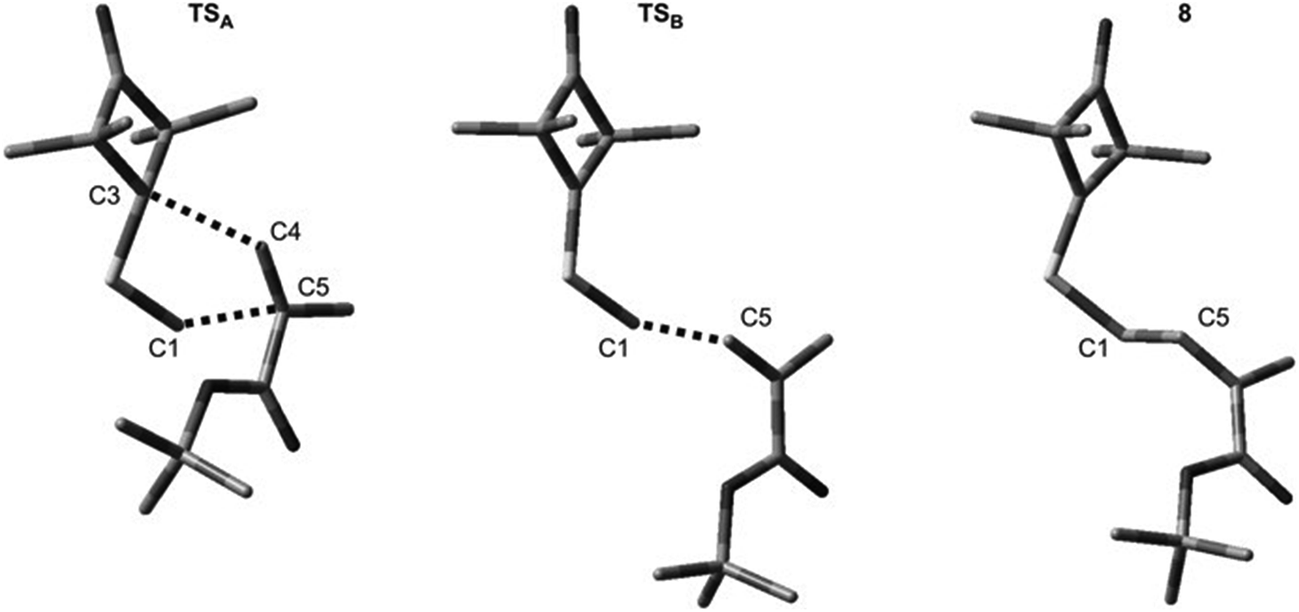 | ||
| Fig. 2 Key structures for reaction between ylide 1 and tert-butyl methacrylate 2 according to B3LYP/6-31G(d) calculations. | ||
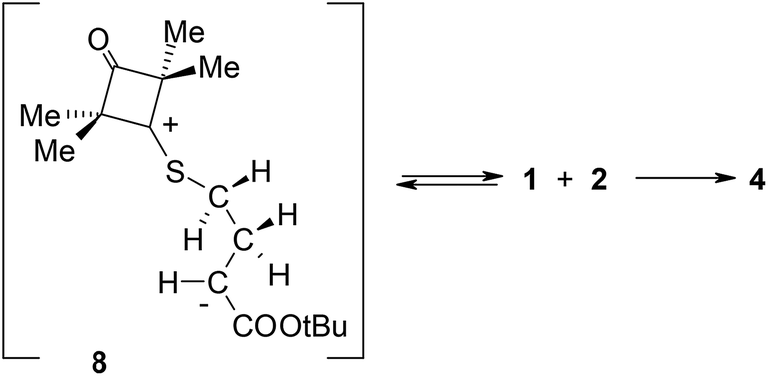 | ||
| Scheme 2 Reaction between ylide 1 and tert-butyl methacrylate 2 according to DFT calculations. | ||
DFT calculations show that a similar cycloaddition reaction of nitroethene 5 as dipolarophile, in toluene, takes place according to a completely different mechanism (Fig. 1). The first stage of the reaction includes formation of the pre-reaction complex (LMA). This is related to a certain drop in enthalpy of the reacting system. No chemical bonds are yet formed within LMA, but reaction centres are oriented in a manner ensuring maximum favourable interactions during the cycloaddition process. The next critical point along the path of reagent conversion includes the TS1A transition state. Reaching this transition state is a much less energetic process than in the case of the 1 + 2 reaction. Only one new bond is formed within TS1A σ – namely the bond between the terminal carbon atom of ylide 1 and the carbon atom of β nitroethene 5. Further conversion of the reacting system along the reaction coordinate leads to formation of adduct 6. It has a zwitterionic nature, indicated by the value of the GEDT index (0.5e). Zwitterion 6 cyclisation takes place by overcoming a small activation barrier and leads through the TS2A transition state. The second bond required for formation of the heterocyclic ring is formed within this transition state (Fig. 3).
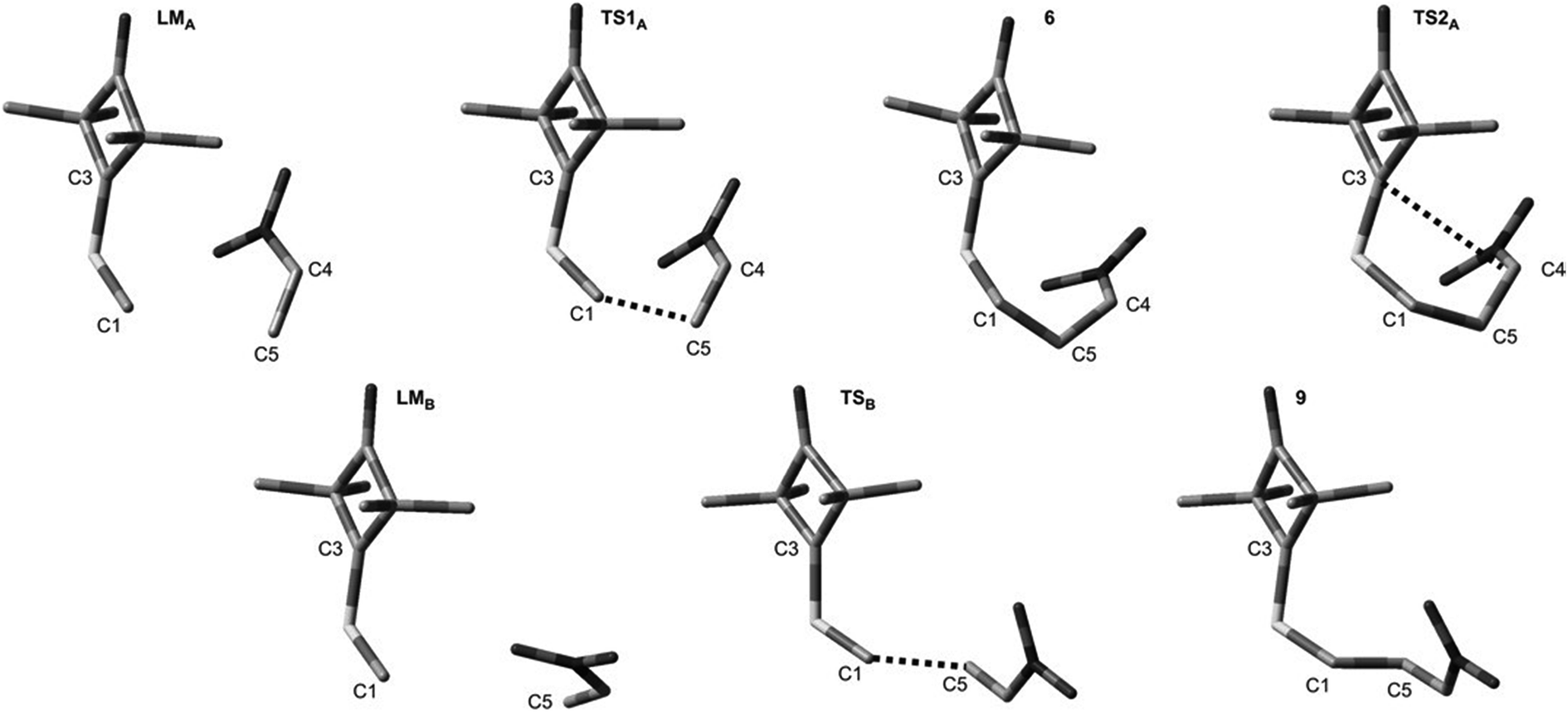 | ||
| Fig. 3 Key structures for reaction between ylide 1 and nitroethene 5 according to B3LYP/6-31G(d) calculations. | ||
DFT calculations show that zwitterions with the “extended” conformation (9) may also be formed in the 1 + 5 reaction. This process should be considered as kinetically allowed. In general, the mechanism of transformations taking place during the 1 + 5 reaction should be illustrated as presented in Scheme 3.
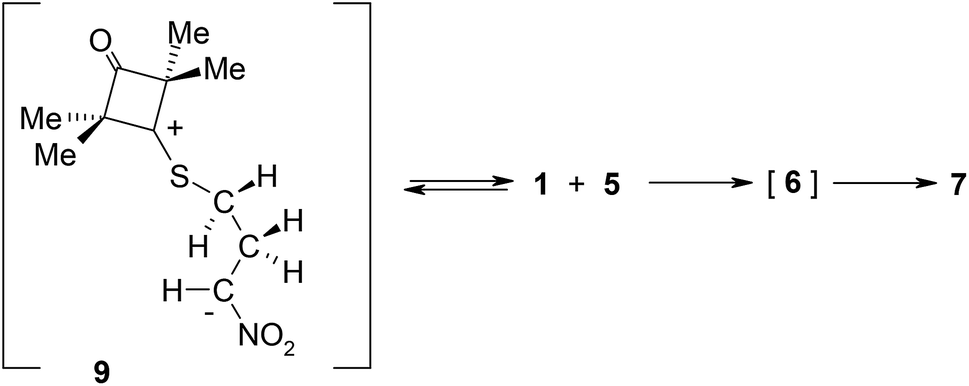 | ||
| Scheme 3 Reaction between ylide 1 and nitroethene 5 according to DFT calculations. | ||
A similar image of the analysed cycloaddition reaction is provided by DFT calculations taking into account the presence of a strongly polar solvent (nitromethane). In particular, the geometries of critical structures are virtually identical to those observed in toluene. Only the quantitative description of energy profiles is slightly different (see ESI†).
Finally, it should be noted, that to verify of potentially possible, alternative biradical mechanism, for both reactions UB3LYP/6-31G(d) calculations has been also performed. These calculations suggest identical reaction mechanism as in the case of B3LYP/6-31G(d) calculations. Additionally, calculations at UB3LYP/6-31G(d) theory level confirmed, that all transition states as well as intermediates have non-radical character. This is confirmed by 〈S2〉 values which in all cases are equal 0.00.
Conclusion
DFT calculations for various levels in theory all indicate that during the reaction of 2,2,4,4-tetramethyl-3-thiocyclobutanone S-methylide (1) with electrophilic olefins, zwitterionic structures may be formed. However, in case of reaction with the less electrophilic t-butyl methacrylate (2), only the “extended” conformation compounds may be formed, which do not undergo conversion into cycloadducts. On the other hand, a zwitterion is an intermediate along the path of reagents conversion into adducts in the reaction of the less electrophilic nitroethene.Acknowledgements
The generous allocation of computing time by the regional computer center “Cyfronet” in Cracow (Grant No. MNiSW/Zeus_lokalnie/PK/009/2013) are gratefully acknowledged.Notes and references
- T. Hashimoto and K. Maruoka, Chem. Rev., 2015, 115, 5366–5412 CrossRef CAS PubMed.
- Chemistry of heterocyclic compounds, ed. A. Padwa and W. H. Pearson, Wiley, New York, 2003, vol. 59 Search PubMed.
- Chemistry of heterocyclic compounds, ed. A. Padwa, Wiley, New York, 1990, vol. 49 Search PubMed.
- H. Wójtowicz-Rajchel and H. Koroniak, J. Fluorine Chem., 2012, 135, 225–230 CrossRef.
- A. F. Khlebnikov, A. S. Koneva, A. A. Virtseva, D. S. Yufit, G. Mlostoń and H. Heimgartner, Helv. Chim. Acta, 2014, 97, 453–470 CrossRef CAS.
- R. Huisgen, P. Pöchlauer, G. Mlostoń and K. Polsborn, Helv. Chim. Acta, 2007, 90, 983–998 CrossRef CAS.
- S. Krompiec, P. Bujak, J. Malarz, M. Krompiec, Ł. Skórka, T. Pluta, W. Danikiewicz, M. Kania and J. Kusz, Tetrahedron, 2012, 68, 6018–6031 CrossRef CAS.
- R. Huisgen, J. Penelle, G. Mlostoń, A. Buyle-Padias and H. K. Hall Jr, J. Am. Chem. Soc., 1992, 114, 266–274 CrossRef CAS.
- All calculations reported in this work were performed on an SGI-Altix 3700 computer in the CYFRONET regional computational centre in Cracow. Hybrid functional B3LYP with the 6-31G(d) basis set included in the GAUSSIAN 09 package was used. Reports published recently show that the B3LYP/6-31G(d) theoretical level successfully was used for the analysis of mechanistic aspects of one-step as well as two-step [3 + 2] cycloadition reactions – see: (a) R. Jasiński, M. Ziółkowska, O. M. Demchuk and A. Maziarka, Cent. Eur. J. Chem., 2014, 12, 586–593 CrossRef; (b) A. Szczepanek, E. Jasińska, A. Kącka and R. Jasiński, Curr. Chem. Lett., 2015, 4, 33–44 CrossRef; (c) R. Jasiński, Tetrahedron, 2013, 69, 927–932 CrossRef; (d) L. R. Domingo, M. J. Aurell and P. Perez, Tetrahedron, 2015, 71, 1050–1057 CrossRef CAS; (e) L. R. Domingo, M. J. Aurell and P. Perez, Tetrahedron, 2014, 70, 4519–4525 CrossRef CAS; (f) S. Emamian, J. Fluorine Chem., 2015, 178, 165–172 CrossRef CAS . Subsequently, similar simulations using more advanced B3LYP/6-31+G(d) theoretical level were performed. The calculations were carried out in the simulated presence of toluene (as model weak polar solvent) and nitromethane (as model strongly polar solvent). For the calculations of solvent effect on the reaction paths the polarizable continuum model (PCM) was used. Detailed informations about calculation procedure are available in ESI.†.
- For this purpose, reactivity indices theory was used (see ESI for computational details†). Similar approach was recently used successfully for interpretation of reaction course for many cycloaddition reactions – for example: (a) R. Jasiński, J. Fluorine Chem., 2015, 176, 35–39 CrossRef; (b) L. R. Domingo, M. Ríos-Gutiérreza and P. Pérez, Tetrahedron, 2015, 71, 2421–2427 CrossRef CAS; (c) S. Emamian, RSC Adv., 2015, 5, 72959–72970 RSC.
- L. R. Domingo, RSC Adv., 2014, 4, 32415–32428 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra20747a |
| This journal is © The Royal Society of Chemistry 2015 |