
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed carbon–carbon bond cleavage of primary alcohols: decarbonylative coupling of acetylenic aldehydes with haloarenes†
Zewei Jina,
Qiang Lia,
Maoshuai Zhua,
Yanqiong Zhanga,
Xufei Yan*b and
Xiangge Zhou *a
*a
aCollege of Chemistry, Sichuan University, 29 Wangjiang Road, Chengdu 610064, P. R. China. E-mail: zhouxiangge@scu.edu.cn
bWest China School of Public Health and West China Fourth Hospital, Sichuan University, Chengdu 610041, P. R. China. E-mail: yanxf92@scu.edu.cn
First published on 11th March 2025
Abstract
In the current work, a palladium-catalyzed C–C bond cleavage reaction of primary alcohols has been developed. This transformation was characterized by a broad substrate scope, superior functional group tolerance, and high efficiency for selective C–C bond cleavage and was then followed by alkynyl-aryl cross coupling. Mechanism studies indicated that the propargyl alcohols underwent β-H elimination to form aldehydes rather than having undergone β-C elimination. The corresponding aldehyde intermediates then proceeded through a decarbonylation and coupling reaction with haloarenes to yield diarylacetylenes.
Introduction
In recent years, transition-metal-catalyzed C–C bond cleavage reactions have garnered widespread attention. There generally exist two main pathways to achieve the C–C bond cleavage mechanistically: (a) oxidative addition via insertion of a low-valence metal into the C–C bonds; (b) β-C elimination driven by the release of small-molecule compounds.1 Transition-metal catalysis of β-C elimination of non-strained non-primary alcohols occurs on the metal alkoxide species, thus resulting in the extrusion of carbonyl compounds and formation of C-M species (Scheme 1a).2 In contrast, β-H elimination is more favoured than β-C elimination for primary alcohols as a consequence of the more thermodynamically preferred M–H bond forming. Hence, the cleavage of such C–C bonds faces more challenges due to the greater tendency of the more accessible β−H elimination occurring. | ||
| Scheme 1 C–C bond cleavage of non-strained alcohols: β-C or β-H elimination strategies. | ||
Considering that primary alcohols can smoothly undergo β-H elimination under transition-metal catalysis, we envisaged the feasibility of combining β-H elimination with decarbonylation, which would be expected to lead to successful C–C bond cleavage of primary alcohols.3 For instance, Jun disclosed a formal dechlorination esterification reaction of aryl chlorides through the cleavage of C–C bonds of primary alcohols under palladium catalysis (Scheme 1b).4 The corresponding aldehyde was initially formed via β-H elimination on the palladium alkoxide species; then the reaction proceeded through a sequence of decarbonylation and esterification with another alcohol molecule to deliver the ester product. Also, propargyl alcohols have exhibited solid reliability in serving as surrogates in allene and alkyne formation as well as ring expansion reactions.5 In this context, Jang reported a copper-catalyzed oxidative decarbonylation reaction of propargyl alcohols for the synthesis of triazole molecules. Including an additional amine was necessary to promote the cleavage of the C(sp3)–C(sp) bond via nucleophilic addition and the subsequent β-C elimination (Scheme 1c).6 Our group has contributed to the field of activation of non-strained C–C bonds.7 We have realized such C(sp3)–C(sp) bond cleavage in propargyl alcohols and propargyl amines, towards the synthesis of 2-arylindoles, in a rhodium-catalyzed/copper-mediated annulation manner (Scheme 1d).8 We have developed a new method for synthesizing diarylalkynes, with our method specifically neither requiring strict control of an inert atmosphere nor needing copper as a co-catalyst—and hence differing from the traditional Sonogashira cross-coupling reaction. In the current work, we attempted to exploit the feasibility of using primary propargyl alcohols as arylacetylene precursors in the coupling with haloarenes, in which a sequence of β-H elimination and decarbonylation would take place,9 and it ultimately afforded the corresponding diarylacetylenes (Scheme 1e). This strategy has successfully enabled the efficient synthesis of diarylalkynes, offering a new route for the synthesis of internal alkynes.
Results and discussion
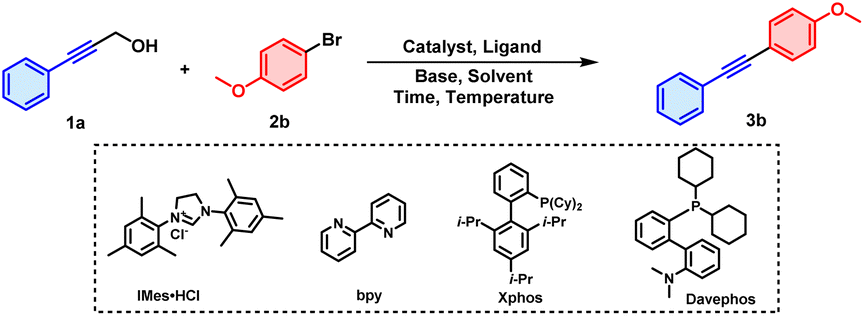
3-Phenyl-2-propyn-1-ol (1a) and 1-bromo-4-methoxy-benzene (2b) were selected as the model substrates for optimizing conditions (Table 1). We initially screened commercially available metal catalysts, and found that RhCl(PPh3)3, Cu(OTf)2 and Pd(OAc)2 were all capable of catalysing the reaction to obtain product 3b, albeit in low yields (entries 1–3). A more electron-rich palladium species, namely Pd(t-Bu3P)2, exhibited a slightly better catalytic efficiency, with the yield increased to 20% with XPhos (2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl) as the ligand (entry 4). Then, different types of N-ligands, P-ligands and NHC-ligands were investigated, and of them, DavePhos (2-dicyclohexylphosphino-2'-(N,N-dimethylamino)biphenyl) gave the best results, with a 25% yield for 3b (entries 5–8). The β-H elimination process is more favourable when a bulky monophosphine ligand like DavePhos is coordinated to the palladium catalyst, since an unoccupied coordination site probably exists at the palladium centre.10 In addition, inclusion of bases have been found to be necessary to facilitate the cleavage of C–C bonds in some cases.11 In our current work, screening different bases revealed KOtBu to be the better than Et3N, K2CO3 and K3PO4, producing 3b in a 30% yield (entries 9–12). Other types of solvents were investigated as well, and using mesitylene instead increased the yield to 43% (entries 13–16). However, it was challenging to purify 3b from residual mesitylene using column chromatography due to the strenuous post-treatment of the high-boiling-point mesitylene and due to the similar polarities of 3b and mesitylene. Therefore, THF was selected as the solvent for further optimizations despite its having given a somewhat lower yield of 40%. In addition, reaction time, temperature, and loading of palladium catalyst were screened systematically in the presence of DavePhos and KOtBu as ligand and base (see the ESI† for details). Ultimately, Pd(t-Bu3P)2 (2.5 mol%), DavePhos (10 mol%), and KOtBu (2.5 equiv.) in THF (2.0 mL) at 120 °C for 14 h under air were selected as the optimal reaction conditions, delivering 3b in 72% yield (entry 17).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Ligand | Base | Solvent | Yieldsb |
| a Unless otherwise noted, the reactions were carried out under air atmosphere with 1a (0.3 mmol), 2b (0.2 mmol), catalyst (10 mol%), ligand (20 mol%), and base (2.0 equiv.) in solvent (2.0 mL) at 130 °C for 12 h.b Isolated yields.c Pd(t-Bu3P)2 (2.5 mol%), DavePhos (10 mol%), KOtBu (2.5 equiv.), 120 °C, 14 h. | |||||
| 1 | RhCl(PPh3)3 | XPhos | Cs2CO3 | CH3CN | <5% |
| 2 | Cu(OTf)2 | XPhos | Cs2CO3 | CH3CN | 12% |
| 3 | Pd(OAc)2 | XPhos | Cs2CO3 | CH3CN | 10% |
| 4 | Pd(t-Bu3P)2 | XPhos | Cs2CO3 | CH3CN | 20% |
| 5 | Pd(t-Bu3P)2 | PCy3 | Cs2CO3 | CH3CN | 10% |
| 6 | Pd(t-Bu3P)2 | bpy | Cs2CO3 | CH3CN | 12% |
| 7 | Pd(t-Bu3P)2 | IMes·HCl | Cs2CO3 | CH3CN | 8% |
| 8 | Pd(t-Bu3P)2 | DavePhos | Cs2CO3 | CH3CN | 25% |
| 9 | Pd(t-Bu3P)2 | DavePhos | K2CO3 | CH3CN | 18% |
| 10 | Pd(t-Bu3P)2 | DavePhos | K3PO4 | CH3CN | 10% |
| 11 | Pd(t-Bu3P)2 | DavePhos | Et3N | CH3CN | <5% |
| 12 | Pd(t-Bu3P)2 | DavePhos | KOtBu | CH3CN | 30% |
| 13 | Pd(t-Bu3P)2 | DavePhos | KOtBu | Mesitylene | 43% |
| 14 | Pd(t-Bu3P)2 | DavePhos | KOtBu | Toluene | 24% |
| 15 | Pd(t-Bu3P)2 | DavePhos | KOtBu | PhCl | 35% |
| 16 | Pd(t-Bu3P)2 | DavePhos | KOtBu | THF | 40% |
| 17c | Pd(t-Bu3P)2 | DavePhos | KOtBu | THF | 72% |
Once the optimal reaction conditions were established, an investigation into the substrate scope for aryl bromides was initiated (Scheme 2). Steric hindrance was found to exert a slight inhibitory effect on the reaction yields—where para-OCH3-substituted phenyl bromide showed a slightly higher reaction efficiency than did those with the ortho and meta substituents, and provided a 72% yield for 3b compared to 63% and 58% yields for 3c and 3d, respectively. A more favoured oxidation addition process with palladium catalysis on the less sterically hindered position might account for these variations.12 Next, a range of electron-donating substituents were investigated, and the corresponding products were obtained in yields ranging from 25 to 80% (3b, 3e, 3f, 3g, 3h, 3i, 3j). Of them, the strongly electron-donating groups NH2 and NH(CH3)2 apparently caused distinct decreases in the yields, as yields of 30% for 3i and 25% for 3j were observed. These two highly nucleophilic amine substrates are prone to oxidation and overconsumption during the reaction, thus apparently resulting in the severe decrease in the corresponding yield.13 Aryl bromides bearing electron-withdrawing substituents, –NO2, –CF3, –CN, and –Cl for instance, were all viable in the reaction, and moderate yields of the corresponding target products (3k, 3l, 3m, 3n) were observed. In addition, multiply substituted substrates also participated in the reaction successfully, as products 3h, 3o, 3p and 3t were obtained in yields ranging from 40% to 70%. A biphenyl substituent was employed as well, and the product 3q was obtained with a yield of 60%. Polycyclic substrates containing naphthyl and phenanthryl also underwent these transformations to reach the corresponding diarylacetylenes, albeit in moderate yields, specifically of 51% and 45% for 3r and 3s. In addition, we investigated the use of aryl iodides as coupling partners in our substrate studies and found that they afforded moderate to good yields. However, compared to bromobenzene, their performance was slightly inferior.
 | ||
| Scheme 2 Scope of aryl bromides.a a1a (0.3 mmol), 2 (0.2 mmol), Pd(t-Bu3P)2 (2.5 mol%), DavePhos (10 mol%) and KOtBu (2.5 equiv.) were stirred in THF (2.0 mL) at 120 °C for 14 h under air. | ||
The scope of aryl-substituted propargyl alcohols was subsequently investigated with para-methoxy-substituted phenyl bromide as the partner reactant (Scheme 3). Reactions with aryl-substituted propargyl alcohols bearing electron-donating groups, including methyl, ethyl, methoxy, ethoxy, tert-butyl, N,N-dimethyl and amino substituents, produced the target diarylacetylenes in moderate to good yields (3u, 3v, 3w, 3x, 3z, 3aa, 3ab). The amino group, despite being relatively reactive,14 was found to be compatible with the reaction, as 3ab was afforded in 38% yield. Substrates containing electron-withdrawing groups, para-CF3 and –CO2Me for instance, delivered the corresponding products 3ac and 3ad—but in relatively low yields, of 20% and 25% yield, respectively, partially due to the competitive homocoupling of arylacetylene detected using gas chromatography-mass spectrometry (GC-MS). As for halogen substituents, fluoro and chloride were tolerated as well, resulting in considerable yields of 3ae and 3af. Consistent with expectations from principles of electronic effects, substrates bearing electron-donating groups performed better than did those bearing electron-withdrawing groups. Steric effects for propargyl alcohols were also examined, and did not notably influence the reaction efficiency, as use of substrates with ortho, meta and para-OCH3 substituents led to the products 3w, 3ak and 3al in 70%, 64% and 62% yields, respectively. For polycyclic, heterocyclic and biphenyl substrates, the yields for the corresponding diarylacetylenes 3ag, 3ah, 3ai and 3aj were acceptable, ranging from 40% to 60%. Finally, we demonstrated the capability of multi-substituted substrates to undergo the reactions and the target 3y, 3am and 3an products were obtained in moderate yields.
 | ||
| Scheme 3 Scope of propargyl alcohols a a 1 (0.3 mmol), 2b (0.2 mmol), Pd(t-Bu3P)2 (2.5 mol%), DavePhos (10 mol%) and KOtBu (2.5 equiv.) were stirred in THF (2.0 mL) at 120 °C for 14 h under air. | ||
To validate the practicality of the reaction, a series of application studies were conducted. First, this transformation could be successfully scaled up to a gram level, and a mass of 0.87 g of the anticipated product 3g was obtained in 75% yield (Scheme 4a). The derivatizations of diarylacetylene were also implemented (Scheme 4b). Diarylacetylene compounds are widely employed in organic synthesis,15 medicinal chemistry,16 and materials science,17 largely due to their distinctive skeletal rigidity and rich π-electron properties.18 The potential of the developed reaction for the synthesis of pharmaceutical molecules was initially demonstrated by the access in 50% yield to 2,3-diphenylquinoxaline 4b,19 a precursor to the antituberculosis drug pyrazinamide. Benzamide and ortho-chloroaniline underwent cyclization and aromatization reactions with diarylacetylene, resulting in the synthesis of quinolone 4d,20 in 65% yield and indole compound 4c,21 in 45% yield. In addition, ruthenium-catalyzed decarboxylative hydroarylation of diarylacetylene with benzoic acid was conducted, and led to a 60% yield of tri-aryl-substituted alkene 4a,22 which serves as a crucial synthetic intermediate in the fields of fine chemicals and materials.
 | ||
| Scheme 4 Synthetic applications. | ||
A series of control experiments were carried out to shed light on the reaction mechanism. Initially, a radical scavenging experiment was conducted in the presence of TEMPO or BHT. The reaction did not give a severely decreased yield of 3b, probably ruling out a radical process (Scheme 5a). Notably, using GC-MS, we could detect 3-phenyl-2-propynal IM-1 with a yield of 30% within the first minute of the reaction (Scheme 5b). The formation of 3-phenyl-2-propynal was consistent with our hypothesis that the reaction involved a β-H elimination process. 3-Phenyl-2-propyn-1-ol was then replaced by 3-phenyl-2-propynal, and an 80% yield of 3b was observed under the standard reaction conditions, thus ultimately validating its role as an intermediate in the reaction (Scheme 5c).
 | ||
| Scheme 5 Mechanistic studies. | ||
Based on the mechanism experiments and relevant literature,3,7 a catalytic cycle was proposed (Scheme 6). According to this proposal, the reaction was initiated by oxidative addition of Pd(0) with haloarenes to generate intermediate IM-2, followed by ligand exchange of IM-2 with substrate 1 to form IM-6. A β-H elimination of IM-6 occurred, and was accompanied by the generation of IM-1 and the Pd–H species.23 Initiated by Pd(0), IM-1 underwent oxidative addition to generate IM-7.24 IM-3 was obtained as a result of decarbonylation of IM-7, and later underwent ligand exchange with the Pd–H species to yield IM-4 and IM-5, respectively.25 Finally, reductive elimination of IM-4 yielded the target diaryl acetylene 3 along with regeneration of the Pd(0) species. Another reductive elimination, of IM-5, also delivered Pd(0), and thus the overall catalytic cycle was realized.
 | ||
| Scheme 6 Proposed catalytic cycle. | ||
Conclusions
In summary, a novel method for the palladium-catalyzed C–C bond cleavage of primary propargyl alcohols has been developed. This method, operating through a β-H elimination mechanism, was shown to achieve the decarbonylative coupling of alkynols with haloarenes, offering a new route for the synthesis of diarylacetylenes. The reaction was found to be characterized by a broad substrate scope, good functional group tolerance, and high efficiency for C–C bond cleavage and re-coupling. Furthermore, the practicality of this reaction was further validated by the synthesis of heterocycles derived from diarylacetylenes, including quinoxalines, isoquinolines and indoles. We plan to utilize this strategy in combination with photocatalysis to achieve the cleavage of C–C bonds of primary alcohols in our further studies.Data availability
The data underlying this study are available in the published article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (Grant No. 22071159 and 22301194) and the Sichuan Science and Technology Program (No. 2023YFS0418) for financial support. Additionally, Ximing Li in Public Health and Preventive Medicine Provincial Experiment Teaching Center at Sichuan University is acknowledged for supporting instrument management.Notes and references
- (a) M. D. R. Lutz and B. Morandi, Chem. Rev., 2021, 121, 300–326 CrossRef CAS PubMed; (b) F. Song, T. Gou, B. Wang and Z. Shi, Chem. Soc. Rev., 2018, 47, 7078–7115 RSC; (c) M. Muruakami and N. Ishida, J. Am. Chem. Soc., 2016, 138, 13759–13769 CrossRef PubMed; (d) Y. Liang, M. Bilal, L. Tang, Y. Guan, Z. Cheng, M. Zhu, J. Wei and N. Jiao, Chem. Rev., 2023, 123, 12313–12370 CrossRef CAS PubMed; (e) F. Song, B. Wang and Z. Shi, Acc. Chem. Res., 2023, 56, 2867–2886 CrossRef CAS PubMed; (f) D. Kim, W. Park and C. Jun, Chem. Rev., 2017, 117, 8977–9015 CrossRef CAS PubMed; (g) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed.
- (a) H. Chow, C. Wan, K. Low and Y. Yeung, J. Org. Chem., 2001, 66, 1910–1913 CrossRef CAS PubMed; (b) T. Nishimura, H. Araki, Y. Maeda and S. Uemura, Org. Lett., 2003, 5, 2997–2999 CrossRef CAS PubMed; (c) A. Funayama, T. Satoh and M. Miura, J. Am. Chem. Soc., 2005, 127, 15354–15355 CrossRef CAS PubMed; (d) K. Choo and M. Lautens, Org. Lett., 2018, 20, 1380–1383 CrossRef CAS PubMed; (e) K. Yasui, N. Chatani and M. Tobisu, Org. Lett., 2018, 20, 2108–2111 CrossRef CAS PubMed; (f) J. R. Bour, J. C. Green, V. J. Winton and J. B. Johnson, J. Org. Chem., 2013, 78, 1665–1669 CrossRef CAS PubMed; (g) L. Mahendar, J. Krishna, A. G. K. Reddy, B. V. Ramulu and G. Satyanarayana, Org. Lett., 2012, 14, 628–631 CrossRef CAS PubMed; (h) L. Mahendar and G. Satyanarayana, J. Org. Chem., 2014, 79, 2059–2074 CrossRef CAS PubMed; (i) T. Li, Z. Wang, M. Zhang, H. Zhang and T. Wen, Chem. Commun., 2015, 51, 6777–6780 RSC.
- (a) L. Wen, J. Ding, L. Duan, S. Wang, Q. An, H. Wang and Z. Zuo, Science, 2023, 382, 458–464 CrossRef CAS PubMed; (b) W. Wang, H. Wang, R. Dai, Y. Wang, Z. Li, X. Yang, B. Lu, N. Jiao and S. Song, ACS Catal., 2023, 13, 9033–9040 CrossRef CAS; (c) M. Valencia, A. D. Merinero, C. L. Aparicio, M. G. Gallego, M. A. Sieera, B. Eguillor, M. A. Esteruelas, M. Olivan and E. onate, Organometallics, 2020, 39, 312–323 CrossRef CAS; (d) F. Gao, J. D. Webb, H. Sorek, D. E. Wemmer and J. F. Hartwig, ACS Catal., 2016, 6, 7385–7392 CrossRef CAS; (e) E. P. K. Olsen, T. Singh, P. Harris, P. G. Andersson and R. Madsen, J. Am. Chem. Soc., 2015, 137, 834–842 CrossRef CAS PubMed; (f) Z. Zhang, D. S. Zijlstra, C. W. Lahive and P. J. Deuss, Green Chem., 2020, 22, 3791–3801 RSC; (g) A. Mazziotta and R. Madsen, Eur. J. Org Chem., 2017, 36, 5417–5420 CrossRef; (h) E. P. K. Olsen and R. Madsen, Chem.–Eur. J., 2012, 18, 16023–16029 CrossRef CAS PubMed; (i) P. P. Bui, S. T. Oyama, A. Takagaki, B. P. Carrow and K. Nozaki, ACS Catal., 2016, 6, 4549–4558 CrossRef CAS; (j) H. W. Cheung, T. Y. Lee, H. Y. Lui, C. H. Yeung and C. P. Lau, Adv. Synth. Catal., 2008, 350, 2975–2983 CrossRef CAS.
- H. Park, D. Kim and C. Jun, ACS Catal., 2015, 5, 397–401 CrossRef CAS.
- H. Qian, D. Huang, Y. Bi and G. Yan, Adv. Synth. Catal., 2019, 361, 3240–3280 CrossRef CAS.
- Y. Kang, Y. Cho, K. Ko and H. Jang, Catal. Sci. Technol., 2015, 5, 3931–3934 RSC.
- (a) Y. Long, W. Zhou, Q. Li and X. Zhou, Org. Biomol. Chem., 2021, 19, 9809–9828 RSC; (b) Q. Tao, Y. Zheng, Q. Li, Y. Long, J. Wang, Z. Jin and X. Zhou, Org. Lett., 2024, 26, 11224–11229 CrossRef CAS PubMed; (c) Q. Tao, H. Zhang, R. Ye, Y. Zhang, Y. Long and X. Zhou, J. Org. Chem., 2024, 89, 13208–13214 CrossRef CAS PubMed; (d) Q. Li, Y. Xu, Y. Long, S. Li, B. Xu, Y. Xia and X. Zhou, ACS Catal., 2023, 13, 7795–7801 CrossRef CAS; (e) Y. Long, Y. Zheng, Y. Xia, L. Qu, Y. Yang, H. Xiang and X. Zhou, ACS Catal., 2022, 12, 4688–4695 CrossRef CAS; (f) Q. Li, M. Zhu, X. Yan, Y. Xia and X. Zhou, J. Organomet. Chem., 2021, 948, 121930 CrossRef CAS; (g) Y. Long, Z. Su, Y. Zheng, S. He, J. Zhong, H. Xiang and X. Zhou, ACS Catal., 2020, 10, 3398–3403 CrossRef CAS.
- (a) X. Yan, R. Ye, H. Sun, J. Zhong, H. Xiang and X. Zhou, Org. Lett., 2019, 21, 7455–7459 CrossRef CAS PubMed; (b) S. He, X. Yan, Y. Lei, H. Xiang and X. Zhou, Chem. Commun., 2020, 56, 2284–2287 RSC.
- (a) J. Zhou, R. Liu, C. Wang and Y. Zhu, J. Org. Chem., 2020, 85, 14149–14157 CrossRef CAS PubMed; (b) H. Lu, T. Yu, P. Xu and H. Wei, Chem. Rev., 2021, 121, 365–411 CrossRef CAS PubMed.
- M. K. Bogdos, O. Stepanovic, A. Bismuto, M. G. Luraschi and B. Morandi, Nat. Synth., 2022, 1, 787–793 CrossRef CAS.
- (a) A. Lennox and G. Lloyd-jones, Angew. Chem., Int. Ed., 2013, 52, 7362 CrossRef CAS PubMed; (b) P. A. Cox, M. Reid, A. G. Leach, A. D. Campbell, E. J. King and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2017, 139, 13156 CrossRef CAS PubMed; (c) C. A. Malapit, J. R. Bour, C. E. Brigham and M. S. Sanford, Nature, 2018, 563, 100–104 CrossRef CAS PubMed; (d) Z. Guo, H. Xu, X. Wang, Z. Wang, B. Ma and H. Dai, Chem. Commun., 2021, 57, 9716 RSC; (e) Z. Liu, C. Liang, Z. Luo, Y. Wu, C. Hong, Q. Li and T. Liu, ACS Catal., 2022, 12, 7030–7036 CrossRef CAS.
- J. Yao, Y. Xiao, H. Li, X. Yang, J. Du, Y. Yin, L. Feng, W. Duan and L. Yu, Org. Lett., 2024, 26, 7307–7312 CrossRef CAS PubMed.
- Y. Jin, Y. Jing, C. Li, M. Li, W. Wu, Z. Ke and H. Jiang, Nat. Chem., 2022, 14, 1118–1125 CrossRef CAS PubMed.
- A. M. Dreis and C. J. Douglas, J. Am. Chem. Soc., 2009, 131, 412–413 CrossRef CAS PubMed.
- (a) S. Zhang, H. Ma, H. E. Ho, Y. Yamamoto, M. Bao and T. Jin, Org. Biomol. Chem., 2018, 16, 5236 RSC; (b) J. Cai, B. Wu, G. Rong, C. Zhang, L. Qiu and X. Xu, Org. Lett., 2018, 20, 2733 CrossRef CAS PubMed; (c) J. Matsuoka, Y. Matsuda, Y. Kawada, S. Oishi and H. Ohno, Angew. Chem., Int. Ed., 2017, 56, 7444 CrossRef CAS PubMed; (d) S. J. Hein, D. Lehnherr, H. Arslan, F. J. UribeRomo and W. R. Dichtel, Acc. Chem. Res., 2017, 50, 2776 CrossRef CAS PubMed; (e) I. T. Trotus, T. Zimmermann and F. Schüth, Chem. Rev., 2014, 114, 1761 CrossRef CAS PubMed; (f) B. Godoi, R. F. Schumacher and G. Zeni, Chem. Rev., 2011, 111, 2937 CrossRef CAS PubMed.
- (a) V. M. Sviripa, W. Zhang, L. M. Kril, A. X. Liu, Y. Yuan, C. G. Zhan, C. Liu and D. S. Watt, Bioorg. Med. Chem. Lett., 2014, 24, 3638 CrossRef CAS PubMed; (b) C. P. Kordik, C. Luo, M. Gutherman, A. H. Vaidya, D. I. Rosenthal, J. J. Crooke, S. L. Mckenney, C. R. Plata-Salaman and A. B. Reitz, Bioorg. Med. Chem. Lett., 2006, 16, 3065 CrossRef CAS PubMed; (c) Q. H. Chen, P. N. P. Rao and E. E. Knaus, Bioorg. Med. Chem., 2005, 13, 6425 CrossRef CAS PubMed.
- (a) T. Sakaguchi, Y. Hayakawa, R. Ishima and T. Hashimoto, Synth. Met., 2012, 162, 64 CrossRef CAS; (b) I. Bylińska, M. Wierzbicka, C. Czaplewski and W. Wiczk, Photochem. Photobiol. Sci., 2016, 15, 45 CrossRef PubMed; (c) F. Li, Z. An, X. Chen and P. Chen, Liq. Cryst., 2015, 42, 1654 CAS.
- F. Diederich, P. J. Stang and R. R. Tykwinski, Acetylene Chemistry: Chemistry, Biology And Material Science, WileyVCH, Weinheim, 2005 Search PubMed.
- C. Chen, W. Hu, M. Liu, P. Yan, J. Wang and M. Chung, Tetrahedron, 2013, 69, 9735–9741 CrossRef CAS.
- Z. Shu, Y. Guo, W. Li and B. Wang, Catal. Today, 2017, 297, 292–297 CrossRef CAS.
- M. Shen, G. Li, B. Z. Lu, A. Hossain, F. Roschangar, V. Farina and C. H. Senanayake, Org. Lett., 2004, 6, 4129–4132 CrossRef CAS PubMed.
- L. Huang, A. Biafora, G. Zhang, V. Bragoni and L. J. Gooβen, Angew. Chem., Int. Ed., 2016, 55, 6933–6937 CrossRef CAS PubMed.
- D. Balcells, A. Nova, E. Clot, D. Gnanamgari, R. H. Crabtree and O. Eisenstein, Organometallics, 2008, 27, 2529–2535 CrossRef CAS.
- A. Modak, S. Rana, A. K. Phukan and D. Maiti, Eur. J. Org Chem., 2017, 28, 4168–4174 CrossRef.
- A. Dermenci, R. E. Whittaker, Y. Gao, F. A. Cruz, Z. Yu and G. Dong, Chem. Sci., 2015, 6, 3201–3210 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00357a |
| This journal is © The Royal Society of Chemistry 2025 |