Subtleties in asymmetric catalyst structure: the resolution of a 6-phospha-2,4,8-trioxa-adamantane and its applications in asymmetric hydrogenation catalysis†‡
Jonathan
Hopewell
,
Piotr
Jankowski
,
Claire L.
McMullin
,
A. Guy
Orpen
and
Paul G.
Pringle
*
School of Chemistry, University of Bristol, Cantock’s Close, Bristol, UK BS8 1TS. E-mail: paul.pringle@bristol.ac.uk; Fax: +44 (0)117 929 0509; Tel: +44 (0)117 928 8114
First published on 2nd November 2009
Abstract
An efficient, classical resolution of the versatile P-ligand intermediate 6-phospha-2,4,8-trioxa-adamantane (CgPH) is described and the rhodium complex of the optically pure secondary phosphine β-CgPH is an active and moderately selective asymmetric hydrogenation catalyst.
The racemic phospha-adamantane cage (denoted CgPH) was first reported in 19611 as the product of the high-yielding hydrophosphination–condensation reaction shown in eqn (1). CgPH is readily available in 100 g quantities and is an atypical secondary phosphine in that it is air stable.2 The unusual stereoelectronic environment that the cage confers on the P atom in CgP-containing ligands has led to a surge of interest in this class of ligands in recent years.2–11
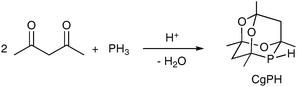 | (1) |
The compound CgPH has been shown to be a useful intermediate for tertiary monophosphines and diphosphines featuring the CgP group.2–5 CgP-containing ligands have found applications in many catalyses, notably alkene carbonylation6 and hydroformylation,7,8 C–C coupling,3,5,9 C–N coupling10 and hydrogenation.11 CgPH has C1-symmetry and thus exists in enantiomeric forms; the α- and β-enantiomers are structurally related to each other by interchanging the two O atoms, which are coloured red in Fig. 1, with the two CH2 groups.
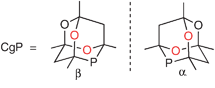 | ||
| Fig. 1 Enantiomeric forms of the CgP unit. | ||
In view of the rigid, diamondoid shape of the CgP unit and the similarity in size of the O and CH2 groups, it was concluded that any enantiodiscrimination by CgP-containing species would have to be subtle. Nevertheless, we show here that classical resolution of CgPH is readily achieved and moreover that optically pure CgPH and ligands derived from it perform surprisingly well in asymmetric hydrogenation catalysis.
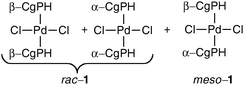
The resolution of CgPH was achieved via its phosphinic acid by the route shown in Scheme 1.12 The 31P NMR spectrum of the mixture of diastereomeric quininium salts [CgPO2][QH] in CD2Cl2 showed two, clearly resolved singlets, presumably because of ion-pairing in solution. After seven, high-recovery crystallisations from toluene, there was only one diastereoisomer of the salt detected by 31P NMR spectroscopy. The optical purity of the resolved CgPH was determined as follows. Treatment of [PdCl2(NCPh)2] with racemic CgPH gave a 1 : 1 mixture of the previously reported2 diastereomeric complexes, rac-1 and meso-1 which gave two, well-separated 31P NMR signals at δ 3.53 and 3.20 ppm. With the resolved CgPH, only one signal (at δ 3.53) was observed and the er was calculated to be over 100 : 1. The overall yield of optically pure CgPH after the five-step procedure (Scheme 1) was ca. 10% and its [α]295D in CHCl3 was +42.2.13
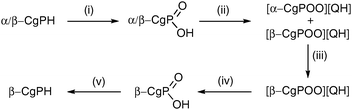 | ||
| Scheme 1 Reagents and conditions (yields): (i) H2O2 in MeOH (97%); (ii) quinine in boiling toluene (98%); (iii) 7× recrystallisations from boiling toluene (81% total recovery); (iv) extraction of CH2Cl2 solution with 2 M aqueous NaOH and then acidification with 12 M HCl (72%); (v) LiAlH4 in Et2O (17%). | ||
Crystal structures for the resolved phosphinic acid and secondary phosphine were obtained (Fig. 2) which showed that the β-enantiomer had been separated.
![Thermal ellipsoid (50% probability) plots of structures of (a) β-CgPOOH and (b) β-CgPH, omitting all carbon-bound hydrogen atoms. Selected geometrical data: bond lengths [Å] and angles [°] for (a) P1–C2 1.8263(16), P1–C9 1.8201(15), P1–O4 1.4942(11), P1–O5 1.5483(11), C2–P1–C9 97.74(7), O4–P1–O5 114.83(6). (b) P1–C2 1.871(4), P1–C9 1.877(4), P1–H21 1.32(3), C2–P1–C9 98.5(16).](/image/article/2010/CC/b916359j/b916359j-f2.gif) | ||
| Fig. 2 Thermal ellipsoid (50% probability) plots of structures of (a) β-CgPOOH and (b) β-CgPH, omitting all carbon-bound hydrogen atoms. Selected geometrical data: bond lengths [Å] and angles [°] for (a) P1–C2 1.8263(16), P1–C9 1.8201(15), P1–O4 1.4942(11), P1–O5 1.5483(11), C2–P1–C9 97.74(7), O4–P1–O5 114.83(6). (b) P1–C2 1.871(4), P1–C9 1.877(4), P1–H21 1.32(3), C2–P1–C9 98.5(16). | ||
Secondary phosphines are rarely used as ligands for catalysis,14 understandably because the P–H bond is normally reactive and their complexes are susceptible to formation of μ-PR2 oligomers. Addition of 2 equiv. of β-CgPH to [Rh(cod)2]BF4 gave [Rh(cod)(β-CgPH)2]BF4 (2) which was tested for the asymmetric hydrogenation of methyl acetamidocinnamate (MAC) and methyl acetamidoacrylate (MAA) and the results are given in Table 1, entry 1. The observed enantiomeric excesses appear modest especially when compared to recently reported monodentate P-ligands15 but we would argue from the following reasoning that the enantioselectivities obtained with the β-CgPH–Rh catalyst are still impressive.
Rationalising the enantioselectivity of an asymmetric hydrogenation catalyst has been a challenge ever since Knowles’ original discovery.16 Enantioselectivity is known to be influenced17 by electronic effects but discussions of catalytic asymmetric induction normally centre on the chiral shape of the metal–ligand moiety leading to discrimination between the diastereomeric intermediates. Using steric models, the sense of asymmetric induction can be reliably predicted18 and the success of ligands such as BINAP and DuPhos has been ascribed to the chiral array of P-substituents blocking diagonal quadrants.19 However, as the space filling model given in Fig. 3(a) shows, the diamondoid structure of CgPH would be expected to make the chiral space created by the enantiomers of CgPH ligands very similar. Fig 3(b) shows a schematic of a CgPH–Rh fragment and while the Me groups on the carbons α to the P (marked in blue) are sterically demanding, they are almost identically so on both sides of the P atom. The main asymmetry lies in the groups β to the P being O or CH2 which are normally regarded as isosteric.20 Therefore a rationale of the enantioselectivity of β-CgPH–Rh catalysts based on steric arguments has to be very delicate.
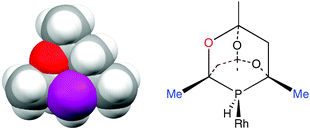 | ||
| Fig. 3 (a) Space filling model of β-CgPH (from the crystal structure shown in Fig. 2a); (b) β-CgPH coordinated to Rh. | ||
To explore the influence the β-CgP group may have on a diphosphine, the diastereomeric ligands La and Lb, featuring R,R- and S,S-3,5-dimethylphospholane moieties, respectively, were prepared by the routes shown in Scheme 2. The 31P NMR data for La and Lb are distinctive and the spectra obtained are consistent with the optical purity of the β-CgPH being over 100 : 1 er. Treatment of [Rh(cod)2]BF4 (cod = 1,5-cyclooctadiene) with La and Lb gave the corresponding chelates [Rh(La)(cod)]BF4 (3a) and [Rh(Lb)(cod)]BF4 (3b) which have been fully characterised. The crystal structure of 3b (Fig. 4) confirms the absolute stereochemistry of the CgP and phospholane units and also shows the presence of the third chiral element: the λ-conformation of the five-membered chelate ring.21
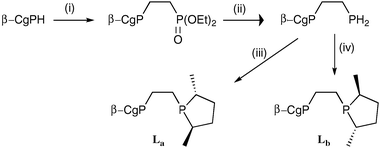 | ||
| Scheme 2 Reagents and conditions: (i) vinyl diethyl phosphonate; (ii) LiAlH4; (iii) 2 equiv. nBuLi followed by the cyclic sulfate of 2R,5R-2,5-pentanediol; (iv) 2 equiv. nBuLi followed by the cyclic sulfate of 2S,5S-2,5-pentanediol. | ||
![Thermal ellipsoid (50% probability) plot of the [Rh(Lb)(cod)]+ cation in 3b, omitting hydrogen atoms and the BF4− counter-anion. Selected geometrical data: bond lengths [Å] Rh1–P1 2.2989(11), Rh1–P2 2.2705(13), P1–C2 1.882(5), P1–C9 1.876(4), P1–C11 1.839(5), P2–C12 1.823(5), P2–C14 1.873(5), P2–C17 1.854(5), C11–C12 1.530(7); angles [°] P1–Rh1–P2 83.01(4), C2–P1–C9 93.9(2), C14–P2–C17 95.1(2); torsion angle [°] P1–C11–C12–P2 −47.9(4).](/image/article/2010/CC/b916359j/b916359j-f4.gif) | ||
| Fig. 4 Thermal ellipsoid (50% probability) plot of the [Rh(Lb)(cod)]+ cation in 3b, omitting hydrogen atoms and the BF4− counter-anion. Selected geometrical data: bond lengths [Å] Rh1–P1 2.2989(11), Rh1–P2 2.2705(13), P1–C2 1.882(5), P1–C9 1.876(4), P1–C11 1.839(5), P2–C12 1.823(5), P2–C14 1.873(5), P2–C17 1.854(5), C11–C12 1.530(7); angles [°] P1–Rh1–P2 83.01(4), C2–P1–C9 93.9(2), C14–P2–C17 95.1(2); torsion angle [°] P1–C11–C12–P2 −47.9(4). | ||
The enantioselectivities obtained with 3a and 3b (entries 2 and 3, Table 1) show, as expected, the effects of matching and mismatching of the chiral elements, with the matched isomer yielding 90% ee. The absolute stereochemistry of the hydrogenated products is controlled by the phospholane moiety.22 Strikingly, the matched catalyst diastereomer is different for MAA and MAC which implies that the substrate influences the intimate structure of the catalyst. In other words, there is a matching of the substrates with the diastereoisomeric catalysts. Recently an interesting example of substrate–diastereoisomer matching was reported for a palladium–carbene catalysed asymmetric arylation.23
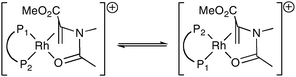
The substrate might exert its influence on the catalyst by influencing the ratio of the geometric isomers of the Rh–diphos intermediates (as exemplified in eqn (2) for MAA binding to Rh(I)) or the substrate may dictate the shape of the catalyst, i.e. an allosteric effect. It has been shown24 that the λ/δ conformation adopted by the five-membered ring in [Rh(chiraphos)(diene)]BF4 in the solid state depends on the ligated diene. Moreover we have previously reported evidence supporting the notion that MAC and MAA influence the λ/δ conformational ratio.25 Computational studies are in progress to determine the source and generality of this substrate effect in asymmetric hydrogenation catalysis.
 | (2) |
In conclusion, the enantiomers of 6-phospha-2,4,8-trioxa-adamantane (CgPH), which are ostensibly closely similar, have been readily resolved and give catalysts which yield surprisingly good enantioselectivities in asymmetric hydrogenation. This opens up the opportunity for the application of ligands derived from optically pure CgPH in asymmetric versions of the many catalyses in which monodentate and bidentate CgP-containing ligands excel.6–10 An example of substrate-matching with diastereomeric catalysts has been uncovered which illustrates the subtleties in asymmetric catalyst design.
We thank the EPSRC, the Royal Society, COST action CM0802 “PhoSciNet” and the CCDC for supporting this work and Johnson-Matthey for a loan of precious metal compounds.
Notes and references
- M. Epstein and S. H. Buckler, J. Am. Chem. Soc., 1961, 83, 3279 CrossRef CAS.
- J. H. Downing, J. Floure, K. Heslop, M. F. Haddow, J. Hopewell, M. Lusi, H. Phetmung, A. G. Orpen, P. G. Pringle, R. I. Pugh and D. Zambrano-Williams, Organometallics, 2008, 27, 3216 CrossRef CAS and references therein.
- G. M. Adjabeng, T. Brenstrum, J. Wilson, C. S. Frampton, A. Robertson, J. Hillhouse, J. McNulty and A. Capretta, Org. Lett., 2003, 5, 953 CrossRef CAS.
- (a) T. J. Cunningham, M. R. J. Elsegood, P. F. Kelly, M. B. Smith and P. M. Staniland, Eur. J. Inorg. Chem., 2008, 2326 CrossRef CAS; (b) G. M. Brown, M. R. J. Elsegood, A. J. Lake, N. M. Sanchez-Ballester, M. B. Smith, T. S. Varley and K. Blann, Eur. J. Inorg. Chem., 2007, 1405 CrossRef CAS.
- M. Guinó, A. C. Sullivan and J. R. H. Wilson, J. Mol. Catal. A: Chem., 2008, 293, 25 CrossRef CAS.
-
(a)
J. C. L. J. Suykerbuyk, E. Drent and P. G. Pringle, US Pat., WO9
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 842717 (to Shell), 1998;
(b) V. Gee, A. G. Orpen, H. Phetmung, P. G. Pringle and R. I. Pugh, Chem. Commun., 1999, 901 RSC;
(c) R. I. Pugh, P. G. Pringle and E. Drent, Chem. Commun., 2001, 1476 RSC;
(d)
E. Drent, P. G. Pringle and R. I. Pugh, US Pat., WO2001028972 (to Shell), 2001;
(e)
M. Schaefer, M. Slany, E. Zeller and M. Röper, US Pat., WO2002026382 (to BASF), 2002;
(f)
E. Drent, R. H. van der Made and R. I. Pugh, US Pat., WO2003070679 (to Shell), 2003;
(g)
E. Drent, R. H. van der Made, P. G. Pringle and R. I. Pugh, US Pat., WO2003070370 (to Shell), 2003;
(h)
X. Sava, M. Slany and M. Röper, US Pat., WO2003031457 (to BASF), 2003;
(i)
M. Slany, M. Schaefer and M. Röper, Ger Pat., DE10228293 (to BASF), 2003;
(j) I. R. Butler, P. K. Baker, G. R. Eastham, K. M. Fortune, P. N. Horton and M. B. Hursthouse, Inorg. Chem. Commun., 2004, 7, 1049 CrossRef CAS;
(k)
E. Drent, R. Ernst and W. W. Jager, US Pat., WO2004103948 (to Shell), 2004;
(l)
G. Eastham, US Pat., WO2004024322 (to Lucite), 2004;
(m)
G. Eastham, US Pat., WO2005118519 (to Lucite), 2005;
(n)
G. Eastham and N. Tindale, US Pat., WO2005079981 (to Lucite), 2005;
(o)
G. R. Eastham and N. Tindale, US Pat., WO2007119079 (to Lucite), 2007;
(p)
G. R. Eastham, M. Waugh and P. I. Richards, US Pat., WO2009057640 (to Lucite), 2007;
(q)
G. R. Eastham and I. Butler, US Pat., WO2008065448 (to Lucite), 2008;
(r)
G. R. Eastham and P. I. Richards, US Pat., WO2009010782 (to Lucite), 2009.
842717 (to Shell), 1998;
(b) V. Gee, A. G. Orpen, H. Phetmung, P. G. Pringle and R. I. Pugh, Chem. Commun., 1999, 901 RSC;
(c) R. I. Pugh, P. G. Pringle and E. Drent, Chem. Commun., 2001, 1476 RSC;
(d)
E. Drent, P. G. Pringle and R. I. Pugh, US Pat., WO2001028972 (to Shell), 2001;
(e)
M. Schaefer, M. Slany, E. Zeller and M. Röper, US Pat., WO2002026382 (to BASF), 2002;
(f)
E. Drent, R. H. van der Made and R. I. Pugh, US Pat., WO2003070679 (to Shell), 2003;
(g)
E. Drent, R. H. van der Made, P. G. Pringle and R. I. Pugh, US Pat., WO2003070370 (to Shell), 2003;
(h)
X. Sava, M. Slany and M. Röper, US Pat., WO2003031457 (to BASF), 2003;
(i)
M. Slany, M. Schaefer and M. Röper, Ger Pat., DE10228293 (to BASF), 2003;
(j) I. R. Butler, P. K. Baker, G. R. Eastham, K. M. Fortune, P. N. Horton and M. B. Hursthouse, Inorg. Chem. Commun., 2004, 7, 1049 CrossRef CAS;
(k)
E. Drent, R. Ernst and W. W. Jager, US Pat., WO2004103948 (to Shell), 2004;
(l)
G. Eastham, US Pat., WO2004024322 (to Lucite), 2004;
(m)
G. Eastham, US Pat., WO2005118519 (to Lucite), 2005;
(n)
G. Eastham and N. Tindale, US Pat., WO2005079981 (to Lucite), 2005;
(o)
G. R. Eastham and N. Tindale, US Pat., WO2007119079 (to Lucite), 2007;
(p)
G. R. Eastham, M. Waugh and P. I. Richards, US Pat., WO2009057640 (to Lucite), 2007;
(q)
G. R. Eastham and I. Butler, US Pat., WO2008065448 (to Lucite), 2008;
(r)
G. R. Eastham and P. I. Richards, US Pat., WO2009010782 (to Lucite), 2009. - R. A. Baber, M. L. Clarke, K. M. Heslop, A. C. Marr, A. G. Orpen, P. G. Pringle, A. Ward and D. E. Zambrano-Williams, Dalton Trans., 2005, 1079 RSC.
-
(a)
J. F. Deffner, E. R. Tucci, J. V. Ward, H. I. Thayer and H. J. Elder, Fr. Pat., FR1534510 (to Gulf), 1968;
(b)
W. Ahlers, US Pat., WO2001085661 (to BASF), 2001;
(c)
W. Ahlers and M. Slany, US Pat., WO2001085662 (to BASF), 2001;
(d)
K. Q. Almeida Lenero, E. Drent, R. van Ginkel and R. I. Pugh, US Pat., WO2005058788 (to Shell), 2005;
(e) M. L. Clarke and G. J. Roff, Chem.–Eur. J., 2006, 12, 7979;
(f) M. L. Clarke and G. J. Roff, Green Chem., 2007, 12, 792 Search PubMed.
- (a) T. Brenstrum, D. A. Gerristma, G. M. Adjabeng, C. S. Frampton, J. Britten, A. J. Robertson, J. McNulty and A. Capretta, J. Org. Chem., 2004, 69, 7635 CrossRef CAS; (b) S. A. Ohnmacht, T. Brenstrum, K. H. Bleicher, J. McNulty and A. Capretta, Tetrahedron Lett., 2004, 45, 5661 CrossRef CAS; (c) G. M. Adjabeng, T. Brenstrum, C. S. Frampton, A. J. Robertson, J. Hillhouse, J. McNulty and A. Capretta, J. Org. Chem., 2004, 69, 5082 CrossRef CAS.
-
(a) D. Gerristma, T. Brenstrum, J. McNulty and A. Capretta, Tetrahedron Lett., 2004, 45, 8319 CrossRef CAS;
(b)
T. P. Bender and J. A. Coggan, US Pat., US20080318 (to Xerox), 2008;
(c)
J. A. Coggan, US Pat., US2009131720 (to Xerox), 2009.
- A. Hadovic, A. J. Lough, R. H. Morris, P. G. Pringle and D. E. Zambrano-Williams, Inorg. Chim. Acta, 2006, 359, 2864 CrossRef.
- The procedure is a modification of that used to resolve 2,5-diphenylphospholane, see: F. Guillen, M. Rivard, M. Toffano, J. Y. Legros, J. C. Daran and J. C. Fiaud, Tetrahedron, 2002, 58, 5895 Search PubMed.
- From the filtrates of the fractional crystallisations of the [β-CgPO2][QH], samples rich in the α-enantiomer of CgPOOH (er 10 : 1) were obtained (see ESI‡).
- M. Ostermeier, J. Priess and G. Helmchen, Angew. Chem., Int. Ed., 2002, 41, 612 CrossRef CAS.
- M. T. Reetz, Angew. Chem., Int. Ed., 2008, 47, 2556 CrossRef CAS and references therein.
- W. S. Knowles, Acc. Chem. Res., 1983, 16, 106 CrossRef CAS.
- Y.-Y. Yan and T. V. RajanBabu, Org. Lett., 2000, 26, 4137 CrossRef.
- (a) J. Halpern, D. P. Riley, A. S. C. Chan and J. J. Pluth, J. Am. Chem. Soc., 1977, 99, 8055 CrossRef CAS; (b) W. S. Knowles, Acc. Chem. Res., 1983, 16, 106 CrossRef CAS; (c) S. K. Armstrong, J. M. Brown and M. J. Burk, Tetrahedron Lett., 1993, 34, 879 CrossRef CAS; (d) T. Imamoto, J. Watanabe, Y. Wada, H. Masuda, H. Yamada, H. Tsuruta, S. Matsukawa and K. Yamaguchi, J. Am. Chem. Soc., 1998, 120, 1635 CrossRef CAS; (e) I. D. Gridnev and T. Imamoto, Acc. Chem. Res., 2004, 37, 633 CrossRef CAS; (f) I. D. Gridnev, N. Higashi, K. Asakura and T. Imamoto, J. Am. Chem. Soc., 2000, 122, 7183 CrossRef CAS.
- T. Ohkuma, M. Kitamura and R. Noyori, in New Frontiers in Asymmetric Catalysis, ed. K. Mikami and M. Lautens, Wiley-Interscience, Hoboken (USA), 2007 Search PubMed.
- J. H. Block, in Wilson and Gisvold’s Textbook of Organic Medicinal and Pharmaceutical Chemistry, ed. J. H. Block and J. M. Beale Jr., Lippincott, Williams and Wilkins Publishers, Philadelphia, 1998 Search PubMed.
- L. Dahlenburg, Coord. Chem. Rev., 2005, 249, 2962 CrossRef CAS and references therein.
- M. J. Burk, Acc. Chem. Res., 2000, 33, 363 CrossRef CAS.
- X. Luan, R. Mariz, C. Robert, M. Gatti, S. Blumentritt, A. Linden and R. Dorta, Org. Lett., 2008, 10, 5569 CrossRef CAS.
- H. Tsuruta, T. Imamoto, K. Yamaguchi and I. D. Gridnev, Tetrahedron Lett., 2005, 46, 2879 CrossRef CAS.
- (a) R. A. Baber, J. G. de Vries, A. G. Orpen, P. G. Pringle and K. von der Luehe, Dalton Trans., 2006, 4821 RSC; (b) D. W. Norman, C. A. Carraz, D. J. Hyett, P. G. Pringle, J. B. Sweeney, A. G. Orpen, H. Phetmung and R. L. Wingad, J. Am. Chem. Soc., 2008, 130, 6840 CrossRef CAS.
Footnotes |
| † This article is part of a ChemComm ‘Catalysis in Organic Synthesis’ web-theme issue showcasing high quality research in organic chemistry. Please see our website (http://www.rsc.org/chemcomm/organicwebtheme 2009) to access the other papers in this issue. |
| ‡ Electronic supplementary information (ESI) available: Full experimental details for the synthesis and characterisation of the ligands and complexes, conditions used for the catalysis and X-ray structural data. CCDC 743916–743918. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b916359j |
| This journal is © The Royal Society of Chemistry 2010 |