
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegration of RNAseq transcriptomics and N-glycomics reveal biosynthetic pathways and predict structure-specific N-glycan expression†
Michael Russelle S.
Alvarez
 a,
Xavier A.
Holmes
a,
Armin
Oloumi
a,
Sheryl Joyce
Grijaldo-Alvarez
a,
Ryan
Schindler
a,
Qingwen
Zhou
a,
Anirudh
Yadlapati
a,
Atit
Silsirivanit
c and
Carlito B.
Lebrilla
*ab
a,
Xavier A.
Holmes
a,
Armin
Oloumi
a,
Sheryl Joyce
Grijaldo-Alvarez
a,
Ryan
Schindler
a,
Qingwen
Zhou
a,
Anirudh
Yadlapati
a,
Atit
Silsirivanit
c and
Carlito B.
Lebrilla
*ab
aDepartment of Chemistry, University of California, Davis, Davis, California, USA. E-mail: cblebrilla@ucdavis.edu
bDepartment of Chemistry, Biochemistry, Molecular, Cellular and Developmental Biology Graduate Group, University of California, Davis, Davis, California, USA
cDepartment of Biochemistry, Faculty of Medicine, Khon Kaen University, Khon Kaen, Thailand
First published on 4th April 2025
Abstract
The processes involved in protein N-glycosylation represent new therapeutic targets for diseases but their stepwise and overlapping biosynthetic processes make it challenging to identify the specific glycogenes involved. In this work, we aimed to elucidate the interactions between glycogene expression and N-glycan abundance by constructing supervised machine-learning models for each N-glycan composition. Regression models were trained to predict N-glycan abundance (response variable) from glycogene expression (predictors) using paired LC-MS/MS N-glycomic and 3′-TagSeq transcriptomic datasets from cells derived from multiple tissue origins and treatment conditions. The datasets include cells from several tissue origins – B cell, brain, colon, lung, muscle, prostate – encompassing nearly 400 N-glycan compounds and over 160 glycogenes filtered from an 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-gene transcriptome. Accurate models (validation R2 > 0.8) predicted N-glycan abundance across cell types, including GLC01 (lung cancer), CCD19-Lu (lung fibroblast), and Tib-190 (B cell). Model importance scores ranked glycogene contributions to N-glycan predictions, revealing significant glycogene associations with specific N-glycan types. The predictions were consistent across input cell quantities, unlike LC-MS/MS glycomics which showed inconsistent results. This suggests that the models can reliably predict N-glycosylation even in samples with low cell amounts and by extension, single-cell samples. These findings can provide insights into cellular N-glycosylation machinery, offering potential therapeutic strategies for diseases linked to aberrant glycosylation, such as cancer, and neurodegenerative and autoimmune disorders.
000-gene transcriptome. Accurate models (validation R2 > 0.8) predicted N-glycan abundance across cell types, including GLC01 (lung cancer), CCD19-Lu (lung fibroblast), and Tib-190 (B cell). Model importance scores ranked glycogene contributions to N-glycan predictions, revealing significant glycogene associations with specific N-glycan types. The predictions were consistent across input cell quantities, unlike LC-MS/MS glycomics which showed inconsistent results. This suggests that the models can reliably predict N-glycosylation even in samples with low cell amounts and by extension, single-cell samples. These findings can provide insights into cellular N-glycosylation machinery, offering potential therapeutic strategies for diseases linked to aberrant glycosylation, such as cancer, and neurodegenerative and autoimmune disorders.
Introduction
Glycosylation is a common form of post-translation modification of proteins. Major changes in protein glycosylation correlate to the progression of diseases such as Alzheimer's disease,1–3 autoimmune disease,4,5 and cancer.6–10 Protein N-glycosylation involves the action of the whole machinery of glycosidases, glycosyltransferases, transport proteins, and chaperones that work in conjunction with each other to enact post-translational modification on glycoproteins.11–13 These enzymes and proteins are coded into the transcriptome by over 400 glycogenes.11 As such, the cellular machinery that regulates the expression of these glycogenes in conjunction with relative quantification of the end-products, N-glycans, and glycoproteins, has been an interest as a potential target for therapeutics.7,8,14,15Efforts have been made in the past to correlate glycogene expression with the abundance of these glycogene products.16–18 Novel methods such as SUGAR-seq,19 scGlycan-seq,20 and scGR-seq21 enabled simultaneous quantification of glycogene transcripts and glycans through the use of lectin-based glycan profiling. In addition to these novel methods, computational tools such as GlycoMaple,22 SHAP,23 and Glcopacity,24 have aided in analyzing RNAseq and lectin-based glycan profiling data from these methods to comprehensively study glycosylation. Lectin-based glycomic profiling methods are preferred when complexed with other methods such as RNAseq, due to the convenient and rapid analyses that require no additional complicated or large instruments as well as the wide availability of fluorescently-labeled lectins able to characterize the glycan structural motifs.25 However, these approaches have been limited by the recognition capacity of lectin-based glycan profiling, which fails to capture the complete glycan structures or composition, and inability to differentiate between the classes of glycoconjugates – N-glycan, O-glycan, or glycosphingolipid. For example, the SNA lectin can recognize α-2,6-linked sialic acid to galactose residues, but it is unable to distinguish whether the structural motif came from an N-glycan, O-glycan, or glycosphingolipid.26 More critically, lectins do not allow quantitation between different structures. Thus, it has so far been difficult to correlate the transcriptomic expression of glycogenes with individual glycan structures or compositions, or with the main classes of glycans.
On the other hand, LC-MS-based glycomic methods provide more comprehensive structural analyses of cellular glycosylation, allowing the detection and quantification of individual N-glycan compositions or structures, especially when performed on a stationary phase that yields isomeric separation such as PGC.25 As such, LC-MS/MS-based methods surpass lectin-based limitations,25 and complementing results of LC-MS/MS-based methods with RNAseq data may provide a better elucidation of glycosylation pathways.
In order to address the efforts of correlating glycogene expression with quantitative glycomic abundance, we developed a method to integrate RNAseq transcriptomic and LC-MS/MS N-glycomic data, correlating glycogene expression with protein N-glycosylation abundances across cells from diverse tissue origins and conditions. Non-linear regression models were constructed to predict N-glycan abundances from glycogene expression profiles and identify key genes associated with specific N-glycans. The approach was validated by accurately predicting N-glycosylation in cell lines (GLC01, CCD19-Lu, Tib-190) regardless of cell sample amount. This method provides a platform to identify glycogenes implicated in cancer and N-glycan biosynthesis, enabling the development of targeted therapeutics for these pathways.
Results and discussion
glycoPATH integration of glycogene transcriptomics with LC-MS N-glycomics
We developed the glycoPATH workflow, which employs comprehensive characterization of the N-glycome using LC-MS/MS to quantify the abundances of each N-glycan structure in the cell glycocalyx for the various cell lines (Fig. 1).25 The cell lines were selected to provide a broad dataset of tissue origins to train the method. We then incorporated transcriptomic information obtained using standard 3′-TagSeq quantification methods for each cell line.27,28 To obtain N-glycan abundances we employed analytical methods previously developed in this laboratory.25,29 With LC-MS, we quantified more than 360 N-glycan compounds and categorized based on type: high-mannose, undecorated, fucosylated, sialylated, and sialofucosylated. We observed significant differences in the abundances of high-mannose, sialylated, and sialofucosylated N-glycans between B cells, brain, colon, lung, muscle, and prostate cells (Fig. 2A and C). We also observed significant differences in the expression of abundant N-glycan compositions between the cells (Fig. 2B). For example, the bi-antennary fucosylated N-glycan H5N4F1 were most abundant in brain and lung cells, with B cells and colon cells expressing less of it. The bi-antennary sialofucosylated N-glycan H5N4F1S1 was likewise abundant in brain, lung, and muscle cells, with B cells and colon cells containing less of this N-glycan composition. Interestingly, the bisected sialofucosylated N-glycan H5N5F1S1 was abundantly expressed only in B cells, with other cell types expressing negligible amounts (<5% by abundance). | ||
| Fig. 1 The glycoPATH workflow for correlating N-glycan and site-specific glycosylation with glycogene expression in cells. Cells were harvested for RNA and N-glycans, which underwent 3′-TagSeq transcriptomic and Chip-QToF N-glycomic analyses, respectively. Data gathered were used to train regression models using MATLAB to calculate glycogene correlations and N-glycan abundance predictions. | ||
 | ||
| Fig. 2 N-glycomic profiles significantly differ by tissue origin. The abundances of N-glycan types differ significantly between cells (A). Similarly, abundant N-glycan structures, such as H5N4F1, H5N4F1S1, and H5N5F1S1, significantly differ between cell lines obtained from different tissue origins (B). PLS-DA clustering methods show drastic N-glycomic differences between tissue types, particularly between tissues and B cells (C). | ||
To quantify the expression of glycogenes, we performed 3′-TagSeq RNAseq analysis for quantifying gene expression using tag abundance and then normalized using TMM normalization.27 To reduce noise and highlight differences in glycogene expression, we filtered the ∼18000 transcriptome data to include the genes relevant to N-glycan expression (nearly 170 glycogenes)11 that encompasses sugar transporters, nucleotide sugar synthesis, dolichol pathway proteins, mannosyltransferases, lysosomal targeting and degradation, mannosidases, GlcNactransferases, galactosyltransferases, fucosyltransferases, and sialyltransferases (Fig. 3A). With the glycogenes alone, we observed stark differences in expression amongst the cells we assayed (Fig. 3B and D). For example, the mannosidase MAN1A1 was observed to be most abundant in B cells followed by prostate cells. Similarly, MGAT3, which adds an N-acetylglucosamine to the chitobiose core to synthesize bisected N-glycans, is most abundant in B cells compared to the rest of the tissue cell types. We also quantified several fucosyltransferases and sialyltransferases; for example, FUT11 (which adds a terminal α1,3-fucose) and FUT8 (which adds a core α1,6-fucose) are expressed significantly differently between tissue types, with FUT11 being consistently higher than FUT8 expression within the same tissue type. ST6Gal1, which adds a terminal α2,6-sialic acid, are expressed differently between tissue types (Fig. 3C).
 | ||
| Fig. 3 Glycogene transcriptomic profiles significantly differ by tissue origin. Over 160 N-glycan biosynthesis-related genes were previously annotated and quantified, spanning sugar transproters, nucleotide sugar synthesis, dolichol pathway genes, glycosyltransferases and glycosidases (A). These glycogenes significantly differ in expression between tissues (B). Similarly, mannosidases (MAN1A1), GlcNActransferases (MGAT3), fucosyltransferases (FUT7), and sialyltransferases (ST6Gal1) differ significantly between tissues (C). PLS-DA clustering methods show drastic transcriptomic differences between tissue types, particularly between tissues and B cells (D). | ||
Constructing regression models for N-glycan abundances using supervised machine learning
From the transcriptomic and N-glycomic results we observed that the N-glycan abundances reflect the differences in glycogene expression. As such, we aimed to construct supervised machine-learning models to explain how these tissue-dependent differences came about as well as create a tool to predict N-glycan abundances from transcriptomic information. Regression models were constructed using the Regression Learner app in MATLAB. For each N-glycan composition abundance (response variable), we utilized the glycogene expression values (predictor variables) and then screened the app's repertoire of models to identify the best-performing model for each composition (Fig. 4). To train the models, we used the collected paired datasets from 50 unique cell samples; each paired dataset contained normalized gene expressions of 167 annotated glycogenes and normalized abundances of 138 N-glycan structures having abundances >0.05%. To evaluate the predictive models and protect against over-fitting, we performed 5-fold cross-validation and calculated the model performance (RMSE and R2 after validation, Table 1 and ESI Table 1†) and ranked the best model per N-glycan. The model selected for each glycan structure were those with the best-performing validation metrics: lowest RMSE and highest R2 (Table 1). A summary of the performance of all models tested for each N-glycan is available in ESI Table 1†. | ||
| Fig. 4 Model construction and 5-fold cross-validation of supervised ML models (A) for predicting N-glycan (e.g. H8N2, H5N4, H5N4F1, H5N4S1, H5N4F1S1) abundances from glycogene expression data (B). | ||
| N-Glycan type | N-Glycan composition | Putative N-glycan structure | Regression model | Average abundance (%) | R 2 | RMSE |
|---|---|---|---|---|---|---|
| High-mannose | H9N2 |

|
Trilayered neural network | 5.28 ± 2.18 | 0.56 | 30.817 |
| High-mannose | H8N2 |

|
Linear SVM | 6.41 ± 2.78 | 0.48 | 1.5606 |
| High-mannose | H7N2 |

|
Cubic SVM | 5.17 ± 2.24 | 0.41 | 1.4336 |
| High-mannose | H6N2 |

|
Boosted tree ensemble | 4.33 ± 1.98 | 0.54 | 1.2465 |
| High-mannose | H5N2 |

|
Rational quadratic GPR | 3.83 ± 1.53 | 0.46 | 1.0583 |
| Undecorated | H5N4 |
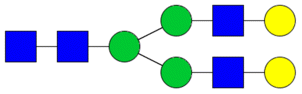
|
Bilayered neural network | 1.99 ± 1.38 | 0.86 | 0.48265 |
| Undecorated | H6N5 |
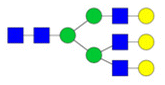
|
Squared exponential GPR | 0.79 ± 0.93 | 0.79 | 0.36207 |
| Undecorated | H7N6 |

|
Trilayered neural network | 0.64 ± 1.05 | 0.82 | 0.40917 |
| Fucosylated | H5N4F1 |

|
Squared exponential GPR | 2.48 ± 2.06 | 0.85 | 0.71845 |
| Fucosylated | H6N5F1 |

|
Quadratic SVM | 1.81 ± 1.52 | 0.58 | 0.85673 |
| Fucosylated | H7N6F1 |

|
Trilayered neural network | 1.07 ± 0.96 | 0.8 | 0.40341 |
| Sialylated | H5N4S1 |
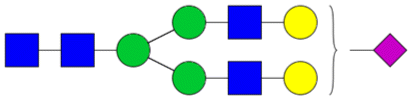
|
Medium neural network | 2.32 ± 2.57 | 0.9 | 0.74494 |
| Sialylated | H6N5S1 |
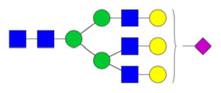
|
Trilayered neural network | 0.90 ± 1.30 | 0.83 | 0.49163 |
| Sialylated | H6N5S2 |

|
Trilayered neural network | 0.76 ± 0.86 | 0.75 | 0.43418 |
| Sialylated | H7N6S1 |

|
Linear SVM | 0.62 ± 1.28 | 0.22 | 0.92776 |
| Sialofucosylated | H5N4F1S1 |

|
Medium neural network | 4.04 ± 3.37 | 0.88 | 1.2381 |
| Sialofucosylated | H5N4F1S2 |
|
Rational quadratic GPR | 2.45 ± 2.30 | 0.9 | 0.77661 |
| Sialofucosylated | H6N5F1S1 |

|
Linear SVM | 1.78 ± 1.16 | 0.78 | 0.60771 |
| Sialofucosylated | H7N6F1S1 |

|
Boosted tree ensemble | 1.68 ± 1.31 | 0.82 | 0.54195 |
Based on the results, we created models with good performance (validation R2 > 0.7) especially for undecorated (H5N4, H7N6), fucosylated (H5N4F1, H5N4F2, H7N6F1), sialylated (H5N4S1, H6N5S1) and sialofucosylated N-glycans (H5N4F1S1, H5N4F1S2, H7N6F1S1). On average, we observed good model performance in predicting the most abundant N-glycans per category (Table 1): undecorated (average validation R2 = 0.82), fucosylated (average validation R2 = 0.74), sialylated (average validation R2 = 0.83), and sialofucosylated (average validation R2 = 0.85) N-glycans. We found that N-glycans with models having poor performance (R2 < 0.3) tended to have very low abundance (<0.05%). Hence, after filtering out the N-glycans with very low abundance (<0.05%), we obtained several models with good performance (R2 > 0.7): high-mannose (n = 1), undecorated (n = 9), fucosylated (n = 22), sialylated (n = 6), sialofucosylated (n = 22) (ESI Fig. 1–5†).
Furthermore, we observed that each N-glycan composition necessitated different regression models. For example, the best model for the fucosylated bi-antennary compound H5N4F1 was the squared exponential GPR (R2 = 0.85, RMSE = 0.71845) whereas the best model for the sialylated bi-antennary compound H5N4S1 was the Medium Neural Network (R2 = 0.9, RMSE = 0.74494) (Table 1). A likely explanation for the differences in model characteristics between N-glycan structures is the differences in glycogene interactions upon biosynthesis; for example, fucosylated N-glycan structures would not necessitate the involvement of sialyltransferases in its biosynthesis, while sialylated structures would not involve fucosyltransferases in its biosynthesis. Likewise, we observed differences in model characteristics between undecorated multiple-branched N-glycans: the bi-antennary H5N4 utilized a Bilayered neural network (R2 = 0.86, RMSE = 0.48265), the tri-antennary H6N5 used a Squared exponential GPR (R2 = 0.79, RMSE = 0.36207), and the tetra-antennary structure H7N6 performed best under a Trilayered neural network (R2 = 0.82, RMSE = 0.40917). Due to the numerous possible GlcNActransferases that could catalyze the branching reactions of N-glycans (e.g. MGAT1, MGAT2, MGAT4A/B/C, MGAT5/5B), it is likely that each additional N-glycan branch (e.g. tri-, tetra-antennary) necessitates the involvement of more GlcNAc-transferase than the bi-antennary structure; hence, more complicated glycogene interactions are likely to occur with highly-branched structures compared to bi-antennary ones.
There were N-glycan compositions with relatively poor model performance: H6N5F1 (validation R2 = 0.58), H5N4S2 (validation R2 = 0.3), and H7N6S1 (validation R2 = 0.22); we attribute these low predictability values to potential structural variations arising from differences in linkages, especially that of fucose (α1,3 vs. α1,6), sialic acid (α2,3 vs. α2,6), and galactose (α1,3 vs. α1,4). We also observed fair to poor model performances with our high-mannose predictive models, for example: H9N2 (trilayered neural network, R2 = 0.56, RMSE = 30.817), H8N2 (linear SVM, R2 = 0.48, RMSE = 1.5606), and H7N2 (cubic SVM, R2 = 0.41, RMSE = 1.4336). Although the low model performance warrants further exploration, a probable explanation is the structural separation of N-glycans with similar compositions but different glycosidic linkages, thereby decreasing model resolution.30 As such, refining the N-glycomic structural characterization method with linkage information to obtain precise structures will aid in refining the current predictive models in a future study. Another potential reason highlighted above is low abundance of some glycan structures; wherein we observed poor performance of models (R2 < 0.3) for structures with less than 0.05% relative abundance. We summarized the model parameters for each N-glycan structure into MATLAB workspaces, including the training and validation datasets into Github (https://github.com/MichRussAlv/glycoPATH).
Predicting glycan abundances of an independent test set with glycoPATH
To further demonstrate the capability to predict N-glycan abundances from RNAseq, we characterized the N-glycome of an immortalized lung cancer cell line, GLC01, in comparison to lung fibroblast cells CCD19-Lu and B cells Tib-190 (Fig. 5). GLC01 was recently immortalized by CDK4-insertion into primary cells derived from a patient with stage III lung cancer.29 CCD19-Lu cells were purchased from ATCC and derived from the lung fibroblasts of a non-cancer patient. We also included B cells Tib-190 in the comparison, owing to the large functional differences between B cells and lung cells (Fig. 1 and 2). Comparing the experimental and predicted results of N-glycans (high-mannose, undecorated, fucosylated, sialylated, sialofucosylated) showed highly accurate predicted values for the three cell lines (Fig. 5B). Deconvoluting the N-glycan types into the most abundant compounds per type showed the method's ability to predict N-glycosylation at the structural level (Fig. 5C). As such, the predicted relative abundances matched that of experimentally-determined abundances of high-mannose (H9N2, H8N2, H7N2, H6N2), undecorated (H5N4, H6N5, H7N6), fucosylated (H5N4F1, H6N5F1, H7N6F1), sialylated (H5N4S1, H6N5S1, H7N6S1), and sialofucosylated (H5N4F1S1, H6N5F1S1, H7N6F1S1) N-glycans. | ||
| Fig. 5 Using RNAseq and glycoPATH to characterize the N-glycome of novel lung cancer cell line GLC01, lung fibroblast cell CCD19-Lu, and B cells Tib-190 (A). Comparing the experimental and predicted abundances per N-glycan type (B) and individual N-glycan compositions of the most abundant N-glycans per type (C). | ||
Beyond predicting the bulk N-glycome of cells, we aimed to determine whether decreasing the number of cellular starting material will affect the RNAseq data and thus, the N-glycomic predictions (Fig. 6). The rationale for this experimental design is to circumvent the inherent dependency of the N-glycomic LC-MS/MS method with the starting amount of cell sample. For example, the bulk N-glycome obtained from LC-MS/MS glycomics decreases by a factor of ten with each ten-fold dilution of the starting cell suspension consisting of five million cells (5 M) (Fig. 6B). Furthermore, we start to observe skewed results from LC-MS/MS glycomics upon decreasing the amount of starting material. In contrast, transcripts can be prepared from relatively lower amounts of starting material and still obtain consistent results; hence, low-input transcriptomics.27,28 Using transcriptomics and our predictive models (RNAseq + models), we observed that the predicted N-glycomes were more consistent in abundances when calculated from RNAseq (Fig. 6C). Moreso, the predicted N-glycomes from were similar to the LC-MS/MS profile obtained from bulk cells. In addition, the predicted profiles of 5 M, 500 K, and 50 K cells were found to be more consistent compared to the corresponding profiles obtained from LC-MS/MS glycomics. For example, with LC-MS/MS glycomics we observed sialylated abundances of 13.59% (from 5 M cells), 11.46% (from 500 K cells), and 5.60% (from 50 K cells), showing a decrease in observable sialylated compounds with lower amount of starting cell material. In contrast with RNAseq + ML-models, we predicted sialylated abundances of 13.59% (from 5 M cells), 12.91% (from 500 K cells), and 13.89% (from 50 K cells). Thus, we can obtain accurate representations of N-glycome of bulk cell populations even when extracted from low-input samples.
 | ||
| Fig. 6 Demonstration of low-input N-glycomics from small amount of cell material using RNAseq and glycoPATH. Low-input cell samples were prepared from dilutions of GLC01 cell suspension harvested from flasks to obtain five million (5 M), five hundred thousand (500 K), and fifty thousand (50 K) cells (A). With LC-MS/MS N-glycomics, amount of starting material (number of cells) have a huge impact on the N-glycome profiles and subsequent quantification by relative abundance (B). On the other hand, the N-glycome of cells predicted using glycoPATH reflect that of the bulk cell population at highest concentration (5 M) (C). | ||
Constructing putative N-glycan biosynthesis pathways of each structure with glycoPATH
After model construction using the Regression Learner app (MATLAB R2022a), we calculated the importance of individual glycogenes to the models using an F test. From there, we discerned patterns in glycogene correlation with individual N-glycan compositions based on type: high-mannose, undecorated, fucosylated, sialylated, and sialofuosylated (Fig. 7). For high-mannose N-glycans, we observed type I mannosidases MAN1A1, MAN1A2, and MAN2A2 to have high importance scores compared to the other mannosidases such as MAN12, MAN1C1, MAN2B1, and MAN2B2 (Fig. 7A). Undecorated N-glycans are interesting in that we observed the patterns for branched N-glycans such as biantennary H5N4, tri-antennary H6N5, and tetra-antennary H7N6 corresponding to galactosyltransferases and GlcNActransferases (Fig. 7B). In particular, galactosyltransferases B3Galt4, B3Galt5, B4Galt1, B4Galt2, and B4Galt5 and the GlcNActransferases MGAT1, MGAT3, MGAT4A, and MGAT4B were found to be important (i.e., have high importance scores) in undecorated N-glycan structures. It is interesting to note that some of these enzymes do not directly catalyze the reaction to produce the corresponding N-glycan; such as MGAT3 (catalyzes production of bisected N-glycans) having high importance score in the abundance of H5N4 and H6N5 (Fig. 7B). However, there is an inverse relationship between MGAT3 and biantennary N-glycans, wherein cells with high MGAT3 expression (e.g. B cells, Fig. 3C) have lower abundance of bi-antennary glycans (Fig. 2B). | ||
| Fig. 7 Ranked feature importance of glycogenes with specific N-glycan compositions in the high-mannose (A), undecorated (B), fucosylated (C), sialylated (D), and sialofucosylated (E) types. The feature importance scores were calculated from MATLAB Regression Learner app using F test. | ||
For fucosylated glycans, the most abundant compositions were found to be biantennary structures H5N4F1 and H5N4F2, and monofucosylated structures H6N5F1 and H7N6F1. The fucosyltransferases with high importance scores for these catalyze the addition of antennary fucose (FUT7, FUT11) as well as core-fucose (FUT8), with the exception of the bi-fucosylated H5N4F2 with lower importance score compared to the other structures (Fig. 5C). We believe these results indicate that for N-glycan compositions with several fucose residues, the combination of terminal and core-fucosyltransferases can be used to calculate its abundance. In determining correlations with sialylated N-glycans, ST3Gal6 have the highest importance scores, followed by ST3Gal3 and ST6Gal1 (Fig. 7D). In particular, we observed that ST3Gal6, which catalyzes the addition of terminal sialic acid in an α2,3-linkage, have the highest importance score with bi-, tri-, and tetra-antennary structures with mono- or bi-sialylation. ST3Gal3, which catalyzes the same reaction, have higher importance score to bi-sialylated structures H5N4S2 and H6N5S2. Finally, ST6Gal1, which adds α2,6-linked sialic acid, has highest importance score with monosialylated biantennary compound H5N4S1.
Finally, we sought to identify the fucosyltransferases and sialyltransferases with the highest importance scores for sialofucosylated structures (Fig. 7E). We observed similar trends with the fucosyltransferases and fucosylated N-glycans, wherein the enzymes catalyzing the addition of terminal (FUT7, FUT11) and core fucose (FUT8) had high importance scores. With sialyltransferases, ST3Gal3 lost importance except for the monosialylated monofucosylated triantennary structure H6N5F1S1, whereas ST3Gal6 remained to have higher importance scores across the N-glycan compositions surveyed. ST6Gal1 and ST6Gal2 both have high importance scores, having both catalyze the addition of α2,6-linked sialic acid. Altogether, these results indicate that multiple enzymes can catalyze the same reaction to produce the same N-glycan composition as well as enzymes having specific substrate preferences. With this method, we are able to discern the specific glycogenes that play important roles in the synthesis of specific N-glycan compositions.
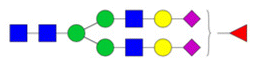
An interesting application for the glycogene feature importance calculation is that we can rank glycogenes that catalyze the reaction pathway from precursor high-mannose structures to sialofucosylated structures, after overlaying the enzymatic rules catalyzed by every glycogene enzyme in the pathway. For example, in order to synthesize the sialofucosylated structure H5N4F1S1 from the high-mannose structure H9N2 it has to go through mannose trimming by mannosidases (α1,2-linked: MAN1A1, MAN1A2, MAN1B1, MAN1C1; α1,3-/α1,6-linked (MAN2A1, MAN2A2, MAN2B1, MAN2B2), addition of GlcNAc (β1,2-linked: MGAT1, MGAT2; β1,4-linked: MGAT4A, MGAT4B, MGAT4C; β1,6-linked: MGAT5, MGAT5B; bisecting: MGAT3), addition of fucose (α1,2-linked: FUT1, FUT2; α1,3-linked: FUT10, FUT11, FUT4, FUT4, FUT5, FUT6, FUT7, FUT9; α1,6-linked: FUT8), and addition of sialic acid (α2,3-linked: ST3Gal1, ST3Gal2, ST3Gal3, ST3Gal4; α2,6-linked: ST6Gal1, ST6Gal2).11 Ranking the importance of these glycogenes in the H5N4F1S1 model shows the following most important genes per reaction: α1,2-mannosidase: MAN1A1 (score = 8.2625); α1,3/α1,6-mannosidase: MAN2A1 (score = 11.416), MAN2B1 (score = 7.187); β1,2-GlcNActransferase: MGAT1 (score = 5.0537); β1,4-GlcNActransferase: MGAT5B (score = 4.6258), MGAT4B (score = 4.4976); galactosyltransferase: B4Galt2 (score = 9.3097), B4Galt1 (score = 7.5278); α1,3-fucosyltransferase: FUT7 (score = 9.1033), FUT11 (score = 8.4866); and α2,6-sialyltransferase: ST6Gal1 (score = 5.9686), ST6Gal2 (score = 6.4552) (Fig. 8).
 | ||
| Fig. 8 Ranking the glycogenes for each reaction can identify the putative biosynthetic pathway. For example, the putative biosynthetic pathway from the high-mannose H9N2 up to the sialofucosylated structure H5N4F1S1, showing the glycogenes with the highest importance scores at each step (A). The pathway was validated by quantifying the H5N4F1S1 final structure in brain and colon cells, where brain cells significantly have higher abundance (B). Correspondingly, several important glycogenes in the pathway are significantly increased in brain cells compared to colon cells, too (C). | ||
Validating the pathway analysis with N-glycan inhibitors
To test our N-glycan biosynthetic pathway model, we selected specific structures. We quantified the sialofucosylated N-glycan H5N4F1S1 and corresponding glycogene expression levels of the most important glycogenes in the H5N4F1S1 model (Fig. 8A). We quantified the H5N4F1S1 N-glycan and found that there were drastically higher amounts in brain cells (approximately four times as much) compared to colon cells (Fig. 8B). Based on the pathway, the following glycogenes were important: mannosidases (MAN1A1, MAN2A1), GlcNActransferases (MGAT1, MGAT5B), galactosyltransferases (B4Galt1, B4Galt2), fucosyltransferases (FUT7, FUT11), and sialyltransferases (ST6Gal1, ST6Gal2). Among these glycogenes, we found significantly higher expression in brain cells compared to colon cells of MAN2A1, MGAT1, B4Galt1, FUT11, and ST6Gal2 (Fig. 8C). We also observed higher expression (albeit not statistically significant) of MGAT5B and B4Galt2 in brain cells compared to colon cells. On the other hand, we observed that the expression levels of MAN1A1 and ST6Gal1 were not significantly different between colon and brain cells. Collectively, these results suggest that several glycogenes correlate highly with N-glycan structures, wherein higher glycogene expression leads to higher expression of N-glycans and lower glycogene expression leads to lower N-glycan abundance.To further validate the pathway analysis method, we determined whether the model can detect perturbations in the N-glycan biosynthesis pathways caused by glycosylation inhibitors. We recently characterized the effect of glycosylation inhibitors such as kifunensine (type-I mannosidase inhibitor) and 2-deoxy-2-fluorofucose (fucosylation inhibitor) on the N-glycan profiles of cells.32,33 Kifunensine inhibits the activity of type-I mannosidases (e.g. MAN1A1),34 thereby preventing the maturation of N-glycans from high-mannose types (e.g. H9N2) into undecorated, fucosylated, sialylated, and sialofucosylated types. Similarly, 2-deoxy-2-fucose is a pan-inhibitor of fucosylation, through inhibition of GDP-fucose synthesis and fucosyltransferase activities.35 Thus, 2-deoxy-2-fucose is an effective inhibitor of both fucosylated (e.g. H5N4F1, H5N5F1) and sialofucosylated(e.g. H5N4F1S1, H5N5F1S1) N-glycan structures.
To test this notion, we treated B cells with these inhibitors and subsequently extracted the transcriptomic profiles for 3′-TagSeq analysis. We then predicted the resulting glycomic data with the predictive models to determine the specific glyco-enzymes affected by the inhibitors (Fig. 9A and B). With kifunensine, we expect type-I mannosidases (MAN1A1) to be inhibited resulting in higher abundance of H9N2 in the kifunensine-treated cells (Fig. 9C). Interestingly we also found slightly higher abundance (albeit less drastic) of H8N2 in the kifunensine-treated cells; such a result may suggest that the kifunensine treatment to be very effective in inhibiting the first step in the pathway – the trimming of H9N2 into H8N2. Furthermore, we observe a slight decrease in abundance of the smaller high-mannose N-glycan H5N2 in the kifunensine-treated cells; such a result indicates that kifunensine is less active in inhibiting α1,3/6-linked mannosidases (type-II mannosidases). Unsurprisingly, we predicted the abundance of mature N-glycans (e.g. H5N5, H5N5F1, H5N5S1, H5N5F1S1) to be lower in kifunensine-treated cells due to inhibition of high-mannose trimming. The predicted N-glycan abundances followed the trend of experimentally-derived kifuneninse-treated cells (Fig. 9C). With kifunensine, there is an increase in the abundance of H9N2 and H8N2 N-glycans and a decrease in abundance of mature N-glycans (e.g. H5N5, H5N5F1, H5N5S1, H5N5F1S1).
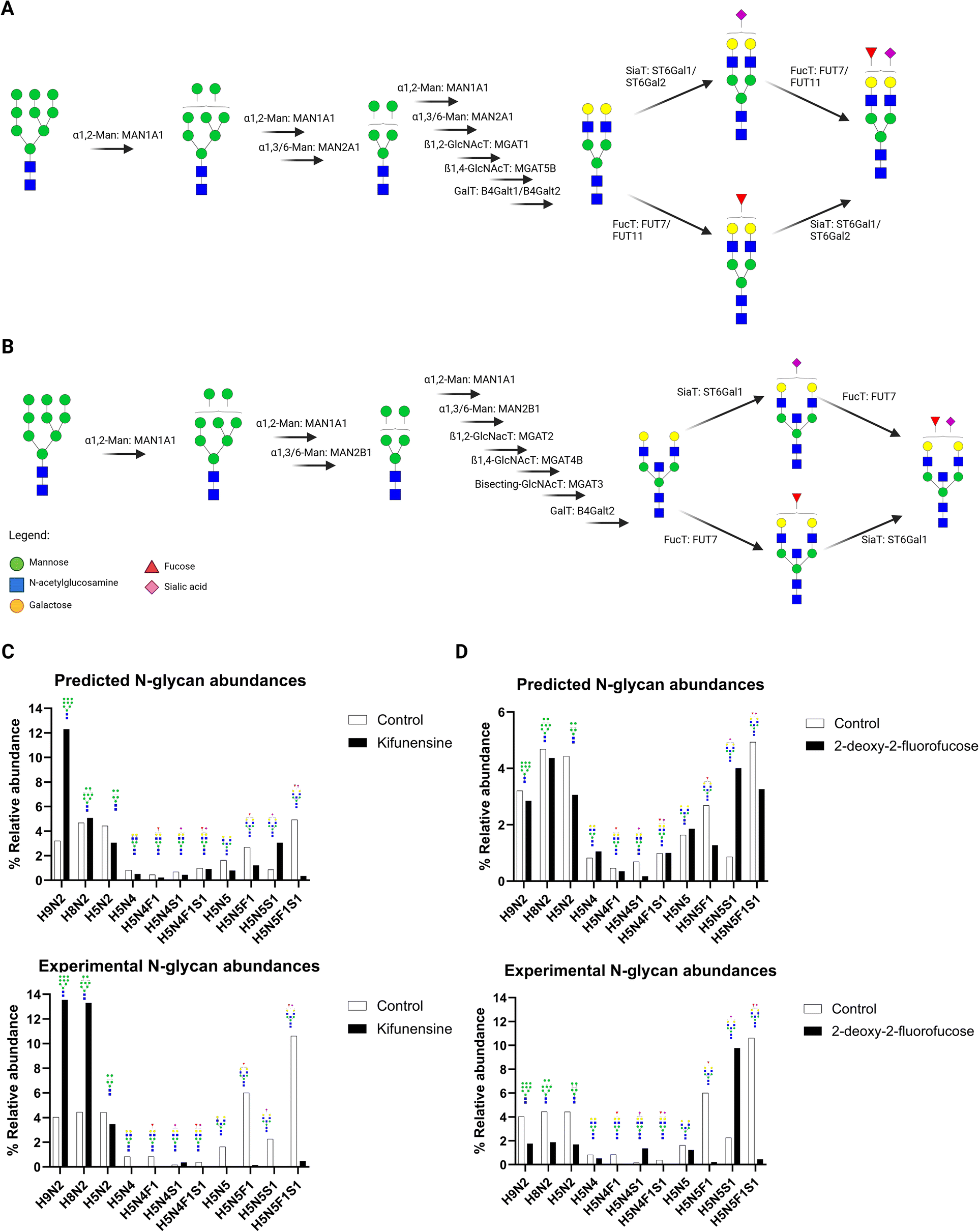 | ||
| Fig. 9 Predicting the glycosylation pattern of cells treated with glycosylation inhibitors. B cells (Tib-190) were treated with control (DMSO), kifunensine (mannosidase I inhibitor), and 2-deoxy-2-fluorofucose (GDP-fucose synthesis inhibitor). Pathway prediction of bi-antennary sialofucosylated structure H5N4F1S1 (A) and bisected sialofucosylated structure H5N5F1S1 (B) identified the important glycogenes involved in biosynthesis. Comparing the experimental and predicted of N-glycan abundances of these structures show the altered glycosylation profile caused by these kifunensine (C) and 2-deoxy-2-fluorofucose (D). | ||
With 2-deoxy-2-fluorofucose-inhibited cells, we expected fucosyltransferases (FUT7, FUT11) to be inhibited (Fig. 9A and B). Indeed, we observed a drastic decrease in abundance of both fucosylated (H5N5F1) and sialofucosylated (H5N5F1S1) N-glycans in the treated-cells. We also observed a corresponding drastic increase in abundance of sialylated H5N5S1 N-glycan in the treated cells. Based on the pathway (Fig. 9D), wherein the undecorated substrate H5N5 could be decorated by either fucose (H5N5F1), sialic acid (H5N5S1), or both (H5N5F1S1). This result corresponds to 2-deoxy-2-fluorofucose being a fucosylation inhibitor; in that the H5N5 precursor is shunted onto the sialic acid decoration step to form H5N5S1, instead of the fucose decoration step to form H5N5F1. Thus, we observe drastically higher abundance of H5N5S1 compared to both H5N5F1 and H5N5F1S1. Similarly, we compared the predicted N-glycan abundances with experimentally-derived 2-deoxy-2-fluorofucose-treated cells and found a similar trend (Fig. 9D). In particular, the abundances of fucosylated N-glycans H5N5F1 and H5N5F1S1 decreased, while the abundance of sialylated H5N5S1 increased.
An interesting outcome of integrating both pathway analysis and N-glycan abundance prediction is the ability to identify which glycogene is highly correlated with the biosynthesis pathway. For example, there are several fucosyltransferases expressed by B cells (e.g. FUT7, FUT11, FUT8) but the glycogene that had the highest importance score was FUT7; thus, specific targeting of FUT7 could lead to more precise knock-down of fucosylated N-glycans in these cells as opposed to using pan-inhibitors.
Discussion
Aberrant glycosylation has been well-documented in diseases such as cancer,14–16,31 Alzheimer's disease,1,2 and autoimmune diseases.4,5 In lung cancer specifically, previous reports have shown aberrant expressions of N-glycans, such as increased high-mannose and sialofucosylated structures, both in serum31–34 and tissues30,35,36 of cancer patients compared to non-cancer samples. These alterations were observed to be focused on cancer-associated glycoproteins, such as integrins,35,37–40 EGFR,41,42 and cell-adhesion molecules.43–45 These results were found to be concomitant with dysregulated glycogene expression.43,46 As such, there is much interest in correlating glycogene expression with N-glycosylation.Previous methods such as SUGAR-seq,19 scGlycan-seq,20 and scGR-seq21 used lectin-based glycan profiling coupled with RNAseq methods to simultaneously quantify glycogene and N-glycan expression. Software, such as GlycoMaple,22 SHAP,23 and Glcopacity,24 have been developed to recapitulate the data obtained from these methods. However, lectin-based glycan profiling methods are known to have several limitations. Due to lectin binding being specific to glycan epitopes and not individual structures, it is unable to distinguish between specific N-glycan compounds.25 For example, the fucose-binding lectin such as AAL47 can bind to fucose in α1,2-, α1,3-, α1,4-, and α1,6-linkages, regardless of being present in N- or O-glycans.25 Thus, lectin-based profiling may lack specificity for defined N-glycan structures. LC-MS/MS methods can provide the necessary resolution required to quantify specific N-glycan structures, and integrating its analysis with RNAseq transcriptomics can further provide insights into the relationship between the glycome and transcriptome.
Protein N-glycosylation involves the action of the whole machinery of glycosidases, glycosyltransferases, transport proteins, and chaperones that work in conjunction with each other to enact post-translational modification on glycoproteins.11,12 These enzymes and proteins are coded into the transcriptome by over 160 glycogenes: 14 mannosidases, 18 galactosyltransferases, 35 N-acetylglucosaminyltransferases, 13 fucosyltransferases, and 21 sialyltransferases.11 Members of the mannosidase family catalyze the removal of mannose residues either in an α1,2-, α1,3-, or α1,6-linkage, essentially processing high-mannose type N-glycans into other types.43,48 The action of both N-acetylglucosaminyltransferases and galactosyltransferases signal the transition of N-glycans from high-mannose types into either hybrid- or complex-type N-glycans. Once GlcNAc has been added to the antenna of the N-glycan, galactosyltransferases can subsequently act on it to catalyze the transfer of UDP-Gal to the antenna GlcNAc either in a β1,3- or β1,4-linkage.49 Upon biosynthesis of either hybrid- or complex-type N-glycans, further decoration with fucose and/or sialic acid residues is acted upon by fucosyltransferases and sialyltransferases, respectively. Fucosyltransferases add fucose residues from GDP-Fuc to either the antenna GlcNAc in an α1,3-linkage, or to the core-GlcNAc in an α1,6-linkage; the latter reaction is known to be catalyzed only by FUT8.50 On the other hand, sialyltransferases can catalyze the addition of sialic acid from CMP-NeuAc to antenna GlcNAc residues, either in α2,3- or α2,6-linkage.51
In the multi-step biosynthetic process such as the production of N-glycans, multiple glycogenes interact and work together to synthesize the eventual N-glycan structure conjugated to the glycoprotein. Hence, this process necessitates the use of robust predictive models and algorithms, such as machine-learning algorithms, to holistically incorporate these multi-gene interactions.52 Based on the modeling results, each N-glycan necessitated a different machine-learning algorithm to construct. Among the best performing models were gaussian processes most frequently with a rational quadratic kernel. This model type was specifically developed to suit modelling tasks based on smaller datasets where inputs have widely varying degrees of correlation with predicted outputs.53,54 In the context of biochemical networks, Gaussian Process Regression has been shown to be an effective method for modelling systems in which external pathways have a significant influence on the subsystem in question.53 With this modelling approach the influence of specific glycosylation genes as a subset of the total transcriptome were pinpointed as factors in N-glycan abundance expression. We identified that N-glycan linkage diversity may contribute to model performance. For example, an N-glycan with composition H5N5 can have at least three possible structures: bisected bi-antennary, and two tri-antennary structures. Before the addition of a third GlcNAc residue, given the known rules of N-glycan biosynthesis, the linkages of the nine residues in a bi-antennary glycan H5N4 can inferred based on composition. This involves addition of two GlcNAc residues to the chitobiose core common to all N-glycan which is then extended by β1,2 linkage forming MGAT1 and MGAT2. Following the putative biosynthetic pathways presented in Fig. 8, an addition of a fifth GlcNAc implicates either MGAT3, MGAT4, or MGAT5 to form a β1,4-linked bisected structure, a β1,4-linked tri-antennary structure, or a β1,6-linked tri-antennary structure, respectively. Thus, the biosynthetic pathways to create H5N5 structures are naturally linked with each other due to having common reaction precursors being acted on by MGAT3, MGAT4, or MGAT5.
Using our predictive models, we successfully predicted the N-glycome for both bulk cell samples and low-input cell samples. Notably, we found that LC-MS/MS N-glycomics is highly sensitive to the amount of starting cell material, which led to skewed abundances of high-mannose, undecorated, fucosylated, sialylated, and sialofucosylated N-glycan structures. In contrast, transcriptomics combined with machine-learning models produced more consistent results regardless of the starting cell population size, enabling us to predict protein glycosylation even with a smaller cell sample (i.e. low-input N-glycomics) or from single cells (single-cell N-glycomics). By correlating N-glycomics with glycogene expression data, we can see patterns in N-glycan biosynthesis of lung cells that coincided with reports of aberrant glycosylation in cancer. High-mannose N-glycan structures were observed to have significant negative correlations with MAN1A1 (removes α1,2-linked mannose) and MAN2A1 (which removes both α1,6- and α1,3-linked mannose) among other mannosidases. MAN1A1 in particular, is known to be correlated with impaired survival in breast54 and bone43 cancers. Fucosylated N-glycans significantly correlated with fucosyltransferases FUT7 and FUT11, which adds fucose to antenna GlcNAc in an α1,3-linkage, as well as FUT8, which adds fucose to the core GlcNAc in an α1,6-linkage; these are interestingly associated with invasion and metastatic potentials of tumors through hypoxic conditions.55–57 Finally, sialylated N-glycans correlated with ST6Gal2 and ST3Gal3, which add sialic acid residues to antenna galactose in an α2,6-linkage and α2,3-linkage, respectively. Overexpression of α2-3 sialyltransferase III (ST3Gal-III) in pancreatic cancer has been implicated in pancreatic tumor progression. Overexpression of α2-6 sialyltransferase I (ST6GalNAc-I) was related to poor patient survival in colorectal carcinoma patients.58 In lung cancer patients, aberrant glycosylation has been found to be correlated to aberrant expression of glycosylation enzymes as well. Gene-expression analysis of lung tissue sections from smokers and never-smokers found significantly upregulated MAN1A2, MAN2A1, MGAT2, MGAT4B, B4GALT2, FUT2, FUT3, FUT6, and FUT8 while several enzymes, MAN1A1, MAN1C1, MAN2A2, MGAT1, MGAT3, and FUT1 were significantly down-regulated.30,46 In addition to these enzymes, FUT7 has also been observed to play a role in lung cancer. The expression of FUT7 and/or FUT4 has been positively associated with significantly shorter survival in lung cancer patients compared to the patients that did not express these genes.59 FUT7 expression was consistent with sLex expression level; basing on the biosynthetic pathway of sLex, this was to be expected. L-selectin ligands, which are synthesized by FUT7, was found to also play a role in the metastasis mechanism of leukocyte L-selectin action, with attenuated metastasis observed in FUT7−/− mice. FUT7 was also found to have a role in the metastasis of human colorectal carcinoma cells (LOVO), with increased metastatic potential, sLex and glycoprotein CD24 expressions after transfection with FUT7, implying the role of FUT7 in glycosylation of CD24 and its enhancement of metastatic potential.60 Likewise, overexpression of CD15S epitopes were observed after transfection of SEBTA-001 (biopsy-derived brain metastatic NSCLC) and NCI–H1299 (metastatic NSCLC from cervical lymph node) with FUT7.61 This led to enhanced cell adhesion of these cells to an endothelial cell monolayer of hCMEC/D3 (human cerebral microvascular endothelial cell line), with knockdown of FUT7 expression leading to decreased cell adhesion. In addition to metastatic pathways, FUT7 also plays a role in other oncogenic pathways. FUT7 overexpression in A549 (NSCLC) cells led to increased sLex expression, which correlated to the activation of the EGFR/AKT/mTOR pathway, triggering cell proliferation.62 In human hepatocarcinoma cell lines, FUT7 overexpression led to increased the sLex expression of the InR (insulin receptor)-α subunit, enhancing autophosphorylation of InR-β and further phosphorylation of insulin receptor substrate-1 (IRS-1), protein kinase B (PKB/Akt), MAPK, MEK, PDK-1, PKN, c-Raf-1 and β-catenin.63 Overexpression of FUT7 in heptocarcinoma cell line also downregulated the protein expression and activity of the cyclin-dependent kinase inhibitor p27Kip1 protein, an inhibitor of CDK2.64 By decreasing p27Kip1, the increased CDK2 activity stimulated the phosphorylation (and deactivation) of the retinoblastoma protein and stimulated G1/S transition and cell proliferation. The reduced p27Kip1, enhanced CDK2 and Rb phosphorylation, and cell proliferation, were correlated with the amount of sLex, the biosynthetic product of FUT7.
Materials and methods
Materials
A549 (cat.# CCL-185), NCI–H23 (cat.# CRL-5800), Calu-3 (cat.# HTB-55), BEAS (cat.# CRL-9609), CCD19-Lu (cat.# CCL-210), HMC3 (cat.# CRL-3304), DAOY (cat.# HTB-186), A204 (cat.# HTB-82), SJCH30 (cat.# CRL-2061), HCT116 (cat.# CCL-247), HT29 (cat.# HTB-38), PNT2 (cat.# CVCL-2164), TIB (cat.# TIB-190), DB (cat.# CRL-2289), and SKW (cat.# TIB-215) cell lines were obtained from ATCC. The cell line GLC01 was provided by the Lung Center of the Philippines through Dr Francisco M. Heralde III. Protease inhibitor cocktail set V (EDTA-free, cat.# 539137), sucrose (cat.# S7903), sodium carbonate (Na2CO3, cat.# S7795), and ammonium bicarbonate (NH4HCO3, cat.# A6141) were from Sigma-Aldrich. Dithiothreitol (DTT, cat.#V3151) was from Promega. PNGase F (cat.#P0704L) was purchased from New England Biolabs. LC-MS-grade trifluoroacetic acid (cat.# A116-50), formic acid (cat.# A117-50), and acetonitrile (ACN, cat.# A955-4) were from FisherScientific. RNeasy Mini kit (cat.# 74104), QIAshredder (cat.# 79656), and RNASe-free DNAse set (cat.# 79254) were purchased from Qiagen. Graphitized carbon (PGC) SPE plates (cat.# FNSCAR800) were purchased from Glygen. iSPE-HILIC® SPE catridges (cat.# 200.001.0100) were obtained from Hilicon.Cell line culture and glycan extraction
All cells were cultured in RPMI media supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin, according to manufacturer instructions. Cells were incubated in 37 °C and 5% CO2. For all assays, cells were cultured in at least two separate replicates. Extraction of cell membrane components, primarily glycoproteins, and glycoconjugates, was performed following previously described protocols.25,65 After reaching 80% confluency, cells were washed three times with PBS, then harvested using cell scrapers in PBS. The cells were harvested by centrifuging at 200×g for 5 minutes at 4 °C, aspirating the excess PBS to obtain the cell pellets. The cell pellets were resuspended in homogenization buffer (0.25 M sucrose, 20 mM HEPES buffer, 1:100 protease inhibitor cocktail set V, pH 7.4), followed by sonication using a probe-tip (Q700; QSonica, cat. no. Q700-110) set to 25 amplitude and a 5 s on-10 s off cycle. The lysed cell suspensions were cleared off cellular debris and nuclei by centrifuging at 2000×g for 10 minutes at 4 °C. Then, the supernatant containing the membrane fractions was further ultracentrifuged at 200000×g for 45 minutes, 4 °C. The resulting membrane pellet was washed using 0.2 M Na2CO3, followed by another round of ultracentrifugation. Excess inorganic salts were washed from the resulting pellet with Milli-Q water and another round of ultracentrifugation. The final product, cell membrane pellets, were stored at −20 °C for further sample preparation and LC-MS analysis.
N-Glycomics using nLC-QToF LC-MS/MS
The cell membrane pellets containing glycoproteins were processed further to release the intact N-glycans. To do so, the pellets were resuspended in N-glycan release solution (100 mM NH4HCO3, 5 mM DTT) and put in a boiling water bath for 2 minutes, cycling between 10 s on and 10 s off the bath. Afterward, 2 μL of PNGase F was added to the samples, followed by incubation at 37 °C for 18 hours. Milli-Q water was added to quench the reaction, followed by ultracentrifugation (200000×g, 45 minutes, 4 °C) to obtain a supernatant containing the release of N-glycans. The N-glycans were cleaned up using PGC-SPE by following the gradient: 80% ACN (0.1% v/v TFA), Milli-Q water, sample loading, washing with Milli-Q water, then elution with 40% ACN (0.05% v/v TFA). The solvent was removed from the cleaned-up N-glycans in vacuo and then stored at −20 °C until LC-MS/MS analysis.
N-Glycomics was performed following previously defined methods.25,66 The dried and cleaned-up N-glycans were reconstituted in Milli-Q water and then transferred into LC-MS/MS vials for injection into the instrument. The samples were injected into an Agilent 1200 series liquid chromatography system with an Agilent PGC-II chip (40-nL enrichment column, 5 μm; 75 μm × 43 mm separation column; Agilent Technologies, cat. no. G4240-64010). Analytical separation was performed using a gradient of mobile phase A (3% ACN, 0.1% v/v formic acid) and B (90% ACN, 1% v/v formic acid): 0–2 min: 0–0%, 2–20 min: 0–16%, 20–40 min: 16–72%, 40–42 min: 72–100%, 42–52 min: 100–100%, 52–54 min: 100–0%, 54–65 min: 0–0%.
Mass spectra were acquired using an Accurate mass QToF (Agilent Technologies, model no. 6520) over the 600–2000 m/z range in positive-ion mode. The Vcap was kept at 1850 V throughout the run. MS scans were acquired at 0.8 spectra per s and MS/MS scans were acquired at 1.0 spectra per s. Fragments were obtained in CID, with collision energies calculated using Vcollision = 1.8 × (m/z)/100–2.4. Mass spectra were acquired using data-dependent acquisition, selecting the top 5 precursors per scan for fragmentation.
Acquired LC-MS/MS data were processed using MassHunter Qualitative Analysis Software B.07.00 (Agilent Technologies). N-Glycan structures were identified using the MassHunter's Find by Molecular Feature algorithm using previously defined parameters, with matching to our in-house database of previously-identified N-glycans from lung cancer cells, which were subsequently manually validated using MS/MS spectra.35,65,67 Relative quantification of N-glycans was achieved by measuring the area under the curve (XIC) of each N-glycan structure. The XICs were further processed by normalizing to the TIC and subsequent classification and summation based on: high-mannose (containing >3 mannose residues), undecorated (containing <4 mannose residues, >3 N-acetylglucosamine residues, and no fucose nor sialic acid residues), fucosylated (containing <4 mannose residues, >3 N-acetylglucosamine residues, at least 1 fucose and no sialic acid residues), sialylated (containing <4 mannose residues, >3 N-acetylglucosamine residues, no fucose, and at least 1 sialic acid residue), or sialofucosylated (containing <4 mannose residues, >3 N-acetylglucosamine residues, at least 1 each of fucose and sialic acid residues).
RNA extraction and 3′-TagSeq RNAseq analysis
Total RNA was extracted from cultured cells following the RNeasy Mini kit manufacturer instructions. Cells were harvested by trypsinization, then resuspended in buffer RLT to obtain cell lysates. These were further homogenized using the QiAshredder spin columns, which were centrifuged at 21000×g for 2 minutes. To the flow-through, 70% ethanol solution was added, which was transferred to the RNeasy spin column, centrifuging afterward at 8000×g for 30 s. The spin columns were further washed with buffer RW1. To the spin columns, DNAse I incubation mix (1:7 DNAse I solution in buffer RDD) was added, followed by incubation at room temperature for 15 minutes. The spin columns were further washed with buffer RW1 by centrifuging at 8000×g for 30 s. This was followed by twice centrifugation with buffer RPE at 8000×g for 30 s and then 2 minutes. Finally, cleaned-up RNA samples were collected by adding sterile RNAse-free H2O to the spin-columns and centrifuging for 8000×g for 1 minute. The samples were stored at −80 °C until submission to the UC Davis DNAtech Core Facility. The samples were analyzed using Batch 3′ Tag-Seq analysis, with a read number of 4 M. Upon receipt of RNAseq data, raw reads were processed using a custom script based on the Tag-seq script (https://github.com/ben-laufer/Tag-seq). Data were processed using quantification mode to obtain normalized and annotated gene counts. Glycogene expression data was obtained by filtering the transcriptome based on annotated genes relevant to glycan biosynthesis and processing.11
Statistical analyses and regression model construction
Comparisons of glycomic and transcriptomic profiles between cells were performed using 2-way ANOVA (GraphPad Prism v10.4). To correlate the transcriptomic data with LC-MS/MS N-glycomic data we utilized the Regression Learner app in MATLAB R2022a (ver. 9.12). For each N-glycan composition abundance (response variable), we used the glycogene expression data as predictor variables. These datasets were used to train several models in the app using a 5-fold cross-validation scheme. After training, the models were ranked based on performance using RMSE and R2 metrics, with the best-performing models (low RMSE and high R2 values) per N-glycan were selected for further testing in the several use-cases outlined above. For each glycan, we trained several supervised machine-learning regression models to identify the best fitting model: linear regression models (linear, interactions linear, robust linear, stepwise linear), tree models (fine, medium, coarse), SVM models (linear, quadratic, cubic, fine Gaussian, medium Gaussian, coarse Gaussian), ensemble models (boosted trees, bagged trees), Gaussian process regression models (squared exponential, Matern 5/2, exponential, rational quadratic), neural networks (narrow, medium, wide, bilayered, trilayered), and kernels (SVM, least squares regression).Conclusions
Herein, we present methods for integrating data analysis for LC-MS/MS glycomics and glycoproteomics with RNAseq transcriptomics, to elucidate N-glycosylation pathways in lung cells. Specifically, we report here novel insights into the N-glycan biosynthetic pathway, obtained from correlating glycogene expression with N-glycosylation abundance, both in overall N-glycosylation using N-glycomics and in a site-specific manner using glycoproteomics. We observed that certain mannosidases (MAN1A1, MAN2A1), GlcNActransferases (MGAT1, MGAT3), galactosyltransferases (B4GALT1/2), fucosyltransferases (FUT7/8/11), and sialyltransferases (ST3Gal6, ST6Gal1, ST6Gal2) held high importance scores in the regression models of high-mannose, undecorated, fucosylated, sialylated, and sialofucosylated N-glycans.Numerous bioinformatic tools have been developed to aid glycoinformatics, such as in predicting glycan structures from transcriptomic data,22,68 glycosites from genomic data,69,70 protein-glycan interactions,71,72 and lectin-based glycan signatures from RNAseq.23 As of writing, there is still no methodology to predict protein N-glycosylation abundance from glycogene expression data. Hence, this is the motivation for our method glycoPATH, which can correlate information from both RNAseq transcriptomics and LC-MS/MS glycomics to predict glycosylation. From the correlations, we were able to construct prediction models to predict the N-glycome of cells derived from multiple tissue origins (CCD19-Lu, GLC01, Tib-190), from cells with low amount of starting material, and cells with perturbed glycosylation profiles due to inhibitor treatment. Although we were able to obtain accurate results of our predictions using the glycosylation enzymes, additional refinement of the models could be used to improve the accuracy and precision of predictions. Specifically, incorporation of glycosylation enzyme protein abundance, activity, and localization, may be beneficial.73–75 Additionally, the models can be further refined by incorporating linkage information (e.g. α2,3- vs. α2,6-linked sialic acid) of the N-glycan structures into the N-glycan dataset. Finally, the models were trained on global glycosylation profiles obtained using nLC-QToF methods; as such, does not contain glycosite- and glycoprotein-specific information. Future work involving quantifying glycoprotein-specific glycan abundances using nLC-Orbitrap methods will benefit from the ML-based modeling presented here. While compositional abundances are the natural starting point for this form of correlation study, there is strong potential for the incorporation of linkage information in future work. In depth structural analysis of glycans falls into two broad categories of teasing apart structural features by tandem MS as well as distinguishing linkage information with retention time. Ongoing projects are seeking to delve into the next layer of glycan structural information, involving knock-outs of specific glycosyltransferase genes to identify specific peaks corresponding to specific glycan linkages. Altogether, the method presented here can provide comprehensive information on the glycogenes involved in protein glycosylation.
Data availability
Raw and processed mass spectrometry data are freely available and can be found on the MassIVE repository (Glycomics: DOI: https://10.25345/C50K26N61, MSV000092941). Training and validation data, and MATLAB workspace are available on GitHub (https://github.com/MichRussAlv/glycoPATH).Author contributions
M. R. S. A. designed and performed experiments, analyzed data, created the figures and wrote the manuscript. X. A. H. performed experiments and analyzed data. A. O., S. J. A., R. S., Q. Z., and A. S. performed experiments. A. S. analyzed data and wrote the manuscript. C. B. L conceived the idea, supervised the study, and co-wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research reported in this report was supported by General Medicine of the National Institutes of Health under the award numbers RO1GM049077 and RO1AG062240.Notes and references
- X. Tang, J. Tena, J. Di Lucente, I. Maezawa, D. J. Harvey, L.-W. Jin, C. B. Lebrilla and A. M. Zivkovic, Sci. Rep., 2023, 13, 7816 CrossRef CAS PubMed.
- J. Tena, X. Tang, Q. Zhou, D. Harvey, M. Barajas-Mendoza, L. Jin, I. Maezawa, A. M. Zivkovic and C. B. Lebrilla, Alz & Dem Diag Ass & Dis Mo, 2022, 14(1), e12309 Search PubMed.
- L. Yu, Z. Huo, J. Yang, H. Palma-Gudiel, P. A. Boyle, J. A. Schneider, D. A. Bennett and J. Zhao, Front. Aging Neurosci., 2021, 13, 765259 CrossRef CAS PubMed.
- K. Flevaris and C. Kontoravdi, Int. J. Mol. Sci., 2022, 23, 5180 CrossRef CAS PubMed.
- X. Zhou, M. J. Kailemia, Y. Sun, Z. Shuai, G.-X. Yang, S. Dhaliwal, L. Cristoferi, P. S. C. Leung, P. Invernizzi, C. L. Bowlus, C. B. Lebrilla, A. A. Ansari, W. M. Ridgway, W. Zhang and M. E. Gershwin, J. Autoimmun., 2020, 113, 102503 CrossRef CAS PubMed.
- S. S. Pinho and C. A. Reis, Nat. Rev. Cancer, 2015, 15, 540–555 CrossRef CAS PubMed.
- D. H. Dube and C. R. Bertozzi, Nat. Rev. Drug Discov., 2005, 4, 477–488 CrossRef CAS PubMed.
- M. M. Fuster and J. D. Esko, Nat. Rev. Cancer, 2005, 5, 526–542 CrossRef CAS PubMed.
- D. Thomas, A. K. Rathinavel and P. Radhakrishnan, Biochim. Biophys. Acta Rev. Canc, 2021, 1875, 188464 CrossRef CAS PubMed.
- A. Peixoto, M. Relvas-Santos, R. Azevedo, L. L. Santos and J. A. Ferreira, Front. Oncol., 2021, 1875(1), 188464 Search PubMed.
- T. Groth, A. D. Diehl, R. Gunawan and S. Neelamegham, Bioinformatics, 2022, 38(24), 5413–5420 CrossRef CAS PubMed.
- P. Hossler, B. C. Mulukutla and W.-S. Hu, PLoS One, 2007, 2(8), e713 CrossRef PubMed.
- P. Stanley, K. W. Moremen, N. E. Lewis, N. Taniguchi and M. Aebi, in Essentials Of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, D. Mohnen, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 4th edn, 2022 Search PubMed.
- F. Diniz, P. Coelho, H. O. Duarte, B. Sarmento, C. A. Reis and J. Gomes, Cancers, 2022, 14, 911 CrossRef CAS PubMed.
- A. F. Costa, D. Campos, C. A. Reis and C. Gomes, Trends Cancer Res., 2020, 6, 757–766 CrossRef CAS PubMed.
- W. Peng, R. zhu, S. Zhou, P. Mirzaei and Y. Mechref, Sci. Rep., 2019, 9, 17361 CrossRef PubMed.
- L. Bezjak, V. Erklavec Zajec, Š. Baebler, T. Stare, K. Gruden, A. Pohar, U. Novak and B. Likozar, Biotechnol. Bioeng., 2021, 118, 1476–1490 CrossRef CAS PubMed.
- C. Blöchl, D. Wang, O. A. Mayboroda, G. S. M. Lageveen-Kammeijer and M. Wuhrer, Cell Biosci., 2023, 13, 31 CrossRef PubMed.
- C. J. Kearney, S. J. Vervoort, K. M. Ramsbottom, I. Todorovski, E. J. Lelliott, M. Zethoven, L. Pijpers, B. P. Martin, T. Semple, L. Martelotto, J. A. Trapani, I. A. Parish, N. E. Scott, J. Oliaro and R. W. Johnstone, Sci. Adv., 2021, 7, eabe3610 CrossRef CAS PubMed.
- F. Minoshima, H. Ozaki, H. Odaka and H. Tateno, iScience, 2021, 24, 102882 CrossRef CAS.
- H. Odaka, H. Ozaki and H. Tateno, STAR Protoc., 2022, 3, 101179 CrossRef CAS PubMed.
- Y.-F. Huang, K. Aoki, S. Akase, M. Ishihara, Y.-S. Liu, G. Yang, Y. Kizuka, S. Mizumoto, M. Tiemeyer, X.-D. Gao, K. F. Aoki-Kinoshita and M. Fujita, Dev. Cell, 2021, 56, 1195–1209.e7 CrossRef CAS PubMed.
- R. Qin, L. K. Mahal and D. Bojar, iScience, 2022, 25, 105163 CrossRef CAS PubMed.
- L. A. Dworkin, H. Clausen and H. J. Joshi, iScience, 2022, 25, 104419 CrossRef CAS PubMed.
- Q. Li, Y. Xie, M. Wong, M. Barboza and C. B. Lebrilla, Nat. Protoc., 2020, 15, 2668–2704 CrossRef CAS PubMed.
- D. Bojar, L. Meche, G. Meng, W. Eng, D. F. Smith, R. D. Cummings and L. K. Mahal, ACS Chem. Biol., 2022, 17, 2993–3012 CrossRef CAS PubMed.
- R. S. McClure, Y. Rericha, K. M. Waters and R. L. Tanguay, Front. Bioinform., 2023, 3, 1234218 CrossRef PubMed.
- J. Wang, S. A. Rieder, J. Wu, S. Hayes, R. A. Halpin, M. de los Reyes, Y. Shrestha, R. Kolbeck and R. Raja, Sci. Rep., 2019, 9, 8445 CrossRef PubMed.
- M. R. S. Alvarez, P. G. Moreno, S. J. B. Grijaldo-Alvarez, A. Yadlapati, Q. Zhou, M. P. Narciso, G. C. Completo, R. C. Nacario, J. F. Rabajante, F. M. Heralde III and C. B. Lebrilla, Glycobiology, 2024, cwae030 CrossRef CAS PubMed.
- L. R. Ruhaak, S. L. Taylor, C. Stroble, U. T. Nguyen, E. A. Parker, T. Song, C. B. Lebrilla, W. N. Rom, H. Pass, K. Kim, K. Kelly and S. Miyamoto, J. Proteome Res., 2015, 14, 4538–4549 CrossRef CAS PubMed.
- M. R. Alvarez, J. Tena, Q. Zhou, M. Cabanatan, S. Tomacas, L. M. Serrano, S. J. B. Grijaldo, J. Rabajante, M. T. Barzaga, N. Tan-Liu, F. I. Heralde, C. B. Lebrilla, G. C. Completo and R. Nacario, ACS Omega, 2022, 7(44), 40230–40240 CrossRef CAS PubMed.
- L. R. Ruhaak, C. Stroble, J. Dai, M. Barnett, A. Taguchi, G. E. Goodman, S. Miyamoto, D. Gandara, Z. Feng, C. B. Lebrilla and S. Hanash, Cancer Prev. Res., 2016, 9, 317–323 CrossRef CAS PubMed.
- Y. Liang, P. Han, T. Wang, H. Ren, L. Gao, P. Shi, S. Zhang, A. Yang, Z. Li and M. Chen, Clin. Proteomics, 2019, 16, 20 CrossRef PubMed.
- B. Mészáros, G. Járvás, A. Farkas, M. Szigeti, Z. Kovács, R. Kun, M. Szabó, E. Csánky and A. Guttman, J. Chromatogr. B, 2020, 1137, 121913 CrossRef PubMed.
- M. R. Alvarez, Q. Zhou, J. Tena, M. Barboza, M. Cabanatan, S. Tomacas, L. M. Serrano, M. T. Barzaga, N. Tan-Liu, C. B. Lebrilla, G. C. Completo, F. I. Heralde and R. Nacario, Cancers, 2022, 15(5), 1559 CrossRef PubMed.
- H. J. An, P. Gip, J. Kim, S. Wu, K. W. Park, C. T. McVaugh, D. V. Schaffer, C. R. Bertozzi and C. B. Lebrilla, Mol. Cell. Proteomics, 2012, 11, M111.010660 CrossRef PubMed.
- V. Adorno-cruz and H. Liu, Genes Dis., 2019, 6, 16–24 CrossRef CAS PubMed.
- G. Bendas and L. Borsig, Int. J. Cell Biol., 2012, 2012(1), 676731 Search PubMed.
- H. Hamidi and J. Ivaska, Nat. Rev. Cancer, 2018, 18, 533–548 CrossRef CAS PubMed.
- G. Marsico, L. Russo, F. Quondamatteo and A. Pandit, Trends Cancer Res., 2018, 4, 537–552 CrossRef CAS PubMed.
- Y.-C. Liu, H.-Y. Yen, C.-Y. Chen, C.-H. Chen, P.-F. Cheng, Y.-H. Juan, C.-H. Chen, K.-H. Khoo, C.-J. Yu, P.-C. Yang, T.-L. Hsu and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11332–11337 CrossRef CAS PubMed.
- H.-Y. Yen, Y.-C. Liu, N.-Y. Chen, C.-F. Tsai, Y.-T. Wang, Y.-J. Chen, T.-L. Hsu, P.-C. Yang and C.-H. Wong, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 6955–6960 CrossRef CAS PubMed.
- D. D. Park, C. Phoomak, G. Xu, L. P. Olney, K. A. Tran, S. S. Park, N. E. Haigh, G. Luxardi, W. Lert-itthiporn, M. Shimoda, Q. Li, N. Matoba, F. Fierro, S. Wongkham, E. Maverakis and C. B. Lebrilla, Proc. Natl. Acad. Sci. U.S.A., 2020, 117, 7633–7644 CrossRef CAS PubMed.
- Y. Bai, W. Huang, L. Ma, J. L. Jiang and Z. Chen, Int. J. Mol. Sci., 2014, 15(4), 6356–6377 CrossRef PubMed.
- S. D. Lim, A. N. Young, G. P. Paner and M. B. Amin, Virchows Archiv : an international journal of pathology., 2008, 452, 49–55 CrossRef CAS PubMed.
- M. T. Landi, T. Dracheva, M. Rotunno, J. D. Figueroa, H. Liu, A. Dasgupta, F. E. Mann, J. Fukuoka, M. Hames, A. W. Bergen, S. E. Murphy, P. Yang, A. C. Pesatori, D. Consonni, P. A. Bertazzi, S. Wacholder, J. H. Shih, N. E. Caporaso and J. Jen, PLoS One, 2008, 3, e1651 CrossRef PubMed.
- P. Norton, M. A. Comunale, H. Herrera, M. Wang, J. Houser, M. Wimmerova, P. R. Romano and A. Mehta, Proteomics, 2016, 16, 3126–3136 CrossRef CAS PubMed.
- K. W. Moremen and A. V. Nairn, in Handbook Of Glycosyltransferases And Related Genes, ed. N. Taniguchi, K. Honke, M. Fukuda, H. Narimatsu, Y. Yamaguchi and T. Angata, Springer Japan, Tokyo, 2014, pp. 1297–1312 Search PubMed.
- T. Hennet, Cell. Mol. Life Sci., 2002, 59, 1081–1095 CrossRef CAS PubMed.
- A. García-García, L. Ceballos-Laita, S. Serna, R. Artschwager, N. C. Reichardt, F. Corzana and R. Hurtado-Guerrero, Nat. Commun., 2020, 11, 973 CrossRef PubMed.
- F. Dall'Olio and M. Chiricolo, Glycoconj. J., 2001, 18, 841–850 CrossRef PubMed.
- W. Zhou, Z. Yan and L. Zhang, Sci. Rep., 2024, 14, 5905 CrossRef CAS PubMed.
- P. Gao, A. Honkela, M. Rattray and N. D. Lawrence, Bioinformatics, 2008, 24, i70–i75 CrossRef PubMed.
- K. Legler, R. Rosprim, T. Karius, K. Eylmann, M. Rossberg, R. M. Wirtz, V. Müller, I. Witzel, B. Schmalfeldt, K. Milde-Langosch and L. Oliveira-Ferrer, Br. J. Cancer, 2018, 118, 847–856 CrossRef CAS PubMed.
- I. C. Ye, E. J. Fertig, J. W. DiGiacomo, M. Considine, I. Godet and D. M. Gilkes, Mol. Cancer Res., 2018, 16, 1889–1901 CrossRef CAS PubMed.
- E. Zodro, M. Jaroszewski, A. Ida, T. Wrzesiński, Z. Kwias, H. Bluyssen and J. Wesoly, Tumor Biol., 2013, 35, 2607–2617 CrossRef PubMed.
- W. Ruan, Y. Yang, Q. Yu, T. Huang, Y. Wang, L. Hua, Z. Zeng and R. Pan, Cell Biol. Int., 2021, 45, 2275–2286 CrossRef CAS PubMed.
- F. Schneider, W. Kemmner, W. Haensch, G. Franke, S. Gretschel, U. Karsten and P. M. Schlag, Cancer Res., 2001, 61, 4605–4611 CAS.
- J. Ogawa, H. Inoue and S. Koide, Cancer Res., 1996, 56, 325–329 CAS.
- J. Tomlinson, J. L. Wang, S. H. Barsky, M. C. Lee, J. Bischoff and M. Nguyen, Int. J. Oncol., 2000, 16, 347–353 CAS.
- S. A. Jassam, Z. Maherally, K. Ashkan, G. J. Pilkington and H. L. Fillmore, J. Neuro Oncol., 2019, 143, 405–415 CrossRef CAS PubMed.
- J.-X. Liang, W. Gao and L. Cai, OncoTargets Ther., 2017, 10, 3971–3978 CrossRef PubMed.
- Q. Yuan, X. Chen, Y. Han, T. Lei, Q. Wu, X. Yu, L. Wang, Z. Fan and S. Wang, Int. J. Cancer, 2018, 143, 2319–2330 CrossRef CAS PubMed.
- X. Wang, Z. Deng, C. Huang, T. Zhu, J. Lou, L. Wang and Y. Li, J. Proteonomics, 2018, 172, 1–10 CrossRef CAS PubMed.
- M. R. S. Alvarez, Q. Zhou, S. J. B. Grijaldo, C. B. Lebrilla, R. C. Nacario, F. M. Heralde, J. F. Rabajante and G. C. Completo, Molecules, 2022, 27, 3834 CrossRef CAS PubMed.
- Q. Li, Y. Xie, M. Wong and C. Lebrilla, Cells, 2019, 8, 882 CrossRef CAS PubMed.
- Q. Zhou, M. R. Alvarez, K. Solakyildirim, J. Tena, L. M. Serrano, M. Lam, C. Nguyen, F. Tobias, A. B. Hummon, R. Nacario and C. Lebrilla, Glycobiology, 2022, 33(1), 2–16 CrossRef PubMed.
- S. Kawano, K. Hashimoto, T. Miyama, S. Goto and M. Kanehisa, Bioinformatics, 2005, 21, 3976–3982 CrossRef CAS PubMed.
- T. Pitti, C.-T. Chen, H.-N. Lin, W.-K. Choong, W.-L. Hsu and T.-Y. Sung, Sci. Rep., 2019, 9, 15975 CrossRef PubMed.
- S. Sun, B. Zhang, P. Aiyetan, J.-Y. Zhou, P. Shah, W. Yang, D. A. Levine, Z. Zhang, D. W. Chan and H. Zhang, J. Proteome Res., 2013, 12, 5609–5615 CrossRef CAS PubMed.
- E. J. Carpenter, S. Seth, N. Yue, R. Greiner and R. Derda, Chem. Sci., 13, 6669–6686 RSC.
- O. C. Grant, X. Xue, D. Ra, A. Khatamian, B. L. Foley and R. J. Woods, Glycobiology, 2016, 26, 1027–1028 CrossRef CAS PubMed.
- E. Reynders, F. Foulquier, W. Annaert and G. Matthijs, Glycobiology, 2011, 21, 853–863 CrossRef CAS PubMed.
- K. J. Colley, A. Varki, R. S. Haltiwanger and T. Kinoshita, in Essentials Of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, D. Mohnen, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Cold Spring Harbor Laboratory Press, Cold Spring Harbor (NY), 4th edn, 2022 Search PubMed.
- A. S. Opat, C. van Vliet and P. A. Gleeson, Biochimie, 2001, 83, 763–773 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00467e |
| This journal is © The Royal Society of Chemistry 2025 |