
Cyclometallated ruthenium(II) complexes with ditopic thienyl–NHC ligands: syntheses and alkyne annulations†‡
Xiaoke
Xie
and
Han Vinh
Huynh
*
Department of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore 117543, Republic of Singapore. E-mail: chmhhv@nus.edu.sg; Fax: +65 6779 1691; Tel: +65 6516 2670
First published on 12th October 2015
Abstract
A series of six [RuX(2-thienyl–NHC)(p-cymene)] complexes (9a–c and 10–14; X = halido) have been prepared via NHC directed C–H activation. All complexes have been fully characterized, and the molecular structures of four complexes are reported. The rotational barrier of the N-benzyl substituent in the bromido complex 9b has been measured by variable temperature 1H NMR spectroscopy. Preliminary reactivity studies of complex 12 with alkynes provided four new annulated aza-heterocyclic thiophenes.
Introduction
In the past few decades, transition metal-mediated C–H functionalizations have emerged as powerful tools for the synthesis of valuable products from inexpensive starting materials.1 Generally, the C–H activation is initiated by the binding of the transition metal to a directing group so that the adjacent C–H bond can be selectively activated to form metallacycles. The metallacycles then undergo subsequent reductive eliminations upon reaction with various substrates to produce the functionalized products.1i,2Commonly, weak σ and π donors are selected as directing groups for the C–H functionalizations in order to facilitate the reductive elimination.3 Catalytic C–H activation/alkyne annulations assisted by weaker donors have also been developed previously.23 On the other hand, strong carbon donors, such as N-heterocyclic carbenes (NHCs), are seldom used as directing groups due to their innate strong donating abilities, which would make the respective C–M bonds more inert toward reductive eliminations.4 As a result, only a few NHC-based metallacycles have been reported in a long period of time.5 A few research groups reported examples of NHC-directed alkyne annulations assisted by stoichiometric or catalytic amounts of ruthenium or rhodium complexes. Using this methodology functionalized azoles or azolium salts can be synthesized.6,7
Meanwhile, interest in the C–H functionalizations of heterocycles (Het-H functionalization)8 has increased tremendously as functionalized heterocycles have a plethora of applications in the fields of agrochemicals, functional materials and drugs.9 Among these, annulated thiophenes incorporated into aza-heterocycles have demonstrated unique medicinal properties. For example, annulated thiophenes that bear benzimidazole moieties exhibit promising cytotoxicity against cancer cells,10 and have potential as DNA intercalators. Furthermore, these compounds could have potential optical applications due to their planar structure and enhanced chemical stabilities.11 Therefore, it is desirable to explore new and facile synthetic approaches to annulated aza-heterocyclic thiophenes.
Herein, we report our preliminary study on thiophene annulation through C–H bond functionalization. Several cyclometalated Ru–thienyl–NHC complexes (NHC = imy or bimy) were synthesized and characterized. Subsequently, four annulated aza-heterocyclic thiophenes (aza-heterocyclic = imidazolium or benzimidazolium) were easily obtained from the metallacycles via reductive eliminations on treatment with different alkynes.
Results and discussion
Synthesis of the ligand precursors
The ligand precursors 3–8 were obtained by a two-step sequence that is summarized in Scheme 1. The Cu(I) catalyzed Ullmann-type coupling of imidazole or benzimidazole with 2-bromothiophene in the presence of L-proline afforded the mono-arylated compounds 1 and 2 in good yields (Scheme 1).12 The copper salts generated as byproducts in this procedure can be easily separated from the organic products by a modified extractive work-up. Accordingly, these salts were converted into water-soluble copper–ammine complexes by addition of aqueous ammonia to the reaction mixture.13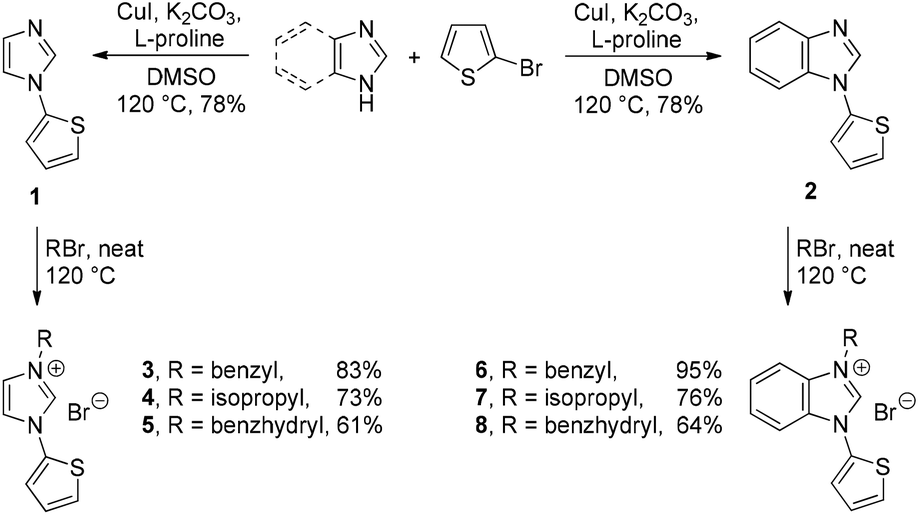 | ||
| Scheme 1 Two-step synthesis of ligand precursors 3–8. | ||
Subsequent N-alkylations of compounds 1 and 2 with excess alkyl bromides give rise to the functionalized precursor salts 3–8 in moderate to good yields (Scheme 2). All these compounds are soluble in chlorinated solvents such as dichloromethane and chloroform. The stepwise decrease in yields going from 3 to 5 can be explained by the increasing bulkiness of the alkylation reagent, and the same trend was observed for the yields of 6 to 8, respectively.
 | ||
| Scheme 2 Synthesis of ruthenium metallacycles 9a, 10–14. | ||
Synthesis and characterization of the Ru(II) NHC metallacycles
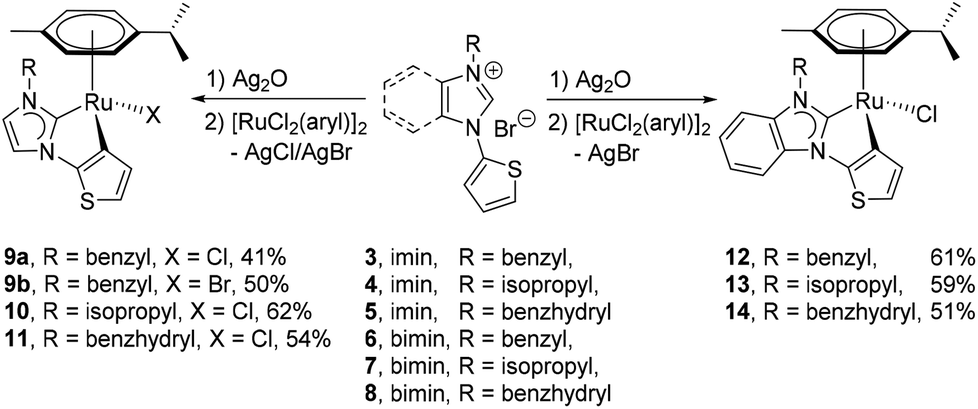
The preparation of complexes 9a, 9b and 10–14 were carried out by treating the precursor salts 3–8 with Ag2O and subsequent addition of [Ru(p-cymene)Cl2]2 at ambient temperature (Scheme 2). The in situ Het-H activation is anticipated to occur after the silver–carbene mediated ruthenation.14 Since the formation constant for AgBr is larger than that for AgCl, the chlorido ligand is expected to be retained at the ruthenium centre, while the carbene transfer is expected to be driven by AgBr precipitation. Surprisingly, the reaction involving salt 3 yielded the bromido complex 9b as the major product in 50% yield, while the expected chlorido complex 9a was obtained in only 41% due to halide scrambling (vide infra). In order to complete the halido series, the iodido complex 9c was also prepared in quantitative yield by a chlorido–iodido ligand exchange reaction by treating 9a with excess lithium iodide (Scheme 3).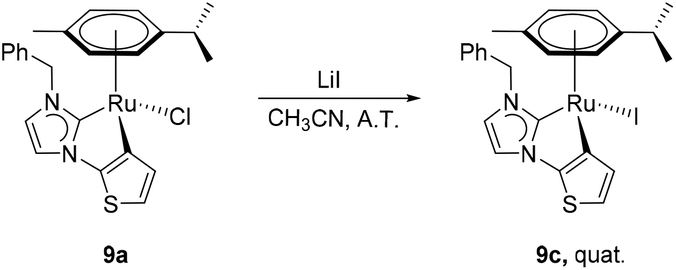 | ||
| Scheme 3 Synthesis of iodido complex 9c. | ||
In contrast, halide scrambling did not pose a problem in the complexation of salts 4 and 5 that bear more bulky N-substituents. Here, the chlorido complexes were formed predominantly, and only trace amounts of the bromido complexes were detected. A similar observation was made for benzimidazolium salts 6–8, where the chlorido products 12–14 were obtained as major products regardless of the N-substituents.
Complexes 9–14 were isolated in moderate to good yields as microcrystalline, yellow solids, which are readily soluble in most organic solvents such as MeOH, DMSO, DMF and CH3CN, but insoluble in nonpolar solvents, such as hexane, diethyl ether and toluene. They are stable for months in solutions under aerobic conditions at ambient temperature. However, these complexes were found to slowly decompose during purification attempts of the crude products by column chromatography on silica gel. Furthermore, when pure samples of the complexes were dissolved in unstabilized, deuterated chloroform, a color change of the respective solutions from yellow to green was observed after several hours. Both observations indicate the sensitivity of these complexes towards slightly acidic materials, where easy acidolysis of the Ru–Cthio bonds can be anticipated.2b,15 To circumvent this process, all the crude complexes were subsequently purified using neutral alumina, and all the NMR samples were prepared by using CDCl3 that was passed through a short pad of alumina to remove traces of hydrochloric acid.
The formation of complexes 9–14 was confirmed by multinuclear NMR spectroscopy and mass spectrometry. In the 1H NMR spectra, the absence of downfield signals characteristic of the azolium salts and the presence of resonances for both the N-substituents and the bimy/imi ligands indicated the formation of the expected complexes. In each complex, the loss of symmetry upon cyclometalation results in the diastereotopy of the p-cymene protons, which resonate as four doublets ranging from 6.0 to 5.0 ppm with a vicinal coupling constant of around 3J(H,H) = 6 Hz.
The two benzylic protons in the chlorido complex 9a give rise to a singlet at 5.73 ppm in the 1H NMR spectrum. In contrast, the respective spectra of the bromido and iodido complexes 9b and 9c, respectively, show two sets of doublets with resonances at 5.74 and 5.67 ppm (for 9b) or at 5.73 and 5.68 ppm (for 9c), with geminal coupling constants of 2J(H,H) = 17 Hz indicative of diastereotopic benzylic protons.
This observation supports the view that the rotational freedom of the N-benzyl substituent is largely affected by the nature of the halido ligands. In complex 9a, the rotational barrier is anticipated to be low due to the presence of the smaller chlorido ligand. The successive replacement of the latter by the larger bromido and iodido ligands leads to an increasingly difficult rotation around the N–CH2 single bond in complexes 9b and 9c, respectively, as a result of steric crowding around the metal center. This in turn is manifested in the diastereotopic splitting of the initial singlet observed for 9a into two doublets in the spectra of 9b/c each with half integral values.
To get more insights into the relative rotational barriers for complexes 9a–c, a series of variable temperature (VT) 1H NMR experiments were carried out in CDCl3. Even at a low temperature of 233 K, fast exchange between the two benzylic protons was still observed for the chlorido complex 9a, indicating that the activation energy for the rotation for the benzyl group in 9a is very low.16 Another VT 1H NMR study was carried out for the bromido complex 9b (Fig. 1). Here, a minimal proton exchange was noted at 268 K with a maximum chemical shift separation of Δv = 45 Hz. Upon slow warming of the sample, the initial separate signals start to merge at ∼328 K, and coalescence for the diastereotopic benzylic protons occurs at ∼343 K allowing for an estimation of the rotational barrier as 16.9 kcal mol−1.17 In contrast, the benzylic proton signals for iodido complex 9c did not coalesce in this temperature range, providing evidence that proton exchange in this case is much slower even at higher temperatures.16 Thus, a higher rotational barrier is to be expected for 9c.
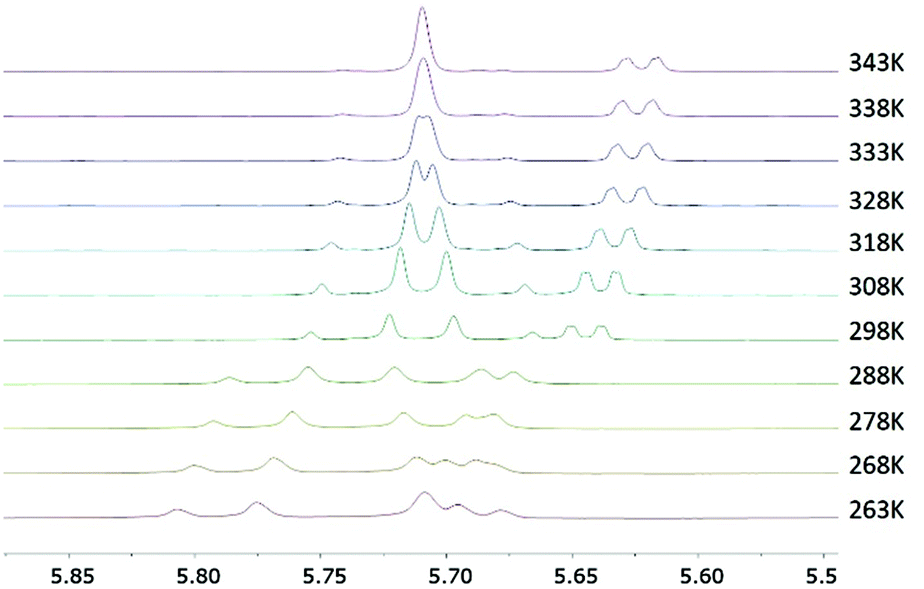 | ||
| Fig. 1 Section of the variable-temperature 1H NMR spectral plot of complex 9b in CDCl3. | ||
In the 13C NMR spectra for complexes 9a–14, the carbene signals resonate at 188.9, 188.4, 188.3, 186.5, 189.6, 202.6, 200.8 and 203.3 ppm, respectively. These chemical shifts are notably more downfield compared to 13Ccarbene signals of related [RuCl2(η6-p-cymene)(NHC)] analogues, which are not cyclometalated.18 The formal replacement of a chlorido ligand by a supposedly stronger anionic carbon donor results in a less Lewis acidic ruthenium centre leading to an expected downfield shift of the carbene carbon.19 Moreover, the carbon donors of the thienyl moiety of complexes 9a–14 were identified at 155.7, 154.8, 154.9, 155.2, 155.7, 153.7, 154.3 and 154.6 ppm, respectively, by means of 2D NMR spectroscopy.
Molecular structures
X-ray diffraction analyses were carried out on single crystals of 9a, 9b, 11 and 12, which were obtained by slow diffusion of diethyl ether into their concentrated solutions in dichloromethane (Fig. 2). Although all complexes are chiral-at-metal, they expectedly crystallized in racemic forms due to the lack of chiral induction from the achiral ligands.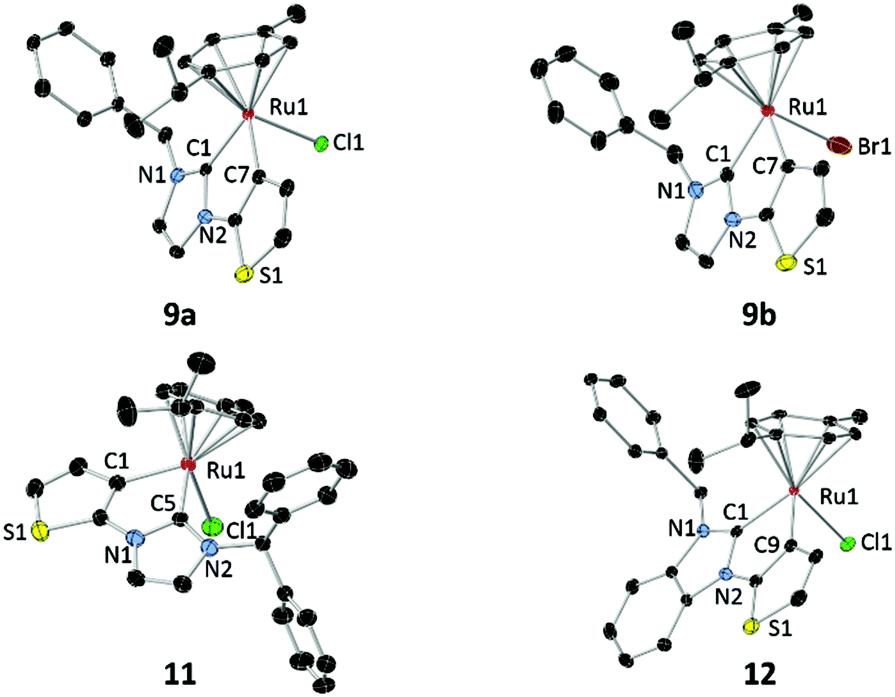 | ||
| Fig. 2 Molecular structures of 9a, 9b, 11 and 12 showing 50% probability ellipsoids. Solvent molecules and hydrogen atoms are omitted for clarity. | ||
In each of the molecular structures, the Ru atom lies in a pseudo-octahedral environment. The half sandwich complexes adopt a piano-stool configuration with the p-cymene ligand as the “stool” and the “tripod” is composed of the chlorido, NHC and thienyl donors. The N-substituents point away from the p-cymene ligand in order to circumvent steric repulsion. Cyclometalation leads to formation of planar tricyclic (imy) or tetracyclic (bimy) metallacycles incorporating both NHC and thienyl rings in each of the structures. The dihedral angles defined by the metallacycles and the p-cymene planes are 55.3° (α1) for 9a, 55.8° (α2) for 9b, 50.9° (α3) for 11 and 55.5° (α4) for 12. The α3 angle of 11 is significantly smaller than α1 of 9a, indicating the greatest repulsion between the more bulky N-benzhydryl substituent and the p-cymene ligand in 11, which forces the metallacyclic plane closer to the p-cymene ligand. On the other hand, α1 and α2 are comparable despite the presence of different halido ligands in complexes 9a and 9b. Although the halido ligands have a great influence on the rotation of the N-benzyl group (vide supra), their impact on the p-cymene is expectedly minimal as they point away from the aryl ligand. Similarly, benzannulation of the NHC ring does not result in any significant distinction between the α1 and α4 angles in 9a and 12, respectively.
Selected bond lengths and angles are listed in Table 1. The Ru–Cthio bond lengths fall in the range of 2.0555–2.0646 Å. These values do not deviate significantly from other Ru complexes bearing cyclometalated thienyl moieties.20 The Ru–Ccarbene bond lengths for complexes 9a and 9b averaging to 2.046 Å are identical within 3σ. However, these bonds for complexes 11 (2.0353(18) Å) and 12 (2.024(2) Å) are slightly shorter, making their ruthenium centers sterically more congested.
| Bond parameter | 9a | 9b | 11 | 12 |
|---|---|---|---|---|
| Ct denotes centroid of the p-cymene ring. | ||||
| Ru–Cthio | 2.0660(16) | 2.0646(19) | 2.0555(19) | 2.0575(19) |
| Ru–Ccarbene | 2.0460(15) | 2.047(2) | 2.0353(18) | 2.024(2) |
| Ru–halide | 2.4301(4) | 2.493(10) | 2.4260(8) | 2.4203(7) |
| Halide–Ru–Cthio | 87.52(4) | 86.23(6) | 90.11(5) | 87.45(6) |
| Halide–Ru–Ccarbene | 87.22(4) | 86.12(6) | 84.23(5) | 87.50(5) |
| Cthio–Ru–Ccarbene | 77.30(6) | 77.07(8) | 76.79(7) | 77.49(8) |
| Ru–Ct | 1.7181(2) | 1.7265(2) | 1.7237(3) | 1.7317(5) |
The ruthenium–centroid (Ru–Ct) distances also reflect the stereoelectronic properties of the respective ligands. The replacement of the chlorido (9a) with a bromido ligand (9b) leads to elongation of this bond parameter, which is consistent with the greater spatial demand of the larger bromido ligand. Similarly, substitution of the N-benzyl (9a) with the bulkier benzhydryl wing-tip (11) also results in a longer ruthenium–centroid distance. However, the most significant difference occurs upon benzannulation of the NHC ligand. Here, electronic factors are at play, since complexes 9a and 12 are sterically very similar. The imidazolin-2-ylidene ligand in 9a is more strongly donating and enhances back donation from the metal to the p-cymene ligand, which in turn results in a shorter Ru–Ct separation.
Proposed mechanism of C–H activation
The ease of C–H activation that did not require the addition of an excessive external base in our one-pot protocol was surprising and called for a more detailed investigation. Thus, 1H NMR tracing of the C–H activation step was attempted (see the ESI‡). For this purpose, salt 4 was chosen due to its simpler NMR spectroscopic signature, and it was reacted with silver(I) oxide in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio to form the respective silver–NHC complex I (Scheme 4, step 1). The compound was isolated as a white powder, and characterized by 1H NMR spectroscopy. Subsequently, a mixture of the pre-isolated silver-species and half-equivalent of the Ru-dimer was taken up in CD2Cl2 in a NMR tube for monitoring (step 2). Fast transmetallation to ruthenium was indicated by an immediate precipitation of silver halide. More importantly, resonances due to the silver–carbene and the ruthenium dimer decreased significantly, and signals including a multiplet at 2.74 ppm, a singlet at 2.00 ppm and two doublets at 1.50 and 1.23 ppm, of a new species bearing both cymene and NHC ligands emerged at ∼10 min into the reaction (Fig. S5‡). We assign this to the formation of the neutral dichlorido–RuII complex II as a major product. In addition, small amounts of the final product 10 can be observed as evidenced by two low intensity doublets at 0.91 and 0.77 ppm, respectively. However, the ratios of the compounds did not change significantly even after 4 h (Fig. S6 and S7‡). Since no external base was added at this stage, one may suppose that complexes II and 10 are in a dynamic equilibrium, which lies predominantly on the side of the dichlorido species II (step 3). In principle, the chlorido ligands could assist in the deprotonation of the thiophene moiety. However, this process is less favourable in an aprotic medium, i.e. CD2Cl2. The addition of 1 equiv. of silver(I) oxide led to an enhancement of the C–H activation process and after 3 h a compelling increase in the amount of 10 was detected (Fig. S8‡). After 7 h, the resonances from intermediate II have almost disappeared, while signals of complex 10 have become predominant (Fig. S9‡). In addition, a set of new signals attributed to free p-cymene appeared at 2.89 ppm (septet), 2.04 ppm (singlet) and 1.25 ppm (doublet) indicating partial decomposition upon prolonged standing in solution.
:1 ratio to form the respective silver–NHC complex I (Scheme 4, step 1). The compound was isolated as a white powder, and characterized by 1H NMR spectroscopy. Subsequently, a mixture of the pre-isolated silver-species and half-equivalent of the Ru-dimer was taken up in CD2Cl2 in a NMR tube for monitoring (step 2). Fast transmetallation to ruthenium was indicated by an immediate precipitation of silver halide. More importantly, resonances due to the silver–carbene and the ruthenium dimer decreased significantly, and signals including a multiplet at 2.74 ppm, a singlet at 2.00 ppm and two doublets at 1.50 and 1.23 ppm, of a new species bearing both cymene and NHC ligands emerged at ∼10 min into the reaction (Fig. S5‡). We assign this to the formation of the neutral dichlorido–RuII complex II as a major product. In addition, small amounts of the final product 10 can be observed as evidenced by two low intensity doublets at 0.91 and 0.77 ppm, respectively. However, the ratios of the compounds did not change significantly even after 4 h (Fig. S6 and S7‡). Since no external base was added at this stage, one may suppose that complexes II and 10 are in a dynamic equilibrium, which lies predominantly on the side of the dichlorido species II (step 3). In principle, the chlorido ligands could assist in the deprotonation of the thiophene moiety. However, this process is less favourable in an aprotic medium, i.e. CD2Cl2. The addition of 1 equiv. of silver(I) oxide led to an enhancement of the C–H activation process and after 3 h a compelling increase in the amount of 10 was detected (Fig. S8‡). After 7 h, the resonances from intermediate II have almost disappeared, while signals of complex 10 have become predominant (Fig. S9‡). In addition, a set of new signals attributed to free p-cymene appeared at 2.89 ppm (septet), 2.04 ppm (singlet) and 1.25 ppm (doublet) indicating partial decomposition upon prolonged standing in solution.
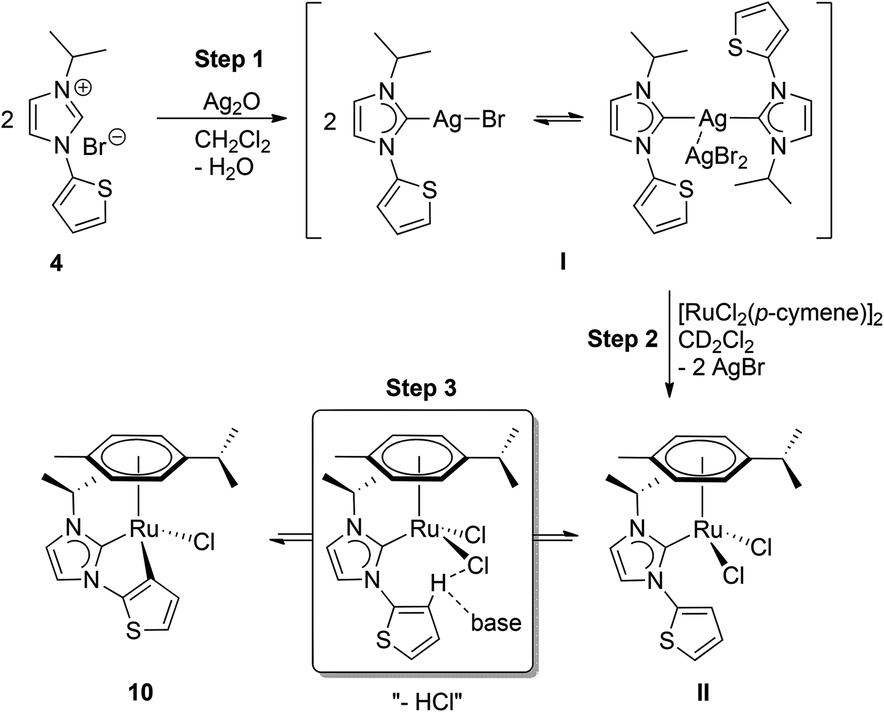 | ||
| Scheme 4 Proposed mechanism of cyclometalation. | ||
Overall, these observations are in line with the proposal that the C–H activation step is rate-determining and occurs likely base-assisted (e.g. CMD mechanism) as suggested in previous studies.23 Moreover, protic media (water is present in the one-pot procedure) and addition of an external base facilitate this step and drive the formation of the cyclometalated species.
Synthesis of aza-heterocyclic thiophenes
In a preliminary study, N-benzylbenzimidazolin-2-ylidene complex 12 was chosen to test its reactivity with alkynes. In boiling methanol, it smoothly reacted with an excess of different alkynes in the presence of excess KPF6 to give the cationic aza-heterocyclic thiophenes 15–18 (Table 2). The reaction of 12 with symmetrical and internal diaryl and dialkyl acetylenes generated 15 and 16 in good yields. For terminal alkynes, two possible isomers can be expected. Notably, a regiospecific annulation took place between 12 and the phenylacetylene, resulting solely in the 5-phenyl product 17 in 75% yield (vide infra).21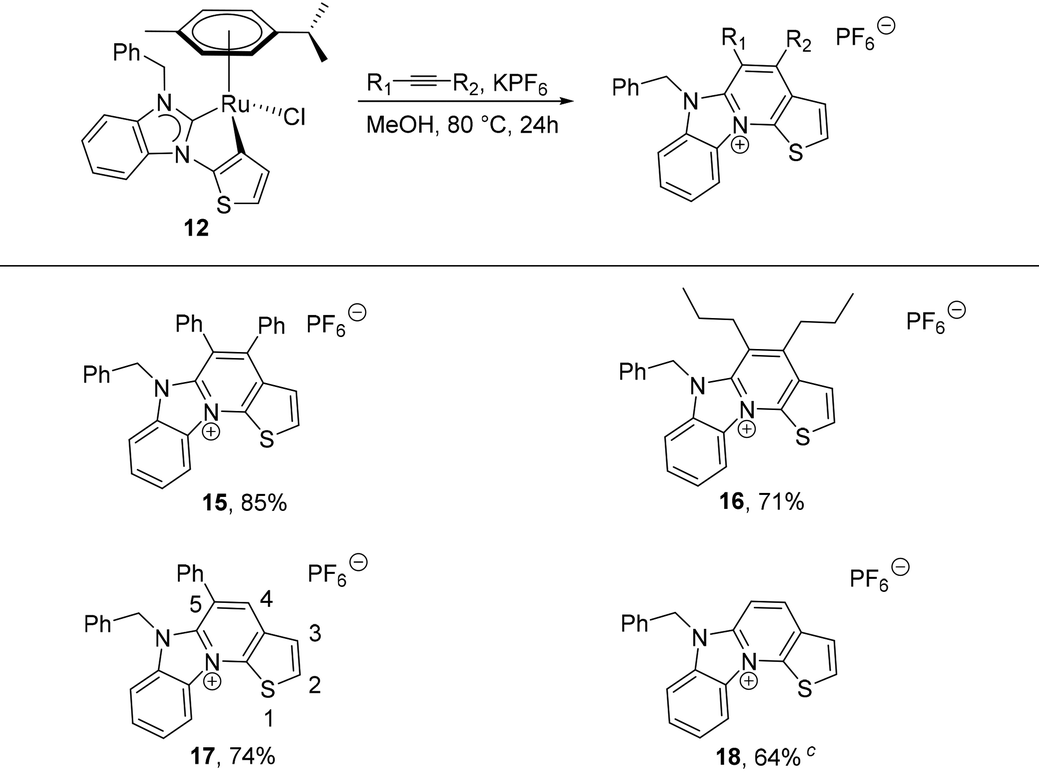
Finally, the 4,5-unsubstituted product 18 was generated in 64% yield from 12 and trimethylsilyl acetylene, which can be conveniently used in place of gaseous acetylene. In this case, a useful in situ desilylation reaction occurs, the mechanism of which is currently unclear, but will be subject to future studies. The proposed reaction pathway for the formation of the aza-heterocyclic thiophenes is similar to that reported by the groups of Pfeffer,22 Urriolabeitia20d and Ackermann23 (Scheme 5), according to which the reaction is initiated by alkyne insertion into the κ2-C,C bound ditopic thienyl–NHC ligand. This is followed by an intramolecular, reductive elimination step, which supposedly generates the intermediate ruthenium(0) adduct IV containing the new heterocycle. The latter is particularly short-lived and quickly collapses under liberation of the new aza-heterocyclic thiophene cation.
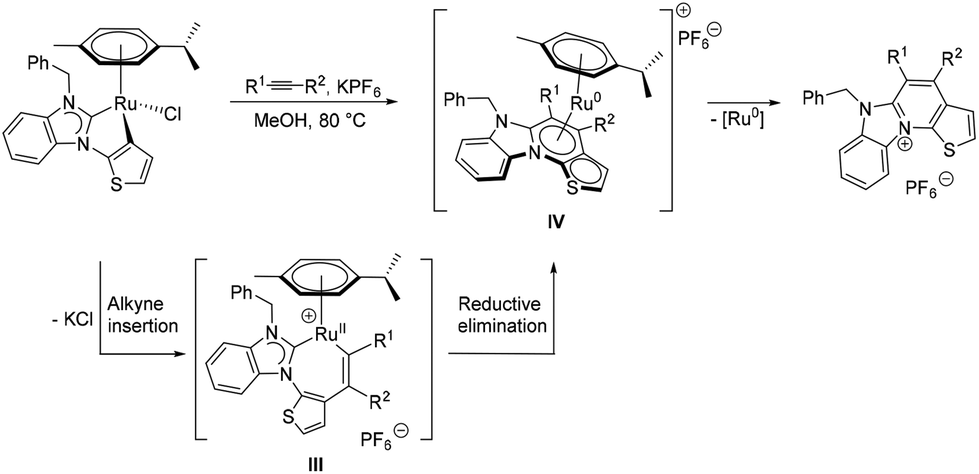 | ||
| Scheme 5 Proposed pathway for the formation of aza-heterocyclic thiophenes. | ||
The intermediacy of ruthenium(0) species can be supported by the observed slow precipitation of ruthenium black during the course of the reaction. Moreover, it should be noted, that complexes reminiscent of II, but derived from cyclometalated N- and O-donors, have been structurally characterized by the aforementioned groups.20d,22,23a In the migratory insertion step of phenylacetylene, the alkyne tends to bind to the ruthenium(II) centre at the more substituted position (i.e. R1 = Ph and R2 = H). This generates a more stable intermediate III containing a sterically better protected metal centre, which eventually results in the regioselectivity observed in the synthesis of 17.
Conclusions
The NHC directed Het-H activation of the thienyl moiety has allowed for a rapid synthesis of RuII–thienyl–NHC metallacycles 9a–14. All complexes are the first examples of cycloruthenated NHC complexes incorporating thienyl moieties and have been fully characterized. The C–H activation of the thienyl substituent is rate determining and occurs base-assisted and favourably in protic media. The rotational barrier of the N-benzyl substituent in complexes 9a–c is halido dependent, and has been determined for the bromido complex 9b by VT 1H NMR spectroscopy. Moreover, alkyne annulations of complex 12 afforded four new annulated aza-heterocyclic thiophenes, which could have potential medical and optical applications. Work is in progress to further extend the substrate scope of the annulation reactions, to provide a more detailed mechanistic understanding and to explore catalytic variations of this reaction.Acknowledgements
The authors thank the National University of Singapore and “GSK-EDB Singapore Partnership for Green and Sustainable manufacturing” for financial support (WBS R143-000-513-592). Technical support from staff at the CMMAC of our department is appreciated.Notes and references
- For selected recent reviews, see: (a) C. Zhao, M. R. Crimmin, F. D. Toste and R. G. Bergman, Acc. Chem. Res., 2014, 47, 517–529 CrossRef CAS PubMed; (b) L. Ackermann, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed; (c) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef CAS PubMed; (d) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (e) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236–10254 CrossRef CAS PubMed; (f) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed; (g) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814–825 CrossRef CAS PubMed; (h) K. M. Engle, T. Mei, M. Wasa and J. Yu, Acc. Chem. Res., 2012, 45, 788–802 CrossRef CAS PubMed; (i) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147–1169 CrossRef CAS PubMed; (j) O. Daugulis, H. Do and D. Shabashov, Acc. Chem. Res., 2009, 42, 1074–1086 CrossRef CAS PubMed.
- (a) J. Dupont, C. S. Consorti and J. Spencer, Chem. Rev., 2005, 105, 2527–2572 CrossRef CAS PubMed; (b) M. Albrecht, Chem. Rev., 2010, 110, 576–623 CrossRef CAS PubMed; (c) Y. Han and G. Jin, Chem. Soc. Rev., 2014, 43, 2799–2823 RSC; (d) S. Rousseaux, S. I. Gorelsky, B. K. W. Chung and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 10692–10705 CrossRef CAS PubMed.
- For selected examples and reviews, see: (a) X. Wang, W. Gong, L. Fang, R. Zhu, S. Li, K. Engle and J. Yu, Nature, 2015, 519, 334–338 CrossRef CAS PubMed; (b) Y. Liu, H. Xu, W. Kong, M. Shang, X. Dai and J. Yu, Nature, 2014, 515, 389–393 CrossRef CAS PubMed; (c) S. Rakshit, C. Grohmann, T. Besset and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2350–2353 CrossRef CAS PubMed; (d) D. Shabashov and O. Daugulis, Org. Lett., 2005, 7, 3657–3659 CrossRef CAS PubMed; (e) G. Chotana, M. Rak and M. Smith III, J. Am. Chem. Soc., 2005, 127, 10539–10544 CrossRef CAS PubMed; (f) D. Colby, R. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624–655 CrossRef CAS PubMed.
- For reviews, see: (a) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS PubMed; (b) S. Diez-Gonzalez and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874–883 CrossRef CAS; (c) J. Vignolle, X. Cattoën and D. Bourissou, Chem. Rev., 2009, 109, 3333–3384 CrossRef CAS PubMed; (d) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS PubMed; (e) T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef PubMed.
- For selected examples, see: (a) K. F. Donnelly, R. Lalrempuia, H. Muller-Bunz and M. Albrecht, Organometallics, 2012, 31, 8414–8419 CrossRef CAS; (b) R. Corberan, M. Sanau and E. Peris, J. Am. Chem. Soc., 2006, 128, 3974–3979 CrossRef CAS PubMed; (c) R. Corberan, M. Sanau and E. Peris, Organometallics, 2006, 25, 4002–4008 CrossRef CAS; (d) R. Dorta, E. D. Stevens and S. P. Nolan, J. Am. Chem. Soc., 2004, 126, 5054–5055 CrossRef CAS PubMed.
- For the synthesis of azolium salts, see: (a) C. Ma, C. An, Z. Li, B. Li, B. Song, S. Xu and B. Wang, Organometallics, 2014, 33, 5164–5172 CrossRef CAS; (b) D. Ghorai and J. Choudhury, Chem. Commun., 2014, 50, 15159–15162 RSC; (c) D. Ghorai and J. Choudhury, ACS Catal., 2015, 5, 2692–2696 CrossRef CAS; (d) R. Q. Thenarukandiyil and R. J. Choudhury, Organometallics, 2015, 34, 1890–1897 CrossRef CAS; (e) Q. Ge, B. Li, H. Song and B. Wang, Org. Biomol. Chem., 2015, 13, 7695–7710 RSC; (f) A. Normand, S. K. Yen, H. V. Huynh, T. S. A. Hor and K. Cavell, Organometallics, 2008, 27, 3153–3160 CrossRef CAS.
- For the synthesis of azole compounds, see: K. Tan, R. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2001, 123, 2685–2686 CrossRef CAS PubMed.
- For reviews and examples, see: (a) L. Ackermann, R. Vicente and A. R. Kapdi, Angew. Chem., Int. Ed., 2009, 48, 9792–9826 CrossRef CAS PubMed; (b) X. Guo, X. D. Gu, Z. Wu and W. Zhang, Chem. Rev., 2015, 115, 1622–1651 CrossRef CAS PubMed; (c) A. Maehara, H. Tsurugi, T. Satoh and M. Miura, Org. Lett., 2008, 10, 1159–1162 CrossRef CAS PubMed; (d) T. Mukai, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2009, 74, 6410–6413 CrossRef CAS PubMed; (e) J. Lewis, R. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2007, 129, 5332–5333 CrossRef CAS PubMed.
- (a) A. F. Pozharskii, A. T. Soldatenkov and A. R. Katrizky, Heterocycles in Life and Society: An Introduction to Heterocyclic Chemistry, Biochemistry and Applications, John Wiley & Sons, Ltd., Chichester, UK, 2nd edn, 2011 CrossRef; (b) L. D. Quin and J. A. Tyrell, Fundamentals of Heterocyclic Chemistry: Importance in Nature and in the Synthesis of Pharmaceuticals, John Wiley & Sons, Inc., New York, 2010 Search PubMed.
- M. Hranjec, I. Piantanida, M. L. Šuman, K. Pavelić and G. J. Karminski-Zamola, J. Med. Chem., 2008, 51, 4899–4910 CrossRef CAS PubMed.
- (a) T. Mori, T. Nishimura, T. Yamamoto, I. Doi, E. Miyazaki, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2013, 135, 13900–13913 CrossRef CAS PubMed; (b) H. Choi, S. Paek, J. Song, C. Kim, N. Cho and J. Ko, Chem. Commun., 2011, 47, 5509–5511 RSC.
- D. Ma and Q. Cai, Org. Lett., 2003, 5, 3799–3802 CrossRef CAS PubMed.
- See the ESI‡ for details.
- H. M. J. Wang and J. B. Lin, Organometallics, 1998, 17, 972 CrossRef CAS.
- (a) A. D. Ryabov, Chem. Rev., 1990, 90, 403 CrossRef CAS; (b) L. Mercs, A. Neels, H. Stoeckli-Evans and M. Albrecht, Inorg. Chem., 2011, 50, 8188–8196 CrossRef CAS PubMed.
- See the ESI‡ for details.
- The following approximate equation was used for the calculation of ΔG‡:ΔG‡ = 1.98Tc (23.76 + ln (Tc/κ)) (calories per mol) where
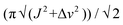 , Tc = 343 K, 2J(H,H) = 17 Hz and Δv = 45 Hz. According to: http://www.chem.wisc.edu/areas/reich/nmr/08-tech-03-dnmr.htm.
, Tc = 343 K, 2J(H,H) = 17 Hz and Δv = 45 Hz. According to: http://www.chem.wisc.edu/areas/reich/nmr/08-tech-03-dnmr.htm. - (a) P. S. Siah, X. Xiaoke, B. Gnanaprakasam, T. T. Dang, B. Ramalingam, H. V. Huynh and A. M. Seayad, RSC Adv., 2015, 5, 4434–4442 RSC; (b) X. Xie and H. V. Huynh, ACS Catal., 2015, 5, 4143–4151 CrossRef CAS.
- (a) D. Yuan and H. V. Huynh, Organometallics, 2012, 31, 405–412 CrossRef CAS; (b) J. C. Bernhammer and H. V. Huynh, Dalton Trans., 2012, 41, 8600–8608 RSC; (c) S. Guo, H. Sivaram, D. Yuan and H. V. Huynh, Organometallics, 2013, 32, 3685–3696 CrossRef CAS; (d) Q. Teng and H. V. Huynh, Inorg. Chem., 2014, 53, 10964–10973 CrossRef CAS PubMed.
- (a) A. J. Deeming, S. N. Jayasuriya, A. J. Arce and Y. D. Sanctis, Organometallics, 1996, 15, 786–793 CrossRef CAS; (b) C. Moorlag, O. W. Wolf, C. Bohne and B. O. Partric, J. Am. Chem. Soc., 2005, 127, 6382–6393 CrossRef CAS PubMed; (c) T. S. Lobana, G. Bawa, R. J. Butcher and M. Zeller, Organometallics, 2008, 27, 175–180 CrossRef CAS; (d) L. Cuesta, T. Soler and E. P. Urriolabeitia, Chem. – Eur. J., 2012, 18, 15178–15189 CrossRef CAS PubMed.
- The structure elucidation was carried out by 2D NMR spectroscopy. See the ESI.‡.
- H. C. L. Abbenhuis, H. M. Pfeffer and J.-P. Sutter, Organometallics, 1993, 12, 4464–4472 CrossRef CAS.
- (a) S. Warratz, C. Kornhaaß, A. Cajaraville, B. Niepötter, D. Stalke and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 5513–5517 CrossRef CAS PubMed; (b) S. Nakanowatari and L. Ackermann, Chem. – Eur. J., 2014, 20, 5409–5413 CrossRef CAS PubMed; (c) F. Yang and L. Ackermann, J. Org. Chem., 2014, 79, 12070–12082 CrossRef CAS PubMed; (d) L. Ackermann, A. V. Lygin and N. Hofmann, Angew. Chem., Int. Ed., 2011, 50, 6379–6382 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor F. Ekkehardt Hahn on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures; characterization data; 1H and 13C{H} NMR spectra; selected crystallographic data. CCDC 1424745 for 9a, 1424802 for 9b, 1424788 for 11 and 1424805 for 12. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qo00292c |
| This journal is © the Partner Organisations 2015 |