
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning covalent bonding in zinc-based hybrid halides towards tunable room-temperature phosphorescence†
Yibo Cuia,
Jiawei Linb,
Kunjie Liua,
Yuhe Shaoa,
Dong Zhaoa,
Zhongnan Guo b,
Jing Zhao*a,
Zhiguo Xia*c and
Quanlin Liu*a
b,
Jing Zhao*a,
Zhiguo Xia*c and
Quanlin Liu*a
aThe Beijing Municipal Key Laboratory of New Energy Materials and Technologies, School of Materials Science and Engineering, University of Science and Technology Beijing, Beijing 100083, P. R. China. E-mail: jingzhao@ustb.edu.cn; qlliu@ustb.edu.cn
bThe Beijing Municipal Key Laboratory of New Energy Materials and Technologies, School of Materials Sciences and Engineering, Chemistry and Biological Engineering, University of Science and Technology Beijing, Beijing 100083, China
cState Key Laboratory of Luminescent Materials and Devices, Guangdong Provincial Key Laboratory of Fiber Laser Materials and Applied Techniques, Guangdong Engineering Technology Research, Development Centre of Special Optical Fiber Materials and Devices, School of Physics and Optoelectronics, South China University of Technology, Guangzhou, 510641, China. E-mail: xiazg@scut.edu.cn
First published on 10th April 2025
Abstract
Organic–inorganic metal halides with tunable and state room-temperature phosphorescence (RTP) properties in advanced luminescent materials have received broad interest. Herein, 2-(methylamino)pyridine (MAP), 2-[(methylamino)methyl]pyridine (MAMP), and 2-(2-methylaminoethyl)pyridine (MAEP) were designed and hybridized with Zn2+ and Cl−/Br−, yielding 11 hybrid materials. MAP-based compounds, with a narrow bandgap (3.57 eV), exhibit limited RTP due to inefficient intersystem crossing (ISC) and unstable triplet excitons. In contrast, MAMP (4.49 eV)- and MAEP (4.50 eV)-based compounds achieve enhanced RTP through bandgap alignment with Zn halides, enabling efficient energy transfer, ISC, and triplet exciton stabilization via strong hydrogen bonding and π-conjugation effects. Covalent bonding in MAMP and MAEP compounds provides greater rigidity and exciton stability than hydrogen-bonded systems, resulting in prolonged afterglow durations, while Br-bonding enhances ISC and spin–orbit coupling (SOC), and the weak interactions increase non-radiative decay, further reducing afterglow duration. Density functional theory calculations confirm the enhanced SOC in MAMP and MAEP compounds, further improving RTP efficiency. This work demonstrates the precise control of RTP properties, highlighting the potential in advanced anti-counterfeiting and emerging photonics applications.
1. Introduction
Room-temperature phosphorescent (RTP) materials have attracted widespread attention due to their potential in optoelectronic technologies.1 Previously, inorganic persistent materials, such as transition metal and rare-earth ion-doped gallates,2 aluminates,3,4 and sulfides,5,6 have achieved afterglow durations of several hours. However, challenges such as difficulties in controlling morphology and particle size hinder further development. In contrast, organic phosphorescent materials offer multiple advantages such as diverse molecular structures and excellent bio-compatibility.7–13 Recently, organic–inorganic metal halides (OIMHs), combining the flexibility of organic components with the stability of inorganic components, have emerged as promising luminescent materials.14 As an example, Zn2+-based OIMHs with RTP properties exhibit diverse photophysical properties influenced by molecular interactions and halide effects. Yan's group reported ZnCl2–BZT, ZnCl2–CBZT, and ZnBr2–BZT (BZT = 1H-benzotriazole; CBZT = 5-methyl-1H-benzotriazole), where strong hydrogen bonding and π–π stacking significantly enhance RTP.15 Lei's group designed (BTPP)2ZnCl4 (BTPP = benzyltriphenylphosphonium), achieving efficient triplet-state activation through heavy-atom effects.16 Zhao's group developed ZnCl2·R/S-2-MP (R/S = 2-methylpiperazine), utilizing N–H⋯Cl hydrogen bonding to induce structural chirality and stabilize triplet excitons.17 TBA2ZnCl4 (TBA = 3,4,5-trimethoxybenzylamine), explored by Fu's group, exhibited tunable RTP driven by halide incorporation ratios, achieving white-light emission via well-ordered molecular arrangements.18 Li's group highlighted the role of strong π–π interactions in [Zn(impc)2(H2O)4]·2H2O (Himpc = 5-(1H-1,2,4-imidazol-1-yl)nicotinic acid) in prolonging phosphorescence lifetimes.19 Kuang's group developed BAPPZn2(ClyBr1−y)8 (BAPP = 1,4-bis(3-ammoniopropyl)piperazinium; y = 0–1), demonstrating tunable afterglow lifetimes via halide-induced heavy-atom effects, enabling simple visual detection.20 These results collectively highlight the versatility of Zn2+-based OIMHs in RTP applications, emphasizing the importance of organic–inorganic interactions and halide substitution.In different OIMH systems, the [ZnX4]2− unit, characterized by its wide bandgap and optical inertness, typically exhibits no photoluminescence (PL). However, incorporating this inorganic unit into hybrids with organic components effectively preserves the photochemical activity of the organic moiety, enabling enhanced luminescence performance. The inorganic unit, through the heavy atom effect, markedly strengthens spin–orbit coupling (SOC),21 thereby substantially improving intersystem crossing (ISC) efficiency.22 Additionally, the formation of strong hydrogen-bonding networks between inorganic and organic components effectively restricts molecular vibrations and rotations, significantly suppressing non-radiative decay of the triplet state.23 This mechanism has been extensively investigated in pure organic RTP materials.24–26 Furthermore, π–π stacking interactions increase molecular rigidity, stabilizing aggregated triplet excitons and contributing to improved RTP performance.23,27 These synergistic effects enable organic–inorganic hybrid systems to exhibit exceptional phosphorescence properties at room temperature, offering valuable insights for the design and optimization of RTP materials. However, from a molecular design perspective, achieving diverse RTP performance through precise structural modulation remains a challenging yet promising research field.
The synthesized OIMHs underscore the pivotal role of coordinated bandgap modulation between organic and inorganic components in achieving RTP properties. Therefore, this work elucidates how halide substitution and bonding states influence RTP performance. By hybridizing 2-(methylamino)pyridine (MAP), 2-[(methylamino)methyl]pyridine (MAMP), and 2-(2-methylaminoethyl)pyridine (MAEP) with Zn2+ and Cl−/Br−, 11 OIMHs were synthesized, including the rare one-dimensional (1D) (MAP)ZnCl3. At 80 K, these cations exhibited afterglow emission but lacked RTP properties. MAP-based compounds, with a narrow bandgap (3.57 eV), displayed limited RTP due to ISC and unstable triplet excitons. In contrast, MAMP- and MAEP (4.50 eV)-based compounds demonstrated significantly enhanced RTP performance through optimal bandgap alignment with Zn halides, enabling efficient energy transfer, ISC, and stabilization of triplet excitons via strong hydrogen bonding and π-conjugation effects. Covalently bonded Type-C structures exhibited greater rigidity and exciton stability compared to hydrogen-bonded Type-H structures, leading to prolonged afterglow durations and superior thermal stability, with (MAMP)ZnCl3 retaining 50% of its initial emission intensity at 423 K. While Br− substitution enhanced ISC and SOC, it also increased non-radiative decay, thereby shortening afterglow durations. Density functional theory (DFT) and time-dependent density functional theory (TD-DFT) calculations reveal that the involvement of inorganic components in band-edge states, coupled with significantly enhanced SOC effects in MAMP and MAEP compounds, substantially improves RTP efficiency. This work underscores the potential of multi-modal modulation in optimizing RTP properties and provides a comprehensive framework for the design of advanced OIMHs tailored for optoelectronic and anti-counterfeiting applications.
2. Experimental
2.1. Chemicals
2-(Methylamino)pyridine (98% purity) was procured from Boer, 2-[(methylamino)methyl]pyridine (98% purity) from Beijing Huawei Ruike Chemical Technology Co., Ltd, and 2-(2-methylaminoethyl)pyridine (98% purity) from Aladdin. The synthesis utilized high-purity chemicals, including zinc(II) chloride (ZnCl2; 99.95%, Aladdin), zinc(II) bromide (ZnBr2; 99.9%, Aladdin), hydrobromic acid (HBr; 48 wt% in H2O, Aladdin), and hydrochloric acid (HCl; 36–38 wt% in H2O, Sinopharm). All chemicals were used as received without further purification.2.2. Synthesis
The raw materials and synthesis conditions for the new compounds are summarized in Table 1. Under reaction conditions A, the mixture was heated to 90 °C with stirring until clear, then naturally cooled to room temperature and stored at −4 °C for 2 days. Under reaction conditions B, after being heated to 90 °C and stirred until clear, the solution was maintained at this temperature for 200 minutes and then cooled to 20 °C at a rate of 1.1 °C h−1.| Compound | Organic | Ligand | Solvent | Crystallization pathway |
|---|---|---|---|---|
| (MAMP)ZnCl4 | 125 μL (1.0 mmol) | 0.136 g ZnCl2 (1.0 mmol) | 0.5 mL HCl | A |
| (MAMP)ZnBr4 | 125 μL (1.0 mmol) | 0.450 g ZnBr2 (1.0 mmol) | 1.0 mL HBr | A |
| (MAP)ZnCl3 | 210 μL (2.0 mmol) | 0.585 g ZnCl2 (4.3 mmol) | 0.5 mL HCl | A |
| (MAP)2ZnBr4 | 210 μL (2.0 mmol) | 0.540 g ZnBr2 (2.4 mmol) | 2.0 mL HBr | A |
| (MAEP)ZnCl4 | 140 μL (1.0 mmol) | 0.272 g ZnCl2 (2.0 mmol) | 0.5 mL HCl | A |
| (MAEP)ZnBr4 | 140 μL (1.0 mmol) | 0.518 g ZnBr2 (2.3 mmol) | 0.8 mL HBr | A |
| (MAP)[(MAP)ZnCl3] | 630 μL (6.0 mmol) | 0.408 g ZnCl2 (3.0 mmol) | 0.5 mL HCl | B |
| (MAMP)ZnCl3 | 575 μL (4.6 mmol) | 0.626 g ZnCl2 (4.6 mmol) | 0.83 mL HCl | B |
| (MAMP)ZnBr3 | 450 μL (3.6 mmol) | 0.810 g ZnBr2 (3.6 mmol) | 0.75 mL HBr | B |
| (MAEP)ZnCl3 | 255 μL (1.8 mmol) | 0.245 g ZnCl2 (1.8 mmol) | 0.33 mL HCl | B |
| (MAEP)ZnBr3 | 570 μL (4.0 mmol) | 0.900 g ZnBr2 (4.0 mmol) | 0.85 mL HCl | B |
Type-H structures typically crystallized at −4 °C and required a lower initial concentration of hydrochloric acid, whereas Type-C structures formed at room temperature and required a higher hydrochloric acid concentration.
2.3. Characterization
Single-crystal X-ray diffraction (SCXRD) measurements were performed using a Bruker APEX-II CCD diffractometer with Mo Kα radiation. The structural analysis of the series of compounds was carried out using Olex2 software.28–31 A PANalytical Empyrean Series 3 diffractometer was employed to carry out powder X-ray diffraction (PXRD) with a Cu Kα radiation source at 45 kV and 40 mA at room temperature. UV-Vis reflectance spectroscopy was performed using a Shimadzu UV-3600 spectrometer with BaSO4 as the reference. Room temperature photoluminescence excitation (PLE), PL spectra, PL decay curves and photoluminescence quantum yield (PLQY) were collected using an FLS920 fluorescence spectrophotometer equipped with a PMT detector and a 150 W Xe lamp. Low-temperature PLE spectra were obtained by cooling the sample to 80 K in liquid nitrogen using an Oxford Instruments attachment. Delayed phosphorescence spectra were measured using a Nanolog spectrometer equipped with a 450 W CW xenon lamp and an R928P photomultiplier tube detector, covering the wavelength range of 200–900 nm.The average decay lifetime is calculated using
| τave = (A1τ12 + A2τ22)/(A1τ1 + A2τ2) | (1) |
PVC was fabricated into the desired shape, and relevant phosphors were deposited at specific grids or locations. The device was subsequently sealed with adhesive tape.
All afterglow images were captured using a Xiaomi 12s Pro smartphone.
2.4. Calculation
Using CrystalExplorer software, the organic components were selected as the primary framework for surface construction. The atomic environment within a radius of 3.80 Å was expanded to enable high-performance computational analysis.33,343. Results and discussion
The detailed structural parameters of the 11 compounds are listed in Tables S1–S4† and crystal images are provided in Fig. S1.† Simulated diffraction patterns derived from SCXRD data align well with experimental PXRD patterns (Fig. S2 and S3†), confirming the phase purity of the compounds. As shown in Fig. 1a–c, protonation plays a critical role in modulating the oxidation state of organic cations, significantly affecting their structures and optical properties. The sp2-hybridized nitrogen in the pyridine ring strongly attracts electron density, making it prone to protonation or coordination with Zn2+ to form hydrogen or covalent bonds.38 Moreover, the protonation behavior of the side-chain nitrogen is influenced by the combined effects of the pyridine ring's electron-withdrawing properties and steric hindrance from the side chain.39 In MAP (C6H8N2), where the side-chain nitrogen is directly attached to the pyridine ring (Fig. 1a), minimal steric hindrance and a pronounced electron-withdrawing effect make the side-chain nitrogen less prone to protonation, resulting in the formation of monoprotonated MAP+. Conversely, in MAMP (C7H10N2) and MAEP (C8H12N2), the side-chain nitrogen is separated from the pyridine ring by a methyl or ethyl group, respectively (Fig. 1b and c). This separation reduces the electron-withdrawing effect and increases steric hindrance, facilitating protonation of both the pyridine and side-chain nitrogens, thereby yielding diprotonated MAMP2+ and MAEP2+. | ||
| Fig. 1 (a)–(c) Protonated organic compounds MAP+, MAMP2+, and MAEP2+, with steric hindrance highlighted in red; (d) representative Type-H compound (MAMP)ZnCl4; (e) representative Type-C compound (MAMP)ZnCl3; (f) rare 1D zinc-based compound (MAP)ZnCl3, with an enlarged view of its inorganic 1D chain. | ||
Zn2+-based hybrid halide compounds containing [ZnX4]2− (X = Cl, Br) are defined as Type-H structures, where organic and inorganic components are connected via hydrogen bonds. In (MAP)2ZnBr4, (MAMP)ZnX4, and (MAEP)ZnX4, the [ZnX4]2− units exist as isolated tetrahedra (Fig. 1d), consistent with previously reported compounds such as (PMA)2ZnBr4 (PMA = phenylmethylammonium).40 Research on zinc-based compounds reveals that organic components can directly coordinate with zinc centers, forming Zn–N bonds in Type-C structures. Here, zinc atoms are coordinated by three halogen atoms and one nitrogen atom from the organic ligand, creating covalently bonded frameworks (Fig. 1e). A similar coordination pattern is observed in [(pipda)4Zn2Br4]·H2O (pipda = 2-piperidylmethylamine).41–43 Additionally, a rare one-dimensional chain structure is identified in (MAP)ZnCl3, where [ZnCl4]2− units are linked via corner-sharing (Fig. 1f). The ordered arrangement of Zn2+ ions and ligands breaks centrosymmetry, as confirmed by second harmonic generation measurements, which show an intensity approximately 1.2 times that of KDP (Fig. S4†). The complete crystal structure details of the obtained compounds are presented in Fig. S5 and S6.†
The organic compounds MAP, MAMP, and MAEP exhibit visible afterglow durations exceeding 3 seconds under 365 nm ultraviolet (UV) excitation at 80 K (Fig. S7a†). Notably, MAP-based compounds also display visible afterglow at 80 K but lack RTP (Fig. S7b†). In contrast, RTP is exclusively observed in zinc-based compounds containing MAMP and MAEP cations, with afterglow durations ranging from 0.1 seconds for (MAMP)ZnBr4 to 1.3 seconds for (MAMP)ZnCl3 at room temperature (Fig. 2a). Furthermore, the consistent RTP durations observed in both crystalline and powdered forms indicate that surface defects have negligible influence on the afterglow behavior (Fig. S8†). Moreover, Type-C compounds demonstrate longer RTP durations compared to Type-H ones, with chlorides generally exhibiting significantly longer afterglow times than bromides. These findings highlight the critical roles of the organic cation, the organic–inorganic bonding mode, and the halide component in determining RTP performance.
 | ||
| Fig. 2 (a) Photographs of Zn-based halides with MAMP and MAEP ligands under 365 nm UV light and their visually observed delayed luminescence after the UV light is turned off. (b) (MAP)[(MAP)ZnCl3] and MAP, (c) (MAMP)ZnCl3 and MAMP, and (d) (MAEP)ZnCl3 and MAEP. (e) Fluorescence lifetimes of (MAP)[(MAP)ZnCl3], (MAMP)ZnCl3, and (MAEP)ZnCl3. | ||
Steady-state optical properties, including UV-visible diffuse reflectance and PL spectra, were systematically analyzed and are shown in Fig. S9 and S10.† Additionally, the PLQY of all compounds was measured, with the results shown in Fig. S11† and all photophysical parameters summarized in Table 2. Type-C compounds generally exhibit higher PLQY than their Type-H counterparts, except that the PLQY of (MAMP)ZnCl3 (Type-C, 6.54%) is slightly lower than that of (MAMP)ZnCl4 (Type-H, 7.10%). This underscores the influence of bonding mode on PLQY. Using Type-C chlorides as an example, the structure–property relationships of their optical performance are analyzed. (MAP)[(MAP)ZnCl3] exhibits a broad absorption peak at 345 nm with a shoulder at 270 nm, corresponding to an optical bandgap of 3.40 eV. Its PL spectrum reveals a single excitation peak at 340 nm, consistent with the absorption spectrum, and an emission peak at 430 nm (Fig. 2b). Further analysis shows that the excitation peak of (MAP)[(MAP)ZnCl3] aligns with that of the organic cation MAP, with a fluorescence lifetime of 1.74 ns (Fig. 2e). In contrast, the absorption spectra of (MAMP)ZnCl3 and (MAEP)ZnCl3 are primarily concentrated in the 200–290 nm range, corresponding to intramolecular electronic transitions of the organic cations. Both compounds exhibit prominent absorption peaks at 255 nm and weaker broad absorption features in the 300–450 nm range, which are attributed to charge transfer (CT) transitions between the organic ligands and Zn2+ centers.44,45 Their optical bandgaps are determined to be 4.39 eV and 4.43 eV, respectively (Fig. S9b†). The PLE spectrum of (MAMP)ZnCl3 exhibited a primary excitation peak at 430 nm and a small excitation peak at 315 nm (Fig. 2c). PL with different excitation wavelengths ranging from 285 nm to 430 nm revealed that the emission peak remained consistently stable at 480 nm (Fig. S12a†). The fluorescence lifetimes measured at 375 nm laser excitation and 430 nm nanosecond pulsed light source excitation are 1.91 ns and 2.79 ns, respectively (Fig. 2e and S12b†). Similarly, the PLE spectrum of (MAEP)ZnCl3 exhibits a primary excitation peak at 420 nm, corresponding to an emission peak at 485 nm, with a secondary excitation peak at 330 nm contributing to this emission (Fig. 2d). The fluorescence lifetime of (MAEP)ZnCl3 is measured to be 1.62 ns (Fig. 2e). Notably, the excitation peaks of (MAMP)ZnCl3 and (MAEP)ZnCl3 exhibit significant overlap with the emission peaks of their respective organic cations, indicating efficient energy transfer between the organic and inorganic components. This is further supported by CT transitions observed in the 300–450 nm range, with subsequent theoretical calculations providing additional confirmation. Additionally, all three compounds exhibit fluorescence lifetimes in the nanosecond range, confirming that the emission primarily originates from the singlet state of the organic components.
| Compound | Absorption peaks (nm) | Band gap (eV) | Ex (nm) | Em (nm) | Stokes shift (eV) | PLQY (%) |
|---|---|---|---|---|---|---|
| MAP | 310 | 3.57 | 375 | 430 | 22.56 | — |
| (MAP)[(MAP)ZnCl3] | 345 (270) | 3.40 | 340 | 430 | 13.79 | 9.59 |
| (MAP)ZnCl3 | 345 | 3.36 | 359 | 390 | 40.02 | 8.00 |
| (MAP)2ZnBr4 | 345 (270) | 3.37 | 352 | 385 | 37.60 | 13.89 |
| MAMP | 250 | 4.49 | 375 | 440 | 19.09 | — |
| (MAMP)ZnCl3 | 255 (330) | 4.39 | 430 (315) | 480 | 24.81 | 18.05 |
| (MAMP)ZnBr3 | 250 (430) | 4.42 | 430 (345) | 510 | 15.51 | 13.99 |
| (MAMP)ZnCl4 | 255 (320) | 4.36 | 395 | 485 | 13.79 | 2.30 |
| (MAMP)ZnBr4 | 260 | 3.96 | 400 (310) | 490 | 13.79 | 1.30 |
| MAEP | 220 (335) | 4.50 | 390 | 430 | 31.02 | — |
| (MAEP)ZnCl3 | 255 (430) | 4.43 | 420 (325) | 485 | 19.09 | 6.54 |
| (MAEP)ZnBr3 | 260 (430) | 4.43 | 420 | 490 | 17.72 | 8.86 |
| (MAEP)ZnCl4 | 255 (320) | 4.33 | 397 | 485 | 14.10 | 7.10 |
| (MAEP)ZnBr4 | 260 | 4.15 | 420 (315) | 515 | 13.06 | 4.30 |
Zn2+ indirectly affects the luminescence behavior of hybrid compounds by modulating the electronic states and excitation energy levels of organic ligands through coordination, thereby influencing their PL properties.46,47 PL spectral analysis under identical conditions shows that pure organic ligands emit as MAMP > MAEP ≫ MAP, while hybrid compounds follow (MAP)[(MAP)ZnCl3] > (MAMP)ZnCl3 > (MAEP)ZnCl3 (Fig. S12c and d†). Although MAMP and MAEP exhibit stronger emission in their pure forms, their hybrid compounds show lower emission intensities compared to (MAP)[(MAP)ZnCl3] due to differences in energy transfer mechanisms and the involvement of triplet states. Analyses indicate that the incorporation of inorganic components in (MAMP)ZnCl3 and (MAEP)ZnCl3 facilitates singlet-to-triplet transitions but also leads to energy dissipation, thereby reducing overall emission intensity.
Steady-state optical data (Table 2) highlight that (1) Type-H compounds exhibit larger Stokes shifts compared to Type-C compounds. The greater structural rigidity in Type-C compounds enhances their resistance to thermal quenching. This is evidenced by (MAMP)ZnCl3 retaining approximately 50% of its room-temperature PL intensity at 432 K, significantly higher than the 365 K observed for (MAMP)ZnCl4 (Fig. S13†) and (2) bromides exhibit a significant red shift in PL emission compared to chlorides, attributed to bandgap narrowing and enhanced spin–orbit coupling, consistent with the heavy-atom effect.18,48
The MAP-based compounds lack RTP due to their narrow bandgap (3.57 eV), which restricts ISC efficiency and prevents the formation of stable triplet excitons. In contrast, the optical bandgaps of MAMP (4.49 eV) and MAEP (4.50 eV) exhibit better alignment with those of the inorganic components (ZnCl2: 3.98 eV and ZnBr2: 3.92 eV), promoting enhanced energy transfer and facilitating efficient ISC (Fig. S9†). This alignment enables the stabilization of triplet excitons, significantly improving RTP performance. The matching relationship between the organic and inorganic components in Zn-based compounds has been extensively discussed by Zhou's Group.49 Overall, the narrower bandgap of MAP restricts ISC efficiency, hindering the formation of stable triplet excitons. In contrast, the optimized bandgap alignment in MAMP- and MAEP-based compounds facilitates efficient energy transfer and stabilizes triplet excitons.
The difference in afterglow duration between Type-C and Type-H structures is linked to rigidity and exciton stability. Type-C structures with strong covalent bonds stabilize triplet excitons, extending afterglow duration, while weaker hydrogen bonds in Type-H structures allow easier exciton relaxation and faster decay. Although Br− promotes SOC through a heavy-atom effect, its larger atomic radius and weaker ion–lattice interactions reduce exciton stability, increasing non-radiative decay and limiting its impact on phosphorescence intensity and afterglow duration. Previous research consistently shows that these factors shorten the afterglow in bromides.46,50
Taking (MAMP)ZnCl3 as an example, its phosphorescence emission behavior was thoroughly researched. Excitation at 375 nm (the optimal excitation wavelength for the organic component) resulted in an emission spectrum featuring a minor peak at 430 nm and a major peak at 480 nm (Fig. 3a). The lifetimes of these peaks were 1.75 ns and 1.91 ns, respectively, indicating their origin from the singlet state of the organic component. Further research revealed that excitation at 430 nm (the optimal excitation wavelength for the compound) allowed energy absorption by the inorganic component, which facilitated emission from both the singlet and triplet states via a CT mechanism. In the delayed phosphorescence spectrum (0.5 ms), two prominent emission peaks were observed at 470 nm and 537 nm (Fig. 3b), with lifetimes of 0.46 s and 0.42 s (Fig. 3c), characteristic of triplet state emissions with significantly longer decay times. Fig. S13a† shows that the relative intensity of the 537 nm peak increases with decreasing temperature, further validating this observation. Notably, the normalized fluorescence and phosphorescence spectra overlap significantly in the 470–480 nm region, likely due to the small energy gap (ΔEST) between the singlet (S1) and triplet (T1) states, which facilitates efficient energy transfer and emission processes.
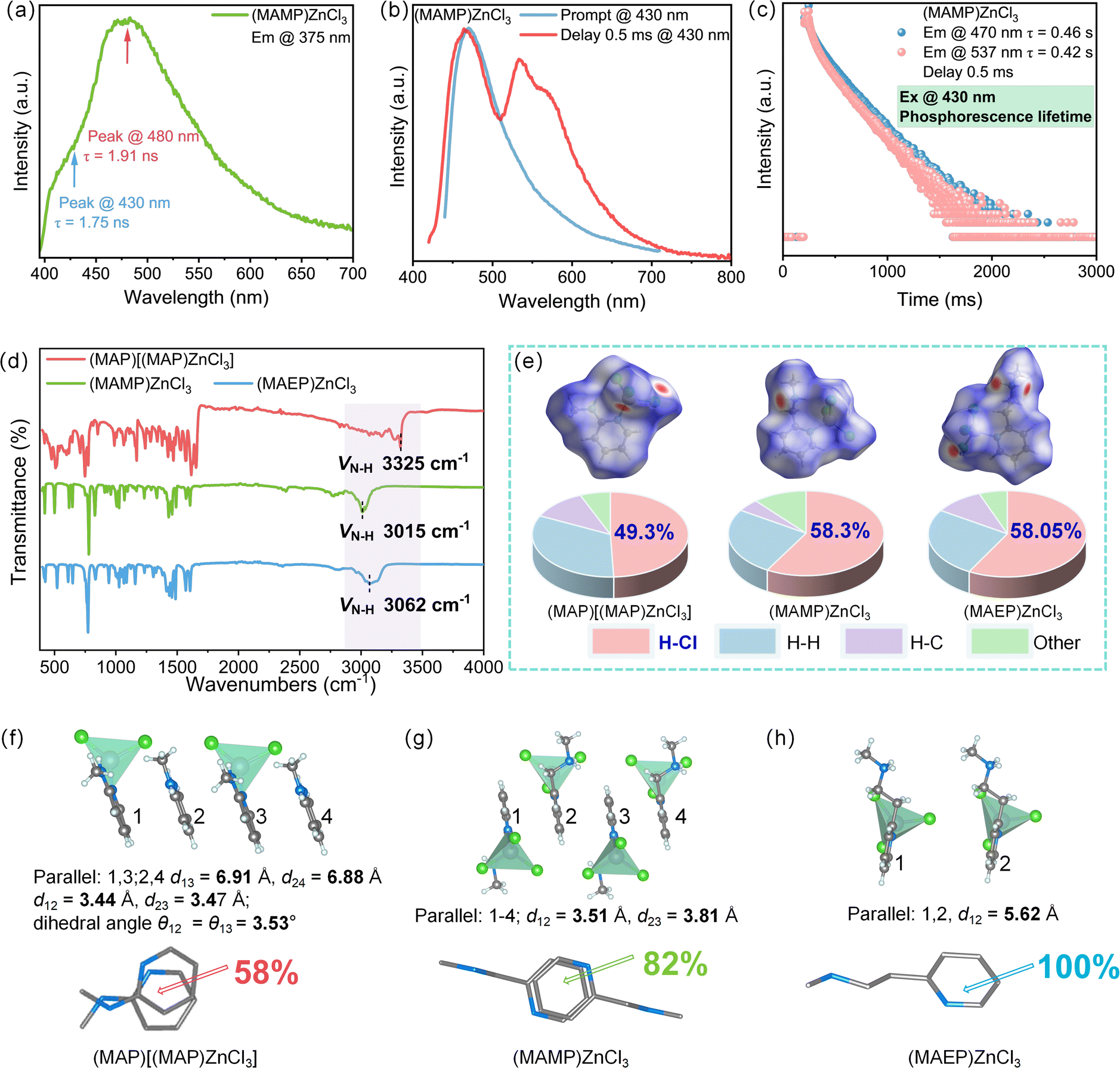 | ||
| Fig. 3 (a) PL spectrum of (MAMP)ZnCl3 under 375 nm excitation. (b) Delayed PL spectrum of (MAMP)ZnCl3. (c) Phosphorescence lifetime of (MAMP)ZnCl3. (d) FTIR spectra of (MAP)[(MAP)ZnCl3], (MAMP)ZnCl3, and (MAEP)ZnCl3. (e) Analysis of intermolecular interactions. (f)–(h) π–π distances and stacking overlap area analysis for (MAP)[(MAP)ZnCl3], (MAMP)ZnCl3, and (MAEP)ZnCl3, respectively. | ||
Moreover, strong hydrogen bonding and significant π-conjugation effects in MAMP and MAEP compounds further stabilized triplet excitons, promoting phosphorescence behavior. Fourier-transform infrared spectroscopy revealed characteristic vibrational modes of organic components, including N–H, C–H, and C–N stretching vibrations at 3200–3500 cm−1 and 2800–3000 cm−1, while Zn–Cl and Zn–N vibrations appeared in the 400–600 cm−1 and 500–700 cm−1 regions, respectively. In (MAP)[(MAP)ZnCl3], the N–H stretching vibration peak was observed at 3325 cm−1, while in (MAMP)ZnCl3 and (MAEP)ZnCl3, these peaks shifted to 3015 cm−1 and 3062 cm−1, respectively, indicating that protonation enhanced hydrogen bond strength and reduced vibrational frequency (Fig. 3d).51 CrystalExplorer analysis revealed that the proportion of intermolecular H–Cl interactions increased from 49.3% in (MAP)[(MAP)ZnCl3] to 58.3% in (MAMP)ZnCl3 and 58.0% in (MAEP)ZnCl3, further confirming the enhancement of hydrogen bonding strength (Fig. 3e). The specific contributions of different chemical bonds to intermolecular interactions are detailed in Table S5,† while the corresponding two-dimensional fingerprint plots are presented in Fig. S14.†
For π–π stacking, the overlap area of adjacent π rings increased from 58% in (MAP)[(MAP)ZnCl3] to 82% and 100% in (MAMP)ZnCl3 and (MAEP)ZnCl3, respectively. In terms of stacking distances, adjacent π rings in MAP were non-parallel, with a dihedral angle of 3.53° and distances of 3.44–3.47 Å, while the closest parallel π rings had a much larger separation of 6.88–6.91 Å, indicating weak stacking interactions. In contrast, (MAMP)ZnCl3 and (MAEP)ZnCl3 exhibited parallel stacking, with stacking distances of 3.51–3.81 Å and 5.62 Å, respectively (Fig. 3f–h).52 These results indicate that in the MAMP and MAEP systems, stronger hydrogen bonding and enhanced π–π conjugation effectively suppress non-radiative decay, significantly enhance SOC efficiency, and promote ISC, thereby stabilizing triplet excitons. These synergistic effects ultimately enable RTP in MAMP- and MAEP-based systems.21–23,52
To gain a deeper understanding of the PL behavior, DFT and TD-DFT calculations were conducted. The results reveal that the band gaps of (MAP)[(MAP)ZnCl3], (MAMP)ZnCl3, and (MAEP)ZnCl3 are 2.80 eV, 3.48 eV, and 3.83 eV, respectively, slightly smaller than their corresponding optical band gaps (Fig. S15a–c†). This underestimation issue primarily arises from discontinuities present in the exchange correlation energy.53–57 In (MAP)[(MAP)ZnCl3], the valence band maximum (VBM) and conduction band minimum (CBM) are primarily governed by the organic component, indicating that the optical properties are dominated by the organic moiety. In contrast, in (MAMP)ZnCl3 and (MAEP)ZnCl3, the CBM is mainly derived from the organic component, while the VBM is primarily contributed by the p-orbitals of Cl, highlighting the synergistic interaction between the organic and inorganic components in determining the optical behavior (Fig. S15d–f†).
Additional TD-DFT calculations and SOC coefficient analyses demonstrate that the ΔEST values between the S1 and T1 states are minimal, measuring 0.0022 eV, 0.0058 eV, and 0.0064 eV for (MAP)[(MAP)ZnCl3], (MAMP)ZnCl3, and (MAEP)ZnCl3, respectively, which facilitates efficient ISC. The ξ(S1, T1) coefficients for these compounds are 0.36 cm−1, 1.19 cm−1, and 2.00 cm−1, while the ξ(T1, S0) coefficients are 1.29 cm−1, 19.51 cm−1, and 12.45 cm−1, respectively. These findings indicate that MAMP-based and MAEP-based compounds exhibit significantly stronger SOC effects compared to MAP-based compounds, particularly in the ξ(T1, S0) transition. The enhanced SOC in MAMP-based and MAEP-based compounds increases the probability of phosphorescence emission by improving the transition efficiency from the triplet state to the ground state (Fig. 4a–c).58,59 As a result, MAMP-based and MAEP-based compounds display pronounced afterglow at room temperature, owing to their superior SOC effects that effectively promote phosphorescence emission.
 | ||
| Fig. 4 (a)–(c) The TD-DFT-calculated SOC coefficients and energy level diagrams for the compounds (MAP)[(MAP)ZnCl3], (MAMP)ZnCl3, and (MAEP)ZnCl3; insets display the charge distributions of the LUMO and HOMO. (d) Summary of the ΔEST values and the SOC coefficients for transitions between S1 and T1, and T1 and S0 for this series of compounds. (e) The proposed emission mechanism for (MAMP)ZnCl3. | ||
A comparison of the SOC results for the Type-C structure of (MAMP)ZnCl3 and the Type-H structure of (MAMP)ZnCl4 reveals that the Type-H structure exhibits stronger SOC effects during the singlet-to-triplet transition, whereas the Type-C structure demonstrates stronger SOC effects during the triplet-to-ground state transition. This distinction results in enhanced phosphorescence emission in the Type-C structure (Fig. 4b and S16a†). Although (MAMP)ZnBr3 exhibits a stronger SOC effect, the heavier atomic effect of the bromide ion compared to chloride enhances non-radiative transitions, thereby suppressing phosphorescence emission and significantly shortening its afterglow duration (Fig. 4b and S16b†). The configurations of singlet and triplet excited states for five representative compounds, determined through TD-DFT calculations, are summarized in Tables S6–S10.†
Using (MAMP)ZnCl3 as an example, its RTP mechanism is proposed as follows: upon excitation at 375 nm, electrons transition from the ground state to the excited singlet state (S*), followed by non-radiative relaxation to a lower excited state. The system then relaxes radiatively to the ground state, emitting fluorescence at 430 nm with a lifetime of 1.75 ns. This 430 nm emission subsequently excites the inorganic component, transferring energy via CT to the singlet state of the organic component, resulting in fluorescence emission at 480 nm with a lifetime of 1.91 ns. This process further facilitates ISC, populating the triplet state. After a 0.5 ms delay, significant phosphorescence emissions are observed at 470 nm and 537 nm, with lifetimes of 0.46 s and 0.42 s, respectively. The small ΔEST allows for the simultaneous observation of fluorescence and phosphorescence within the 470–480 nm range (Fig. 4e).
The phosphorescence-based encryption model enables multi-layered anti-counterfeiting by encoding information under UV excitation, where emitted light signifies “1” and its absence denotes “0”. Fig. 5a and b show the system's structure and material arrangement for encoding. Upon removing the light source, the device decodes information sequentially, displaying corresponding characters or data. Using ASCII encoding, long-RTP (MAMP)ZnCl3, short-RTP (MAMP)ZnBr3, non-RTP (MAP)[(MAP)ZnCl3] and BaSO4 (control) are precisely positioned on the substrate (Fig. 5c). When UV is turned on/off at intervals, decoded outputs include “wwuj,” “ustb,” and “USTB” and numerical data such as “888,” “463,” and “151” (Fig. 5d). This work highlights the potential of phosphorescent materials in anti-counterfeiting and offers insights into optimizing phosphorescence-based encryption for advanced photonic materials and authentication solutions.
 | ||
| Fig. 5 (a) and (b) Structural diagram of the system, illustrating the integration of various phosphorescent materials and the operational logic. (c) Decoding process displaying ASCII-encoded characters at different time intervals with the UV on and UV off. (d) Anti-counterfeiting application for numerical data. | ||
4. Conclusion
In this work, we investigate the critical influence of organic ligands, organic–inorganic bonding states, and halide substitution on modulating the photophysical properties of Zn2+-based hybrid halides, thereby achieving efficient RTP with tunable afterglow durations. Our findings show that MAP-based compounds lack RTP due to smaller optical bandgaps and inefficient ISC, leading to the absence of stable triplet excitons. In contrast, MAMP- and MAEP-based compounds, with larger bandgaps better aligned with the inorganic components, improve energy transfer and ISC efficiency, enabling pronounced RTP. Furthermore, Type-C structures, characterized by covalent Zn–N bonds, enhance exciton stability and prolong afterglow durations, while Type-H structures, connected via hydrogen bonds, exhibit faster exciton relaxation and shorter afterglow times. Halide substitution also significantly influences RTP; bromides, while enhancing SOC, suffer from increased non-radiative decay, which limits afterglow intensity. By integrating the experimental results and theoretical analyses, this work establishes robust structure–property relationships and demonstrates the potential of these materials for multi-layered anti-counterfeiting applications, providing valuable insights for the design of advanced photonic materials with customizable optical properties.Data availability
Detailed crystallographic information and supplementary figures and tables can be found in the ESI.† Crystallographic data are available via the Cambridge Crystallographic Data Centre (CCDC): 2409875, 2409876, 2409996, 2409998, 2410002, 2413621, 2413624, 2413627, 2413629, 2413633, and 2413733. The data supporting this article have been included as part of the ESI.†Author contributions
Yibo Cui was responsible for data curation, investigation, writing – original draft, conceptualization, formal analysis, and visualization. Jiawei Lin and Zhongnan Guo were involved in software development. Kunjie Liu contributed to methodology. Dong Zhao provided resources. Yuhe Shao supervised the project (supervision). Jing Zhao contributed to conceptualization and was responsible for funding acquisition. Zhiguo Xia and Quanlin Liu also contributed to conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key Research and Development Program of China (No. 2021YFA0718900). This work was also supported by the National Natural Science Foundation of China (52272133) and the Fundamental Research Funds for the Central Universities FRF-EYIT-23-04.Notes and references
- X. Li, Y. Wang, Z. Zhang, S. Cai, Z. An and W. Huang, Recent Advances in Room-Temperature Phosphorescence Metal–Organic Hybrids: Structures, Properties, and Applications, Adv. Mater., 2024, 36, 2308290 CrossRef CAS PubMed.
- S. Liu, J. Du, Z. Song, C. Ma and Q. Liu, Intervalence charge transfer of Cr3+-Cr3+ aggregation for NIR-II luminescence, Light: Sci. Appl., 2023, 12, 181 CrossRef CAS PubMed.
- M. Pellerin, E. Glais, T. Lecuyer, J. Xu, J. Seguin, S. Tanabe, C. Chanéac, B. Viana and C. Richard, LaAlO3: Cr3+, Sm3+: nano-perovskite with persistent luminescence for in vivo optical imaging, J. Lumin., 2018, 202, 83–88 CrossRef CAS.
- Y. Katayama, H. Kobayashi and S. Tanabe, Deep-red persistent luminescence in Cr3+-doped LaAlO3 perovskite phosphor for in vivo imaging, Appl. Phys. Express, 2014, 8, 012102 CrossRef.
- D. Jia, J. Zhu and B. Wu, Improvement of persistent phosphorescence of Ca0. 9Sr0. 1S: Bi3+ by codoping Tm3+, J. Lumin., 2000, 91, 59–65 CrossRef CAS.
- W. Hoogenstraaten and H. Klasens, Some properties of zinc sulfide activated with copper and cobalt, J. Electrochem. Soc., 1953, 100, 366 CrossRef CAS.
- H. Wang, H. Shi, W. Ye, X. Yao, Q. Wang, C. Dong, W. Jia, H. Ma, S. Cai and K. Huang, Amorphous Ionic Polymers with Color-Tunable Ultralong Organic Phosphorescence, Angew. Chem., Int. Ed., 2019, 58, 18776–18782 CrossRef CAS PubMed.
- X. Wang, H. Shi, H. Ma, W. Ye, L. Song, J. Zan, X. Yao, X. Ou, G. Yang and Z. Zhao, Organic phosphors with bright triplet excitons for efficient X-ray-excited luminescence, Nat. Photonics, 2021, 15, 187–192 CrossRef CAS.
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Molecular afterglow imaging with bright, biodegradable polymer nanoparticles, Nat. Biotechnol., 2017, 35, 1102–1110 CrossRef CAS PubMed.
- L. Bian, H. Shi, X. Wang, K. Ling, H. Ma, M. Li, Z. Cheng, C. Ma, S. Cai and Q. Wu, Simultaneously enhancing efficiency and lifetime of ultralong organic phosphorescence materials by molecular self-assembly, J. Am. Chem. Soc., 2018, 140, 10734–10739 CrossRef CAS PubMed.
- Z. Zhao, A. Li and W. Z. Yuan, Nonconventional Luminophores: Emission Mechanism, Regulation, and Applications, Acc. Chem. Res., 2025, 12667–12673 Search PubMed.
- T. Yang, Y. Li, Z. Zhao and W. Z. Yuan, Clustering-triggered phosphorescence of nonconventional luminophores, Sci. China: Chem., 2023, 66, 367–387 CrossRef CAS.
- B. Liu, B. Chu, L. Zhu, H. Zhang, W.-Z. Yuan, Z. Zhao, W.-M. Wan and X.-H. Zhang, Clusteroluminescence: a gauge of molecular interaction, Chin. Chem. Lett., 2023, 34, 107909 CrossRef CAS.
- M. Li and Z. Xia, Recent progress of zero-dimensional luminescent metal halides, Chem. Soc. Rev., 2021, 50, 2626–2662 RSC.
- B. Zhou and D. Yan, Color-tunable persistent luminescence in 1D zinc–organic halide microcrystals for single-component white light and temperature-gating optical waveguides, Chem. Sci., 2022, 13, 7429–7436 RSC.
- J.-Q. Zhao, D.-Y. Wang, T.-Y. Yan, Y.-F. Wu, Z.-L. Gong, Z.-W. Chen, C.-Y. Yue, D. Yan and X.-W. Lei, Synchronously Improved Multiple Afterglow and Phosphorescence Efficiencies in 0D Hybrid Zinc Halides with Ultrahigh Anti-Water Stabilities, Angew. Chem., Int. Ed., 2024, e202412350 CAS.
- Y. Wang, C. Wang, M. Sun, P. Zhao, X. Fang, J. Zhang, Y. Guo and G. Zhao, Room Temperature Phosphorescence in Chiral 0D Zn(II) Metal Halide Crystals for Multiple Anti-Counterfeiting, Adv. Opt. Mater., 2024, 12, 2301843 CrossRef CAS.
- Z. Hua, H. Yu, H. Gong, P. Li, T. Zhang, Z. Deng, W. Sun, Y. Tian and H. Fu, Heavy-Atom Effect Regulating Room-Temperature Phosphorescence of Organic–Inorganic Zn-Based Halides for White-Light Emission, J. Phys. Chem. Lett., 2024, 15, 4729–4736 CrossRef CAS PubMed.
- J. Liu, Z. Chen, J. Hu, H. Sun, Y. Liu, Z. Liu and J. Li, Time-resolved color-changing long-afterglow for security systems based on metal–organic hybrids, Inorg. Chem. Front., 2022, 9, 584–591 RSC.
- J. H. Wei, W. T. Ou, J. B. Luo and D. B. Kuang, Zero-Dimensional Zn-Based Halides with Ultra-Long Room-Temperature Phosphorescence for Time-Resolved Anti-Counterfeiting, Angew. Chem., Int. Ed., 2022, 61, e202207985 CrossRef CAS PubMed.
- S. Koseki, M. W. Schmidt and M. S. Gordon, MCSCF/6-31G (d, p) calculations of one-electron spin-orbit coupling constants in diatomic molecules, J. Phys. Chem., 1992, 96, 10768–10772 CrossRef CAS.
- J. Yu, H. Ma, W. Huang, Z. Liang, K. Zhou, A. Lv, X.-G. Li and Z. He, Purely organic room-temperature phosphorescence endowing fast intersystem crossing from through-space spin–orbit coupling, JACS Au, 2021, 1, 1694–1699 CrossRef CAS PubMed.
- L. Zhou, K. Li, Y. Chang, Y. Yao, Y. Peng, M. Li and R. He, High-efficiency color-tunable ultralong room-temperature phosphorescence from organic–inorganic metal halides via synergistic inter/intramolecular interactions, Chem. Sci., 2024, 15, 10046–10055 RSC.
- Z. Chai, C. Wang, J. Wang, F. Liu, Y. Xie, Y.-Z. Zhang, J.-R. Li, Q. Li and Z. Li, Abnormal room temperature phosphorescence of purely organic boron-containing compounds: the relationship between the emissive behavior and the molecular packing, and the potential related applications, Chem. Sci., 2017, 8, 8336–8344 RSC.
- M. S. Kwon, D. Lee, S. Seo, J. Jung and J. Kim, Tailoring intermolecular interactions for efficient room-temperature phosphorescence from purely organic materials in amorphous polymer matrices, Angew. Chem., 2014, 126, 11359–11363 CrossRef.
- J. Yang, X. Zhen, B. Wang, X. Gao, Z. Ren, J. Wang, Y. Xie, J. Li, Q. Peng and K. Pu, The influence of the molecular packing on the room temperature phosphorescence of purely organic luminogens, Nat. Commun., 2018, 9, 840 CrossRef PubMed.
- Y. H. Liu, B. L. Zhang, Y. J. Wang, X. Y. Zhang, Y. B. Shang, T. C. Liu, X. W. Lei, Z. W. Chen and C. Y. Yue, Cationic Engineering Strategy to Achieve Controllable Room-Temperature-Phosphorescence in Hybrid Zinc Halides, Adv. Opt. Mater., 2024, 2303285 CrossRef CAS.
- G. M. Sheldrick and T. R. Schneider, in Methods in enzymology, Elsevier, 1997, vol. 277, pp. 319–343 Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- J. Lübben, C. M. Wandtke, C. B. Hübschle, M. Ruf, G. M. Sheldrick and B. Dittrich, Aspherical scattering factors for SHELXL–model, implementation and application, Acta Crystallogr., Sect. A: Found. Adv., 2019, 75, 50–62 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- S. Kurtz and T. Perry, A powder technique for the evaluation of nonlinear optical materials, J. Appl. Phys., 1968, 39, 3798–3813 CrossRef CAS.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, CrystalExplorer: a program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- W. Kohn and L. J. Sham, Self-Consistent Equations Including Exchange and Correlation Effects, Phys. Rev., 1965, 140, A1133–A1138 CrossRef.
- P. Hohenberg and W. Kohn, Inhomogeneous Electron Gas, Phys. Rev., 1964, 136, B864–B871 CrossRef.
- E. Klingsberg, Pyridine and Its Derivatives, John Wiley & Sons, 2009, part 1, vol. 14 Search PubMed.
- B. S. Schafman and P. G. Wenthold, Regioselectivity of pyridine deprotonation in the gas phase, J. Org. Chem., 2007, 72, 1645–1651 CrossRef CAS PubMed.
- S. He, S. Hao, L. Fan, K. Liu, C. Cai, C. Wolverton, J. Zhao and Q. Liu, Efficient Solar Spectrum-Like White-Light Emission in Zinc-Based Zero-Dimensional Hybrid Metal Halides, Adv. Opt. Mater., 2023, 11, 2300218 CrossRef CAS.
- Y. Wang, T. Zhou, J. Chen, H. Qin, J. Wu, Q. Zhang, J. Zheng, X. Li, Y. Y. Sun and Y. He, Zero-Dimensional Organic–Inorganic Hybrid Zinc Halides for Multiple Applications in Anti-Counterfeiting, X-Ray Imaging and White LEDs, Adv. Opt. Mater., 2024, 12, 2301864 CrossRef CAS.
- J. Chen, H.-Y. Wu, M.-B. Xu, M.-C. Wang, Q.-Q. Chen, B.-X. Li, C.-L. Hu and K.-Z. Du, Halide-driven polarity tuning and optimized SHG-bandgap balance in (C4H11N2)ZnX3 (X= Cl, Br, I), Inorg. Chem. Front., 2024, 11, 5587–5597 RSC.
- H. Zhang, X. Jiang, Y. Zhang, K. Duanmu, C. Wu, Z. Lin, J. Xu, J. Yang, Z. Huang and M. G. Humphrey, Toward Strong UV-Vis-NIR Second-Harmonic Generation by Dimensionality Engineering of Zinc Thiocyanates, J. Am. Chem. Soc., 2024 Search PubMed.
- Y.-P. Du, Q. Wang, M.-Y. Zhu, Y.-J. Ma, J.-H. Li and G.-M. Wang, Halogen Engineering Strategy-Induced Color-Tunable Room Temperature Phosphorescence in Metal–Organic Halides, Inorg. Chem., 2024, 63, 17127–17133 CrossRef CAS PubMed.
- X. Zhang, Y. Xiong, K. Liu, N. Wang, L. Fan, W. Li, X. Zhao, J. Zhao and Q. Liu, Thermal stable zinc-based hybrid halides with high external quantum efficiency as temperature detectors, J. Mater. Chem. C, 2022, 10, 13137–13142 RSC.
- L. J. Xu, A. Plaviak, X. Lin, M. Worku, Q. He, M. Chaaban, B. J. Kim and B. Ma, Metal Halide Regulated Photophysical Tuning of Zero-Dimensional Organic Metal Halide Hybrids: From Efficient Phosphorescence to Ultralong Afterglow, Angew. Chem., 2020, 132, 23267–23271 CrossRef.
- A. Yangui, R. Roccanova, T. M. McWhorter, Y. Wu, M.-H. Du and B. Saparov, Hybrid organic–inorganic halides (C5H7N2)2MBr4 (M = Hg, Zn) with high color rendering index and high-efficiency white-light emission, Chem. Mater., 2019, 31, 2983–2991 CrossRef CAS.
- Y.-J. Ma, Z. Qi, G. Xiao, X. Fang and D. Yan, Metal-Halide Coordination Polymers with Excitation Wavelength-and Time-Dependent Ultralong Room-Temperature Phosphorescence, Inorg. Chem., 2022, 61, 16477–16483 CrossRef CAS PubMed.
- Q. Ren, G. Zhou, Y. Mao, N. Zhang, J. Zhang and X.-M. Zhang, Optical activity levels of metal centers controlling multi-mode emissions in low-dimensional hybrid metal halides for anti-counterfeiting and information encryption, Chem. Sci., 2024, 15, 16536–16545 RSC.
- E. Lucenti, A. Forni, C. Botta, L. Carlucci, C. Giannini, D. Marinotto, A. Pavanello, A. Previtali, S. Righetto and E. Cariati, Cyclic triimidazole derivatives: intriguing examples of multiple emissions and ultralong phosphorescence at room temperature, Angew. Chem., Int. Ed., 2017, 56, 16302–16307 CrossRef CAS PubMed.
- D. Y. Liu, L. Y. Xiong, X. Y. Dong, Z. Han, H. L. Liu and S. Q. Zang, Reversible Local Protonation-Deprotonation: Tuning Stimuli-Responsive Circularly Polarized Luminescence in Chiral Hybrid Zinc Halides for Anti-Counterfeiting and Encryption, Angew. Chem., Int. Ed., 2024, 63, e202410416 CrossRef CAS PubMed.
- L. Zhou, K. Li, Y. Chang, Y. Yao, Y. Peng, M. Li and R. He, High-efficiency color-tunable ultralong room-temperature phosphorescence from organic–inorganic metal halides via synergistic inter/intramolecular interactions, Chem. Sci., 2024, 15, 10046 RSC.
- J. P. Perdew and M. Levy, Physical Content of the Exact Kohn-Sham Orbital Energies: Band Gaps and Derivative Discontinuities, Phys. Rev. Lett., 1983, 51, 1884–1887 CrossRef CAS.
- R. Godby, M. Schlüter and L. Sham, Trends in self-energy operators and their corresponding exchange-correlation potentials, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 36, 6497 CrossRef CAS PubMed.
- M. I. Houchati, H. Ferjani, Y. B. Smida, A. Oueslati, N. Chniba-Boudjada, O. Fabelo, J.-F. Bardeau, W. Paulus, M. Ceretti and A. H. Hamzaoui, Green luminescence in zero-dimensional lead bromide hybrid material (C5H9N3)2PbBr6: experimental and theoretical studies, J. Mol. Struct., 2024, 1295, 136687 CrossRef CAS.
- X. Wen, J. Cheng, P. Qian, Z. Zhang, H. Zeng, L. Huang, G. Zou and Z. Lin, Synthesis and structure-dependent optical properties of two new organic–inorganic hybrid antimony(III) chlorides, Dalton Trans., 2024, 53, 260–266 RSC.
- K. Liu, S. Hao, J. Cao, J. Lin, L. Fan, X. Zhang, Z. Guo, C. Wolverton, J. Zhao and Q. Liu, Antimony doping to enhance luminescence of tin(IV)-based hybrid metal halides, Inorg. Chem. Front., 2022, 9, 3865–3873 RSC.
- G. Zhou, Y. Mao, J. Zhang, Q. Ren, M. S. Molokeev, Z. Xia and X. M. Zhang, Dynamic Phosphorescence/Fluorescence Switching in Hybrid Metal Halides Toward Time-Resolved Multi-Level Anti-Counterfeiting, Adv. Funct. Mater., 2024, 2413524 Search PubMed.
- J. Q. Zhao, D. Y. Wang, T. Y. Yan, Y. F. Wu, Z. L. Gong, Z. W. Chen, C. Y. Yue, D. Yan and X. W. Lei, Synchronously Improved Multiple Afterglow and Phosphorescence Efficiencies in 0D Hybrid Zinc Halides With Ultrahigh Anti-Water Stabilities, Angew. Chem., Int. Ed., 2024, e202412350 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2409875, 2409876, 2409996, 2409998, 2410002, 2413621, 2413624, 2413627, 2413629, 2413633 and 2413733. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00931f |
| This journal is © The Royal Society of Chemistry 2025 |