Aza-dibenzocyclooctynes for fast and efficient enzyme PEGylation via copper-free (3+2) cycloaddition†
Marjoke F.
Debets‡
,
Sander S.
van Berkel‡
,
Sanne
Schoffelen
,
Floris P. J. T.
Rutjes
,
Jan C. M.
van Hest
* and
Floris L.
van Delft
*
Institute for Molecules and Materials, Radboud University Nijmegen, Heyendaalseweg 135, NL-6525 AJ Nijmegen, The Netherlands. E-mail: J.vanHest@science.ru.nl; F.vanDelft@science.ru.nl; Fax: (+31) 24 3653393; Tel: (+31) 24 3653204; (+31) 24 3652373
First published on 6th November 2009
Abstract
A strained aza-dibenzocyclooctyne was prepared via a high-yielding synthetic route. Copper-free, strain-promoted click reaction with azides showed excellent kinetics, and a functionalised aza-cyclooctyne was applied in fast and efficient PEGylation of enzymes.
The process of PEG conjugation to either peptides or proteins, known as PEGylation, is a procedure of growing interest in both biotechnology and therapeutical science.1 PEGylation of proteins improves their in vivo applicability by reducing aggregation, shielding critical protein binding sites and improving the water solubility.2 As a result, a large variety of methods for the conjugation of PEG to either peptides or proteins is currently available.3 In most cases, however, a large excess of PEG and prolonged reaction times are required, and nevertheless generally result in suboptimal conversions.4 With the recent discovery of the Cu(I)-catalysed Huisgen cycloaddition reaction,5 a powerful new methodology was added to the field of bioconjugation6 with the particular benefits that the reaction is selective, fast and high-yielding. The Cu(I)-catalysed 1,3-dipolar cycloaddition reaction has become of great use in labelling studies, the development of new therapeutics and in protein modification.7 In addition, the Cu(I)-catalysed 1,3-dipolar cycloaddition reaction has also been applied in the synthesis of PEG–proteinbioconjugates .8 A disadvantage of this reaction in PEGylation is the potential presence of residual copper in the product. Not only is Cu(I) toxic to cells, it can also bind to the active site of enzymes, thereby blocking or reducing biological activity.9,10 Furthermore, Cu(I) is easily disproportionated in an aqueous environment, thus reducing the rate of the reaction. Therefore, interest is growing in methods involving Cu-free 1,3-dipolar cycloaddition reactions.11 Several strain-promoted systems, such as oxanorbornadienes,12 cyclooctynes,10 and dibenzocyclooctynes13 (Fig. 1) have recently been developed for the fast and selective reaction with azide-containing biomolecules and have found application in e.g. tumour imaging,14glycanlabelling,10,13in vivo imaging,15 and surface modification.16 However, to the best of our knowledge, these systems have not yet been employed in the PEGylation of proteins.
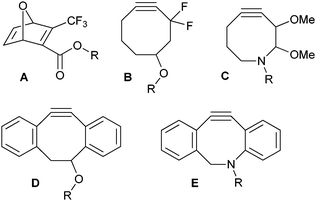 | ||
| Fig. 1 Strain-promoted systems for Cu-free click reactions. | ||
Inspired by the dibenzocyclooctyne derivative (DIBC, structure D in Fig. 1) developed by Boons et al.13 and aza-dimethoxycyclooctyne (DIMAC, structure C in Fig. 1) synthesised by Bertozzi et al.,17 we set out to develop the hybrid structure aza-dibenzocyclooctyne (DIBAC, structure E in Fig. 1). The aza-dibenzocyclooctyne was designed to combine the favourable kinetics of DIBC (D)13 with the increased hydrophilicity of DIMAC (C).17 With respect to the latter, the nitrogen atom in our designed analogue should allow straightforward functionalisation of the aniline moiety and modification of the system, e.g. by sulfonation. At the same time, we set ourselves the goal to develop a straightforward, high-yielding and fast synthetic route. Herein we report the facile synthesis of aza-dibenzocyclooctyne and derivatives thereof for the quantitative PEGylation of proteins.
The synthetic route towards key-intermediate dihydro-dibenzo-azocine (5) is shown in Scheme 1, and is based on a Sonogashira cross-coupling reaction and a reductive amination ring-closing step. Whereas the Sonogashira reaction under standard inert atmosphere initially resulted in Glaser coupling, performing the reaction under an N2/H2-atmosphere18 effectively nihilated Glaser coupling and led to the Sonogashira coupling product 1 in quantitative yield. Next, Boc-protection, generating compound 2, was found to be essential, in order to make partial hydrogenation of the acetylene to the Z-alkene possible, yielding 3 (95%). Next, Dess–Martin oxidation led to aldehyde4, the precursor for the reductive amination. Boc-deprotection under acidic conditions resulted in immediate and exclusive formation of a cyclic imine, which was not isolated but subjected to NaBH4 reduction, generating the free secondary amine (5) in quantitative yield over the two steps. Via this high-yielding protocol the key aza-cyclooctene intermediate 5 was synthesised in an excellent yield of 70% over five steps.
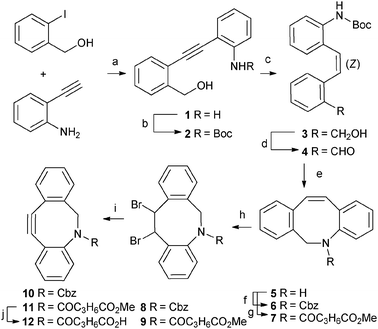 | ||
| Scheme 1 Reagents and conditions: (a) PdCl2(PPh3)2, CuI, Et3N, THF, N2/H2-atmosphere, r.t., 4 h (99%); (b) Boc2O, THF, 70 °C, 2 d (83%); (c) 10% Pd/BaSO4, quinoline, H2, MeOH, r.t., 1.5 h (95%); (d) Dess–Martin periodinane, NaHCO3, CH2Cl2, r.t., 40 min (90%); (e) (1) 2 M HCl in EtOAc, r.t., 1 h; (2) NaBH4, H2O, r.t., o.n. (100%); (f) CbzCl, Na2CO3, H2O, CH2Cl2, r.t., 3 h (86%); (g) ClCOC3H6CO2Me, Et3N, CH2Cl2, r.t., 1.5 h (94%); (h) Br2, CH2Cl2, 0 °C, 2 h (8: 67%, 9: 81%); (i) KOtBu, THF, 0 °C → r.t., o.n. (10), −40 °C, 2 h (11) (10: 87%, 11: 84%); (j) LiOH, THF, H2O, r.t., 3 h (92%). | ||
Unfortunately, direct bromination of dihydrodibenzo-azocine 5 resulted in the intramolecular substitution of a bromide, leading to an indoline, thus requiring protection of the amine. Consequently, a Cbz-group was introduced generating compound 6 in good yield (86%). The alkene moiety could then be smoothly converted into an alkynevia successive bromination (67%) and elimination (87%). Elimination was performed using KOtBu since other bases such as LDA, n-BuLi, NaOH (6 or 12 M) all failed to give the desired aza-dibenzocyclooctyne 10. Compound 10, although fulfilling our aim to prepare an aza-cyclooctyne, is clearly lacking a handle for further functionalisation. Unfortunately, deprotection of the Cbz-group, using either acidic or basic conditions, did not yield the free amine (i.e.E, R = H, Fig. 1). Instead, 6H-isoindolo[2,1-a]indole (13) was formed, presumably via an 5-endo-dig cyclisation, thereby relieving ring-strain with formation of the indole. Consequently, N-functionalisation was required prior to alkyne formation. To this end, 5 was equipped with a small functionalised linker, leading to compound 7 in 94% yield (Scheme 1). Subsequent bromination (81%) and elimination with KOtBu (84%) cleanly resulted in alkyne11. A 1 M solution of KOtBu in THF was used since the use of solid KOtBu resulted in hydrolysis of the methyl ester, which gave rise to undesired side-reactions. Finally, a functionalizable probe (12) was prepared viahydrolysis of the methyl ester. The above reported reaction sequence yielded the desired strained aza-cyclooctyne probe, with a functional handle, in a good overall yield of 41% over 9 steps.
Next, the kinetics in the 1,3-dipolar cycloaddition of the Cbz-protected and acid-functionalised DIBACs (10 and 12, respectively) with benzyl azide were determined in CD3OD, giving rate constants of 0.29 M−1 s−1 and 0.31 M−1 s−1, respectively. Cycloaddition of DIBAC 12 and 2-azidopropanoic acid was also investigated by performing the reaction in basic D2O, since addition of a small amount of 2 M NaOH was necessary to dissolve 12. In D2Ocycloaddition was calculated to proceed with a rate constant of 0.36 M−1 s−1, slightly faster than in CD3OD. The results of the kinetic experiments are summarised in Table 1. Much to our satisfaction, the kinetic parameters of our new system proved slightly better than DIBC (k = 0.17 M−1 s−1)13 and DIFO (k = 0.076 M−1 s−1)10 and it reacted approximately 100-fold faster than the hydrophilic DIMAC-system (k = 3×10−3 M−1 s−1).17
To investigate the applicability of the Cu-free 1,3-dipolar cycloaddition in the conjugation of polyethylene glycol to proteins, the aza-dibenzocyclooctyne analogue 12 was functionalised with H2N-PEG2000-OMe via an EDC-coupling, yielding DIBAC-mPEG2000 (15, Scheme 2). For comparison, DIBC-mPEG2000 (16) was also prepared via conversion of the alcohol into a 4-nitrophenyl carbonate13 followed by substitution with H2N-PEG2000-OMe. Initially, azide-containing CalB (AHA-CalB)19 was used for the conjugation studies. This modified enzyme, obtained by recombinant expression in auxotrophic E. coli, contains five azidohomoalanine residues, four of which are concealed inside the protein. Consequently, only one azido residue is exposed on the exterior of the enzyme, readily available for ligation. We have previously reported that this enzyme underwent PEGylation selectively with the most accessible residue by applying the Cu(I)-catalysed (3+2) cycloaddition reaction. However, full conversion could never be reached, despite the use of 20 equivalents of acetylene-functionalised acetylene-PEG5000 and stirring for 1-3 days.19 Now, PEGylation of AHA-CalB was pursued by mixing AHA-CalB (1 μg/μL, corresponding to ~30 μM) with DIBAC-mPEG200015 or DIBC-mPEG200016 (one, two and five equiv). After three hours the reaction was quenched with benzyl azide and analysed by SDS-PAGE (Fig. 2). As becomes clear, with five equivalents of DIBAC-mPEG2000 (15) full conversion was observed within three hours (Fig. 2, lane 2). Interestingly, not only was the enzyme fully functionalised, but it appears that one of the less accessible azides also reacted, generating di-PEGylated CalB, confirming the excellent reactivity of aza-dibenzocyclooctyne towards azides. The higher reactivity of the DIBAC system is further demonstrated by the fact that a higher degree of PEGylation is observed than with DIBC-mPEG2000. In both cases, the small amount of ‘unreacted’ CalB observed is not caused by failure to react, but can be ascribed to the fact that AHA-CalB always contains a small amount of non-modified enzyme.19 Performing the PEGylation with either one or two equivalents of DIBAC 15, approximately 50% conversion to the single PEGylated product was observed (see lanes 3 and 4). Using DIBC 16 under the same conditions resulted in somewhat lower conversions (see lanes 6 and 7).
 | ||
| Scheme 2 Reagents and conditions: (a) H2N-PEG2000-OMe, EDC, DMAP, CH2Cl2, 2 d, r.t.; (b) (1) (4-NO2C6H4)OCOCl, pyridine, CH2Cl2, 4 h, r.t.; (2) H2N-PEG2000-OMe, CH2Cl2, 1 d, r.t.; (c) PBS-buffer (pH 8.5, 30 μM), 3 h, r.t. | ||
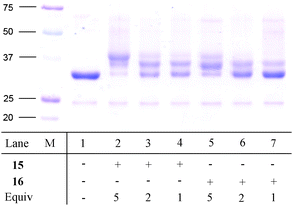 | ||
| Fig. 2 SDS-PAGE gel of PEGylated AHA-CalB (1 μg/μL) using various equivalents of cyclooctyne 15 or 16. | ||
The fast and quantitative ligation of both reagents to AHA-CalB shows the high potential of these systems. To investigate the reactivity and efficiency of these reagents towards other enzymes, HRP was modified via diazotransfer as recently developed in our group.20 The resulting HRP-N3 was then reacted with either 15 or 16 following the same procedure as applied to AHA-CalB, giving almost identical results (See ESI, Fig. S1† ). The aza-dibenzocyclooctyne is in both cases more reactive than the dibenzocyclooctyne, especially when only one or two equivalents were used.
Nevertheless, in comparison to the Cu(I)-catalysed cycloaddition reaction, both DIBCs show fast and efficient modification of both enzymes, clearly showing the power and applicability of these copper-free systems.
In conclusion, we have developed a new, highly efficient reagent for copper-free (3+2) cycloaddition reactions. The aza-dibenzocyclooctyne is easily accessible via a versatile and high-yielding synthetic route, and allows straightforward functionalisation via the nitrogen atom. In addition, the outstanding reaction rate constant (k = 0.3 M−1 s−1) marks the suitability of this probe for bioconjugation purposes, as was underlined by the effective PEGylation of enzymes. The functionalisation, further derivatisation and application of aza-dibenzocyclooctynes in bioconjugation are topics currently under investigation in our laboratories.
Notes and references
- F. M. Veronese and J. M. Harris, Adv. Drug Delivery Rev., 2008, 60, 1–2 CrossRef CAS; G. Pasut, M. Morpurgo and F. M. Veronese, in Delivery of Protein and Peptide Drugs in Cancer, ed. V. P. Torchilin, Imperial College Press, London, 2006, pp. 85–110 Search PubMed.
- G. Pasut, A. Mero, F. Caboi, S. Scaramuzsza, L. Sollai and F. M. Veronese, Bioconjugate Chem., 2008, 19, 2427–2431 CrossRef CAS; J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS.
- F. M. Veronese, Biomaterials, 2001, 22, 405–417 CrossRef CAS.
- W. H. Binder and R. Sachsenhofer, Macromol. Rapid Commun., 2008, 29, 952–981 CrossRef CAS; A. Deiters, T. A. Cropp, D. Summerer, M. Mukherji and P. G. Schultz, Bioorg. Med. Chem. Lett., 2004, 14, 5743–5745 CrossRef CAS; A. J. Dirks, J. J. L. M. Cornelissen and R. J. M. Nolte, Bioconjugate Chem., 2009, 20, 1129–1138 CrossRef CAS.
- C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 41, 2596–2599; V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596–2599 CrossRef CAS.
- G. E. Means and R. E. Feeney, Bioconjugate Chem., 1990, 1, 2–12 CrossRef CAS; J. Rademann, Angew. Chem., Int. Ed., 2004, 43, 4554–4556 CrossRef CAS; C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030–10074 CrossRef CAS; J. M. Baskin and C. R. Bertozzi, QSAR Comb. Sci., 2007, 26, 1211–1219 Search PubMed.
- B. Le Droumaguet and K. Velonia, Macromol. Rapid Commun., 2008, 29, 1073–1089 CrossRef; A. J. Dirks, J. J. L. M. Cornelissen, F. L. van Delft, J. C. M. van Hest, R. J. M. Nolte, A. E. Rowan and F. P. J. T. Rutjes, QSAR Comb. Sci., 2007, 26, 1200–1210 Search PubMed; J.-F. Lutz and Z. Zarafshani, Adv. Drug Delivery Rev., 2008, 60, 958–970 CrossRef CAS.
- M. A. Gauthier and H. A. Klok, Chem. Commun., 2008, 2591–2611 RSC; M. van Dijk, D. T. S. Rijkers, R. M. J. Liskamp, C. F. van Nostrum and W. E. Hennink, Bioconjugate Chem., 2009, 20 DOI:10.1021/bc900087a.
- E. Lallana, E. Fernandez-Megia and R. Riguera, J. Am. Chem. Soc., 2009, 131, 5748–5750 CrossRef CAS.
- J. M. Baskin, J. A. Prescher, S. T. Laughlin, N. J. Agard, P. V. Chang, I. A. Miller, A. Lo, J. A. Codelli and C. R. Bertozzi, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 16793–16797 CrossRef CAS.
- C. R. Becer, R. Hoogenboom and U. S. Schubert, Angew. Chem., Int. Ed., 2009, 48, 4900–4908 CrossRef CAS; E. M. Sletten and C. R. Bertozzi, Angew. Chem., Int. Ed., 2009, 48, 6974–6998 CrossRef CAS.
- S. S. van Berkel, A. J. Dirks, M. F. Debets, F. L. van Delft, J. J. L. M. Cornelissen, R. J. M. Nolte and F. P. J. T. Rutjes, ChemBioChem, 2007, 8, 1504–1508 CrossRef CAS.
- X. Ning, J. Guo, M. A. Wolfert and G.-J. Boons, Angew. Chem., Int. Ed., 2008, 47, 2253–2255 CrossRef CAS.
- S. S. van Berkel, A. J. Dirks, S. A. Meeuwissen, D. L. L. Pingen, O. C. Boerman, P. Laverman, F. L. van Delft, J. J. L. M. Cornelissen and F. P. J. T. Rutjes, ChemBioChem, 2008, 9, 1805–1815 CrossRef CAS; P. Laverman, S. A. Meeuwissen, S. S. van Berkel, W. J. G. Oyen, F. L. van Delft, F. P. J. T. Rutjes and O. C. Boerman, Nucl. Med. Biol., 2009, 36, 749–757 CrossRef CAS.
- S. T. Laughlin, J. M. Baskin, S. L. Amacher and C. R. Bertozzi, Science, 2008, 320, 664–667 CrossRef CAS.
- L. A. Canalle, S. S. van Berkel, L. T. de Haan and J. C. M. van Hest, Adv. Funct. Mater., 2009, 19, 3464–3470 CrossRef CAS.
- E. M. Sletten and C. R. Bertozzi, Org. Lett., 2008, 10, 3097–3099 CrossRef CAS.
- A. Elangovan, Y.-H. Wang and T.-I. Ho, Org. Lett., 2003, 5, 1841–1844 CrossRef CAS.
- S. Schoffelen, M. H. L. Lambermon, M. B. van Eldijk and J. C. M. van Hest, Bioconjugate Chem., 2008, 19, 1127–1131 CrossRef CAS.
- S. F. M. van Dongen, R. L. M. Teeuwen, M. Nallani, S. S. van Berkel, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Bioconjugate Chem., 2009, 20, 20–23 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic procedures for compounds 1–16, kinetic experiments and plots, enzyme functionalisation. See DOI: 10.1039/b917797c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2010 |