
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A review of machine learning methods for imbalanced data challenges in chemistry
Jian
Jiang
*ab,
Chunhuan
Zhang
a,
Lu
Ke
a,
Nicole
Hayes
 b,
Yueying
Zhu
a,
Huahai
Qiu
a,
Bengong
Zhang
a,
Tianshou
Zhou
c and
Guo-Wei
Wei
*bde
b,
Yueying
Zhu
a,
Huahai
Qiu
a,
Bengong
Zhang
a,
Tianshou
Zhou
c and
Guo-Wei
Wei
*bde
aResearch Center of Nonlinear Science, School of Mathematical and Physical Sciences, Wuhan Textile University, Wuhan, 430200, P R. China. E-mail: jjiang@wtu.edu.cn
bDepartment of Mathematics, Michigan State University, East Lansing, Michigan 48824, USA
cKey Laboratory of Computational Mathematics, Guangdong Province, School of Mathematics, Sun Yat-sen University, Guangzhou, 510006, P R. China
dDepartment of Electrical and Computer Engineering, Michigan State University, East Lansing, Michigan 48824, USA
eDepartment of Biochemistry and Molecular Biology, Michigan State University, East Lansing, Michigan 48824, USA. E-mail: weig@msu.edu
First published on 22nd April 2025
Abstract
Imbalanced data, where certain classes are significantly underrepresented in a dataset, is a widespread machine learning (ML) challenge across various fields of chemistry, yet it remains inadequately addressed. This data imbalance can lead to biased ML or deep learning (DL) models, which fail to accurately predict the underrepresented classes, thus limiting the robustness and applicability of these models. With the rapid advancement of ML and DL algorithms, several promising solutions to this issue have emerged, prompting the need for a comprehensive review of current methodologies. In this review, we examine the prominent ML approaches used to tackle the imbalanced data challenge in different areas of chemistry, including resampling techniques, data augmentation techniques, algorithmic approaches, and feature engineering strategies. Each of these methods is evaluated in the context of its application across various aspects of chemistry, such as drug discovery, materials science, cheminformatics, and catalysis. We also explore future directions for overcoming the imbalanced data challenge and emphasize data augmentation via physical models, large language models (LLMs), and advanced mathematics. The benefit of balanced data in new material design and production and the persistent challenges are discussed. Overall, this review aims to elucidate the prevalent ML techniques applied to mitigate the impacts of imbalanced data within the field of chemistry and offer insights into future directions for research and application.
 Jian Jiang | Jian Jiang received his B.S and M.S. degrees in Theoretical Physics at Central China Normal University in 2005 and 2007, respectively. He received his PhD degree in Theoretical Physics from University of Le Mans in France in 2011. He is currently a professor in the School of Mathematical and Physical at Wuhan Textile University in China. His research interest includes AI based topological data analysis on molecular science, drug design and discovery, data mining, and modelling and analysis of complex networks. |
 Chunhuan Zhang | Chunhuan Zhang obtained her B.S. degree in Applied Mathematics in 2023 from Anyang Normal University. She is currently a M.S. candidate at Wuhan Textile University under Dr Huahai Qiu and Dr Jian Jiang. |
 Lu Ke | Lu Ke obtained her B.S. degree in Applied Mathematics in 2022 from Wuhan Textile University. She is currently a M.S. candidate at Wuhan Textile University under Dr Bengong Zhang and Dr Jian Jiang. |
 Nicole Hayes | Nicole Hayes is a PhD candidate in applied mathematics at Michigan State University. There, she focuses on novel machine learning methods for molecular property prediction, with broad applications in drug discovery. Her work has included the integration of deep learning and spectral graph methods for the prediction of scarcely labeled and imbalanced molecular data, as well as the application of topological tools for protein flexibility analysis. |
 Yueying Zhu | Yueying Zhu obtained her PhD degree in Physics from Le Mans University and Central China Normal University under the mentorship of Profs. Qiuping Alexandre Wang, Xu Cai, and Wei Li. Her PhD study focused on the uncertainty and sensitivity analysis of nonlinear dynamical systems, and the modeling and simulations of spreading dynamics on complex network. Now, she is an associate professor in Prof. Jie Liu’s group at Wuhan Textile University. Her current research concerns the application of uncertainty and sensitivity analysis to spreading dynamics, especially epidemic and opinion spreading on a complex network. |
 Huahai Qiu | Huahai Qiu received his PhD degree in Applied Mathematics from Sun Yat-sen University in 2012 and completed his postdoctoral studies at The Shanghai Institutes for Biological Sciences in 2018 under the guidance of Prof. Luonan Chen. Now he is the university-appointed professor of Wuhan Textile University, Wuhan, China. His current research focuses on computational systems biology. |
 Bengong Zhang | Bengong Zhang received his PhD degree in Applied Mathematics from South China University of Technology in 2010 and completed his postdoctoral studies at The University of Tokyo in 2013 under the guidance of Prof. Kazuyuki Aihara and Luonan Chen. His postdoctoral studies focused on computational systems biology. Now he is the professor of Wuhan Textile University, Wuhan, China. His current research concerns scRNA-seq data analysis and machine learning. |
 Tianshou Zhou | Tianshou Zhou received his PhD degree in Academy of Mathematics and System Science, CAS in 2001 and completed his postdoctoral Research at The Tsinghua University in 2003 under the guidance of Prof. Yun Tang. His postdoctoral studies focused on dynamics of comlex systems. Now he is a professor of Sun Yatsen University, Guangzhou, China. His current research interest is in computational systems biology. |
 Guo-Wei Wei | Guo-Wei Wei received his PhD degree from the University of British Columbia and is currently an MSU Research Foundation Professor at Michigan State University. His research focuses on the mathematical foundations of bioscience and artificial intelligence (AI). Dr Wei pioneered mathematical AI paradigms, such as topological deep learning (TDL), that integrate profound mathematical structures with AI to tackle biological challenges. His mathematical AI has led to victories in D3R Grand Challenges, a worldwide annual competition series in computer-aided drug design. Using TDL, genotyping, and computational biophysics, the Wei team unveiled the mechanisms of SARS-CoV-2 evolution and successfully predicted emerging dominant SARS-CoV-2 variants. |
1 Introduction
The awarding of the 2024 Nobel Prize in Chemistry to David Baker for computational protein design and to Demis Hassabis and John M. Jumper for protein structure prediction underscores the growing influence of artificial intelligence (AI) in scientific discovery. As AI and machine learning (ML) become integral to advancing chemical research,1 one of the most pressing challenges is the issue of imbalanced data. In many chemical datasets, the disproportionate distribution of classes poses significant obstacles to the development of reliable and accurate models, particularly when applied to complex chemical phenomena.Imbalanced data, a common phenomenon in data science, refers to significant disparities in the number of samples from different categories in classification tasks. The emergence of imbalanced data in chemistry is primarily attributed to the complexity and diversity of molecular data due to several factors. Naturally occurring biases in molecular distributions, where certain structures are more abundant than others, lead to a skew in data availability. Additionally, “selection bias” in sample collection processes can further exacerbate the imbalance. For instance, datasets may over-represent specific types of molecules or reactions due to experimental priorities or technical limitations. In drug discovery,2 active drug molecules are often significantly outnumbered by inactive ones due to the constraints of cost, safety, and time. Similarly, in molecular property prediction,3 models designed to assess toxicity often predict toxic outcomes more frequently, as toxic substances comprise a significant portion of the data. The study of protein–protein interactions also suffers from this imbalance, with experimentally validated interactions being much rarer than non-interactions.4
The presence of imbalanced data has direct implications for the performance of ML models. Most algorithms, such as random forests (RF) and support vector machines (SVM),5 assume a uniform distribution of data across categories.6 When trained on imbalanced datasets, models tend to focus on classes with more abundant data, often neglecting the minority classes. This bias results in models that are less sensitive to underrepresented features, which can critically undermine the accuracy of predictions in real-world applications. Consequently, overcoming the limitations imposed by imbalanced data is essential for the advancement of ML in chemical research.
Addressing the issue of imbalanced data in chemistry has become a major area of interest for researchers. Various strategies have been proposed, including resampling techniques like oversampling and undersampling, data augmentation, and ensemble algorithms. Feature engineering and selection methods have also been explored to mitigate the negative effects of data imbalance. However, despite the increasing body of work in this area, the existing reviews provide a general overview of these methods without specifically addressing their applications within chemistry. This review aims to fill this gap by offering a comprehensive overview of the imbalanced data challenge and solutions in chemical research, with a particular focus on recent advancements and their practical implications. Through this examination, we seek to provide researchers with a deeper understanding of the challenge posed by imbalanced data and to stimulate further progress in developing effective solutions.
The rest of this article is organized as follows. Sections 2.1–2.4 provide a detailed description of current technologies and algorithms for handling imbalanced data and demonstrate their applications in distinct fields of chemistry. Section 2.5 lists some indicators for evaluating model performance. In Section 3, we discuss new trends and challenges in the study of imbalanced data in chemistry and highlight future perspectives.
2 Current approaches and techniques
2.1 Resampling techniques
 | ||
| Fig. 1 (a) A schematic diagram of an oversampling method, demonstrating the approach of the oversampling technique to balance the dataset. (b) This example demonstrates the application of Borderline-SMOTE method in properties prediction of polymer materials.7 Firstly, experimental data of 23 rubber materials were collected, and the nearest neighbor interpolation (NNI) algorithm was used to expand the dataset, resulting in a total of 483 datasets. Then, the K-means algorithm was used to cluster these datasets into two categories. Finally, based on the clustering results, Borderline-SMOTE was used to interpolate along the boundaries of the minority samples, generating two clusters with sample sizes of 314 and 396, respectively. (c) This illustration showcases the utilization of the SMOTE technique in the domain of catalyst development.8 126 heteroatoms doped arsenenes were collected as the original dataset, and the absolute value of Gibbs free energy changes (|ΔGH|) of 0.2 eV was selected as the threshold to divide the original data into two categories (88 with |ΔGH| > 0.2 eV and 38 with |ΔGH| < 0.2 eV). Then, SMOTE was applied to solve the problem of data imbalance and obtain two types of evenly distributed data. | ||
One of the most prominent oversampling methods is the Synthetic Minority Over-sampling Technique (SMOTE), first introduced by Chawla et al. in 2002.14 SMOTE generates new minority class samples by synthesizing them from the existing data, which helps preserve the original feature distribution and mitigates overfitting. Its ability to enhance model generalization has led to widespread adoption across various chemistry domains. For instance, in materials design, SMOTE has been used to resolve class imbalance when integrated with Extreme Gradient Boosting (XGBoost) and nearest neighbor interpolation, improving the prediction of mechanical properties of polymer materials.7 The illustration of this process of balancing data is shown in Fig. 1b. Similarly, as part of an ML method trained on molecular dynamics data to predict the tensile stress of natural rubber, SMOTE is used to interpolate at a few sample boundaries to solve the problem of sample imbalance.15 In catalyst design, the authors used SMOTE to solve the problem of uneven data distribution in the original dataset, improving the predictive performance of ML models and promoting candidate screening of hydrogen evolution reaction catalysts.8 The illustration of SMOTE for balancing data for this catalyst design example is displayed in Fig. 1c.
However, SMOTE has limitations, such as introducing noisy data, struggling with complex decision boundaries, failing to account for internal distribution differences within the minority class, and requiring high computational costs. To address these issues, advanced oversampling techniques have been developed, including Borderline-SMOTE,16 SVM-SMOTE,17 RF-SMOTE,18 Safe-level-SMOTE,19 SMOTE-NC,20 and ADASYN.21 These methods refine SMOTE's approach by better handling class overlap, decision boundary complexity, and minority class distribution, expanding its applicability to more complex datasets. In drug discovery, the uneven distribution of active and inactive compounds affects the prediction accuracy of ML models. Therefore, in a search for new histone deacetylase 8 (HDAC8) inhibitors, Nurani et al. used SMOTE to construct a balanced dataset.22 They further selected an RF model, which demonstrated the best predictive performance on their training set compared to other tested ML methods, and the resulting RF-SMOTE prediction model was indicated to be helpful in identifying new HDAC8 inhibitors. In protein engineering, sample imbalance is a major challenge for predicting protein–protein interaction sites. As traditional SMOTE methods focus equally on every minority class sample, while Borderline-SMOTE methods are more sensitive to boundary samples, Jiang et al. used a CNN model with Borderline-SMOTE to predict protein–protein interaction sites, which is helpful for protein design and mutation analysis.23 Furthermore, a combination of the most distant undersampling and Safe-level-SMOTE oversampling techniques has been used to address data imbalance issues, demonstrating its excellent performance in balancing the number of lysine formylation sites and non-formylation sites and enabling the prediction of lysine formylation sites when paired with ML.24
 | ||
Fig. 2 (a) A schematic diagram of undersampling method, demonstrating the approach of undersampling technique to balance the dataset. (b) This example demonstrates the application of a new method based on RUS technology in the realm of drug discovery.25 The majority samples in the drug target dataset are clustered using K-means clustering method and divided into different clusters. After that, the RUS method is used to randomly select a cluster from these clusters, repeat multiple times, and combine the selected cluster with minority samples in the original dataset to form a new balanced set. (c) This instance illustrates the use of the Tomek-Links approach for addressing imbalance in data within the realm of materials design.26 Initially, SMOTE is used to generate minority samples, making the dataset roughly balanced. Then, Tomek Links is used to identify and remove the majority samples in Tomek-Links (samples near the classification boundary) to clean the data, thereby refining the roughly balanced dataset into a finer one. (d) This example uses the NearMiss-2 method to address data imbalance within the domain of protein-ligand binding.27 Firstly, a training dataset of peptide sequences is constructed, containing 4242 minority samples with malonylation sites and 71![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 809 majority samples without malonylation sites. Next, the NearMiss-2 method is used to calculate the distance between each majority sample and each minority sample, and then the k farthest minority samples are selected to calculate the average distance to these k minority samples. Finally, the majority sample with the smallest average distance is retained to achieve data balance. 809 majority samples without malonylation sites. Next, the NearMiss-2 method is used to calculate the distance between each majority sample and each minority sample, and then the k farthest minority samples are selected to calculate the average distance to these k minority samples. Finally, the majority sample with the smallest average distance is retained to achieve data balance. | ||
RUS randomly removes a portion of majority class samples to balance the dataset. The sampling rate is typically determined by the ratio of majority to minority class samples. After removing the excess majority samples, the resulting dataset is better balanced, allowing the model to learn from both classes in an unbiased manner. RUS has been successfully applied in various chemistry domains, including the prediction of anti-parasitic peptides,28 drug-target interaction (DTI) prediction,30 and compound–protein interaction prediction.31 In drug discovery,25 due to the greater number of non-interacting (negative samples) drug–target pairs than interacting (positive samples) drug–target pairs, this imbalanced dataset reduces prediction accuracy. Therefore, a new RUS-based method was used to process the data, as shown in Fig. 2b. While RUS is simple to implement and can reduce training time by decreasing the dataset size, it has potential drawbacks. Removing too many majority class samples can lead to the loss of important information, which may negatively affect model performance, particularly in drug discovery and genomics. In areas such as protein engineering and quantum chemistry, where intricate patterns and subtle variations in data are crucial, careful consideration is required to avoid discarding valuable information.
The NearMiss algorithm reduces the number of majority class samples while preserving key distribution characteristics, improving classifier performance, particularly in binary classification tasks. Its core principle is to select majority class samples that are closest to the minority class in the feature space for undersampling.
NearMiss is widely used due to its efficiency in handling high-dimensional data. Its robustness against noisy data and outliers, combined with scalability and ease of integration with other algorithms, makes it suitable for various applications in chemistry. For example, in 2022, Wang et al. applied the NearMiss-2 method to address imbalanced data in protein acetylation site prediction, significantly improving the Malsite-Deep model's accuracy in protein engineering.27 Their workflow is shown in Fig. 2d. Similarly, in molecular dynamics simulations, NearMiss is used to address data imbalance, which facilitates the identification of different conformational states of protein receptors.32
Despite its advantages, NearMiss may lead to the loss of valuable information due to undersampling, particularly in fields like drug discovery, catalyst design, and genomics. Additionally, due to its reliance on proximity in the feature space, NearMiss can struggle with capturing complex, nonlinear relationships, limiting its effectiveness in highly imbalanced or intricate datasets in protein–ligand binding or quantum chemistry.
The Tomek Links method reduces the number of majority class samples by identifying and removing those that are close to minority class samples in the feature space. This approach improves the model's ability to focus on the minority class by reducing class overlap. It works by identifying pairs of majority and minority class samples that are nearest neighbors, called Tomek Links, and removing the majority class samples from these pairs. This enhances the distinction between classes for model training.
Tomek Links has been applied in various chemical domains, including identifying glutarylation sites29 and pharmacophoric fragments of DYRK1A inhibitors,33 boosting the efficiency of experimental parameter optimization of nanometric solid solution alloys design26 (an illustration of the specific process is shown in Fig. 2c), and predicting compound–protein interactions.34 This method is particularly effective for noise reduction while preserving the overall data structure, improving model performance in fields like genomics, materials, and drug discovery. However, its reliance on identifying noise points based on proximity can risk removing valuable data. Additionally, its efficiency declines with larger datasets, which limits its applicability in certain large-scale contexts.
2.1.3.1 SMOTE-Tomek links. The SMOTE-Tomek Links technique combines oversampling and undersampling to enhance dataset balance and classification performance.35 SMOTE synthesizes new minority class samples, while Tomek Links removes overlapping boundary samples, refining the dataset and reducing class overlap. This approach effectively mitigates data imbalance and overfitting, leading to clearer class boundaries and improved classifier accuracy and generalization. Widely applied in fields such as protein engineering,29 genomics, and transcriptomics,36 SMOTE-Tomek Links has demonstrated its ability to improve classification models, particularly for high-dimensional gene expression data, by facilitating the identification of key biomarkers. However, it can be computationally expensive, especially for large datasets, and excessive oversampling may still risk overfitting. Therefore, careful parameter tuning is essential to maximize the method's effectiveness, particularly in drug discovery and catalyst design.
2.1.3.2 SMOTE-edited nearest neighbor (SMOTE-ENN). SMOTE-ENN (edited nearest neighbor) is a hybrid resampling method by combining SMOTE's oversampling with the ENN technique to remove noisy majority class samples.37 This approach rebalances class distributions, enhancing the representativeness of minority classes while improving the model's robustness by reducing overfitting. SMOTE-ENN has been successfully applied in diverse chemical fields, such as protein–ligand binding38 and DTI prediction.39 Specifically, considering the large number of non-interaction class samples and the low proportion of interaction class samples in the DTI dataset, there is a significant class imbalance problem. Therefore, SMOTE-ENN technology was adopted to solve this problem, helping to improve the accuracy of drug–target interaction prediction.40 Although highly effective in improving model performance on imbalanced datasets, SMOTE-ENN is computationally intensive and sensitive to parameter selection. Careful parameter tuning is needed to mitigate risks such as generating poor-quality samples, particularly in noisy or unevenly distributed datasets.
2.1.4.1 Density-based spatial clustering of applications with noise-SMOTE (DBSCAN-SMOTE). DBSCAN-SMOTE (DBSM) is a hybrid method that combines the density-based clustering algorithm DBSCAN with SMOTE,41 as shown in Fig. 3a. In DBSM, DBSCAN identifies core, boundary, and noise points within clusters by using parameters like neighborhood radius and minimum sample size. SMOTE is then applied to the core points of these clusters, increasing the representation of minority class samples. This approach effectively reduces data imbalance and optimizes sample distribution, enhancing both the performance and generalization of classification models. DBSM is well-suited for imbalanced datasets with noise or irregular cluster shapes. It has broad applicability in chemistry. In predicting cervical cancer,43 Gowri and colleagues employed DBSCAN to tackle the data imbalance in cervical cancer datasets. The selection of DBSCAN was due to its capability to detect anomalous samples by examining the density of data points, obviating the requirement for predefined parameters. In the context of drug screening, Koh et al. used the DBSCAN method to process imbalanced data due to the fact that it can identify and classify different antagonists based on the density distribution of compound structures.44
 | ||
| Fig. 3 (a) The schematic diagram of DBSM algorithm flow.41 The process of DBSM includes two parts: undersampling and oversampling. For the undersampling part, apply DBSCAN to create clusters from all training sets. Then a portion of the majority samples is deleted from each cluster. The output of the undersampling technique is only majority samples. For the oversampling part, SMOTE is used to add synthetic sampless of minority samples to the training set. Therefore, the final output of the DBSM algorithm is a new training set consisting only of the majority samples from the undersampling part and the minority samples from the oversampling part. (b) This example demonstrates the application of the K-means SMOTE method in predicting bioluminescent proteins to address imbalanced data.42 Firstly, K-means is used to cluster the majority and minority samples separately to solve the problem of intra-class imbalance. Secondly, SMOTE is used for oversampling a small number of samples (luminescent proteins) to increase the number of minority samples and form a new balanced dataset with the majority samples. | ||
DBSCAN-SMOTE handles noisy data and outliers well, making it effective in fields like genomics and protein–ligand binding, but its performance depends heavily on parameter selection and can be computationally expensive for large datasets.
2.1.4.2 K-means SMOTE. K-means SMOTE is a hybrid technique created by integrating K-means clustering with SMOTE.45 It first partitions the data into clusters using K-means, then focuses on those with a higher proportion of minority class samples for targeted oversampling. Minority samples are generated between selected clusters to improve distribution, with sample density guiding the oversampling process. This approach enhances both the quantity and representativeness of minority class samples, improving model performance.
In the biomedical context, due to the diversity of disease subtypes or drug responses leading to uneven class distribution in the data, the K-means SMOTE method, by combining clustering and oversampling techniques, can effectively balance the imbalanced dataset while preserving its intrinsic structure, thereby enhancing the drug prediction model's ability to identify minority class samples.46 In the field of protein engineering, Nath et al. employed the K-means SMOTE method to manage imbalanced data, which is attributable to its capability to efficiently refine the class distribution in the dataset, catering to the challenge of low sequence similarity among bioluminescent proteins.42 An illustration of K-means SMOTE method is shown in Fig. 3b.
K-means SMOTE effectively improves model performance by generating realistic minority class samples, but its two-step process, consisting of clustering and oversampling, costs many computational resources and requires careful parameter optimization.
2.2 Data augmentation
This approach has proven effective in handling imbalanced data in various chemical applications. For example, in protein–ligand binding prediction, Lu et al. applied Gaussian noise addition to training data, since Gaussian noise can improve the model's adaptability to large-scale protein conformational changes and the ability of the model to identify hidden binding sites.47 Similarly, in drug discovery, Chakraborty et al. added Gaussian noise to the latent representation of autoencoders as the noise addition increased molecular diversity and complexity while maintaining the rationality of molecular structure.48
Gaussian noise addition improves model generalization on imbalanced data by simulating random disturbances, but its effectiveness depends on the careful tuning of noise levels to avoid distorting minority class features or compromising interpretability.
2.2.2.1 Generative adversarial networks (GANs). GANs, first proposed by Goodfellow et al. in 2014,49 are a class of DL models consisting of two neural networks: a generator and a discriminator. The generator's goal is to create realistic synthetic data, while the discriminator tries to distinguish between real and fake data. In the context of imbalanced data, GANs can be used to generate synthetic samples for the minority class. By training the generator to produce new, realistic samples of the underrepresented class, GANs help to increase the quantity and diversity of minority class data. This approach allows models to learn better representations of the minority class, reducing the bias towards the majority class. GANs have been successfully applied in many fields to handle class imbalance, such as drug design,50–53 materials design,54 protein engineering,55,56 catalyst design,57 and others. Due to the limitations of classical GANs in training stability and exploring certain regions of chemical space, Li et al. proposed a novel quantum GAN in 2021, which had a hybrid generator (QGAN-HG) for discovering small drug molecules.58 In predicting antiviral peptides, Lin et al. used GAN to address the issue of imbalanced antiviral peptide datasets,59 due to its ability to produce new samples that closely matched the distribution of real data. An illustration of balancing data process is given in Fig. 4a.
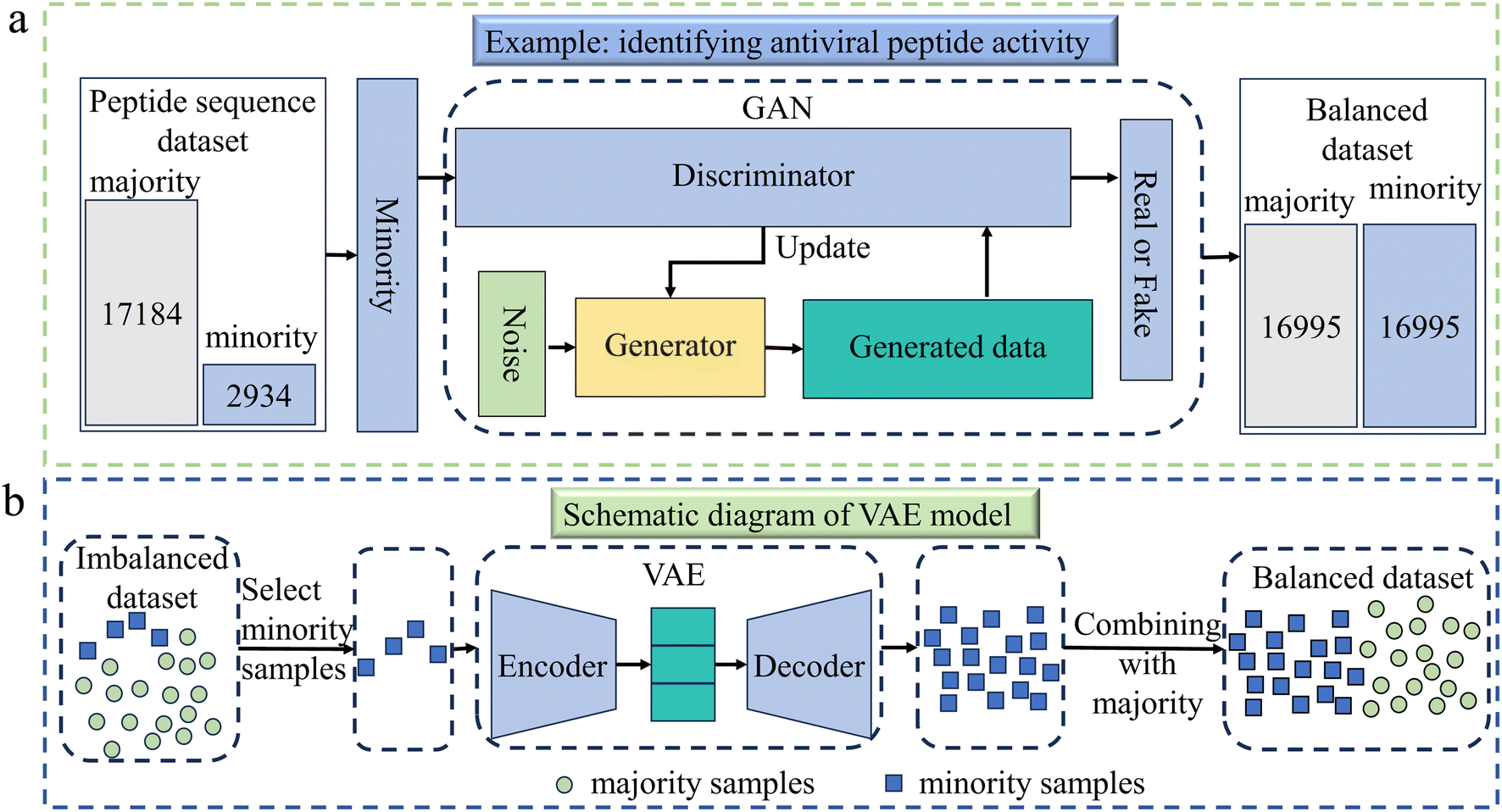 | ||
| Fig. 4 (a) This example demonstrates the application of generative adversarial network (GAN) in identifying antiviral peptide activity.59 Firstly, an imbalanced dataset was constructed, consisting of 2934 antiviral peptides (AVPs) and 17184 non-antiviral peptides. The AVPs were used as input data to train the GAN model and then many AVP-like data were generated. Finally, the generated data were added to the original AVP data to achieve balance between the majority and minority samples. (b) The illustration of the variational autoencoder (VAE) algorithm for balancing data.60 It is divided into two parts: encoder and decoder. The former compresses the input into probabilistic latent representations, while the latter reconstructs data from latent space, the part between the encoder and the decoder. When applied to imbalanced data, VAE achieves balance between majority and minority classes by generating new samples for the minority class. | ||
GANs effectively balance imbalanced data by generating diverse, high-quality minority class samples, but their unstable training and risk of mode collapse may limit their ability to fully capture minority class features.
2.2.2.2 Variational autoencoders (VAEs). A VAE is a type of generative model that learns to map input data to a continuous latent space, from which it can generate new data samples.61,62 It consists of two main components: an encoder that compresses the input into a probabilistic latent representation, and a decoder that reconstructs the data from the latent space. When applied to imbalanced data, VAEs can generate synthetic data by producing new samples for the minority class, as shown in Fig. 4b. The encoder–decoder structure of VAEs allows them to learn meaningful latent representations of the minority class, and by sampling from the latent space, new realistic data points can be generated. This helps mitigate the bias towards the majority class by enriching the diversity and quantity of minority class samples, making VAEs effective for handling class imbalance in fields such as drug discovery,63,64 protein engineering,65 molecular dynamics,66 and materials design.67
Recently, Schilter et al. used the VAE method in catalyst design to handle imbalanced data, based on the ability of VAE to autonomously learn meaningful structural features from the data. Compared to other existing advanced methods, VAE performed better in handling imbalanced datasets, as it not only maintained data distribution but also generated effective and innovative datasets.68 In protein–ligand binding, Ngo et al. used the VAE method, as it can learn information about the entire protein structure and utilize its powerful generative ability to generate ligands with high binding affinity and synthetic feasibility.69
VAEs effectively augment minority classes by generating new samples from a continuous latent space; however, their reliance on fixed distributions and tendency to produce blurry samples can constrain their ability to capture complex data features.
In imbalanced datasets, the minority class often lacks sufficient diversity, making it harder for the model to learn its patterns. By augmenting the features, new dimensions of variation can be introduced to the minority class, providing more informative and diverse data points. This allows the model to better distinguish the minority class from the majority class, reducing bias and improving classification accuracy. Feature augmentation works well when combined with other imbalance-handling techniques such as oversampling, enhancing the model's ability to generalize across both minority and majority classes and providing a more balanced representation of the data.
This method has been applied in many chemical fields, such as in DTI prediction78 and DDI prediction.79 In protein function prediction, Wan et al. proposed the FFPred-GAN method in 2020 and used feature augmentation to handle imbalanced data,80 which can effectively simulate the complex features of proteins in organisms without changing the distribution of the original data, while generating high-quality synthetic protein feature samples. Hayes et al. proposed the BTDT-MBO algorithm in 2024, which transformed molecular structures into informative feature vectors and employed a feature augmentation strategy, significantly improving the recognition capability of minority classes within molecular datasets.75 Additionally, in protein–ligand binding, Akbar et al. used feature augmentation techniques to handle imbalanced data owing to their capability to integrate information from multiple feature vectors and improve the model's recognition ability when parsing complex biological data.81
While feature augmentation can improve model performance on imbalanced data, it risks adding irrelevant features, overfitting, or noise, and does not directly address the core imbalance between majority and minority classes.
2.3 Algorithmic approaches
2.3.1.1 Boosting. The boosting algorithm constructs a powerful model by concatenating multiple simple weak learners,82 as shown in Fig. 5a. It iteratively updates weights to make each subsequent learner focus more on misclassified minority class samples, thus balancing the attention to both minority and majority classes. Since the proposal of the boosting algorithm, many extensions have been proposed, such as Adaptive Boosting,85 Extreme Gradient Boosting (XGBoost),86 Gradient Boosting Decision Tree,87etc.
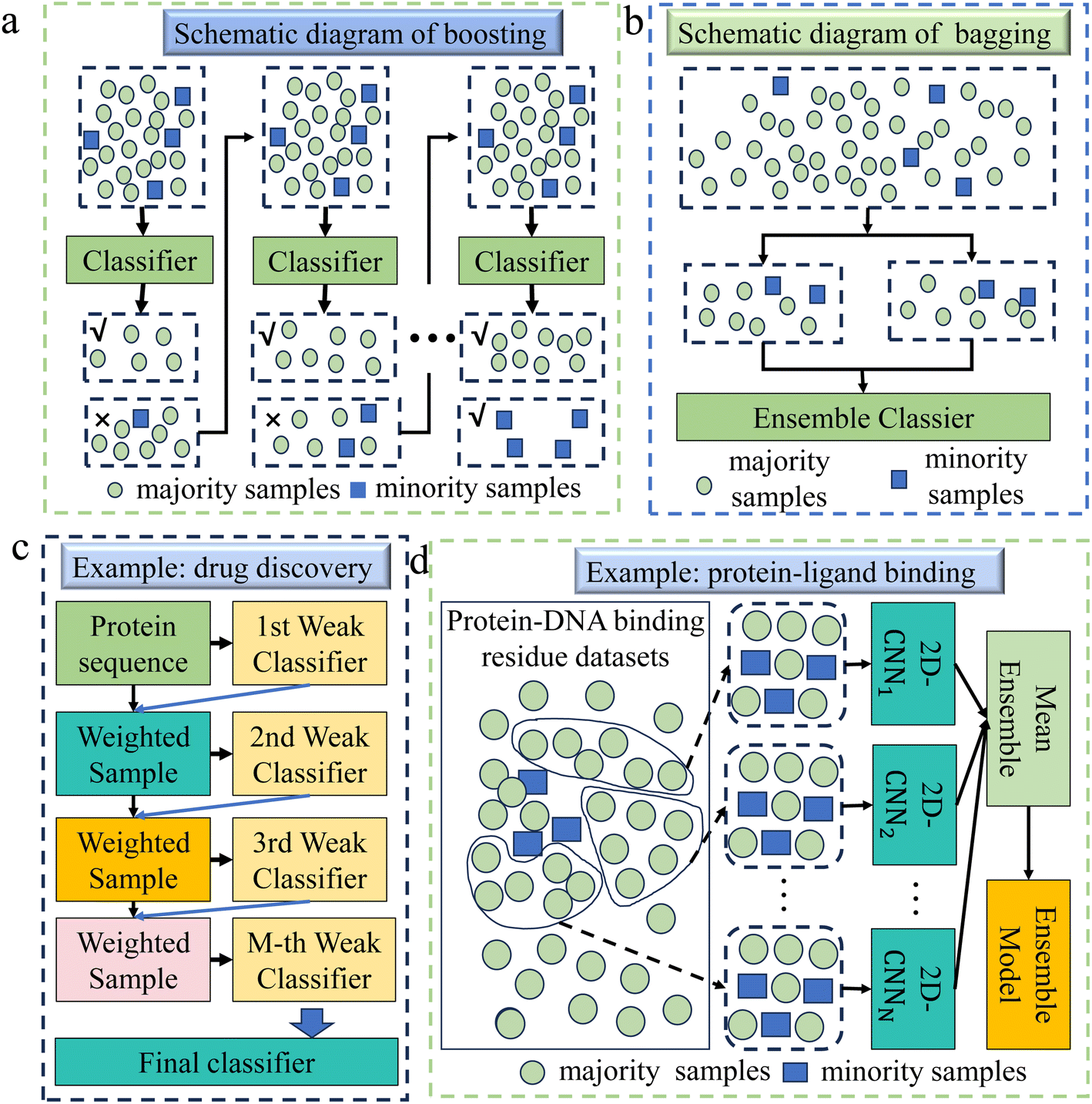 | ||
| Fig. 5 (a) A schematic diagram of the boosting algorithm. This method constructs a powerful classifier by connecting multiple weak classifiers. It uses an iterative process to make each subsequent classifier focus more on the misclassified minority class samples in the previous classifier's classification results, thus balancing the attention to minority and majority classes. (b) A schematic diagram of the bagging algorithm. It creates multiple subsets through random sampling and substitution, and it improves the recognition of minority classes by increasing the presence of minority samples in the subsets. (c) This example demonstrates the application of boosting in drug discovery.83 Firstly, an imbalanced dataset was constructed including proteins that can interact with drugs and proteins that cannot interact with drugs. The model then randomly selects samples with the same weight and chance from the dataset to train the first classifier model. Then, each classifier is tested on all samples in the dataset, and the weights of misclassified samples are updated iteratively to generate the final classification model from several individual weak classifiers. (d) This example demonstrates the application of bagging methods in the field of protein–ligand binding.84 Firstly, the majority samples and minority samples are separated from the original training set. Then, a certain number of samples are randomly selected from the majority samples and merged with the minority samples to form a new subset, which is repeated multiple times. Using the two-dimensional convolutional neural network (2D-CNN) framework to learn on each subset, an ensemble model is finally formed according to the mean ensemble strategy. | ||
Boosting has many applications in chemical fields, such as drug discovery,30 catalyst design,88 protein engineering,89 protein–ligand binding,90 biomaterials design,91etc. Xue et al. selected the Gradient Boosting method for the design of biomaterials,91 due to its capability to effectively address data imbalance by incrementally building and refining models. This approach, in contrast to others, enabled a more precise identification of the intricate features influencing biomaterial properties. In genomics and transcriptomics, Liu et al. chose the XGBoost method to handle imbalanced data,92 thanks to its excellent generalization ability and higher prediction accuracy in handling high-dimensional problems, as well as its effectiveness in dealing with imbalanced data and categories. In drug discovery, Sikander et al. used the XGBoost method for accurate prediction of druggable proteins.83 This method demonstrated an excellent ability to handle high-dimensional data and strong resistance to overfitting, and its working principle diagram is shown in Fig. 5c.
Boosting enhances minority class classification by increasing sample weights, but its growing complexity with iterations requires careful tuning, and it often needs to be combined with techniques like data sampling or feature selection to improve efficiency.
2.3.1.2 Bagging. Bagging is an ensemble method that creates multiple training subsets through random sampling with replacement, training independent models on each subset,93 as shown in Fig. 5b. The final output is obtained by aggregating predictions via voting or averaging. In imbalanced datasets, bagging can reduce bias by increasing the presence of minority class samples in some subsets, improving recognition of minority classes and reducing the influence of the majority class. While it stabilizes models like decision trees and mitigates overfitting, bagging alone does not well solve sample imbalance and often requires additional techniques like oversampling or undersampling for better minority class performance.
Bagging has a wide range of chemical applications, such as drug discovery,94,95 genomics and transcriptomics,96 catalyst design,97etc. In the study of drug toxicity detection,98 Gupta used an ensemble model based on bagging because the bagging method can effectively reduce the misjudgment of minority class samples by the model. Compared with a single classifier, it was more suitable for complex data classification in the field of biochemistry. Gong et al. employed the bagging method for addressing imbalanced data in druggable protein prediction due to its efficacy in mitigating model bias that arose from such imbalance. This method, in contrast to a single SVM classifier, offered a superior ability to integrate the significance of various features.99 In terms of protein–ligand binding, Hu et al. developed a method termed PredDBR to predict protein–DNA binding residues,84 as depicted in Fig. 5d, and employed the bagging method to address imbalanced data, since bagging was more adept at handling complex features in bioinformatics compared to a single model.
 | ||
| Fig. 6 (a) A schematic diagram of the cost-sensitive learning (CSL) method. It assigns different weights to differently misclassified samples, focusing the model more on high-cost minority sample errors, thereby reducing the likelihood of misclassification. (b) This example demonstrates the application of the cost-sensitive XGBoost method in genomics and transcriptomics.101 The imbalanced genomic data (minority:majority = 1:55) is input into the cost-sensitive XGBoost framework for processing, using the CSL method to assign weights to the samples. Then, the XGBoost classifier is used for processing to obtain a balanced dataset for subsequent analysis or modeling processes. | ||
CSL has been widely applied in multiple fields of chemistry. For example, due to the highly imbalanced data in the DTI dataset, Aleb adopted a CSL method to improve model performance, which can assign higher weights to minority class samples in biological contexts, thereby more effectively identifying and predicting drug compound and protein interactions in drug design.102 In genomics and transcriptomics, Hazan et al. developed an advanced ML model called INFLAMeR for the identification of novel functional long non-coding RNAs (lncRNAs). This model employed a cost-sensitive XGBoost classifier to tackle the imbalance of training data, as depicted in Fig. 6b, with the rationale that the CSL allowed for the allocation of higher weights to minority categories, thereby enhancing the model's capability to detect key lncRNAs.101
CSL is efficient for large-scale molecular data and can addresses class imbalance by balancing performance across categories, improving predictive accuracy, especially in drug screening. However, improper cost settings may lead to overfitting minority classes and reduce generalization, and uncertainty in cost function design may impact model performance.
2.4 Feature engineering and selection strategies
Feature engineering is a key technology in data preprocessing, which involves extracting, processing, or creating new features from raw data to optimize the performance of ML models.77 It can not only help identify key features of minority classes, but also enhance the sensitivity of the model to minority class samples by designing specific features or transformations, improving the model's performance on imbalanced data.Feature selection, as an important component of feature engineering, can remove redundant features and enhance the recognition accuracy of minority categories by filtering out the most relevant and important feature subsets in the dataset. Feature selection—including filter, wrapper, and embedded technologies as well as random feature selection—has various applications in fields such as drug discovery,103 protein engineering,104–106 genomics and transcriptomics,107etc.
 | ||
| Fig. 7 (a) The filter method sorts the six input samples (each with four features, different colors represent different features) directly based on different performance evaluation indicators and selects the feature with the highest score. (b) This example demonstrates the application of the wrapper feature selection method in the field of drug discovery.108 Firstly, through evaluating the extracted features, different weights are assigned for features. Then, a subset is selected from the feature set, and the wrapper method is used to choose the features that are most beneficial for model performance. (c) The schematic diagram of the embedded method, which combines feature selection with model training to ultimately obtain an optimal feature subset. (d) The workflow diagram of the random feature selection method, which randomly selects a subset of features from the entire feature set as the final feature subset. | ||
For instance, in 2023, Le et al. used the filter technology method for feature selection in protein engineering, both because the method can perform feature selection independently of the model and because it was more suitable for qualitatively evaluating feature importance in biomaterials.110 In the realm of catalyst design, Benavides-Hernández et al. opted for filter technology on the grounds that it successfully delineated the features with the most significant influence on catalyst efficacy while obviating the need for an augmented experimental workload.111
Filter technology has some limitations. It may not adequately capture the significance of certain features within a particular model or the intricate interactions among features. This technology may require further adjustments to ensure that the model does not lean towards the majority of class.
Case in point: in drug discovery, Mesrabadi et al. introduced a model for predicting DTI in 2023, using wrapper technology to remove irrelevant features. Compared to other methods, it was more suitable for predicting complex drug–target relationships in bioinformatics, ensuring that the selected features had a direct positive impact on model performance,108 as shown in Fig. 7b. In catalyst design, Shi et al. used wrapper technology to select the most critical subset for predicting adsorption energy from a large number of candidate features,112 as this technology ensured that the selected features contributed the most to the performance of the prediction model.
The wrapper method has high computational costs for large imbalanced datasets due to repeated model training, especially with few minority class samples, and is often combined with filtering or embedding techniques to optimize feature selection.
The utility of embedded feature selection techniques spans various domains. For instance, in catalyst design, Ma et al. developed an ML-driven model to identify catalysts,113 which used embedded recursive feature elimination (RFE) to remove redundant features. A key benefit of this method is that it can dynamically select features during model training, enabling greater synchronization. Similarly, in drug discovery, Zhao et al. introduced a method based on convolutional neural networks (CNN) to embed relationship path features. Compared to other methods, embedded technology can better extract drug disease relationship path features.114
Although the embedded method can help the model better focus on minority categories, it may not fully reflect the importance of features or the complex interactions between features, which may require additional strategies to supplement when dealing with imbalanced data.
Recently, in DTI prediction, RFS was used in the training process of the model.115 In protein recognition research, Qiang et al. constructed a recognition model based on the RF model,116 where each tree was constructed based on a randomly selected subset of features. The use of random feature selection was to enhance feature representation, aiming to extract information from different perspectives of numerous feature descriptors and eliminate redundant and irrelevant features through the optimization of the feature space.
While RFS saves time and reduces overfitting, its randomness may cause accuracy fluctuations, especially in correlated chemical data, requiring optimization or combination with other methods for stable minority class predictions.
2.5 Evaluation metrics suitable for imbalanced datasets
When dealing with imbalanced data, traditional metrics in ML models like accuracy and precision are not suitable, as they can be skewed by the majority class, giving a false sense of high performance even when the minority class is poorly predicted. In contrast, balanced accuracy,117 the F1 score,118 Area Under the Receiver Operating Characteristic Curve (AUC-ROC),119 and Matthews correlation coefficient (MCC)120 are more suitable.The F1 Score focuses on the minority class by addressing false positives and false negatives, making it useful when false negatives are costly. The mathematical formula for the F1 score is given as follows:
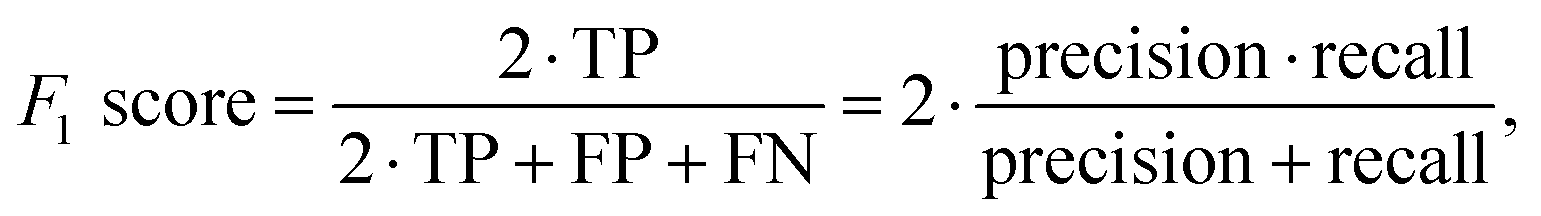 | (1) |
The AUC-ROC curve evaluates a classifier's ability to distinguish between classes by measuring true positive and false positive rates across classification thresholds. The definitions of true positive and false positive rates are given by:
 | (2) |
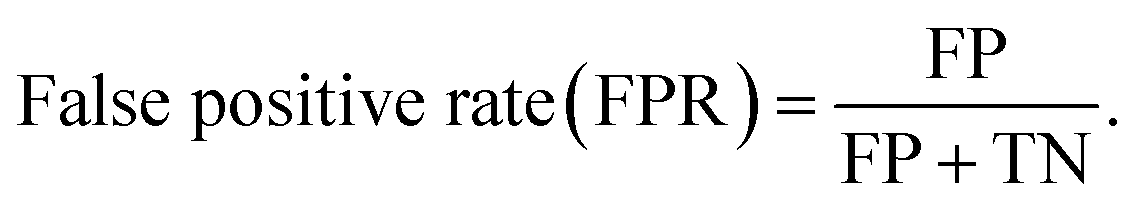 | (3) |
MCC offers a balanced evaluation, accounting for true/false positives and negatives and providing a comprehensive performance view even with imbalanced classes. The mathematical formula is given in the following form:
 | (4) |
3 Perspectives for future directions and challenges
In reviewing techniques for addressing imbalanced data, we have analyzed their strengths, weaknesses, and applications in chemistry, as summarized in Table 1. In this chapter, we briefly give the rules of thumb for selecting suitable methods to address data imbalance issues, explore future research directions, offer unique perspectives, and discuss new approaches to better tackle the imbalanced data challenge in chemistry.| Methods | Strengths | Weaknesses | Ref | |
|---|---|---|---|---|
| Resampling techniques | Oversampling techniques | Generate new minority class samples, while preserving the original feature distribution and mitigating overfitting | Introduce noisy data, struggle with complex decision boundaries, fail to account for internal distribution difference within the minority class, and high computational costs | 6 and 7 |
| Undersampling techniques | Reduce training time by decreasing the dataset size, and robustness against noisy data and outliers | The loss of important information, and its efficiency declines with large datasets, which limits its applicability in certain large-scale contexts | 28, 29 and 32 | |
| Hybrid techniques | Synthesize new minority class samples and remove overlapping boundary samples, effectively mitigating data imbalance and overfitting | Computationally expensive for large datasets, and sensitive to parameter selection, particularly in noisy or unevenly distributed datasets | 35, 37, 38 and 40 | |
| Cluster-based techniques | Effectively reduce data imbalance and optimize sample distribution, enhancing both the performance and generalization of classification models | Depend on parameter optimization and can be computationally expensive for large datasets | 41–45 | |
| Data augmentation | Noise addition | Make the model less sensitive to superficial trends in the data and better equipped to handle unpredictable environments | Need careful tuning of noise levels to avoid distorting minority class features or compromising interpretability | 47 and 48 |
| Deep generative models | Learn better representations of the minority class, reducing the bias towards the majority class | Unstable training and risk of mode collapse, and reliance on fixed distributions and tendency to produce blurry samples | 49, 50, 61, 62 and 68 | |
| Feature augmentation | Provide more informative and diverse data points to better distinguish the minority class from the majority class, reducing bias and improving classification accuracy | Risk adding irrelevant features, overfitting, or noise, and does not directly address the core imbalance between majority and minority classes | 70 and 70–75 | |
| Ensemble methods | Boosting | Adaptively emphasize misclassified minority instances, effectively reducing bias toward majority classes | The growing complexity with iterations requires careful tuning and often needs to be combined with techniques like data sampling or feature selection to improve efficiency | 82–87 |
| Bagging | Reduce variance and improve stability by aggregating predictions from multiple independently trained models | Less effective at handling severe class imbalance due to reliance on random sampling, potentially neglecting minority classes | 93–95 | |
| Cost-sensitive learning | Cost-sensitive learning | Enable the model to pay more attention to high-cost minority sample errors by reweighting majority and minority samples | Improper cost settings may lead to overfitting minority classes and reduce generalization, and uncertainty in cost function design may impact model performance | 100 and 101 |
| Feature engineering and selection strategies | Filter technology | Recognize minority classes with high-dimensional data processing capabilities | It may not adequately capture the significance of certain features within a particular model or the intricate interactions among features | 109–111 |
| Wrapper technology | Accurately identify key features that have significant predictive effects on minority categories | High computational costs for large imbalanced datasets due to repeated model training | 108 and 112 | |
| Embedded technology | Automatically identify and select feature subsets that are crucial for minority class prediction during training | Feature selection may be biased toward majority class due to intrinsic optimization criteria | 113 and 114 | |
| Random feature selection | Simple, computationally inexpensive, and reduce variance by decorrelating selected features | Its randomness may overlook critical features vital for accurately predicting minority classes, leading to suboptimal classification performance | 115 and 116 | |
3.1 Rules of thumb for selecting suitable methods
Selecting suitable methods for addressing data imbalance issues requires the consideration of several key factors, including the severity of imbalance, dataset size, computational resources, and data complexity.Firstly, for mild or moderate data imbalance cases (e.g., minority-to-majority ratio is equal to or less than 1:10), simpler techniques such as oversampling or undersampling can be considered. For datasets with relatively small sizes and low dimensinality, oversampling like SMOTE can effectively enhance minority class representation without significantly increasing complexity. However, oversampling may inadvertently introduce noise or exacerbate overfitting, especially in cases where minority class samples are noisy or contain outliers. To mitigate this, combining oversampling with noise-filtering techniques or employing advanced variants such as Borderline-SMOTE is recommended.
Conversely, if the dataset is large, significantly imbalanced, or noisy, undersampling techniques often become the method of choice due to their computational efficiency. They streamline model training by reducing majority class samples, improving model responsiveness, and reducing resource consumption. However, undersampling should be performed carefully to avoid discarding valuable information. Techniques such as Tomek Links are recommended to selectively remove redundant majority class samples while preserving important data points.
In scenarios with severe imbalance (e.g., minority-to-majority ratio of 1:50 or higher), traditional sampling methods alone may be insufficient. Leveraging more sophisticated approaches such as deep generative models like GANs becomes advantageous. Nevertheless, such methods require substantial computational resources and careful tuning, making them more suitable for projects where high accuracy and predictive performance justify the resource investment.
For datasets characterized by significant noise, feature redundancy, or high dimensionality, feature selection or augmentation methods should be employed. Cost-sensitive learning methods are particularly suitable for contexts where different misclassification costs are explicitly defined or can be quantified precisely. Adjusting misclassification costs allows models to prioritize correctly classifying minority class instances, directly addressing imbalance at the algorithmic level without additional sampling.
Finally, ensemble-based methods, such as Random Forest, AdaBoost, or gradient boosting like XGBoost, are consistently robust and effective. They inherently manage data imbalance by aggregating multiple weak classifiers, providing strong predictive accuracy and reduced variance. Combining ensemble methods with advanced sampling or feature-selection approaches can further enhance predictive performance, particularly in complex real-world applications.
3.2 Emerging trends in imbalanced data research in chemistry
In chemistry, recent research on the imbalanced data challenge reflects the convergence of interdisciplinary approaches and the development of novel methodologies.3.3 Physical-model-based data augmentation
One future research trend on data augmentation strategies is focusing on integrating physical models to generate virtual data with meaningful physical properties, thereby enriching datasets and improving model performance.In this research field, molecular dynamics (MD) simulation plays a crucial role by simulating molecular motion and interactions, revealing conformations that experimental data may miss. This is especially relevant in areas like protein–ligand interactions126 and reaction mechanisms, where rare but important configurations are underrepresented. By generating such scenarios, MD could help build richer datasets, addressing data imbalance and improving models' ability to predict molecular properties and reactions.
Density functional theory (DFT) and molecular docking are pivotal in data augmentation strategies. DFT, a quantum mechanical approach, can generate critical data on chemical reactivity, catalytic activity, photophysical properties, nuclear magnetic resonance spectra,127 binding free energy,128 and molecular electronic structures,129 enhancing ML models' ability to predict molecular properties. Its broad applicability across molecular systems can help balance datasets and improve generalization. Through predicting interactions between molecules and receptors,130 molecular docking could provide binding patterns and affinity data. Both techniques can enrich datasets, especially for imbalanced data, improving the accuracy of models in predicting molecular interactions and properties.
The integration of physical models with ML is emerging as a key trend. By incorporating physical laws into ML frameworks, models can enhance prediction accuracy and interpretability. Advances in computational power enable physics-based data augmentation, creating more diverse training datasets and fostering more generalizable chemical models. For example, thermodynamic and statistical mechanics simulations can generate data on equilibrium constants, free energy changes, and reaction rates, enriching datasets, addressing imbalances, and boosting model performance.
3.4 Large-language-model-based data augmentation
As chemistry faces the challenge of imbalanced datasets, the rise of deep learning (DL), especially Large Language Models (LLMs), such as ChatGPT and Gemini, offers promising solutions. LLMs excel in data augmentation, as demonstrated by Sarker et al., who showed that models like ChatGPT improve accuracy in drug identification and classification.131 Furthermore, models like Chemformer, introduced by Ross Irwin in 2022,132 can handle a range of chemical tasks, including sequence-to-sequence and discriminative tasks, showcasing the potential of LLMs in chemical research.LLMs could offer innovative solutions for addressing imbalanced chemical data by learning from diverse representations, such as textual descriptions and chemical structures. For example, generating molecular structures from textual representations demonstrates how LLMs can enrich datasets and mitigate the data imbalance challenge, advancing chemical data analysis.133
3.5 Mathematics-enabled data augmentation
A trending topic in data science is the integration of AI with advanced mathematics, such as differential geometry, algebraic topology, combinatorics, geometric topology, etc.70,71 Recently, mathematical AI has become an emerging paradigm in molecular data sciences, including drug design competitions,134,135 the discovery of viral evolution mechanisms,136 the forecasting of emerging dominant variants,137,138 protein engineering,139 protein mutation prediction,140 toxicity prediction,141 drug addiction analysis,142 polymer property,143etc. The multiscale topology-enabled transformer by Chen et al. gives rise to the best prediction of protein–ligand binding affinities.144 This mathematical approach captures stereochemistry,145 which is missing in typical sequence-based molecular language models. As such, mathematics-enabled generative models will be a new approach for data augmentation.Additionally, the graph-based Merriman–Bence–Osher method has been utilized to handle imbalanced data.75 It leverages its diffusion process to propagate label information across the graph, enabling better representation of minority classes in semi-supervised learning tasks.
3.6 New materials design and manufacture
While the experimental synthesis of the minority class offers the ultimate solution to imbalanced data, balanced datasets play a key role in AI-assisted active design and manufacture of new materials. Enhanced computational power and refined algorithms are accelerating breakthroughs in new materials development by ensuring more equitable data distribution.Balancing datasets significantly improves the accuracy of ML models in predicting new materials. Recent studies using techniques like feature engineering146 have led to more reliable models that excel in predicting material properties and optimizing synthesis pathways, accelerating material research and development. The use of balanced datasets is driving the integration of high-throughput experimentation and computational simulations in material innovation.147 By reducing bias and improving data efficiency, these datasets guide experimental design and, when combined with simulations, enable researchers to explore a wider range of parameters, accelerating the discovery and development of new materials.
Balanced datasets are crucial for fostering material diversity. By creating and utilizing these datasets, researchers can more effectively explore existing material databases, uncovering unique materials and driving innovative material design.
3.7 Persistent challenges and areas for further investigation
In chemical research, the management of imbalanced data continues to encounter significant challenges. Although various solutions have been proposed in recent years, several fundamental issues remain unresolved and demand urgent attention. First, the combination of small data148 and imbalanced data poses one of the most significant challenges in molecular science. Data scarcity and imbalance are especially pronounced across numerous chemical applications. Due to the inherent difficulty in producing large volumes of balanced and high-quality data during experiments, particularly when studying new materials or rare compounds, or when performing toxicity evaluations, finding efficient methods to collect, share, and integrate data has become a pressing concern.Additionally, the absence of standardized processes for data handling and dissemination between different research projects and laboratories further complicates the resolution of imbalanced data issues. The current mechanisms for data sharing require substantial improvement. Although a growing number of academic institutions and journals advocate for open data practices, operational challenges persist. For example, inconsistent data repository formats and incomplete or insufficiently detailed data documentation hinder effective reuse. Thus, establishing a standardized platform, spearheaded by relevant organizations or funding bodies, to regulate data submission and validation processes is crucial for enhancing data transparency and quality.
While several approaches have been applied to mitigate the imbalanced data challenge,2,149 the resilience and generalizability of these methods still require significant refinement. Many existing techniques struggle when applied to small sample sizes,148 making it difficult to address the complex molecular structures and reaction pathways inherent to chemical research. Consequently, future studies should prioritize the development of more precise and field-specific data augmentation and modeling techniques. Notably, enhancement methods based on physical models may hold considerable promise in rectifying data imbalance, although these approaches are still in the nascent stages of exploration.
To drive progress in this area, forthcoming research should aim to create robust frameworks that facilitate the widespread implementation of imbalanced data technologies. Moreover, researchers should emphasize the thorough validation of new algorithms, particularly through testing across various chemical application scenarios, to ensure their practical utility and consistency. These efforts will provide a strong foundation and theoretical backing for addressing the imbalanced data problem in the chemical sciences.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Author contributions
CZ contributed to writing the Section 2 and the preparation of figures; LK contributed to writing the Section 3; JJ contributed to writing the remainder and the preparation of figures and table; NH, YZ, HQ, BZ, TZ, and GW revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported in part by NIH grants R01AI164266, and R35GM148196, National Science Foundation grants DMS2052983, and IIS-1900473, and MSU Foundation. The work of Huahai Qiu and Bengong Zhang was supported by the National Natural Science Foundation of China under Grant No. 12271416 and No. 12371500, respectively.References
- J. Jiang, L. Ke, L. Chen, B. Dou, Y. Zhu and J. Liu, et al., Transformer technology in molecular science, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2024, 14(4), e1725 Search PubMed.
- S. Korkmaz, Deep learning-based imbalanced data classification for drug discovery, J. Chem. Inf. Model., 2020, 60(9), 4180–4190 CrossRef CAS PubMed.
- S. B. Gunturi and N. Ramamurthi, A novel approach to generate robust classification models to predict developmental toxicity from imbalanced datasets, SAR QSAR Environ. Res., 2014, 25(9), 711–727 CrossRef CAS PubMed.
- A. Deng, H. Zhang, W. Wang, J. Zhang, D. Fan and P. Chen, et al., Developing computational model to predict protein-protein interaction sites based on the XGBoost algorithm, Int. J. Mol. Sci., 2020, 21(7), 2274 CrossRef CAS PubMed.
- D. A. Pisner and D. M. Schnyer, Support vector machine, in Machine learning, Elsevier, 2020, pp. 101–121 Search PubMed.
- S. J. Basha, S. R. Madala, K. Vivek, E. S. Kumar and T. Ammannamma, A review on imbalanced data classification techniques, in 2022 International Conference on Advanced Computing Technologies and Applications (ICACTA), IEEE, 2022, pp. 1–6 Search PubMed.
- D. Li, J. Liu and J. Liu, NNI-SMOTE-XGBoost: A Novel Small Sample Analysis Method for Properties Prediction of Polymer Materials, Macromol. Theory Simul., 2021, 30(5), 2100010 CrossRef CAS.
- A. Chen, J. Cai, Z. Wang, Y. Han, S. Ye and J. Li, An ensemble learning classifier to discover arsenene catalysts with implanted heteroatoms for hydrogen evolution reaction, J. Energy Chem., 2023, 78, 268–276 CrossRef CAS.
- Q. Gao, X. Jin, E. Xia, X. Wu, L. Gu and H. Yan, et al., Identification of orphan genes in unbalanced datasets based on ensemble learning, Front. Genet., 2020, 11, 820 CrossRef CAS PubMed.
- S. Bej, A. M. Galow, R. David, M. Wolfien and O. Wolkenhauer, Automated annotation of rare-cell types from single-cell RNA-sequencing data through synthetic oversampling, BMC Bioinf., 2021, 22, 1–17 CrossRef PubMed.
- C. Kumari, M. Abulaish and N. Subbarao, Using SMOTE to deal with class-imbalance problem in bioactivity data to predict mTOR inhibitors, SN Comput. Sci., 2020, 1, 1–7 CrossRef.
- A. K. Azlim Khan and N. H. Ahamed Hassain Malim, Comparative Studies on Resampling Techniques in Machine Learning and Deep Learning Models for Drug-Target Interaction Prediction, Molecules, 2023, 28(4), 1663 CrossRef CAS PubMed.
- N. Mohanty, B. K. Behera and C. Ferrie, A Quantum Approach to Synthetic Minority Oversampling Technique (SMOTE), Quantum Machine Intelligence, 2025 DOI:10.1007/s42484-025-00248-6.
- N. V. Chawla, K. W. Bowyer, L. O. Hall and W. P. Kegelmeyer, SMOTE: synthetic minority over-sampling technique, J. Artif. Intell. Res., 2002, 16, 321–357 CrossRef.
- Y. Huang, Q. Chen, Z. Zhang, K. Gao, A. Hu and Y. Dong, et al., A machine learning framework to predict the tensile stress of natural rubber: Based on molecular dynamics simulation data, Polymers, 2022, 14(9), 1897 CrossRef CAS PubMed.
- H. Han, W. Y. Wang and B. H. Mao, Borderline-SMOTE: a new over-sampling method in imbalanced data sets learning, in International conference on intelligent computing, Springer, 2005, pp. 878–887 Search PubMed.
- X. Zheng, SMOTE variants for imbalanced binary classification: heart disease prediction, University of California, Los Angeles, 2020 Search PubMed.
- Z. Fan, Z. Sun, A. Wang, Y. Yin, G. Jin and C. Xin, Machine Learning Classification Model for Screening of Infrared Nonlinear Optical Crystals, J. Electron. Mater., 2023, 52(6), 4162–4168 CrossRef CAS.
- C. Bunkhumpornpat, K. Sinapiromsaran and C. Lursinsap, Safe-level-smote: Safe-level-synthetic minority over-sampling technique for handling the class imbalanced problem, in Advances in knowledge discovery and data mining: 13th Pacific-Asia conference, PAKDD 2009 Bangkok, Thailand, April 27-30, 2009 proceedings 13, Springer, 2009, pp. 475–482 Search PubMed.
- H. G. Gozukara Bag, F. H. Yagin, Y. Gormez, P. P. González, C. Colak and M. Gülü, et al., Estimation of obesity levels through the proposed predictive approach based on physical activity and nutritional habits, Diagnostics, 2023, 13(18), 2949 CrossRef PubMed.
- H. He, Y. Bai, E. A. Garcia and S. Li, ADASYN: Adaptive synthetic sampling approach for imbalanced learning, in 2008 IEEE international joint conference on neural networks (IEEE world congress on computational intelligence), Ieee, 2008, pp. 1322–1328 Search PubMed.
- A. Nurani, Y. Yamashita, Y. Taki, Y. Takada, Y. Itoh and T. Suzuki, Identification of a Histone Deacetylase 8 Inhibitor through Drug Screenings Based on Machine Learning, Chem. Pharm. Bull., 2024, 72(2), 173–178 CrossRef CAS PubMed.
- C. Jiang, W. Lv and J. Li, Protein-protein interaction sites prediction using batch normalization based CNNs and oversampling method borderline-SMOTE, IEEE/ACM Trans. Comput. Biol. Bioinf., 2023, 20(3), 2190–2199 CAS.
- C. Jia, M. Zhang, C. Fan, F. Li and J. Song, Formator: predicting lysine formylation sites based on the most distant undersampling and safe-level synthetic minority oversampling, IEEE/ACM Trans. Comput. Biol. Bioinf., 2019, 18(5), 1937–1945 Search PubMed.
- S. H. Mahmud, W. Chen, H. Meng, H. Jahan, Y. Liu and S. M. Hasan, Prediction of drug-target interaction based on protein features using undersampling and feature selection techniques with boosting, Anal. Biochem., 2020, 589, 113507 CrossRef CAS PubMed.
- Y. Hu, H. Lan, B. Hu, J. Gong, D. Wang, W. D. Zhang, et al., Machine Learning Boosted Entropy-Engineered Synthesis of stable Nanometric Solid Solution CuCo Alloys for Efficient Nitrate Reduction to Ammonia, arXiv, 2024, preprint, arXiv:240800142, DOI:10.48550/arXiv.2408.00142.
- M. Wang, L. Song, Y. Zhang, H. Gao, L. Yan and B. Yu, Malsite-deep: prediction of protein malonylation sites through deep learning and multi-information fusion based on NearMiss-2 strategy, Knowl.-Based Syst., 2022, 240, 108191 CrossRef.
- W. Zhang, E. Xia, R. Dai, W. Tang, Y. Bin and J. Xia, PredAPP: predicting anti-parasitic peptides with undersampling and ensemble approaches, Interdiscip. Sci.:Comput. Life Sci., 2022, 1–11 Search PubMed.
- Q. Ning, X. Zhao and Z. Ma, A novel method for Identification of Glutarylation sites combining Borderline-SMOTE with Tomek links technique in imbalanced data, IEEE/ACM Trans. Comput. Biol. Bioinf., 2021, 19(5), 2632–2641 Search PubMed.
- S. H. Mahmud, W. Chen, Y. Liu, M. A. Awal, K. Ahmed and M. H. Rahman, et al., PreDTIs: prediction of drug–target interactions based on multiple feature information using gradient boosting framework with data balancing and feature selection techniques, Briefings Bioinf., 2021, 22(5), bbab046 CrossRef PubMed.
- X. Bai and Y. Yin, Exploration and augmentation of pharmacological space via adversarial auto-encoder model for facilitating kinase-centric drug development, J. Cheminf., 2021, 13, 1–15 Search PubMed.
- M. A. Gutiérrez-Mondragón, C. König and A. Vellido, Recognition of Conformational States of a G Protein-Coupled Receptor from Molecular Dynamic Simulations Using Sampling Techniques, in International Work-Conference on Bioinformatics and Biomedical Engineering, Springer, 2023, pp. 3–16 Search PubMed.
- M. Bi, Z. Guan, T. Fan, N. Zhang, J. Wang and G. Sun, et al., Identification of Pharmacophoric Fragments of DYRK1A Inhibitors Using Machine Learning Classification Models, Molecules, 2022, 27(6), 1753 CrossRef CAS PubMed.
- A. R. Purnajaya, W. A. Kusuma and M. K. D. Hardhienata, Performance comparison of data sampling techniques to handle imbalanced class on prediction of compound-protein interaction, Biogenesis: Jurnal Ilmiah Biologi, 2020, 8(1), 41–48 Search PubMed.
- G. E. Batista, R. C. Prati and M. C. Monard, A study of the behavior of several methods for balancing machine learning training data, ACM SIGKDD Explorations Newsletter, 2004, vol. 6, 1, pp. 20–29 Search PubMed.
- J. H. Wang, C. Y. Liu, Y. R. Min, Z. H. Wu and P. L. Hou, Cancer Diagnosis by Gene-Environment Interactions via Combination of SMOTE-Tomek and Overlapped Group Screening Approaches with Application to Imbalanced TCGA Clinical and Genomic Data, Mathematics, 2024, 12(14), 2209 CrossRef.
- M. Muntasir Nishat, F. Faisal, I. Jahan Ratul, A. Al-Monsur, A. M. Ar-Rafi and S. M. Nasrullah, et al., A Comprehensive Investigation of the Performances of Different Machine Learning Classifiers with SMOTE-ENN Oversampling Technique and Hyperparameter Optimization for Imbalanced Heart Failure Dataset, Sci. Program., 2022, 2022(1), 3649406 Search PubMed.
- M. Kumari and N. Subbarao, A hybrid resampling algorithms SMOTE and ENN based deep learning models for identification of Marburg virus inhibitors, Future Med. Chem., 2022, 14(10), 701–715 CrossRef CAS.
- M. Zhang, H. Gao, X. Liao, B. Ning, H. Gu and B. Yu, DBGRU-SE: predicting drug–drug interactions based on double BiGRU and squeeze-and-excitation attention mechanism, Briefings Bioinf., 2023, 24(4), bbad184 CrossRef.
- A. Puri, M. K. Gupta and K. Sachdev, An ensemble-based approach using structural feature extraction method with class imbalance handling technique for drug-target interaction prediction, Multimed. Tools Appl., 2022, 81(26), 37499–37517 CrossRef.
- Y. Sanguanmak and A. Hanskunatai, DBSM: The combination of DBSCAN and SMOTE for imbalanced data classification, in 2016 13th International joint conference on computer science and software engineering (JCSSE), IEEE, 2016, pp. 1–5 Search PubMed.
- A. Nath and K. Subbiah, Unsupervised learning assisted robust prediction of bioluminescent proteins, Comput. Biol. Med., 2016, 68, 27–36 CrossRef CAS PubMed.
- G. Kannan, et al., Cervical cancer prediction using outlier deduction and over sampling methods, ScienceOpen Preprints, 2022 Search PubMed.
- D. H. Koh, W. S. Song and Ey Kim, Multi-step structure-activity relationship screening efficiently predicts diverse PPARγ antagonists, Chemosphere, 2022, 286, 131540 CrossRef CAS PubMed.
- G. Douzas, F. Bacao and F. Last, Improving imbalanced learning through a heuristic oversampling method based on k-means and SMOTE, Inf. Sci., 2018, 465, 1–20 CrossRef.
- X. Song, Y. Li and H. Wu, Research on random forest drug classification prediction model based on KMeans-SMOTE, in International Conference on Biomedical and Intelligent Systems (IC-BIS 2022), SPIE, 2022, vol. 12458, pp. 402–408 Search PubMed.
- W. Lu, J. Zhang, W. Huang, Z. Zhang, X. Jia and Z. Wang, et al., DynamicBind: Predicting ligand-specific protein-ligand complex structure with a deep equivariant generative model, Nat. Commun., 2024, 15(1), 1071 CrossRef CAS PubMed.
- R. Chakraborty and Y. Hasija, Utilizing deep learning to explore chemical space for drug lead optimization, Expert Syst. Appl., 2023, 229, 120592 CrossRef.
- I. Goodfellow, J. Pouget-Abadie, M. Mirza, B. Xu, D. Warde-Farley, S. Ozair, et al., Generative adversarial nets, Advances in Neural Information Processing Systems, 2014, vol. 27 Search PubMed.
- M. Abbasi, B. P. Santos, T. C. Pereira, R. Sofia, N. R. Monteiro and C. J. Simões, et al., Designing optimized drug candidates with Generative Adversarial Network, J. Cheminf., 2022, 14(1), 40 Search PubMed.
- Y. Bian, J. Wang, J. J. Jun and X. Q. Xie, Deep convolutional generative adversarial network (dcGAN) models for screening and design of small molecules targeting cannabinoid receptors, Mol. Pharmaceutics, 2019, 16(11), 4451–4460 CrossRef CAS.
- O. Prykhodko, S. V. Johansson, P. C. Kotsias, J. Arús-Pous, E. J. Bjerrum and O. Engkvist, et al., A de novo molecular generation method using latent vector based generative adversarial network, J. Cheminf., 2019, 11, 1–13 Search PubMed.
- Ł. Maziarka, A. Pocha, J. Kaczmarczyk, K. Rataj, T. Danel and M. Warchoł, Mol-CycleGAN: a generative model for molecular optimization, J. Cheminf., 2020, 12(1), 2 Search PubMed.
- T. Hu, H. Song, T. Jiang and S. Li, Learning representations of inorganic materials from generative adversarial networks, Symmetry, 2020, 12(11), 1889 CrossRef.
- E. Lin, C. H. Lin and H. Y. Lane, De novo peptide and protein design using generative adversarial networks: an update, J. Chem. Inf. Model., 2022, 62(4), 761–774 CrossRef CAS PubMed.
- M. Karimi, S. Zhu, Y. Cao and Y. Shen, De novo protein design for novel folds using guided conditional wasserstein generative adversarial networks, J. Chem. Inf. Model., 2020, 60(12), 5667–5681 CrossRef CAS PubMed.
- A. Ishikawa, Heterogeneous catalyst design by generative adversarial network and first-principles based microkinetics, Sci. Rep., 2022, 12(1), 11657 CrossRef CAS PubMed.
- J. Li, R. O. Topaloglu and S. Ghosh, Quantum generative models for small molecule drug discovery, IEEE Trans. Quantum Eng., 2021, 2, 1–8 Search PubMed.
- T. T. Lin, Y. Y. Sun, W. C. Cheng, I. H. Lu, S. H. Chen and C. Y. Lin, Developing an Antiviral Peptides Predictor with Generative Adversarial Network Data Augmentation, bioRxiv, 2021, preprint, DOI:10.1101/2021.11.29.470292.
- S. Wei, Z. Chen, S. K. Arumugasamy and I. M. L. Chew, Data augmentation and machine learning techniques for control strategy development in bio-polymerization process, Environ. Sci. Ecotechnology, 2022, 11, 100172 CrossRef CAS.
- R. Wei and A. Mahmood, Recent advances in variational autoencoders with representation learning for biomedical informatics: A survey, IEEE Access, 2020, 9, 4939–4956 Search PubMed.
- R. Wei, C. Garcia, A. El-Sayed, V. Peterson and A. Mahmood, Variations in variational autoencoders-a comparative evaluation, IEEE Access, 2020, 8, 153651–153670 Search PubMed.
- T. Li, X. M. Zhao and L. Li, Co-VAE: Drug-target binding affinity prediction by co-regularized variational autoencoders, IEEE Trans. Pattern Anal. Mach. Intell., 2021, 44(12), 8861–8873 Search PubMed.
- H. Dong, J. Xie, Z. Jing and D. Ren, Variational autoencoder for anti-cancer drug response prediction, arXiv, 2020, preprint, arXiv:200809763, DOI:10.48550/arXiv.2008.09763.
- A. Hawkins-Hooker, F. Depardieu, S. Baur, G. Couairon, A. Chen and D. Bikard, Generating functional protein variants with variational autoencoders, PLoS Comput. Biol., 2021, 17(2), e1008736 CrossRef CAS.
- H. Tian, X. Jiang, F. Trozzi, S. Xiao, E. C. Larson and P. Tao, Explore protein conformational space with variational autoencoder, Front. Mol. Biosci., 2021, 8, 781635 CrossRef PubMed.
- A. J. Lew and M. J. Buehler, Encoding and exploring latent design space of optimal material structures via a VAE-LSTM model, Forces Mech., 2021, 5, 100054 CrossRef.
- O. Schilter, A. Vaucher, P. Schwaller and T. Laino, Designing catalysts with deep generative models and computational data. A case study for Suzuki cross coupling reactions, Digital Discovery, 2023, 2(3), 728–735 RSC.
- N. K. Ngo and T. S. Hy, Multimodal protein representation learning and target-aware variational auto-encoders for protein-binding ligand generation, Mach. Learn.: Sci. Technol., 2024, 5(2), 025021 Search PubMed.
- D. D. Nguyen, Z. Cang and G. W. Wei, A review of mathematical representations of biomolecular data, Phys. Chem. Chem. Phys., 2020, 22(8), 4343–4367 RSC.
- J. Townsend, C. P. Micucci, J. H. Hymel, V. Maroulas and K. D. Vogiatzis, Representation of molecular structures with persistent homology for machine learning applications in chemistry, Nat. Commun., 2020, 11(1), 3230 CrossRef CAS PubMed.
- D. Chen, K. Gao, D. D. Nguyen, X. Chen, Y. Jiang and G. W. Wei, et al., Algebraic graph-assisted bidirectional transformers for molecular property prediction, Nat. Commun., 2021, 12(1), 3521 CrossRef CAS.
- A. Goyal, A. Zafar, M. Kumar, S. Bharadwaj, B. Tejas and J. Malik, Cirrhosis disease classification by using polynomial feature and XGBoosting, in 2022 13th International Conference on Computing Communication and Networking Technologies (ICCCNT), IEEE, 2022, pp. 1–5 Search PubMed.
- D. Zhang, H. Zhang, J. Tang, M. Wang, X. Hua and Q. Sun, Feature pyramid transformer, in Computer Vision–ECCV 2020: 16th European Conference, Glasgow, UK, August 23–28, 2020, Proceedings, Part XXVIII 16, Springer, 2020, pp. 323–339 Search PubMed.
- N. Hayes, E. Merkurjev and G. W. Wei, Graph-Based Bidirectional Transformer Decision Threshold Adjustment Algorithm for Class-Imbalanced Molecular Data, J. Comput. Biophys. Chem., 2024, 23(10), 1339–1358 CrossRef.
- W. Cheng, Y. Shen and L. Huang, Adaptive factorization network: Learning adaptive-order feature interactions, in Proceedings of the AAAI Conference on Artificial Intelligence, 2020, vol. 34, pp. 3609–3616 Search PubMed.
- T. Verdonck, B. Baesens, M. Óskarsdóttir and S. vanden Broucke, Special issue on feature engineering editorial, Mach. Learn., 2024, 113(7), 3917–3928 CrossRef.
- G. Li, W. Sun, J. Xu, L. Hu, W. Zhang and P. Zhang, GA-ENs: A novel drug–target interactions prediction method by incorporating prior Knowledge Graph into dual Wasserstein Generative Adversarial Network with gradient penalty, Appl. Soft Comput., 2023, 139, 110151 CrossRef.
- S. Lin, Y. Wang, L. Zhang, Y. Chu, Y. Liu and Y. Fang, et al., MDF-SA-DDI: predicting drug–drug interaction events based on multi-source drug fusion, multi-source feature fusion and transformer self-attention mechanism, Briefings Bioinf., 2022, 23(1), bbab421 CrossRef PubMed.
- C. Wan and D. T. Jones, Protein function prediction is improved by creating synthetic feature samples with generative adversarial networks, Nat. Mach. Intell., 2020, 2(9), 540–550 CrossRef.
- S. Akbar, A. Raza, T. Al Shloul, A. Ahmad, A. Saeed and Y. Y. Ghadi, et al., PAtbP-EnC: Identifying anti-tubercular peptides using multi-feature representation and genetic algorithm-based deep ensemble model, IEEE Access, 2023, 11, 137099–137114 Search PubMed.
- J. Tanha, Y. Abdi, N. Samadi, N. Razzaghi and M. Asadpour, Boosting methods for multi-class imbalanced data classification: an experimental review, J. Big Data, 2020, 7, 1–47 CrossRef.
- R. Sikander, A. Ghulam and F. Ali, XGB-DrugPred: computational prediction of druggable proteins using eXtreme gradient boosting and optimized features set, Sci. Rep., 2022, 12(1), 5505 CrossRef CAS PubMed.
- J. Hu, Y. S. Bai, L. L. Zheng, N. X. Jia, D. J. Yu and G. J. Zhang, Protein-DNA binding residue prediction via bagging strategy and sequence-based cube-format feature, IEEE/ACM Trans. Comput. Biol. Bioinf., 2021, 19(6), 3635–3645 Search PubMed.
- R. E. Schapire, et al., A brief introduction to boosting, in Ijcai, Citeseer, 1999, vol. 99. pp. 1401–1406 Search PubMed.
- T. Chen and C. Guestrin, Xgboost: A scalable tree boosting system, in Proceedings of the 22nd acm sigkdd international conference on knowledge discovery and data mining, 2016, pp. 785–794 Search PubMed.
- J. H. Friedman, Greedy function approximation: a gradient boosting machine, Ann. Stat., 2001, 1189–1232 Search PubMed.
- H. Mai, T. C. Le, D. Chen, D. A. Winkler and R. A. Caruso, Machine learning for electrocatalyst and photocatalyst design and discovery, Chem. Rev., 2022, 122(16), 13478–13515 CrossRef CAS PubMed.
- X. Liu, Y. Luo, P. Li, S. Song and J. Peng, Deep geometric representations for modeling effects of mutations on protein-protein binding affinity, PLoS Comput. Biol., 2021, 17(8), e1009284 CrossRef CAS PubMed.
- R. Özçelik, H. Öztürk, A. Özgür and E. Ozkirimli, Chemboost: A chemical language based approach for protein–ligand binding affinity prediction, Mol. Inf., 2021, 40(5), 2000212 CrossRef PubMed.
- K. Xue, F. Wang, A. Suwardi, M. Y. Han, P. Teo and P. Wang, et al., Biomaterials by design: Harnessing data for future development, Mater. Today Bio, 2021, 12, 100165 CrossRef CAS PubMed.
- Q. Liu, M. Reed, H. Zhu, Y. Cheng, J. Almeida and G. Fruhbeck, et al., Epigenome-wide DNA methylation and transcriptome profiling of localized and locally advanced prostate cancer: Uncovering new molecular markers, Genomics, 2022, 114(5), 110474 CrossRef CAS PubMed.
- T. Zhang, Q. Fu, H. Wang, F. Liu, H. Wang and L. Han, Bagging-based machine learning algorithms for landslide susceptibility modeling, Nat. Hazards, 2022, 110(2), 823–846 CrossRef.
- S. Aluvala, J. N. Kalshetty, S. O. Husain, J. Kaur and H. K. Thind, An Evolution of Hybrid Bagging Technique in Chemoinformatic and Drug Discovery, in 2024 Second International Conference on Data Science and Information System (ICDSIS), IEEE, 2024, pp. 1–4 Search PubMed.
- Y. Hao, B. Li, D. Huang, S. Wu, T. Wang and L. Fu, et al., Developing a Semi-Supervised Approach Using a PU-Learning-Based Data Augmentation Strategy for Multitarget Drug Discovery, Int. J. Mol. Sci., 2024, 25(15), 8239 CrossRef CAS.
- M. Sassi, J. Bronsard, G. Pascreau, M. Emily, P. Y. Donnio and M. Revest, et al., Forecasting Staphylococcus aureus infections using genome-wide association studies, machine learning, and transcriptomic approaches, Msystems, 2022, 7(4), e00378 CrossRef PubMed.
- Tr Wang, Jc Li, W. Shu, S. Hu, R. Ouyang and W. Li, Machine-learning adsorption on binary alloy surfaces for catalyst screening, Chin. J. Chem. Phys., 2020, 33(6), 703–711 CrossRef CAS.
- V. K. Gupta, Toxicity detection of small drug molecules of the mitochondrial membrane potential signalling pathway using bagging-based ensemble learning, Int. J. Data Min. Bioinform., 2022, 27(1–3), 201–220 CrossRef.
- Y. Gong, B. Liao, P. Wang and Q. Zou, DrugHybrid_BS: Using hybrid feature combined with bagging-SVM to predict potentially druggable proteins, Front. Pharmacol., 2021, 12, 771808 CrossRef CAS PubMed.
- A. N. Tarekegn, M. Giacobini and K. Michalak, A review of methods for imbalanced multi-label classification, Pattern Recognit., 2021, 118, 107965 CrossRef.
- J. M. Hazan, R. Amador, T. Ali-Nasser, T. Lahav, S. R. Shotan and M. Steinberg, et al., Integration of transcription regulation and functional genomic data reveals lncRNA SNHG6's role in hematopoietic differentiation and leukemia, J. Biomed. Sci., 2024, 31(1), 27 CrossRef CAS.
- N. Aleb, Cost-Sensitive Deep Learning Models for Drug-Target Interaction Prediction, Int. J. Adv. Soft Comput. Appl., 2021, 13(2), 25 Search PubMed.
- M. Ali and T. Aittokallio, Machine learning and feature selection for drug response prediction in precision oncology applications, Biophys. Rev., 2019, 11(1), 31–39 CrossRef CAS.
- F. J. Veredas, D. Urda, J. L. Subirats, F. R. Cantón and J. C. Aledo, Combining feature engineering and feature selection to improve the prediction of methionine oxidation sites in proteins, Neural Comput. Appl., 2020, 32(2), 323–334 CrossRef.
- S. Gupta, J. Baudry and V. Menon, Using big data analytics to “back engineer” protein conformational selection mechanisms, Molecules, 2022, 27(8), 2509 CrossRef CAS PubMed.
- S. Liu, C. Cui, H. Chen and T. Liu, Ensemble learning-based feature selection for phage protein prediction, Front. Microbiol., 2022, 13, 932661 CrossRef PubMed.
- Q. Yang, B. Li, J. Tang, X. Cui, Y. Wang and X. Li, et al., Consistent gene signature of schizophrenia identified by a novel feature selection strategy from comprehensive sets of transcriptomic data, Briefings Bioinf., 2020, 21(3), 1058–1068 CrossRef CAS PubMed.
- H. Abbasi Mesrabadi, K. Faez and J. Pirgazi, Drug–target interaction prediction based on protein features, using wrapper feature selection, Sci. Rep., 2023, 13(1), 3594 CrossRef CAS.
- R. Zebari, A. Abdulazeez, D. Zeebaree, D. Zebari and J. Saeed, A comprehensive review of dimensionality reduction techniques for feature selection and feature extraction, J. Appl. Sci. Technol. Trends, 2020, 1(1), 56–70 CrossRef.
- N. Q. K. Le, W. Li and Y. Cao, Sequence-based prediction model of protein crystallization propensity using machine learning and two-level feature selection, Briefings Bioinf., 2023, 24(5), bbad319 CrossRef.
- J. Benavides-Hernández and F. Dumeignil, From Characterization to Discovery: Artificial Intelligence, Machine Learning and High-Throughput Experiments for Heterogeneous Catalyst Design, ACS Catal., 2024, 14(15), 11749–11779 CrossRef.
- T. T. Shi, G. Y. Liu and Z. X. Chen, Machine Learning Prediction of CO Adsorption Energies and Properties of Layered Alloys Using an Improved Feature Selection Algorithm, J. Phys. Chem. C, 2023, 127(20), 9573–9583 CrossRef CAS.
- T. Ma, J. Wang, L. Ban, H. He, Z. Lu and J. Zhu, et al., A machine-learning-based composition design of ternary Cu-based Rochow-Müller catalyst with high M2 selectivity, Appl. Catal., A, 2024, 675, 119592 CrossRef CAS.
- D. Zhao, J. Wang, S. Sang, H. Lin, J. Wen and C. Yang, Relation path feature embedding based convolutional neural network method for drug discovery, BMC Med. Inf. Decis. Making, 2019, 19, 121–130 CrossRef.
- Y. Chu, A. C. Kaushik, X. Wang, W. Wang, Y. Zhang and X. Shan, et al., DTI-CDF: a cascade deep forest model towards the prediction of drug-target interactions based on hybrid features, Briefings Bioinf., 2021, 22(1), 451–462 CrossRef PubMed.
- X. Qiang, C. Zhou, X. Ye, P. Du, R. Su and L. Wei, CPPred-FL: a sequence-based predictor for large-scale identification of cell-penetrating peptides by feature representation learning, Briefings Bioinf., 2020, 21(1), 11–23 Search PubMed.
- V. García, R. A. Mollineda and J. S. Sánchez, Index of balanced accuracy: A performance measure for skewed class distributions, in Iberian conference on pattern recognition and image analysis, Springer, 2009, pp. 441–448 Search PubMed.
- D. Chicco and G. Jurman, The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation, BMC Genomics, 2020, 21, 1–13 CrossRef PubMed.
- G. Naidu, T. Zuva and E. M. Sibanda, A review of evaluation metrics in machine learning algorithms, in Computer Science On-line Conference, Springer, 2023, pp. 15–25 Search PubMed.
- D. Chicco and G. Jurman, The Matthews correlation coefficient (MCC) should replace the ROC AUC as the standard metric for assessing binary classification, BioData Min., 2023, 16(1), 4 CrossRef PubMed.
- A. Talukder, Multimodal Data Fusion and Machine Learning for Deciphering Protein-Protein Interactions, Texas A&M University, 2021 Search PubMed.
- A. Huang, X. Xie, X. Wang and S. Peng, A multimodal data fusion-based deep learning approach for drug-drug interaction prediction, in International Symposium on Bioinformatics Research and Applications, Springer, 2022, pp. 275–285 Search PubMed.
- P. Chourasia, Z. Tayebi, S. Ali and M. Patterson, Empowering pandemic response with federated learning for protein sequence data analysis, in 2023 International Joint Conference on Neural Networks (IJCNN), IEEE, 2023, pp. 01–08 Search PubMed.
- S. Chen, D. Xue, G. Chuai, Q. Yang and Q. Liu, FL-QSAR: a federated learning-based QSAR prototype for collaborative drug discovery, Bioinformatics, 2020, 36(22–23), 5492–5498 CAS.
- A. Xie, Z. Zhang, J. Guan and S. Zhou, Self-supervised learning with chemistry-aware fragmentation for effective molecular property prediction, Briefings Bioinf., 2023, 24(5), bbad296 CrossRef PubMed.
- R. Shukla and T. Tripathi, Molecular dynamics simulation of protein and protein–ligand complexes, Comput.-Aided Drug Des., 2020, 133–161 CrossRef PubMed.
- B. Guan, H. Jiang, Y. Wei, Z. Liu, X. Wu and H. Lin, et al., Density functional theory researches for atomic structure, properties prediction, and rational design of selective catalytic reduction catalysts: Current progresses and future perspectives, Mol. Catal., 2021, 510, 111704 CrossRef CAS.
- L. Gundelach, T. Fox, C. S. Tautermann and C. K. Skylaris, Protein–ligand free energies of binding from full-protein DFT calculations: convergence and choice of exchange–correlation functional, Phys. Chem. Chem. Phys., 2021, 23(15), 9381–9393 RSC.
- J. Liu, Z. Wang, L. Kou and Y. Gu, Mechanism exploration and catalyst design for hydrogen evolution reaction accelerated by density functional theory simulations, ACS Sustain. Chem. Eng., 2023, 11(2), 467–481 CrossRef CAS.
- C. J. Morris and D. D. Corte, Using molecular docking and molecular dynamics to investigate protein-ligand interactions, Mod. Phys. Lett. B, 2021, 35(08), 2130002 CrossRef CAS.
- S. Sarker, L. Qian and X. Dong, Medical data augmentation via chatgpt: A case study on medication identification and medication event classification, arXiv, 2023, preprint, arXiv:230607297, DOI:10.48550/arXiv.2306.07297.
- R. Irwin, S. Dimitriadis, J. He and E. J. Bjerrum, Chemformer: a pre-trained transformer for computational chemistry, Mach. Learn.: Sci. Technol., 2022, 3(1), 015022 Search PubMed.
- M. Moret, I. Pachon Angona, L. Cotos, S. Yan, K. Atz and C. Brunner, et al., Leveraging molecular structure and bioactivity with chemical language models for de novo drug design, Nat. Commun., 2023, 14(1), 114 CrossRef CAS PubMed.
- D. D. Nguyen, K. Gao, M. Wang and G. W. Wei, MathDL: mathematical deep learning for D3R Grand Challenge 4, J. Comput.-Aided Mol. Des., 2020, 34, 131–147 CrossRef CAS PubMed.
- D. D. Nguyen, Z. Cang, K. Wu, M. Wang, Y. Cao and G. W. Wei, Mathematical deep learning for pose and binding affinity prediction and ranking in D3R Grand Challenges, J. Comput.-Aided Mol. Des., 2019, 33, 71–82 CrossRef CAS PubMed.
- J. Chen, R. Wang, M. Wang and G. W. Wei, Mutations strengthened SARS-CoV-2 infectivity, J. Mol. Biol., 2020, 432(19), 5212–5226 CrossRef CAS PubMed.
- J. Chen and G. W. Wei, Omicron BA. 2 (B. 1.1. 529.2): high potential for becoming the next dominant variant, J. Phys. Chem. Lett., 2022, 13(17), 3840–3849 CrossRef CAS PubMed.
- J. Chen, Y. Qiu, R. Wang and G. W. Wei, Persistent Laplacian projected Omicron BA. 4 and BA. 5 to become new dominating variants, Comput. Biol. Med., 2022, 151, 106262 CrossRef CAS PubMed.
- Y. Qiu and G. W. Wei, Persistent spectral theory-guided protein engineering, Nat. Comput. Sci., 2023, 3(2), 149–163 CrossRef CAS PubMed.
- M. Wang, Z. Cang and G. W. Wei, A topology-based network tree for the prediction of protein–protein binding affinity changes following mutation, Nat. Mach. Intell., 2020, 2(2), 116–123 CrossRef PubMed.
- K. Wu, Z. Zhao, R. Wang and G. W. Wei, TopP–S: Persistent homology-based multi-task deep neural networks for simultaneous predictions of partition coefficient and aqueous solubility, J. Comput. Chem., 2018, 39(20), 1444–1454 CrossRef CAS PubMed.
- Z. Zhu, B. Dou, Y. Cao, J. Jiang, Y. Zhu and D. Chen, et al., Tidal: topology-inferred drug addiction learning, J. Chem. Inf. Model., 2023, 63(5), 1472–1489 CrossRef CAS PubMed.
- Y. Zhang, C. Shen and K. Xia, Multi-Cover Persistence (MCP)-based machine learning for polymer property prediction, Briefings Bioinf., 2024, 25(6), bbae465 CrossRef PubMed.
- D. Chen, J. Liu and G. W. Wei, Multiscale topology-enabled structure-to-sequence transformer for protein–ligand interaction predictions, Nat. Mach. Intell., 2024, 6(7), 799–810 CrossRef.
- L. Shen, H. Feng, F. Li, F. Lei, J. Wu and G. W. Wei, Knot data analysis using multiscale Gauss link integral, Proc. Natl. Acad. Sci. U. S. A., 2024, 121(42), e2408431121 CrossRef CAS PubMed.
- J. Cai, X. Chu, K. Xu, H. Li and J. Wei, Machine learning-driven new material discovery, Nanoscale Adv., 2020, 2(8), 3115–3130 RSC.
- E. Ren, P. Guilbaud and F. X. Coudert, High-throughput computational screening of nanoporous materials in targeted applications, Digital Discovery, 2022, 1(4), 355–374 RSC.
- B. Dou, Z. Zhu, E. Merkurjev, L. Ke, L. Chen and J. Jiang, et al., Machine learning methods for small data challenges in molecular science, Chem. Rev., 2023, 123(13), 8736–8780 CrossRef CAS PubMed.
- S. Tsukiyama, M. M. Hasan, S. Fujii and H. Kurata, LSTM-PHV: prediction of human-virus protein–protein interactions by LSTM with word2vec, Briefings Bioinf., 2021, 22(6), bbab228 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |