
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTandem activated caged galactoside prodrugs: advancing beyond single galactosidase dependence†
Yunying
Tan
a,
Jie
Liu
a,
Dianya
Yong
a,
Jing
Hu
 b,
Peter H.
Seeberger
c,
Junjie
Fu
*ae and
Jian
Yin
*ade
b,
Peter H.
Seeberger
c,
Junjie
Fu
*ae and
Jian
Yin
*ade
aKey Laboratory of Carbohydrate Chemistry and Biotechnology, Ministry of Education, School of Biotechnology & School of Life Sciences and Health Engineering, Jiangnan University, Wuxi 214122, PR China. E-mail: jfu@jiangnan.edu.cn; jianyin@jiangnan.edu.cn
bWuxi School of Medicine, Jiangnan University, Wuxi 214122, PR China
cBiomolecular Systems Department, Max Planck Institute of Colloids and Interfaces, Potsdam 14476, Germany
dSchool of Life Sciences and Health Engineering, Jiangnan University, Wuxi 214122, PR China
eInnovation Center for Vaccine Engineering, Jiangnan University, Wuxi 214122, PR China
First published on 18th March 2025
Abstract
β-Galactoside prodrugs, activated by β-galactosidase (β-gal) highly expressed in some cancer cells, have been explored as anticancer agents for three decades. However, the distribution of β-gal lacks sufficient specificity to ensure precise drug release at cancer sites. By utilizing the highly stringent substrate specificity of β-gal, we chose the naturally occurring hydroxyl group of galactose as a prodrug modification site and developed a new class of tandem activated caged galactoside (TACG) prodrugs that require an additional trigger for more controlled on-demand drug release. We demonstrated that attaching various masking groups to the 6-hydroxyl group of galactose renders the galactosides resistant to β-gal hydrolysis. Focusing on the photosensitive mask 4,5-dimethoxy-2-nitrobenzyl (DMNB), we synthesized O6-DMNB modified galactosides of combretastatin A4 and 8-hydroxyquinoline, showcasing their UV/β-gal-dependent anticancer activities. We further established synthetic routes for O2-, O3-, and O4-DMNB modified TACGs. Comparative intracellular studies highlighted the O2-DMNB modified TACG as the most effective positional isomer, offering superior light-dependent selectivity. This insight led to the discovery of the O2-DMNB modified galactoside of combretastatin A4 as a potent UV-dependent microtubule assembly inhibitor. Our work provides a straightforward, effective, and universally applicable strategy for constructing dual-stimulus responsive galactoside prodrugs, extendable to various glycoside prodrugs, advancing carbohydrate-based drug discovery.
1. Introduction
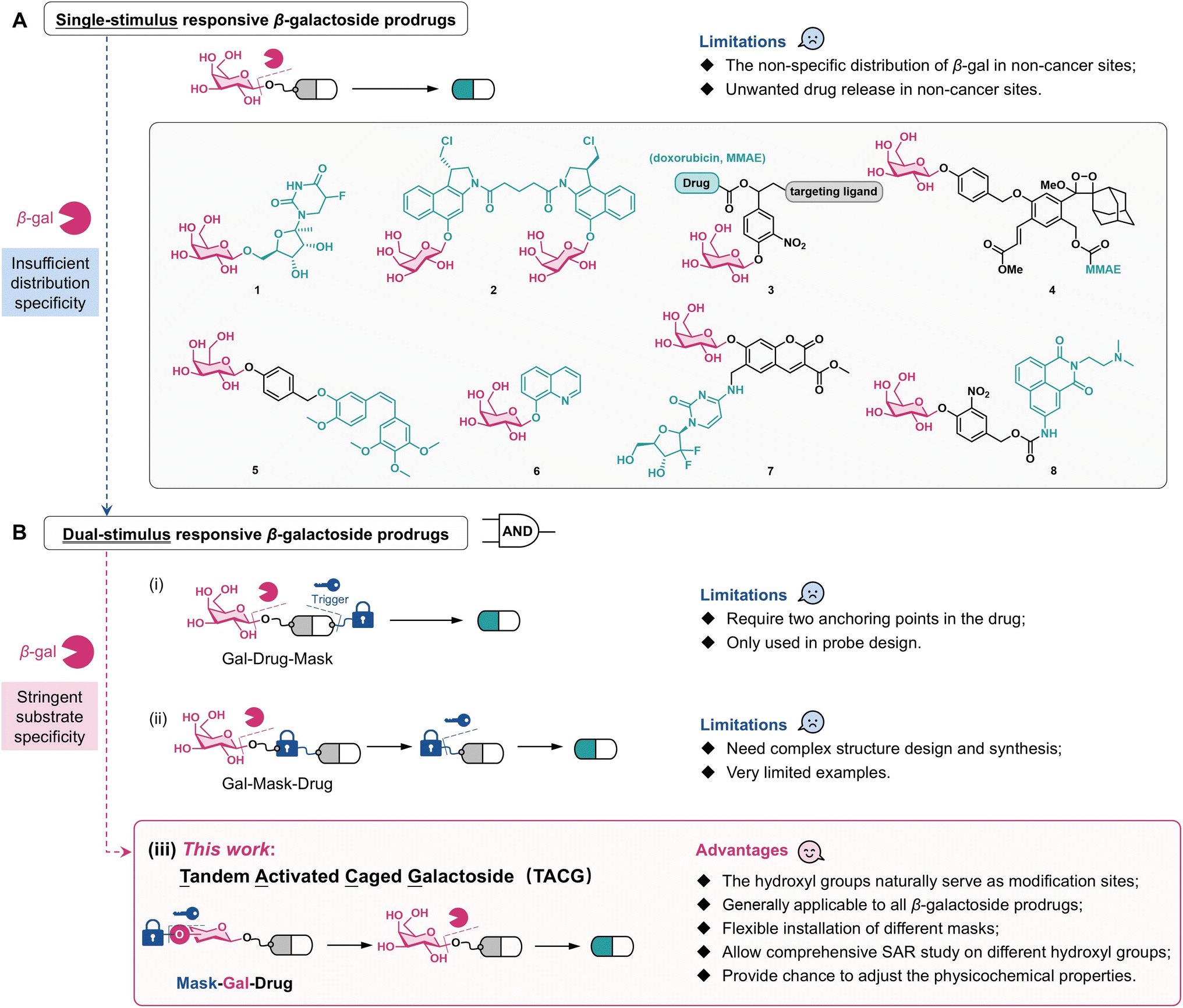
Caged prodrugs, where essential functional groups such as hydroxyl or amino groups of drug molecules are chemically modified with masking groups, play pivotal roles in medicinal chemistry and chemical biology.1 These masks function as locks that deactivate or significantly reduce the activity of the parent drugs. Upon exposure to specific stimuli, termed triggers, these masks can be removed, thereby releasing the parent drugs with restored bioactivity. Traditional stimuli for prodrug activation include light, oxidation–reduction reactions, low pH environments, enzymatic cleavage, and bioorthogonal reactions.2 Recent advancements have introduced novel activation methods involving mechanical force,3 ultrasound,4 and X-rays.5,6Due to their aberrant metabolism, cancer cells often exhibit significantly elevated levels of specific enzymes compared with normal cells. Consequently, prodrugs designed to respond to these cancer-specific enzymes offer the potential for controlled activation and release of cytotoxic drugs within cancer cells, which is crucial in chemotherapy. Enzymes currently exploited for targeted prodrug activation in cancer cells include alkaline phosphatase,7 matrix metalloproteinases,8 tyrosinase,9 cathepsin B,10 NAD(P)H quinone dehydrogenase 1,11 and glycosidases.12,13 Notably, β-galactosidase (β-gal) activity is elevated in certain cancer cells, particularly ovarian cancer cells, making β-galactoside prodrugs widely applicable in anticancer therapy.12–15 These prodrugs are constructed by linking the drug's active group to D-galactose via a β-galactosidic bond (Scheme 1A), which can be cleaved by β-gal-catalyzed hydrolysis to release the parent drug. The 5-fluorouracil β-galactoside prodrug (1) was reported in 1994.16 Since 2002, a series of duocarmycin β-galactoside prodrugs (2) have been developed.17–19 Since 2011, β-galactoside prodrugs capable of releasing drugs such as doxorubicin and monomethyl auristatin E (MMAE) and functionalized with targeting ligands such as folates and monoclonal antibodies (3) have been reported.20–23 By integrating MMAE with the dioxetane luminophore, the β-galactoside theranostic prodrug (4) is monitored in chemiluminescence diagnostic mode.24 In addition, β-galactoside prodrugs of combretastatin A4 (5),25 8-hydroxyquinoline (6),26 gemcitabine (7),27 and amonafide (8)28 have been reported.
 | ||
| Scheme 1 Design of novel dual-stimulus responsive β-galactoside prodrugs. | ||
All these prodrugs (1–8) release parent drugs solely in response to β-gal and hence can be categorized as single-stimulus responsive β-galactoside prodrugs (Scheme 1A). However, studies have shown elevated β-gal activity not only in cancer cells but also in M2 polarized macrophages, glucose-induced macrophages, neurons, osteoclasts, and other specific cells.29,30 Immunohistochemical analysis revealed the widespread distribution of β-gal in tissues such as liver, intestinal mucosa, and placenta.15,31,32 This suggests a potential risk of premature release of toxic drugs in non-cancerous sites. Recent findings indicate increased content and activity of β-gal in lysosomes of senescent cells, making β-galactoside prodrugs responsive to senescence-associated β-gal, an important class of senolytics.33 This further raises concerns about the systemic toxicity of β-galactoside prodrugs given the widespread distribution of senescent cells. Therefore, despite its high expression in some cancer cells, the distribution of β-gal is insufficiently specific to ensure precise drug release from single-stimulus responsive β-galactoside prodrugs. Utilizing antibody-directed enzyme-prodrug therapy (ADEPT) to deliver β-gal to cancer cells is an effective strategy to address this issue.16,19 However, ADEPT requires precise design of antibodies and enzymes, increasing therapeutic complexity. Another strategy involves targeted delivery systems based on nanomaterials to enrich β-gal within cancer cells.34 Compared with small molecule prodrugs, nanomaterial-based delivery systems present uncertainties in pharmacokinetic properties and quality uniformity. Furthermore, these strategies may elicit immune reactions and do not fundamentally prevent β-galactoside prodrugs from being activated by β-gal before reaching their target sites.
One promising strategy to enhance the controlled activation of small molecule prodrugs involves advancing single-stimulus responsive prodrugs to dual-stimulus responsive prodrugs, which require activation by two different stimuli to release the parent drugs. This “AND gate” logic significantly enhances the controllability of drug release (Scheme 1B). Typical examples of dual-stimulus responsive prodrugs for cancer treatment include those activated by histone deacetylase/cathepsin L,35 phosphatase/trans-cyclooctene,36 and reactive oxygen species (ROS)/tyrosinase,37 but have been applied rarely for glycoside prodrugs.
Theoretically, two strategies can be employed to construct dual-stimulus responsive galactoside prodrugs. The first strategy involves simultaneously conjugating galactose and another masking group to the drug molecule. The locked drug is released upon activation by the specific trigger of the masking group and β-gal (Scheme 1B(i)). This strategy requires two anchoring points within the drug molecule38 and has not been applied to glycoside prodrugs. The second strategy introduces another masking group between galactose and the drug molecule, resulting in prodrugs sequentially activated by β-gal and the specific trigger (Scheme 1B(ii)). Here, careful selection of the additional masking group, which itself usually needs to provide an anchoring point for galactose linkage, makes the design and synthesis challenging and less versatile. To date, only one galactoside prodrug, which sequentially responds to β-gal and green light to release nitrogen mustard, has been reported.39 No general and facile strategy to construct dual-stimulus responsive galactoside prodrugs is currently available.
Carbohydrates are characterized by multiple hydroxyl groups that render their structures much more complex than nucleotide or peptide chains and pose substantial challenges in chemical synthesis. In glycosylation reactions, the numerous hydroxyl groups are often considered “troublemakers” and typically need to be protected first. However, from a medicinal chemistry perspective, these hydroxyl groups are also “opportunity providers.” Since the hydroxyl groups on carbohydrates determine their type and biological function, they naturally serve as sites for prodrug modification. For instance, modification of the 4- and 6-hydroxyl groups of the gene expression inducer isopropyl β-D-thiogalactopyranoside (IPTG) with 4-nitrobenzyl resulted in an IPTG precursor that releases IPTG under hypoxic conditions and induces target gene expression.40 Modification of trehalose-6-phosphate (T6P), a crucial signaling molecule in plant growth, led to a photocaged T6P prodrug. This prodrug is more easily absorbed by plants than T6P and releases T6P under light exposure to promote plant growth.41N-Azidoacetylmannosamine (ManNAz) is widely used for carbohydrate metabolic labeling. By modifying the hydroxyl group of ManNAz, precursor molecules can be obtained to release ManNAz in response to stimuli such as light and ROS.42,43
This study presents a general strategy for constructing dual-stimulus responsive galactoside prodrugs by introducing a masking group onto the hydroxyl group of β-galactosides. The constructed prodrug requires sequential activation by the specific trigger and β-gal to release the parent drug (Scheme 1B(iii)). The rationale for this design stems from the stringent substrate specificity of β-gal: even subtle structural variations in the hydroxyl groups of galactoside render the β-galactosidic bond unresponsive to β-gal.31,44 In fact, in some galactoside prodrugs, the 2-, 3-, 4-, and 6-hydroxyl groups of galactose are acetylated for physicochemical property considerations.39,45,46 These prodrugs need to undergo esterase hydrolysis to expose the hydroxyl groups of galactose before being hydrolyzed by β-gal. Since esterase is widely distributed in the body, such prodrugs cannot be considered as authentic dual-stimulus responsive galactoside prodrugs. Chen et al. introduced masks with azido groups onto the four hydroxyl groups of coumarin β-galactosides.34 The resulting molecule requires removal of the masking groups under reducing conditions to expose the hydroxyl groups before responding to β-gal, but this strategy has not been applied to prodrugs. Hou et al. confirmed that introducing a simple methyl or isopropyl group onto the 6-hydroxyl group of β-galactosides is sufficient to make the galactosidic bond resistant to natural β-gal, but responsive only to artificial β-gal engineered via the bump-and-hole approach.31 This observation further demonstrates that even a minor modification of a single hydroxyl group of galactose can lock the galactosidic bond.
The newly designed prodrugs are named Tandem Activated Caged Galactosides (TACGs) that remain resistant to β-gal until the removal of the masking group from the hydroxyl groups. In principle, all single-stimulus responsive galactoside prodrugs can be converted into TACGs by introducing masking groups onto galactose, making it a generally applicable strategy. A wide and flexible range of masking groups can be employed to design highly versatile TACGs. The 2-, 3-, 4-, and 6-hydroxyl groups of galactose can in principle all be used for linking the masking group, allowing for comprehensive structure–activity relationship (SAR) studies and prodrug optimization. The modification of the hydroxyl groups also provides opportunities for adjusting the physicochemical properties and cellular uptake efficiency of the prodrugs.
2. Results
2.1. Initial validation of the TACG concept
To validate the TACG design, we used 4-methylumbelliferone (MU) as a fluorescent reporter and chose the 6-hydroxyl group of galactose as the locking site due to its ease of selective modification. We selected common masking groups used in prodrug design, including 2-nitrobenzyl (ONB), 4,5-dimethoxy-2-nitrobenzyl (DMNB), allyl (ALL), tert-butyldiphenylsilyl (TBDPS), 4-boronic acid benzyl (BAB), and 4-azidobenzyl (AZB), to modify the 6-hydroxyl group of MU-β-D-galactoside (Gal-MU), constructing TACG model compounds 6-ONB-Gal-MU, 6-DMNB-Gal-MU, 6-ALL-Gal-MU, 6-TBDPS-Gal-MU, 6-BAB-Gal-MU, and 6-AZB-Gal-MU, respectively (Fig. 1A). ONB and DMNB are classic UV-responsive groups,47 while ALL and TBDPS are common bioorthogonal groups responsive to Pd(0)48 and fluoride ions,49 respectively. BAB and AZB groups are responsive to hydrogen peroxide (H2O2)50 and reducing species such as hydrogen sulfide (H2S),51 respectively. | ||
| Fig. 1 (A) Design of TACG model compounds, 6-MASK-Gal-MU. (B) The docking of Gal-MU and representative TACG model compounds with A. oryzae β-gal (PDB: 4IUG). β-Gal is shown as a grey surface, key amino acid residues interacting with the hydroxy groups are shown in yellow, and ligand molecules are shown as stick models with the galactose moiety in red, MU in green, and the masking group in blue. (C) HPLC study of the dual-stimulus responsiveness of TACG model compounds. (D) On-demand release of MU from 6-DMNB-Gal-MU solution containing β-gal under UV irradiation (five seconds each). (E) β-Gal concentration- and time-dependent release of MU from 6-DMNB-Gal-MU after UV irradiation. The green dashed lines in (D) and (E) indicate the theoretical maximum fluorescence intensity corresponding to the complete release of MU from 6-DMNB-Gal-MU. | ||
Two synthetic routes were developed for these TACG model compounds (see synthesis details in the ESI†). The first route involves selectively introducing the masking group onto the 6-hydroxyl group, followed by glycosylation to conjugate MU, and finally deprotection to obtain the target molecule. The second route involves first glycosylating MU with an O-protected galactose and then selectively exposing the 6-hydroxyl group through an orthogonal protection strategy and finally introducing the masking group and removing the protecting groups on galactose to obtain the target molecule. Since the glycosylation step requires bromination under acidic conditions, the second route is more suitable for acid-sensitive masking groups such as ALL and AZB.
The Schrödinger Maestro docking tool was used to predict the binding of Gal-MU and TACG model compounds with β-gal.52,53 The results showed that Gal-MU fits well into the active pocket of β-gal from both Aspergillus oryzae (A. oryzae) (Fig. 1B) and humans (Fig. S1†). Notably, the hydroxyl groups of galactose form hydrogen bonds with amino acid residues in the active pocket of β-gal, similar to the binding mode of free galactose.54,55 For example, in the binding of Gal-MU with A. oryzae β-gal, the 2-, 3-, 4-, and 6-hydroxyl groups form hydrogen bonds with Glu298, Tyr96/Ala141, Asn140/Glu142, and Glu142/Tyr364, respectively. In contrast, the galactose moiety of TACG model compounds could not enter the active pocket of β-gal due to the steric hindrance of the O6 masking group (Fig. 1B, S2 and S3†). Consistent with the docking results, ultraviolet-visible (UV-vis) spectroscopy, fluorescence spectroscopy, and high-performance liquid chromatography (HPLC) demonstrated that Gal-MU rapidly responds to β-gal to release MU (Fig. S4†). In contrast, after incubating the six TACG model compounds with β-gal for 24 hours, HPLC showed no significant changes, indicating that they do not respond to β-gal (Fig. 1C and S5†). We next investigated the dual-stimulus responsiveness of the TACG model compounds using HPLC. Taking 6-DMNB-Gal-MU as an example, it was almost completely converted to Gal-MU after UV irradiation (365 nm, 6 W, and 3 minutes) in the absence of β-gal, and a distinct MU peak was observed after UV irradiation in the presence of β-gal (Fig. 1C). Therefore, 6-DMNB-Gal-MU requires sequential activation by UV/β-gal to release MU, echoing our design. The dual responsiveness of 6-ONB-Gal-MU, 6-ALL-Gal-MU, and 6-TBDPS-Gal-MU was similar to that of 6-DMNB-Gal-MU (Fig. 1C and S5†). However, for 6-BAB-Gal-MU and 6-AZB-Gal-MU, only trace amounts of MU were detected under UV/β-gal. Mass spectrometry showed that after responding to H2O2, the BAB group of 6-BAB-Gal-MU was converted to 4-hydroxybenzyl (HB), which hardly underwent 1,6-elimination to release Gal-MU as expected. A similar situation was observed for 6-AZB-Gal-MU (Fig. S6†). We speculate that this is due to the high pKa value of the 6-hydroxyl group, which has a weak leaving ability and hinders the 1,6-elimination process.56 Photoactivation as an external stimulus offers advantages including precise spatiotemporal controllability, remote triggering, tunable excitation of power and wavelength, and high biocompatibility.47 Therefore, we chose DMNB, a widely used photolabile group in chemical biology research, as the masking group for subsequent TACG studies.
Utilizing the fluorescent properties of MU, we further studied the MU-releasing behavior of 6-DMNB-Gal-MU. Fluorescence spectrophotometry (Fig. S7†) and high-resolution mass spectrometry (HRMS) (Fig. S8†) clearly indicated that 6-DMNB-Gal-MU does not respond to β-gal but converts to Gal-MU upon UV irradiation, which is then hydrolyzed by β-gal to release MU. Importantly, we can precisely control the total amount of MU released by adjusting the UV irradiation time. In a solution of 6-DMNB-Gal-MU containing β-gal, fluorescence detection showed a gradual increase in MU release with each five-second UV irradiation, while almost no MU was released without UV, even with extended incubation time (Fig. S9† and 1D). After UV irradiation (three minutes) of a solution of 6-DMNB-Gal-MU containing β-gal, the amount of MU released showed a clear dependence on β-gal concentration and β-gal incubation time (Fig. 1E).
2.2. Establishment of a synthetic route to O6-photocaged TACGs
After validating the design concept using TACG model molecules, we aimed to synthesize authentic TACG prodrugs responsive to UV/β-gal. The microtubule assembly inhibitor combretastatin A-4 (CA4) and metal ion chelator 8-hydroxyquinoline (HQ), both previously applied in single-stimulus responsive galactoside prodrugs,25,26 were selected as the drug cargos. The phenolic hydroxyl groups essential for the bioactivity of CA4 and HQ were linked to O6-DMNB masked D-galactose via a para-hydroxy-benzyl alcohol-based self-immolative spacer (SIS),56,57 resulting in 6-DMNB-Gal-CA4 and 6-DMNB-Gal-HQ (Fig. 2A). The SIS was introduced to minimize steric hindrance from different drug cargos, ensuring efficient β-gal recognition and hydrolysis after DMNB removal by UV. Additionally, the SIS enables modular expansion to a broader range of drug molecules, including amine-containing drugs, which require a carbamate linkage for conjugation. | ||
| Fig. 2 (A) Synthetic route to the O6-DMNB modified TACG. (B) Docking results of the O6-DMNB modified TACG and the corresponding uncaged galactosides with A. oryzae β-gal. | ||
We established a three-step synthetic protocol (Fig. 2A and see synthesis details in the ESI†): selective O6-DMNB modification, SIS linking and drug conjugation, and galactose deprotection. Starting with D-galactose, the 1,2- and 3,4-hydroxyl groups were protected with isopropylidene, yielding intermediate 9 with an exposed 6-hydroxyl group. O6-Alkylation with DMNB-Br under the catalysis of silver oxide and tetrabutylammonium iodide (TBAI)58 produced compound 10. Cleavage of the isopropylidene groups with trifluoroacetic acid followed by acetylation of the hydroxyl groups yielded intermediate 11. Bromination of 11 with hydrobromic acid followed by glycosylation with para-hydroxy-3-nitrobenzaldehyde produced glycoside 12. The aldehyde group was reduced with sodium borohydride to afford compound 13, which was linked to drugs via the Mitsunobu reaction59 or Williamson etherification, resulting in compound 14. Finally, deprotection of the acetyl groups on galactose with sodium methoxide yielded 6-DMNB-Gal-CA4 and 6-DMNB-Gal-HQ. To facilitate intracellular studies, we also selected a widely used fluorescent molecule with a dicyanomethylene-4H-benzopyran (DCP)60 scaffold as a model cargo, preparing 6-DMNB-Gal-DCP via the same route.
As expected, Maestro docking revealed that Gal-CA4, Gal-HQ, and Gal-DCP bind to the active pocket of A. oryzae β-gal in a similar manner, forming hydrogen bonds through the hydroxyl groups of galactose with Tyr96, Asn140, Ala141, Glu142, Glu298, Tyr342, and Tyr364 surrounding the active pocket (Fig. 2B and S10†). This indicates that these galactosides can be efficiently hydrolyzed by β-gal to release the cargos. The introduction of the DMNB group at the 6-hydroxyl group significantly affected the binding mode of 6-DMNB-Gal-CA4, 6-DMNB-Gal-HQ, and 6-DMNB-Gal-DCP with β-gal, preventing the caged galactose moiety from entering the active pocket, suggesting that they cannot respond to β-gal. Docking studies with human β-gal suggested similar results (Fig. S11 and S12†).
2.3. Intracellular tandem activation behaviour of 6-DMNB-Gal-DCP
DCP has been successfully utilized for the intracellular detection of various species, including β-gal.60 Consistent with docking predictions, we confirmed that Gal-DCP can be effectively hydrolyzed by β-gal to release DCP (Fig. S13†). To verify the UV/β-gal dual-stimulus responsiveness of 6-DMNB-Gal-DCP (Fig. 3A), we conducted fluorescence spectrophotometry (Fig. 3B), HPLC (Fig. 3C), and LC-HRMS (Fig. S14†) studies. These studies clearly demonstrated that upon UV irradiation, 6-DMNB-Gal-DCP converts to Gal-DCP, which is subsequently hydrolyzed by β-gal to release DCP. For intracellular studies, we selected human ovarian cancer cell lines OVCAR3 and SKOV3 as β-gal high-expression cells and human lung cancer cells A549 as β-gal low-expression cells. X-Gal staining46 (Fig. 3D) and western blot (Fig. 3E) confirmed the higher β-gal activity and level in OVCAR3 and SKOV3 cells compared with A549 cells. Flow cytometry was employed to analyze the release of DCP from 6-DMNB-Gal-DCP in the three cell lines (Fig. 3F). Quantitative analysis of the mean fluorescence intensity (MFI) indicated that, compared with the blank groups, the fluorescence intensity in OVCAR3, SKOV3, and A549 cells increased by 4.6, 7.0, and 4.1 times, respectively, after incubation with 6-DMNB-Gal-DCP without UV irradiation. Upon UV irradiation (365 nm, 6 W, and 3 minutes), the fluorescence intensity in β-gal high-expression OVCAR3 and SKOV3 cells increased dramatically by 25.6 and 27.1 times, respectively, while in β-gal low-expression A549 cells, it increased by only 7.6 times. Confocal laser scanning microscope (CLSM) images corroborated the flow cytometry trends (Fig. 3G). These results clearly demonstrate the intracellular tandem activation behavior of 6-DMNB-Gal-DCP.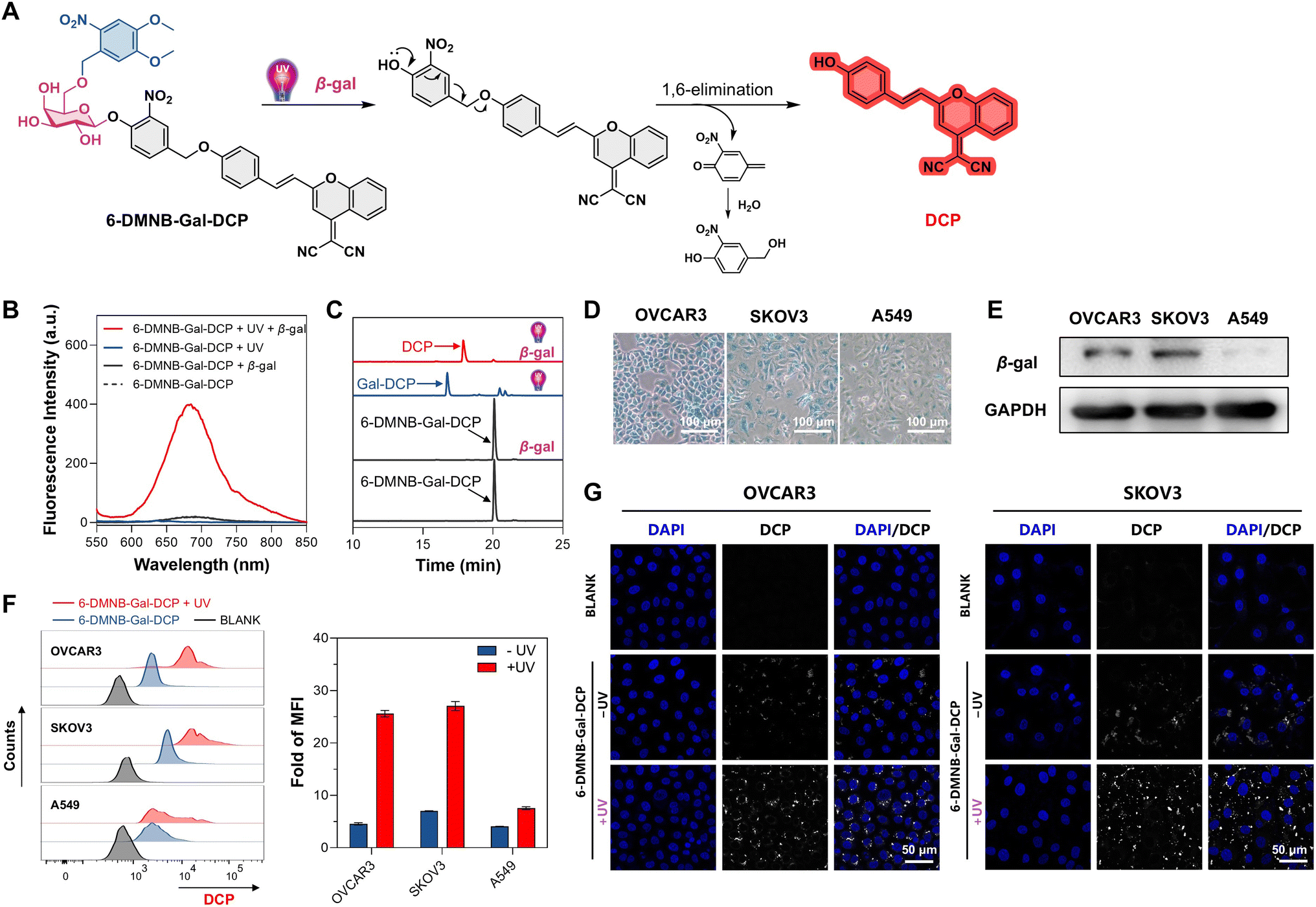 | ||
| Fig. 3 (A) The process of DCP release from 6-DMNB-Gal-DCP in response to UV/β-gal. (B) Fluorescence spectrophotometry and (C) HPLC detection of the dual-stimulus responsiveness of 6-DMNB-Gal-DCP. (D) Detection of β-gal activity in indicated cells by X-Gal staining. (E) Detection of the β-gal level in indicated cells by western blot. (F) Flow cytometry analysis of intracellular DCP release and quantitative analysis of mean fluorescence intensity (MFI) fold changes after UV irradiation, mean ± SD, and n = 4. (G) Confocal laser scanning microscope (CLSM) imaging showing intracellular DCP release from 6-DMNB-Gal-DCP (λex = 535 nm and λem = 680 nm). | ||
2.4. 6-DMNB-Gal-CA4 as a tandem activated microtubule assembly inhibitor
CA4 is a natural product with a cis-stilbene structure that binds to the colchicine binding site of β-tubulin, inhibiting αβ dimerization, arresting the cell cycle at the G2/M phase, and ultimately leading to cell death. However, the non-selective toxicity of CA4 to normal cells limits its clinical application.61,62 The Suzuki group modified the essential phenolic hydroxyl group of CA4 to prepare a galactoside prodrug of CA4, demonstrating its β-gal-dependent microtubule assembly inhibitory activity.25 Using the TACG design, we synthesized 6-DMNB-Gal-CA4 as a photocaged galactoside (Fig. 4A). HPLC and HRMS studies confirmed that 6-DMNB-Gal-CA4 converts to Gal-CA4 upon UV irradiation, which is further hydrolyzed by β-gal to release CA4 (Fig. S15 and S16†). The cytotoxicity of 6-DMNB-Gal-CA4 on OVCAR3 cells was evaluated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. The results showed that UV irradiation alone does not affect the cell viability but significantly increases the cytotoxicity of 6-DMNB-Gal-CA4, particularly in the 50–100 nM range. For example, at a concentration of 70 nM, the cell survival rate remained close to 70% after 48 hours without UV irradiation, whereas it decreased to 35% with UV irradiation during the 48 hour incubation (Fig. 4B). At a concentration of 100 nM, the cytotoxicity of 6-DMNB-Gal-CA4 with UV exposure was comparable to that of Gal-CA4 (Fig. S17†), with a cell survival rate of less than 20%, while the group without UV exposure had a survival rate close to 40% under the same conditions. Live/dead cell staining further confirmed that the number of dead cells in the UV-irradiated 6-DMNB-Gal-CA4 group was significantly higher than in the non-irradiated group (Fig. 4C and S18†). Next, we investigated the microtubule assembly inhibitory activity of 6-DMNB-Gal-CA4 via immunofluorescence.61,62 OVCAR3 cells were treated with 6-DMNB-Gal-CA4 (100 nM) or Gal-CA4 (50 nM) for nine hours, with the UV-irradiated group exposed to light for three minutes during incubation. After fixation, the cells were sequentially treated with an anti-α-tubulin primary antibody and a fluorescently labeled secondary antibody, followed by CLSM observation (Fig. 4D). The results showed that microtubule filaments were still clearly visible in the non-irradiated 6-DMNB-Gal-CA4 group. In contrast, in the UV-irradiated 6-DMNB-Gal-CA4 group, microtubule assembly was significantly inhibited, with tubulin primarily existing in a punctate distribution, similar to the Gal-CA4 group. | ||
| Fig. 4 (A) The release of CA4 from 6-DMNB-Gal-CA4 in response to UV/β-gal and its anticancer mechanism through microtubule assembly inhibition. (B) MTT assay and (C) live/dead staining showing the cytotoxicity of 6-DMNB-Gal-CA4 on OVCAR3 cells with and without UV irradiation, mean ± SD, and n = 3. (D) CLSM observation of the inhibitory activity of 6-DMNB-Gal-CA4 on microtubule assembly in OVCAR3 cells, with green indicating microtubules and blue indicating nuclei. | ||
2.5. 6-DMNB-Gal-HQ as a tandem activated metal chelator
HQ complexes with metal ions like Cu(II) and Fe(III) through its phenolic hydroxyl group and quinoline nitrogen, promoting intracellular ROS generation, activating apoptotic pathways, and inducing cell apoptosis.63 Various HQ prodrugs, including galactoside prodrugs, have been developed by modifying the phenolic hydroxyl group. We synthesized 6-DMNB-Gal-HQ as a photocaged galactoside (Fig. 5A). Upon β-gal activation, Gal-HQ released HQ (Fig. S19†), which chelated Cu(II), showing UV absorption at 258 nm (Fig. S20†). Adding Cu(II) to a 6-DMNB-Gal-HQ solution with β-gal showed no significant UV-vis spectrum change. After three minutes of UV irradiation, an absorption peak at 258 nm confirmed HQ release upon UV/β-gal activation (Fig. S21†). The color change of HQ solution upon Fe(III) chelation visually demonstrated HQ release from 6-DMNB-Gal-HQ in response to UV/β-gal (Fig. S22†). Furthermore, in a 6-DMNB-Gal-HQ solution with β-gal, progressive on-demand HQ release was achieved with each five-second UV irradiation (Fig. S23†). Conversely, Gal-HQ solution continuously released HQ in the presence of β-gal (Fig. S24†). HPLC and HRMS further confirmed that 6-DMNB-Gal-HQ requires sequential UV and β-gal activation to release HQ (Fig. S25 and S26†). | ||
| Fig. 5 (A) The release of HQ from 6-DMNB-Gal-HQ in response to UV/β-gal and its anticancer mechanism through metal ion chelation. (B) MTT assay measuring the toxicity of 6-DMNB-Gal-HQ on OVCAR3 and A549 cells with and without UV irradiation, mean ± SD, and n = 3. (C) The ability of 6-DMNB-Gal-HQ to inhibit OVCAR3 cell colony formation with and without UV irradiation. (D) DCFH-DA probe detecting ROS generation in OVCAR3 cells. (E) GreenNuc™ Caspase-3 probe detecting caspase 3 activation levels in OVCAR3 cells. (F) Annexin V-FITC/PI apoptosis assay evaluating the ability of 6-DMNB-Gal-HQ to induce apoptosis of OVCAR3 cells with and without UV irradiation. | ||
MTT assays showed that the cytotoxicity of Gal-HQ is primarily dependent on β-gal levels, with significantly higher toxicity towards β-gal overexpressing OVCAR3 and SKOV3 cells compared with β-gal low-expressing A549 cells (Fig. S27†). The cytotoxicity of 6-DMNB-Gal-HQ, however, also depends on UV irradiation. For instance, in OVCAR3 cells, nearly all cells died at a drug concentration of 2 μM with UV, while the survival rate remained at 77.4% without UV (Fig. 5B). Similar results were observed in SKOV3 cells (Fig. S28†). In β-gal low-expressing A549 cells, even with UV irradiation, the survival rate remained at 62.4% at a drug concentration of 2 μM (Fig. 5B). Live/dead staining (Fig. S29 and S30†) and colony formation assays (Fig. 5C and S31†) further confirmed that 6-DMNB-Gal-HQ exerts cytotoxicity and inhibits ovarian cancer cell proliferation upon UV/β-gal activation. Using the DCFH-DA probe, we found that ROS levels in ovarian cancer cells significantly increased after UV irradiation in the 6-DMNB-Gal-HQ group, whereas almost no green fluorescence was observed without UV (Fig. 5D and S32†). In contrast, no significant ROS generation was observed in 6-DMNB-Gal-HQ-treated A549 cells, regardless of UV irradiation (Fig. S33†). Using the caspase 3 probe, we observed significant green fluorescence representing activated caspase 3 after UV irradiation in 6-DMNB-Gal-HQ-treated OVCAR3 cells, while almost no green fluorescence was seen without UV (Fig. 5E). Flow cytometry analysis of 6-DMNB-Gal-HQ-induced apoptosis in OVCAR3 cells showed that UV irradiation increased the apoptosis rate from 6.5% to 48.3% (Fig. 5F). Similar trends were observed in SKOV3 cells (Fig. S34†). Additionally, wound healing assays indicated that 6-DMNB-Gal-HQ effectively inhibits ovarian cancer cell migration upon dual activation (Fig. S35 and S36†).
2.6. Expansion to O2-, O3-, and O4-photocaged TACGs
The above study demonstrated the effectiveness of the O6-photocaged TACG. We further investigated whether other hydroxyl groups on galactose, besides the 6-hydroxyl group, could serve as sites for constructing TACGs. To this end, we established a synthetic method for introducing the DMNB group into the 2-, 3-, and 4-hydroxyl groups of galactose, respectively (Fig. 6A and see synthesis details in the ESI†). | ||
| Fig. 6 (A) Synthetic route to O2, O3, and O4-DMNB modified TACGs. (B) 1H–13C HSQC spectra of DMNB-Gal-CA4 isomers. | ||
The synthesis of the O2-DMNB TACG began with IPTG.40 Selective protection of the 6-hydroxyl group with TBDPS and the 3,4-hydroxyl groups with isopropylidene yielded the key intermediate 15 with an exposed 2-hydroxyl group. After screening the alkylation conditions (Table S2†), DMNB was linked to the 2-hydroxyl group using DMNB-Br under the catalysis of TBAI and silver oxide in CH2Cl2, resulting in compound 16. For the subsequent glycosylation reaction (Table S3†), we initially tried a one-step procedure using N-iodosuccinimide (NIS) and trimethylsilyl trifluoromethanesulfonate (TMSOTf), which, however, failed. We then hydrolyzed the isopropyl thioglycoside with N-bromosuccinimide (NBS) and converted it to an N-phenyltrifluoroacetimidate donor, followed by coupling with 3-nitro-4-hydroxybenzaldehyde under TMSOTf. However, the product was predominantly in the α-configuration. Finally, we used iodine monobromide in anhydrous CH2Cl2 to brominate the thioglycoside, yielding the α-configured brominated product that was then etherified with 3-nitro-4-hydroxybenzaldehyde via an SN2 reaction, resulting in a predominantly β-configured glycosylation product. Under conditions similar to those in Fig. 2A, the aldehyde group of the linker was reduced, and the cargo molecule was introduced to obtain compound 17. Finally, in situ generated HCl in MeOH was used to simultaneously remove the TBDPS and isopropylidene protecting groups, yielding the target product.
The synthesis of the O3-DMNB TACG also began with IPTG. Selective protection of the 4,6-hydroxyl groups with benzylidene afforded intermediate 18 with free 2- and 3-hydroxyl groups. Under the catalysis of dibutyltin oxide and cesium fluoride, DMNB was introduced at the 3-hydroxyl group with high regioselectivity,64 resulting in compound 19. Using the above-described method of iodine monobromide-mediated bromination followed by glycosylation, the linker and cargo were introduced, and the final product was obtained after removing the acetyl protection.
We initially attempted to use IPTG again for the synthesis of the O4-DMNB TACG. To this end, the 2,3-hydroxyl groups were protected using 2,3-butanedione, and the 6-hydroxyl group was protected with tert-butyldimethylsilyl (TBDMS), yielding an intermediate with an exposed 4-hydroxyl group.40 This hydroxyl group is a poor nucleophile, and the steric hindrance from the O6-TBDMS further reduced its activity. Despite trying various alkylation conditions, we were unable to introduce the DMNB group (Table S4†). To enhance the reactivity of the 4-hydroxyl group, we switched to using D-galactose as the starting material. By dehydrative cyclization, we protected the 1,6-hydroxyl groups, followed by protecting the 3,4-hydroxyl groups with isopropylidene and the 2-hydroxyl group with a benzoyl group, yielding compound 24. Under acidic conditions, the isopropylidene was removed, exposing the 3,4-hydroxyl groups to obtain intermediate 25.65 At this stage, the 4-hydroxyl group was in an equatorial position with minimal steric hindrance, allowing the selective introduction of the DMNB group to the 4-hydroxyl group under the catalysis of dibutyltin oxide, resulting in compound 26. The ring was opened in a 2% H2SO4 acetic anhydride solution to obtain compound 27. After introducing the linker and cargo molecules, the acetyl protection was removed to yield the target product.
We selected CA4 and DCP as cargo and synthesized positional isomers of DMNB-Gal-CA4 and DMNB-Gal-DCP. Their structures were confirmed by HRMS, 1H NMR, and 13C NMR. Additionally, HSQC spectra clearly showed distinct carbon–hydrogen correlation signals on the galactose moiety of the positional isomers of DMNB-Gal-CA4 and DMNB-Gal-DCP (Fig. 6B and S37†). To further explore the versatility of this route, we also synthesized isomers of DMNB-Gal-MU and ONB-Gal-MU, where galactose is directly linked to MU without a SIS. With these four series of positional isomers in hand, we proceeded to SAR studies.
To determine whether the O2, O3, and O4 positional isomers also do not respond to β-gal, we conducted preliminary validation using positional isomers of ONB-Gal-MU and DMNB-Gal-MU. Docking revealed that the galactose moiety of these isomers could not enter the active pocket of β-gal (Fig. S38 and S39†). HPLC confirmed that the positional isomers were not degraded after 24 hours of incubation with β-gal in solution. Upon UV irradiation, these caged galactosides were converted to Gal-MU, which was further enzymatically hydrolyzed to release MU (Fig. S40 and S41†). Quantitative analysis showed that ONB-Gal-MU and DMNB-Gal-MU isomers release MU at rates of 72–87% and 96–98%, respectively (Fig. S42†). These results suggest that the 2, 3, and 4-hydroxyl groups can also serve as modification sites for TACGs.
2.7. Intracellular comparative analysis of DMNB-Gal-DCP positional isomers
Next, we conducted a comparative analysis of the four positional isomers of DMNB-Gal-DCP. Fluorescence spectroscopy (Fig. S43†) and HPLC detection (Fig. S44†) showed that, similar to ONB-Gal-MU and DMNB-Gal-MU, the four positional isomers of DMNB-Gal-DCP exhibited no significant differences in their UV/β-gal-responsive DCP release behavior in solution, consistent with the docking results (Fig. S45†). However, in ovarian cancer cells, the efficiency of DCP release varied significantly among the isomers. For example, in OVCAR3 cells, after incubating the cells with DMNB-Gal-DCP isomers for 12 hours, followed by three minutes of UV irradiation and an additional six hours of incubation (Fig. 7A), flow cytometry analysis revealed that the MFI value for the 2-DMNB-Gal-DCP and 6-DMNB-Gal-DCP groups reached 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 976 and 17884 a.u., respectively, significantly higher than the 1401 a.u. for the 4-DMNB-Gal-DCP group. The 3-DMNB-Gal-DCP group showed an MFI value of only 994 a.u., even after UV irradiation (Fig. 7B). The ratio of MFI values with and without UV irradiation was used to evaluate the UV-dependent DCP release performance of each isomer. The ratio for the 2-DMNB-Gal-DCP group was 6.5, higher than the 4.8 for the 6-DMNB-Gal-DCP group, suggesting that 2-DMNB-Gal-DCP may achieve higher cargo release controllability through dual activation. CLSM further visually confirmed that significant red fluorescence was observed in the 2-DMNB-Gal-DCP and 6-DMNB-Gal-DCP groups after UV irradiation, while the 4-DMNB-Gal-DCP group showed weaker fluorescence, and the 3-DMNB-Gal-DCP group showed almost no fluorescence (Fig. 7C).
976 and 17884 a.u., respectively, significantly higher than the 1401 a.u. for the 4-DMNB-Gal-DCP group. The 3-DMNB-Gal-DCP group showed an MFI value of only 994 a.u., even after UV irradiation (Fig. 7B). The ratio of MFI values with and without UV irradiation was used to evaluate the UV-dependent DCP release performance of each isomer. The ratio for the 2-DMNB-Gal-DCP group was 6.5, higher than the 4.8 for the 6-DMNB-Gal-DCP group, suggesting that 2-DMNB-Gal-DCP may achieve higher cargo release controllability through dual activation. CLSM further visually confirmed that significant red fluorescence was observed in the 2-DMNB-Gal-DCP and 6-DMNB-Gal-DCP groups after UV irradiation, while the 4-DMNB-Gal-DCP group showed weaker fluorescence, and the 3-DMNB-Gal-DCP group showed almost no fluorescence (Fig. 7C).
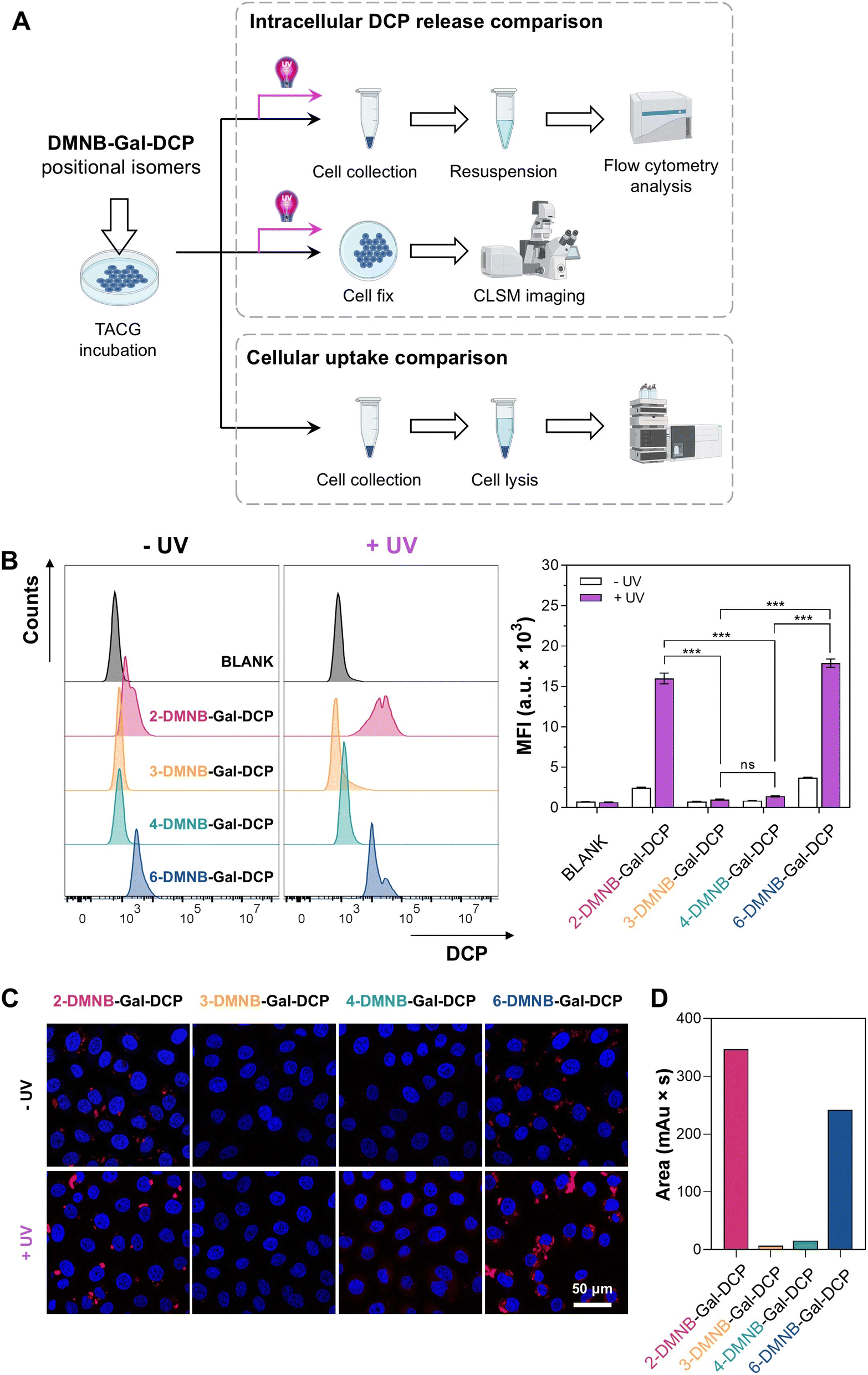 | ||
| Fig. 7 (A) Schematic diagram of the intracellular comparative study of DMNB-Gal-DCP positional isomers. (B) Flow cytometry and (C) CLSM imaging comparing the ability of DMNB-Gal-DCP isomers to release DCP in OVCAR3 cells, mean ± SD, n = 3, ns = not significant (P > 0.05), and ***P < 0.001. (D) HPLC analysis comparing the uptake efficiency of DMNB-Gal-DCP isomers by OVCAR3 cells after a 3 hour incubation. | ||
We hypothesized that the differences in DCP release efficiency among the positional isomers in cells were primarily due to differences in their uptake efficiency by cells.66,67 To verify this hypothesis, we quantitatively measured the uptake efficiency of DMNB-Gal-DCP positional isomers by ovarian cancer cells using HPLC. After incubating OVCAR3 cells with different DMNB-Gal-DCP isomers for three hours, live cells were collected and lysed and the intracellular DMNB-Gal-DCP content was quantified by HPLC. The results showed that after three hours, the intracellular content of 2-DMNB-Gal-DCP was the highest, followed by 6-DMNB-Gal-DCP, while only small amounts of 4-DMNB-Gal-DCP and 3-DMNB-Gal-DCP were detected inside the cells (Fig. 7D and S46†). The trend after 24 hours of incubation (Fig. S47†) was consistent with that after three hours and aligned with the final DCP release efficiency observed in the flow cytometry analysis. The uptake efficiency of DMNB-Gal-DCP positional isomers by SKOV3 cells followed the same pattern (Fig. S48†), as confirmed by CLSM imaging (Fig. S49†). These results suggest that although the 2-, 3-, 4-, and 6-hydroxyl groups of galactose can all be modified to construct TACGs that do not respond to β-gal, the modification position can significantly affect the cellular uptake efficiency of TACGs and ultimately influence the intracellular delivery efficiency of the cargo.
2.8. 2-DMNB-Gal-CA4 outperforms 6-DMNB-Gal-CA4
Since 2-DMNB-Gal-DCP and 6-DMNB-Gal-DCP demonstrated significantly higher efficiency in releasing DCP within cells upon dual activation compared to 3-DMNB-Gal-DCP and 4-DMNB-Gal-DCP, and 2-DMNB-Gal-DCP exhibited greater light dependency than 6-DMNB-Gal-DCP, we systematically compared the anticancer activities of 2-DMNB-Gal-CA4 and 6-DMNB-Gal-CA4 to determine if the SAR of DMNB-Gal-DCP could guide us in identifying a superior anticancer galactoside prodrug.CA4 exerts its anticancer activity by inhibiting microtubule assembly, thereby affecting mitosis, arresting the cell cycle, inducing apoptosis, and inhibiting cell migration. We evaluated the cytotoxicity (Fig. 8A and S50†), cell cycle arrest capability (Fig. 8B and S51†), apoptosis-inducing ability (Fig. 8C and S52†), and cell migration inhibition activity (Fig. 8D and S53†) of 2-DMNB-Gal-CA4 and 6-DMNB-Gal-CA4 in OVCAR3 cells using MTT assays, live/dead staining, flow cytometry, and wound healing assays, respectively. We quantitatively calculated the IC50 values, dead cell ratios, G2/M phase cell ratios, apoptotic cell ratios, and cell migration rates with and without UV irradiation, defining these ratios as the light-dependent selectivity index (LDSI) (Table 1). The results indicated that both 2-DMNB-Gal-CA4 and 6-DMNB-Gal-CA4 effectively killed OVCAR3 cells, arrested the cell cycle at the G2/M phase, induced apoptosis, and inhibited cell migration upon UV irradiation. The LDSI values for various biological activities showed that 2-DMNB-Gal-CA4 had a higher light dependency for its bioactivity compared to 6-DMNB-Gal-CA4, particularly in terms of cytotoxicity and G2/M phase cell cycle arrest. These results suggest that, as expected, 2-DMNB-Gal-CA4 exhibits superior anticancer potential compared with 6-DMNB-Gal-CA4, especially in terms of its light-dependent toxicity, which is a crucial characteristic for anticancer prodrugs. The microtubule assembly inhibitory activity of 2-DMNB-Gal-CA4 in response to UV/β-gal was also validated in OVCAR3 cells via immunofluorescent analysis (Fig. 8E). The anticancer activity and mechanisms of action of 2-DMNB-Gal-CA4 were also demonstrated in SKOV3 cells (Fig. S54–58†).
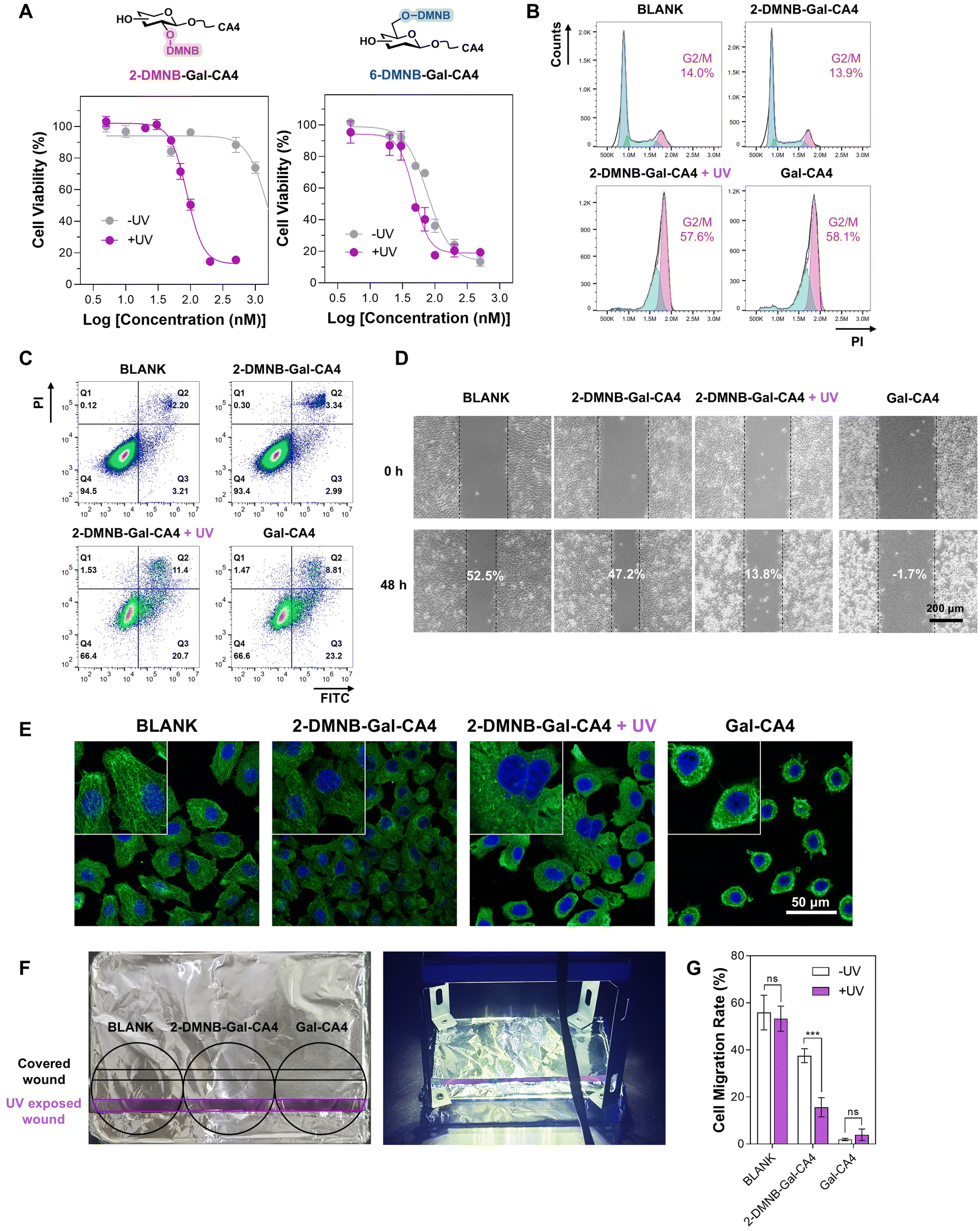 | ||
| Fig. 8 (A) MTT assay comparing the cytotoxicity of 2-DMNB-Gal-CA4 and 6-DMNB-Gal-CA4 on OVCAR3 cells, mean ± SD, and n = 3. (B) Cell cycle arrest effects, (C) apoptosis inducing capability, (D) cell migration inhibitory ability, and (E) microtubule assembly inhibitory activity of 2-DMNB-Gal-CA4 in OVCAR3 cells. (F) Setup of a one-well two-wound experiment. (G) Migration rates of OVCAR3 cells with indicated treatments for 48 h, mean ± SD, n = 3, ns = not significant (P > 0.05), and ***P < 0.001. | ||
| Bioactivity values | 2-DMNB-Gal-CA4 | 6-DMNB-Gal-CA4 | ||||
|---|---|---|---|---|---|---|
| −UV | +UV | LDSI | −UV | +UV | LDSI | |
| a Values without UV/values with UV. b Values with UV/values without UV. | ||||||
| IC50 (nM) | 1674.9 | 137.6 | 12.2a | 108.1 | 59.2 | 1.8a |
| Dead cells (%) | 1.1 | 35.2 | 32.0b | 15.3 | 37.0 | 2.4b |
| G2/M cells (%) | 13.6 | 59.0 | 4.3b | 80.6 | 78.6 | 1.0b |
| Apoptotic cells (%) | 7.6 | 28.7 | 3.8b | 23.5 | 28.7 | 1.2b |
| Migration rate (%) | 48.7 | 12.3 | 4.0a | 21.1 | 11.4 | 1.9a |
To demonstrate that the spatiotemporally controlled release of 2-DMNB-Gal-CA4 by UV/β-gal enables precise and localized bioactivity, we conducted a tailored wound healing assay using OVCAR3 cells. Two wounds were created in a single well, which was then covered with aluminum foil, leaving only one wound exposed to UV light (Fig. 8F).68 The results indicated that UV irradiation had minimal effects on the migration rates of cells in the blank and Gal-CA4 groups. However, cells treated with 2-DMNB-Gal-CA4 exhibited a significant decrease in the migration rate, from 35% without UV to 12% with UV, reflecting a LDSI of 3.1 (Fig. 8G and S59†).
Finally, we evaluated the anticancer efficacy of 2-DMNB-Gal-CA4 using a zebrafish xenograft model, a prevalent method for assessing photocaged prodrugs.69,70 CM-DiI-labeled OVCAR3 cells were implanted into the yolk sacs of zebrafish embryos at two days post-fertilization (dpf). By three dpf, the zebrafish received microinjections of either saline, 2-DMNB-Gal-CA4 (100 nM), or Gal-CA4 (100 nM). Additionally, a subgroup of the 2-DMNB-Gal-CA4 treated zebrafish was exposed to UV irradiation for three minutes, 12 hours post-injection. The proliferation of cancer cells, indicated by fluorescence intensity, was assessed at six dpf (Fig. 9). The fluorescence intensity in the 2-DMNB-Gal-CA4 group without UV exposure only slightly decreased when compared with the vehicle control. Conversely, UV irradiation resulted in a significant reduction in fluorescence intensity in the 2-DMNB-Gal-CA4 group, comparable to that observed with Gal-CA4 treatment. These findings confirm the in vivo anticancer activity of 2-DMNB-Gal-CA4 upon UV/β-gal activation.
 | ||
| Fig. 9 (A) Representative images of zebrafish with different treatments. (B) Quantitative fluorescence density. Mean ± SD, n = 5, ns = not significant (P > 0.05), **P < 0.01, and ***P < 0.001. | ||
3. Discussion and conclusion
Caged prodrugs hold promise for on-demand drug release. However, achieving precise disease targeting with single-stimulus responsive prodrugs remains challenging. This is because the stimuli used to activate the prodrugs, although elevated in certain disease microenvironments, lack sufficient specificity. Many enzymes that are abnormally elevated in cancer cells are also expressed to varying degrees in non-cancer cells, illustrating a relative “more and less” relationship rather than a binary “presence and absence”.71β-Gal-responsive galactoside prodrugs have been explored as anticancer candidates for three decades. In the past decade, research on galactoside prodrugs as senolytics has also emerged. However, β-gal faces similar issues of insufficient distribution specificity. Therefore, there is a need for further improvement of galactoside prodrugs from a medicinal chemistry perspective. Dual-stimulus responsive prodrugs offer greater control over drug release compared with single-stimulus responsive prodrugs. However, there is currently a lack of effective and universal methods to construct dual responsive galactoside prodrugs.
This study focuses on the hydroxyl groups of galactose for prodrug modification by primarily leveraging the high substrate specificity of glycosidases. By installing masking groups on the hydroxyl group of galactose, we upgrade single-stimulus responsive galactoside prodrugs to dual-stimulus responsive galactoside prodrugs, TACGs. After constructing TACG model molecules with different O6 masking groups, studying their cargo release controllability, and conducting virtual docking studies, we selected a TACG modified with the photosensitive group DMNB as our primary research focus. Photosensitive groups are widely used in the study of photocaged prodrugs due to the high spatiotemporal controllability of light, making these prodrugs highly promising for precise drug release applications. DMNB and other photolabile groups have also been used as linkers in solid-phase synthesis of oligosaccharides72 and as hydroxyl group protecting groups73 of carbohydrates, but their application in glycoside prodrugs is unprecedented. We demonstrated the UV/β-gal dual responsiveness of 6-DMNB-Gal-DCP within cells. Furthermore, we selected two drug molecules, CA4 and HQ, and synthesized 6-DMNB-Gal-CA4 and 6-DMNB-Gal-HQ, proving their UV/β-gal-dependent anticancer activity.
Considering that β-galactoside prodrugs have four exposed hydroxyl groups, we further established a synthetic method for O2-, O3-, O4-, and O6-DMNB-modified TACGs based on orthogonal protection strategies in carbohydrate chemistry. We found that these TACG positional isomers exhibited almost identical dual-responsive cargo releasing behavior in solution but showed significant differences at the cellular level. Through intracellular studies of the four positional isomers of DMNB-Gal-DCP, we found that 2-DMNB-Gal-DCP and 6-DMNB-Gal-DCP had significantly higher efficiency in releasing DCP within cells compared to 3-DMNB-Gal-DCP and 4-DMNB-Gal-DCP, likely due to differences in the cellular uptake efficiency of the isomers. Based on this SAR, we systematically compared 2-DMNB-Gal-CA4 and 6-DMNB-Gal-CA4, finding that both could exert bioactivity through dual activation, but 2-DMNB-Gal-CA4 exhibited significantly higher light-dependent selectivity than 6-DMNB-Gal-CA4. Future studies are needed to further explore the general SAR patterns of positional isomers and their impact on dual activation efficiency. While molecular docking has provided insights into their interactions with β-gal, its static nature limits the understanding of dynamic enzymatic recognition and cellular uptake. To address this, molecular dynamics simulations could be employed to better characterize the interactions of different isomers with glycosidases and carbohydrate transporters, offering deeper mechanistic insights for rational prodrug design.
The highly stringent substrate specificity of glycosidase is not limited to β-galactosidase, but also applies to β-glucosidase,74 β-N-acetylglucosaminidase,75 β-glucuronidase,76etc. Therefore, we believe that the TACG strategy will have a broad range of applications in the field of prodrug development.
Ethical statement
All animal procedures were performed in accordance with the Institutional Animal Care and Use Committee (IACUC) Guidelines of Hunter Biotechnology, Inc. (Hangzhou, China) and approved by the Experimental Animal Ethics Committee of Hunter Biotechnology, Inc. (IACUC-2024-9245-01).Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Yunying Tan: conceptualization, methodology, writing – original draft, investigation, and formal analysis. Jie Liu: formal analysis and data curation. Dianya Yong: formal analysis and data curation. Jing Hu: writing – review & editing and funding acquisition. Peter H. Seeberger: writing – review & editing and funding acquisition. Junjie Fu: conceptualization, writing – review & editing, writing – original draft, resources, and funding acquisition. Jian Yin: writing – review & editing, supervision, resources, funding acquisition, and conceptualization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the National Natural Science Foundation of China (22325803, 22277042, and 22077052), the 111 Project (111-2-06), and the China Scholarship Council (CSC). P. H. S. thanks the Max Planck Society for generous financial support.References
- Z. Fralish, A. Chen, S. Khan, P. Zhou and D. Reker, Nat. Rev. Drug Discovery, 2024, 23, 365–380 CrossRef CAS PubMed
.
- R. Bargakshatriya and S. K. Pramanik, ChemBioChem, 2023, 24, e202300155 CrossRef CAS PubMed
- X. Hu, T. Zeng, C. C. Husic and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 15018–15023 CrossRef CAS PubMed
- G. Liu, Y. Zhang, H. Yao, Z. Deng, S. Chen, Y. Wang, W. Peng, G. Sun, M. K. Tse, X. Chen, J. Yue, Y. K. Peng, L. Wang and G. Zhu, Sci. Adv., 2023, 9, eadg5964 CrossRef CAS PubMed
- Q. Fu, S. Zhang, S. Shen, Z. Gu, J. Chen, D. Song, P. Sun, C. Wang, Z. Guo, Y. Xiao, Y. Q. Gao, Z. Guo and Z. Liu, Nat. Biomed. Eng., 2024, 8, 1425–1435 CrossRef CAS PubMed
- Q. Fu, Z. Gu, S. Shen, Y. Bai, X. Wang, M. Xu, P. Sun, J. Chen, D. Li and Z. Liu, Nat. Chem., 2024, 16, 1348–1356 CrossRef CAS PubMed
- K. Tyagi, R. Kumari and V. Venkatesh, Org. Biomol. Chem., 2023, 21, 4455–4464 RSC
- Q. Yao, L. Kou, Y. Tu and L. Zhu, Trends Pharmacol. Sci., 2018, 39, 766–781 CrossRef CAS PubMed
- M. Bai, J. Huang, X. Zheng, Z. Song, M. Tang, W. Mao, L. Yuan, J. Wu, X. Weng and X. Zhou, J. Am. Chem. Soc., 2010, 132, 15321–15327 CrossRef CAS PubMed
- M. Poreba, FEBS J., 2020, 287, 1936–1969 CrossRef CAS PubMed
- X. Li, Z. Liu, A. Zhang, C. Han, A. Shen, L. Jiang, D. A. Boothman, J. Qiao, Y. Wang, X. Huang and Y. X. Fu, Nat. Commun., 2019, 10, 3251 CrossRef PubMed
- Y. Kim, H. Li, J. Choi, J. Boo, H. Jo, J. Y. Hyun and I. Shin, Chem. Soc. Rev., 2023, 52, 7036–7070 RSC
- H. Martin, L. R. Lazaro, T. Gunnlaugsson and E. M. Scanlan, Chem. Soc. Rev., 2022, 51, 9694–9716 RSC
- S. K. Chatterjee, M. Bhattacharya and J. J. Barlow, Cancer Res., 1979, 39, 1943–1951 CAS
- H. B. Bosmann and T. C. Hall, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 1833–1837 CrossRef CAS PubMed
- R. Abraham, N. Aman, R. von Borstel, M. Darsley, B. Kamireddy, J. Kenten, G. Morris and R. Titmas, Cell Biochem. Biophys., 1994, 24–25, 127–133 CrossRef CAS PubMed
- L. F. Tietze, J. M. von Hof, M. Muller, B. Krewer and I. Schuberth, Angew. Chem., Int. Ed., 2010, 49, 7336–7339 CrossRef CAS PubMed
- L. F. Tietze, F. Major and I. Schuberth, Angew. Chem., Int. Ed., 2006, 45, 6574–6577 CrossRef CAS PubMed
- L. F. Tietze, T. Feuerstein, A. Fecher, F. Haunert, O. Panknin, U. Borchers, I. Schuberth and F. Alves, Angew. Chem., Int. Ed., 2002, 41, 759–761 CrossRef CAS PubMed
- B. Renoux, F. Raes, T. Legigan, E. Peraudeau, B. Eddhif, P. Poinot, I. Tranoy-Opalinski, J. Alsarraf, O. Koniev, S. Kolodych, S. Lerondel, A. Le Pape, J. Clarhaut and S. Papot, Chem. Sci., 2017, 8, 3427–3433 RSC
- R. Barat, T. Legigan, I. Tranoy-Opalinski, B. Renoux, E. Peraudeau, J. Clarhaut, P. Poinot, A. E. Fernandes, V. Aucagne, D. A. Leigh and S. Papot, Chem. Sci., 2015, 6, 2608–2613 RSC
- T. Legigan, J. Clarhaut, I. Tranoy-Opalinski, A. Monvoisin, B. Renoux, M. Thomas, A. Le Pape, S. Lerondel and S. Papot, Angew. Chem., Int. Ed., 2012, 51, 11606–11610 CrossRef CAS PubMed
- M. Thomas, J. Clarhaut, P. O. Strale, I. Tranoy-Opalinski, J. Roche and S. Papot, ChemMedChem, 2011, 6, 1006–1010 CAS
- S. Gnaim, A. Scomparin, S. Das, R. Blau, R. Satchi-Fainaro and D. Shabat, Angew. Chem., Int. Ed., 2018, 57, 9033–9037 CAS
- T. Doura, K. Takahashi, Y. Ogra and N. Suzuki, ACS Med. Chem. Lett., 2017, 8, 211–214 CAS
- V. Oliveri, M. Viale, C. Aiello and G. Vecchio, J. Inorg. Biochem., 2015, 142, 101–108 CrossRef CAS PubMed
- M. Maiti, K. Kikuchi, K. K. Athul, A. Kaur and S. Bhuniya, Chem. Commun., 2022, 58, 6413–6416 RSC
- E. Calatrava-Pérez, L. A. Marchetti, G. J. McManus, D. M. Lynch, R. B. P. Elmes, D. C. Williams, T. Gunnlaugsson and E. M. Scanlan, Chem.–Eur. J., 2022, 28, e202103858 CrossRef PubMed
- J. Behmoaras and J. Gil, J. Cell Biol., 2020, 220, e202010162 CrossRef PubMed
- Y. Gao, Y. Hu, Q. Liu, X. Li, X. Li, C. Y. Kim, T. D. James, J. Li, X. Chen and Y. Guo, Angew. Chem., Int. Ed., 2021, 60, 10756–10765 CrossRef CAS PubMed
- J. Hou, Y. Pan, D. Zhu, Y. Fan, G. Feng, Y. Wei, H. Wang, K. Qin, T. Zhao, Q. Yang, Y. Zhu, Y. Che, Y. Liu, J. Cheng, D. Kong, P. G. Wang, J. Shen and Q. Zhao, Nat. Chem. Biol., 2019, 15, 151–160 CrossRef CAS PubMed
- J. A. de Mera-Rodríguez, G. Álvarez-Hernán, Y. Gañán, G. Martín-Partido, J. Rodríguez-León and J. Francisco-Morcillo, Front. Cell Dev. Biol., 2021, 9, 623175 CrossRef PubMed
- J. García-Fleitas, A. García-Fernández, V. Martí-Centelles, F. Sancenón, A. Bernardos and R. Martínez-Máñez, Acc. Chem. Res., 2024, 57, 1238–1253 CrossRef PubMed
- J. Chen, K. Li, J. S. L. Shon and S. C. Zimmerman, J. Am. Chem. Soc., 2020, 142, 4565–4569 CrossRef CAS PubMed
- N. Ueki, S. Lee, N. S. Sampson and M. J. Hayman, Nat. Commun., 2013, 4, 2735 CrossRef PubMed
- Q. Yao, F. Lin, X. Fan, Y. Wang, Y. Liu, Z. Liu, X. Jiang, P. R. Chen and Y. Gao, Nat. Commun., 2018, 9, 5032 CrossRef PubMed
- G. Li, Y. Yang, Y. Zhang, P. Huang, J. Yan, Z. Song, Q. Yuan and J. Huang, CCS Chem., 2022, 4, 1654–1670 CrossRef CAS
- M. Chen, C. Wang, Z. Ding, H. Wang, Y. Wang and Z. Liu, ACS Cent. Sci., 2022, 8, 837–844 CrossRef CAS PubMed
- S. Chen, Y. Tang, Y. Li, M. Huang, X. Ma, L. Wang, Y. Wu, Y. Wang, W. Fan and S. Hou, Biosens. Bioelectron., 2023, 236, 115401 CrossRef CAS PubMed
- S. L. Collins, J. Saha, L. C. Bouchez, E. M. Hammond and S. J. Conway, ACS Chem. Biol., 2018, 13, 3354–3360 CrossRef CAS PubMed
- C. A. Griffiths, R. Sagar, Y. Geng, L. F. Primavesi, M. K. Patel, M. K. Passarelli, I. S. Gilmore, R. T. Steven, J. Bunch, M. J. Paul and B. G. Davis, Nature, 2016, 540, 574–578 CrossRef CAS PubMed
- B. Cheng, Y. Wan, Q. Tang, Y. Du, F. Xu, Z. Huang, W. Qin and X. Chen, Chin. J. Chem., 2022, 40, 806–812 CrossRef CAS
- J. Wang, W. Cao, W. Zhang, B. Dou, X. Ding, M. Wang, J. Ma and X. Li, J. Med. Chem., 2024, 67, 8296–8308 CrossRef CAS PubMed
- X. Li, W. Qiu, J. Li, X. Chen, Y. Hu, Y. Gao, D. Shi, X. Li, H. Lin, Z. Hu, G. Dong, C. Sheng, B. Jiang, C. Xia, C.-Y. Kim, Y. Guo and J. Li, Chem. Sci., 2020, 11, 7292–7301 RSC
- M. Chang, F. Gao, G. Gnawali, H. Xu, Y. Dong, X. Meng, W. Li, Z. Wang, B. Lopez, J. S. Carew, S. T. Nawrocki, J. Lu, Q. Y. Zhang and W. Wang, J. Med. Chem., 2024, 67, 7301–7311 CrossRef CAS PubMed
- Y. Cai, H. Zhou, Y. Zhu, Q. Sun, Y. Ji, A. Xue, Y. Wang, W. Chen, X. Yu, L. Wang, H. Chen, C. Li, T. Luo and H. Deng, Cell Res., 2020, 30, 574–589 CrossRef CAS PubMed
- R. Weinstain, T. Slanina, D. Kand and P. Klan, Chem. Rev., 2020, 120, 13135–13272 CrossRef CAS PubMed
- J. Konc, V. Sabatino, E. Jimenez-Moreno, E. Latocheski, L. R. Perez, J. Day, J. B. Domingos and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2022, 61, e202113519 CrossRef CAS PubMed
- C. Wang, H. Hong, M. Chen, Z. Ding, Y. Rui, J. Qi, Z. C. Li and Z. Liu, Angew. Chem., Int. Ed., 2021, 60, 19750–19758 CrossRef CAS PubMed
- X. Yao, W. Sun, Y. Yuan, J. Hu, J. Fu and J. Yin, Bioorg. Chem., 2024, 150, 107560 CrossRef CAS PubMed
- V. S. Lin, W. Chen, M. Xian and C. J. Chang, Chem. Soc. Rev., 2015, 44, 4596–4618 RSC
- X. Xiang, C. Dong, L. Zhou, J. Liu, Z. M. Rabinowitz, Y. Zhang, H. Guo, F. He, X. Chen, Y. Wang, L. Cui and X. Ma, J. Med. Chem., 2024, 67, 5924–5934 CrossRef CAS PubMed
- J. A. Chen, W. Guo, Z. Wang, N. Sun, H. Pan, J. Tan, Z. Ouyang, W. Fu, Y. Wang, W. Hu and X. Gu, Anal. Chem., 2020, 92, 12613–12621 CrossRef CAS PubMed
- U. Ohto, K. Usui, T. Ochi, K. Yuki, Y. Satow and T. Shimizu, J. Biol. Chem., 2012, 287, 1801–1812 CrossRef CAS PubMed
- M. M. Maksimainen, A. Lampio, M. Mertanen, O. Turunen and J. Rouvinen, Int. J. Biol. Macromol., 2013, 60, 109–115 CrossRef CAS PubMed
- E. Ermini, A. Brai, E. Cini, F. Finetti, G. Giannini, D. Padula, L. Paradisi, F. Poggialini, L. Trabalzini, P. Tolu and M. Taddei, Chem. Sci., 2024, 15, 6168–6177 RSC
- S. Gnaim and D. Shabat, Acc. Chem. Res., 2019, 52, 2806–2817 CrossRef CAS PubMed
- L. Wang, Y. Hashidoko and M. Hashimoto, J. Org. Chem., 2016, 81, 4464–4474 CrossRef CAS PubMed
- H. Takeuchi, Y. Fujimori, Y. Ueda, H. Shibayama, M. Nagaishi, T. Yoshimura, T. Sasamori, N. Tokitoh, T. Furuta and T. Kawabata, Org. Lett., 2020, 22, 4754–4759 CrossRef CAS PubMed
- K. Gu, Y. Xu, H. Li, Z. Guo, S. Zhu, S. Zhu, P. Shi, T. D. James, H. Tian and W. H. Zhu, J. Am. Chem. Soc., 2016, 138, 5334–5340 CrossRef CAS PubMed
- S. Hua, F. Chen and S. Gou, Eur. J. Med. Chem., 2020, 187, 111949 CrossRef CAS PubMed
- X. Yang, B. Cheng, Y. Xiao, M. Xue, T. Liu, H. Cao and J. Chen, Eur. J. Med. Chem., 2021, 213, 113058 CrossRef CAS PubMed
- Y. Zhang, J. Yang, T. Meng, Y. Qin, T. Li, J. Fu and J. Yin, Eur. J. Med. Chem., 2021, 212, 113153 CrossRef CAS PubMed
- X. Zou, C. Qin, C. L. Pereira, G. Tian, J. Hu, P. H. Seeberger and J. Yin, Chem.–Eur. J., 2018, 24, 2868–2872 CrossRef CAS PubMed
- C. Liang, D. W. Lee, M. G. Newton and C. K. Chu, J. Org. Chem., 1995, 60, 1546–1553 CrossRef CAS
- W. Ye, Q. Tang, T. Zhou, C. Zhou, C. Fan, X. Wang, C. Wang, K. Zhang, G. Liao and W. Zhou, Eur. J. Med. Chem., 2024, 264, 115988 CrossRef CAS PubMed
- M. Patra, S. G. Awuah and S. J. Lippard, J. Am. Chem. Soc., 2016, 138, 12541–12551 CrossRef CAS PubMed
- J. Qi, A. S. Amrutha, S. Ishida-Ishihara, H. M. Dokainish, P. K. Hashim, R. Miyazaki, M. Tsuda, S. Tanaka and N. Tamaoki, J. Am. Chem. Soc., 2024, 146, 18002–18010 CrossRef CAS PubMed
- K. Zhang, M. Ji, S. Lin, S. Peng, Z. Zhang, M. Zhang, J. Zhang, Y. Zhang, D. Wu, H. Tian, X. Chen and H. Xu, J. Med. Chem., 2021, 64, 7331–7340 CrossRef CAS PubMed
- D. Liu, B. Yu, X. Guan, B. Song, H. Pan, R. Wang, X. Feng, L. Pan, H. Huang, Z. Wang, H. Wu, Z. Qiu, Z. Li and J. Bian, Chem. Sci., 2023, 14, 4174–4182 RSC
- J. Xiong, J. C. H. Chu, W.-P. Fong, C. T. T. Wong and D. K. P. Ng, J. Am. Chem. Soc., 2022, 144, 10647–10658 CrossRef CAS PubMed
- C. J. Crawford and P. H. Seeberger, Chem. Soc. Rev., 2023, 52, 7773–7801 RSC
- G. Liu, Y. Feng, Q. Zhang and Y. Chai, Org. Lett., 2024, 26, 5746–5751 CrossRef CAS PubMed
- X. Cao, X. Du, H. Jiao, Q. An, R. Chen, P. Fang, J. Wang and B. Yu, Acta Pharm. Sin. B, 2022, 12, 3783–3821 CrossRef CAS PubMed
- F. Liu, H.-M. Chen, Z. Armstrong and S. G. Withers, ACS Cent. Sci., 2022, 8, 656–662 CrossRef CAS PubMed
- M. de Graaf, T. J. Nevalainen, H. W. Scheeren, H. M. Pinedo, H. J. Haisma and E. Boven, Biochem. Pharmacol., 2004, 68, 2273–2281 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00722d |
| This journal is © The Royal Society of Chemistry 2025 |