
Direct borylation of benzyl alcohol and its analogues in the absence of bases†
Zhi-Chao
Cao
a,
Fei-Xian
Luo
a,
Wen-Juan
Shi
a and
Zhang-Jie
Shi
*ab
aBeijing National Laboratory of Molecule Science (BNLMS) and Key Laboratory of Bioorganic Chemistry and Molecular Engineering of Ministry of Education, College of Chemistry and Green Chemistry Center, Peking University, Beijing, 100871, China. E-mail: zshi@pku.edu.cn
bState Key Laboratory of Organometallic Chemistry, Chinese Academy of Science, Shanghai, 200032, China
First published on 10th September 2015
Abstract
Direct borylation of arylmethanols to synthesize important and useful benzylboronates was carried out through Pd(OAc)2-catalyzed sp3 C–O activation. This borylation is compatible with various functional groups under mild conditions in the absence of any bases, offering an atom- and step-economical way to produce benzylboron compounds.
Alcohol is not only one of the most popular and abundant structural motifs in the natural and synthetic world,1 but also used as a versatile building block in organic synthesis.2 Conventional direct applications of alcohols are limited in C–C bond formation, majorly focused on Friedel–Crafts alkylation which featured harsh conditions.3 In recent decades, cross-coupling reactions by employing C–O electrophiles partially updated the application of alcohols in C–C bond formation. Various alcohol derivatives, such as carbamates,4 sulfonates,5 ethers,6 and so on, have been successfully applied in cross couplings. However, direct catalytic transformation of alcohols, other than relatively active allyl,7 propargyl,8 and allenyl9 alcohols, remained challenging due to the inertness of their C–O bonds and the Brønsted acidity of OH, resulting in the protonation of the active R–M intermediate in desirable transformations. As one of the most important alcohols, benzyl alcohol exhibited its limited reactivity in few transformations recently reported by Yi10 and us.11 Herein, we reported a reliable and simple method to synthesize arylmethylboronic esters from easily and commercially available arylmethanols via Pd catalysis under mild conditions.
Organoboron compounds are featured as one of the most important ingredients in Suzuki-Miyaura coupling12 and other important transformations13 due to their high functional group tolerance, easy work-up, non-toxicity, as well as their increasing commercial availability. They became the most versatile nucleophiles in organic synthesis, providing powerful ways to construct C–C, C–O and other functionalities.12,13 As one of the common and important organoboron compounds, not only do benzylboron compounds show their great application potential in traditional cross-couplings and organic synthesis,14 but they also exhibit special features. For example, Molander and co-workers discovered a new reaction, in which benzylic trifluoroborates released benzyl radicals, followed by carbon–carbon bond formation through a single-electron transfer pathway.15
Obviously, extensive methods to synthesize benzylboron compounds have a significant synthetic value. Indeed, many efforts have been carried out to meet such a goal.16 The most common method for preparation of benzylboron compounds is nucleophilic borylation with Grignard or lithium reagents, which held poor compatibility with functionalities and presented complicated manipulation.16a The development of Miyaura borylation starting from benzyl halides and sulfonic benzyl esters partially solved the problems (Scheme 1a).16f–l However, the use of the irritative benzyl halides and preparation of sulfonates limited their practical applications. Direct borylation of benzyl C–H bonds was ideal, unfortunately with poor functional group compatibility and limited substrate scope.16m,n Another beautiful pathway to produce the benzylboronic esters has been recently carried out by Wang and co-workers starting from the tosylhydrazone and diboron reagents without transition-metal catalysis.16o Reliable methods to furnish such important organoboron compounds from simple and easily available chemicals under mild conditions are still appealing. During the preparation of this manuscript, Martin and coworkers reported an elegant example to produce benzylboronates from benzyl ethers through C–O activation,16p Hartwig and co-workers realized the iridium catalyzed direct borylation of benzylic C–H bonds with high selectivity and broad substrate scope.16q Considering the importance of benzylboron compounds and easy availability of benzyl alcohols, we conceived a Pd catalytic system to carry out the direct borylation of benzyl alcohols to produce benzylboron reagents under mild conditions (Scheme 1b).
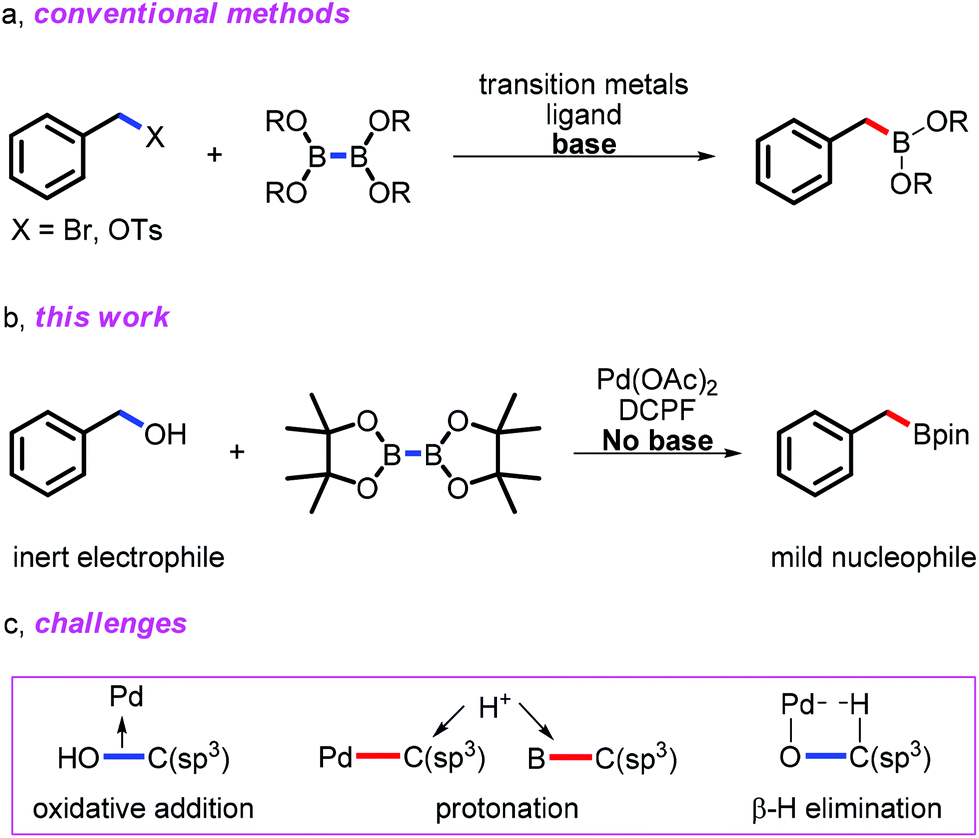 | ||
| Scheme 1 Construction of benzylic boronates via C–O bond activation. | ||
Only a few examples of the direct borylation via cleavage of the active C–O bond have been reported.17 However, to the best of our knowledge, there is no existent way to carry out the direct borylation of benzyl alcohols. In fact, direct borylation of inactivated benzyl alcohols through transition-metal catalysis faces three challenges (Scheme 1c): (1) the Brønsted acidity of benzyl alcohols potentially accelerated the protonation of the key R–M species and final benzylboron compounds, leading to the formation of toluene derivatives (pKa of benzyl alcohol: 15.4, pKa of allylic alcohol: 15.52);18 (2) high bond dissociation energy (BDE) of the unactivated benzyl C–OH bond increased the difficulty of oxidative addition to transition metals.19 (Bond dissociation energy: the C–OH bond in benzyl alcohol: 340 kJ mol−1, the C–Cl bond in benzyl chloride: 300 kJ mol−1, the C–Br bond in benzyl bromide: 251 kJ mol−1, the C–I bond in benzyl iodide: 183 kJ mol−1); (3) last but not least, the coordination of benzoxyl toward transition-metals (ArCH2O–M) induced the potential oxidation through β-hydride elimination, competing with the desired borylation process.20
Due to the unique reactivity of naphthylmethanol, 1a was selected as the model substrate. The other important reason to choose 1a as an objective was that the desired product was also easily isolated due to the good fluorescence of 2a (Table 1). We first chose Pd(OAc)2 in THF as the catalyst set due to its stability, commercial availability and broad application in conventional Miyaura borylation. The ligand played a crucial role in this borylation and the desired product (2a) was obtained in 48% NMR yield when the bidentate phosphine ligand DCPF21 was employed (entry 9), while all the other tested monodentate phosphine ligands failed (entries 1–4). Notably, other bidentate phosphine ligands, such as DPPB (entry 5), DPPM (entry 6), DCPE (entry 7), DPPF (entry 8) and DtBuPF (entry 10), were also not suitable, probably due to their different electron densities or unsuitable bite angle.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Catalyst | Ligand (loading/mol%) | Yb (%) | Entry | Catalyst | Ligand (loading/mol%) | Yb (%) |
| a The reaction was carried out on the 0.2 mmol scale. b NMR yield, numbers in parenthesis are the ratios of 1a, 2-methylnaphthalene, and diarylethane. c The temperature was 100 °C. d Benzene was used as the solvent. e Et2O was used as the solvent. f Benzene/Et2O (0.1 mL/0.9 mL) was used as the solvent. g HBpin was used as the borylation reagent. h B2cat2 was used as the borylation reagent. | |||||||
| 1 | Pd(OAc)2 | PPh3 (40) | 0 | 11 | Pd(PPh3)4 | DCPF (20) | 0 |
| 2 | Pd(OAc)2 | PCy3 (40) | 0 | 12 | PD(OTf)2 | DCPF (20) | 0 |
| 3 | Pd(OAc)2 | PtBu3 (40) | 0 | 13 | PDCl2 | DCPF (20) | 0 |
| 4 | Pd(OAc)2 | XPhos (40) | 0 | 14c | Pd(OAc)2 | DCPF (20) | 50 |
| 5 | Pd(OAc)2 | DPPB (20) | 0 | 15c,d | Pd(OAc)2 | DCPF (20) | 57 |
| 6 | Pd(OAc)2 | DPPM (20) | 0 | 16c,e | Pd(OAc)2 | DCPF (20) | <10 |
| 7 | Pd(OAc)2 | DCPE (20) | 0 | 17c,f | Pd(OAc)2 | DCPF (20) | 68 |
| 8 | Pd(OAc)2 | DPPF (20) | 0 | 18c,f | Pd(OAc) 2 | DCPF (15) | 78 (0, 15, 1) |
| 9 | Pd(OAc)2 | DCPF (20) | 48 | 19c,f,g | Pd(OAc)2 | DCPF (15) | 0 |
| 10 | Pd(OAc)2 | DtBuPF (20) | 0 | 20c,f,h | Pd(OAc)2 | DCPF (15) | 0 |
We further investigated different Pd catalysts and found that Pd catalysts were also critical to this borylation. For example, Pd(PPh3)4, PdCl2 and Pd(OTf)2 all failed. By screening the temperature we found that the full conversion of the naphthylmethanol (1a) was reached at 100 °C after 3 h. Unfortunately, 30% of the protonated product 2-methylnaphthalene was observed, lowering the efficiency of this borylation. As demonstrated as one of the challenges, this by-product might be generated by protonating the final benzylic boronates or active benzyl–Pd species.
To avoid this by-product, the solvent's effect was systematically investigated. With the use of non-polar benzene as the solvent, the best result was 57% NMR yield of (2a) while the by-product was still formed. To our satisfaction, only the desired product was observed with diethyl ether as the solvent.22 Due to the volatility of diethyl ether, we finally selected benzene/diethyl ether as the co-solvent and the highest yield was obtained when 15 mol% DCPF was used with 15% of the by-product, which can easily be isolated (entry 18).23
Different borylation reagents were further studied with 2-naphthylmethanol (1a) (entries 19 and 20). To our interest, only bis(pinacolato)diboron showed a credible reactivity. Other borylation reagents, for example, HBpin and B2cat2, completely failed in this transformation.
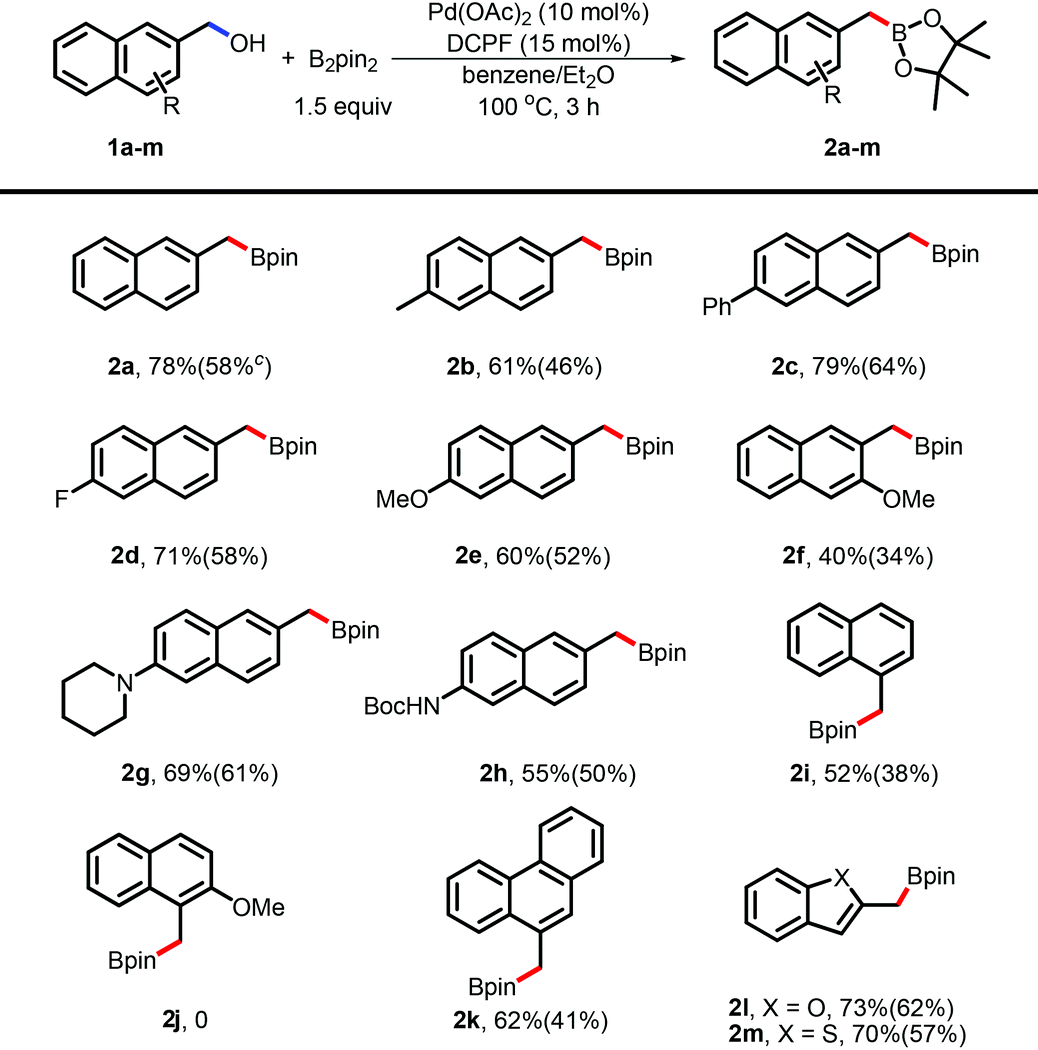
With the optimized conditions in hand, we applied this borylation to a variety of functionalized naphthylmethanols (Table 2). Substituted naphthylmethanols, such as alkyl- and aryl-substituents, were suitable for this borylation and the desired naphthylmethylboron compounds (2a and 2b) were obtained in moderate to good yields. Both electron-rich (2e, 2g, and 2h) and electron-poor (2d) substituents did not obviously affect the efficacy and the desired products were produced in credible yields. It was important to note that the heterocycle-containing substrates, such as 2-benzofuranmethanol (2l) and benzo[b]thiophene-2-methanol (2m), were also suitable and the corresponding arylmethylboronates were obtained in good yields. To test the effect of the steric hindrance, the sterically demanding substrates 1i, 1j, as well as 1f, were subjected to the reaction system to test the reactivity. Both 1f and 1i gave the desired products (2f and 2i) albeit in the relatively poor yields. To our interest, 1j completely failed in this borylation, demonstrating that the increased steric hindrance is critical by combining these two steric effects. The diol 2,6-naphthalenedimethanol 1n was also investigated for this borylation under slightly changed conditions (Scheme 2). Notably, product 2b was obtained as the major one in 55% isolated yield. This result was also in concordance with the hypothesis of challenge in protonation.
 | ||
| Scheme 2 Investigation of the reactivity of 2,6-naphthalenedimethanol. | ||
| a Reaction conditions: benzyl alcohol (0.2 mmol), B2pin2 (0.3 mmol), Pd(OAc)2 (10 mol%), DCPF (15 mol%), benzene and diethyl ether as the mixed solvents, 100 °C oil bath, 3 h. b NMR yield and isolated yield in parentheses, decomposition occurs through column chromatography. c The reaction was performed on the 1.0 mmol scale. |
|---|
|
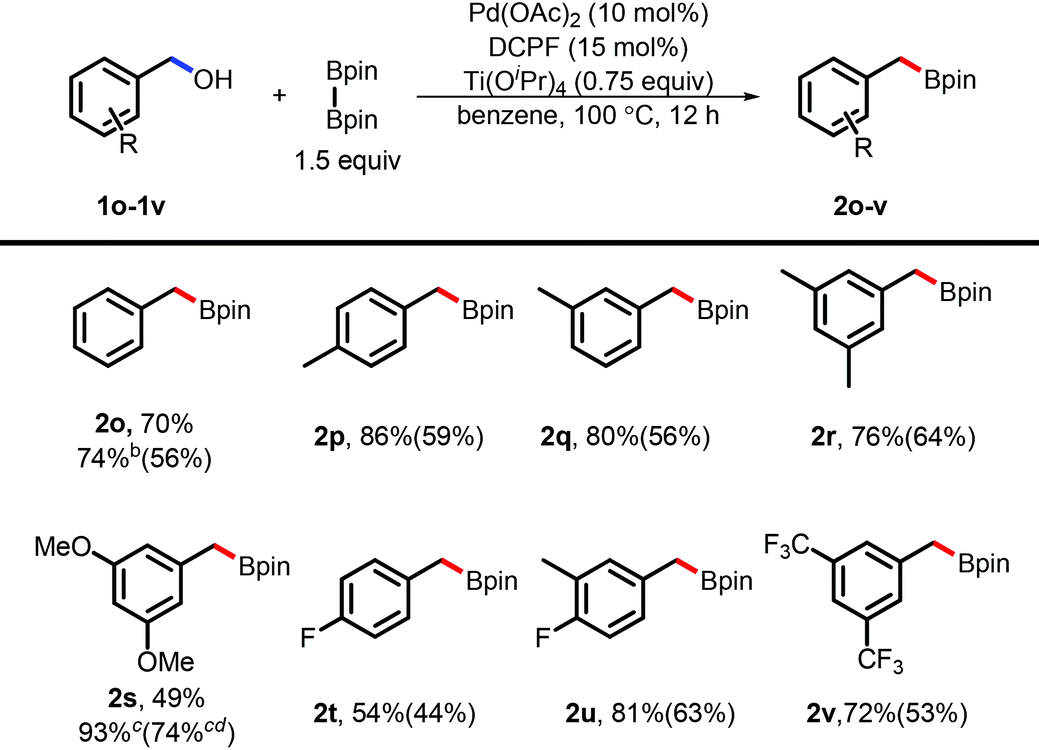
The success of the borylation of naphthylmethanols inspired us to apply such a borylation to simple benzyl alcohols. Unfortunately, no desired products were observed under the standard conditions. To our delight, a variation of the standard conditions by adding 0.75 equivalent amount of Ti(OiPr)4 promoted such a borylation reaction in high efficiency (Table 3). Such an effect induced by Ti(OiPr)4 was also observed in the previous studies in the transformation of alcohols.24 To further explore the applications, benzyl alcohols bearing mono- or multi-substituent groups were investigated and the desired products were obtained in good to excellent yields (by NMR). Both the electron-rich and electron-poor ones gave the desired products in high efficiency. It is noteworthy that the product (2t) was obtained in a relatively low yield, probably arising from the electron-withdrawing effect and/or coordinating effect of the F-atom without the steric hindrance.
| a Reaction conditions: benzyl alcohol (0.2 mmol), B2pin2 (0.3 mmol), Pd(OAc)2 (10 mol%), Ti(OiPr)4 (0.75 equiv.), benzene (1 mL), 100 °C oil bath, 12 h, NMR yield and isolated yield in parentheses, decomposition occurs through column chromatography. b Benzene (0.2 mL) and Et2O (0.8 mL) as the solvents. c n[Ti(OiPr)4] = 0.2 mmol. d The reaction was performed on the 1.0 mmol scale. |
|---|
|
To solidify the possibility of the application, we conducted the borylation of both naphthylmethanol (1a) and 3,5-dimethoxybenzyl alcohol (1s) on the mmol scale under the corresponding developed conditions. To our delight, the desired boron reagents were isolated in comparable yields although these benzylboron esters were considered very reactive and difficult to isolate (see the ESI†). Moreover, the transformation of the desired benzylic boronates to stable and versatile benzylic trifluoroborates was also conducted (Scheme 3). To our delight, the catalytic system was treated with KHF2 in one-pot and the desired product was isolated in 43% isolated yield. This development also extended the application potential of this borylation.
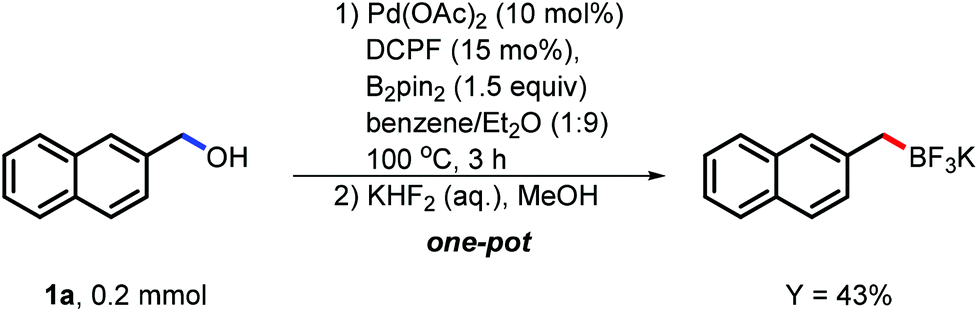 | ||
| Scheme 3 Transformation of benzylic boronate to benzylic trifluoroborate. | ||
Based on the previous reports11b,25 and our observations, the catalytic cycle was proposed as shown in Scheme 4. Arylmethanols 1 might have a weak and reversible interaction with B2pin2 to form the key intermediate I. However, this weak interaction influences the catalytic process in two aspects: (1) such a coordination between B2pin2 and benzyl alcohols weakened the benzylic C–O bond, facilitating the oxidative addition of benzylic C–O toward Pd(0) species; (2) the alcohols could act as an inner-base to activate the B–B bond of B2pin2, further promoting the transmetallation. The intermediate I further undergoes oxidative addition to Pd(0) to generate the Pd(II) species II, which subsequently undergoes either intramolecular or intermolecular transmetallation to form intermediate III. The reductive elimination of III fulfilled the catalytic cycle by releasing the desired product and regenerated the active Pd(0) species. For the relatively inert benzyl alcohol, the Lewis acidity of B2pin2 might not be strong enough to activate it. Thus, Ti(OiPr)4 was required to activate the benzylic C–O bonds by alkoxide exchange to form BnO–Ti species IV to facilitate the desired borylation. The formation of by-products in the borylation system could also be well documented based on this catalytic cycle, such as the toluene derivatives may be formed via the protonation of either the desired product 2 or active intermediates II and III.
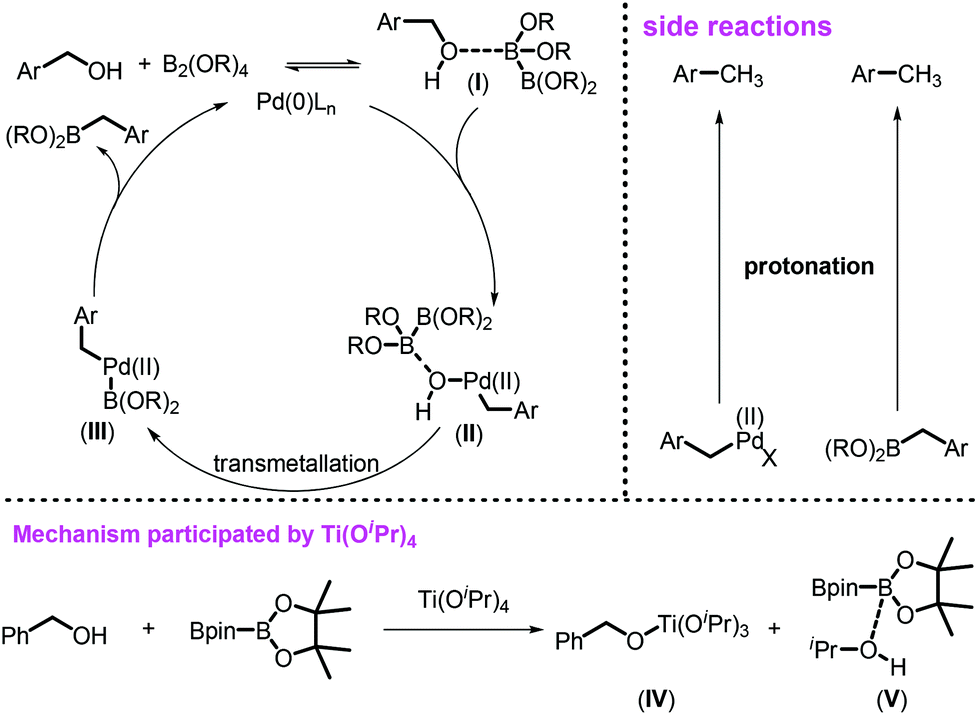 | ||
| Scheme 4 Proposed mechanism. | ||
In principle, this proposed catalytic mechanism might also be suitable for the borylation of phenol and its derivatives. Unfortunately, both conditions have been used for the direct borylation of phenol derivatives via Pd catalysis but failed. After the hard-work we found that, the borylation of 2-naphthol took place with Ni catalysis and 45% yield of 2w was isolated (Scheme 5). Further efforts to promote the efficacy of this transformation are still being made in our lab.
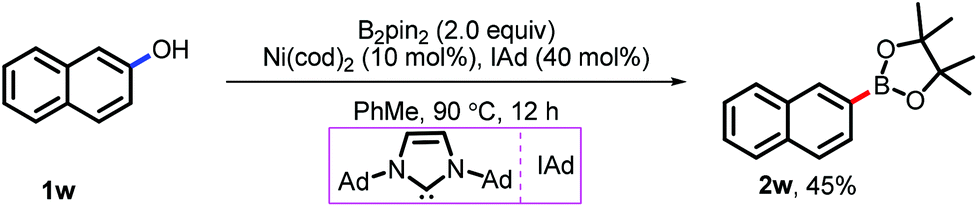 | ||
| Scheme 5 Direct borylation of phenol. | ||
Conclusions
In summary, we developed a simple and efficient borylation of arylmethanols with B2pin2 through palladium-catalyzed sp3 C–O bond activation for the first time. Most importantly, such a transformation is atom- and step-economical to construct benzylboron compounds from arylmethanols in the absence of any bases under mild conditions. This method can be a privileged alternative to construct benzylboron derivatives, thereby complementing the earlier catalytic methods typically from benzyl halides. Further investigations to extend the substrate scope, to promote the efficiency and to study the detailed mechanism are underway.Financial support by MOST (2015CB856600 and 2013CB228102) and NSFC (no. 21332001 and 21431008) is gratefully acknowledged.
Notes and references
- (a) E. Lamb, Pharmacy Times, 2009, 5, 26–28 Search PubMed; (b) C. W. Lindsley, ACS chem. neurosci., 2011, 2, 276–277 CrossRef CAS PubMed.
- (a) T. W. Greene and P. G. Wuts, Protective groups in organic synthesis, Wiley, New York, 1991 Search PubMed; (b) T. Weber and J. Brunner, J. Am. Chem. Soc., 1995, 117, 3084–3095 CrossRef CAS; (c) A. Haines, Methods for Oxidation of Organic Compounds V2: Alcohols, Alcohol Derivatives, Alky Halides, Nitroalkanes, Alkyl Azides, Carbonyl Compounds Hydroxyarenes and Aminoarenes, Elsevier, 2012 Search PubMed.
- (a) G. A. Olah, S. Kobayashi and M. Tashiro, J. Am. Chem. Soc., 1972, 94, 7448–7461 CrossRef CAS; (b) M. Rueping and B. J. Nachtsheim, Beilstein J. Org. Chem., 2010, 6, 6 Search PubMed.
- (a) A. Antoft-Finch, T. Blackburn and V. Snieckus, J. Am. Chem. Soc., 2009, 131, 17750–17752 CrossRef CAS PubMed; (b) K. W. Quasdorf, M. Riener, K. V. Petrova and N. K. Garg, J. Am. Chem. Soc., 2009, 131, 17748–17749 CrossRef CAS PubMed; (c) N. Yoshikai, H. Matsuda and E. Nakamura, J. Am. Chem. Soc., 2009, 131, 9590–9599 CrossRef CAS PubMed.
- (a) W. J. Scott, G. Crisp and J. Stille, J. Am. Chem. Soc., 1984, 106, 4630–4632 CrossRef CAS; (b) S. Sengupta, M. Leite, D. S. Raslan, C. Quesnelle and V. Snieckus, J. Org. Chem., 1992, 57, 4066–4068 CrossRef CAS; (c) J. F. Hartwig, Angew. Chem., Int. Ed., 1998, 37, 2046–2067 CrossRef CAS; (d) J. Terao, H. Watanabe, A. Ikumi, H. Kuniyasu and N. Kambe, J. Am. Chem. Soc., 2002, 124, 4222–4223 CrossRef CAS PubMed; (e) B.-T. Guan, X.-Y. Lu, Y. Zheng, D.-G. Yu, T. Wu, K.-L. Li, B.-J. Li and Z.-J. Shi, Org. Lett., 2010, 12, 396–399 CrossRef CAS PubMed; (f) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2010, 111, 1346–1416 CrossRef PubMed; (g) B.-J. Li, D.-G. Yu, C.-L. Sun and Z.-J. Shi, Chem. – Eur. J., 2011, 17, 1728–1759 CrossRef CAS PubMed.
- (a) M. Tobisu, T. Shimasaki and N. Chatani, Angew. Chem., Int. Ed., 2008, 47, 4866–4869 CrossRef CAS PubMed; (b) B.-T. Guan, S.-K. Xiang, T. Wu, Z.-P. Sun, B.-Q. Wang, K.-Q. Zhao and Z.-J. Shi, Chem. Commun., 2008, 1437–1439 RSC; (c) J. W. Dankwardt, Angew. Chem., Int. Ed., 2004, 43, 2428–2432 CrossRef CAS PubMed; (d) T. Hayashi, Y. Katsuro and M. Kumada, Tetrahedron Lett., 1980, 21, 3915–3918 CrossRef CAS; (e) F. Zhao, D.-G. Yu, R.-Y. Zhu, Z. Xi and Z.-J. Shi, Chem. Lett., 2011, 40, 1001–1003 CrossRef CAS.
- (a) C. Chuit, H. Felkin, C. Frajerman, G. Roussi and G. Swierczewski, Chem. Commun., 1968, 1604–1605 RSC; (b) D. E. Bergbreiter and D. A. Weatherford, J. Chem. Soc., Chem. Commun., 1989, 883–884 RSC; (c) M. Kimura, Y. Horino, R. Mukai, S. Tanaka and Y. Tamaru, J. Am. Chem. Soc., 2001, 123, 10401–10402 CrossRef CAS; (d) Y. Kayaki, T. Koda and T. Ikariya, Eur. J. Org. Chem., 2004, 4989–4993 CrossRef CAS PubMed; (e) B. M. Trost and J. Quancard, J. Am. Chem. Soc., 2006, 128, 6314–6315 CrossRef CAS PubMed; (f) Y.-X. Li, Q.-Q. Xuan, L. Liu, D. Wang, Y.-J. Chen and C.-J. Li, J. Am. Chem. Soc., 2013, 135, 12536–12539 CrossRef CAS PubMed; (g) J. Ye, J. Zhao, J. Xu, Y. Mao and Y. J. Zhang, Chem. Commun., 2013, 49, 9761–9763 RSC; (h) H.-B. Wu, X.-T. Ma and S.-K. Tian, Chem. Commun., 2014, 50, 219–221 RSC.
- Y. Nishibayashi, I. Wakiji and M. Hidai, J. Am. Chem. Soc., 2000, 122, 11019–11020 CrossRef CAS.
- M. Yoshida, T. Gotou and M. Ihara, Chem. Commun., 2004, 1124–1125 RSC.
- D.-H. Lee, K.-H. Kwon and C. S. Yi, Science, 2011, 333, 1613–1616 CrossRef CAS PubMed.
- (a) D.-G. Yu, X. Wang, R.-Y. Zhu, S. Luo, X.-B. Zhang, B.-Q. Wang, L. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2012, 134, 14638–14641 CrossRef CAS PubMed; (b) Z.-C. Cao, D.-G. Yu, R.-Y. Zhu, J.-B. Wei and Z.-J. Shi, Chem. Commun., 2015, 51, 2683–2686 RSC.
- (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (b) G. A. Molander and L. Jean-Gérard, in Organic Reactions, John Wiley & Sons, Inc., 2004 Search PubMed; (c) A. Suzuki, in Metal-Catalyzed Cross-Coupling Reactions, Wiley-VCH Verlag GmbH, 2007, pp. 48–97 Search PubMed; (d) F. Jäkle, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, 2011 Search PubMed; (e) D. G. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, John Wiley & Sons, 2012 Search PubMed; (f) K. Smith, in Organometallics in Synthesis: A Manual, John Wiley &Sons, Inc, 2013, pp. 465–533 Search PubMed.
- E.-I. Negishi and M. J. Idacavage, in Organic Reactions, John Wiley & Sons, Inc., 2004 Search PubMed.
- (a) G. A. Molander and N. Ellis, Acc. Chem. Res., 2007, 40, 275–286 CrossRef CAS PubMed; (b) J. D. St. Denis, C. C. G. Scully, C. F. Lee and A. K. Yudin, Org. Lett., 2014, 16, 1338–1341 CrossRef CAS PubMed; (c) B. W. Glasspoole, K. Ghozati, J. W. Moir and C. M. Crudden, Chem. Commun., 2012, 48, 1230–1232 RSC; (d) H. Koyama, H. Doi and M. Suzuki, Int. J. Org. Chem., 2013, 03, 220–223 CrossRef; (e) R. D. Grigg, J. W. Rigoli, R. Van Hoveln, S. Neale and J. M. Schomaker, Chem. – Eur. J., 2012, 18, 9391–9396 CrossRef CAS PubMed; (f) S. Sueki and Y. Kuninobu, Org. Lett., 2013, 15, 1544–1547 CrossRef CAS PubMed; (g) K. Smith, M. C. Elliott and D. H. Jones, J. Org. Chem., 2013, 78, 9526–9531 CrossRef CAS PubMed.
- (a) K. Miyazawa, Y. Yasu, T. Koike and M. Akita, Chem. Commun., 2013, 49, 7249–7251 RSC; (b) Y. Yasu, T. Koike and M. Akita, Adv. Synth. Catal., 2012, 354, 3414–3420 CrossRef CAS PubMed; (c) J. C. Tellis, D. N. Primer and G. A. Molander, Science, 2014, 345, 433–436 CrossRef CAS PubMed.
- (a) I. Beletskaya and A. Pelter, Tetrahedron, 1997, 53, 4957–5026 CrossRef CAS; (b) K. M. Sadhu and D. S. Matteson, Organometallics, 1985, 4, 1687–1689 CrossRef CAS; (c) D. S. Matteson, J. Organomet. Chem., 1999, 581, 51–65 CrossRef CAS; (d) R. Martin and J. B. Jones, Tetrahedron Lett., 1995, 36, 8399–8402 CrossRef CAS; (e) J. R. Falck, M. Bondlela, J. Ye and S.-D. Cho, Tetrahedron Lett., 1999, 40, 5647–5650 CrossRef CAS; (f) T. Ishiyama, Z. Oohashi, T.-a. Ahiko and N. Miyaura, Chem. Lett., 2002, 780–781 CrossRef CAS; (g) A. Giroux, Tetrahedron Lett., 2003, 44, 233–235 CrossRef CAS; (h) C.-T. Yang, Z.-Q. Zhang, H. Tajuddin, C.-C. Wu, J. Liang, J.-H. Liu, Y. Fu, M. Czyzewska, P. G. Steel, T. B. Marder and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 528–532 CrossRef CAS PubMed; (i) T. C. Atack, R. M. Lecker and S. P. Cook, J. Am. Chem. Soc., 2014, 136, 9521–9523 CrossRef CAS PubMed; (j) S. K. Bose, K. Fucke, L. Liu, P. G. Steel and T. B. Marder, Angew. Chem., Int. Ed., 2014, 53, 1799–1803 CrossRef CAS PubMed; (k) A. Bej, D. Srimani and A. Sarkar, Green Chem., 2012, 14, 661–667 RSC; (l) R. B. Bedford, P. B. Brenner, E. Carter, T. Gallagher, D. M. Murphy and D. R. Pye, Organometallics, 2014, 33, 5940–5943 CrossRef CAS; (m) T. Ishiyama, K. Ishida, J. Takagi and N. Miyaura, Chem. Lett., 2001, 1082–1083 CrossRef CAS; (n) S. Shimada, A. S. Batsanov, J. A. K. Howard and T. B. Marder, Angew. Chem., Int. Ed., 2001, 40, 2168–2171 CrossRef CAS; (o) H. Li, L. Wang, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2012, 51, 2943–2946 CrossRef CAS PubMed; (p) C. Zarate, R. Manzano and R. Martin, J. Am. Chem. Soc., 2015, 137, 6754–6758 CrossRef CAS PubMed; (q) M. A. Larsen, C. V. Wilson and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 8633–8643 CrossRef CAS PubMed.
- (a) S. Sebelius, V. J. Olsson and K. J. Szabó, J. Am. Chem. Soc., 2005, 127, 10478–10479 CrossRef CAS PubMed; (b) N. Selander and K. l. n. J. Szabó, J. Org. Chem., 2009, 74, 5695–5698 CrossRef CAS PubMed; (c) D. A. Wilson, C. J. Wilson, C. Moldoveanu, A.-M. Resmerita, P. Corcoran, L. M. Hoang, B. M. Rosen and V. Percec, J. Am. Chem. Soc., 2010, 132, 1800–1801 CrossRef CAS PubMed; (d) N. Selander, J. R. Paasch and K. J. Szabó, J. Am. Chem. Soc., 2010, 133, 409–411 CrossRef PubMed; (e) K. Huang, D.-G. Yu, S.-F. Zheng, Z.-H. Wu and Z.-J. Shi, Chem. – Eur. J., 2011, 17, 786–791 CrossRef CAS PubMed; (f) J. M. Larsson and K. J. Szabó, J. Am. Chem. Soc., 2012, 135, 443–455 CrossRef PubMed; (g) T. S. N. Zhao, Y. Z. Yang, T. Lessing and K. J. Szabo, J. Am. Chem. Soc., 2014, 136, 7563–7566 CrossRef CAS PubMed; (h) H. Kinuta, M. Tobisu and N. Chatani, J. Am. Chem. Soc., 2015, 137, 1593–1600 CrossRef CAS PubMed.
- (a) H. C. Brown and G. Zweifel, J. Am. Chem. Soc., 1960, 82, 4708–4712 CrossRef CAS; (b) H. C. Brown and G. Zweifel, J. Am. Chem. Soc., 1960, 82, 1504–1505 CrossRef CAS; (c) H. C. Brown and K. J. Murray, Tetrahedron, 1986, 42, 5497–5504 CrossRef CAS; (d) K. Endo, T. Ohkubo and T. Shibata, Org. Lett., 2011, 13, 3368–3371 CrossRef CAS PubMed.
- Y.-R. Luo, Handbook of bond dissociation energies in organic compounds, CRC Press, 2002 Search PubMed.
- The aldehyde product was observed during the optimization experiment.
- DCPF = bis(dicyclohexylphosphino)ferrocene, DtBuPF = bis(di-tert-butylphosphino)ferrocene.
- The catalytic system was not soluble in diethyl ether and resulted in low yield.
- 10 mol% ligand loading leads to incomplete conversion of alcohols.
- Direct activation of alcohols assisted by Ti(OiPr)4: (a) K. Itoh, N. Hamaguchi, M. Miura and M. Nomura, J. Chem. Soc., Perkin Trans. 1, 1992, 2833–2835 Search PubMed; (b) T. Satoh, M. Ikeda, M. Miura and M. Nomura, J. Org. Chem., 1997, 62, 4877–4879 Search PubMed; (c) Y.-X. Li, Q.-Q. Xuan, L. Liu, D. Wang, Y.-J. Chen and C.-J. Li, J. Am. Chem. Soc., 2013, 135, 12536–12539 Search PubMed.
- Interaction between the O-atom and B-atom, see: (a) D.-G. Yu and Z.-J. Shi, Angew. Chem., Int. Ed., 2011, 50, 7097–7100 Search PubMed; (b) T. P. Blaisdell, T. C. Caya, L. Zhang, A. Sanz-Marco and J. P. Morken, J. Am. Chem. Soc., 2014, 136, 9264–9267 Search PubMed; (c) Y. Nagashima, K. Hirano, R. Takita and M. Uchiyama, J. Am. Chem. Soc., 2014, 136, 8532–8535 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5qo00243e |
| This journal is © the Partner Organisations 2015 |