 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel approach to high pressure flow digestion
Helmar
Wiltsche
*,
Paul
Tirk
,
Herbert
Motter
,
Monika
Winkler
and
Günter
Knapp
Graz, University of Technology, Institute of Analytical Chemistry and Food Chemistry, Graz, Austria. E-mail: helmar.wiltsche.tugraz.at
First published on 7th November 2013
Abstract
A new high pressure flow digestion system has been developed for sample digestion at a pressure of up to 40 bar and a temperature of about 230 °C. The reaction with acids takes place in a PFA tube and is heated by microwave radiation in a multimode cavity. As the PFA tube cannot withstand the harsh digestion conditions without support, it is placed inside a coiled glass tube pressurized by 40 bar nitrogen thus forming an autoclave. Corrosion of system components by acid fumes and related sample contamination is circumvented by establishing a slow but steady flow of the high pressure nitrogen countercurrent to the sample flow. The presented system does not constrain the selection of the digestion reagent. Acid cocktails of nitric acid with hydrochloric and/or hydrofluoric acid as well as hydrogen peroxide were successfully used for the digestion of various samples. The method accuracy was validated with five certified reference materials (BCR 62, DORM-2, NIST SRM 1515, NIST SRM 1567, NIST SRM 1568) and good agreement between the determined and the certified values was obtained for Al, Ca, Cr, Cu, Fe, Mg, Mn, Ni, Pb, and Zn using inductively coupled plasma optical emission spectrometry (ICP-OES) for analyte quantification. The flow digestion of the CRMs resulted in clear solutions with residual carbon concentrations (RCC) between 11 and 40%. Spike recoveries of Al, As, Ba, Be, Bi, Cd, Co, Cr, Cu, Fe, Mg, Mn, Mo, Ni, Pb, Sb, Se, Sr, Ti, V, and Zn were between 94 and 105%. For Hg the spike recovery was 89%. The fully automated high pressure flow digestion system is capable of digesting up to 6 samples per hour.
Introduction
Flow digestion is an attractive alternative to closed vessel microwave assisted digestion due to the ease of automation, the reduced risk of contamination and the capability of direct coupling to analyte quantification techniques. In general the sample is first mixed with the digestion acid (usually HNO3 or acid cocktails). Then the sample–acid mixture is heated by passing through a heating zone. In continuous flow systems the sample is pumped through the heating zone continuously by a carrier solution1–3 while in stopped flow systems the sample is pushed into the heating zone by the carrier solution, then the carrier flow is stopped and the sample is heated.4–6 At the end of the digestion period, the sample is pushed out of the digestion zone by turning on the carrier solution again. In most flow digestion systems reported in the literature microwave heating was used but in some cases conductive heating was employed. After the heating zone, gases that evolved during the digestion (e.g. nitrous oxides, CO2) can be removed by a gas/liquid separator.Flow digestion systems can be classified as already noted by their mode of operation (continuous flow or stopped flow), by their means of heating the sample–acid mixture in the digestion zone (conductive heating, microwave heating) or by the pressure inside the digestion system. The pressure inside the heated digestion coil is of great importance as it determines the maximum temperature of the acid mixture and thereby the efficiency of the digestion.7 Three pressure regimes may be distinguished: ambient, medium (<25 bar) and high pressure (>25 bar) flow digestion systems. It is well known that the digestion efficiency of any acid digestion system increases with the temperature of the digestion acid. As the boiling point of the acid limits the maximum attainable digestion temperature it is highly desirable to increase the pressure inside the digestion system.
Ambient pressure systems dominate the literature as they are relatively easy to build. Burguera8–10 pioneered these systems using microwave assisted sample heating. Ambient pressure flow digestion systems are capable of operating with highly corrosive acids like HCl or mixtures of HCl and HNO3 as the entire flow path is usually made of either inert polymers or glass.10,11 The main disadvantage of ambient pressure flow digestion systems is that gaseous reaction products eject the sample–acid mixture quickly from the heated dissolution zone reducing the effective digestion time and causing undesired peak broadening. The maximum digestion acid concentration12,13 and the maximum power level14–16 in the heated digestion zone are therefore limited by the gas evolution. Moreover, the digestion acid boils at about 120 °C, causing low efficient oxidation of organic substances.
In a medium pressure digestion system the digestion acid is pressurized up to about 25 bar. Thereby the acid's boiling point is considerably increased (e.g. 10% HNO3: 230 °C) and the solubility of gaseous reaction products in the digestion mixture is enhanced significantly, reducing dispersion effects.2 The pressure limit of 25 bar is somewhat arbitrarily chosen as the pressure limit of fiber reinforced PTFE tubes.17 In medium pressure flow digestion systems the oxidation efficiency of HNO3 is significantly higher than in ambient pressure systems.
High pressure flow digestion systems operate at pressures above 25 bar. This pressure region is comparable with contemporary closed vessel microwave assisted batch digestion systems. The main difference between batch digestion systems and flow systems in this pressure region is the shorter digestion time in flow systems. Typical digestion times in flow systems are between 2 and 5 min. Haiber and Berndt18 developed a high pressure system operating at up to 360 °C and 300 bar pressure. In this pressure range all reaction products which are gaseous at ambient pressure remain in a liquid phase.19 The high digestion temperature resulted in extremely low residual carbon concentrations (≪1% RCC). A Pt/Ir (80/20) tube was used as the heated pressurized digestion tube20 as this material showed excellent resistance to nitric and hydrofluoric acid.18,21 Nevertheless, mixtures of nitric and hydrochloric acid drastically reduced the digestion tube lifetime.21 The digestion tube was directly heated by clamping a supply voltage to the two ends of the digestion coil making use of its inherent resistance.18 It is interesting to note, that Bian et al.22 encountered losses of Ag, Ga and Sb during high pressure digestion in the Pt/Ir tube that increased with rising digestion temperature.
Another high pressure flow digestion system using microwave heating is the pressure equilibrium system described by Knapp et al.7,23 The underlying principle is that microwave energy is dominantly absorbed by the liquid phase, whereas steam and gaseous reaction products are not significantly heated. Thereby, a boiling equilibrium is formed much in the same way as in reflux boiling. In the pressure equilibrium system the PTFE dissolution coil is placed in a pressurized vessel, which in turn is located in a microwave field resulting in nearly equal pressure inside and outside of the PTFE tubing. This reduces the mechanical stress on the tubing significantly. Nevertheless, the pressure equilibrium system had several shortcomings: the length of the digestion tube – and as a consequence the actively heated volume – is restricted by the size of the waveguide of the used focused microwave oven. Moreover, cross-contamination between successive samples was observed and delicate optimization of the restrictor length, system pressure and carrier flow was necessary.
The aim of this work was to develop a radical new design of a pressure equilibrium system, overcoming the previous shortcomings. A high degree of automation was considered necessary to ensure reproducible experimental conditions.
Experimental
Flow digestion system
A high pressure, continuous flow, pressure equilibrium7 flow digestion system was constructed for this work. Briefly, diluted nitric acid (1% v/v) was continuously pumped through the system by an acid resistant all Ti HPLC pump (Knauer, Germany – Fig.1A). If not stated otherwise a carrier flow of 2.0 mL min−1 was used. A 10 mL sample loop (Fig. 1C) connected to a 6-port valve (Knauer, Germany – Fig. 1B, fluorinated polymer sealed, wide bore channel) was used to inject the samples (Fig. 1L) into the high pressure stream of the flow system. Automated digestion operation was attained by using an autosampler for sample handling (ASX-1400, Cetac, USA; not shown in Fig. 1 for clarity). Two autosampler needles were used: one for sample uptake and one for collecting the digests. Slurries were stirred for 10 seconds by a polyetheretherketone (PEEK) paddle prior to sampling into the flow digestion system in order to overcome problems associated with settling of solids. The PEEK paddle and the needles were rinsed with water in a separate washing position of the autosampler before sample uptake. The sample volume introduced into the sample loop was controlled by a high precision dispenser (1-Channel MultiDispenser, ProLiquid, Germany – Fig. 1D) connected to the 6-port valve. Many systems reported in the literature used a simpler arrangement, in which the sample loop was completely filled with sample by e.g. a peristaltic pump. Using a high precision dispenser for sample uptake allowed us to modify the volume introduced into the digestion system from 0.5 to 10 mL (maximum volume of the sample loop). Drawing the sample through the sample loop avoided potential contamination of the dispenser as the sample never entered the dispenser. Moreover, this mode of dispenser operation allowed embedding the sample between two 2 mL segments of 30% (v/v) nitric acid.18 After injecting the sample into the high pressure stream the autosampler needle was moved to the rinsing position and 10 mL rinsing solution (Fig. 1M) were pumped by the dispenser through the needle for cleaning purposes. The sample digestion was performed in a perfluoroalkoxy (PFA) tube (1.5 mm inner diameter, 2.5 mm outer diameter, 4.5 m length) placed inside a coiled borosilicate glass tube (4 mm inner diameter, 8 mm outer diameter, 270 mm coil diameter, Fig. 1G). By pressurizing the glass tube with nitrogen (40 bar), the pressure inside and outside of the PFA tubing was almost equal during digestion, significantly reducing the mechanical stress on the tubing during the digestion process. The glass coil therefore formed a pressurized autoclave for the PFA digestion tube. Nitrogen was introduced into the system through the gas/liquid separator (Fig. 1H) and left the glass coil through an exit port (Fig. 1E). A restrictor capillary (2 m 0.15 mm inner diameter PEEK tube – Fig. 1F) connected to this exit port maintained the pressure inside the glass coil and limited the flow of N2 to about 1 L min−1. This countercurrent flow of N2 removed traces of water and acid from the glass digestion coil that otherwise would have accumulated there causing corrosion of the stainless steel tubes and sample contamination. It is important to note, that these traces of liquid resulted not from leakage but from diffusion of steam through the PFA tube.24 Similar behavior was also observed in other pressure equilibrium systems.7 The digestion coil was installed vertically18 inside a commercial microwave oven (Multiwave 3000, Anton Paar, Austria) designed for pressurized batch digestion. This microwave oven does not employ pulse width modulation (PWM) for regulating the power level but is of a constant power type. Thereby overheating and violent reactions associated with PWM modulated microwave ovens could be avoided. Moreover, the oven can be operated under conditions of high reflected microwave power as the magnetron cooling is sufficiently strong, eliminating the need for an absorbing water ballast and enabling fully automated system operation over prolonged time. After leaving the microwave irradiated zone the digested samples were cooled (Fig. 1K) and gaseous reaction products were removed in a gas/liquid separator (Fig. 1H). This gas/liquid separator was constructed from a 20 mm outer diameter quartz tube installed in a PEEK casing. After depressurization in a restrictor capillary (12 m 0.5 mm inner diameter PTFE tube – Fig. 1I) the digests were collected in 50 mL sample tubes (Fig. 1J) using the autosampler's second sampling needle. All system parameters and procedures including the autosampler were computer controlled thus allowing unattended system operation.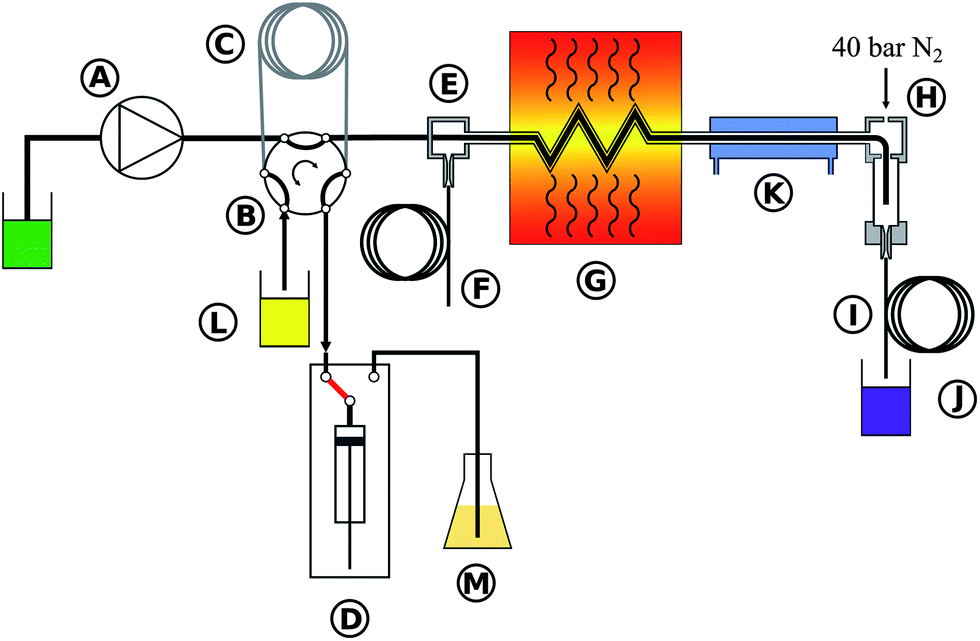 | ||
| Fig. 1 Schematic of the flow system. A: high pressure pump for carrier solution, B: 6-port high pressure valve, C: sample loop, D: high precision dispenser for controlled sample uptake, E: exit port of the N2 pressurizing system, F: N2 restrictor capillary, G: microwave heated zone, H: gas/liquid separator and nitrogen inlet, I: sample restrictor capillary, J: collection of the digested sample, K: cooling unit, L: undigested sample, and M: rinsing solution. | ||
To suppress microwave leakage from the oven, the glass coil (Fig. 2C) was connected to a grounded stainless steel tube (Fig. 2D). This glass/steel connection was of crucial importance to the entire system. Both, the glass tube and the steel tube had to stay in place at a pressure of 40 bar to avoid leakage. Nonetheless, the connection had to be flexible enough to compensate for thermal expansion of both tubes. A design similar to a packing gland was used to meet both requirements: the glass tube (Fig. 2C) was fixated by a PEEK ferrule (Fig. 2F) using a tightening screw (Fig. 2E). A 1 mm gap between the glass and the steel tube allowed thermal expansion of both tubes. Over both tubes a 20 mm long silicone rubber tube (Fig. 2G) of 8 mm inner diameter was slipped evenly. A lantern ring (2 mm long, 8 mm inner diameter, not shown in Fig. 2) was then installed on the steel tube and the silicone rubber tube was compressed by tightening the packing screw (Fig. 2H, formed by an 8 mm inner diameter Swagelok tube fitting, part number SS-8M0-6). The metal tube (Fig. 2D) itself was also locked in place by this fitting, a metal ferrule (Fig. 2I) and the corresponding tightening nut (Fig. 2J). The PFA digestion tube (Fig. 2K) could be moved freely inside this glass-to-metal interface leaving plenty of space for the high pressure nitrogen to flow around the PFA tube.
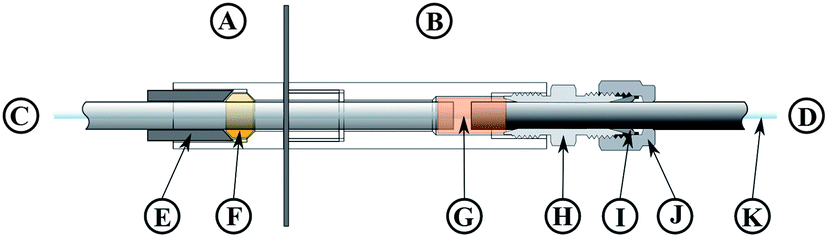 | ||
| Fig. 2 Glass-to-metal interface. A: part inside of microwave oven, B: outside part, C: glass tube, D: metal tubing, E: tightening screw, F: PEEK ferrule, G: silicon rubber tube, H: Swagelok tube fitting, I: metal ferrule, J: tightening nut, and K: PFA digestion tube. | ||
The optimized operating conditions are listed in Table 1.
| Parameter | Value |
|---|---|
| Carrier flow rate | 2.0 mL min−1 of 1% v/v HNO3 |
| Sample volume introduced | 5 mL |
| Pressure within the digestion coil | 40 bar |
| Heated volume of the digestion coil | 6 mL |
| Microwave power | 400 W |
| Final volume after digestion | 20 mL |
Instrumentation
Analytes were quantified with an axially viewed ICP-OES (Ciros Vision EOP, Spectro, Germany) using a cross-flow nebulizer, a Scott type spray chamber and a standard ICP torch with a 2.5 mm inner diameter injector. 1350 W RF power, 12.5 L min−1 outer gas flow, 0.6 L min−1 intermediate gas flow and 0.83 L min−1 nebulizer gas flow were used. The following emission lines were used for analyte quantification: Al 167.078 nm, As 189.042 nm, B 249.773 nm, Ba 455.404, Be 234.861, Bi 190.241 nm, Ca 317.933 nm, Cd 214.438, Co 228.616 nm, Cr 267.716 nm, Cu 219.226 nm, Fe 238.204 nm, Mg 280.270 nm, Mn 257.611 nm, Mo 202.030 nm, Na 588.995 nm, Ni 231.604 nm, Pb 220.353 nm, Sb 206.833 nm, Se 196.090 nm, Sr 407.771 nm, Ti 334.941 nm, V 292.464 nm, and Zn 213.856 nm.Using the conditions reported in Table 1 and 1% (m/v) sample slurries, the LOQs were 4, 12, 8, 16, 8, 11, 10, 4, 12, 16, 20, and 8 mg kg−1 for Al, B, Ca, Cr, Cu, Fe, Mg, Mn, Na, Ni, Pb, and Zn respectively. The LOQs were calculated from calibration functions (0–1 mg L−1; 5 concentration levels, 5 replications; 95% confidence level) and corrected for the sample dilution. LOQs are only given for elements analyzed in CRMs or real samples. RSDs were below 3% in all ICP-OES measurements. Scandium (Sc 361.384 nm) was used as an internal standard at a concentration of 1 mg L−1 throughout.
Residual carbon content (RCC) in digests was determined using a total organic carbon analyzer (TOC-5050, Shimadzu, Japan) or ICP-OES (C 193.091 nm emission line).
Closed vessel microwave assisted digestions were performed in standard ceramic supported PFA vessels using a commercial microwave digestion system (Multiwave 3000, Anton Paar GmbH, Austria). About 0.5 g solid sample or 5 g liquid sample were mixed with the digestion acids (HNO3, or cocktails of HNO3 and HCl or HF). For 5 minutes the microwave power was ramped to 1400 W and thereafter the sample was digested for additional 15 minutes at the maximum permissible vessel pressure (40 bar).
The acid concentration in the digested samples was determined by manual titration with 0.1 mol L−1 NaOH (Roth, Germany) using phenolphthalein as an indicator.
Reagents, certified reference materials and samples
Purified water (18 MΩ cm−1, Barnstead Nanopure, Thermo Fisher Scientific, USA) and high purity acids (HCl and HF Suprapur, Merck, Germany; HNO3 purified by subboiling in a quartz still) were used throughout. Calibration standards were prepared from a 100 mg L−1 multi element solution (Roth, Germany) in 3% HNO3 (v/v). Calibration solutions for the determination of the residual carbon content were prepared from potassium hydrogen phthalate (pa quality, Merck, Germany).Six certified reference materials (CRMs) were used in this work: BCR 62 (olive leaves), CNRC DORM-2 (dogfish muscle), NIST SRM 1515 (apple leaves), NIST SRM 1547 (peach leaves), NIST SRM 1567 (wheat flour), and NIST SRM 1568 (rice flour).
For initial system characterization commercially available milk powder (Aptamil Folgemilch, Milupa, Austria), apple juice (Spar, Austria) and orange juice (Spar, Austria) were used.
Slurries with 1% solids were prepared by thoroughly mixing the solid sample with water, adding concentrated acids and making up to volume with water afterwards. Thereby sample clotting was avoided. Liquid samples were diluted with the relevant concentrated acids.
Results and discussion
Optimization of the microwave field uniformity
A common problem in multimode microwave cavities is the inhomogeneous distribution of microwave radiation. To investigate this effect 12 glass vials (volume: 14 mL) filled with 10 mL 3% HNO3 were placed in a circular arrangement (equally spaced, radius: 250 mm, height above cavity floor: 170 mm) inside the microwave cavity. This setup matched the position of the glass digestion coil inside the cavity. After heating for 60 seconds with 400 W the temperature of each vessel was quickly determined with a digital thermometer (300 K, Voltcraft, Taiwan). The error of the temperature measurement was found to be dominated by the cooling of the liquid in the vials during the measurement as the vial temperature was measured consecutively rather than simultaneously. Based on repeated measurements of the same vial an error of 3 °C was estimated. The experiment was repeated with a mode stirrer (140 mm diameter aluminum disc with six wings bent 80 mm upwards) installed inside the microwave cavity. This mode stirrer was spun by the digestion vessel rotation mechanism of the Multiwave 3000.Without the mode stirrer the temperature in the glass vials ranged from 44 °C to 74 °C with a median of 51 °C. By using the mode stirrer the temperature profile inside the microwave cavity flattened as shown in Fig. 3. The temperature of the diluted nitric acid ranged from 49 °C to 66 °C with a median of 55 °C. Moreover, the homogeneity of the microwave radiation in the region of the digestion coil could be improved. Consequently the mode stirrer was used throughout the remaining experiments.
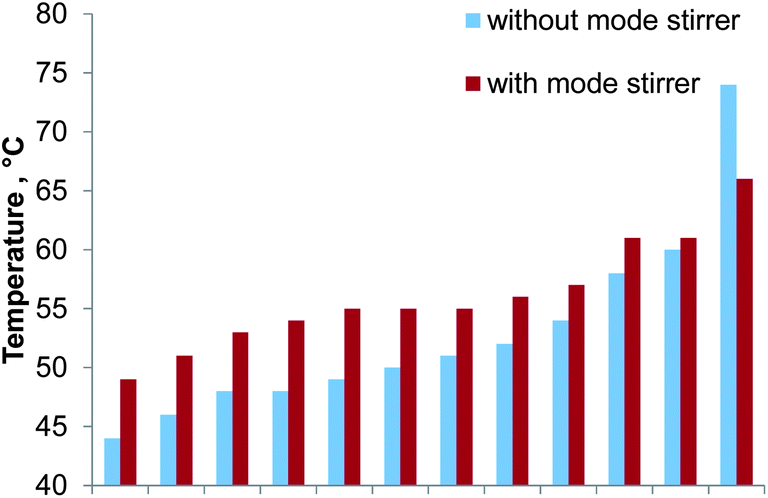 | ||
| Fig. 3 Temperature distribution inside the microwave cavity with and without a spinning mode stirrer. Values arranged with increasing temperature; error bars not shown for clarity. | ||
Optimization of the digestion parameters
The residual carbon content (RCC) is commonly used to assess the completeness of a digestion. Based on previous experience glucose and glycine were selected as test substances for RCC determination as their behavior during acid digestion is known to be very different.7 Glucose tends to react violently with nitric acid upon reaching the digestion temperature and serves as a “stress test” for the flow digestion system. Glycine on the other hand is difficult to digest completely.The effect of microwave power on the completeness of the digestion was evaluated using solutions of 25 g L−1 glucose or 28 g L−1 glycine in 30% (v/v) nitric acid and the instrument conditions reported in Table 1. As shown in Fig. 4 the decomposition of glucose starts at a power level of 300 W resulting in a RCC of 24 ± 1% (mean value ± standard uncertainty). With increasing power the RCC decreased almost linear to 14.4 ± 0.8% at 800 W. For glycine the digestion conditions were not aggressive enough: only at a power level of 800 W some of the glycine was completely decomposed resulting in a RCC of 96 ± 1%. This behavior can be traced to a digestion temperature significantly below the expected 250 °C within the digestion zone. As shown by Pichler et al.7 glycine is digested only at temperatures above 235 °C for a residence time in the heated digestion zone of 3 minutes. From these results it can be estimated that the digestion temperature in the presented system was between 230 and 235 °C. This temperature is also in good agreement with the data reported by Pichler et al. for glucose. A precise measurement of the temperature within the digestion zone was not possible due to potential microwave leakage and geometrical constraints. We believe that the reason for the somewhat low digestion temperature in the presented flow digestion system is the relatively small overall sample volume of 6 mL in the microwave cavity. This results in reduced microwave energy coupling. Depending on the digested sample matrix, all further experiments were conducted with either 400 or 600 W.
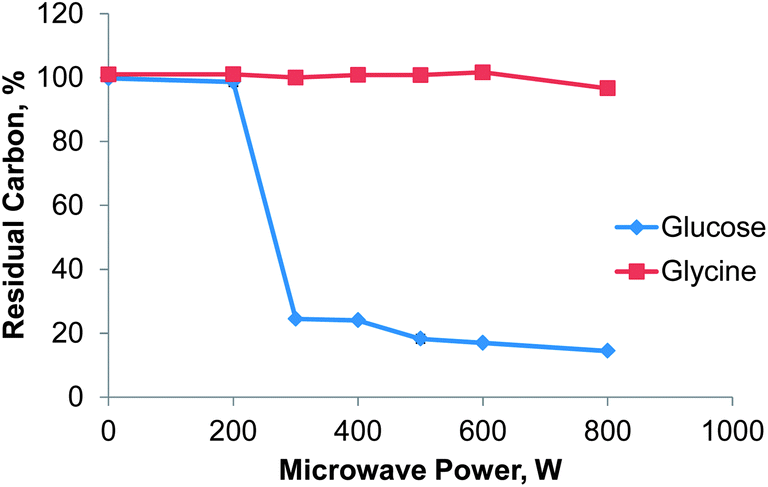 | ||
| Fig. 4 Effect of microwave power on the digestion efficiency of glucose and glycine expressed as RCC, n = 3, error bars smaller than the dots. | ||
It is important to note that even at 800 W the magnetron temperature remained below 60 °C. An experiment with 1000 W was also attempted, but the magnetron temperature increased quickly tripping the microwave digestion ovens over-temperature control circuit. According to the instrument manufacturer, below 1000 W both magnetrons share the power load, each providing one half. Above 1000 W one magnetron operates at full rated power whereas the second one is used for power regulation. Compared to pressurized flow digestion systems employing mono mode cavities (typically about 100 W),7 the power level in this setup was far higher.
Due to the relatively large multimode cavity the microwave coupling to the liquid phase inside the digestion coil was low. Consequently, a reduction of the chamber height by a factor of 1.6 was investigated. A tight fitting grounded aluminum sheet was installed inside the microwave cavity reducing its height from 350 mm to 225 mm but leaving length and width unchanged. In a similar experiment to the one above, no significant change in the RCC was encountered for glucose and glycine. The aluminum sheet was therefore not used further on.
The carrier flow rate defines the residence time of the sample in the microwave heated zone. Decreasing the carrier flow rate is not only known to improve the digestion efficiency but also to lengthen the overall time needed to process the sample. The effect of the carrier flow rate on the RCC was investigated using a test solution of 28 g L−1 glycine in 30% (v/v) nitric acid and the instrument conditions reported in Table 1. Reducing the carrier flow rate from 2 mL min−1 to 0.5 mL min−1 led to a near linear decrease of the RCC from 99 ± 1 to 89 ± 2% for glycine as shown in Fig. 5. It is important to note that by reducing the carrier flow rate the digestion time for one sample increased from 10 to 40 minutes. This was deemed impractical and unjustified by the small reduction in RCC despite the high degree of automation in the presented system. Thus the carrier flow rate was set to 2 mL min−1 for all further experiments. A stopped flow mode of digestion was not attempted with the presented system.
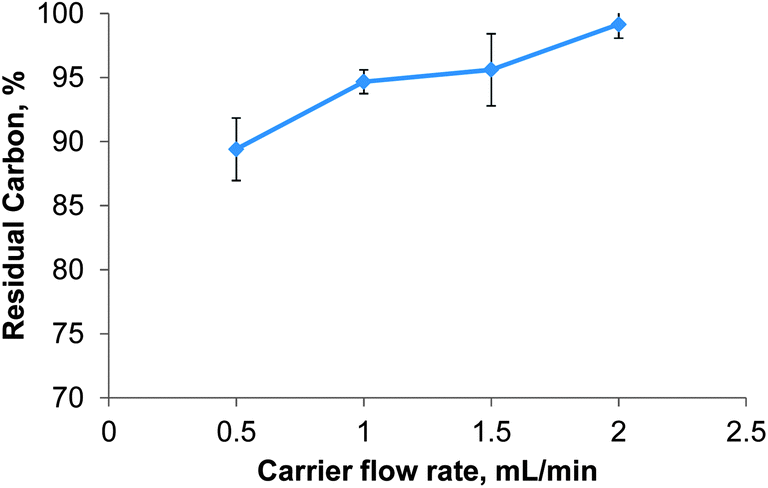 | ||
| Fig. 5 Effect of the carrier flow rate on the digestion efficiency of glycine at 400 W microwave power, n = 5. | ||
Increasing the sample volume inside the microwave cavity might be another approach to improve the RCC. Thereby the total volume of liquid inside the cavity would be increased. It was found that regardless of the large radius of the glass coil the introduction of the PFA tube into the coil was not possible for glass coils with more than three turns. During initial tests it was attempted to introduce the PFA tube into a six turn coil. This failed even with the ample use of ethanol as a lubricating agent (PTFE spray or low viscosity oil proved to be inferior to ethanol) after about three and a half turns. The radius of the glass coil was not further altered, as a smaller radius would have worsened the above mentioned problems with the PFA tube and a much larger coil radius was not possible due to the cavity size.
Comparison between flow digestion and closed vessel batch digestion
Typical samples processed in flow digestion systems are fruit juices and milk powder. Two juice samples were investigated in this work: clear apple juice and orange juice with pulp. Each sample was acidified with nitric acid to a final concentration of about 30% HNO3 (v/v) prior to high pressure flow digestion. For the digestion of commercial milk powder a slurry (1% m/v) was prepared. All samples were clear and colorless after digestion. The same samples were also digested in a common closed vessel batch digestion procedure.The data presented in Table 2 show that generally there is no significant difference between high pressure flow digestion and closed vessel batch digestion in all three matrices on the 95% confidence level. For Ca, Mg and Na in orange juice a Student t-test indicated differences between the two digestion principles with lower values attained by flow digestion. We attribute these findings in closed vessel digestion to contamination during sample transfer.
| Apple juice | Orange juice | Commercial milk powder | ||||
|---|---|---|---|---|---|---|
| Flow digestion | Closed vessel digestion | Flow digestion | Closed vessel digestion | Flow digestion | Closed vessel digestion | |
| mg kg−1 | mg kg−1 | mg kg−1 | mg kg−1 | mg g−1 | mg g−1 | |
| B | 2.2 ± 0.2 | 2.16 ± 0.06 | 0.99 ± 0.04 | 1.06 ± 0.03 | ND | ND |
| Ca | 130 ± 2 | 132 ± 3 | 154 ± 2 | 162 ± 3 | 3.90 ± 0.08 | 4.1 ± 0.2 |
| Cu | <LOQ (0.2) | <LOQ (0.2) | 0.18 ± 0.01 | 0.19 ± 0.02 | ND | ND |
| Fe | 3.9 ± 0.2 | 4.1 ± 0.3 | 0.8 ± 0.2 | 1.0 ± 0.2 | 0.062 ± 0.002 | 0.073 ± 0.007 |
| Mg | 59 ± 4 | 62 ± 1 | 111 ± 2 | 121 ± 3 | 0.32 ± 0.01 | 0.33 ± 0.02 |
| Mn | 0.50 ± 0.03 | 0.52 ± 0.02 | 0.23 ± 0.01 | 0.26 ± 0.01 | ND | ND |
| Na | 31.6 ± 0.5 | 32 ± 1 | 17.6 ± 0.2 | 20 ± 1 | 1.17 ± 0.08 | 1.21 ± 0.04 |
| Zn | 0.08 ± 0.01 | 0.06 ± 0.01 | 0.15 ± 0.01 | 0.15 ± 0.01 | 0.032 ± 0.002 | 0.034 ± 0.002 |
Effect of the digestion acid cocktail
The efficiency of a digestion is often described by the RCC. As already shown, this parameter is heavily affected by the sample matrix. Moreover, no information is provided on the tolerance of the flow digestion system towards the acid cocktail. Using a CRM (NIST SRM 1547, peach leaves) the effect of different digestion acid cocktails on both the flow digestion system itself and the analytical characteristics of the digests was investigated. In each run 5 mL of a 1% (m/v) slurry were introduced into the flow digestion system.The effect of different acid cocktails on the digestion of NIST SRM 1547 (peach leaves) is shown in Table 3. For Ca and Mg all acid cocktails resulted in good agreement between the determined and the certified values. The obtained values of B and Mn were slightly lower and the results of Na are slightly higher than the certified values irrespectively of the digestion acid mixture indicating other processes than the sample digestion. It is important to note, that for all acid cocktails used, the analyte concentration in the blank digestions (only acid) was below the respective limit of quantification. For Al only about 28% of the certified value was obtained if just nitric acid was used for digestion. If on the other hand HF was present in the digestion acid cocktail, close agreement between the obtained and the certified concentrations was found. This behavior is well known from closed vessel batch digestion.
| Determined | Certified | |||||
|---|---|---|---|---|---|---|
| 30% HNO3 | 30% HNO3–3.6% HCl | 30% HNO3–3% H2O2 | 30% HNO3–3.6% HCl 1.3% HF | |||
| Al | 70 ± 3 | 132 ± 18 | 160 ± 20 | 240 ± 10 | 249 ± 8 | mg kg−1 |
| B | 27.2 ± 0.9 | 23 ± 1 | 25.1 ± 0.9 | ND | 29 ± 2 | mg kg−1 |
| Ca | 15 ± 1 | 14.8 ± 0.1 | 14.8 ± 0.1 | 11.4 ± 0.4 | 15.6 ± 0.2 | g kg−1 |
| Mg | 3.94 ± 0.2 | 3.88 ± 0.06 | 3.88 ± 0.02 | 3.18 ± 0.06 | 4.32 ± 0.08 | g kg−1 |
| Mn | 93 ± 3 | 91.2 ± 0.6 | 91.9 ± 0.6 | 91 ± 1 | 98 ± 3 | mg kg−1 |
| Na | ND | 39 ± 2 | 31 ± 3 | ND | 24 ± 2 | mg kg−1 |
In general, the composition of the digestion acid cocktail is of great importance for complete digestion. Only by means of that, low analyte results can be avoided. Consequently, a digestion system should not pose limits on the composition of the acid cocktail. Until now this goal has not been reached for high pressure flow digestion systems. Even dissolution coils made of Pt/Ir (80% Pt, 20% Ir) – despite their excellent stability towards HNO3 and HF18 – are reported to be attacked by HNO3–HCl mixtures.21 One clear advantage of the present flow digestion system is the absence of metals in any part of the system which is in contact with concentrated acids. As a result, a high degree of freedom is given to the selection of the digestion acid cocktail composition.
Method validation
Five certified reference materials of biological samples were analyzed in order to verify the accuracy of the flow digestion procedure. The acid cocktails were selected based on previous experience in closed vessel batch digestion of these materials. In each run 5 mL of a 1% (m/v) slurry were introduced into the flow digestion system.The results of the CRM analysis listed in Table 4 are in good agreement with the certified values. It should be noted that regardless of the large number of stainless steel components in the presented high pressure flow digestion system no significant contamination of the samples with Fe, Cr or Mn was encountered. Moreover, the titanium components in the HPLC pump used for carrier flow generation didn't result in significant contamination either. The concentration of Fe, Cr, Mn and Ti in blank solution digests prepared by injecting the diluted acid rather than the sample slurry was below the respective LOQ (11, 16, 4, and 8 mg kg−1 for Fe, Cr, Mn and Ti; values corrected for the sample dilution caused by the digestion). This clearly demonstrates the effectiveness of the countercurrent flow of nitrogen that is used to pressurize the digestion coil.
| Determined | Certified | |||
|---|---|---|---|---|
| BCR 62 (olive leaves) 30% HNO3, 3.6% HCl, 0.8% HF | Al | 415 ± 9 | 450 ± 20 | mg kg−1 |
| Cu | 42 ± 1 | 46.6 ± 1.8 | mg kg−1 | |
| Mn | 53.9 ± 0.9 | 57.0 ± 2.4 | mg kg−1 | |
| Pb | 22 ± 1 | 25.0 ± 1.5 | mg kg−1 | |
| Zn | 13.5 ± 0.6 | 16.0 ± 0.7 | mg kg−1 | |
| DORM-2 (dogfish muscle) 20% HNO3, 6% HCl | Al | 5 ± 1 | 10.9 ± 1.7 | mg kg−1 |
| Cr | 37.2 ± 0.6 | 34.7 ± 5.5 | mg kg−1 | |
| Fe | 153.2 ± 0.6 | 142 ± 10 | mg kg−1 | |
| Mn | 3.99 ± 0.02 | 3.66 ± 0.34 | mg kg−1 | |
| Ni | 23 ± 2 | 19.4 ± 3.1 | mg kg−1 | |
| Zn | 19.7 ± 0.7 | 25.6 ± 2.3 | mg kg−1 | |
| NIST SRM 1515 (apple leaves) 30% HNO3, 3% HF | Al | 290 ± 10 | 286 ± 9 | mg kg−1 |
| Ca | 14.5 ± 0.2 | 15.26 ± 0.15 | g kg−1 | |
| Fe | 68 ± 1 | 83 ± 5 | mg kg−1 | |
| Mg | 2.49 ± 0.04 | 2.71 ± 0.08 | g kg−1 | |
| Mn | 49.7 ± 0.7 | 54 ± 3 | mg kg−1 | |
| Zn | 11.9 ± 0.7 | 12.5 ± 0.3 | mg kg−1 | |
| NIST SRM 1567 (wheat flour) 30% HNO3 | Ca | 186 ± 6 | 190 ± 10 | mg kg−1 |
| Fe | 20 ± 1 | 18.3 ± 1.0 | mg kg−1 | |
| Zn | 17 ± 4 | 10.6 ± 1.0 | mg kg−1 | |
| NIST SRM 1568 (rice flour) 30% HNO3 | Ca | 136 ± 7 | 140 ± 20 | mg kg−1 |
| Mn | 17.5 ± 0.9 | 20.1 ± 0.4 | mg kg−1 | |
| Zn | 21.4 ± 0.9 | 19.4 ± 1.0 | mg kg−1 | |
The RCC (mean value ± standard uncertainty, n = 3, 95% confidence level) values after high pressure flow digestion were 35 ± 2%, 40 ± 2%, 11.6 ± 0.6%, and 10.6 ± 0.6% for BCR 62 (olive leaves), NIST SRM 1515 (apple leaves), NIST SRM 1567 (wheat flour), and NIST SRM 1568 (rice flour), respectively. It is important to note that all digests were clear and without any visual particles.
Spike recoveries were obtained from 1% slurries of commercial milk powder in 30% HNO3 (v/v) fortified prior to flow digestion with Al, As, Ba, Be, Bi, Cd, Co, Cr, Cu, Fe, Hg, Mg, Mn, Mo, Ni, Pb, Sb, Se, Sr, Ti, V, and Zn. The final concentration in the digested samples was 1 mg L−1 for each element. The spike recoveries for all elements listed were between 94 and 105% (RSD's below 3% for all elements; n = 4), with the exception of Hg. For Hg the spike recovery was 89% indicating some losses in the PFA digestion tube.
The acid concentration after flow digestion in these milk power samples was 3.8 ± 0.1 mol L−1 (mean ± standard uncertainty; 95% confidence level). The high acidity in the digested samples is typical for conventional acid digestion. It is interesting to note that recently new digestion techniques for lower sample acidity adverted: they involve the use of UV radiation during digestion and allow a significant reduction of acid consumption.25
Conclusion
With the presented high pressure flow digestion system it was possible to digest samples fully automated at a pressure of up to 40 bar. The reason for the relatively high RCC values for glycine can be expected to originate from the inefficient coupling of microwave energy into the small sample volume (6 mL) situated in the rather large multimode cavity. This is consistent with data obtained at a very low carrier flow of 0.5 mL min−1. The reduction of the RCC under these conditions can be explained by the long residence time of the sample in the microwave irradiated zone. Nevertheless, good agreement with certified values was achieved and the results were similar to closed vessel batch digestion. In contrast to most published pressurized flow digestion systems the present system did not limit the selection of the digestion reagents. Even prolonged system operation with acid cocktails of HNO3, HF and HCl did neither result in sample contamination by dissolved structural materials nor in significant corrosion of the system. Furthermore, no signs of element specific losses22 were encountered. Another advantage of the presented system is the fully automated processing of samples. After loading the sample slurries to the autosampler the digestion process is performed autonomously and without the risk of contamination due to various sample handling steps commonly necessary in closed vessel batch digestion protocols. The presented high pressure flow digestion system is capable of digesting up to six samples per hour using the operating conditions listed in Table 1. A comparison with other flow digestion systems reported in the literature is shown in Table 5.| Presented high pressure flow digestion system | Ambient pressure – continuous flow digestion systems | Ambient pressure – closed flow and stopped flow digestion systems | Medium pressure continuous flow digestion systems | Medium pressure stopped flow digestion systems | Conductively heated high pressure flow digestion systems | |
|---|---|---|---|---|---|---|
| Reference | 1,8,10,26 and 27 | 28–30 | 2 and 31 | 4,5 and 32 | 19–21 | |
| System pressure | 40 bar | 1 bar | 2–25 bar | up to 200 bar | ||
| Digestion temperature | ∼230 °C | ∼120 °C | 180–200 °C | up to 300 °C | ||
| System tolerates HF, HCl, and/or H2O2 | Yes | Yes | Yes | No | No | No |
Acknowledgements
The authors acknowledge the help of Armin Bacher for his support in the initial optimization experiments. Günter Fellner is thanked for machining most of the components of the presented system.References
- M. Burguera, J. L. Burguera, C. Rondon, C. Rivas, P. Carrero, M. Gallignani and M. R. Brunetto, J. Anal. At. Spectrom., 1995, 10, 343–347 RSC.
- S. J. Haswell and D. Barclay, Analyst, 1992, 117, 117–120 RSC.
- R. E. Sturgeon, S. N. Willie, B. A. Methven, J. W. H. Lam and H. Matusiewicz, J. Anal. At. Spectrom., 1995, 10, 981–986 RSC.
- T. J. Gluodenis and J. F. Tyson, J. Anal. At. Spectrom., 1993, 8, 697–704 RSC.
- G. Légère and E. D. Salin, Appl. Spectrosc., 1995, 49, 14A–22A CrossRef.
- T. J. Gluodenis and J. F. Tyson, J. Anal. At. Spectrom., 1992, 7, 301–306 RSC.
- U. Pichler, A. Haase, G. Knapp and M. Michaelis, Anal. Chem., 1999, 71, 4050–4055 CrossRef CAS.
- J. L. Burguera and M. Burguera, J. Anal. At. Spectrom., 1993, 8, 235–241 RSC.
- J. L. Burguera, M. Burguera, P. Carrero, C. Rivas, M. Gallignani and M. R. Brunetto, Anal. Chim. Acta, 1995, 308, 349–356 CrossRef CAS.
- M. Burguera, J. L. Burguera and O. M. Alarcón, Anal. Chim. Acta, 1986, 179, 351–357 CrossRef CAS.
- M. Bettinelli, S. Spezia, A. Ronchi and C. Minoia, Rapid Commun. Mass Spectrom., 2002, 16, 1432–1439 CrossRef CAS PubMed.
- S. Hinkamp and G. Schwedt, Anal. Chim. Acta, 1990, 236, 345–350 CrossRef CAS.
- A. Meyer and G. Schwedt, LaborPraxis, 1993, 4, 44–48 Search PubMed.
- M. L. Balconi, M. Borgarello, R. Ferraroli and F. Realini, Anal. Chim. Acta, 1992, 261, 295–299 CrossRef CAS.
- M. De la Guardia, V. Carbonell, A. Morales-Rubio and A. Salvador, Talanta, 1993, 40, 1609–1617 CrossRef CAS PubMed.
- H. Matusiewicz and R. E. Sturgeon, Fresenius' J. Anal. Chem., 1994, 349, 428–433 CrossRef CAS.
- T. Dülcks, LaborPraxis, 1996, 9, 68–69 Search PubMed.
- S. Haiber and H. Berndt, Fresenius' J. Anal. Chem., 2000, 368, 52–58 CrossRef CAS PubMed.
- P. Jacob and H. Berndt, J. Anal. At. Spectrom., 2002, 17, 1615–1620 RSC.
- C. Gräber and H. Berndt, J. Anal. At. Spectrom., 1999, 14, 683–691 RSC.
- Q. Z. Bian, P. Jacob, H. Berndt and K. Niemax, Anal. Chim. Acta, 2005, 538, 323–329 CrossRef CAS.
- Q. Z. Bian, PhD thesis, Universität Dortmund, 2005.
- G. Knapp, USA Patent 5,672,316, 1995.
- M. Lafer, PhD thesis, Graz University of Technology, Austria, 2010.
- J. S. F. Pereira, R. S. Picoloto, L. S. F. Pereira, R. C. L. Guimarães, R. A. Guarnieri and E. M. M. Flores, Anal. Chem., 2013, 85(22), 11034–11040 CrossRef CAS PubMed.
- M. A. Z. Arruda, M. Gallego and M. Valcarcel, J. Anal. At. Spectrom., 1995, 10, 501–504 RSC.
- S. Recknagel, P. Brätter, A. Tomiak and U. Rösick, Fresenius' J. Anal. Chem., 1993, 346, 833–836 CrossRef CAS.
- V. Carbonell, M. de la Guardia, A. Salvador, J. L. Burguera and M. Burguera, Anal. Chim. Acta, 1990, 238, 417–421 CrossRef CAS.
- V. Carbonell, A. Morales-Rubio, A. Salvador, M. de la Guardia, J. L. Burguera and M. Burguera, J. Anal. At. Spectrom., 1992, 7, 1085–1089 RSC.
- E. R. Pereira-Filho and M. A. Z. Arruda, Analyst, 1999, 124, 1873–1877 RSC.
- H. Staufenberg, LaborPraxis, 1993, 11, 58–59 Search PubMed.
- V. Karanassios, F. H. Li, B. Liu and E. D. Salin, J. Anal. At. Spectrom., 1991, 6, 457–463 RSC.
| This journal is © The Royal Society of Chemistry 2014 |