
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGraphendofullerene: a novel molecular two-dimensional ferromagnet†
Diego
López-Alcalá
 a,
Ziqi
Hu
b and
José J.
Baldoví
*a
a,
Ziqi
Hu
b and
José J.
Baldoví
*a
aInstituto de Ciencia Molecular, Universitat de València, Catedrático José Beltrán 2, 46980 Paterna, Spain. E-mail: j.jaime.baldovi@uv.es
bKey Laboratory of Precision and Intelligent Chemistry, Collaborative Innovation Center of Chemistry for Energy Materials (iChEM), Department of Materials Science and Engineering, University of Science and Technology of China, Hefei 230026, China
First published on 24th March 2025
Abstract
Carbon chemistry has attracted a lot of attention from chemists, physicists and material scientists in the last few decades. The recent discovery of graphullerene provides a promising platform for many applications due to its exceptional electronic properties and the possibility to host molecules or clusters inside the fullerene units. Herein, we introduce graphendofullerene, a novel molecular-based two-dimensional (2D) magnetic material formed by trimetallic nitride clusters encapsulated on graphullerene. Through first-principles calculations, we demonstrate the successful incorporation of the molecules into the 2D network formed by C80 fullerenes, which leads to robust long-range ferromagnetic order with a Curie temperature (TC) of 38 K. Additionally, we achieve a 45% and 18% increase in TC by strain engineering and electrostatic doping, respectively. These findings open the way for a new family of molecular 2D magnets based on graphendofullerene for advanced technologies.
Introduction
Since the mid 80s carbon chemistry has emerged as a prominent research field due to the discovery of synthetic carbon allotropes such as fullerenes1,2 and carbon nanotubes,3 and the isolation of graphene in 2004,4,5 thus offering countless possibilities for exploiting different physical phenomena.6 This is due to the different orbital hybridizations and arrangements that provide them with unique optical and electronic properties. In particular, graphene pioneered the field of 2D materials that nowadays covers a wide range of functionalities,7,8 from insulators9 to superconductors10 and magnetic systems,11 leading to applications in catalysis,12 gas sensing,13 valleytronics14 and spintronics.15 Moreover, they can be assembled into van der Waals (vdW) heterostructures,16 and their layers can be twisted with respect to each other, to create novel multifunctional materials and devices.17Fullerenes stand out due to their advanced applications in photovoltaics,18 biomedicine19,20 or catalysis.21 These arise from their structural morphology, which allows them to harbor small molecules or clusters inside the carbon backbone,22–24 forming the so-called endohedral fullerenes or metallofullerenes.25,26 Due to their heterogeneous nature, these systems have enormous potential in cutting-edge technologies such as information storage27–29 or spintronics.30–34 Very recently, a 2D fullerene-based material equivalent to graphene, the so-called graphullerene, has been synthesized.35,36 Graphullerene's structure consists of a 2D network of fullerenes covalently interconnected, which provides the system with robust structural stability,37,38 and remarkable electronic,39–42 mechanical43–46 and optical47–49 properties. This makes graphullerene well suited as an exciting platform for many different technological applications.50–52 However, the capability of the fullerene building blocks as host structures within the 2D network still remains unexplored, even from a theoretical point of view, which would provide infinite possibilities emerging from the versatility of chemistry, in the 2D limit.
In this work, we report a first-principles study on a novel 2D material that we named ‘graphendofullerene’ by analogy to graphullerene and endohedral fullerene. The graphullerene-based material that we propose as a proof of concept is formed by using magnetic endohedral metallofullerene cages (V3N@C80) as building blocks. First, we demonstrate the thermodynamic stability of graphendofullerene and calculate its electronic structure, magnetic properties and spin dynamics. Interestingly, our microscopic analysis reveals that intermolecular magnetic interactions in the network lead to long-range ferromagnetic (FM) order. Finally, we apply strain engineering and electrostatic doping to enhance the magnetic behavior of the system. Our results pave the way for the preparation of magnetic graphullerene-based materials and their future optimization for emerging applications, since a wide range of possibilities arise from the host capabilities of fullerene building blocks.
Computational details
Structural relaxations and AIMD simulations were performed using the Vienna ab initio Simulation Package (VASP).53,54 The generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhof (PBE) parametrization was used to account for exchange-correlation energy.55 van der Waals interactions were considered using the Grimme D3 method approximation.56 The projector-augmented wave (PAW) method was used with an energy cutoff of 530 eV. A 4 × 5 × 1 k-point grid was used to sample reciprocal space (Fig. S9†). The lattice constant and atomic coordinates were relaxed until forces were less than 0.03 eV Å−1. AIMD simulations were performed using the NVT canonical ensemble for 9 ps with a time step of 2 fs, using the optimized lattice parameters and structure. Charge transfer analysis was performed using Bader charge partition as proposed by the Henkelman group.57 Total energies for the FM and three different AFM configurations (single-stripe, double-stripe and zig-zag, see Fig. 2), and magnetic properties were calculated using the SIESTA code,58,59 in order to take advantage of the localized pseudoatomic orbital approach. We used norm-conserving scalar relativistic pseudo-potentials taken from the Pseudo-Dojo database60 in the psml format.61 The same k-point grid was used in combination with a real-space mesh cutoff of 700 Ry, with a double-ζ polarized basis set for all atoms. We fed the TB2J package62 with SIESTA wavefunctions to calculate magnetic exchange couplings. We used as a spin Hamiltonian, where Si and Sj are distinct magnetic moments present in the lattice and Jij is the magnetic exchange interaction between them. Atomistic spin dynamics simulations were carried out using Vampire software,63 where a 30 nm × 30 nm sample was considered in combination with 500
as a spin Hamiltonian, where Si and Sj are distinct magnetic moments present in the lattice and Jij is the magnetic exchange interaction between them. Atomistic spin dynamics simulations were carried out using Vampire software,63 where a 30 nm × 30 nm sample was considered in combination with 500![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 equilibration and loop time steps.
000 equilibration and loop time steps.
Results and discussion
C60 is the smallest fullerene which obeys the isolated pentagonal ring (IPR) rule,64 but its reduced inner space limits the incorporation of a wide variety of guest molecules. This limitation is overcome by C80, the next member that follows the IPR rule in the fullerene family with the same point group symmetry (Ih), due to its larger volume.65 Graphullerene has been reported to exist in several polymorph forms.36 Among them, the quasi-hexagonal phase (qHP) (Fig. S1a†) has shown higher stability and robustness since it maximizes the number of sp3 C atoms bridging the fullerene cages, which creates a more dense arrangement of cages and releases surface tension, thus stabilizing the entire system.38,66 Therefore, we selected a 2D qHP Ih-C80 graphullerene network in which each fullerene cage is surrounded by six neighboring cages (Fig. S1a†). This phase corresponds to a hexagonal lattice with a Pmna (No. 53) space group, where the fully optimized lattice parameters are a = 17.84 Å and b = 10.29 Å. The chemical bonds between adjacent cages are based on [2 + 2] cycloaddition along a and b lattice vectors and diagonal lines of the rectangular unit cell. Covalent intramolecular C–C σ bonds have a length of 1.42 Å, whereas intermolecular bonds are 1.59 Å, owing to the novel sp3 character of the C attaching C80 cages. Our calculations show that the fullerene units are slightly distorted after polymerization, where the cages adopt an ellipsoidal shape, as we can observe in Fig. S1b.† The calculated out-of-plane diameter of the cages is 8 Å, whereas in the plane parallel to the lattice we can observe that the presence of covalent bonds between C cages generates a larger diameter of 8.9 Å.Encapsulation of trimetallic nitrides has been demonstrated to be a powerful methodology to stabilize highly symmetric icosahedral fullerenes,67 since the isolation of monometallofullerene is still challenging because of their minimal charge transfer to the C atoms that limits the stability of the host–guest system.22 Previous studies have reported the successful synthesis of VxSc3–xN@C80 (x = 1, 2);68 nevertheless the incorporation of V3N remains challenging from an experimental point of view but it has been theoretically predicted to be feasible.69 This guest molecule would represent the best candidate to induce magnetism in the 2D network of graphullerene since it has a higher presence of V magnetic atoms. Given this, we explore the feasibility of incorporating V3N into fullerene units that form a 2D graphullerene network. We selected a V3N@C80 conformation where the guest clusters adopt a configuration with V3+ atoms pointing towards half of the sp3 bridges in a C3-symmetry fashion (Fig. 1a), as the linking C atoms have shown a pronounced electronic localization (Fig. S2†). We can see that the clusters present a slight distortion from planarity, due to unpaired electrons in N, but they mostly lay in the network plane. Also, the incorporation of the clusters barely distorts the fullerene cages, where the average host–guest distance is ∼2.5 Å. Then, we calculate the formation energy (EF), that is defined as EF = EV3N@C80 − (EV3N + EC80. We found an EF of −10.73 eV/molecule that is compatible with similar theoretical studies for the endohedral metallofullerene cages.70 This indicates a favorable formation of the V3N@C80 network, because the system is stabilized with respect to its separate V3N and C80 units. A charge transfer of 2.4 electrons per V3N cluster to the graphullerene network is revealed by Bader charge analysis, where the V atoms are responsible to donate charge density to the C skeleton (Fig. 1c). We do not observe any further redistribution of the charge density after encapsulation, which prevents the exhibition of radical character in the system, as has been observed in endohedral metallofullerenes.71 Furthermore, in order to corroborate the stability of the herein proposed graphendofullerene, we performed ab initio molecular dynamics (AIMD) simulations and phonon calculations to monitor the dynamic stability of the system at a given temperature. Our calculations unveil that the structure is thermodynamically stable at room temperature and up to 600 K since the structure is retained under the inclusion of thermal fluctuations (ESI† Sections 2.1 and 2.2).
 | ||
| Fig. 1 (a) V3N@C80 graphendofullerene top view. Color code: carbon (grey), vanadium (red) and nitrogen (blue). (b) Electronic band structure (left) and projected density of states (PDOS) (right) for the V3N@C80 monolayer. Blue (red) color in (a) represents spin up (down) electrons. (c) Charge density difference after V3N encapsulation. Yellow (blue) regions represent charge accumulation (depletion). (d) Energy level polarization of C80. | ||
The electronic band structure and density of states of the V3N@C80 network are shown in Fig. 1b. One can observe a clear influence of the host–guest interaction due to strong spin-dependent hybridization between energy levels around the Fermi energy level due to the charge transfer interaction, leading to half-metallicity of the system as it shows metallic and semiconductor behavior for spin up and down components, respectively. The half-metal ground state can provide the system with 100% polarized spin currents, which can lead to an enormous interest in spintronics for spin-dependent tunnel junctions and magnetoresistance devices.72,73 Consequently, the energy levels of C atoms on the network are polarized due to the interaction with magnetic clusters, as illustrated in Fig. 1d. This interaction is responsible for redistributing the charge density of the system after encapsulation and harnessing the stabilization of the network.
The electronic polarization around the Fermi level arising from the half-metallic band structure of V3N@C80 makes this system a promising candidate for spintronics. Therefore, we systematically study the magnetic properties of graphendofullerene as a novel molecule-based 2D magnet. The calculated magnetic moments are 2.7 μB for V atoms, whereas the long-range magnetic order present in the monolayer is predicted to be ferromagnetic. We computed the energy difference between the possible antiferromagnetic (AFM) and the FM configurations, namely EAFM and EFM, respectively (Fig. 2). We observe that the FM order is the most stable magnetic configuration in V3N@C80 (Table S1†). As depicted in Fig. 1b, around the Fermi level the energy levels are completely polarized, which stabilizes the FM order in the network. Regarding magnetic exchange interactions (J), the arrangement of C80 cages and the morphology of the guest clusters allow several Js in the network (Fig. 3a). We categorize them into two different groups, namely (i) intramolecular interactions, i.e. those interactions between V atoms from the same molecule (J1) and, (ii) intermolecular interactions, which are those magnetic interactions between adjacent molecules in the network (J2). Due to the close distance between V atoms forming guest clusters (∼3.2 Å), J1 interactions have a high intensity of ∼50 meV and short-range intrinsic character, mainly due to a robust hybridization of d orbitals around the Fermi level that enhances magnetic intramolecular interactions (Fig. 3c) but prevents the participation of J1 in the magnetic reciprocity of the network. These intramolecular interactions stabilize a ferromagnetic coupling between the V atoms inside the guest molecules. On the other hand, the main factor in the stabilization of a long-range magnetic order in graphendofullerene is intermolecular ferromagnetic exchange interactions between adjacent V3N molecules. The orientation of guest clusters inside C80 cages provides two different first neighbor intermolecular interactions, namely, J21, that arises from the V atoms directly oriented along the x axis of the network, and J22, which is aligned with the y axis. In the case of J21, the V atoms point directly towards them, and the resulting interaction at 6.7 Å is 2.28 meV, whereas J22 is equal to 0.33 meV due to a longer distance of interaction (7.8 Å). J21 (J22) are one (two) order of magnitude lower than J1 but connect magnetic molecules and provides the V3N@C80 network with a robust long-range magnetic order, that opens the way to observe critical properties of a ferromagnet such as Curie temperature (TC) or coercivity on this molecule-based 2D network. We applied the same methodology to V2ScN@C80 (Fig. S10 and S11†), where we observe that the magnetic intermolecular interactions are preserved despite the lower presence of magnetic V atoms.
 | ||
| Fig. 2 Spin density of considered magnetic configurations for V3N@C80. (a) Ferromagnetic (FM), (b) zig-zag, (c) single-stripe and (d) double-stripe antiferromagnetic (AFM) configurations. Color code: spin up (blue) and down (red) electrons. | ||
 | ||
| Fig. 3 (a) Schematic representation of intra/intermolecular magnetic interactions in the V3N@C80 network. (b) Orbital contribution to J21 from interacting first neighbor V atoms. (c) PDOS of d orbitals of V atoms in J21 and C atoms in linking moieties of V3N@C80. | ||
In order to check the role of C80 graphullerene in the mediation of intermolecular magnetic exchange coupling, we recomputed the magnetic exchange interactions in a free-standing molecular array (i.e. removing all C atoms from the calculation). We found that intermolecular interactions are suppressed, whereas J1 remains present (see Fig. S5†). This result is a useful hint that points to the carbon skeleton as a mediator of the magnetic interactions that stabilize ferromagnetism in the network. Magnetic interactions between two atoms are governed by all the possible interactions of their orbitals. In our case we considered the interactions between d orbitals of V atoms in the network, so we can decompose the magnetic exchange interactions into all the pairing possibilities between these orbitals. Our calculations reveal that intermolecular interactions are mainly governed by a ferromagnetic dyz–dyz contribution, with moderate dxz–dyz and dxz–dxz ferromagnetic and dz2–dz2 antiferromagnetic pathways between adjacent V atoms (Fig. 3b). This can be rationalized since a positive contribution to the magnetic exchange coupling tends to favor FM interactions and a negative contribution has an AFM character as depicted in Fig. 3b in red and blue colors for FM and AFM characters, respectively. The influence of these d orbitals is mainly governed by their hybridization with the p orbitals of C atoms in the linking moieties. Around the Fermi level, the contributions of dyz, dxz and dz2 are dominant and their hybridization with the energy levels of C atoms that link the C80 cages is deduced (Fig. 3c). One can note that this is the main factor in the mediation of intermolecular magnetic interactions. This analysis highlights the critical role of d orbital occupancy of magnetic transition metal atoms in intermolecular exchange, as reported for similar magnetic molecular systems.74
Subsequently, we performed well-converged spin–orbit calculations in V3N@C80 graphendofullerene to elucidate the preferential spin orientation. We computed the magnetic anisotropy energy (MAE) as the difference in energy in the different possible spin orientations along the Cartesian axis. We found that the in-plane spin configurations are more stable –only differing by 0.03 meV per atom along the x and y directions–, with 0.18 meV per atom MAE with respect to the off-plane direction z. These results indicate a favorable in-plane spin orientation, similar to those observed in the MPX3 (M = Mn, Ni, Fe; X = S, Se) family.75
To further understand the magnetic behavior of graphendofullerene we performed atomistic spin dynamics simulations. This approach offers a deeper investigation of the more complex magnetic interactions at play, revealing dynamic behaviors that extend beyond the static properties previously described. We predict a TC of 38 K for V3N@C80 graphendofullerene (Fig. 4a), which is similar to that of the first 2D magnetic materials studied (CrI3 or Cr2Ge2Te6)76,77 but still far from that of the recently synthesized above-room-temperature Fe3GaTe2 or VSe2.78,79 Although this value is still far from room temperature, the interplay between molecular design and intricate magnetic phenomena presents a fertile ground for future enhancements. Analogous to other 2D materials, the magnetic response has been proven to be particularly tunable using tensile strain,80 electrostatic doping81 and proximity effects.82 We also simulated TC by only considering J1 interactions, which result in a suppression of ferromagnetism above 0 K (Fig. S6†). This unveils the critical role of intermolecular interactions in the stabilization of long-range magnetic order in the network, as graphullerene enables a pathway to connect nearest neighbor magnetic molecules via polarized charge transfer between host and guest species in the network.
 | ||
| Fig. 4 Simulated (a) Curie temperature, (b) hysteresis loop at different temperatures and (c) magnon dispersion for the V3N@C80 graphendofullerene network. | ||
We next focus on two key aspects that further highlight the magnetic performance for future applications of V3N@C80 graphendofullerene: the hysteresis loop and magnon dispersion. First, we focus on the magnetic hysteresis behavior of the network, which indicates the relationship between an applied magnetic field (B) and the magnetization of the system. We compute it at different temperatures to simulate the effect of thermal fluctuations until TC is exceeded (Fig. 4b). These findings support the results observed in critical temperature simulations, as the evolution of temperature is closing the loop revealing the transition from the ferromagnetic to paramagnetic order at temperatures above TC. Besides, we found a coercive field (Hc) of 2 T at 0 K, which is consistent with the expected value of 2.34 T following the classical Hc limit as Hc ≈ 2K/μ0Ms, where K is the magnetic anisotropy, μ0 is the vacuum permeability and Ms is the saturation magnetization.
From the calculated magnetic exchange couplings, we simulate the spin-wave spectrum of V3N@C80 graphendofullerene using the Holstein–Primakoff transformation as introduced by linear spin-wave theory (LSWT) (Fig. 4c). At high frequencies, we can observe four magnon bands (between ∼90 and ∼115 meV) dominated by J1, which are the strongest magnetic interactions in the material and localized inside the guest molecules. On the other hand, the two magnon bands that have more contribution of J2 interactions are lying at lower energies (between 0 and ∼3 meV) due to the less intense character of these intermolecular interactions. Arising from the anisotropy of the magnetic exchange interactions, we observe a higher dispersion of spin excitations in the Г–X and S–Y k-paths (corresponding to the x axis in real space). This observation matches the magnetic configuration described above, as we predict that J21 interactions stabilize the ferromagnetic order in the network and this magnetic interaction takes place along the x axis.
Additionally, we explore the effect of mechanical deformation and electrostatic doping on the magnetic properties of the V3N@C80 monolayer. Fig. 5a shows the variation of first neighbor inter/intramolecular interactions in a reasonable range of ± 4% of biaxial strain, since graphullerene has been predicted to have a fracture strain in the range of 6.5–8%.44 As any mechanical deformation of the lattice will affect the distance between the C80 cages, J21 is critically affected by strain, since this interaction mainly depends on the communication between fullerene units. On the other hand, one may observe that intramolecular interactions are affected to a much lesser extent. According to our calculations, a biaxial compression of the lattice parameters up to 4% would enhance the strength of magnetic exchange interactions by 8% and 37% for J1 and J21, respectively. With respect to the calculated trend of J21 upon strain, we can observe a deviation from linearity at elongations close to 4%, owing to a competition between FM and AFM orders that appears due to the larger distance between magnetic molecules. We can conclude that the enhancement of J21 is mainly due to a higher overlap between dxz and dyz with the C sp3 linkers between cages, as any other competing pathway of interaction is playing a role. We corroborate this fact by looking at the orbital contributions to magnetic exchange couplings, where the same picture is observed compared to the former case (Fig. S7†). In the case of J1, the increased intensity is driven by stronger intramolecular interactions, as the reduced volume of fullerene brings the atoms inside the cage closer. By contrast, when positive strain is applied, we observe a distinct shift in the magnetic behavior of the system. As the lattice expands, the intermolecular distances between C80 cages increase, weakening the magnetic interactions. This reduction in coupling strength leads to a noticeable decrease in both J1 and J21, highlighting the sensitivity of these interactions to tensile strain. Obviously, this change in magnetic interactions has a direct impact on the TC of the system. Fig. 5b shows the evolution of TC of V3N@C80 graphendofullerene as a function of strain. As magnetic interactions weaken under elongation, TC is critically diminished, indicating a loss of magnetic ordering at lower temperatures. However, the opposite result comes from the application of negative strain. Our results show that compressive strain leads to a significant increase in TC, which increases up to 55 K within a 4% compression of the 2D molecular ferromagnetic network. This enhancement in TC is compatible with those observed for a similar range of strain in inorganic 2D ferromagnets, such as Cr2Ge2Te6 (ref. 83) and CrI3.84
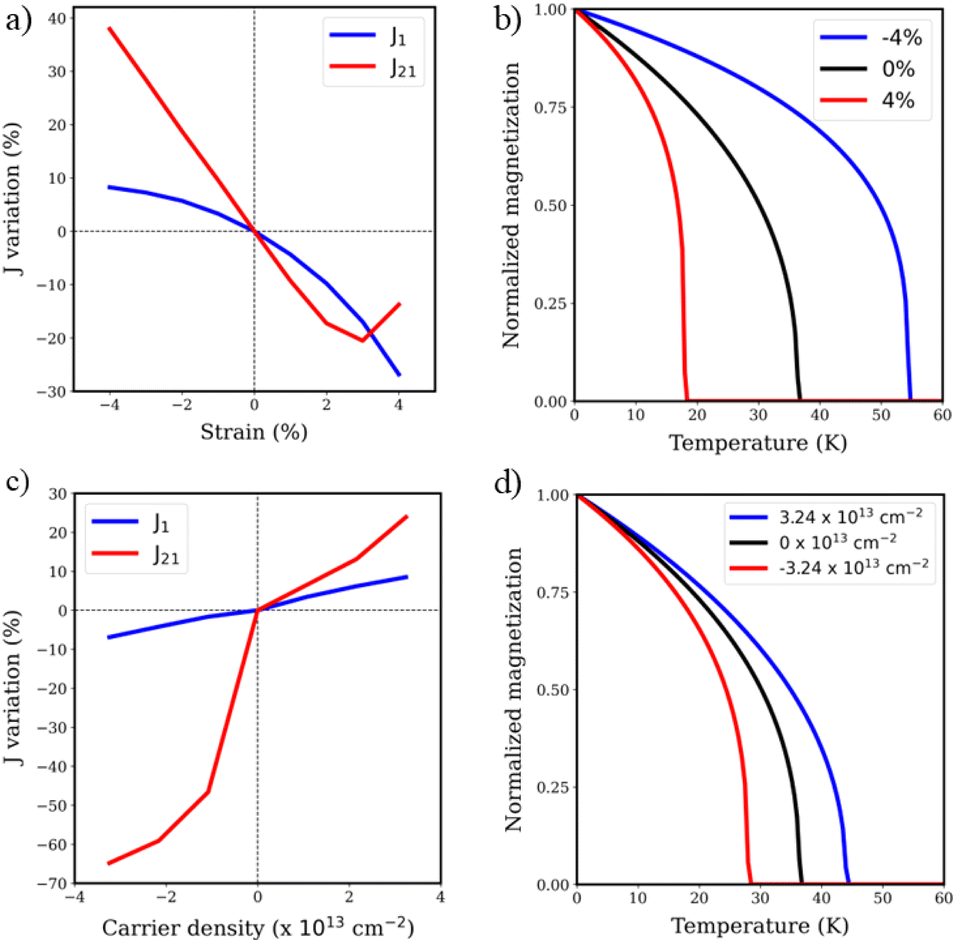 | ||
| Fig. 5 Evolution with strain of (a) intra/intermolecular magnetic exchange couplings and (b) Curie temperature and evolution with electrostatic doping of (c) intra/intermolecular magnetic exchange couplings and (d) Curie temperature of V3N@C80 graphendofullerene. A positive (negative) sign of the carrier density is equivalent to an excess of holes (electrons). | ||
Analogously, the manipulation of the magnetic properties of V3N@C80 graphendofullerene could be achieved by external manipulation using electrostatic doping, which has been shown to represent a powerful tool to tune the properties of 2D materials due to their reduced dimensionality.85Fig. 5c shows how intra/intermolecular magnetic interactions change with the introduction of carrier density in the network. One can see that the introduction of holes enhances the magnetic exchange couplings present in the system, which increases the Curie temperature of the system up to 45 K (Fig. 5d). On the other hand, the introduction of electrons in the systems drastically suppresses the intermolecular interactions, resulting in a diminution of the Curie temperature of V3N@C80. The drastic diminution of J21 upon the introduction of electron carriers could be explained with a remarkable suppression of the FM dxz–dyz and dxz–dxz pathways (Fig. S8†), which has an enormous impact on the strength of the intermolecular magnetic interactions.
The present work represents the first investigation of magnetism in a single layer of graphullerene. By using endohedral metallofullerenes, we induce and tailor magnetic properties in this carbon-based 2D covalent network. The intrinsic properties of the fullerene units can provide a plethora of graphendofullerene networks with a wide range of emerging possibilities, since fullerenes (i) are able to host different molecules inside them, and (ii) have different shapes and geometries. Recent advances in chemistry have allowed the incorporation of a wide range of molecules on endohedral fullerenes, from mono-, di- and tri-metallofullerenes to molecules with a higher degree of complexity such as nitride-, carbide- and methanoclusterfullerenes, mainly containing transition metals and rare-earth ions.24 These unique structures combine the electronic and geometric properties of fullerenes with the specific characteristics of guest species, which permits the application of these structures for a wide range of applications in optoelectronics, nanomagnetism, or energy harvesting and conversion.22 Indeed, graphendofullerene is not constrained to the network investigated in this work, but it will also transfer the infinite possibilities of endohedral fullerenes to the 2D limit, thus generating a new family of functional materials which represents a fertile playground to explore many interesting phenomena for applications in optoelectronics, spintronics, magnonics and quantum technologies, among others. Furthermore, the highly organized covalent network presents superior stability to conventional 2D magnets, facilitating its transferability to other substrates or matrices. This significantly enhances the practical integration of 2D magnetic materials into scalable spintronic devices for applications in next-generation technologies, and their tunability by using different metallic endofullerenes, whose properties are preserved, as building blocks.
Conclusions
In summary, we introduce graphendofullerene, a two-dimensional magnetic network based on a V3N cluster encapsulated on C80 graphullerene, using first principles calculations. Our systematic study provides a detailed understanding of the magnetic properties of V3N@C80 graphendofullerene, which shows a robust ferromagnetic behavior sustained by long-range magnetic interactions enabled by the carbon network. Our atomistic spin simulations reveal that magnetic order can persist up to 38 K and we also determine the hysteresis loop and magnon dispersion of graphendofullerene. Finally, we apply strain engineering and electrostatic doping that allows us to achieve a 45% increase in TC under the application of 4% compressive strain, evidencing the successful improvement of the magnetic properties of V3N@C80 graphendofullerene with external manipulation. This work not only bridges molecular chemistry and carbon chemistry but also provides a versatile platform for the future development of tunable, high-performance next generation molecular-based 2D magnets.Data availability
Data is available upon request to the corresponding author.Author contributions
This work is part of the PhD thesis of D. L. A. D. L. A. performed the DFT calculations. Z. H. provided experimental insights and contributed to the interpretation of results. J. J. B. conceived and supervised the work. The manuscript was written by D. L. A. and J. J. B. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the European Union (ERC-2021-StG 101042680 2D-SMARTiES), the Generalitat Valenciana (grant CIDEXG/2023/1), the National Natural Science Foundation of China (52302052) and the Anhui Provincial Natural Science Foundation (2308085MB33).Notes and references
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- F. Diederich, R. Ettl, Y. Rubin, R. L. Whetten, R. Beck, M. Alvare, S. Anz, D. Sensharma, F. Wudl, K. C. Khemani and A. Koch, Science, 1991, 252, 548–551 CrossRef CAS.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 Search PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 Search PubMed.
- A. Hirsch, Nat. Mater., 2010, 9, 868–871 CrossRef CAS.
- G. R. Bhimanapati, Z. Lin, V. Meunier, Y. Jung, J. Cha, S. Das, D. Xiao, Y. Son, M. S. Strano, V. R. Cooper, L. Liang, S. G. Louie, E. Ringe, W. Zhou, S. S. Kim, R. R. Naik, B. G. Sumpter, H. Terrones, F. Xia, Y. Wang, J. Zhu, D. Akinwande, N. Alem, J. A. Schuller, R. E. Schaak, M. Terrones and J. A. Robinson, ACS Nano, 2015, 9, 11509–11539 CrossRef CAS PubMed.
- S. Z. Butler, S. M. Hollen, L. Cao, Y. Cui, J. A. Gupta, H. R. Gutiérrez, T. F. Heinz, S. S. Hong, J. Huang, A. F. Ismach, E. Johnston-Halperin, M. Kuno, V. V. Plashnitsa, R. D. Robinson, R. S. Ruoff, S. Salahuddin, J. Shan, L. Shi, M. G. Spencer, M. Terrones, W. Windl and J. E. Goldberger, ACS Nano, 2013, 7, 2898–2926 CrossRef CAS.
- Y. Yu. Illarionov, T. Knobloch, M. Jech, M. Lanza, D. Akinwande, M. I. Vexler, T. Mueller, M. C. Lemme, G. Fiori, F. Schwierz and T. Grasser, Nat. Commun., 2020, 11, 3385 CrossRef CAS.
- Y. Saito, T. Nojima and Y. Iwasa, Nat. Rev. Mater., 2016, 2, 16094 CrossRef.
- M. Gibertini, M. Koperski, A. F. Morpurgo and K. S. Novoselov, Nat. Nanotechnol., 2019, 14, 408–419 CrossRef CAS PubMed.
- D. Deng, K. S. Novoselov, Q. Fu, N. Zheng, Z. Tian and X. Bao, Nat. Nanotechnol., 2016, 11, 218–230 Search PubMed.
- S. Yang, C. Jiang and S. Wei, Appl. Phys. Rev., 2017, 4, 021304 Search PubMed.
- J. R. Schaibley, H. Yu, G. Clark, P. Rivera, J. S. Ross, K. L. Seyler, W. Yao and X. Xu, Nat. Rev. Mater., 2016, 1, 16055 CrossRef CAS.
- E. C. Ahn, npj 2D Mater. Appl., 2020, 4, 17 CrossRef CAS.
- K. S. Novoselov, A. Mishchenko, A. Carvalho and A. H. Castro Neto, Science, 2016, 353, aac9439 CrossRef CAS.
- Y. Cao, V. Fatemi, S. Fang, K. Watanabe, T. Taniguchi, E. Kaxiras and P. Jarillo-Herrero, Nature, 2018, 556, 43–50 CrossRef CAS.
- C.-Z. Li, H.-L. Yip and A. K.-Y. Jen, J. Mater. Chem., 2012, 22, 4161 RSC.
- S. H. Friedman, D. L. DeCamp, R. P. Sijbesma, G. Srdanov, F. Wudl and G. L. Kenyon, J. Am. Chem. Soc., 1993, 115, 6506–6509 CrossRef CAS.
- A. Montellano, T. Da Ros, A. Bianco and M. Prato, Nanoscale, 2011, 3, 4035 RSC.
- Y. Pan, X. Liu, W. Zhang, Z. Liu, G. Zeng, B. Shao, Q. Liang, Q. He, X. Yuan, D. Huang and M. Chen, Appl. Catal., B, 2020, 265, 118579 Search PubMed.
- A. A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989–6113 CrossRef CAS.
- S. Yang, T. Wei and F. Jin, Chem. Soc. Rev., 2017, 46, 5005–5058 RSC.
- Z. Hu and S. Yang, Chem. Soc. Rev., 2024, 53, 2863–2897 RSC.
- H. Kato, A. Taninaka, T. Sugai and H. Shinohara, J. Am. Chem. Soc., 2003, 125, 7782–7783 CrossRef CAS PubMed.
- C.-R. Wang, T. Kai, T. Tomiyama, T. Yoshida, Y. Kobayashi, E. Nishibori, M. Takata, M. Sakata and H. Shinohara, Nature, 2000, 408, 426–427 CrossRef CAS PubMed.
- R. Westerström, J. Dreiser, C. Piamonteze, M. Muntwiler, S. Weyeneth, H. Brune, S. Rusponi, F. Nolting, A. Popov, S. Yang, L. Dunsch and T. Greber, J. Am. Chem. Soc., 2012, 134, 9840–9843 CrossRef PubMed.
- F. Liu, C.-L. Gao, Q. Deng, X. Zhu, A. Kostanyan, R. Westerström, S. Wang, Y.-Z. Tan, J. Tao, S.-Y. Xie, A. A. Popov, T. Greber and S. Yang, J. Am. Chem. Soc., 2016, 138, 14764–14771 Search PubMed.
- F. Liu, D. S. Krylov, L. Spree, S. M. Avdoshenko, N. A. Samoylova, M. Rosenkranz, A. Kostanyan, T. Greber, A. U. B. Wolter, B. Büchner and A. A. Popov, Nat. Commun., 2017, 8, 16098 Search PubMed.
- T. Wang, J. Wu, W. Xu, J. Xiang, X. Lu, B. Li, L. Jiang, C. Shu and C. Wang, Angew. Chem., Int. Ed., 2010, 49, 1786–1789 CrossRef CAS.
- Y. Feng, T. Wang, Y. Li, J. Li, J. Wu, B. Wu, L. Jiang and C. Wang, J. Am. Chem. Soc., 2015, 137, 15055–15060 CrossRef CAS PubMed.
- Y. Ma, T. Wang, J. Wu, Y. Feng, L. Jiang, C. Shu and C. Wang, Chem. Commun., 2012, 48, 11570 RSC.
- B. Wu, T. Wang, Y. Feng, Z. Zhang, L. Jiang and C. Wang, Nat. Commun., 2015, 6, 6468 CrossRef CAS PubMed.
- H. Kurihara, Y. Iiduka, Y. Rubin, M. Waelchli, N. Mizorogi, Z. Slanina, T. Tsuchiya, S. Nagase and T. Akasaka, J. Am. Chem. Soc., 2012, 134, 4092–4095 CrossRef CAS.
- E. Meirzadeh, A. M. Evans, M. Rezaee, M. Milich, C. J. Dionne, T. P. Darlington, S. T. Bao, A. K. Bartholomew, T. Handa, D. J. Rizzo, R. A. Wiscons, M. Reza, A. Zangiabadi, N. Fardian-Melamed, A. C. Crowther, P. J. Schuck, D. N. Basov, X. Zhu, A. Giri, P. E. Hopkins, P. Kim, M. L. Steigerwald, J. Yang, C. Nuckolls and X. Roy, Nature, 2023, 613, 71–76 CrossRef CAS.
- L. Hou, X. Cui, B. Guan, S. Wang, R. Li, Y. Liu, D. Zhu and J. Zheng, Nature, 2022, 606, 507–510 CrossRef CAS.
- G. Shen, L. Li, S. Tang, J. Jin, X.-J. Chen and Q. Peng, Crystals, 2023, 13, 224 CrossRef CAS.
- B. Peng, Nano Lett., 2023, 23, 652–658 CrossRef CAS.
- R. M. Tromer, L. A. Ribeiro and D. S. Galvão, Chem. Phys. Lett., 2022, 804, 139925 CrossRef CAS.
- U. Argaman and G. Makov, npj Comput. Mater., 2023, 9, 211 CrossRef CAS.
- A. Capobianco, J. Wiktor, A. Landi, F. Ambrosio and A. Peluso, Nano Lett., 2024, 24, 8335–8342 CrossRef CAS PubMed.
- T. S. A. Cassiano, M. L. Pereira, G. M. e Silva, P. H. de Oliveira Neto and L. A. Ribeiro, Nanoscale, 2024, 16, 2337–2346 RSC.
- S. Zhao, X. Zhang, Y. Ni, Q. Peng and Y. Wei, Carbon, 2023, 202, 118–124 CrossRef CAS.
- P. Ying, H. Dong, T. Liang, Z. Fan, Z. Zhong and J. Zhang, Extreme Mech. Lett., 2023, 58, 101929 CrossRef.
- L. Yu, J. Xu, B. Peng, G. Qin and G. Su, J. Phys. Chem. Lett., 2022, 13, 11622–11629 CrossRef CAS.
- P. Ying, O. Hod and M. Urbakh, Nano Lett., 2024, 24, 10599–10604 CrossRef CAS.
- A. Champagne, M. Camarasa-Gómez, F. Ricci, L. Kronik and J. B. Neaton, Nano Lett., 2024, 24, 7033–7039 CrossRef CAS PubMed.
- D. Yuan, H. Pi, Y. Jiang, Y. Hu, L. Zhou, Y. Jia, G. Su, Z. Fang, H. Weng, X. Ren and W. Zhang, Sci. China Phys. Mech. Astron., 2023, 66, 247211 CrossRef CAS.
- G.-Y. Li, S.-C. Mo, X.-Y. Qiu, J. Nie, J.-X. Li, P.-Y. Liang, F. Ning and S.-Z. Chen, ACS Appl. Nano Mater., 2025, 8, 649–658 CrossRef CAS.
- B. Peng, J. Am. Chem. Soc., 2022, 144, 19921–19931 CrossRef CAS PubMed.
- Y. Tong, H. Liu, S. Dai and D. Jiang, Nano Lett., 2023, 23, 7470–7476 CrossRef CAS PubMed.
- X. Chang, X. Liu, W. Zheng, L. Zhou and J. Zhang, Appl. Surf. Sci., 2023, 637, 157909 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B:Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, J. Phys.: Condens. Matter, 2002, 14, 2745–2779 CrossRef CAS.
- A. García, N. Papior, A. Akhtar, E. Artacho, V. Blum, E. Bosoni, P. Brandimarte, M. Brandbyge, J. I. Cerdá, F. Corsetti, R. Cuadrado, V. Dikan, J. Ferrer, J. Gale, P. García-Fernández, V. M. García-Suárez, S. García, G. Huhs, S. Illera, R. Korytár, P. Koval, I. Lebedeva, L. Lin, P. López-Tarifa, S. G. Mayo, S. Mohr, P. Ordejón, A. Postnikov, Y. Pouillon, M. Pruneda, R. Robles, D. Sánchez-Portal, J. M. Soler, R. Ullah, V. W. Yu and J. Junquera, J. Chem. Phys., 2020, 152, 204108 CrossRef.
- M. J. van Setten, M. Giantomassi, E. Bousquet, M. J. Verstraete, D. R. Hamann, X. Gonze and G.-M. Rignanese, Comput. Phys. Commun., 2018, 226, 39–54 CrossRef CAS.
- A. García, M. J. Verstraete, Y. Pouillon and J. Junquera, Comput. Phys. Commun., 2018, 227, 51–71 CrossRef.
- X. He, N. Helbig, M. J. Verstraete and E. Bousquet, Comput. Phys. Commun., 2021, 264, 107938 CrossRef CAS.
- R. F. L. Evans, W. J. Fan, P. Chureemart, T. A. Ostler, M. O. A. Ellis and R. W. Chantrell, J. Phys.: Condens. Matter, 2014, 26, 103202 CrossRef CAS.
- H. W. Kroto, Nature, 1987, 329, 529–531 CrossRef CAS.
- A. R. Khamatgalimov and V. I. Kovalenko, Fullerenes, Nanotubes Carbon Nanostruct., 2011, 19, 599–604 CrossRef CAS.
- A. R. Khamatgalimov, A. V. Luzhetskii and V. I. Kovalenko, Int. J. Quantum Chem., 2008, 108, 1334–1339 CrossRef CAS.
- S. Stevenson, G. Rice, T. Glass, K. Harich, F. Cromer, M. R. Jordan, J. Craft, E. Hadju, R. Bible, M. M. Olmstead, K. Maitra, A. J. Fisher, A. L. Balch and H. C. Dorn, Nature, 1999, 401, 55–57 CrossRef CAS.
- T. Wei, S. Wang, X. Lu, Y. Tan, J. Huang, F. Liu, Q. Li, S. Xie and S. Yang, J. Am. Chem. Soc., 2016, 138, 207–214 CrossRef CAS PubMed.
- M. Li, Y. Zhao, K. Yuan, R. Zhao, P. Zhao and X. Zhao, Carbon, 2018, 132, 312–322 CrossRef CAS.
- S. Bhusal, T. Baruah, Y. Yamamoto and R. R. Zope, Int. J. Quantum Chem., 2018, 118, e25785 CrossRef.
- M. K. Singh, P. Shukla, M. Khatua and G. Rajaraman, Chem.–Eur. J., 2020, 26, 464–477 CrossRef CAS PubMed.
- X. Li and J. Yang, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2017, 7, e1314 CrossRef.
- M. Ashton, D. Gluhovic, S. B. Sinnott, J. Guo, D. A. Stewart and R. G. Hennig, Nano Lett., 2017, 17, 5251–5257 CrossRef CAS.
- S. Ghosh, S. K. Singh, S. Tewary and G. Rajaraman, Dalton Trans., 2013, 42, 16490 RSC.
- M. Rybak, P. E. Faria Junior, T. Woźniak, P. Scharoch, J. Fabian and M. Birowska, Phys. Rev. B:Condens. Matter Mater. Phys., 2024, 109, 054426 CrossRef CAS.
- B. Huang, G. Clark, E. Navarro-Moratalla, D. R. Klein, R. Cheng, K. L. Seyler, D. Zhong, E. Schmidgall, M. A. McGuire, D. H. Cobden, W. Yao, D. Xiao, P. Jarillo-Herrero and X. Xu, Nature, 2017, 546, 270–273 CrossRef CAS PubMed.
- C. Gong, L. Li, Z. Li, H. Ji, A. Stern, Y. Xia, T. Cao, W. Bao, C. Wang, Y. Wang, Z. Q. Qiu, R. J. Cava, S. G. Louie, J. Xia and X. Zhang, Nature, 2017, 546, 265–269 CrossRef CAS.
- Y. Deng, Y. Yu, Y. Song, J. Zhang, N. Z. Wang, Z. Sun, Y. Yi, Y. Z. Wu, S. Wu, J. Zhu, J. Wang, X. H. Chen and Y. Zhang, Nature, 2018, 563, 94–99 CrossRef CAS.
- W. Yu, J. Li, T. S. Herng, Z. Wang, X. Zhao, X. Chi, W. Fu, I. Abdelwahab, J. Zhou, J. Dan, Z. Chen, Z. Chen, Z. Li, J. Lu, S. J. Pennycook, Y. P. Feng, J. Ding and K. P. Loh, Adv. Mater., 2019, 31, 1903779 CrossRef CAS.
- F. Miao, S.-J. Liang and B. Cheng, npj Quantum Mater., 2021, 6, 59 CrossRef.
- S. Jiang, L. Li, Z. Wang, K. F. Mak and J. Shan, Nat. Nanotechnol., 2018, 13, 549–553 CrossRef CAS.
- Z. Wang, C. Tang, R. Sachs, Y. Barlas and J. Shi, Phys. Rev. Lett., 2015, 114, 016603 CrossRef CAS.
- X.-J. Dong, J.-Y. You, B. Gu and G. Su, Phys. Rev. Appl., 2019, 12, 014020 CrossRef CAS.
- A. M. León, J. W. González, J. Mejía-López, F. Crasto de Lima and E. Suárez Morell, 2D Mater., 2020, 7, 035008 CrossRef.
- Y. Wang, J. Xiao, H. Zhu, Y. Li, Y. Alsaid, K. Y. Fong, Y. Zhou, S. Wang, W. Shi, Y. Wang, A. Zettl, E. J. Reed and X. Zhang, Nature, 2017, 550, 487–491 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc01278c |
| This journal is © The Royal Society of Chemistry 2025 |