
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal–silicon triple bonds: reactivity of the silylidyne complexes [Cp*(CO)2M![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) Si–Tbb] (M = Cr – W)†
Si–Tbb] (M = Cr – W)†
Kanishk
Tomer
 ,
Gregor
Schnakenburg
,
Ujjal
Das
and
Alexander C.
Filippou
*
,
Gregor
Schnakenburg
,
Ujjal
Das
and
Alexander C.
Filippou
*
Institut für Anorganische Chemie, Universität Bonn, Gerhard-Domagk-Str. 1, Bonn, 53121, Germany. E-mail: filippou@uni-bonn.de
First published on 24th March 2025
Abstract
The different reactivity pattern of M![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si and MC bonds (M = transition metal) is illustrated by a series of reactions of the silylidyne complexes [Cp*(CO)2MSi–Tbb] (1-M) (M = Cr – W; Cp* = η5-pentamethylcyclopentadienyl; Tbb = 4-tert-butyl-2,6-bis(bis(trimethylsilyl)methyl)phenyl)). Complexes 1-M were obtained selectively from Li[Cp*M(CO)3] and the 1,2-dibromodisilene (E)-Tbb(Br)Si
Si and MC bonds (M = transition metal) is illustrated by a series of reactions of the silylidyne complexes [Cp*(CO)2MSi–Tbb] (1-M) (M = Cr – W; Cp* = η5-pentamethylcyclopentadienyl; Tbb = 4-tert-butyl-2,6-bis(bis(trimethylsilyl)methyl)phenyl)). Complexes 1-M were obtained selectively from Li[Cp*M(CO)3] and the 1,2-dibromodisilene (E)-Tbb(Br)Si![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Si(Br)Tbb. The reaction of 1-Mo and 1-W with two equivalents of mesityl isocyanate leads selectively to complex 2-Mo and 2-W, respectively, featuring a novel κ2O,O-imidocarbonatosilyl ligand. Ring opening of ethyloxirane occurs rapidly with 1-Mo and leads to the hydrido–enolatosilylidene complex 3-Mo illustrating the Si-centered electrophilicity of the silylidyne complex. Trimethylsilyldiazomethane induces a cleavage of the MoSi bond of 1-Mo after a rapid double [2 + 1] cycloaddition of the terminal N-atom, resulting in the first silaamidinato complex 4-Mo. In comparison, the reaction of 1-Mo with mesityl azide gives, after N2 elimination, the Mo–silaiminoacyl complex 5-Mo. All compounds were fully characterized and the isomerism and dynamics of 3-Mo in solution were analysed by a combination of spectroscopic and quantum-chemical studies.
Si(Br)Tbb. The reaction of 1-Mo and 1-W with two equivalents of mesityl isocyanate leads selectively to complex 2-Mo and 2-W, respectively, featuring a novel κ2O,O-imidocarbonatosilyl ligand. Ring opening of ethyloxirane occurs rapidly with 1-Mo and leads to the hydrido–enolatosilylidene complex 3-Mo illustrating the Si-centered electrophilicity of the silylidyne complex. Trimethylsilyldiazomethane induces a cleavage of the MoSi bond of 1-Mo after a rapid double [2 + 1] cycloaddition of the terminal N-atom, resulting in the first silaamidinato complex 4-Mo. In comparison, the reaction of 1-Mo with mesityl azide gives, after N2 elimination, the Mo–silaiminoacyl complex 5-Mo. All compounds were fully characterized and the isomerism and dynamics of 3-Mo in solution were analysed by a combination of spectroscopic and quantum-chemical studies.
1. Introduction
Transition metal carbyne complexes are an important class of organometallic compounds displaying a diverse reactivity highlighted in many review articles,1–9 and a monography.10 Numerous stoichiometric transformations of carbyne complexes have been accomplished since the landmark discovery of the first examples by E. O. Fischer et al. in 1973 (Fig. 1, A),11 the most prominent one involving the metathetical exchange of triple bonds, which culminated in the design of well-defined catalysts for the metathesis of alkynes,7–9 with applications in total synthesis, polymer chemistry and supramolecular chemistry.12–15 In comparison to carbyne complexes, isolation of group 14 heavier element analogs of the general formula LnME–R (M = transition metal, E = Si – Pb) is far more challenging given the reluctance of the Si – Pb to form one and even more so two π-bonds at the expense of σ-bonds for thermodynamic reasons. It requires fine stereoelectronic tuning of the metal fragment LnM and kinetic stabilization of the ME functionality by a bulky substituent R to prevent uncontrollable follow-up reactions of the targeted compounds, such as head-to-tail cyclooligomerizations or unselective σ-bond activations.16–18 Therefore it is not surprising that it took more than 20 years after E. O. Fischer's publication for the first germylidyne complex to be isolated by P. Power et al. taking advantage of the specific steric protection provided by a m-terphenyl substituent (Fig. 1, C).19–21 Following this report, numerous group 6 germylidyne complexes22–26 and first complexes featuring metal–tin27–29 and metal–lead triple bonds30–32 (Fig. 1, D) were obtained in our group using a N2 or PMe3 elimination method that proved to be significant for the development of this field.18,33–35 Isolation of compounds featuring a metal–silicon triple bond was an uphill task due to the anomalous low electronegativity of silicon36 and the lack of suitable sources for the SiR (silylidyne) fragment.17
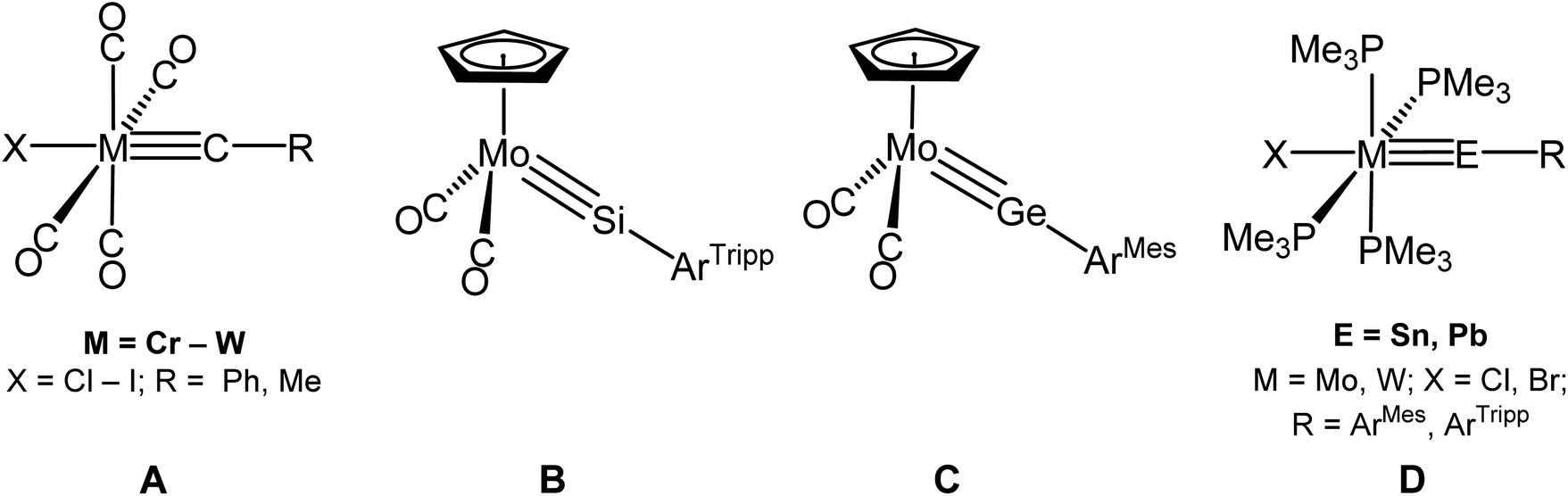 | ||
| Fig. 1 First reported complexes featuring metal–tetrel triple bonds; ArTripp = C6H3-2,6-Tripp2, Tripp = 2,4,6-triisopropylphenyl; ArMes = C6H3-2,6-Mes2, Mes = 2,4,6-trimethylphenyl. | ||
Pioneering work of T. D. Tilley et al. on transition metal silyl and silylene complexes led to complexes containing base-stabilized silylidyne ligands.37,38 The breakthrough came with the isolation of NHC-stabilized silicon(II) halides39–43 and 1,2-dihalodisilenes44–47 which were used in our group as selective electrophiles in reactions with electron-rich metal complexes to access stepwise or directly first neutral and cationic group 6 silylidyne complexes48–52 (Fig. 1, B; Fig. 2, G and H) and then to transfer this approach to other transition metals16,18,53–56 expanding considerably the scope of this chemistry (Fig. 2, E, F, I and J).
 | ||
| Fig. 2 Silylidyne complexes of various transition metal groups isolated and fully characterized in our group; SIDipp = C[N(Dipp)CH2]2, Dipp = 2,6-diisopropylphenyl; Tbb = C6H2-2,6-(CH(SiMe3)2)2-4-tBu; Eind = 1,1,3,3,5,5,7,7-Et8-s-hydrindacene-4-yl. | ||
Another method to form metal-silicon triple bonds was developed by T. D. Tilley and H. Tobita and H. Hashimoto et al. involving a stepwise metal-centered dehydrogenation of trihydridosilanes, which led to a first group 8 silylidyne complex, [Cp*(PiPr3)(H)OsSi–Tripp]+,57 and the group 6 metal silylidyne complexes [Cp*(CO)2MSi–R] (M = Cr – W; R = C(SiMe3)3, Eind), respectively.58–61
Silylidyne complexes have a similar electronic structure as Fischer-type carbyne complexes according to quantum chemical analyses.18,62,63 However, the charge distribution along the ME bond differs significantly from that of Fischer carbyne complexes with the silicon atom carrying a high positive partial charge, whereas the carbyne–carbon is nearly electroneutral or carries a slight negative partial charge.18 Also the MSi triple bonds are more polar than the MC bonds.64 The increased polarity of the MSi triple bonds (M(δ−)–Si(δ+)) and the high silicon-centered electrophilicity provide a rationale for the high and distinct reactivity of the silylidyne complexes compared to that of the isovalent carbyne complexes. This is illustrated in Fig. 3 using a series of reactions studied in our group shortly after the synthesis of the silylidyne complex B in 2010.49,51,65,66 For example, fast single and double additions of anionic nucleophiles at the electrophilic Si atom gave anionic silylidene complexes K and dianionic silyl complexes L respectively,66 and regioselective 1,2-addition reactions of B with H2O, NH3 and HCl yielded the silylidene–hydrido complexes M featuring a M–H⋯Si bonding interaction (Fig. 3).49 In comparison, reactions of the isovalent carbyne complexes [(η5-C5R15)(CO)2MC–R2] (M = Mo, W; R1 = H, Me; R2 = Me, Ph, NEt2) with hydrogen halides show the opposed regioselectivity67 leading to secondary carbene or η2-acyl complexes, after single or double protonation of the carbyne carbon, respectively,68–70 and reactions of Fischer type carbyne complexes with H2O or NH3 have not been reported so far.
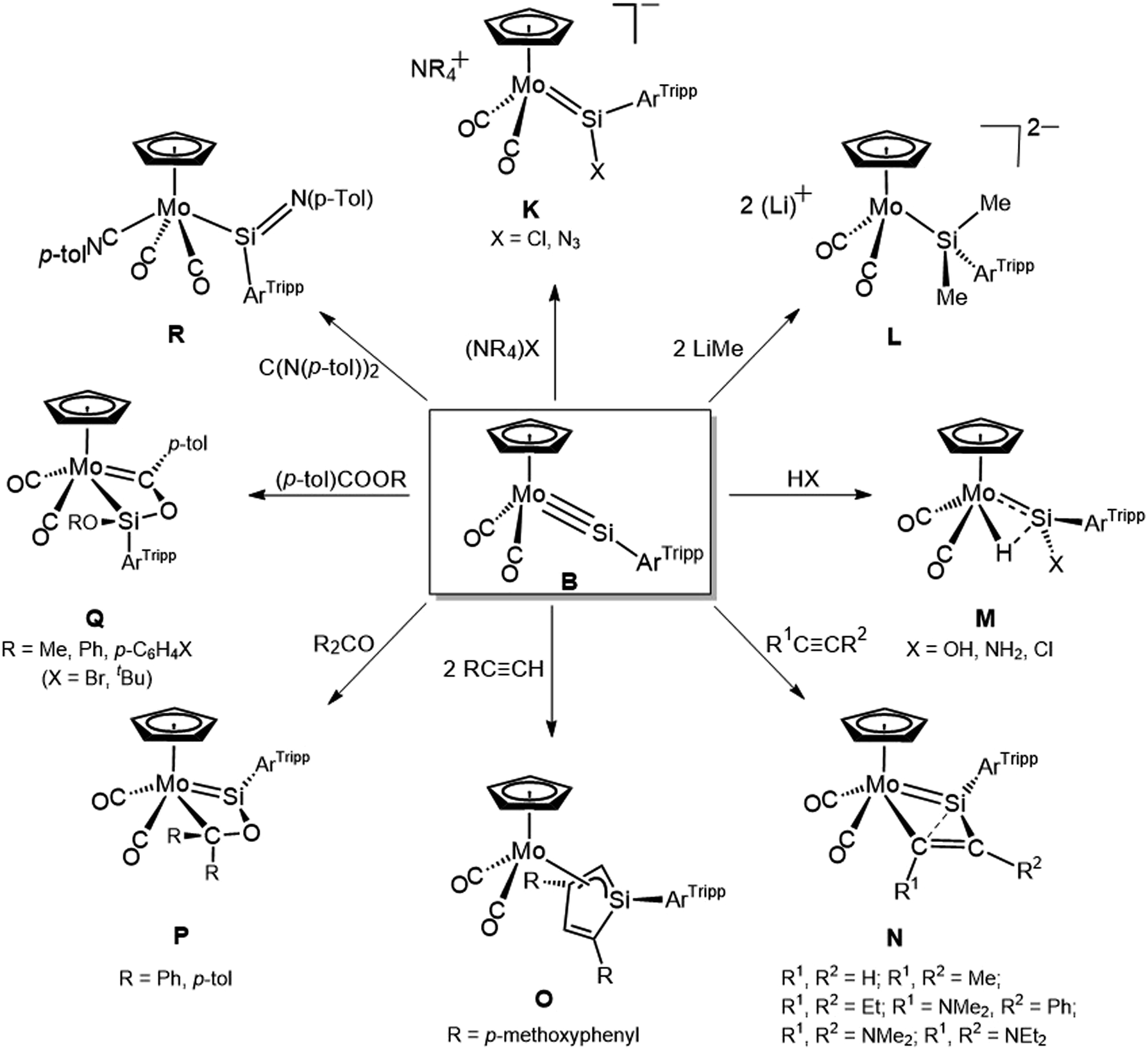 | ||
| Fig. 3 Various reactions of the silylidyne complex B studied from 2010 to 2012 in our group. | ||
Remarkably, complex B rapidly undergoes [2 + 2] cycloadditions with various alkynes affording selectively the 1,2-metallasilacyclobutadienes N, which can further react with alkynes to give the η3-Si,C,C-silacyclopentadienyl complexes O (Fig. 3). In comparison, reactions of the isovalent carbyne complexes [(η5-C5R15)(CO)2MC–R2] (M = Mo, W; R1 = H, Me; R2 = Me, Ph, NEt2) with alkynes are not known, and in general the outcome of reactions of Fischer-type carbyne complexes with alkynes is unpredictable leading to different products such as alkyne polymers,71 phenols,72 alkyne(carbyne) complexes73 or carbyne–alkyne–carbonyl coupling products.74,75
Complex B also reacts rapidly with carbonyl compounds. The [2 + 2] cycloadditions products P are formed regioselectively with ketones, whereas with esters the metalacyclic carbene complexes Q are obtained presumably via [2 + 2] cycloaddition followed by migration of the OR substituent from the carbonyl–carbon to the silicon atom (Fig. 3). In comparison, no reactions of Fischer-type carbyne complexes with carbonyl compounds have been reported so far, and Schrock-type carbyne complexes, which contain a nucleophilic carbyne–carbon and an electrophilic metal center, undergo Wittig-type reactions with aldehydes, ketones and esters yielding oxo–vinyl complexes.76 It is noteworthy, that the latter reactions exhibit the opposite regioselectivity to those of B and lead to Ccarbyne–Ccarbonyl coupling products.
Finally, reaction of silylidyne complex B with the carbodiimide (p-Tol)NCN(p-Tol) was found to give the iminosilyl (silaiminoacyl) complex R after [2 + 2] cycloaddition followed by cycloreversion (Fig. 3). In this case, the behaviour of B also differs from that of carbyne complexes77 due to the metal(δ−)−silicon(δ+) bond polarity.
Interesting reactivity patterns as presented above for B have been encovered later for the osmium complex [Cp*(PiPr3)(H)OsSi–Tripp]+ by T. D. Tilley et al.57 and the group 6 metal silylidyne complexes [Cp*(CO)2MSi–R] by H. Tobita and H. Hashimoto et al.,58–60,78 and recently even the photochemical H–H and benzene C–H bond activation by [Cp*(CO)2CrSi–Eind] was reported confirming the high reactivity of silylidyne complexes.61
In the present work, the different reactivity pattern of MSi and MC bonds is exemplified by a series of reactions of the silylidyne complexes [Cp*(CO)2MSi–Tbb] (1-M: M = Cr – W) with isocyanates, oxiranes, diazoalkanes and organic azides leading to complexes with novel Si-based ligands.
2. Results and discussion
2.1 Synthesis and characterization of the silylidyne complexes 1-M (1-M: M = Cr – W)
Complex 1-Mo was obtained upon heating of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mixture of the 1,2-dibromodisilene (E)-Tbb(Br)SiSi(Br)Tbb and Li[Cp*Mo(CO)3] in toluene at 110 °C for 4 hours. Monitoring of the reaction by 1H NMR and IR spectroscopy revealed the selective formation of the silylidyne complex 1-Mo, which after workup was isolated in 68% yield as an air-sensitive, orange-red, thermally stable solid melting at 174 °C (eqn (1)).
:2 mixture of the 1,2-dibromodisilene (E)-Tbb(Br)SiSi(Br)Tbb and Li[Cp*Mo(CO)3] in toluene at 110 °C for 4 hours. Monitoring of the reaction by 1H NMR and IR spectroscopy revealed the selective formation of the silylidyne complex 1-Mo, which after workup was isolated in 68% yield as an air-sensitive, orange-red, thermally stable solid melting at 174 °C (eqn (1)). | (1) |
Heating of (E)-Tbb(Br)SiSi(Br)Tbb with the metallate Li[Cp*Mo(CO)2(PMe3)] in toluene at 110 °C also yielded 1-Mo after PMe3 elimination. In this case, conversion is faster and is completed in 30 min, but is less selective leading according to IR and NMR spectroscopy to a 10:1 mixture of 1-Mo and the silylidyne complex [Cp*(CO)(PMe3)MoSi–Tbb] (1-Mo-PMe3) (eqn (2)). The two products were separated after several crystallizations from n-pentane and 1-Mo isolated in pure form, albeit in low yield (23%).
 | (2) |
For comparison reasons, the chromium and tungsten silylidyne complexes [Cp*(CO)2MSi–Tbb] (1-Cr (M = Cr); 1-W (M = W)) were also obtained selectively from (E)-Tbb(Br)SiSi(Br)Tbb and Li[Cp*M(CO)3] in toluene at 110 °C (eqn (1)). 1-Cr and 1-W were isolated as very air-sensitive, dark-brown and orange-red solids in 66% and 56% yields, respectively (eqn (1)). Like 1-Mo, both compounds are thermally stable solids and melt at 168 and 182 °C, respectively. Complexes 1-M were fully characterized and their molecular structures determined by single-crystal X-ray diffraction (Fig. 4).
 | ||
| Fig. 4 DIAMOND plot of the molecular structure of 1-Mo. Thermal ellipsoids are set at 50% probability, hydrogen atoms are omitted and the substituents of the Tbb group are presented as wireframe for the sake of clarity. Selected bond lengths (pm) and bond angles (°) for 1-Mo (bond lengths and bond angles of 1-W are given in square brackets): Mo–Si1 223.2(1) [224.55(6)], Mo–C35 197.0(4) [195.7(3)], Mo–C36 196.2(4) [196.7(3)], Si1–C1 185.6(3) [185.1(2)], Mo–Si1–C1 174.3(1) [174.88(8)], Si1–Mo–C35 89.1(1) [89.42(7)], Si1–Mo–C36 86.8(1) [86.97(7)], C35–Mo–C36 87.9(2) [88.5(1)], Si1–Mo–Cg 135.86(7) [135.86(2)], C35–Mo–Cg 122.2(1) [120.74(7)], C36–Mo–Cg 121.2(1) [120.98(7)]; Cg is the Cp* ring centroid. | ||
Selected bonding parameters and spectroscopic data of 1-M and related group 6 metal silylidyne complexes previously characterized in our group,49,51,52,79,80 are summarized in Table 1 for comparison with those of analogous carbyne complexes.81–87
| Silylidyne complexes | ||||||
|---|---|---|---|---|---|---|
| a A− = B(C6H3-3,5-(CF3)2)4−. b In toluene. c In PhF. d In (D6)benzene. e In (D5)chlorobenzene. f Bonding parameters of the two independent molecules found in the crystal lattice. g In CH2Cl2. h In n-hexane. i In n-pentane. j In thf. k In CD2Cl2 at −40 °C. l In CD2Cl2 at −50 °C. m In CD2Cl2 at −20 °C. n In CD2Cl2 at −30 °C. o In CD2Cl2/CH2Cl2 at r.t. p In CDCl3 at r.t. q In CD2Cl2 at r.t. | ||||||
| Entry | Complex | ν(CO) (cm−1)b,c | δ(29Si) (ppm)d,e |
d(MSi) (pm) |
∠(MSi–C) (°) |
Ref. |
| 1 | [Cp*(CO)2CrSi–Tbb] (1-Cr) |
1906, 1847b | 299.9d | 210.3(2) | 170.4(2) | This work |
| 2 | [Cp*(CO)2MoSi–Tbb] (1-Mo) |
1916, 1854b | 308.9d | 223.2(1) | 174.3(1) | This work |
| 3 | [Cp*(CO)2WSi–Tbb] (1-W) |
1911, 1848b | 314.0d (1J(183W,29Si) = 316 Hz) | 224.55(6) | 174.88(8) | This work |
| 4 | [Cp*(CO)2CrSi–SIDipp]+A−a |
1966, 1912c | 127.8e | 212.20(9) | 169.76(9) | 49 and 51 |
| 5 | [Cp*(CO)2MoSi–SIDipp]+A−a |
1973, 1915c | 148.3e | 222.12(9) | 174.2(1) | 49 and 51 |
| 6 | [Cp*(CO)2WSi–SIDipp]+A−a |
1968, 1906c | 178.2e (1J(183W,29Si) = 420 Hz) | 222.4(3) | 172.1(3) | 51 |
| 7 | [Tp′(CO)2MoSi–Tbb] |
1912, 1836b | 258.5d | 226.14(9)f | 160.8(1)f | 52 |
| 225.43(9) | 158.5(1) | |||||
| 8 | [Tp′(CO)2WSi–Tbb] |
1901, 1823b | 259.8d (1J(183W,29Si) = 272 Hz) | 227.06(8)f | 161.7(1)f | 79 and 80 |
| 226.37(8) | 159.5(1) | |||||
|
||||||
| Carbyne complexes | ||||||
| Entry | Complex | ν(CO) (cm−1)g,h,i,j |
δ(MC) (ppm)k,l,m,n,o,p |
d(MC) (pm) |
∠(MC–C) (°) |
Ref. |
| 9 | [Cp(CO)2CrC–Ph] |
1991, 1922g | 325.7k | 170.5(2) | 175.5(2) | 81 |
| 10 | [Cp(CO)2MoC–Ph] |
1996, 1922g | 309.4l | — | — | 82 |
| 11 | [Cp(CO)2WC–Ph] |
1984, 1905g | 299.3m | — | — | 83 |
| 12 | [Cp(CO)2WC–(p-tol)] |
1990, 1919h | 300.1n | 182(2) | 176(2) | 84 |
| 13 | [Cp*(CO)2CrC–Ph] |
1984, 1921i | 323.7k | — | — | 81 |
| 14 | [Cp*(CO)2WC–(p-tol)] |
1981, 1910h | 301.3o | — | — | 85 |
| 15 | [Tp′(CO)2MoC–(p-tol)] |
1982, 1899j | 288.9p | 180.4(4) | 163.1(3) | 86 |
| 16 | [Tp′(CO)2WC–(p-tol)] |
1974, 1888h | 279.6q | 182.9(3) | 163.2(3) | 87 |
The three-legged piano–stool complexes 1-M are essentially Cs symmetric with the symmetry plane bisecting the Cp*M(CO)2 fragment and passing through the aryl plane of the Tbb group, as evidenced by the twist angle θ between the aryl plane of the Tbb substituent and the plane defined by the atoms Si, Mo and Cg (θ = 1.7(1)° (1-Mo), 1.91(7)° (1-W); Cg is the Cp* ring centroid) (Fig. 4).
All complexes feature very short metal–silicon bonds and almost linearly coordinated silicon centers with M–Si–CTbb bond angles of 170.4–174.9° (Fig. 4 and Table 1). In fact, the Cr–Si distance of 1-Cr (210.3(2) pm) is the shortest reported so far for a Cr–Si bond,42 and is slightly shorter than those of the neutral silylidyne complexes [Cp*(CO)2CrSi–Eind] (211.51(4) pm) and [Cp*(CO)(PMe3)CrSi–Eind] (212.0(1) pm)61 or the cationic silylidyne complex [Cp*(CO)2CrSi–SIDipp]B(ArF)4 (d(CrSi) = 212.20(9) pm; SIdipp = C[N(Dipp)CH2]2 (Dipp = 2,6-diisopropylphenyl), ArF = C6H3-3,5-(CF3)2).50 The MSi bond lengths of 1-Mo (223.2(1) pm) and 1-W (224.55(6) pm) are slightly longer than those of the neutral silylidyne complexes [Cp(CO)2MoSi–ArTripp] (222.41(7) pm)48 and [Cp*(CO)2WSi–C(SiMe3)3] (222.97(9) pm) respectively,58 and also slightly longer than those of the cationic silylidyne complexes [Cp*(CO)2MSi–SIDipp]B(ArF)4 (d(MoSi) = 222.12(9) pm; d(WSi) = 222.4(3) pm), but shorter than those of the Tp′ analogs (Table 1).49,51 A further comparison shows that the CrSi bond lengths are 10–14 pm shorter than the MSi bonds (M = Mo, W), the value comparing well with the difference of the triple-bond covalent radii r3 of the elements (r3(Cr) = 103 pm, r3(Mo) = 113 pm; r3(W) = 115 pm), reported by P. Pyykkö.88 Notably, the CrSi bond length of 1-Cr (210.7 pm) and the MoSi bond length of 1-Mo (223.2 pm) are considerably shorter than the sum of the triple bond radii rCr + rSi (219.95 pm) and rMo + rSi (229.45 pm), which are derived from the experimental MM bond lengths of Cp*2M2(CO)4 (d(CrCr) = 228.0(2) pm;89d(MoMo) = 248.8(3) pm90) and the SiSi bond length of Si2Tbb2 (d(SiSi) = 210.1(1) pm)91 using the equations rM = d(MM)/2 and rSi = d(SiSi)/2, respectively. This bond shortening can be explained by the increased polarity and strength of the MSi bonds of 1-M (M = Cr, Mo).
The IR spectra of complexes 1-Cr – 1-W display two intense absorption bands, which are typical for cis-dicarbonyl complexes and are assigned to the in-phase (A′ symmetric) and out-of-phase (A′′ symmetric) CO stretching modes of the Cs-symmetric complexes (Fig. 5). The ν(CO) absorption bands of the silylidyne complexes appear at considerably lower wavenumbers than those of the isovalent carbyne complexes (cf. entry 1 with 13, 3 with 14 or 7 with 15 in Table 1) indicating the much higher σ-donor/π-acceptor ratio of the silylidyne ligand SiR (R = Tbb) than that of the carbyne ligand CR (R = Ph, p-Tol). It should also be noted, that the ν(CO) absorption bands of the silylidyne complexes 1-Mo and 1-W appear even at lower wavenumbers than those of the aminocarbyne complexes Cp*(CO)2MCNEt2 (M = Mo, W).92,93 In general, the ν(CO) bands of the Cp* containing silylidyne complexes appear at higher wavenumbers than those of the Tp′analogues (cf. entry 2 with 7 or 3 with 8 in Table 1) and the ν(CO) bands of the molybdenum silylidyne complexes at higher wavenumbers than those of their tungsten congeners (cf. entry 2 with 3 or 7 with 8 in Table 1). Notably, the same trends are observed in the isovalent carbyne complexes (cf. entry 14 with 16, 10 with 11 or 15 with 16 in Table 1) illustrating the stronger electron-donating ability of the Tp′ than the Cp* ligand in these systems94 and the stronger W–CO than Mo–CO π-back bonding.95
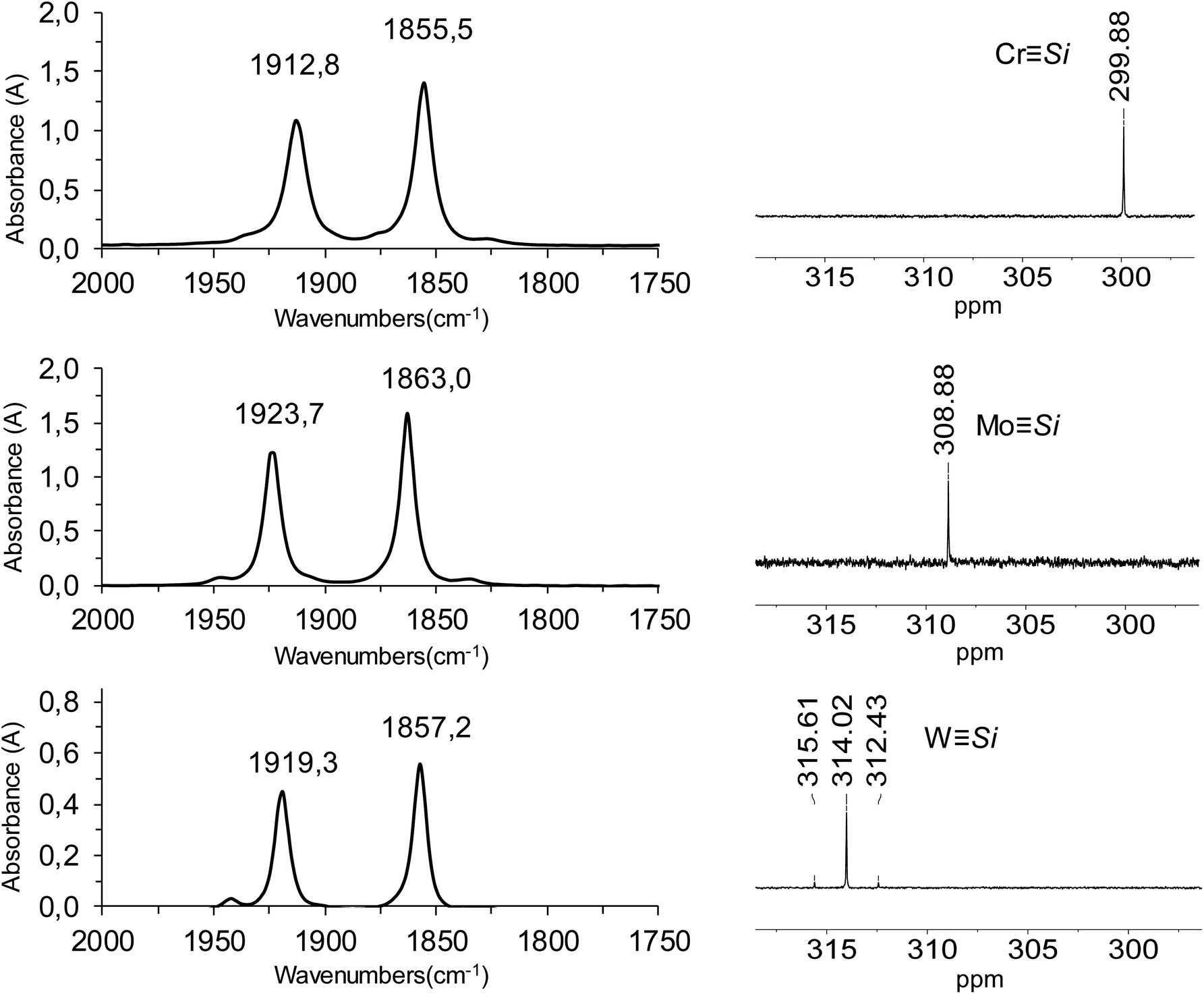 | ||
| Fig. 5 (left) FT-IR spectra of the silylidyne complexes 1-Cr – 1-W in n-hexane in the spectral range 2000–1750 cm−1 showing the two ν(CO) absorption bands; (right) 29Si{1H} NMR spectra (99.36 MHz) of 1-Cr – 1-W in (D6)benzene at 298 K showing the MSi signals. | ||
The most distinctive NMR spectroscopic feature of the silylidyne complexes 1-M (M = Cr – W) is the markedly deshielded 29Si (MSi) NMR signal, which moves from Cr (δ = 299.9 ppm) → Mo (δ = 308.9 ppm) → W (δ = 314.0 ppm) stepwise to lower field (Fig. 5). It is noteworthy, that the isovalent carbyne complexes Cp(CO)2MCPh do also exhibit a very deshielded 13C (MC) NMR signal, which, however, moves from Cr (δ = 325.7 ppm) → Mo (δ = 309.4 ppm) → W (δ = 299.3 ppm) in the opposite direction, i.e. to higher field (Table 1). Interestingly, the 29Si (MSi) NMR signals of 1-Mo and 1-W appear at lower field than those of their Tp′ analogs (cf. entry 2 with 7 and 3 with 8 in Table 1) and the same trend is observed for the related Cp/Cp* and Tp′ containing carbyne complexes (cf. entry 10 with 15 and 14 with 16 in Table 1). All tungsten silylidyne complexes have much larger 1J(183W–29Si) coupling constants than tungsten silyl complexes including 2-W (vide infra).
2.2 Reactions of silylidyne complexes with isocyanates and oxiranes
Complexes 1-Mo and 1-W react slowly with two equivalents of mesityl isocyanate in toluene at 80 °C to exclusively give complexes 2-Mo and 2-W (eqn (3)). No intermediates were detected by IR or NMR spectroscopy in these reactions leading exclusively to the trans isomers. In comparison, complex 1-Cr does not react with MesNCO even after prolonged heating in toluene at 110 °C, the shorter CrSi bond being apparently too sterically protected by the Cp* ligand and the Tbb substituent. | (3) |
Complexes 2-Mo and 2-W were isolated as air-sensitive, creamy-white and white solids in 85 and 89% yields, respectively. Both compounds are thermally stable and decompose upon melting at 227 and 228 °C, respectively. Complexes 2-Mo and 2-W were fully characterized and their molecular structures determined by X-ray diffraction (Fig. 6).
 | ||
| Fig. 6 DIAMOND plot of the molecular structure of 2-Mo. Thermal ellipsoids are set at 30% probability, hydrogen atoms were omitted and the substituents of the Tbb group are presented as wireframe for the sake of clarity. Selected bond lengths (pm) and bond angles (°): for 2-Mo (bond lengths and bond angles of 2-W are given in square brackets): Mo–Si1 253.01(7) [253.5(1)], Mo–C35 198.8(3) [198.8(5)], Mo–C36 197.8(3) [197.2(5)], Mo–C37 204.2(3) [204.2(5)], Si1–O1 174.8(2) [175.4(3)], Si1–O2 176.0(2) [175.9(3)], Si1–C1 188.8(2) [187.9(4)], O1–C25 135.9(3) [135.6(5)], O2–C25 136.0(3) [136.6(5)], N1–C25 125.9(3) [125.7(5)], Si1–Mo–C35 74.11(7) [75.2(1)], Si1–Mo–C37 127.7(1) [128.4(1)], C35–Mo–C36 110.3(1) [111.6(2)], O1–Si1–O2 75.69(8) [75.7(1)], O1–C25–O2 104.6(2) [104.8(3)], Si1–O1–C25 90.1(1) [90.0(2)], Si1–O2–C25 89.5(1) [89.5(2)]. | ||
The “four-legged piano-stool” complexes feature a novel silyl ligand containing a κ2O,O-bonded imidocarbonato substituent. Complexes 2-M can therefore be regarded as cyclic silyl esters of the unknown N-mesityl-imidocarbonic acid C(NMes)(OH)2. Imidocarbonates {C(NR)(OR)2} are in general less known96–100 than their structural isomers C(O)(NR2)(OR) (carbamates),101 and only two silyl esters related to 2-M have been reported so far.102,103 The 1,3-dioxasiletane ring formed by the imidocarbonato substituent is planar and the Si–O, C–O, CN bond lengths and inner angles of the ring (O–Si–O, O–C–O, Si–O–C) compare very well with those of the compound Si(O2CNDipp){C(SiMe3)2CH2}2 obtained from the silylene Si{C(SiMe3)2CH2}2 and DippNCO.102 The M–Si bond lengths of 2-Mo (253.01(7) pm) and 2-W (253.5(1) pm) are ca. 30 pm longer than the MSi bond lengths of 1-Mo and 1-W, but are in good agreement with the median value of the Mo–Si (254.2 pm) and W–Si single bond lengths (255.6 pm) of all structurally characterized complexes to date.104
The IR spectra of 2-Mo and 2-W in toluene solution display two characteristic absorption bands at 1710/1692 cm−1 and 1710/1694 cm−1 respectively, which are assigned to the ν(O2CN) stretching modes of the imidocarbonato susbstituent (ESI, Fig. S24 and S43†). In addition, the IR spectra show one absorption band for the CN stretching mode of the isocyanide ligand at 2103 cm−1 (1-Mo) and 2097 cm−1 (1-W) and two absorption bands for the in-phase and out-of-phase CO stretching modes of the carbonyl ligands at 1962 cm−1 and 1901 cm−1 (1-Mo) and 1954 cm−1 and 1894 cm−1 (1-W), respectively. The higher frequency ν(CO) band is much less intense than the lower frequency ν(CO) band, confirming the trans arrangement of the two CO ligands.105,106
A salient spectroscopic feature of 2-Mo and 2-W is the unusually deshielded 29Si NMR signal, which appears at considerably lower field (δ(2-Mo): 105.8 ppm, δ(2-W): 88.6 ppm) than that of Si(O2CNDipp){C(SiMe3)2CH2}2 (δ = 53.4 ppm)102 and of all other four-cordinate Si compounds with a four-membered siletane ring Si(X1)(X2)C = Y (X1, X2, Y = NR, O) (δ = −90.9–53.4 ppm).107 The 29Si NMR signal of 2-W is flanked by a pair of 183W satellites resulting in a 1J(W–Si) coupling constant of 79.6 Hz, that is larger than those of other tungsten silyl complexes (1J(W–Si) = 5–64 Hz),108 but much smaller than that of 1-W (316 Hz). The 13C{1H} NMR spectra of 2-Mo and 2-W ((D6)benzene, 298 K) display characteristic NMR signals for the CO2NMes carbons at 149.3 and 149.4 ppm, and for the isocyanide-carbons at 184.8 and 171.2 ppm, respectively (ESI, Chapter 2.4 and 2.5†). Notably, complexes 2-Mo and 2-W contain a stereogenic four-coordinate Si center due to the fixed orientation of the N-bonded Mes group, leading to two 13C NMR resonances for the trans-arranged CO ligands (ESI, Fig. S28 and S48†). In addition, rotation of the Tbb substituent about the Si–CTbb bond is hindered on the NMR time scale and leads to diastereotopic ortho (C2/C6) and meta C3/C5 positions of the Tbb aryl ring and to a double set of 1H, 13C and 29Si NMR signals for the substituents at these positions, which were fully assigned by 2D NMR spectroscopy of 2-Mo in (D8)THF at 243 K (ESI, Fig. S25–S30†). A dynamic process sets on with increasing temperature leading to a site exchange of the diastereotopic CO ligands and the diastereotopic ortho and meta positions of the Tbb ring, as evidenced by the 1H and 13C NMR spectra of 2-Mo in (D6)benzene at 348 K (ESI, Fig. S37–S40†). The process leads to a racemization of 2-Mo and occurs presumably via an inversion of the Nimido-bonded mesityl group. The Gibbs energy of activation (ΔG‡ (298 K)) for this process was found to be approximately 60 kJ mol−1, which compares well with the values obtained for the N-aryl imidocarbonates (MeO)2CN(p-C6H4X) (X = H, Cl, Me) (Ea (in acetone) = 56.1–69.5 kJ mol−1).97
Formation of 2-M (M = Mo, W) can be explained by the three-step reaction sequence outlined in Fig. 7. The first step involves a [2 + 2] cycloaddition to give the 1,3,2-metallaoxasilacyclo-1-buten-4-imine a, which after ring opening gives the η1-silaacyl-isocyanide complex b. Species b reacts with another equivalent of MesNCO to give the final product 2-M. cis–trans isomerization may precede or follow the reaction of b with MesNCO leading finally only to the trans-isomer 2-M.
 | ||
| Fig. 7 Possible products c–d′ of the reactions of 1-Mo and 1-W with MesNCO. Only c is observed showing the high chemo- and regioselectivity of these reactions. | ||
No evidence was found for the formation of the putative products d, c′ and d′ (Fig. 7). This suggests that both [2 + 2] cycloaddition steps are highly chemoselective with the CO bond of MesNCO being exclusively involved in the [2 + 2] cycloaddition with the MSi bond of 1-M and then with the SiO bond of b (Fig. 7). Both [2 + 2] cycloaddition steps are also highly regioselective with the O atom getting exclusively attached to the silicon atom, due to the high M(δ−)Si(δ+) bond polarity of 1-M and the Si(δ+) = O(δ−) bond polarity of b. Notably, the Schrock-type neopentylidyne complex [Cl3(dme)WCtBu] undergoes with cyclohexylisocyanate (CyNCO) a Wittig-type Ccarbyne–Cisocyanate coupling reaction to give after ring-opening a tungsten imido-ketenyl intermediate, which then inserts a second equivalent of CyNCO into the W–Cketenyl bond to yield an oxaazetin tungstenacycle.109 The opposite chemoselectivity and regioselectivity of the cycloaddition step in this reaction reflects the reversed polarity of the metal–carbon bonds of Schrock-type carbyne complexes (M(δ+)C(δ−)) and the metal–silicon bonds (M(δ−)Si(δ+)) of 1-M.
The factors that favour an O- instead of an N-binding to silicon and lead exclusively to the CO cycloaddition product in both steps (1-M → a and b → 2-M) are subtle. In fact, reaction of MeNCO with the silylidene complex cation [Cp*(PMe3)2Ru = SiMe2]+ shows the opposite chemoselectivity and leads exclusively to the [(CN) + (RuSi)] cycloaddition product [Cp*(PMe3)2RuSiMe2N(R)CO]+.110 Both reactants contain in this case sterically less demanding methyl groups at the isocyanate-nitrogen and silicon atoms. In contrast, reaction of the silylidene–hydrido complex [Cp*(CO)(H)RuSi(H)C(SiMe3)3], which contains a sterically demanding trisyl substituent at silicon, with MesNCO yields the CO hydrosilylation product [Cp*(CO)Ru[η2-N,Si–N(Mes) = C(H)OSi(H)C(SiMe3)3].111 In this reaction, a precoordination of MesNCO via the O-atom to the electrophilic silylidene–silicon atom was proposed to be the first step based on quantum chemical studies.112 It should also be noted, that protonation of HNCO in FSO3H/SO2 or MeNCO in the gas-phase occurs at the N-site.113,114 In contrast, silylation of SiMe3NCO with (SiMe3)[B(C6F5)4] occurs at the O-site to give [SiMe3NCOSiMe3][B(C6F5)4], where the N-silylated cation [(SiMe3)2NCO]+ is almost isoenergetic.115
The electrophilicity of the Si center and the high polarity MSi in 1-M let us presume that the silylidyne complexes would promote the ring opening of cyclic ethers. In fact, addition of ethyloxirane to an orange-red solution of 1-Mo in n-hexane at room temperature was accompanied by a rapid color change to brownish-yellow to yield regioselectively the cis-(E-but-1-ene-1-olato)silylidene–hydrido complex 3-Mo along with a minor Cp* containing component, which on the basis of its 1H NMR spectroscopic features is suggested to be the Z-stereoisomer 3-Mo′ (eqn (4)). The E/Z stereoisomers are formed in the molar ratio of 7:3. Conversion of 1-Mo to 3-Mo/Mo′ is an unusual Lewis-acid promoted ring-opening reaction of an oxirane by α-elimination.116 The oxirane is presumably activated upon O-coordination to the Lewis acidic Si center of 1-Mo for the next step, which involves ring-opening after regioselective proton abstraction from the more acidic ring-CH2 group by the basic molybdenum center. The reaction of 1-Mo with ethyloxirane is reminiscent of the reaction of the hydrido–hydrosilylidene complex [Cp*(CO)2(H)WSi(H){C(SiMe3)3}] with oxiranes,117 whereas reactions of Fischer-type carbyne complexes with oxiranes have not yet been reported.
 | (4) |
After work-up 3-Mo was isolated as an analytically pure, very air-sensitive, pale-yellow solid that melts with decomposition at 165 °C. Compound 3-Mo is the first enolatosilylidene complex to be fully characterized and its structure was determined by single-crystal X-ray diffraction (Fig. 8).1183-Mo crystallizes in the non chiral space group Pbca and exists in the solid-state as a racemic mixture of the C (clockwise) and A (anticlockwise) enantiomers due to the presence of a crystallographic inversion center. The molecular structure of the A enantiomer is depicted in Fig. 8a and shows a “four-legged piano-stool” complex with a cis-arrangement of the two CO ligands (∠(CO–Mo–CO) = 84.9(3)°). The silylidene ligand is coordinated to the Mo center via a short Mo–Si bond (d(MoSi) = 235.8(2) pm), which compares well with that of the aminosilylidene complex [Cp(CO)2(H)MoSi(NH2)ArTripp] (d(MoSi) = 237.96(5) pm (M, Fig. 3),49 and lies in the range found for other arylsilylidene complexes (d(MoSi) = 228.8(2)–238.72(7) pm).38,48,60,66,119,120 The MoSi bond of 3-Mo is considerably longer than the MoSi bond of 1-Mo (223.2(1) pm) and shorter than the Mo–Si single bond of 2-Mo (253.01(7) pm). The silicon center is trigonal-planar coordinated with the Tbb ring plane lying orthogonal to the silicon coordination plane. The angles at silicon differ though markedly. The Mo–Si–CTbb angle is widened to 135.4(2)° due to the steric demand of the Tbb substituent, whereas the O–Si–CTbb angle is lowered to 100.0(3)° reflecting the low tendency of silicon to hybridize and the increased s character of the Si hybrid orbital involved in the Mo–Si σ bond. The silylidene ligand adopts a tilted conformation with the buten-1-ene-1-olato substituent pointing towards the Cp* ligand, as evidenced by the dihedral angle Cg–Mo–Si1–O1 of −46.2(3)° (Cg: Cp* ring centroid) (Fig. 8b). The hydrido ligand was localized in the difference Fourier map in the vacant cis-coordination site relative to the silylidene ligand (∠(H1–Mo–Si1) = 58(2)°) at a distance of 181(7) pm and 208(6) pm from the molybdenum and Si1 atoms, respectively.
 | ||
| Fig. 8 (a) DIAMOND plot of the molecular structure of the A (anticlockwise) enantiomer of 3-Mo.Thermal ellipsoids are set at 30% probability, hydrogen atoms were omitted except the Mo-bonded hydrogen atom and the substituents of the Tbb group are presented as wireframe for the sake of clarity. Selected distances (pm) and bond angles (°): for 3-Mo: Mo–Si1 235.8(2), Mo–H1 181(7), Mo–C39 196.1(8), Mo–C40 197.8(8), Si1⋯H1 208(6), Si1–O1 164.7(5), Si1–C1 188.2(6), C25–O1 138(1), C25–C26 133.0(12), C26–C27 148.0(8), C27–C28 153(1), Si1–Mo–C39 75.4(2), Si1–Mo–C40 106.4(2), Si1–Mo–H 58.1(19), C39–Mo–C40 84.9(3), Mo–Si1–C1 135.4(2), Mo–Si1–O1 124.6(2), O1–Si1–C1 100.0(3), O1–C25–C26 121.3(8), C25–C26–C27 124.3(9). (b) View along the Mo–Si bond of 3-Mo (Olex plot) showing the tilted conformation of the silylidene ligand and the anticlinal conformation of the Mo–H and Si–OR bonds (the methyl groups of the Cp* ligand and the substituents of the Tbb group were omitted for clarity). | ||
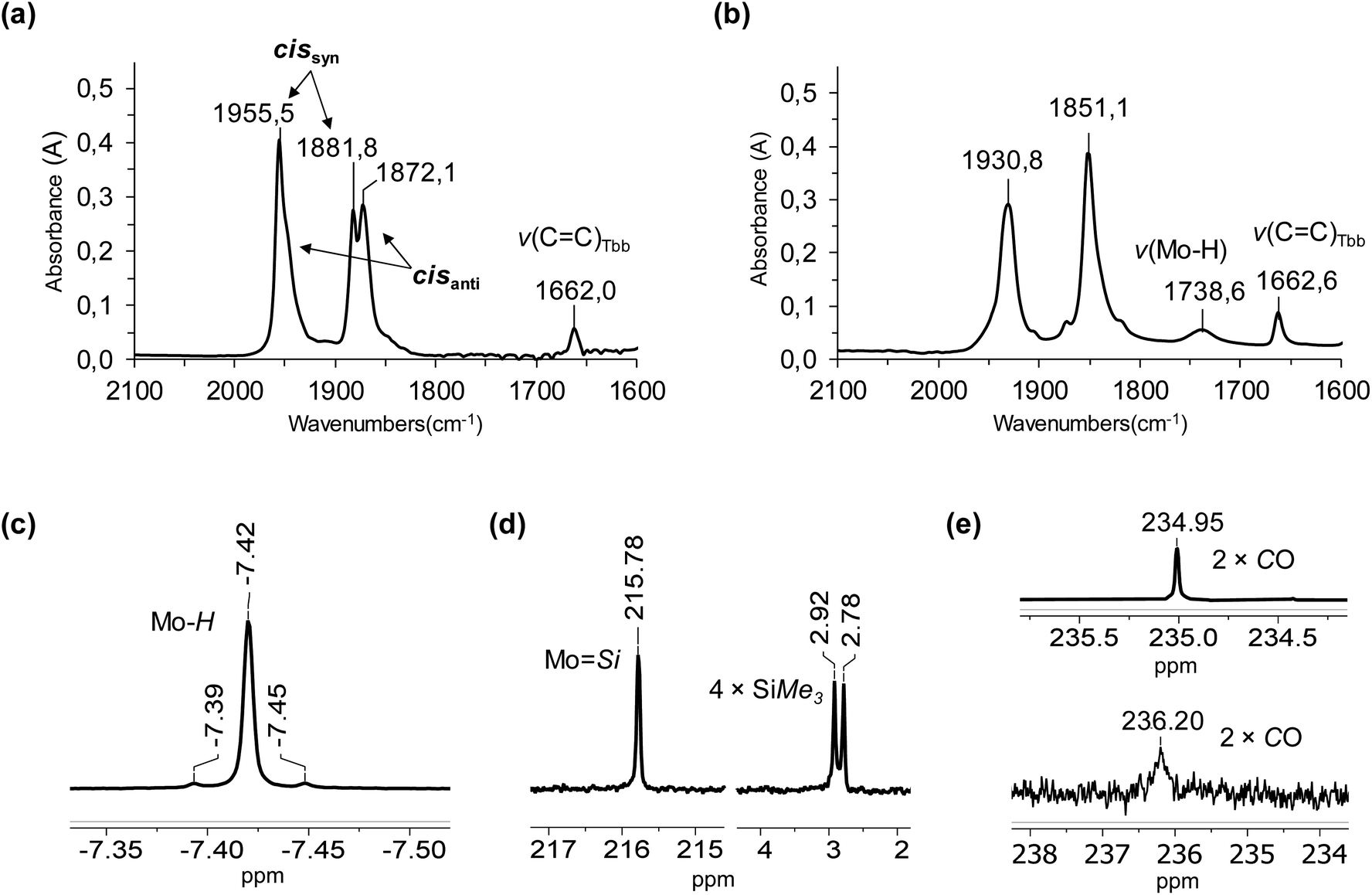 | ||
| Fig. 9 (a) Solution IR spectrum of 3-Mo in n-hexane in the spectral range 2100–1600 cm−1. (b) ATR-IR spectrum of 3-Mo in the spectral range 2100–1600 cm−1. (c) Hydride NMR signal with its 29Si satellites in the 1H NMR spectrum of 3-Mo in (D6)benzene at 298 K. (d) 29Si NMR spectrum of 3-Mo in (D6)benzene at 298 K. (e) (top) CO signal in the 13C{1H} NMR spectrum of 3-Mo in (D6)benzene at 298 K; (bottom) CO signal in the 13C{1H} NMR spectrum of 3-Mo in (D8)toluene at 193 K. | ||
This observation is in line with the results of quantum chemical studies suggesting the presence of two diastereomers at similar low energy, labeled as cisanti and cissyn, respectively. Both isomers have cis-ligated CO ligands, are present in solution as a racemic mixture of the C (clockwise) and A (anticlockwise) enantiomers, and display a tilted conformation of the silylidene ligand, with the Si-bonded OR substituent pointing towards the Cp* ring, as evidenced by the dihedral angle Cg–Mo–Si–O of +48.7/–48.7° (C/A-cisanti) and −55.8°/+55.8° (C/A-cissyn), respectively (Fig. 10a). The isomers C/A-cisanti and C/A-cissyn differ though in the relative orientation of the silylidene and the hydrido ligand. In A-cisanti the Mo–H and Si–OR groups adopt an anticlinal conformation (∠H–Mo–Si–OR = −157.1°), i.e. the Mo–H bond vector points almost in the opposite direction of the Si–OR bond vector, whereas in A-cissyn the Mo–H and Si–OR groups have a synclinal conformation (∠H–Mo–Si–OR = −50.9°) (Fig. 10b). cisanti is the most stable isomer with a slightly lower Gibbs energy of 5.4 kJ mol−1 than the cissyn isomer. In addition, a trans-configurated isomer, labeled as trans, was found to be a local minimum on the PES, whose Gibbs energy is 15.3 kJ mol−1 higher than the most stable isomer cisanti (Fig. 10).
 | ||
| Fig. 10 (a) Side view of the calculated structures (Olex plots) of the isomers A-cisanti, A-cissyn and trans of 3-Mo with their relative zero-point vibrational energy corrected inner energies ΔU(0 K) (kJ mol−1) and standard Gibbs energies ΔG° (kJ mol−1) (values in curly brackets); (b) view along the Mo–Si bond of the calculated structures of the isomers A-cisanti, A-cissyn and trans (Olex plots) with values of the dihedral angles Cg–Mo–Si–O and H–Mo–Si–OR, respectively. The methyl groups of the Cp* ligand and the substituents of the Tbb group were omitted for clarity. | ||
The calculated bond lengths and angles of cisanti agree very well with the experimental bond lengths and bond angles of 3-Mo and even the twist angles describing the twist of the silylidene ligand with respect to the Cp* and the hydrido ligand, respectively, or the Cg–Mo–Si–O dihedral angle, which describes the orientation of the OR substituent with respect to the Cp* ring, compare very well with the experimental values (Table 2).
| Mo–Si | Mo–H | Si⋯H | CO–Mo–CO | H–Mo–Si | Cg–Mo–H | Cg–Mo–Si | ΣSia | TA1b | TA2c | DHA1d | DHA2e | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Sum of the bond angles at the Mo-bonded silicon atom (Si). b Twist angle between the (Cg,Mo,Si)-plane (Cg: Cp* ring centroid) and the (O,Si,CTbb)-plane. c Twist angle between the (H,Mo,Si)-plane and the (O,Si,CTbb)-plane d Dihedral angle Cg–Mo–Si–O. e Dihedral angle H–Mo–Si–O. | ||||||||||||
| A-(3-Mo)exp | 235.8(2) | 181(7) | 208(6) | 84.9(3) | 58(2) | 120(2) | 131.4(1) | 360 | 45.2(2) | 31(3) | −46.2(3) | −150(3) |
| A-cis anti (A-(3-Mo)th) | 236.1 | 173.1 | 209.1 | 85.3 | 59.1 | 121.9 | 127.8 | 360 | 48.5 | 22.0 | −48.7 | −157.1 |
| A-cis syn | 236.6 | 175.0 | 195.9 | 82.4 | 54.4 | 122.3 | 126.5 | 359.8 | 53.6 | 52.2 | 55.8 | −50.9 |
| trans | 239.1 | 170.2 | 361.3 | 107.5 | 123.1 | 111.7 | 125.2 | 359.9 | 47.4 | 45.2 | −48.8 | 132.6 |
Curve fitting and deconvolution of the IR spectrum using a Gaussian profile gave four bands peaking at 1956, 1950, 1883 and 1872 cm−1 and their frequency integrated intensities (ESI, Fig. S88 and Table S5†). After comparison with the calculated harmonic frequencies of the cisanti and cissyn isomers, the bands at 1950 and 1872 cm−1 were assigned to the in-phase and out-of-phase combination of the two CO stretching modes of the cisanti isomer, and those at 1956 and 1883 cm−1 to the in-phase and out-of-phase combination of the two CO stretching modes of the cissyn isomer (Fig. 9a). The equilibrium constant  and the standard Gibbs energy difference
and the standard Gibbs energy difference  (G°(cisanti) − G(cissyn)) at 298 K was estimated assuming that the sum of the frequency integrated intensities of the two ν(CO) bands of the isomer cisanti is equal to that of cissyn. The obtained values (Kexp = 2.6,
(G°(cisanti) − G(cissyn)) at 298 K was estimated assuming that the sum of the frequency integrated intensities of the two ν(CO) bands of the isomer cisanti is equal to that of cissyn. The obtained values (Kexp = 2.6,  (298 K) = −2.4 kJ mol−1) are in reasonable agreement with the calculated values (Kth = 8.8,
(298 K) = −2.4 kJ mol−1) are in reasonable agreement with the calculated values (Kth = 8.8, 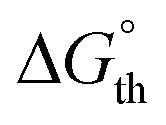 = −5.4 kJ mol−1). From the Gibbs energy difference
= −5.4 kJ mol−1). From the Gibbs energy difference  between the trans isomer and the cisanti isomer at 298 K of 15.3 kJ mol−1, the molar ratio
between the trans isomer and the cisanti isomer at 298 K of 15.3 kJ mol−1, the molar ratio  of 0.002 was obtained, indicating that the trans isomer does not contribute to the IR spectrum of 3-Mo in solution due to its very low concentration.
of 0.002 was obtained, indicating that the trans isomer does not contribute to the IR spectrum of 3-Mo in solution due to its very low concentration.
The IR spectrum of 3-Mo in n-hexane also shows one characteristic band arising from the CC stretching mode of the but-1-ene-1-olato substituent at 1662 cm−1 (Fig. 9a). In comparison, no band could be observed for the Mo–H stretching mode in solution, which is however clearly visible in the ATR-IR spectrum of 3-Mo at 1739 cm−1 (Fig. 9b). To rationalize this observation the relaxed potential energy surface around the minimum structure of cisanti and cissyn was calculated, varying the Mo–H distance from 1.6 to 2.0 Å and the Si–H distance from 1.6–2.2 Å (Fig. 11). The 2-dimensional surface plots show a shallow potential and an enhanced flexibility of the hydride around the respective minimum position. For example, a change of the Si–H distance from 1.9 to 2.2 Å and of the Mo–H distance from 1.7–1.8 Å leads to an increase of the electronic energy of cisanti by maximal 2 kJ mol−1. Therefore, one may assume that several rapidly equilibrating species are present in solution, whose energy differ by less than 2 kJ mol−1 from the minimum structures cisanti and cissyn and whose Mo–H bond lengths and Si–H distances varying in a range of few pm around the values of the minimum structures of cisanti and cissyn, respectively. The positions of the ν(CO) bands of these species are essentially the same as those of cisanti and cissyn, respectively, but their ν(Mo–H) bands vary, as evidenced by a comparison of cisanti and cissyn, which shows that a shortening of the Mo–H bond by 2 pm (cissyn (175.0 pm) → cisanti (173.1 pm)) leads to an increase of the ν(Mo–H) frequency by 56 cm−1 (cissyn (1735 cm−1) → cisanti (1791 cm−1)). One may therefore conclude that the low intensity of the ν(Mo–H) band of cisanti and cissyn is distributed over a larger wavenumber range around the respective band maximum leading to a considerable broadening of the ν(Mo–H) band in solution. This conclusion is confirmed by the close similarity of the Boltzmann corrected IR spectrum of the structural ensemble captured by the two PES scans with the experimental spectrum of 3-Mo in n-hexane solution (ESI, Fig. S90†).
 | ||
| Fig. 11 Two-dimensional representation of the relaxed potential energy surface cisanti (left) and cissyn (right) as a function of the Si–H and Mo–H distance in Å (mesh width: 5 pm). Electronic energies are given in kJ mol−1. The global (fully optimized) minimum (set to 0.0 kJ mol−1) is depicted with a yellow star, respectively. Iso-energy lines at 2, 5, 10, and 20 kJ mol−1 are drawn as black lines. | ||
The computed Mo–H bond lengths of cisanti (173.1 pm) and cissyn (175.0 pm) lie in the range of Mo–H 2c–2e bond lengths (d(Mo–H) = 168.5(3)121–178.9(7)122) obtained from neutron diffraction studies. In comparison, the calculated Si–H distances of cisanti (209.1 pm) and cissyn (195.9 pm) are considerably longer than those obtained for 2c–2e Si–H bonds by neutron diffraction (d(Si–H) = 148.1(5)–150.6(2)).123,124 The Si–H distances of cisanti and cissyn are much shorter than the sum of the van der Waals radii of Si and H (330 pm),125 and appear in the range of distances for which non-classical M–H⋯Si interactions have been discussed.126
A topological analysis of the electron density of both isomers was carried out to address the question, whether a direct Si⋯H bonding interaction is present in cisanti and cissyn. For cisanti, no bond path and no bond critical point was found between the Si and the H atom excluding any direct Si⋯H bonding interaction (Fig. 12, left).
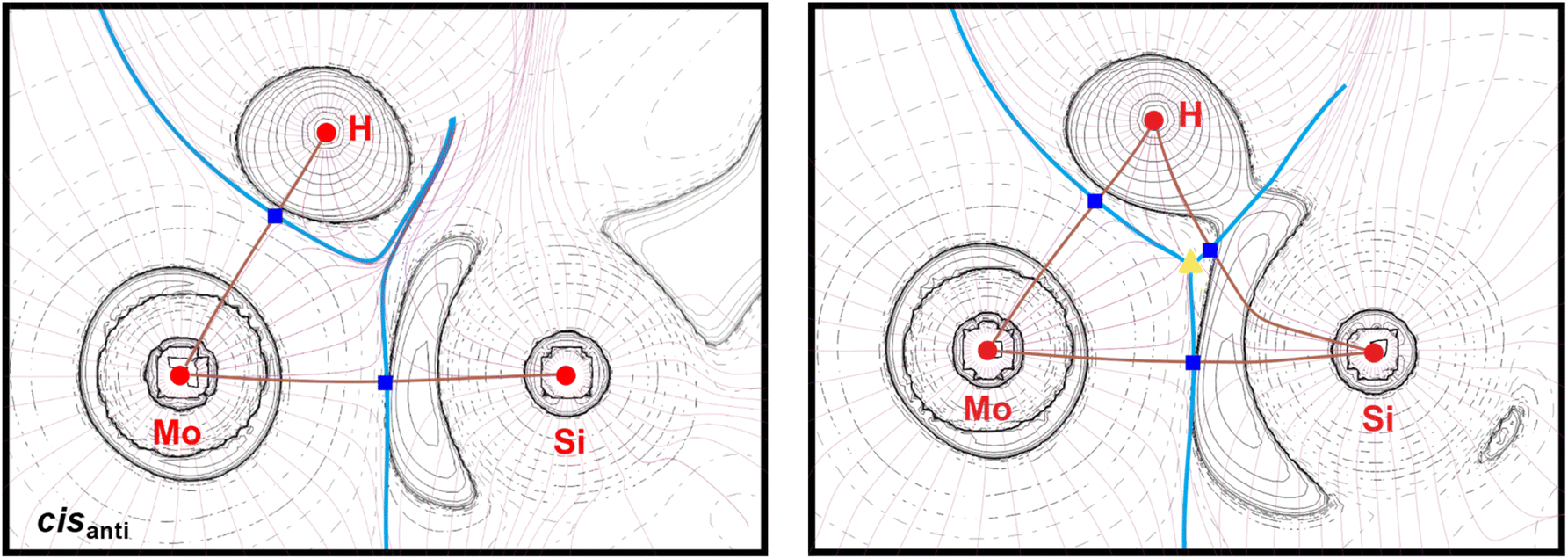 | ||
| Fig. 12 Molecular graph of cisanti (left) and cissyn (right) showing core critical points (red circles), bond critical points (dark blue squares), ring critical points (yellow triangles), bond paths (brown lines), gradient paths (mauve lines, projected into the given plane), areas of charge concentration (solid hairlines) and charge depletion (broken hairlines) as well as interatomic surfaces (light blue lines). | ||
In comparison, for cissyn a bond path with a bond critical point was found between the Si and the H atom. In this case, the interatomic surfaces crossing the Mo–H and the Mo–Si bond paths meet the interatomic surface crossing the Si–H bond path at a ring critical point (rcp), that is closely located to the Si–H bond critical point (Fig. 12, right). The proximity of the ring and bond critical point of the Si–H bond, as well as the similar values of the electron density at these points indicate a very weak direct Si⋯H interaction, which is also reflected in the low value of the energy density (Table 3).
 , relative total energy density
, relative total energy density  and bond ellipticity (ε); the position of the bcp is given in %, whereby a value larger than 50% means that the bcp is further away from atom A
and bond ellipticity (ε); the position of the bcp is given in %, whereby a value larger than 50% means that the bcp is further away from atom A
| A–B | %A | %B | ρ(rbcp)/eÅ−3 | ▽2ρ(rbcp)/eÅ−5 |
|
|
ε |
|---|---|---|---|---|---|---|---|
| a Distance between the rcp and the bcp(Si–H). b Values at the rcp. | |||||||
| cis anti | |||||||
| Mo–H | 65 | 35 | 0.732 | 2.157 | 0.674 | −0.468 | 0.115 |
| Mo–Si | 53 | 47 | 0.598 | 0.113 | 0.478 | −0.465 | 0.279 |
| cis syn | |||||||
| Mo–H | 65 | 35 | 0.686 | 2.917 | 0.727 | −0.429 | 0.185 |
| Mo–Si | 53 | 47 | 0.587 | 0.230 | 0.485 | −0.458 | 0.028 |
| Si–H | 57 | 43 | 0.478 | 0.023 | 0.344 | −0.341 | 1.267 |
| rcp | 17.8 pma | 0.474b | 1.508b | 0.516b | −0.294b | — | |
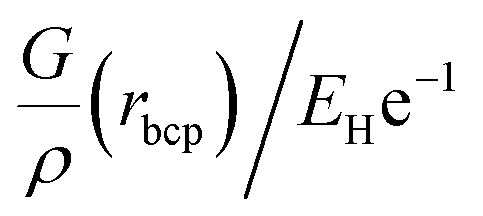

Further information on the Si⋯H interaction in cisanti and cissyn was obtained from the sign and magnitude of the 29Si,1H spin–spin coupling constant. One bond Si,H coupling constants (1J(Si,H)) have negative signs due to the negative gyromagnetic ratio of the 29Si nucleus and range from ca. (−150)–(−400) Hz in silanes and transition metal hydrosilyl complexes, where only 2c–2e [TM-SiHRR′] interactions are present.127,128 In comparison, two-bond (geminal) coupling constants (2J(Si,H)) in silanes127,129 and transition metal silyl hydrides, as [Fe(H)(SiCl3)(CO)4],130,131 usually have positive signs and much smaller values (ca. 5–25 Hz). The calculated 29Si,1H coupling constant of cisanti has a positive sign (+27.7 Hz), as expected for a 2J(Si,H) coupling, and excludes a direct Si⋯H interaction in cisanti in line with the AIM results. In comparison, the calculated 29Si-1H coupling constant of cissyn, where the Si–H distance (195.9 pm) is shorter by 14 pm than in cisanti has a negative value (−7.2 Hz). Notably, the same trend and a sign change of the J(Si,H) coupling from plus to minus was observed in transition metal silyl hydride complexes upon decreasing the Si–H distance leading to an increased direct M–Si⋯H bonding interaction and finally to non classical hydrosilane complexes.131,132 In the case of cissyn the negative sign of the J(Si,H) coupling and its small absolute value suggests in combination with the AIM results a direct, weak Si⋯H bonding interaction. Notably, the experimental value of the J(Si,H) coupling constant of 3-Mo (22 Hz) agrees well with the calculated value of J(Si,H)th (18 Hz) obtained from the following equation,
| J(Si,H)th (3-Mo) = x1 × J(Si,H)th(cisanti) + x2 × J(Si,H)th(cissyn) |
As outlined above, the experimental and theoretical IR studies of 3-Mo indicate the presence of two isomers (cisanti and cissyn) in solution. In comparison, the 1H, 13C{1H} and 29Si{1H} NMR spectra of 3-Mo show only one set of signals, the number and multiplicity of which indicate a time-averaged Cs-symmetric structure in solution with the symmetry plane passing through the silylidene ligand plane and bisecting the Cp*Mo(CO)2 fragment and the Tbb ring plane. For example, the 1H NMR spectrum of 3-Mo in (D6)benzene at 298 K shows only one Mo–H resonance at δ = −7.42 ppm, which is flanked by 29Si satellites due to 29Si-1H spin–spin coupling (Fig. 9c). Furthermore, the 29Si{1H} NMR spectrum of 3-Mo shows only one 29Si resonance for the MoSi group at δ = 215.8 ppm instead of the expected two resonances, and two 29Si resonances for the SiMe3 groups instead of the four signals expected for each of the isomers cisanti and cissyn in case of a hindered rotation of the Tbb substituent around the Si–CTbb bond (Fig. 9d). Finally, the 13C{1H} spectrum of 3-Mo shows only one signal for the two CO ligands at δ = 235.0 ppm (Fig. 9e). This is quite different from what would be expected for the static structures of the C1-symmetric isomers cisanti and cissyn, for which two singlets should be observed for the diastereotopic CO ligands, respectively.
All NMR features of 3-Mo indicate that in solution, a rapid equilibration of the isomers cisanti and cissyn occurs, which proceeds via a Cs-symmetric state. This process is too fast to be resolved on the NMR time scale (101–10−6 s). The two isomers can be observed though by IR spectroscopy due to the faster time scale of this method (10−11–10−14 s). This was verified by quantum chemical studies suggesting a reaction path via two enantiomeric trans intermediates for the interconversion of cisanti and cissyn (Fig. 13).
 | ||
| Fig. 13 Reaction profile for the diastereomerization of A-cisanti to A-cissyn; Olex plots of the calculated structures of A-cisanti, A-cissyn, the transition states (TS1a, TS2 and TS1b) and the intermediates trans and trans* (top views) with their relative zero-point vibrational energy corrected inner energies ΔU(0 K) (kJ mol−1) and standard free energies ΔG° (kJ mol−1) (values in parentheses). | ||
In the first step A-cisanti isomerizes to the trans diastereomer trans by migration of the H ligand from the cis to the trans position to the silylidene ligand (Fig. 13). A closer look at the trajectory connecting A-cisanti to trans via the transition state TS1a shows that the H–Mo–Si angle and the Si–H distance increase continuously along the path from A-cisanti to trans, while the Cg–Mo–H angle increases first from 121.9° (A-cisanti) to a maximum value in a pseudo-trigonal-bipyramidal structure, in which the Cp* and H ligands occupy nearly the apical positions, and then decreases over 153° in the transition state TS1a to 111.7° in the trans product. Remarkably, the tilted conformation of the silylidene ligand is maintained during the isomerization as shown by the dihedral angle Cg–Mo–Si–O making the trans product chiral (Fig. 13).
In the next step the trans isomer transforms to its conformational enantiomer trans* via the Cs symmetric transition state TS2. A look at the trajectory connecting trans to trans* shows that the reaction path involves a rotation of the silylidene ligand around the MoSi bond as shown by the change in the dihedral angle Cg–Mo–Si–O from −48.8° in trans to +48.8° in trans*. The silylidene adopts in the transition state TS2 an upright conformation with the OR substituent pointing towards the Cp* group (∠(Cg–Mo–Si–O) = −0.3°). In the final step trans* converts to the cis diastereomer A-cissyn by migration of the H ligand from the trans to the cis position relative to the silylidene ligand. The reaction path is similar to the one that leads the trans intermediate back to A-cisanti. It is noteworthy, that the isomerization pathway leading to equilibration between A-cisanti and its diastereomer A-cissyn in solution (and in the same way between C-cisanti and C-cissyn) does not involve a change in the metal configuration. The calculated free energies of activation range from 32 to 44.5 kJ mol−1 for the forward reaction and from 29.2 to 41.9 kJ mol−1 for the backward reaction suggesting that the process may be detectable by NMR spectroscopy at low-temperature. However, the 1H NMR (300.1 MHz) spectrum of 3-Mo in (D8)toluene showed no significant change up to 193 K and the 13C{1H} NMR spectrum (75.47 MHz) of 3-Mo at 193 K only showed an incipient broadening of the CO signal at δ = 236.2 ppm (Δν1/2 = 10.3 Hz) (Fig. 9e) indicating that the process at 193 K is still too fast on the NMR time scale (Fig. 14).
2.3 Reactions with 1,3 dipoles
Next, we investigated the reactivity of silylidyne complex 1-Mo with 1,3-dipoles. Diazoalkanes form together with organic azides and nitrous oxide an important class of 1,3-dipoles, the diazonium betaines, according to Huisgen's definition.133 Their 1,3-cycloaddition reactions with alkynes are very important in heterocycle synthesis and have been extensively studied in experiment and theory.134,135 In comparison, reactivity studies towards the heavier alkyne congeners REER (E = Si–Pb) are very scarce136 and reactions of diazoalkanes with transition metal tetrylidyne complexes have not been explored. Earlier work in our group has shown that the germylidyne complex [Cp(CO)2MoGe–ArMes] reacts rapidly and selectively with two equivalents of trimethylsilyldiazomethane to afford a 1-metalla-3-germabicyclo[1.1.0]cyclobutane (eqn (5)).137 The reaction proceeds presumably via two sequential, regioselective [2 + 3] cycloaddition and N2 elimination steps resulting in an overall double carbene transfer to the MoGe bond. | (5) |
The silylidyne complex 1-Mo reacts also rapidly with two equivalents of trimethylsilyldiazomethane. However, no N2 elimination is observed in this reaction leading selectively to the orange-red, highly air sensitive complex 4-Mo (eqn (6)). The reaction has no precedent in alkyne chemistry and probably involves a double [2 + 1] cycloaddition to the terminal N atom of the diazoalkane leading finally to a cleavage of the Mo–Si bond. It is reminiscent of the reaction of the disilene Mes2SiSiMes2138 and phosphasilenes139 with aryldiazoalkanes. Compound 4-Mo is the first silaamidinato complex reported in the literature, whereas metal amidinates are a well-studied class of compounds.140–142
 | (6) |
The molecular structure of 4-Mo shows a planar four-membered metallacycle with an acute angle at the Mo (∠N1–Mo–N3 = 67.0(1)°) and a transannular Mo⋯Si distance of 296.1(1) pm, which is ca. 43 pm longer than the Mo–Si single bond of 2-Mo, suggesting that there is no direct Mo–Si bonding interaction in 4-Mo (Fig. 14). The silicon atom is trigonal planar coordinated and the endocyclic Si–N bond lengths of 165.8 and 165.1 pm are in between those of SiN double bonds (153–163 pm)143 and Si–N single bonds (170–175 pm),144 suggesting a 3c-2π delocalization over the NSiN moiety. The exocyclic N–N and CN bond lengths are in the common range of N–N single and CN double bonds of azines R2CN–NCR2, respectively.1454-Mo displays in the 29Si NMR spectrum a resonance signal at δ = 31.8 ppm.
 | ||
| Fig. 14 DIAMOND plot of the molecular structure of 4-Mo. Thermal ellipsoids are set at 30% probability, hydrogen atoms were omitted and the substituents of the Tbb group are presented as wireframe for the sake of clarity. Selected distances (pm) and bond angles (°): Mo⋯Si1 296.1(1), Mo–N1 224.5(4), Mo–N3 222.7(4), Si1–N1 165.8(4), Si1–N3 165.1(4), Si1–C1 183.6(4), N1–N2 138.2(6), N3–N4 137.4(6), N2–C25 129.2(7), N4–C29 129.0(8), N1–Mo–N3 67.0(1), N1–Si1–N3 96.4(2), N1–Si1–C1 130.8(2), N3–Si1–C1 132.6(2). | ||
Organic azides are also potent 1,3-dipoles and their [3 + 2] cycloaddition with alkynes is one of the most important reactions in click chemistry.134 Few [3 + 2] cycloadditions of organic azides with Fischer-type carbyne complexes were also reported leading exclusively to 2H-1-metalla-2,3,4-triazoles (e, Fig. 15a).82,146 In comparison, reaction of 1-Mo with MesN3 in n-hexane at room temperature yields by N2 elimination the η2-silaiminoacyl complex 5-Mo (eqn (7)).
 | (7) |
 | ||
| Fig. 15 (a) [3 + 2] cycloaddition of organic azides with metal carbyne complexes; (b) proposed pathway for the reaction of 1-Mo with MesN3. | ||
The following reaction pathway can be proposed for the formation of 5-Mo reflecting the Mo(δ−)Si(δ+) bond polarity. In the first step, a [3 + 2] cycloaddition of MesN3 with 1-Mo gives regioselectively the 4H-1-metalla-5-sila-2,3,4-triazole f (Fig. 15b). In this reaction, the internal nucleophilic N atom of MesN3 attacks the electrophilic silicon atom of 1-Mo, whereas the nucleophilic Mo center of 1-Mo gets attached to the electrophilic terminal N atom of MesN3. In the next step, ring opening by cleavage of the NMes–N bond leads to the η1-silaiminoacyl-dinitrogen complex g, which cleaves off N2 by coordination of the imino nitrogen to the metal center, yielding 5-Mo (Fig. 15b). It is noteworthy, that the regioselectivity of the [3 + 2] cycloaddition of the carbyne complexes is opposite to that of the silylidyne complex 1-Mo and that formation of the 4H-1-metalla-2,3,4-triazoles e′, the carbon analogs of f, is kinetically and thermodynamically unfavorable according to quantum chemical calculations.147
5-Mo was isolated as an air sensitive, black, stable solid, that decomposes upon melting at 176 °C. The molecular structure of the “four-legged piano stool” complex 5-Mo features a three membered metallaazasilirene ring (Fig. 16). The Mo–Si (239.77(8)°pm) and Si–N (161.5(3)°pm) bond lengths compare well with those recently reported for the complex [MoCp*(CO)2(κ2Si,N–Si(Eind)NSiMe3)]60 and lie in the upper limit of reported MoSi (228.8(2)–238.72(7) pm)38,48,60,66,119,120 and SiN (153.3(2) pm–162.6(1) pm)143 bond lengths, respectively.
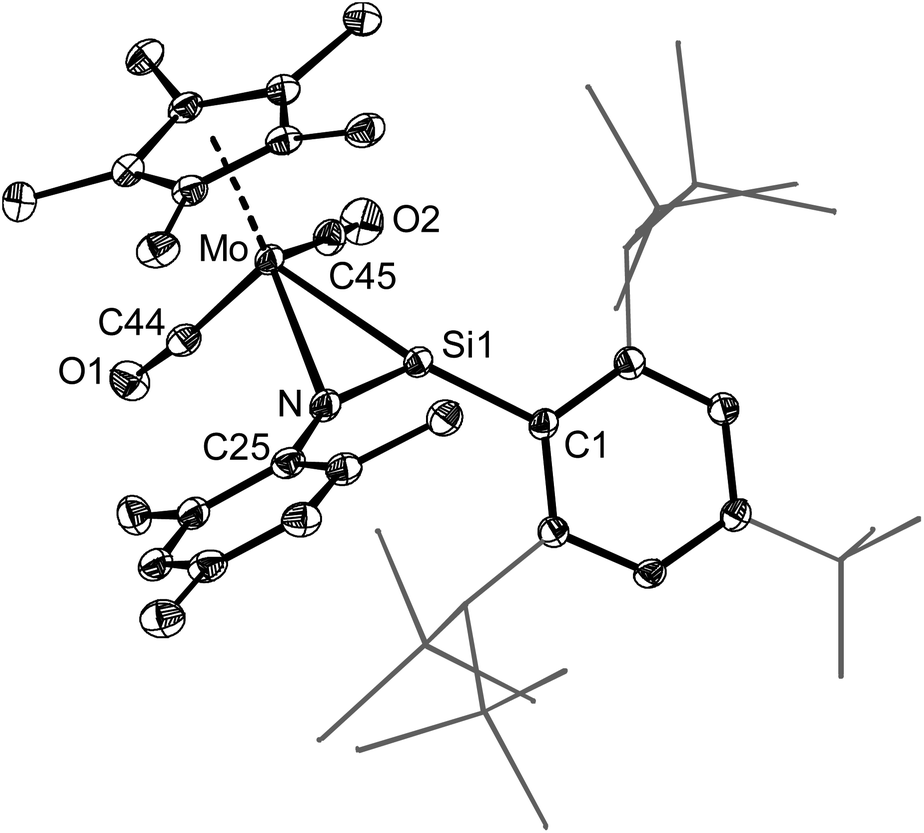 | ||
| Fig. 16 DIAMOND plot of the molecular structure of 5-Mo. Thermal ellipsoids are set at 30% probability, hydrogen atoms were omitted and the substituents of the Tbb group are presented as wireframe for the sake of clarity. Selected bond lengths (pm) and bond angles (°): Mo–Si1 239.77(8), Mo–N 239.1(2), Mo–C44 196.3(3), Mo–C45 195.0(3), Si1–N 161.5(3), Si1–C1 187.0(3), N–C25 140.2(4), Mo–Si1–C1 161.92(9), Mo–Si1–N 70.07(8), Si1–Mo–N 39.41(7), N–Si1–C1 126.4(1), Mo–N–Si1 70.52(9), Mo–N–C25 135.5(2), Si1–N–C25 151.8(2). | ||
These data can be rationalized with the two resonances structures I and II for the silaiminoacyl ligand (Fig. 17). 5-Mo shows a characteristic deshielded 29Si NMR signal at δ = 142.9 ppm. Notably, 5-Mo is stereochemically not rigid in solution as evidenced by the single 13C NMR signal observed for the diastereotopic CO ligands or the single 1H and 13C NMR signals observed for the diastereotopic SiMe3 groups of the Tbb substituent (ESI, Chapter 2.8†). The stereomutation presumably involves a 180° rotation of the silaiminoacyl ligand about an axis connecting the Mo atom with the middle point of the SiN bond and leads to the enantiomer of 5-Mo, in which Si and N atoms have swapped coordination sites. The enantiomerization is too fast on the NMR time scale even at 223 K (1H NMR, 300.1 MHz, (D8)toluene) and has a lower activation barrier than the same process observed for the isoelectronic iminoacyl complexes [MoCp*(CO)2(η2C,N–C(R)NEt)] (R = Me, Et).148
 | ||
| Fig. 17 Resonance structures of 5-Mo. | ||
3. Conclusion
The series of novel reactions of the complexes [Cp*(CO)2MSi–Tbb] (M = Cr – W) presented in this work substantiate the synthetic potential of metal-silicon triple bonds. The different regiochemistry of MSi and MC bonds and the growing number of isolable silylidyne complexes with a variable metal d configuration and coordination number open many perspectives and objectives for a flourishing field of silicon-directed metal chemistry.
Data availability
The ESI† (.pdf) contains the syntheses, the spectroscopic data, the IR and NMR spectra, the crystallographic data and the structure refinement parameters of 1-Cr – 5-Mo, and details of the quantum chemical calculations of 3-Mo. The ESI† (.txt) contains the cartesian coordinates of the calculated structures: deposition numbers CCDC-2421358 (1-Cr), CCDC-2421359 (1-Mo), CCDC-2421360 (1-W), CCDC-2421361 (2-Mo), CCDC-2421362 (2-W·(toluene)) CCDC-2421363 (3-Mo), CCDC-2421364 (4-Mo) and CCDC-2421365 (5-Mo) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.Author contributions
A. C. F. designed and directed the project. K. T. prepared and characterized the compounds, analyzed the data, and wrote the first draft. G. S. performed the theoretical calculations and SCXRD studies. U. D. helped with the data analysis. A. C. F. revised and finalized the manuscript. All authors discussed the results and reviewed the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank the Rheinische Friedrich-Wilhelms Universität Bonn for the financial support of this work. We also thank S. Henn for preliminary experimental work and Mrs K. Kühnel-Lysek, Mrs C. Rödde and Dipl.-Ing. K. Prochnicki for technical support.References
- E. O. Fischer, Angew. Chem., Int. Ed., 1974, 86, 651–663 Search PubMed
.
-
E. O. Fischer, in Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Elsevier, 1976, vol. 14, pp. 1–32 Search PubMed
- F. G. A. Stone, Angew. Chem., Int. Ed. Engl., 1984, 23, 89–99 Search PubMed
- R. R. Schrock, Acc. Chem. Res., 1986, 19, 342–348 Search PubMed
-
H. P. Kim and R. J. Angelici, in Advances in Organometallic Chemistry, ed. B. W. O'Malley, Elsevier, 1987, vol. 27, pp. 51–111 Search PubMed
-
A. Mayr and H. Hoffmeister, in Advances in Organometallic Chemistry, ed. F. G. A. Stone and R. West, Elsevier, 1991, vol. 32, pp. 227–324 Search PubMed
- R. R. Schrock, Chem. Rev., 2002, 102, 145–180 CAS
- R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 3748–3759 Search PubMed
- M. Cui and G. Jia, J. Am. Chem. Soc., 2022, 144, 12546–12566 CAS
-
H. Fischer, E. O. Fischer, P. Hofmann, F. R. Kreissl, R. R. Schrock, U. Schubert and K. Weiss, Carbyne complexes, VCH publishers, New York, 1988, ISBN: 0-89573-849-X Search PubMed
- E. O. Fischer, G. Kreis, C. G. Kreiter, J. Müller, G. Huttner and H. Lorenz, Angew. Chem., Int. Ed. Engl., 1973, 12, 564–565 Search PubMed
- A. Fürstner, Angew. Chem., Int. Ed., 2013, 52, 2794–2819 CrossRef PubMed
- H. Ehrhorn and M. Tamm, Chem.–Eur. J., 2019, 25, 3190–3208 Search PubMed
- S. Huang, Z. Lei, Y. Jin and W. Zhang, Chem. Sci., 2021, 12, 9591–9606 RSC
-
D. Lee, I. Volchkov and S. Y. Yun, in Organic Reactions, ed. S. E. Denmark, Wiley, 2020, pp. 613–931 Search PubMed
- A. C. Filippou, D. Hoffmann and G. Schnakenburg, Chem. Sci., 2017, 8, 6290–6299 Search PubMed
- P. Ghana, M. I. Arz, G. Schnakenburg, M. Straßmann and A. C. Filippou, Organometallics, 2018, 37, 772–780 CrossRef CAS
- L. R. Maurer, J. Rump and A. C. Filippou, Inorganics, 2023, 11, 129 CrossRef CAS
- R. S. Simons and P. P. Power, J. Am. Chem. Soc., 1996, 118, 11966–11967 CrossRef CAS
- L. Pu, B. Twamley, S. T. Haubrich, M. M. Olmstead, B. V. Mork, R. S. Simons and P. P. Power, J. Am. Chem. Soc., 2000, 122, 650–656 CrossRef CAS
- E. Rivard and P. P. Power, Inorg. Chem., 2007, 46, 10047–10064 CAS
- A. C. Filippou, A. I. Philippopoulos, P. Portius and D. U. Neumann, Angew. Chem., Int. Ed., 2000, 39, 2778–2781 CrossRef CAS PubMed
- A. C. Filippou, P. Portius and A. I. Philippopoulos, Organometallics, 2002, 21, 653–661 CrossRef CAS
- A. C. Filippou, A. I. Philippopoulos, P. Portius and G. Schnakenburg, Organometallics, 2004, 23, 4503–4512 Search PubMed
- A. C. Filippou, G. Schnakenburg, A. I. Philippopoulos and N. Weidemann, Angew. Chem., Int. Ed., 2005, 44, 5979–5985 CrossRef CAS PubMed
- A. C. Filippou, N. Weidemann, A. I. Philippopoulos and G. Schnakenburg, Angew. Chem., Int. Ed., 2006, 45, 5987–5991 CrossRef CAS PubMed
- A. C. Filippou, P. Portius, A. I. Philippopoulos and H. Rohde, Angew. Chem., Int. Ed., 2003, 42, 445–447 CrossRef CAS PubMed
- A. C. Filippou, A. I. Philippopoulos and G. Schnakenburg, Organometallics, 2003, 22, 3339–3341 CrossRef CAS
- H. Rohde, M. Menzel, F. Renz and A. C. Filippou, Hyperfine Interact., 2008, 185, 129–132 CrossRef CAS
- A. C. Filippou, H. Rohde and G. Schnakenburg, Angew. Chem., Int. Ed., 2004, 43, 2243–2247 CrossRef CAS PubMed
- A. C. Filippou, N. Weidemann, G. Schnakenburg, H. Rohde and A. I. Philippopoulos, Angew. Chem., Int. Ed., 2004, 43, 6512–6516 CrossRef CAS PubMed
- A. C. Filippou, N. Weidemann and G. Schnakenburg, Angew. Chem., Int. Ed., 2008, 47, 5799–5802 CrossRef CAS PubMed
- S. Saini, A. Agarwal and S. K. Bose, Dalton Trans., 2020, 49, 17055–17075 Search PubMed
- H. Hashimoto and K. Nagata, Chem. Lett., 2021, 50, 778–787 Search PubMed
- T. J. Hadlington, Chem. Soc. Rev., 2024, 53, 9738–9831 CAS
- A. L. Allred and E. G. Rochow, J. Inorg. Nucl. Chem., 1958, 5, 269–288 CrossRef CAS
- S. D. Grumbine, R. K. Chadha and T. D. Tilley, J. Am. Chem. Soc., 1992, 114, 1518–1520 CAS
- B. V. Mork and T. D. Tilley, Angew. Chem., Int. Ed., 2003, 42, 357–360 Search PubMed
- A. C. Filippou, O. Chernov and G. Schnakenburg, Angew. Chem., Int. Ed., 2009, 48, 5687–5690 Search PubMed
- R. S. Ghadwal, H. W. Roesky, S. Merkel, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2009, 48, 5683–5686 CAS
- A. C. Filippou, O. Chernov, B. Blom, K. W. Stumpf and G. Schnakenburg, Chem.–Eur. J., 2010, 16, 2866–2872 Search PubMed
- A. C. Filippou, O. Chernov and G. Schnakenburg, Chem.–Eur. J., 2011, 17, 13574–13583 Search PubMed
- A. C. Filippou, Y. N. Lebedev, O. Chernov, M. Straßmann and G. Schnakenburg, Angew. Chem. Int. Ed., 2013, 125, 7112–7116 Search PubMed
- N. Wiberg, W. Niedermayer, G. Fischer, H. Nöth and M. Suter, Eur. J. Inorg. Chem., 2002, 1066–1070 Search PubMed
- Y. Sugiyama, T. Sasamori, Y. Hosoi, Y. Furukawa, N. Takagi, S. Nagase and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 1023–1031 CAS
- K. Suzuki, T. Matsuo, D. Hashizume and K. Tamao, J. Am. Chem. Soc., 2011, 133, 19710–19713 CrossRef CAS PubMed
- T. Agou, N. Hayakawa, T. Sasamori, T. Matsuo, D. Hashizume and N. Tokitoh, Chem.–Eur. J., 2014, 20, 9246–9249 CrossRef CAS PubMed
- A. C. Filippou, O. Chernov, K. W. Stumpf and G. Schnakenburg, Angew. Chem., Int. Ed., 2010, 49, 3296–3300 CrossRef CAS PubMed
-
O. Chernov, Novel Molecular Si(II) Precursors for Synthesis of the First Compounds with Metal-Silicon Triple Bonds, Dissertation, Rheinische Friedrich-Wilhelms-Universität Bonn, 2012, URN: https://nbn-resolving.org/urn:nbn:de:hbz:5n-29944 Search PubMed
- A. C. Filippou, B. Baars, O. Chernov, Y. N. Lebedev and G. Schnakenburg, Angew. Chem., Int. Ed., 2014, 53, 565–570 CrossRef CAS PubMed
-
B. Baars, Synthese und Reaktivität von kationischen Metallosilylidinen und Metallosilylenen, PhD thesis, Rheinische Friedrich-Wilhelms-Universität Bonn, published in Mensch und Buch Verlag 2017, ISBN: 978-3-86387-825-2
- P. Ghana, M. I. Arz, U. Chakraborty, G. Schnakenburg and A. C. Filippou, J. Am. Chem. Soc., 2018, 140, 7187–7198 Search PubMed
-
N. Wienkenhöver, 1,2-Dibromodisilenes: A Rich Source for Titanium Silylidyne Complexes, Acyclic Silylenes and Disilyne Dianions, PhD thesis, Rheinische Friedrich-Wilhelms-Universität Bonn, published in Mensch und Buch Verlag, 2017, ISBN: 978-3-86387-864-1
-
I. Papazoglou, Unprecedented Tetrylidyne Complexes of Group 6 and 10 Metals, PhD thesis, Rheinische Friedrich-Wilhelms-Universität Bonn, published in Dr Hut Verlag, 2017, ISBN: 978-3-8439-3155-7
- A. Lülsdorf, Tetrylidin-Komplexe des Titans, PhD thesis, Rheinische Friedrich-Wilhelms-Universität Bonn, published in Mensch und Buch Verlag, Berlin, 2021, ISBN: 978-3-96729-134-6.
-
T. Deckstein and A. C. Filippou, (PMe3)3CoSiTbb: A Cobalt Silylidyne Complex via Metathetical Exchange of Cobalt-Tetrel Triple Bonds, in 10th European Silicon Days, Toulouse, France, 2023 Search PubMed
- P. G. Hayes, Z. Xu, C. Beddie, J. M. Keith, M. B. Hall and T. D. Tilley, J. Am. Chem. Soc., 2013, 135, 11780–11783 CrossRef CAS PubMed
- T. Fukuda, T. Yoshimoto, H. Hashimoto and H. Tobita, Organometallics, 2016, 35, 921–924 Search PubMed
- T. Yoshimoto, H. Hashimoto, N. Hayakawa, T. Matsuo and H. Tobita, Organometallics, 2016, 35, 3444–3447 Search PubMed
- H. Hashimoto, K. Watanabe, T. Yoshimoto, N. Hayakawa, T. Matsuo and H. Tobita, Chem.–Eur. J., 2023, 29, e202302470 CrossRef CAS PubMed
- M. Matsuoka, K. Nagata, R. Ohno, T. Matsuo, H. Tobita and H. Hashimoto, Chem.–Eur. J., 2024, 30, e202303765 CrossRef CAS PubMed
- M. Lein, A. Szabó, A. Kovács and G. Frenking, Faraday Discuss., 2003, 124, 365–378 RSC
- N. Takagi, K. Yamazaki and S. Nagase, Bull. Korean Chem. Soc., 2003, 24, 832–836 CrossRef CAS
-
G. Schnakenburg, Quantenchemische Untersuchungen an Tetrela-Ylidin-Komplexen der 6, 8 und 9 Nebengruppe, PhD thesis, Rheinische Friedrich-Wilhelms-Universität Bonn, 2008
-
P. R. Wosniok, Beiträge zur Chemie und Reaktivitätsuntersuchung des ersten Silylidin-Komplexes Cp(CO)2MoSi(C6H3-2,6-Trip2), Rheinische Friedrich-Wilhelms-Universität Bonn, Diplomarbeit, 2012 Search PubMed
- A. C. Filippou, O. Chernov and G. Schnakenburg, Angew. Chem., Int. Ed., 2011, 50, 1122–1126 CrossRef CAS PubMed
- A. Hassner, J. Org. Chem., 1968, 33, 2684–2686 CrossRef CAS
- J. C. Jeffery, J. C. V. Laurie, I. Moore and F. G. A. Stone, J. Organomet. Chem., 1983, 258, C37–C40 CrossRef CAS
- F. R. Kreissl, W. J. Sieber, H. Keller, J. Riede and M. Wolfgruber, J. Organomet. Chem., 1987, 320, 83–90 CrossRef CAS
- A. C. Filippou, P. Portius, J. G. Winter and G. Kociok-Köhn, J. Organomet. Chem., 2001, 628, 11–24 CrossRef CAS
- T. J. Katz, T. H. Ho, N. Y. Shih, Y. C. Ying and V. I. W. Stuart, J. Am. Chem. Soc., 1984, 106, 2659–2668 CrossRef CAS
- T. M. Sivavec and T. J. Katz, Tetrahedron Lett., 1985, 26, 2159–2162 CrossRef CAS
- L. M. Atagi and J. M. Mayer, Organometallics, 1994, 13, 4794–4803 Search PubMed
- H. Fischer and C. Troll, J. Chem. Soc., Chem. Commun., 1994, 457–458 RSC
- A. Mayr, K. S. Lee and B. Kahr, Angew. Chem., Int. Ed. Engl., 1988, 27, 1730–1731 Search PubMed
- J. H. Freudenberger and R. R. Schrock, Organometallics, 1986, 5, 398–400 CrossRef CAS
- R. Goller, U. Schubert and K. Weiss, J. Organomet. Chem., 1993, 459, 229–232 CrossRef CAS
- M. Matsuoka, K. Nagata, R. Ohno, T. Matsuo and H. Hashimoto, Chem. Lett., 2024, 53, upae002 Search PubMed
-
P. Ghana, Synthesis, Characterization and Reactivity of Ylidyne and μ-Ylido Complexes Supported by Scorpionato Ligands, PhD thesis, Rheinische Friedrich-Wilhelms-Universität Bonn, Springer International Publishing AG, 2019, ISBN: 978-3-030-02624-0 978-3-030-02625-7
- P. Ghana, J. Rump, G. Schnakenburg, M. I. Arz and A. C. Filippou, J. Am. Chem. Soc., 2021, 143, 420–432 CrossRef CAS PubMed
- A. C. Filippou, D. Wössner, G. Kociok-Köhn and I. Hinz, J. Organomet. Chem., 1997, 541, 333–343 Search PubMed
- C. M. Stegmair, W. Schütt, W. Ullrich, P. Kiprof, J. Ostermeier and F. R. Kreißl, J. Organomet. Chem., 1993, 447, 251–257 CAS
- E. O. Fischer, T. L. Lindner and F. R. Kreissl, J. Organomet. Chem., 1976, 112, C27–C30 CAS
- E. O. Fischer, T. L. Lindner, G. Huttner, P. Friedrich, F. R. Kreißl and J. O. Besenhard, Chem. Ber., 1977, 110, 3397–3404 CAS
- E. Delgado, J. Hein, J. C. Jeffery, A. L. Ratermann, F. G. A. Stone and L. J. Farrugia, J. Chem. Soc., Dalton Trans., 1987, 1191–1199 CAS
- H. Wadepohl, U. Arnold, H. Pritzkow, M. J. Calhorda and L. F. Veiros, J. Organomet. Chem., 1999, 587, 233–243 CAS
- J. C. Jeffery, F. Gordon, A. Stone and G. K. Williams, Polyhedron, 1991, 10, 215–219 CAS
- P. Pyykkö, S. Riedel and M. Patzschke, Chem.–Eur. J., 2005, 11, 3511–3520 Search PubMed
- J. Potenza, P. Giordano, D. Mastropaolo and A. Efraty, Inorg. Chem., 1974, 13, 2540–2544 CAS
- J.-S. Huang and L. F. Dahl, J. Organomet. Chem., 1983, 243, 57–68 CAS
- T. Sugahara, J.-D. Guo, D. Hashizume, T. Sasamori, S. Nagase and N. Tokitoh, Dalton Trans., 2018, 47, 13318–13322 CAS
- A. C. Filippou, W. Grünleitner, E. O. Fischer, W. Imhof and G. Huttner, J. Organomet. Chem., 1991, 413, 165–179 CrossRef CAS
- A. C. Filippou and W. Grünleitner, Z. Naturforschung B, 1989, 44, 1572–1580 CrossRef CAS
- D. M. Tellers, S. J. Skoog, R. G. Bergman, T. B. Gunnoe and W. D. Harman, Organometallics, 2000, 19, 2428–2432 CrossRef CAS
- G. Frenking, I. Fernández, N. Holzmann, S. Pan, I. Krossing and M. Zhou, JACS Au, 2021, 1, 623–645 CrossRef CAS PubMed
- T. Mukaiyama, T. Fujisawa and T. Hyugaji, Bull. Chem. Soc. Jpn., 1962, 35, 687–690 CrossRef CAS
- N. P. Marullo and E. H. Wagener, J. Am. Chem. Soc., 1966, 88, 5034–5035 CrossRef CAS
- C. R. Flynn and J. Michl, J. Org. Chem., 1974, 39, 3442–3443 CAS
- D. Hoppe and L. Beckmann, Liebigs Ann. Chem., 1980, 1980, 1751–1764 Search PubMed
- J. Kohn and R. Langer, Biomaterials, 1986, 7, 176–182 CAS
- P. Adams and F. A. Baron, Chem. Rev., 1965, 65, 567–602 CAS
- X. Liu, X.-Q. Xiao, Z. Xu, X. Yang, Z. Li, Z. Dong, C. Yan, G. Lai and M. Kira, Organometallics, 2014, 33, 5434–5439 CAS
- H.-W. Lerner, M. Bolte and M. Wagner, Dalton Trans., 2017, 46, 8769–8773 CAS
- A CSD survey gave 81 structures with Mo–Si single bonds and 89 structures with W–Si single bonds (coordination number of Si = 4). The mean and median values of the M–Si bond lengths were 254.2 and 255.6 pm (M = Mo) and 256.4 and 257.4 pm (M = W), respectively.
- W. Beck, A. Melnikoff and R. Stahl, Angew. Chem., Int. Ed. Engl., 1965, 4, 692–693 Search PubMed
- A. C. Filippou, J. G. Winter, M. Feist, G. Kociok-Köhn and I. Hinz, Polyhedron, 1998, 17, 1103–1114 CAS
- A CSD search gave overall 11 structures of four-coordinate Si compounds featuring a siletane ring Si(X1)(X2)C=Y (X1, X2, Y = NR, O)..
- T. Fukuda, H. Hashimoto, S. Sakaki and H. Tobita, Angew. Chem., Int. Ed., 2016, 55, 188–192 CAS
- K. Weiss, U. Schubert and R. R. Schrock, Organometallics, 1986, 5, 397–398 CAS
- G. P. Mitchell and T. D. Tilley, J. Am. Chem. Soc., 1997, 119, 11236–11243 CAS
- M. Ochiai, H. Hashimoto and H. Tobita, Organometallics, 2012, 31, 527–530 Search PubMed
- H. Xie and Z. Lin, Organometallics, 2014, 33, 892–897 Search PubMed
- G. A. Olah, J. Nishimura and P. Kreienbuehl, J. Am. Chem. Soc., 1973, 95, 7672–7680 Search PubMed
- Z. Karpas, W. J. Stevens, T. J. Buckley and R. Metz, J. Phys. Chem., 1985, 89, 5274–5278 Search PubMed
- A. Schulz and A. Villinger, Chem.–Eur. J., 2010, 16, 7276–7281 Search PubMed
- T. Hansen, P. Vermeeren, K. W. J. Zijderveld, F. M. Bickelhaupt and T. A. Hamlin, Chem.–Eur. J., 2023, 29, e202301308 Search PubMed
- H. Hashimoto, M. Ochiai and H. Tobita, J. Organomet. Chem., 2007, 692, 36–43 Search PubMed
- Oxysilylidene complexes are scarce and a CSD search shows that only three tungsten compounds have been structurally characterized..
- B. V. Mork, T. D. Tilley, A. J. Schultz and J. A. Cowan, J. Am. Chem. Soc., 2004, 126, 10428–10440 Search PubMed
- M. Hirotsu, T. Nunokawa and K. Ueno, Organometallics, 2006, 25, 1554–1556 CrossRef CAS
- A. J. Schultz, K. L. Stearley, J. M. Williams, R. Mink and G. D. Stucky, Inorg. Chem., 1977, 16, 3303–3306 CrossRef CAS
- L. Brammer, D. Zhao, R. M. Bullock and R. K. McMullan, Inorg. Chem., 1993, 32, 4819–4824 Search PubMed
- P. P. Gaspar, A. M. Beatty, T. Chen, T. Haile, D. Lei, W. R. Winchester, J. Braddock-Wilking, N. P. Rath, W. T. Klooster, T. F. Koetzle, S. A. Mason and A. Albinati, Organometallics, 1999, 18, 3921–3932 Search PubMed
- M. Grellier, T. Ayed, J.-C. Barthelat, A. Albinati, S. Mason, L. Vendier, Y. Coppel and S. Sabo-Etienne, J. Am. Chem. Soc., 2009, 131, 7633–7640 CrossRef CAS PubMed
- M. Mantina, A. C. Chamberlin, R. Valero, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. A, 2009, 113, 5806–5812 CrossRef CAS PubMed
- J. Y. Corey, Chem. Rev., 2011, 111, 863–1071 Search PubMed
-
J. Schraml and J. M. Bellama, in Determination of Organic Structures by Physical Methods, ed. F. C. Nachod, J. J. Zuckerman and E. W. Randall, Academic Press, New York, vol. 6, 1976, pp. 203–269, ISBN: 978-0-323-14920-4 Search PubMed
- J. Y. Corey and J. Braddock-Wilking, Chem. Rev., 1999, 99, 175–292 Search PubMed
- W. McFarlane, J. Chem. Soc. A, 1967, 1275–1276 Search PubMed
- D. L. Lichtenberger, Organometallics, 2003, 22, 1599–1602 Search PubMed
- P. Meixner, K. Batke, A. Fischer, D. Schmitz, G. Eickerling, M. Kalter, K. Ruhland, K. Eichele, J. E. Barquera-Lozada, N. P. M. Casati, F. Montisci, P. Macchi and W. Scherer, J. Phys. Chem. A, 2017, 121, 7219–7235 Search PubMed
- W. Scherer, P. Meixner, K. Batke, J. E. Barquera-Lozada, K. Ruhland, A. Fischer, G. Eickerling and K. Eichele, Angew. Chem., Int. Ed., 2016, 55, 11673–11677 CrossRef CAS PubMed
-
R. Huisgen, in 1,3 Dipolar Cycloaddition Chemistry, ed. A. Padwa, John Wiley & Sons, New York, vol. 1, 1984, pp. 1–176, ISBN: 978047108364 Search PubMed
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 Search PubMed
- D. H. Ess and K. N. Houk, J. Am. Chem. Soc., 2008, 130, 10187 Search PubMed
- C. Cui, M. M. Olmstead, J. C. Fettinger, G. H. Spikes and P. P. Power, J. Am. Chem. Soc., 2005, 127, 17530–17541 Search PubMed
-
K. W. Stumpf, Germyliden- und Germylidin-Komplexe des Molybdäns, PhD thesis, Rheinische Friedrich-Wilhelms-Universität Bonn, published in Dr Hut Verlag, München, 2014, ISBN: 978-3-8439-1849-7
- H. Piana and U. Schubert, J. Organomet. Chem., 1988, 348, C19–C21 CrossRef CAS
- M. Driess and H. Pritzkow, Angew. Chem. Int. Ed. Engl., 1992, 31, 751–753 Search PubMed
- J. Barker and M. Kilner, Coord. Chem. Rev., 1994, 133, 219–300 Search PubMed
- M. P. Coles, Dalton Trans., 2006, 985 CAS
- S. Collins, Coord. Chem. Rev., 2011, 255, 118–138 CAS
- A CSD survey gave 17 structures of silaimines. The SiN bond lengths ranged from 153.3(2) pm to 162.6(1) pm with a mean and median value of 157.5 and 157.3 pm, respectively..
-
W. S. Sheldrick, in The Chemistry of Organic Silicon Compounds, Part 1, ed. S. Patai and Z. Rappoport, John Wiley & Sons, 1989, pp. 227–303, ISBN: 0-471-91441-X Search PubMed
- D. D. Choytun, L. D. Langlois, T. P. Johansson, C. L. B. Macdonald, G. W. Leach, N. Weinberg and J. A. C. Clyburne, Chem. Commun., 2004, 1842–1843 CAS
- C. M. Stegmair, W. Ullrich, W. Schütt, P. Kiprof and F. R. Kreißl, Chem. Ber., 1992, 125, 1571–1573 CAS
- Q. Zhu, S. Chen, F. Xu and J. Zhu, Inorg. Chem., 2020, 59, 7318–7324 CrossRef CAS PubMed
- A. C. Filippou, W. Grünleitner and C. Völkl, J. Organomet. Chem., 1991, 413, 181–203 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2421358–2421365. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc01063b |
| This journal is © The Royal Society of Chemistry 2025 |