
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA one-pot dilithiation–lithium–zinc exchange–Negishi coupling approach to 2,6-di(hetero)aryl substituted dithienothiazines – a novel class of electronically fine-tunable redox systems†‡
Catherine
Dostert
and
Thomas J. J.
Müller
*
Institut für Organische Chemie und Makromolekulare Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstraße 1, 40225 Düsseldorf, Germany. E-mail: ThomasJJ.Mueller@uni-duesseldorf.de
First published on 23rd March 2015
Abstract
2,6-Di(hetero)aryl and 2-(hetero)aryl substituted dithienothiazines are prepared from N-aryl dithienothiazines by a lithiation–lithium–zinc exchange–Negishi cross-coupling sequence with (hetero)aryl iodides in a one-pot fashion in good to excellent yields. These novel extended π-electron systems can be reversibly oxidized and fine-tuned in their electronic properties as supported by cyclo voltammetric, and absorption and emission spectroscopic studies.
Introduction
Over the past few years, interest in electroactive organic molecules as functional π-systems1 has enormously increased due to important potential technological applications ranging from organic light-emitting diodes2 over organic photovoltaic devices3 to organic field-effect transistors.4 The main advantages of using organic materials are their low production costs, favorable properties such as flexibility, transparency and light weight, tunability of their properties, and their good processability.5 As a consequence, the exploration of novel semiconducting molecular materials has become an ongoing challenge to synthetic organic chemistry. Moreover, small redox-active molecules can be considered as molecular wires6 and, therefore, open new alleys to unimolecular electronics.7 Recently we presented 4H-dithieno[2,3-b:3′,2′-e][1,4]thiazines, congeners to the well-established class of phenothiazines, as novel electron-rich organic π-systems (Scheme 1).8 As a consequence of their unique electronic properties showing two reversible oxidations with Nernstian behavior at low oxidation potentials, dithienothiazines qualify, in principle, well for use as conducting materials or as a donor component in donor–acceptor conjugates. | ||
| Scheme 1 4H-Dithieno[2,3-b:3′,2′-e][1,4]thiazine – a congener of 10H-phenothiazine with increased electron density. | ||
For potential applications, functionalization of the heterocyclic core represents a key step and major challenge. Most interestingly, the thiophene anellation offers an easy entry to thiophene characteristic transformations, such as lithiation at the α-positions with respect to the sulfur atom,9 which is also suitable for sequential one-pot processes.10 This concept was very successfully transposed to the 2,6-difunctionalization of N-substituted dithienothiazines via a dilithiation–electrophilic trapping sequence.11 Therefore, a one-pot dilithiation–cross-coupling strategy to 2,6-di(hetero)aryl substituted dithienothiazines lies at hand. Here, we report a consecutive one-pot dilithiation–transmetallation–Negishi coupling sequence to 2,6-di(hetero)aryl substituted dithienothiazines and 2-(hetero)aryl substituted dithienothiazines and the elucidation of their electronic properties by cyclic voltammetry, absorption and emission spectroscopy, and DFT calculations.
Results and discussion
Synthesis and structure
Interestingly, the electrophile addition to 2,6-dilithio dithienothiazine11 opens up rapid access to highly reactive organometallic species setting the stage for a subsequent cross-coupling reaction in a consecutive one-pot fashion.10 Therefore, we set out to introduce zinc bromide as an electrophile for generating symmetrical bis(organozinc halides), suitable nucleophiles for envisioned Negishi coupling.12The twofold lithiation of N-[4-(n-hexyl)phenyl] 4H-dithieno[2,3-b:3′,2′-e][1,4]thiazine (1) with a slight excess of n-BuLi–TMEDA (N,N,N′,N′-tetramethylethylenediamine) at −78 °C was ensured by reverse addition13 of dithienothiazine 1 to a precooled solution of n-BuLi–TMEDA (Scheme 2). Addition of freshly dried zinc bromide to the 2,6-dilithio dithienothiazine 4 furnished the organozinc bromide species 5. The subsequent addition of (hetero)aryl iodides 2 and a catalytic amount of Pd(PPh3)4 gave rise to the twofold Negishi cross-coupling as a terminating step of this one-pot process, furnishing, after workup and flash chromatography on silica gel, 2,6-di(hetero)aryl substituted dithienothiazines 3 in moderate to excellent yields (Table 1). The structures of all compounds 3 have been unambiguously assigned by spectroscopy (NMR, IR, and MS) and elemental analysis or HRMS. In the NMR spectra, expectedly, characteristic single sets of signals are found for the methine proton singlets around δ 6.2–6.6 and methine carbon nuclei at δ 115–120 of the central dithienothiazine cores.
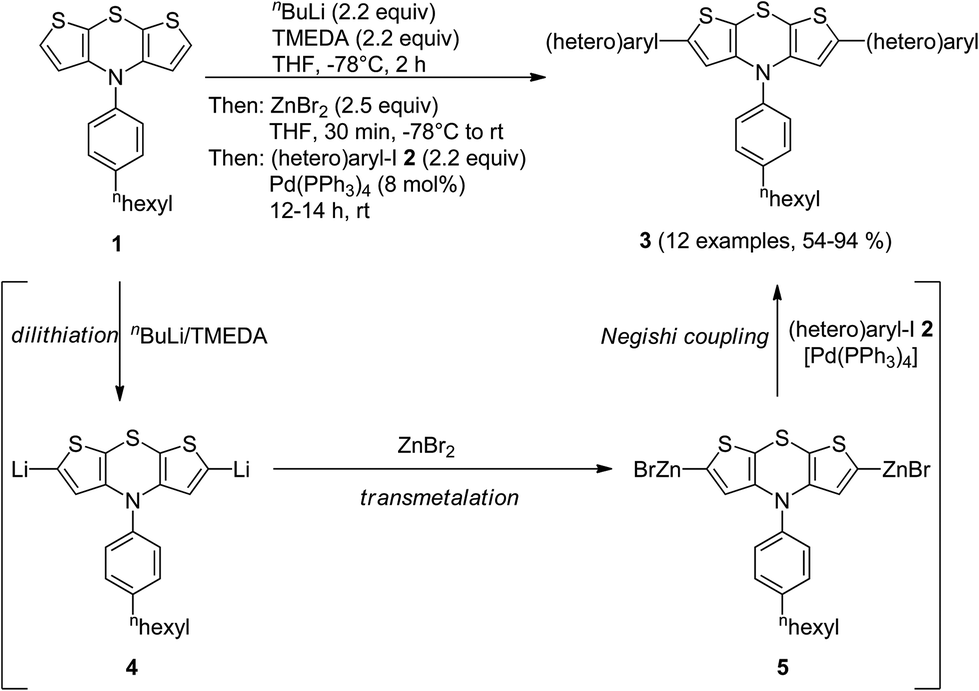 | ||
| Scheme 2 One-pot synthesis of 2,6-di(hetero)aryl substituted dithienothiazines 3 by dilithiation–transmetalation–Negishi coupling sequence. | ||
| Entry | (Hetero)aryl iodide 2 | 2,6-Di(hetero)aryl dithienothiazines 3 (yield) |
|---|---|---|
| 1 | (Hetero)aryl = 4-MeOC6H4 (2a) | 3a (65%) |
| 2 | (Hetero)aryl = 4-MeC6H4 (2b) | 3b (82%) |
| 3 | (Hetero)aryl = Ph (2c) | 3c (87%) |
| 4 | (Hetero)aryl = 4-ClC6H4 (2d) | 3d (78%) |
| 5 | (Hetero)aryl = 4-MeO2CC6H4 (2e) | 3e (80%) |
| 6 | (Hetero)aryl = 4-F3CC6H4 (2f) | 3f (95%) |
| 7 | (Hetero)aryl = 4-NCC6H4 (2g) | 3g (69%) |
| 8 | (Hetero)aryl = 4-O2NC6H4 (2h) | 3h (78%) |
| 9 | (Hetero)aryl = 3-O2NC6H4 (2i) | 3i (77%) |
| 10 | (Hetero)aryl = 2-O2NC6H4 (2j) | 3j (54%) |
| 11 | (Hetero)aryl = 3-pyridyl (2k) | 3k (63%) |
| 12 | (Hetero)aryl = 10-n-hexyl-10H-phenothiazin-3-yl (2l) | 3l (94%) |
The synthesis proceeds smoothly with a variety of aromatic (2a–j) and heteroaromatic iodides (2k and 2l), and the substituents can be electron donating (2a, 2b), electro-neutral (2c), and electron withdrawing substituents (2d–g). Moreover, it is remarkable that the sequence can be conducted with nearly equistoichiometric amounts of reagents to give the targeted structures with excellent efficiency.
Likewise we probed the synthesis of 2-(hetero)aryl substituted dithienothiazines 6 in three representative examples by employing equimolar amounts of reagents. With this adapted stoichiometry under dropwise addition of n-BuLi to a THF solution of 1 and TMEDA, the intermediacy of a monolithiated specimen furnished after workup and chromatography 2-(hetero)aryl substituted dithienothiazines 6 in moderate to good yields (Scheme 3) and the structures of these monosubstituted derivatives 6 have been unambiguously assigned by spectroscopy (NMR, IR, MS) and elemental analysis or HRMS. The lower symmetry of the structures 6 in comparison with the disubstituted derivatives 3 manifests in the appearance of three distinct methine signals, two as doublets at δ 6.1 and 7.15 with coupling constants J = 5.5 Hz and one as singlets between δ 6.2 and 6.6 in the proton NMR spectra, and three resonances for the methine carbon nuclei of the central dithienothiazine cores between δ 116 and 125 in the 13C NMR spectra.
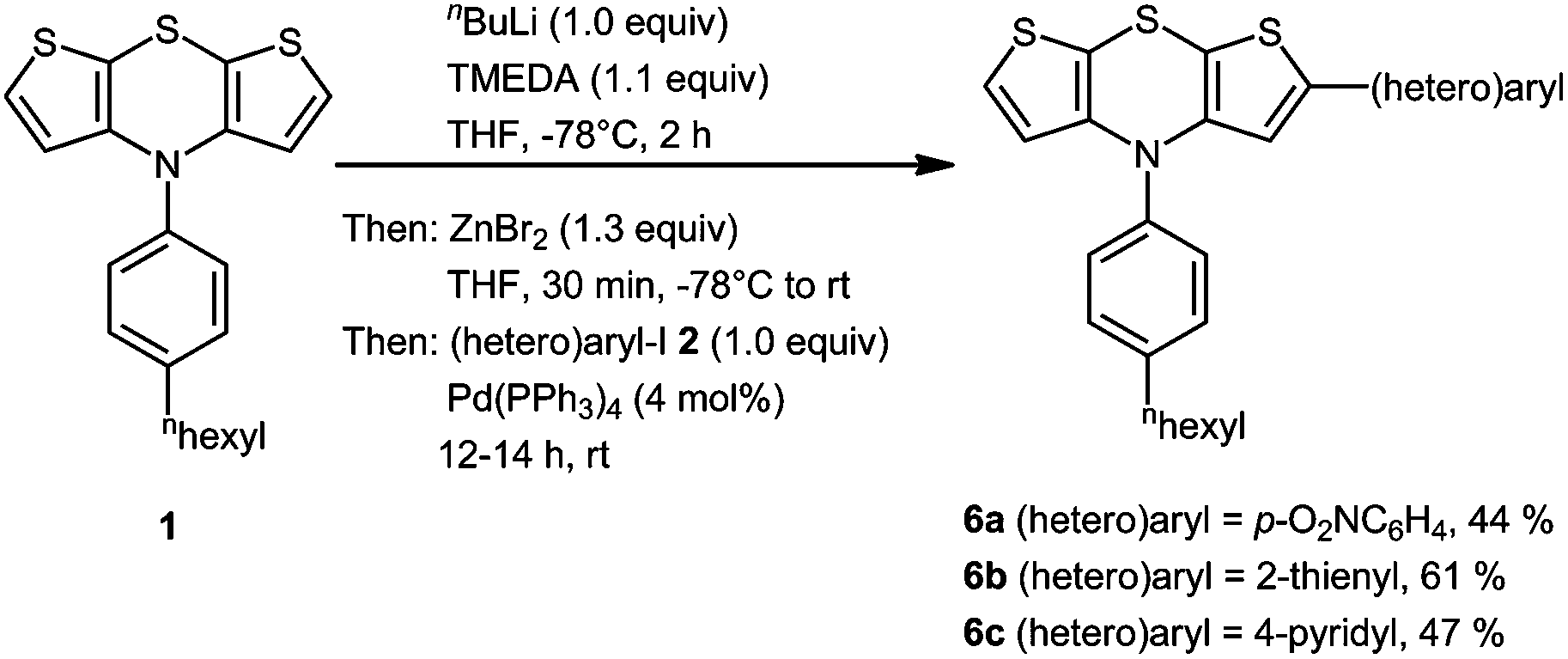 | ||
| Scheme 3 One-pot synthesis of 2-(hetero)aryl substituted dithienothiazines 6 by monolithiation–transmetalation–Negishi coupling sequence. | ||
The structure of the symmetrically disubstituted dithienothiazines 3 was additionally corroborated by studying the DFT-optimized geometries14 (B3LYP functional15 and 6-311G*16 basis set) for the parent compound 1 and selected derivatives of 3, where the n-hexyl substituent has been truncated to an ethyl group for reducing the computational time (Table 2). The para-ethylphenyl substituent on the thiazine core was always oriented in a quasi-axial configuration to ensure global energy minima of the computed structures. The influence of the substituents at positions 2 and 6 on the central 1,4-thiazine folding angle and the thienyl-aryl torsional dihedral angle was studied for electron-releasing (structure 3a), electroneutral (structure 3c), electron-withdrawing (structure 3g), and electron-withdrawing para- (structure 3h), meta- (structure 3i), and ortho-substituents (structure 3j).
| Structurea | Central 1,4-thiazine folding angle | Thienyl-aryl torsional dihedral angle |
|---|---|---|
| a In contrast to the synthesized structures 1 and 3 the n-hexyl substituent has been truncated to an ethyl group. | ||
| 1 | 140.2° | — |
| 3a | 140.3° | 30.8° |
| 3c | 140.7° | 33.4° |
| 3g | 141.1° | 28.2° |
| 3h | 141.1° | 28.0° |
| 3i | 140.6° | 28.1° |
| 3j | 141.1° | 59.8° |
While the central 1,4-thiazine folding angle lies in a narrow margin between 140.2 and 141.1° the thienyl-aryl torsional dihedral angle is diminished to 28° for remote electron-withdrawing substituents (structures 3g–i) in contrast to 30° (structure 3a) and 33° (structure 3c) for electron-releasing and electroneutral substituents. For the former a more efficient overlap of the π-systems can be expected in the electronic ground state. Expectedly, an ortho-nitrophenyl substituent (structure 3j) results in a torsion of 60° from coplanarity.
Electronic properties
The electronic properties of 2,6-di(hetero)aryl substituted dithienothiazines 3 and 2-(hetero)aryl substituted dithienothiazines 6 were experimentally investigated by cyclic voltammetry and by absorption and emission spectroscopy (Table 3), and for elucidating the electronic structure, DFT and TDDFT calculations were performed for selected molecular structures.![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) , oxidation potentials, and semiquinone formation constant KSEM) of dithienothiazine 1, 2,6-di(hetero)aryl and 2-(hetero)aryl substituted dithienothiazines 3 and 6
, oxidation potentials, and semiquinone formation constant KSEM) of dithienothiazine 1, 2,6-di(hetero)aryl and 2-(hetero)aryl substituted dithienothiazines 3 and 6
| Compound | Absorption maxima λmax,abs (ε)a [nm] (L cm−1 mol−1) | Emission λmax,emb [nm] | Stokes-shift Δc [cm−1] |
E
0/+11/2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) d [mV] d [mV] |
E
+1/+21/2d [mV] |
K SEM |
|---|---|---|---|---|---|---|
a Recorded in CH2Cl2 UVASOL at rt.
b Recorded in CH2Cl2 UVASOL at rt with λexc = 310.0 nm.
c Δ = 1/λmax,abs − 1/λmax,em[cm−1].
d Recorded in CH2Cl2, T = 293 K, 0.1 M electrolyte [nBu4N][PF6], Pt working, Ag/AgCl reference, and Pt counter electrodes; potentials are corrected against decamethylferrocene as an external standard with E0/+10 = −95 mV.
e
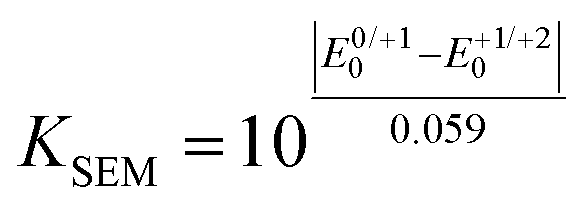 (potentials are inserted in [V] and potential differences are dimensionless).
f
λ
max,exc = 380 nm.
g The oxidation is irreversible.
h
λ
max,exc = 420 nm.
i
E
+1/+31/2.
j
E
+3/+41/2.
k
λ
max,exc = 425 nm.
l
λ
max,exc = 450 nm. (potentials are inserted in [V] and potential differences are dimensionless).
f
λ
max,exc = 380 nm.
g The oxidation is irreversible.
h
λ
max,exc = 420 nm.
i
E
+1/+31/2.
j
E
+3/+41/2.
k
λ
max,exc = 425 nm.
l
λ
max,exc = 450 nm.
|
||||||
| 1 | 240 (22000), 318 (6000) |
— | — | 360 | 1230 | 55.7 × 1013 |
| 3a | 295 (46100), 367sh (6000) |
— | — | 270 | 1000 | 0.21 × 1013 |
| 3b | 294 (48200), 385 (5700) |
— | — | 320 | 1080 | 1.04 × 1013 |
| 3c | 290 (49500), 400 (5300) |
— | — | 340 | 1120 | 1.26 × 1013 |
| 3d | 296 (49550), 409 (6000) |
— | — | 380 | 1130 | 0.44 × 1013 |
| 3e | 309 (49600), 455 (8800) |
— | — | 420 | 1160 | 0.38 × 1013 |
| 3f | 298 (62900), 431 (8500) |
— | — | 440 | 1180 | 0.44 × 1013 |
| 3g | 310 (43600), 457 (7200) |
— | — | 480 | 1210 | 0.26 × 1013 |
| 3h | 254 (31300), 332 (46000), 524 (17900) |
— | — | 500 | 1240 | 0.32 × 1013 |
| 3i | 286 (40100), 440 (4800) |
— | — | 480 | 1230 | 0.68 × 1013 |
| 3j | 264 (30250), 452 (2900) |
— | — | 440 | 1250 | 4.76 × 1013 |
| 3k | 292 (32450), 414 (3900) |
434sh, 560f | 6300 | 410 | —g | — |
| 3l | 239sh (27900), 267sh (30300), 291 (36250), 349 (14300), 422sh (8500) |
497h | 3800 | 290 | 840,i 1090j | 0.07 × 1013 |
| 6a | 250 (25550), 328 (22850), 511 (7300) |
— | — | 430 | 1210 | 1.94 × 1013 |
| 6b | 237sh (12400), 300 (12700), 355sh (3000), 420sh (1900) |
569k | 6200 | 360 | 1130 | 1.37 × 1013 |
| 6c | 238sh (9800), 292 (13300), 367 (1900), 449 (2200) |
606l | 5800 | 440 | —h | — |
The first oxidation E0/+11/2 of 2,6-di(hetero)aryl substituted dithienothiazines 3 and 2-(hetero)aryl substituted dithienothiazines 6 are found between 270 and 500 mV, while the second oxidations E+1/+21/2 from the radical cations to the dications occur between 1000 and 1250 mV. With the exception of compounds 3k and 6c all other 2,6-di(hetero)aryl substituted dithienothiazines 3 and 2-(hetero)aryl substituted dithienothiazines 6 show two clearly separated reversible oxidation waves with Nernstian behavior (Fig. 1).
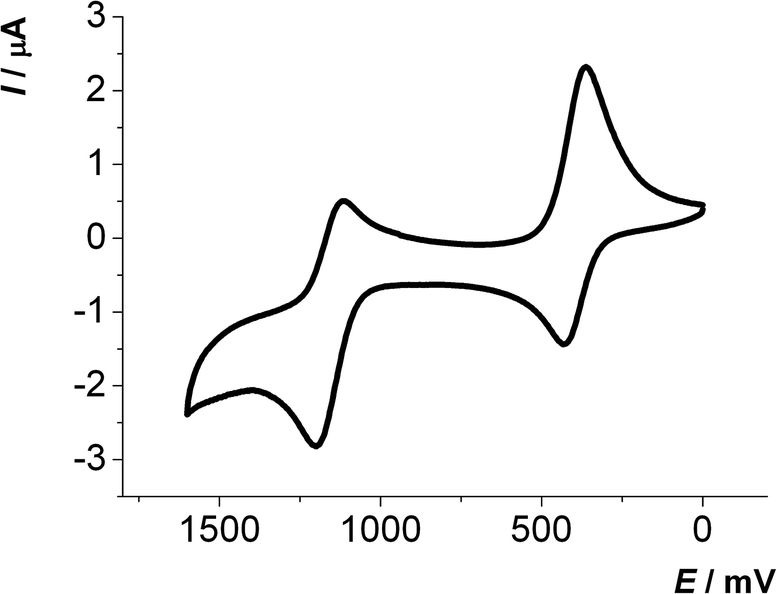 | ||
| Fig. 1 Cyclic voltammogram of compound 3c (recorded in CH2Cl2, T = 293 K, 0.1 M electrolyte [nBu4N][PF6], scan rate ν = 100 mV s−1, Pt working, Ag/AgCl reference and Pt counter electrodes). | ||
Interestingly, the oxidation potentials in the series of the 2,6-diaryl substituted dithienothiazines 3a–3i vary with the electronic nature of the substituent. This strong dependence of the reversible first and second oxidation potentials on the electronic substituent effect is shown in a good linear correlation with the Hammett σp or σm substitution parameters17 (E0/+10, R2 = 0.9889; E+1/+20, R2 = 0.9406) (Fig. 2). Upon oxidation, radical cations 3+˙ and dications 32+ are successively formed and their stabilities are achieved by both inductive and resonance effects. The three nitrosubstituted regioisomers 3h–j nicely illustrate the influence of remote conjugative and inductive substituent effects on the first oxidation potentials as shown by the para-substituted derivative 3h (E0/+11/2 = 500 mV) and the meta-substituted compound 3i (E0/+11/2 = 480 mV) whereas for compound 3j (E0/+11/2 = 440 mV) the lack of overlap by torsion out of coplanarity underlines the stereoelectronic effect of the ortho-substitution.
 | ||
| Fig. 2 Linear correlation plots of the first oxidation potentials E0/+10 [mV] (bottom) and second oxidation potentials E+1/+20 [mV] (top) of 2,6-diaryl-substituted dithienothiazines 3a–i against Hammett σp or σm parameters (E0/+10 = 201 σp/m + 336 [mV], r2 = 0.9889; E+1/20 = 192 σp/m + 1086 [mV], r2 = 0.9406). | ||
The electrochemistry of the 2,6-di(phenothiazine-3-yl) substituted dithienothiazine 3l is more complicated because phenothiazinyl substituents are additionally redox-active. In this case three distinctly separated, reversible oxidations appear in the cyclic voltammogram. The first and third oxidations at E0/+10 = 290 mV and E+3/+40 = 1090 mV clearly show Nernstian behavior, but the second oxidation at E+1/+30 = 840 mV reveals a large difference ΔE1/2 = 112 mV for anodic and cathodic peak potentials with an overall increased intensity of the current density. Therefore, the first and third one-electron oxidations can be assigned as dithienothiazinyl centered oxidation events, whereas the second oxidation rather represents two simultaneously occurring phenothiazinyl centered oxidations as quasi-reversible events.
In addition, the differences between the first and second oxidations are relatively large and indicate considerable stability of the electrochemically generated radical cations 3+˙. By calculation of the semiquinone formation constants KSEM18 for the comproportionation of 3 and 32+ furnishing two moles of 3+˙ the stability can be quantified to lie between 0.07 × 1013 and 4.76 × 1013. In comparison with the 2,6-unsubstituted mother compound 1 the KSEM are approximately 1–2 orders of magnitude lower. Likewise, the KSEM of the 2-substituted derivatives 6 are one order of magnitude lower.
The absorption spectra of all 2,6-di(hetero)aryl substituted dithienothiazines 3 reveal similar absorption characteristics. An intense absorption band appears around 300 nm and the less intensive longest wavelength absorption is found in a range from 367 to 525 nm (Fig. 3). In comparison with the 2,6-unsubstituted mother compound 1 (λmax,abs = 318 nm) the red shift of the longest wavelength absorption bands of the 2,6-di(hetero)aryl substituted derivatives 3 accounts for extended π-electron systems.
 | ||
| Fig. 3 Absorption spectrum of compound 3c (recorded in CH2Cl2, T = 293 K). | ||
The absorption behavior can be plausibly rationalized by a TD-DFT calculation14–16 of compound 3f, where the n-hexyl substituent has been truncated to an ethyl group for reducing the computational time. The experimental UV/vis spectrum of 3f is nicely reproduced by the computation (Fig. 4). While the longest wavelength absorption maximum at 455 nm consists of a 48% contribution of the HOMO–LUMO transition, the strongest absorption band at around 315 nm consists of a 43% contribution from a HOMO–LUMO+2 transition and the shoulder at 350 nm arises from 40% contribution from the HOMO−1–LUMO transition. In addition, the Kohn–Sham frontier molecular orbitals of 3f, i.e. HOMO and LUMO (Fig. 5), which are predominantly involved in constituting the longest wavelength absorption band, display considerable coefficient densities in the central N-aryl dithienothiazine core in the HOMO and in the 2- and 6-aryl substituents in the LUMO. Thereby this transition is accompanied by a considerable charge transfer character from the central donor moiety to the outer acceptor units.
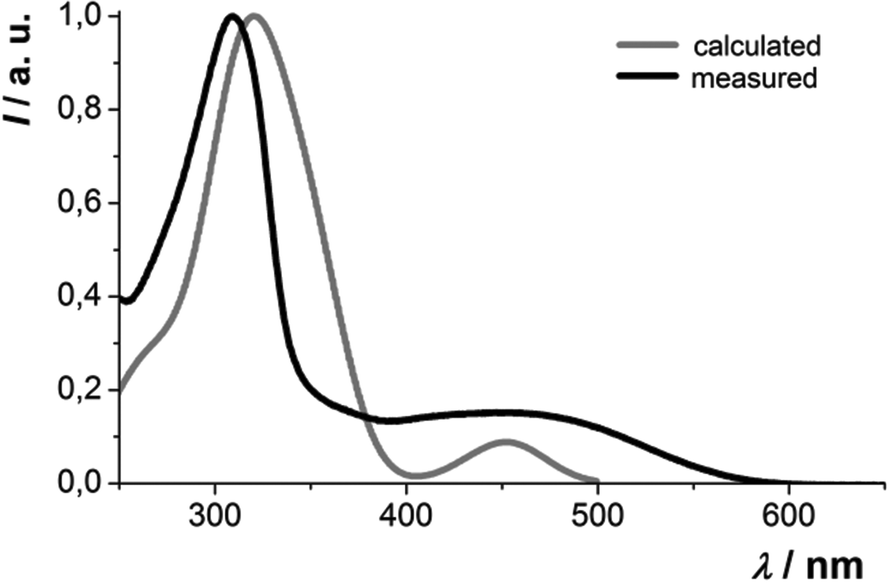 | ||
| Fig. 4 Experimental (black) and calculated (grey) absorption spectra of compound 3f. | ||
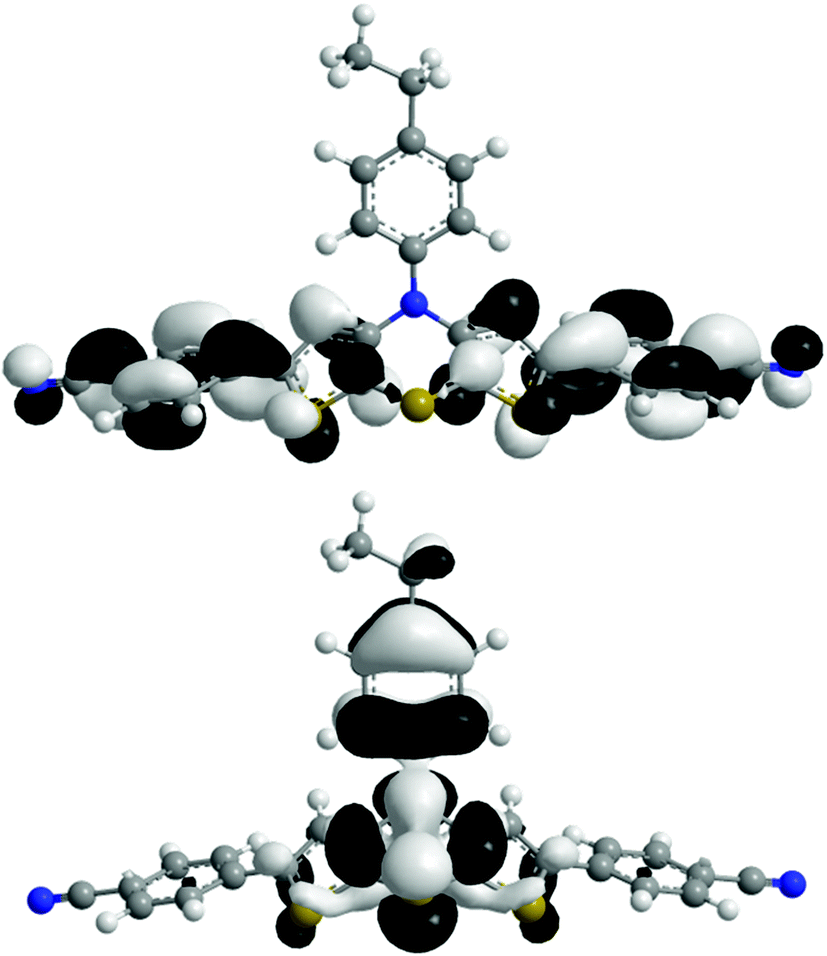 | ||
| Fig. 5 Frontier molecular orbitals (HOMO, bottom, and LUMO, top) of compound 3f (DFT computations with B3LYP functional and 6-311G* basis set). | ||
Furthermore the considerable charge transfer character of the longest wavelength absorption band becomes qualitatively apparent for naked eyes by comparing the bathochromic shift of the color of equimolar dichloromethane solutions of compounds 3c–h with increasing acceptor strength under daylight (Fig. 6).
 | ||
| Fig. 6 Bathochromic shift of equimolar dichloromethane solutions of compounds 3c–h with increasing acceptor strength under daylight. | ||
This observation can be additionally quantified by a good linear correlation of the longest wavelength absorption maxima of compounds 3a–h with the Hammett substitution parameter17σp− (R2 = 0.942) (Fig. 7), whereby σp− indicates the immediate influence of the resonance stabilization of negative charges by mesomeric and inductive substituents.
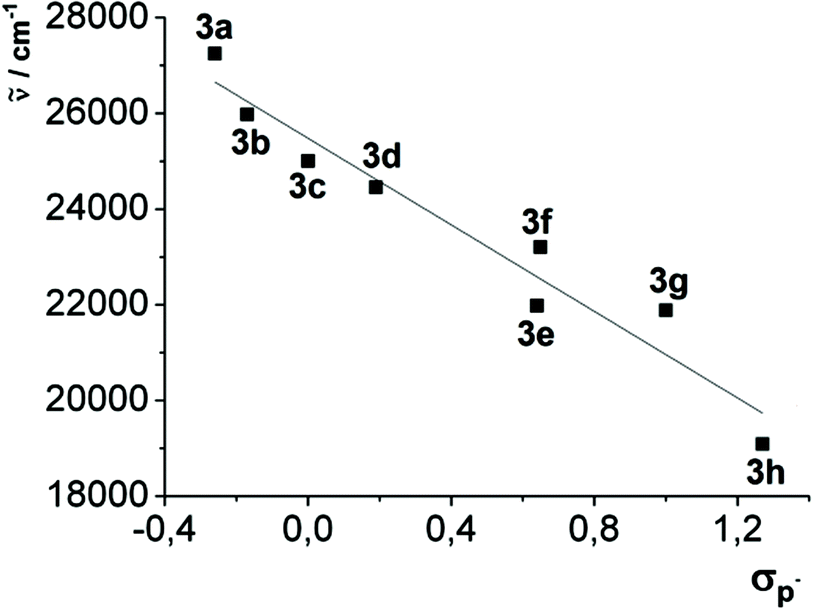 | ||
| Fig. 7 Linear correlation plot of the longest wavelength absorption maxima max,abs [cm−1] of 2,6-diaryl-substituted dithienothiazines 3a–h against the Hammett parameter σp− ( = −4522 × σp− + 25480 [cm−1], r2 = 0.9421). | ||
While the absorption characteristics of 2-(hetero)aryl substituted dithienothiazines 6 indicate three distinct absorption bands, the UV/vis spectrum of 2,6-di(phenothiazine-3-yl) substituted dithienothiazine 3l shows multiple absorption bands and shoulders, indicating the behavior of an extended π-electron system rather than a simple additive behavior of the underlying subchromophores.
Finally, among the 2,6-di(hetero)aryl substituted and 2-(hetero)aryl substituted dithienothiazines 3 and 6 the most peculiar electronic feature is reflected that in contrast to many dithienothiazines, which are essentially nonluminescent (see also Table 3),8,11 the four derivatives show intense luminescence at 560 nm (3k), 497 nm (3l), 569 nm (6b), and 606 nm (6c) with broad unstructured emission bands and large Stokes shifts (Fig. 8).
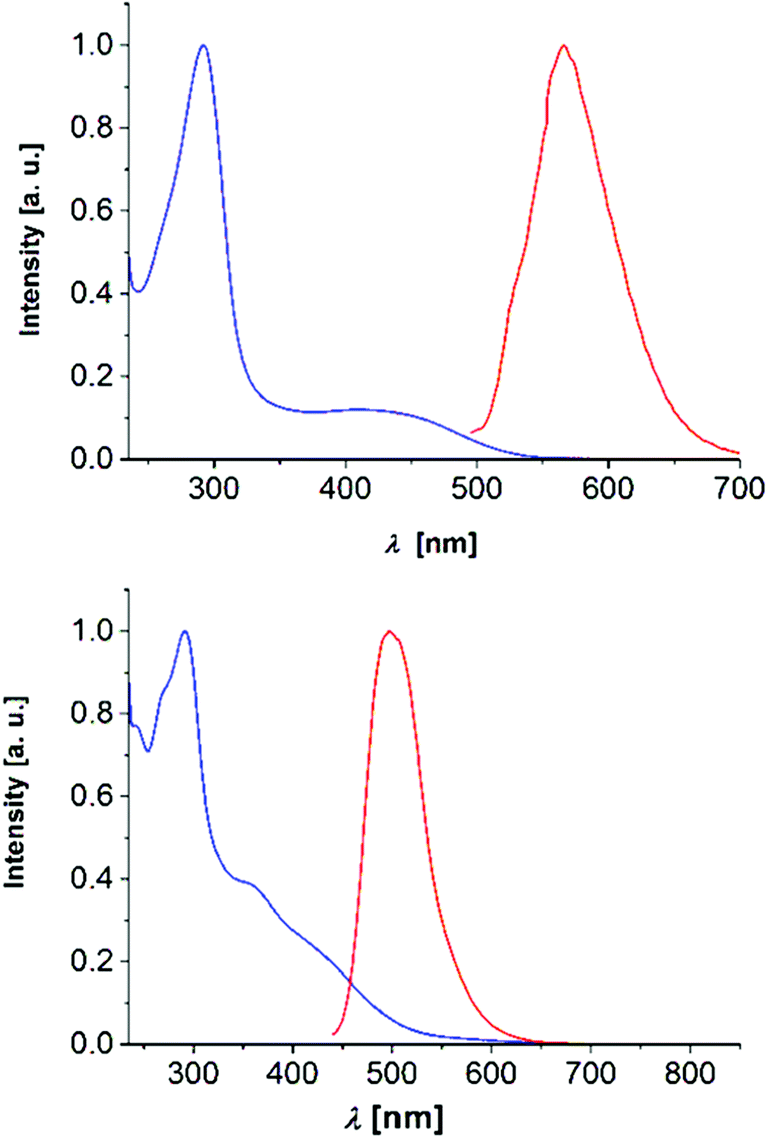 | ||
| Fig. 8 Normalized absorption (blue) and emission (red) spectra of compounds 3k (top, λmax,exc = 380 nm) and 3l (bottom, λmax,exc = 420 nm) (recorded in CH2Cl2, T = 293 K). | ||
Conclusions
The hitherto unknown classes of 2,6-di(hetero)aryl and 2-(hetero)aryl substituted dithienothiazines are readily accessible from N-aryl dithienothiazines by lithiation, lithium–zinc exchange, and subsequent Negishi cross-coupling with (hetero)aryl iodides in good to excellent yields and in a one-pot fashion. The title compounds possess an extended π-electron conjugation and display characteristic electronic features. While the electronic ground state properties, investigated by cyclic voltammetry and UV/vis spectroscopy, indicate a strong influence of the remote substituents that can be semiquantitatively treated by excellent Hammett's linear free enthalpy correlations with σp (oxidation potentials) and σp− (λmax,abs), luminescence as an excited state characteristic is only found for four derivatives with characteristic heterocyclic substitution. In principle this emission behavior is unusual for N-aryl dithienothiazines, but it establishes a novel class of redox active lumophores. The rapid access of 2,6-di(hetero)aryl dithienothiazines with remote substitution furnishing tunable electronic properties renders this class of electron rich reversible multistep redox systems highly intriguing as potential hole conductor materials. Further studies exploiting this expedient synthetic strategy to functional dithienothiazines are currently underway.Experimental
Synthetic procedures
| Entry | (Hetero)aryl iodide 2 [mg] (mmol) | 2,6-Di(hetero)aryl dithienothiazines 3 [mg] (%) |
|---|---|---|
| 1 | 257 (1.10) of 1-iodo-4-methoxybenzene (2a) | 189 (65) of 3a |
| 2 | 240 (1.10) of 1-iodo-4-methylbenzene (2b) | 226 (82) of 3b |
| 3 | 224 (1.10) of iodobenzene (2c) | 227 (87) of 3c |
| 4 | 262 (1.10) of 1-chloro-4-iodobenzene (2d) | 231 (78) of 3d |
| 5 | 288 (1.10) of methyl 4-iodobenzoate (2e) | 257 (80) of 3e |
| 6 | 299 (1.10) of 4-iodobenzotrifluoride (2f) | 313 (95) of 3f |
| 7 | 252 (1.10) of 4-iodobenzonitrile (2g) | 198 (69) of 3g |
| 8 | 274 (1.10) of 1-iodo-4-nitrobenzene (2h) | 241 (78) of 3h |
| 9 | 274 (1.10) of 1-iodo-3-nitrobenzene (2i) | 236 (77) of 3i |
| 10 | 274 (1.10) of 1-iodo-2-nitrobenzene (2j) | 165 (54) of 3j |
| 11 | 226 (1.10) of 3-iodopyridine (2k) | 164 (63) of 3k |
| 12 | 450 (1.10) of 3-iodo-10-n-hexyl-10H-phenothiazine (2l) | 441 (94) of 3l |
4-(4-n-Hexylphenyl)-2,6-bis(4-methoxyphenyl)-4H-dithieno[2,3-b:3′,2′-e][1,4]thiazine (3a). According to the GP1 and after flash chromatography on silica gel (n-hexane–ethyl acetate 20
:1 with 2% of triethylamine) and after crystallization from ethanol–ethyl acetate (1:1), compound 3a (189 mg, 65%) was obtained as yellow needles.
R
f (n-hexane–ethyl acetate 20:1): 0.19, Mp 170 °C. 1H NMR (600 MHz, acetone-d6–CS2 1:1, T = 313 K): δ 0.96 (t, 3J = 7.1 Hz, 3 H), 1.37–1.48 (m, 6 H), 1.75 (p, 3J = 7.4 Hz, 2 H), 2.71–2.77 (m, 2 H), 3.79 (s, 6 H), 6.24 (s, 2 H), 6.80–6.85 (m, 4 H), 7.21–7.29 (m, 4 H), 7.33–7.41 (m, 4 H). 13C NMR (150 MHz, acetone-d6–CS2 1:1, T = 313 K): δ 14.9 (CH3), 23.8 (CH2), 29.9 (CH2), 32.3 (CH2), 32.8 (CH2), 36.6 (CH2), 55.7 (CH3), 102.3 (Cquat), 115.2 (CH), 115.5 (CH), 127.1 (CH), 127.2 (Cquat), 129.2 (CH), 131.1 (CH), 142.1 (Cquat), 142.4 (Cquat), 143.3 (Cquat), 144.7 (Cquat), 160.4 (Cquat). MALDI-TOF MS: m/z 583.2 [M]+. IR (ATR): 602 cm−1 (m), 615 (s), 627 (m), 633 (m), 644 (s), 698 (s), 723 (s), 760 (m), 795 (s), 802 (s), 812 (s), 829 (m), 947 (m), 999 (w), 1018 (w), 1032 (s), 1057 (w), 1076 (w), 1111 (m), 1153 (w), 1177 (s), 1250 (s), 1296 (m), 1375 (m), 1439 (m), 1463 (m), 1501 (s), 1564 (w), 1568 (w), 1607 (w), 2833 (w), 2853 (w), 2922 (w), 2947 (w). UV/Vis (CH2Cl2): λmax (ε) 295 nm (46100), 367 (6000, sh). Anal. calcd for C34H33NO2S3 (583.8): C 69.95, H 5.70, N 2.40, S 16.48; found: C 69.67, H 5.63, N 2.29, S 16.59.
4-(4-Hexylphenyl)-2,6-di-p-tolyl-4H-dithieno[2,3-b:3′,2′-e][1,4]thiazine (3b). According to the GP1 and after flash chromatography on silica gel (n-pentane with 0.5% of triethylamine), compound 3b (134 mg, 78%) was obtained as an orange oil.
R
f (n-hexane): 0.14. 1H NMR (600 MHz, acetone-d6–CS2 1:1): δ 0.93 (t, 3J = 7.0 Hz, 3 H), 1.35–1.44 (m, 6 H), 1.70–1.76 (m, 2 H), 2.31 (s, 6 H), 2.72–2.76 (m, 2 H), 6.37 (s, 2 H), 7.11–7.14 (m, 4 H), 7.24–7.27 (m, 4 H), 7.38–7.43 (m, 4 H). 13C NMR (150 MHz, acetone-d6–CS2 1:1): δ 14.7 (CH3), 21.4 (CH3), 23.6 (CH2), 30.0 (CH2), 32.3 (CH2), 32.7 (CH2), 36.5 (CH2), 102.6 (Cquat), 115.9 (CH), 125.6 (CH), 129.3 (CH), 130.5 (CH), 131.3 (CH), 138.5 (Cquat), 142.0 (Cquat), 142.6 (Cquat), 143.5 (Cquat), 145.0 (Cquat). MALDI-TOF MS: m/z 551.2 [M]+. IR (ATR): 617 cm−1 (w), 638 (w), 667 (w), 710 (w), 760 (m), 804 (s), 945 (w), 999 (w), 1018 (w), 1038 (w), 1057 (w), 1111 (w), 1121 (w), 1182 (w), 1217 (w), 1280 (m), 1310 (w), 1357 (m), 1435 (m), 1504 (s), 1537 (w), 1574 (w), 1611 (w), 2853 (w), 2924 (m), 2953 (w), 3022 (w). UV/Vis (CH2Cl2): λmax (ε) 294 nm (48200), 385 (5700). HRMS (ESI) calcd for C34H33NS3: 551.17751; found: 551.17757.
4-(4-Hexylphenyl)-2,6-diphenyl-4H-dithieno[2,3-b:3′,2′-e][1,4]thiazine (3c). According to the GP1 and after flash chromatography on silica gel (n-hexane with 2% of triethylamine) and after crystallization from ethanol–ethyl acetate (20
:1), compound 3c (227 mg, 87%) was obtained as an orange powder.
R
f (n-hexane): 0.09, Mp 114 °C. 1H NMR (300 MHz, acetone-d6–CS2 1:1): δ 0.96 (t, 3J = 7.0 Hz, 3 H), 1.33–1.50 (m, 6 H), 1.76 (p, 3J = 7.6 Hz, 2 H), 2.70–2.82 (m, 2 H), 6.36 (s, 2 H), 7.18–7.47 (m, 14 H). 13C NMR (75 MHz, acetone-d6–CS2 1:4): δ 15.1 (CH3), 23.8 (CH2), 29.9 (CH2), 32.3 (CH2), 32.8 (CH2), 36.6 (CH2), 103.1 (Cquat), 115.9 (CH), 125.5 (CH), 128.2 (CH), 129.3 (CH), 129.5 (CH), 131.0 (CH), 134.1 (Cquat), 141.5 (Cquat), 141.9 (Cquat), 143.2 (Cquat), 144.4 (Cquat). MALDI-TOF MS: m/z 523.2 [M]+. IR (ATR): 673 cm−1 (w), 685 (s), 712 (w), 748 (s), 810 (w), 822 (m), 889 (w), 945 (w), 1003 (w), 1072 (w), 1098 (w), 1192 (w), 1273 (w), 1368 (w), 1418 (w), 1425 (w), 1451 (m), 1489 (m), 1508 (w), 1528 (w), 1566 (w), 1597 (w), 2359 (w), 2851 (w), 2920 (w), 2053 (w). UV/Vis (CH2Cl2): λmax (ε) 290 nm (49500), 400 (5250). Anal. calcd for C32H29NS3 (522.8): C 73.38, H 5.58, N 2.67; found: C 73.19, H 5.55, N 2.74.
2,6-Bis(4-chlorphenyl)-4-(4-hexylphenyl)-4H-dithieno[2,3-b:3′,2′-e][1,4]thiazine (3d). According to the GP1 and after flash chromatography on silica gel (n-hexane with 2% of triethylamine) and after crystallization from n-hexane, compound 3d (231 mg, 78%) was obtained as light red crystals.
R
f (n-hexane): 0.18, Mp 131 °C. 1H NMR (500 MHz, CD2Cl2): δ 0.91 (t, 3J = 7.0 Hz, 3 H), 1.30–1.41 (m, 6 H), 1.69 (p, 3J = 7.5 Hz, 2 H), 2.67–2.72 (m, 2 H), 6.31 (s, 2 H), 7.24–7.37 (m, 12 H). 13C NMR (125 MHz, CD2Cl2): δ 14.4 (CH3), 23.2 (CH2), 29.6 (CH2), 31.9 (CH2), 32.2 (CH2), 36.2 (CH2), 103.0 (Cquat), 116.2 (CH), 126.6 (CH), 129.0 (CH), 129.5 (CH), 130.9 (CH), 132.7 (Cquat), 133.7 (Cquat), 140.4 (Cquat), 141.2 (Cquat), 143.7 (Cquat), 144.8 (Cquat). MALDI-TOF MS: m/z 591.1 [M]+. IR (ATR): 615 cm−1 (w), 627 (w), 644 (w), 669 (w), 694 (w), 714 (w), 731 (w), 766 (w), 806 (s), 814 (s), 820 (m), 943 (w), 1003 (w), 1059 (w), 1094 (m), 1117 (w), 1179 (w), 1192 (w), 1273 (w), 1371 (m), 1398 (w), 1431 (m), 1485 (m), 1508 (w), 1568 (m), 1888 (w), 2853 (w), 2924 (w), 2053 (w), 3030 (w). UV/Vis (CH2Cl2): λmax (ε) 296 nm (49550), 409 (6000). Anal. calcd for C32H27Cl2NS3 (591.7): C 64.85, H 4.59, N 2.36; found: C 64.80, H 4.81, N 2.26.
Dimethyl 4,4′-(4-(4-hexylphenyl)-4H-dithieno[2,3-b:3′,2′-e][1,4]thiazin-2,6-diyl)dibenzoate (3e). According to the GP1 and after flash chromatography on silica gel (n-hexane–ethyl acetate 10
:1 with 2% of triethylamine) and after crystallization from dichloromethane–n-hexane, compound 3e (257 mg, 80%) was obtained as a red solid.
R
f (n-hexane–ethyl acetate 10:1): 0.28, Mp 199 °C. 1H NMR (300 MHz, acetone-d6–CS2 1:1, T = 293 K): δ 0.96 (t, 3J = 7.0 Hz, 3 H), 1.36–1.50 (m, 6 H), 1.69 (p, 3J = 7.6 Hz, 2 H), 2.74–2.81 (m, 2 H), 3.86 (s, 6 H), 6.51 (s, 2 H), 7.39–7.54 (m, 8 H), 7.89–7.96 (m, 4 H). 13C NMR (150 MHz, acetone-d6–CS2 1:1, T = 313 K): δ 14.9 (CH3), 23.8 (CH2), 30.2 (CH2), 32.3 (CH2), 32.8 (CH2), 36.6 (CH2), 52.3 (CH3), 98.9 (Cquat), 117.4 (CH), 125.3 (Cquat), 126.0 (CH), 128.1 (CH), 130.0 (Cquat), 131.03 (CH), 131.4 (CH), 138.3 (Cquat), 143.9 (Cquat), 144.9 (Cquat), 146.2 (Cquat), 166.2 (Cquat). MALDI-TOF MS: m/z 639.1 [M]+. IR (ATR): 665 cm−1 (w), 694 (m), 725 (w), 766 (s), 818 (m), 851 (w), 947 (w), 962 (w), 1001 (w), 1016 (w), 1063 (w), 1109 (s), 1186 (m), 1192 (w), 1244 (w), 1273 (s), 1317 (w), 1377 (w), 1406 (w), 1420 (w), 1431 (m), 1500 (w), 1539 (w), 1560 (w), 1572 (w), 1603 (m), 1722 (s), 2332 (w), 2361 (w), 2853 (w), 2924 (w). UV/Vis (CH2Cl2): λmax (ε) 309 nm (49600), 455 (8750). Anal. calcd for C36H33NO4S3 (639.9): C 67.58, H 5.20, N 2.19; found: C 67.51, H 5.01, N 2.16.
4-(4-Hexylphenyl)-2,6-bis(4-(trifluormethyl)phenyl)-4H-dithieno[2,3-b:3′,2′-e][1,4]thiazine (3f). According to the GP1 and after flash chromatography on silica gel (n-hexane with 2% of triethylamine) and after crystallization from n-hexane–ethyl acetate, compound 3f (313 mg, 95%) was obtained as fine coral red crystals.
R
f (n-hexane): 0.19, Mp 215 °C. 1H NMR (600 MHz, acetone-d6–CS2 1:1, T = 313 K): δ 0.95 (t, 3J = 7.0 Hz, 3 H), 1.37–1.49 (m, 6 H), 1.75 (p, 3J = 7.6 Hz, 2 H), 2.74–2.79 (m, 2 H), 6.54 (s, 2 H), 7.44 (q, 3J = 8.4 Hz, 4 H), 7.59 (dd, J = 8.3 Hz, 29.7 Hz, 8 H).13C NMR (150 MHz, acetone-d6–CS2 1:1, T = 313 K): δ 14.7 (CH3), 23.7 (CH2), 30.1 (CH2), 32.3 (CH2), 32.7 (CH2), 36.6 (CH2), 105.4 (Cquat), 117.8 (CH), 125.1 (Cquat, 1JCF = 271.6 Hz), 126.0 (CH), 126.9 (CH, 3JCF = 3.8 Hz), 129.5 (CH), 129.9 (Cquat, 2JCF = 32.5 Hz), 131.5 (CH), 137.9 (Cquat), 140.5 (Cquat), 141.6 (Cquat), 144.0 (Cquat), 145.3 (Cquat). MALDI-TOF MS: m/z 659.1 [M]+. IR (ATR): 610 cm−1 (w), 638 (w), 652 (w), 691 (w), 714 (w), 735 (w), 773 (w), 822 (s), 837 (m), 949 (w), 1003 (w), 1013 (m), 1069 (s), 1109 (s), 1121 (m), 1163 (m), 1194 (w), 1246 (w), 1275 (w), 1290 (w), 1325 (s), 1379 (w), 1406 (w), 1435 (m), 1508 (m), 1566 (w), 1611 (w), 2855 (w), 2901 (w), 2926 (w), 2959 (w). UV/Vis (CH2Cl2): λmax (ε) 298 nm (62850), 431 (8500). Anal. calcd for C34H27F6NS3 (659.8): C 61.89, H 4.12, N 2.12; found: C 61.70, H 4.21, N 2.06.
4,4′-(4-(4-Hexylphenyl)-4H-dithieno[2,3-b:3′,2′-e][1,4]thiazin-2,6-diyl)dibenzonitrile (3g). According to the GP1 and after flash chromatography on silica gel (n-hexane–ethyl acetate 10
:1), compound 3g (198 mg, 69%) was obtained as dark red crystals.
R
f (n-hexane–ethyl acetate 10:1): 0.11, Mp 175 °C. 1H NMR (600 MHz, acetone-d6–CS2 1:1, T = 313 K): δ 0.96 (t, 3J = 7.1 Hz, 3 H), 1.39–1.48 (m, 6 H), 1.75 (p, 3J = 7.5 Hz, 2 H), 2.75–2.78 (m, 2 H), 6.53 (s, 2 H), 7.39–7.46 (m, 4 H), 7.53 (d, J = 8.3 Hz, 4 H), 7.54–7.58 (d, J = 8.4 Hz, 4 H). 13C NMR (150 MHz, acetone-d6–CS2 1:1, T = 313 K): δ 14.8 (CH3), 23.7 (CH2), 30.1 (CH2), 32.3 (CH2), 32.7 (CH2), 36.6 (CH2), 111.8 (Cquat), 116.7 (CH), 118.0 (Cquat), 118.8 (Cquat), 126.0 (CH), 127.6 (CH), 131.5 (CH), 133.6 (CH), 138.1 (Cquat), 140.2 (Cquat), 141.8 (Cquat), 144.1 (Cquat), 146.4 (Cquat). MALDI-TOF MS: m/z 573.1 [M]+. IR (ATR): 617 cm−1 (w), 632 (w), 719 (w), 760 (w), 773 (w), 814 (s), 835 (m), 950 (w), 1001 (w), 1065 (w), 1109 (w), 1179 (m), 1248 (w), 1277 (m), 1312 (w), 1381 (m), 1404 (s), 1427 (s), 1497 (s), 1507 (m), 1574 (m), 1599 (s), 2220 (m), 2853 (w), 2924 (w), 2953 (w). UV/Vis (CH2Cl2): λmax (ε) 310 nm (43600), 457 (7200). Anal. calcd for C34H27N3S3 (573.8): C 71.17, H 4.74, N 7.32; found: C 70.94, H 4.73, N 7.06.
4-(4-Hexylphenyl)-2,6-bis(4-nitrophenyl)-4H-dithieno[2,3-b:3′,2′-e][1,4]thiazine (3h). According to the GP1 and after flash chromatography on silica gel (n-hexane–dichloromethane 10
:3 with 2% of triethylamine) and after crystallization from dichloromethane–n-hexane, compound 3h (241 mg, 78%) was obtained as a dark violet powder.
R
f (n-hexane–dichloromethane 10:3): 0.21, Mp 192 °C. 1H NMR (500 MHz, CD2Cl2): δ 0.92 (t, 3J = 7.1 Hz, 3 H), 1.32–1.43 (m, 6 H), 1.71 (p, 3J = 7.5 Hz, 2 H), 2.68–2.76 (m, 2 H), 6.43 (s, 2 H), 7.34 (d, 3J = 8.3 Hz, 2 H), 7.40 (d, 3J = 8.4 Hz, 2 H), 7.45–7.48 (m, 4 H), 8.10–8.13 (m, 4 H). 13C NMR (125 MHz, CD2Cl2): δ 14.4 (CH3), 23.2 (CH2), 29.6 (CH2), 31.9 (CH2), 32.2 (CH2), 36.2 (CH2), 106.0 (Cquat), 117.6 (CH), 124.8 (CH), 125.4 (CH), 129.1 (CH), 131.2 (CH), 139.0 (Cquat), 139.9 (Cquat), 140.7 (Cquat), 144.3 (Cquat), 145.1 (Cquat), 147.0 (Cquat). MALDI-TOF MS: m/z 613.1 [M]+. IR (ATR): 687 cm−1 (w), 746 (m), 822 (w), 833 (w), 849 (m), 1069 (w), 1107 (m), 1188 (w), 1215 (w), 1248 (w), 1273 (w), 1329 (s), 1385 (w), 1404 (w), 1427 (m), 1489 (m), 1514 (m), 1572 (w), 1589 (m), 2855 (w), 2926 (w). UV/Vis (CH2Cl2): λmax (ε) 254 nm (31300), 332 (46000), 524 (17900). Anal. calcd for C32H27N3O4S3 (613.8): C 62.62, H 4.43, N 6.85; found: C 62.43, H 4.44, N 6.57.
4-(4-Hexylphenyl)-2,6-bis(3-nitrophenyl)-4H-dithieno[2,3-b:3′,2′-e][1,4]thiazine (3i). According to the GP1 and after flash chromatography on silica gel (n-hexane–dichloromethane 10
:3 with 2% of triethylamine), compound 3i (236 mg, 77%) was obtained as a chestnut brown solid.
R
f (n-hexane–dichloromethane 10:3): 0.22, Mp 199 °C. 1H NMR (600 MHz, acetone-d6–CS2 1:1, T = 313 K): δ 0.93 (t, 3J = 7.1 Hz, 3 H), 1.37–1.48 (m, 6 H), 1.75 (p, 3J = 7.6 Hz, 2 H), 2.74–2.78 (m, 2 H), 6.63 (s, 2 H), 7.45 (s, 4 H), 7.62 (t, J = 8.0 Hz, 2 H), 7.80 (ddd, J = 0.9 Hz, 1.8 Hz, 7.8 Hz, 2 H), 8.08 (ddd, J = 0.9 Hz, 1.8 Hz, 8.2 Hz, 2 H), 8.17 (t, J = 2.0 Hz, 2 H). 13C NMR (150 MHz, acetone-d6–CS2 1:1, T = 313 K): δ 14.7 (CH3), 23.6 (CH2), 30.1 (CH2), 32.3 (CH2), 32.7 (CH2), 36.5 (CH2), 105.7 (Cquat), 118.2 (CH), 120.0 (CH), 122.9 (CH), 129.4 (CH), 131.3 (CH), 131.45 (CH), 131.53 (CH), 136.0 (Cquat), 139.7 (Cquat), 141.6 (Cquat), 144.1 (Cquat), 145.4 (Cquat), 149.8 (Cquat). MALDI-TOF MS: m/z 613.1 [M]+. IR (ATR): 671 cm−1 (m), 694 (w), 712 (w), 727 (m), 732 (m), 762 (w), 797 (m), 835 (w), 860 (w), 897 (w), 997 (w), 1067 (w), 1073 (w), 1101 (w), 1202 (w), 1281 (m), 1306 (w), 1352 (s), 1385 (w), 1427 (m), 1477 (m), 1510 (m), 1524 (s), 1578 (w), 1614 (w), 2335 (w), 2851 (w), 2922 (w), 2951 (w), 3084 (w). UV/Vis (CH2Cl2): λmax (ε) 286 nm (40100), 440 (4800). Anal. calcd for C32H27N3O4S3 (613.8): C 62.62, H 4.43, N 6.85, S 15.67; found: C 62.40, H 4.51, N 6.72, S 15.44.
4-(4-Hexylphenyl)-2,6-bis(2-nitrophenyl)-4H-dithieno[2,3-b:3′,2′-e][1,4]thiazine (3j). According to the GP1 and after flash chromatography on silica gel (n-hexane–dichloromethane 10
:3 with 2% of triethylamine), compound 3j (165 mg, 54%) was obtained as a chestnut brown oil.
R
f (n-hexane–dichloromethane 10:3): 0.35. 1H NMR (600 MHz, acetone-d6–CS2 1:1): δ 0.91 (t, 3J = 7.1 Hz, 3 H), 1.31–1.42 (m, 6 H), 1.69 (p, 3J = 7.5 Hz, 2 H), 2.68–2.72 (m, 2 H), 6.17 (s, 2 H), 7.29–7.32 (m, 2 H), 7.35–7.38 (m, 2 H), 7.52 (dd, J = 1.3 Hz, 7.8 Hz, 2 H), 7.57 (td, J = 1.4 Hz, 7.6 Hz, 2 H), 7.66 (td, J = 1.3 Hz, 7.6 Hz, 2 H), 7.77 (dd, J = 1.2 Hz, 8.0 Hz, 2 H). 13C NMR (150 MHz, acetone-d6–CS2 1:1): δ 14.8 (CH3), 23.6 (CH2), 30.0 (CH2), 32.3 (CH2), 32.7 (CH2), 36.4 (CH2), 106.0 (Cquat), 120.4 (CH), 124.8 (CH), 127.6 (Cquat), 128.7 (CH), 130.2 (CH), 131.2 (CH), 132.3 (CH), 133.1 (CH), 135.6 (Cquat), 141.3 (Cquat), 143.6 (Cquat), 144.8 (Cquat), 149.8 (Cquat). MALDI-TOF MS: m/z 613.2 [M]+. IR (ATR): 608 cm−1 (w), 652 (m), 681 (w), 696 (w), 710 (s), 721 (m), 748 (s), 775 (s), 824 (m), 831 (w), 837 (w), 853 (m), 949 (w), 988 (w), 999 (w), 1019 (w), 1063 (w), 1113 (w), 1142 (w), 1163 (w), 1188 (w), 1279 (m), 1302 (w), 1358 (s), 1379 (m), 1425 (m), 1481 (m), 1508 (m), 1526 (s), 1566 (w), 1576 (w), 1605 (w), 2855 (w), 2928 (w), 2955 (w). UV/Vis (CH2Cl2): λmax (ε) 264 nm (30250), 452 (2850). Anal. calcd for C32H27N3O4S3 (613.8): C 62.62, H 4.43, N 6.85, S 15.67; found: C 62.24, H 4.61, N 6.57, S 15.27.
4-(4-Hexylphenyl)-2,6-di(pyridin-3-yl)-4H-dithieno[2,3-b:3′,2′-e][1,4]thiazine (3k). According to the GP1 and after flash chromatography on silica gel (n-hexane–ethyl acetate 4
:1 with 2% of triethylamine), compound 3k (164 mg, 63%) was obtained as an intense orange oil.
R
f (n-hexane–ethyl acetate 4:1): 0.15. 1H NMR (300 MHz, acetone-d6–CS2 1:1): δ 0.95 (t, 3J = 7.0 Hz, 3 H), 1.33–1.50 (m, 6 H), 1.74 (p, 3J = 7.5 Hz, 2 H), 2.71–2.79 (m, 2 H), 6.45 (s, 2 H), 7.27 (ddd, J = 0.8 Hz, 4.8 Hz, 8.0 Hz, 2 H), 7.41 (s, 4 H), 7.67 (ddd, J = 1.6 Hz, 2.4 Hz, 8.0 Hz, 2 H), 8.41 (dd, J = 1.5 Hz, 4.7 Hz, 2 H), 8.59 (dd, J = 0.7 Hz, 2.4 Hz, 2 H). 13C NMR (75 MHz, acetone-d6–CS2 1:1): δ 14.9 (CH3), 23.7 (CH2), 30.2 (CH2), 32.3 (CH2), 32.7 (CH2), 36.6 (CH2), 104.5 (Cquat), 117.2 (CH), 124.3 (CH), 129.3 (CH), 130.0 (Cquat), 131.3 (CH), 132.2 (CH), 138.6 (Cquat), 141.4 (Cquat), 143.7 (Cquat), 145.1 (Cquat), 146.6 (CH), 149.4 (CH). MALDI-TOF MS: m/z 525.2 [M]+. IR (ATR): 619 cm−1 (w), 660 (w), 679 (w), 704 (s), 725 (w), 760 (w), 799 (s), 827 (w), 837 (w), 941 (m), 995 (w), 1022 (w), 1063 (w), 1101 (w), 1180 (w), 1234 (w), 1281 (m), 1339 (w), 1379 (m), 1418 (m), 1429 (s), 1477 (s), 1508 (m), 1539 (w), 1574 (m), 1585 (w), 2853 (w), 2924 (m), 2953 (w). UV/Vis (CH2Cl2): λmax (ε) 292 nm (32450), 414 (3900). Anal. calcd for C30H27N3S3 (525.7): C 68.53, H 5.18, N 7.99, S 18.30; found: C 68.13, H 5.26, N 7.75, S 18.51.
3,3′-(4-(4-Hexylphenyl)-4H-dithieno[2,3-b:3′,2′-e][1,4]thiazin-2,6-diyl)bis(10-hexyl-10H-phenothiazine) (3l). According to the GP1 and after flash chromatography on silica gel (n-hexane with 2% of triethylamine) and trituration with n-hexane under ultrasound, compound 3l (441 mg, 94%) was obtained as an orange solid.
R
f (n-hexane): 0.32, Mp 95 °C. 1H NMR (600 MHz, acetone-d6–CS2 1:2): δ 0.90 (t, 3J = 7.1 Hz, 6 H), 0.97 (t, 3J = 7.1 Hz, 3 H), 1.30–1.44 (m, 18 H), 1.75–1.82 (m, 6 H), 2.73-2.79 (m, 2 H), 3.85–3.89 (m, 4 H), 6.25 (s, 2 H), 6.83 (d, J = 8.6 Hz, 2 H), 6.87–6.91 (m, 4 H), 7.04 (dd, J = 1.5 Hz, 6.7 Hz, 2 H), 7.06 (d, J = 2.2 Hz, 2 H), 7.10–7.15 (m, 4 H), 7.34–7.38 (m, 2 H), 7.39–7.43 (m. 2 H). 13C NMR (150 MHz, acetone-d6–CS2 1:2): δ 14.8 (CH3), 14.9 (CH3), 23.7 (CH2), 23.8 (CH2), 27.5 (CH2), 27.7 (CH2), 30.2 (CH2), 32.4 (CH2), 32.5 (CH2), 32.8 (CH2), 36.6 (CH2), 48.1 (CH2), 102.2 (Cquat), 115.5 (CH), 116.4 (2 × CH), 123.4 (CH), 124.1 (CH), 124.78 (Cquat), 124.81 (Cquat), 126.3 (Cquat), 128.0 (CH), 128.2 (CH), 128.8 (CH), 129.3 (CH), 131.2 (CH), 141.4 (Cquat), 141.7 (Cquat), 143.4 (Cquat), 144.8 (Cquat), 145.36 (Cquat), 145.38 (Cquat). MALDI-TOF MS: m/z 933.3 [M]+. IR (ATR): 613 cm−1 (w), 677 (w), 680 (w), 710 (w), 723 (w), 743 (s), 752 (m), 783 (w), 804 (s), 822 (w), 864 (w), 879 (w), 930 (w), 997 (w), 1042 (w), 1059 (w), 1109 (w), 1142 (w), 1167 (w), 1188 (w), 1246 (m), 1285 (w), 1333 (w), 1362 (m), 1373 (m), 1404 (w), 1439 (m), 1466 (s), 1493 (m), 1539 (w), 1572 (w), 1599 (w), 2853 (w), 2868 (w), 2926 (w), 2951 (w). UV/Vis (CH2Cl2): λmax (ε) 239 nm (sh, 27900), 267 (sh, 30250), 291 (36250), 349 (14300), 422 (sh, 8500). Anal. calcd for C56H59N3S5 (933.4): C 71.98, H 6.36, N 4.50, S 17.16; found: C 71.83, H 6.22, N 4.42, S 17.31.
| Entry | (Hetero)aryl iodide 2 [mg] (mmol) | 2-(Hetero)aryl dithienothiazines 6 [mg] (%) |
|---|---|---|
| 1 | 125 (0.50) of 1-iodo-4-nitrobenzene (2h) | 108 (44) of 6a |
| 2 | 105 (0.50) of 2-iodothiophene (2m) | 138 (61) of 6b |
| 3 | 103 (0.50) of 4-iodopyridine (2n) | 105 (47) of 6c |
4-(4-Hexylphenyl)-2-(4-nitrophenyl)-4H-dithieno[2,3-b:3′,2′-e][1,4]thiazine (6a). According to the GP2 and after twofold flash chromatography on silica gel (n-hexane–dichloromethane with 2% of triethylamine and n-hexane–toluene 10
:1 with 1% of triethylamine), compound 6a (108 mg, 44%) was obtained as a dark violet oil.
R
f (n-hexane–dichloromethane 10:1): 0.09. 1H NMR (600 MHz, acetone-d6–CS2 2:1): δ 0.94 (t, 3J = 7.1 Hz, 3 H), 1.36–1.46 (m, 6 H), 1.72 (p, 3J = 7.5 Hz, 2 H), 2.72–2.76 (m, 2 H), 6.11 (d, J = 5.5 Hz, 1 H), 6.64 (s, 1 H), 7.19 (d, J = 5.5 Hz, 1 H), 7.36–7.39 (m, 2 H), 7.41–7.44 (m, 2 H), 7.61–7.65 (m, 2 H), 8.15–8.19 (m, 2 H). 13C NMR (150 MHz, acetone-d6–CS2 2:1): δ 14.7 (CH3), 23.6 (CH2), 30.1 (CH2), 32.4 (CH2), 32.7 (CH2), 36.5 (CH2), 102.7 (Cquat), 107.9 (Cquat), 118.5 (CH), 120.7 (CH), 125.0 (CH), 125.2 (CH), 125.9 (CH), 129.3 (CH), 131.4 (CH), 139.2 (Cquat), 140.3 (Cquat), 141.7 (Cquat), 143.8 (Cquat), 144.4 (Cquat), 146.1 (Cquat), 147.4 (Cquat). MALDI-TOF MS: m/z 492.1 [M]+. IR (ATR): 611 cm−1 (m), 629 (m), 687 (s), 696 (s), 714 (m), 748 (s), 799 (s), 818 (s), 847 (s), 948 (w), 999 (m), 1017 (s), 1061 (m), 1096 (s), 1107 (s), 1175 (w), 1203 (w), 1260 (s), 1329 (s), 1380 (m), 1329 (s), 1380 (m), 1402 (m), 1427 (m), 1491 (m), 1506 (s), 1562 (w), 1589 (m), 2853 (w), 2922 (w), 2957 (w). UV/Vis (CH2Cl2): λmax (ε) 250 nm (25550), 328 (22850), 511 (7300). HRMS (ESI) calcd for C26H24N2O2S3: 492.09999, found: 492.09937.
4-(4-Hexylphenyl)-2-(thiophen-2-yl)-4H-dithieno[2,3-b:3′,2′-e][1,4]thiazine (6b). According to the GP2 and after flash chromatography on silica gel (n-hexane with 2% of triethylamine), compound 6b (138 mg, 61%) was obtained as a pale orange oil.
R
f (n-hexane): 0.19. 1H NMR (600 MHz, acetone-d6–CS2 5:1): δ 0.93 (t, 3J = 7.0 Hz, 3 H), 1.36–1.44 (m, 6 H), 1.72 (p, 3J = 7.6 Hz, 2 H), 2.71–2.75 (m, 2 H), 6.10 (d, J = 5.5 Hz, 1 H), 6.20 (s, 1 H), 6.98 (dd, J = 3.6 Hz, 5.1 Hz, 1 H), 7.04 (dd, J = 1.1 Hz, 3.6 Hz, 1 H), 7.15 (d, J = 5.5 Hz, 1 H), 7.29 (dd, J = 1.1 Hz, 5.1 Hz, 1 H), 7.32–7.35 (m, 2 H). 7.39–7.41 (m, 2 H). 13C NMR (150 MHz, acetone-d6–CS2 2:1): δ 14.7 (CH3), 23.6 (CH2), 29.9 (CH2), 32.4 (CH2), 32.7 (CH2), 36.5 (CH2), 102.7 (Cquat), 103.2 (Cquat), 116.6 (CH), 120.6 (CH), 124.4 (CH), 124.6 (CH), 125.7 (CH), 128.8 (CH), 129.3 (CH), 131.2 (CH), 135.6 (Cquat), 137.2 (Cquat), 141.8 (Cquat), 143.6 (Cquat), 144.6 (Cquat), 145.0 (Cquat). MALDI-TOF MS: m/z 452.9 [M]+. IR (ATR): 627 cm−1 (w), 642 (w), 669 (w), 692 (s), 758 (w), 810 (m), 833 (m), 845 (m), 883 (w), 905 (w), 997 (m), 1016 (w), 1043 (w), 1059 (w), 1078 (w), 1098 (w), 1180 (w), 1225 (w), 1279 (m), 1333 (w), 1350 (w), 1377 (m), 1406 (m), 1459 (m), 1508 (s), 1537 (w), 1570 (w), 1611 (w), 2853 (w), 2924 (m), 2953 (w). UV/Vis (CH2Cl2): λmax (ε) 237 nm (sh, 12400), 300 (12700), 355 (sh, 2950), 420 (sh, 1900). HRMS (ESI) calcd for C24H23NS4: 453.07133, found: 453.07097.
4-(4-Hexylphenyl)-2-(pyridin-4-yl)-4H-dithieno[2,3-b:3′,2′-e][1,4]thiazin (6c). According to the GP2 and after flash chromatography on silica gel (n-hexane–ethyl acetate 5
:1 with 2% of triethylamine), compound 6c (105 mg, 47%) was obtained as a red oil.
R
f (n-hexane–ethyl acetate 5:1): 0.19. 1H NMR (300 MHz, acetone-d6–CS2 5:1): δ 0.96 (t, 3J = 7.1 Hz, 3 H), 1.34–1.47 (m, 6 H), 1.73 (p, 3J = 7.6 Hz, 2 H), 2.70–2.77 (m, 2 H), 6.10 (d, J = 5.5 Hz, 1 H), 6.60 (s, 1 H), 7.15 (d, J = 5.5 Hz, 1 H), 7.24–7.27 (m, 2 H), 7.32–7.37 (m, 2 H), 7.39–7.43 (m, 2 H), 8.43–8.46 (m, 2 H). 13C NMR (75 MHz, acetone-d6–CS2 2:1): δ 14.8 (CH3), 23.7 (CH2), 30.0 (CH2), 32.4 (CH2), 32.7 (CH2), 36.5 (CH2), 102.8 (Cquat), 106.8 (Cquat), 118.0 (CH), 119.2 (CH), 120.6 (CH), 124.8 (CH), 129.3 (CH), 131.3 (CH), 138.8 (Cquat), 140.7 (Cquat), 141.7 (Cquat), 143.7 (Cquat), 144.3 (Cquat), 145.7 (Cquat), 151.2 (CH). MALDI-TOF MS: m/z 448.2 [M]+. IR (ATR): 627 cm−1 (w), 644 (w), 656 (m), 665 (w), 696 (m), 806 (s), 839 (m), 961 (w), 991 (m), 1000 (m), 1016 (w), 1061 (w), 1098 (w), 1117 (w), 1196 (w), 1217 (w), 1279 (m), 1325 (w), 1377 (m), 1404 (s), 1431 (s), 1458 (w), 1491 (s), 1508 (m), 1526 (m), 1545 (w), 1566 (m), 1591 (s), 2853 (w), 2924 (m), 2951 (w), 3028 (w). UV/Vis (CH2Cl2): λmax (ε) 238 nm (sh, 9750), 292 (13250), 367 (1900), 449 (2150). Anal. calcd for C25H24N2S3·⅓ CH3CO2C2H5 (448.7 + 29.4): C 66.17, H 5.62, N 5.86, S 20.12, found: C 66.23, H 5.44, N 5.84, S 20.48.
Acknowledgements
The authors cordially thank the Fonds der Chemischen Industrie for financial support.Notes and references
- For a monograph, see e.g. Functional Organic Materials – Syntheses Strategies, and Applications, ed. T. J. J. Müller and U. H. F. Bunz, Wiley-VCH, Weinheim, 2007 Search PubMed.
- For excellent reviews, see e.g. M. Farinola and R. Ragni, Chem. Soc. Rev., 2011, 40, 3467 RSC; L. Hu and G. Xu, Chem. Soc. Rev., 2010, 39, 3275 RSC; G. Qian, Z. Zhong, M. Luo, D. Yu, Z. Zhang, Z. Y. Wang and D. Ma, Adv. Mater., 2009, 21, 111 CrossRef CAS PubMed; J. Shinar and R. Shinar, J. Phys. D: Appl. Phys., 2008, 41, 133001 CrossRef; J. A. G. Williams, A. J. Wilkinson and V. L. Whittle, Dalton Trans., 2008, 2081 RSC; Organic Light Emitting Devices: Synthesis, Properties and Applications, ed. K. Müllen and U. Scherf, Wiley-VCH, Weinheim, 2006 CrossRef PubMed; J. G. C. Veinot and T. J. Marks, Acc. Chem. Res., 2005, 38, 632 CrossRef PubMed; G. Hughes and M. R. Bryce, J. Mater. Chem., 2005, 15, 94 RSC; C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef PubMed.
- For excellent reviews, see e.g. X. Huang, S. Han, W. Huang and X. Liu, Chem. Soc. Rev., 2013, 42, 173 RSC; M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453 RSC; C. W. Schlenker and M. E. Thompson, Chem. Commun., 2011, 3702 RSC; J. L. Delgado, P.-A. Bouit, S. Filippone, M. A. Herranz and N. Martin, Chem. Commun., 2010, 46, 4853 RSC; A. Mishra, M. K. R. Fischer and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 2474 CrossRef CAS PubMed; Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef PubMed; B. C. Thompson and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2008, 47, 58 CrossRef PubMed; S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324 CrossRef PubMed; Organic Photovoltaics: Concept and Realization, C. J. Brabec, ed., Springer, Berlin, 2003 CrossRef PubMed; P. Peumans, A. Yakimov and S. R. Forrest, J. Appl. Phys., 2003, 93, 3693 CrossRef PubMed.
- For excellent reviews, see e.g. W. Wu, Y. Liu and D. Zhu, Chem. Soc. Rev., 2010, 39, 1489 RSC; H. Klauk, Chem. Soc. Rev., 2010, 39, 2643 RSC; H. Dong, C. Wang and W. Hu, Chem. Commun., 2010, 46, 5211 RSC; J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452 CrossRef CAS PubMed; A. L. Briseno, S. C. B. Mannsfeld, S. A. Jenekhe, Z. Bao and Y. Xia, Mater. Today, 2008, 11, 38 CrossRef; A. Facchetti, Mater. Today, 2007, 10, 28 CrossRef; A. R. Murphy and J. M. J. Fréchet, Chem. Rev., 2007, 107, 1066 CrossRef PubMed; M. Mas-Torrent and C. Rovira, Chem. Soc. Rev., 2008, 37, 827 RSC; M. Mas-Torrent and C. Rovira, Mater. Chem., 2006, 16, 433 RSC; Y. Sun, Y. Liu and D. Zhu, J. Mater. Chem., 2005, 15, 53 RSC; C. D. Dimitrakopoulos and P. R. L. Malenfant, Adv. Mater., 2002, 14, 99 CrossRef; G. Horowitz, Adv. Mater., 1998, 10, 365 CrossRef.
- J. R. Sheats, J. Mater. Res., 2004, 19, 1974 CrossRef CAS; D. J. Gundalch, J. A. Nichols, I. Zhou and T. N. Jackson, Appl. Phys. Lett., 2002, 80, 2925 CrossRef PubMed; A. Hepp, N. von Malm, R. Schmechel and H. von Seggern, Synth. Met., 2003, 138, 201 CrossRef.
- For reviews on molecular wires, see e.g. D. K. James and J. M. Tour, Top. Curr. Chem., 2005, 257, 33 CrossRef CAS; N. Robertson and C. A. McGowan, Chem. Soc. Rev., 2003, 32, 96 RSC.
- For reviews on unimolecular electronics, see e.g. R. M. Metzger, J. Mater. Chem., 2008, 18, 4364 RSC; S. R. Forrest, Nature, 2004, 428, 911 CrossRef CAS PubMed; R. L. Carroll and C. B. Gorman, Angew. Chem., Int. Ed., 2002, 41, 4379 CrossRef; J. M. Tour, Acc. Chem. Res., 2000, 33, 791 CrossRef PubMed.
- C. Dostert, C. Wanstrath, W. Frank and T. J. J. Müller, Chem. Commun., 2012, 48, 7271 RSC.
- S. Gronowitz and A. B. Hörnfeldt, Thiophenes (Best Synthetic Methods), Elsevier Academic Press, Oxford, 1st edn, 2004 RSC; D. J. Chadwick and C. Willbe, J. Chem. Soc., Perkin Trans. 1, 1977, 887 RSC; B. J. Wakefield, The Chemistry of Organolithium Compounds, Pergamon, Oxford, 1974 CrossRef CAS; J. M. Mallan and R. L. Bebb, Chem. Rev., 1969, 69, 693 CrossRef CAS; H. Gilman and D. Shirley, J. Am. Chem. Soc., 1949, 71, 1870 CrossRef.
- C. Muschelknautz, C. Dostert and T. J. J. Müller, Synlett, 2010, 415 CAS.
- C. Dostert, D. Czajkowski and T. J. J. Müller, Synlett, 2014, 371 CAS.
- For recent reviews, see: P. Knochel, M. I. Calaza, E. Hupe, E.-I. Negishi and F. Liu, in Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2004, p. 619 CrossRef CAS; E.-I. Negishi, X. Zeng, Z. Tan, M. Qian, Q. Hu and Z. Huang, in Metal-Catalyzed Cross-Coupling Reactions, ed. A. de Meijere and F. Diederich, Wiley-VCH, Weinheim, 2004, p. 815 CrossRef CAS; E. Negishi, Acc. Chem. Res., 1982, 15, 340 CrossRef CAS; E. Negishi, Q. Hu, Z. Huang, M. Qiang and G. Wang, Aldrichimica Acta, 2005, 38, 71 Search PubMed; E. Negishi, in Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi, Wiley-Interscience, New York, 2002, vol. 2, p. 229 Search PubMed.
- D. Cai, D. L. Hughes and T. R. Verhoeven, Tetrahedron Lett., 1996, 37, 2537 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 03, Revision D.01, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed; A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef PubMed; R. G. Parr and W. Yang, Density-Functional Theory of Atoms and Molecules, Oxford University Press, Oxford, 1989 Search PubMed.
- R. Ditchfield, R. W. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- L. Michaelis, Chem. Rev., 1935, 16, 243 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Ei-ichi Negishi on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: 1H and 13C NMR spectra, UV/Vis and fluorescence spectra of compounds 5, 6, 7, and 8; computed xyz-coordinates of the S0 state of the pyrazoles 5c, 6d, and 8b, computed UV/Vis spectra of TD-DFT calculated structures of 5c, 6d, and 8b, and computed xyz-coordinates of the S1 state of pyrazole 6d. See DOI: 10.1039/c5qo00046g |
| This journal is © the Partner Organisations 2015 |