
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogermylation of alkynes via metal–ligand cooperative catalysis†
Marceline
Humbert
a,
Arnaud
Clerc
a,
Karinne
Miqueu
 b,
Julien
Monot
a,
Blanca
Martin-Vaca
*a and
Didier
Bourissou
*a
b,
Julien
Monot
a,
Blanca
Martin-Vaca
*a and
Didier
Bourissou
*a
aCNRS/Université Paul Sabatier, Laboratoire Hétérochimie Fondamentale et Appliquée (LFA, UMR 5069), 118 Route de Narbonne, 31062 Toulouse Cedex 09, France. E-mail: blanca-maria.martin-vaca@univ-tlse3.fr; didier.bourissou@univ-tlse3.fr
bCNRS/Université de Pau et des Pays de l’Adour, Institut des Sciences Analytiques et Physico-Chimie pour l’Environnement et les Matériaux (IPREM, UMR 5254) Hélioparc, 2 Avenue du Président Angot, 64053 Pau Cedex 09, France
First published on 24th January 2025
Abstract
E–H bond activation (E = B, Ge, Sn) with a (PNS)Pd complex has been investigated theoretically and experimentally. Et3GeH readily adds across the Pd/S bond. Subsequent transfer to C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds enables catalytic hydrogermylation. The reaction is most regio and stereoselective with terminal alkynes. Downstream derivatization of silyl-functionalized vinyl germanes is exemplified.
C bonds enables catalytic hydrogermylation. The reaction is most regio and stereoselective with terminal alkynes. Downstream derivatization of silyl-functionalized vinyl germanes is exemplified.
Organo germanium derivatives are much less developed and used than their silicon, boron and tin congeners in organic synthesis. However, they attract growing interest as complementary substrates for coupling reactions and functionalization processes.1 Most noteworthy is their different and eventually orthogonal reactivity2 which can enable the rapid construction of complex molecules without the need of protection/deprotection steps.
As a matter of fact, it is needed to develop simple, efficient and selective routes to organo germanium derivatives to further explore and fully exploit their synthetic potential. Significant advances have been reported in the field over the last decades,3 but hydrogermylation reactions remain sparse and underdeveloped while they certainly represent the most attractive route to vinyl germanes. Transition metal and Lewis acid catalysed, as well as radical approaches have been documented (Chart S1, ESI†),4 but the transformation is still far from hydrosilylation in terms of scope (with terminal alkynes as substrates essentially) and regio as well as stereo-selectivity (α/β, syn/anti additions).5–8
Our group has recently reported a PNS pincer Pd complex I2 (Scheme 1) that readily activates Si–H bonds thanks to Pd/S cooperativity and that efficiently catalyzes the hydrosilylation of alkynes.9 Taking into account that metal–ligand cooperative catalysis has not been applied yet to hydrogermylation, we became interested in testing Pd/S cooperation for the activation of other E–H bonds, in particular Ge–H bonds. As reported hereafter, the (PNS)Pd complex I2 was found indeed to activate Et3Ge–H stoichiometrically and to catalyze alkyne hydrogermylation. Note that the Ru/S cooperative system developed by Ohki, Tatsumi and Oestreich proved to be highly active towards Si, Sn as well as B–H bonds, and compared to our Pd/S system, it tends to favour dehydrogenative coupling over hydroelementation.10 The activation of Ge–H bonds by this system has only been considered computationally.11
 | ||
| Scheme 1 Stoichiometric reaction of triethylgermane with complex I2. | ||
The feasibility of E–H bond activation via Pd/S cooperation was first probed computationally using Et3EH (E: Si, Ge, Sn) and HBPin as model substrates (Fig. 1).4 The first step is the splitting of the Pd/S dimer to form a σ-complex.12 It is significantly downhill in energy with Ge, about thermoneutral with Sn and endergonic with Si & B (ΔG −6.7, −0.7 and 9.6/17.8 kcal mol−1, respectively). With all the group 14 elements, the σ-complex adopts end-on η1-type coordination. The shortest E⋯Pd contact is observed with Ge (2.473 Å), and it is associated with the most elongated E–H bond (2.136 Å). The noticeable degree of Ge–H bond activation is apparent from the Wiberg bond index (0.246) and the significant contribution of Pd → σ*(Ge–H) backdonation (ΔEorb −24.3 vs. −59.5 kcal mol−1 for the σ(Ge–H) → Pd donation, according to EDA-NOCV calculations).4 Comparatively, the HBpin σ-complex tends more to a side-on η2-type structure, with a short B⋯Pd contact (2.258 Å).
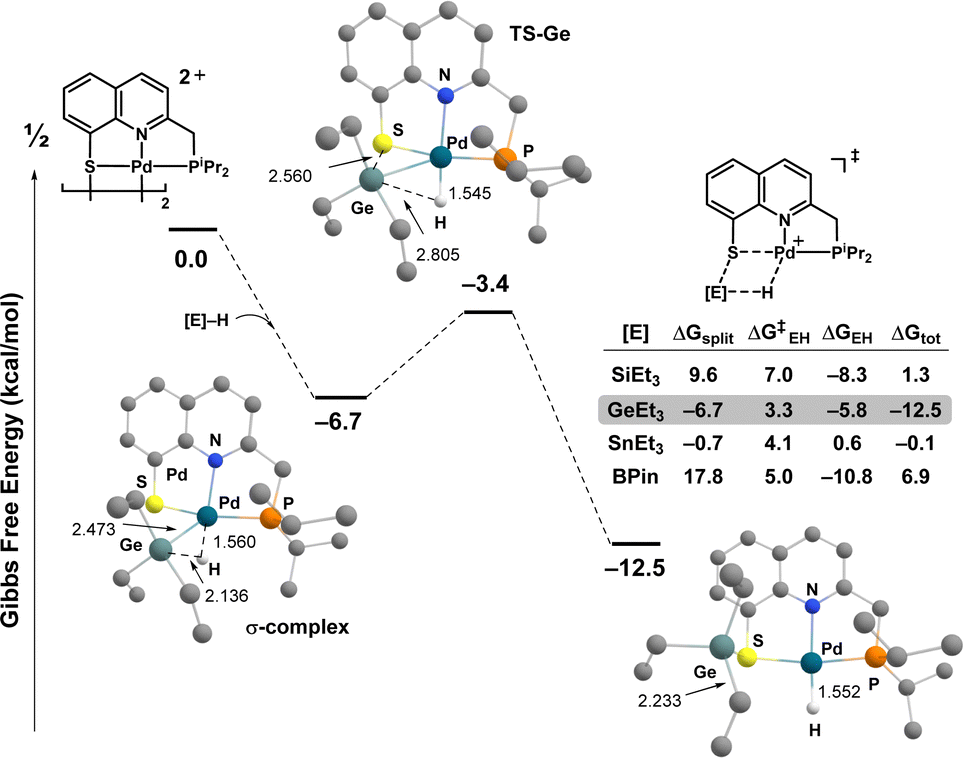 | ||
| Fig. 1 Reaction profiles (ΔG in kcal mol−1) computed for the Pd/S cooperative activation of E–H bond (E: Si, Ge, Sn, B). Only the structures associated with the activation of Et3GeH are shown. For clarity, the hydrogen atoms are omitted, except that at Ge/Pd. Optimizations performed at the SMD(DCM)-B3PW91-D3(BJ)/SDD+f(Pd), SDD+d(Sn), 6-31G** (other atoms) level of theory. Main distances in Å. | ||
Heterolytic cleavage of the E–H bond then proceeds in a concerted manner thanks to Pd/S cooperativity.4 The activation barriers are low and follow the same trend as the Ru/S system:11 Ge < Sn < B < Si (ΔG≠ 3.3, 4.1, 5.0 and 7.0 kcal mol−1, respectively). With the group 14 elements, the cleavage of the E–H bond is asynchronous. At the transition state, the H atom is nearly transferred to Pd (Pd⋯H ∼ 1.55 Å) whereas the EEt3 group is on the way to S with E – S distances of 2.6 to 2.9 Å. IBO analyses confirm this picture, showing a covalent Pd–H bond and a S lone pair starting to mix with E. The H–Bpin activation process is more synchronous. The formation of the Pd–H and S–B bonds are both highly advanced at the TS (Pd⋯H 1.57 Å, B⋯S 2.19 Å). From the σ-complex, the E–H activation step is exergonic for all computed [E]–H bonds except Et3Sn–H. Taking into account the splitting of the Pd/S dimer, the process is thermodynamically favored for Ge, about thermoneutral for Sn & Si, and uphill in energy for B (ΔG −12.5, −0.1/1.3 and 6.9, respectively).
The activation of pinacolborane, tributyltin hydride and triethylgermane by the [(PNS)Pd]2 complex I2 was then investigated experimentally under the same conditions as for hydrosilanes.9 No reaction was observed with HBpin after 48 h at 25 °C, even with 5 equiv.4 In marked contrast, nBu3SnH led to an instantaneous gas evolution (H2), even at low temperature (−78 °C), with concomitant appearance of a 119Sn NMR signal at δ –83 ppm diagnostic of the dehydrogenative coupling product Bu3SnSnBu3.4,13 The best result was actually obtained with Et3GeH (Scheme 1). Here, NMR monitoring revealed rapid and quantitative reaction, with a marginal shift of the 31P{1H} signal (from δ 75.8 to 74.8 ppm), as previously observed upon hydrosilane activation.9 The structure of the obtained complex II was unambiguously established by multi-nuclear NMR spectroscopy. The hydride at Pd resonates as a doublet at δ −12.6 ppm (JPH 4.9 Hz) in the 1H NMR spectrum and germylation of the thiolate moiety is apparent from a 1H–1H NOESY experiment.4
Next, catalytic hydroelementation tests were carried out with diphenylacetylene as benchmark substrate (using 2.5 mol% of I2). No reaction occurred with HBpin at 25 °C, but rising the temperature to 70 °C resulted in 82% alkyne conversion after 4 h and 78% isolated yield of the expected addition products with a syn/anti addition ratio of 28/72.4,14 With nBu3SnH, dehydrogenative Sn–Sn coupling prevailed even under catalytic conditions and in the presence of the alkyne. Only traces of hydrostannylation product were detected in the 119Sn NMR spectrum.4 Once again, the best result was obtained with Et3GeH, for which near full alkyne conversion was achieved in 5 h at 25 °C to yield a 45/55 mixture of the syn/anti addition products (96% isolated yield) (Chart 1).4,15 A good conversion (82%) and similar syn/anti selectivity (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60) was obtained by decreasing the catalyst loading to 0.5 mol% and extending the reaction time (24 h).16 These first catalytic tests suggest that catalytic hydrogermylation via Pd/S cooperativity is indeed feasible. Complex I2 seems to be slightly less reactive in hydrogermylation than hydrosilylation, but substantially more than in hydroboration. We thus focused on hydrogermylation for the catalytic study to assess the synthetic potential of this system to access vinyl germanes.
:60) was obtained by decreasing the catalyst loading to 0.5 mol% and extending the reaction time (24 h).16 These first catalytic tests suggest that catalytic hydrogermylation via Pd/S cooperativity is indeed feasible. Complex I2 seems to be slightly less reactive in hydrogermylation than hydrosilylation, but substantially more than in hydroboration. We thus focused on hydrogermylation for the catalytic study to assess the synthetic potential of this system to access vinyl germanes.
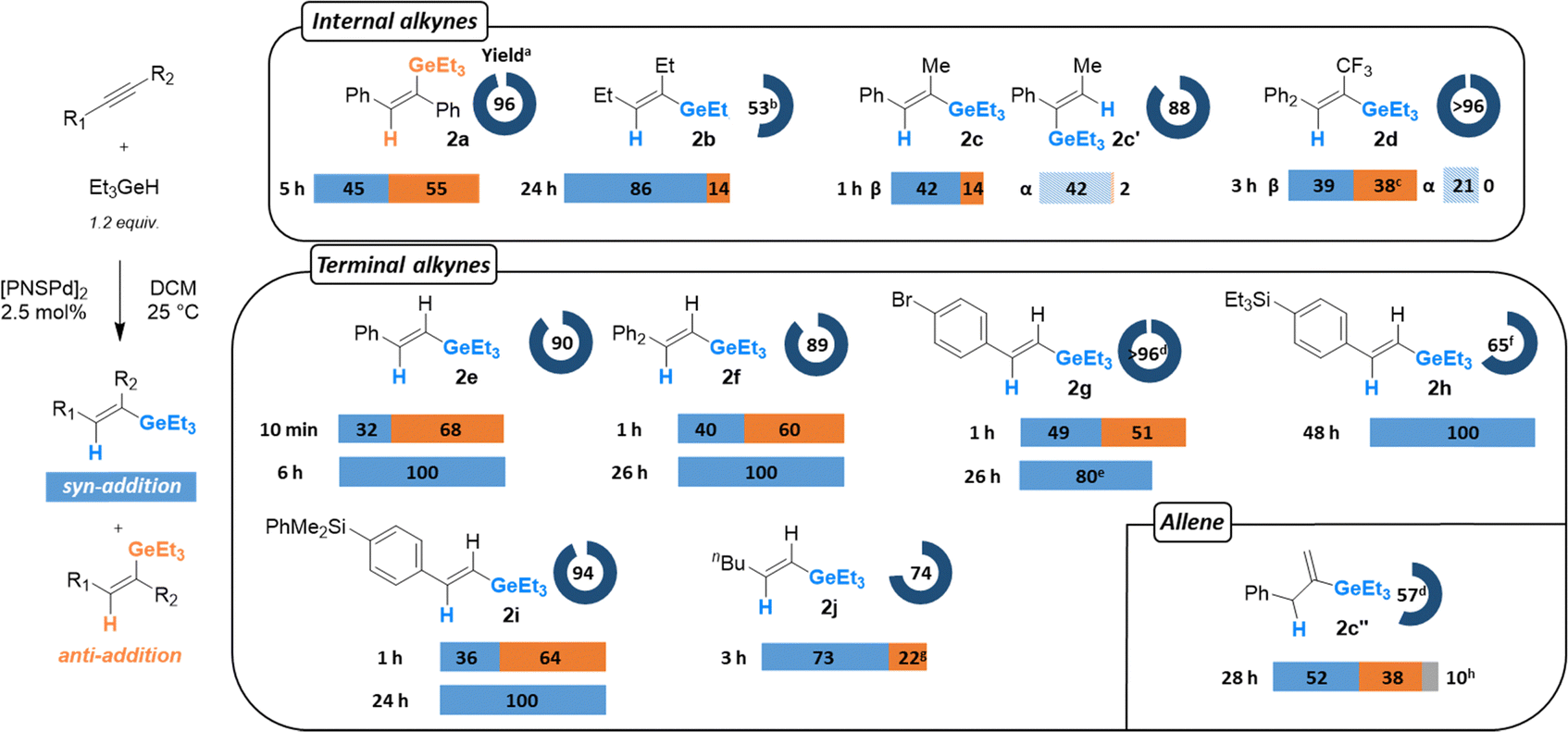 | ||
Chart 1 Hydrogermylation of alkynes catalyzed by the Pd complex I2 at 25 °C. The major vinyl germane product obtained is shown. aIsolated as mixture of isomers. bWith 3 equiv. of Et3GeH. c2% of not identified secondary product. dDetermined by 1H NMR spectroscopy with 1,2,4,5-tetramethylbenzene as internal standard or GS/MS. e20% of secondary product from dehalogenation reaction. fReaction performed at 1.8 mmol scale (400 mg of alkyne). g5% of secondary product from chain-walking. hRatio 2c′′/2c/Z-PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH(CH3)GeEt3. CH(CH3)GeEt3. | ||
First, we confirmed the relevance of the Ge–H activation product to the catalytic transformation. Following the formation of complex II by reaction of I2 with 5 equiv. of Et3GeH, the addition of 5 equiv. of diphenylacetylene led to rapid and quantitative consumption of the substrates and formation of 2a with again 45/55 syn/anti selectivity. A blank reaction performed with diphenylacetylene in the absence of the (PNS)Pd complex showed no reaction, even after 24 h or heating at 70 °C for 6 h. Hydrogermylation of hex-3-yne 1b, a more challenging substrate, was then surveyed (Chart 1). Here, the use of 3 equiv. of Et3GeH was beneficial to maintain a reasonable reaction time (24 h) and an acceptable isolated yield (53%) with 85/15 selectivity in favour of the syn addition product.17 Then, the internal dissymmetric alkynes PhCCMe 1c and Ph–PhCCCF31d were tested. In both cases, only 1.2 equiv. of Et3GeH was sufficient to achieve good yields (>88%) in maximum 3 h. While β addition (the germyl group being introduced in the β position to the aromatic substituent) was largely favoured for 1d (>75/25 vs. 56/44 for 1c), higher syn/anti selectivity was observed for 1c than for 1d (84/16 and 60/40, respectively). The (PNS)Pd complex I2 also proved efficient in promoting the hydrogermylation of terminal alkynes at room temperature, here with complete regioselectivity for β addition (Chart 1). Of note, no C–Ge dehydrogenative coupling was detected. Typically, full conversion of phenylacetylene 1e was achieved within only 10 min. The ensuing vinyl germane 2e then slowly isomerizes so that syn selectivity increases from 40/60 to 100/0 after 6 h at 25 °C. Attempts to selectively obtain the anti addition product by decreasing the catalyst loading to 0.5 mol% or lowering the temperature to −78 °C were unsuccessful. The biphenylene substituted alkyne 1f behaves similarly, except a longer isomerization time (26 h). With 4-bromophenylacetylene 1g, full conversion was reached in 10 min at 25 °C with a high selectivity for β addition. However, a dehalogenation side-product was detected (ca. 20%). Of particular interest is the ability of I2 to catalyze the hydrogermylation of aryl-silylated substrates such as 1h and 1i without any trace of desilylation. Again, a mixture of β-syn/β-anti addition products is initially formed but prolonged reaction times resulted in complete isomerization into the β-syn addition product. Good to excellent yields (65–94%) were obtained for these functionalized vinyl compounds 2h and 2i. Selective β-addition was also achieved with hex-1-yne 1j (73/22 syn/anti addition ratio), although full conversion required longer time (3 h vs. 10 min for 1e). Note that isomerization into the β-syn isomer does not proceed so cleanly in this case, a secondary product resulting from chain-walking of the CC bond forms concomitantly.18 Finally, an allene, PhCCCH2, was also tested. It could be hydrogermylated at 25 °C with good conversion (80% in 28 h), yielding the terminal vinyl germane 2c′′ (resulting from β-addition) as major product.19
Although the hydrogermylation of internal alkynes lacks stereoselectivity, the total β-regioselectivity observed with terminal alkynes and the syn to anti isomerization occurring from aryl alkynes make the transformation synthetically valuable. The hydrogermylation route is particularly attractive and efficient to access silylated products such as 2h and 2i. It nicely complements the known synthetic route involving DiBAl-H reduction of alkynyl germanes (see Scheme 2).2e This prompted us to investigate further skeleton diversification of 2h and 2i.2a,2d Based on a recent report of Schoenebeck et al.,2e the olefination of vinyl germane 2h with n-butyl acrylate was successfully carried out under oxidative Pd-catalyzed conditions, giving the desired product 3 with exclusive E-selectivity. Furthermore, using in situ generated Pd nanoparticles,2d2h and 2i could be efficiently converted to the functionalized stilbenes 4a–d using aryl iodides. The cross-coupling reaction works with electron-enriched and deprived substrates,2e it is fully E-selective and occurs only at the Ge site, leaving the silyl moiety intact for downstream derivatization.
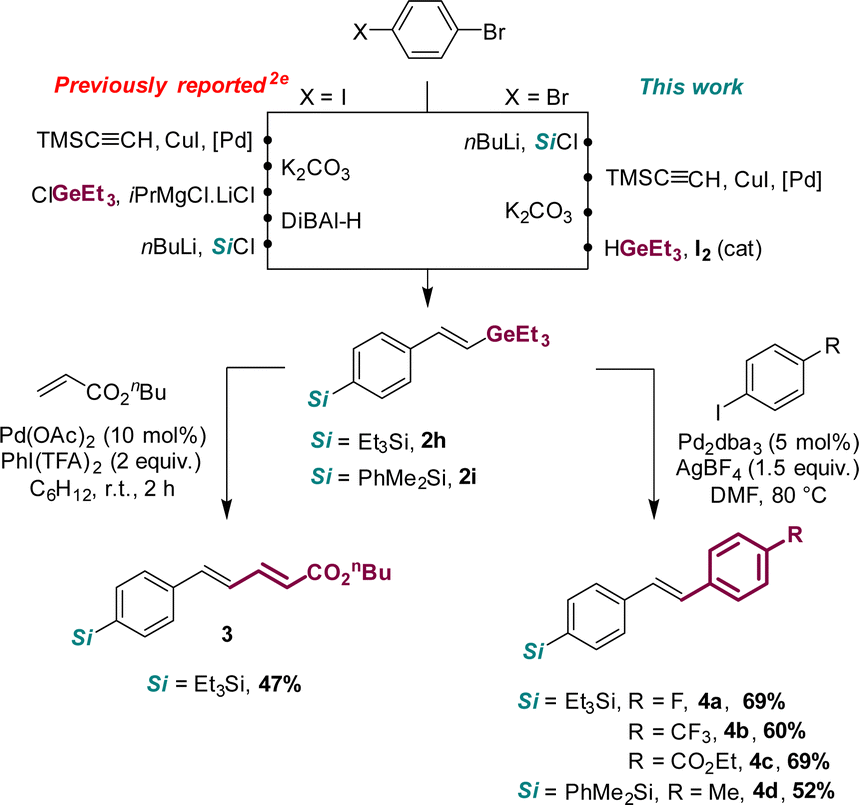 | ||
| Scheme 2 Preparation of Si-functionalized vinylgermanes (comparison of the hydrogermylation route with the known method) and subsequent derivatization of their carbon–germane bond. | ||
In conclusion, complex I2 was found to readily activate Et3Ge–H thanks to Pd/S cooperativity. The synthetic demand for vinyl germane derivatives has driven our investigations towards the catalytic hydrogermylation of alkynes. The PNS pincer Pd complex turned active towards internal and terminal alkynes, as well as allenes under mild conditions (25 °C). Most synthetically useful is the hydrogermylation of aryl alkynes which affords the corresponding vinyl germanes with exquisite regio and stereoselectivity (β syn addition). The compatibility with silyl moieties and selective derivatization of the vinyl germane functionality are also noteworthy.
Financial support from the Centre National de la Recherche Scientifique, the Université de Toulouse, and the Agence Nationale de la Recherche (ANR AAPG2020 CE07 MLC-Photophos project) is gratefully acknowledged. A. C. thanks MESRI (Ministère de l’Enseignement Supérieur, de la Recherche et de l’Innovation) for his PhD fellowship. The “Direction du Numérique” of the Université de Pau et des Pays de l’Adour and the Mésocentre de Calcul Intensif Aquitain (MCIA) are acknowledged for the support of computational facilities. This work was also granted access to the HPC resources of IDRIS under the allocation 2024-[AD010800045R3] made by GENCI.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- C. Fricke and F. Schoenebeck, Acc. Chem. Res., 2020, 53, 2715–2725 Search PubMed.
- (a) C. Fricke, G. J. Sherborne, I. Funes-Ardoiz, E. Senol, S. Guven and F. Schoenebeck, Angew. Chem., Int. Ed., 2019, 58, 17788–17795 Search PubMed; (b) C. Fricke, A. Dahiya, W. B. Reid and F. Schoenebeck, ACS Catal., 2019, 9, 9231–9236 CrossRef CAS PubMed; (c) G. J. Sherborne, A. G. Gevondian, I. Funes-Ardoiz, A. Dahiya, C. Fricke and F. Schoenebeck, Angew. Chem., Int. Ed., 2020, 59, 15543–15548 Search PubMed; (d) A. Selmani and F. Schoenebeck, Org. Lett., 2021, 23, 4779–4784 CrossRef CAS PubMed; (e) A. Dahiya, M. D. Schoetz and F. Schoenebeck, Angew. Chem., Int. Ed., 2023, 62, e202310380 CrossRef CAS PubMed.
- (a) S. Keess and M. Oestreich, Org. Lett., 2017, 19, 1898–1901 CrossRef CAS PubMed; (b) W. Xue, W. Mao, L. Zhang and M. Oestreich, Angew. Chem., Int. Ed., 2019, 58, 6440–6443 CrossRef CAS PubMed.
- See ESI†.
- (a) K. Nozaki, Y. Ichinose, K. Wakamatsu, K. Oshima and K. Utimoto, Bull. Soc. Chim. Jpn, 1990, 63, 2268–2272 CrossRef CAS; (b) S. Bernardoni, M. Lucarini, G. F. Pedulli, L. Valgimigli, V. Gevorgyan and C. Chatgilialoglu, J. Org. Chem., 1997, 62, 8009–8014 CrossRef CAS PubMed; (c) S. Schweizer, C. Tresse, P. Bisseret, J. Lalevée, G. Evano and N. Blanchard, Org. Lett., 2015, 17, 1794–1797 CrossRef CAS PubMed; (d) H. Liang, Y.-X. Ji, R.-H. Wang, Z.-H. Zhang and B. Zhang, Org. Lett., 2019, 21, 2750–2754 CrossRef CAS PubMed.
- T. Schwier and V. Gevorgyan, Org. Lett., 2005, 7, 5191–5194 CrossRef CAS PubMed.
- (a) R. J. P. Corriu and J. J. E. Moreau, J. Chem. Soc. D, 1971, 812–813 RSC; (b) Y. Ichinose, H. Oda, K. Oshima and K. Utimoto, Bull. Soc. Chim. Jpn, 1987, 60, 3468–3470 Search PubMed; (c) F. Wada, S. Abe, N. Yonemaru, K. Kikukawa and T. Matsuda, Bull. Soc. Chim. Jpn, 1991, 64, 1701–1703 CrossRef CAS; (d) T. Matsuda, S. Kadowaki, Y. Yamaguchi and M. Murakami, Org. Lett., 2010, 12, 1056–1058 CrossRef CAS PubMed; (e) M. Itazaki, M. Kamitani and H. Nakazawa, Chem. Commun., 2011, 47, 7854–7856 RSC; (f) M. R. Radzhabov and N. P. Mankad, Org. Lett., 2021, 23, 3221–3226 CrossRef CAS PubMed; (g) M. Bołt, A. Mermela and P. Żak, Chem. Commun., 2023, 59, 11548–11551 RSC.
- S. M. Rummelt, K. Radkowski, D.-A. Roşca and A. Fürstner, J. Am. Chem. Soc., 2015, 137, 5506–5519 CrossRef CAS PubMed.
- A. Clerc, M. Humbert, S. Mallet-Ladeira, K. Miqueu, J. Monot, B. Martín-Vaca and D. Bourissou, ChemistryEurope, 2024, e202400079 Search PubMed.
- (a) H. F. T. Klare, M. Oestreich, J. I. Ito, H. Nishiyama, Y. Ohki and K. Tatsumi, J. Am. Chem. Soc., 2011, 133, 3312–3315 CrossRef CAS PubMed; (b) C. D. Königs, H. F. T. Klare, Y. Ohki, K. Tatsumi and M. Oestreich, Org. Lett., 2012, 14, 2842–2845 CrossRef PubMed; (c) C. D. Königs, H. F. T. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2013, 52, 10076–10079 CrossRef PubMed; (d) T. Stahl, K. Müther, Y. Ohki, K. Tatsumi and M. Oestreich, J. Am. Chem. Soc., 2013, 135, 10978–10981 CrossRef CAS PubMed; (e) T. Stahl, P. Hrobarik, C. D. Konigs, Y. Ohki, K. Tatsumi, S. Kemper, M. Kaupp, H. F. T. Klare and M. Oestreich, Chem. Sci., 2015, 6, 4324–4334 RSC; (f) L. Omann, C. D. Königs, H. F. T. Klare and M. Oestreich, Acc. Chem. Res., 2017, 50, 1258–1269 CrossRef CAS PubMed.
- Y. Li, M. Zhou, S. Park and L. Dang, Inorg. Chem., 2021, 60, 6228–6238 CrossRef CAS PubMed.
- For recent contributions on Pd/Pt σ-complexes with germanes, see: (a) J. Takaya and N. Iwasawa, Eur. J. Inorg. Chem., 2018, 5012–5018 Search PubMed; (b) C. J. Laglera-Gándara, P. Ríos, F. J. Fernández-de-Córdova, M. Barturen, I. Fernández and S. Conejero, Inorg. Chem., 2022, 61, 20848–20859 CrossRef PubMed.
- (a) P. Braunstein and X. Morise, Chem. Rev., 2000, 100, 3541–3552 CrossRef CAS PubMed; (b) For a recent report of dehydrogenative Sn–Sn coupling involving Co/S cooperativity see: S. Elangovan, E. Irran, H. F. T. Klare and M. Oestreich, Organometallics, 2022, 41, 852–857 CrossRef CAS.
- No reaction was observed under the same conditions but in the absence of the (PNS)Pd catalyst I2.
- All the obtained vinyl germanes have been unambiguously authenticated by comparison of their 1H NMR with literature data and selectivities were determined by 2D NOESY NMR experiments.
- Increasing the quantity of triethylgermane did not improve the reaction time neither the syn/anti ratio even with longer reaction time (up to 5 days).
- Attempt to decrease the amount of triethylgermane led to a slowdown of the reaction with only 35% yield after 24 h. Warming the reaction media to 50 °C proved to be detrimental as the triethylgermane degraded.
- Chain-walking of the CC bond was already reported with Pd(PPh3)4, see reference 5a.
- Only few examples were reported with H2PtCl6, Pd(PPh3)4 or Au/TiO2 and mixtures of isomers were also observed, see: Z. Zhao, F. Zhang, D. Wang and L. Deng, Chin. J. Chem., 2023, 41, 3063–3081 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical data, NMR spectra, computational data (PDF). See DOI: https://doi.org/10.1039/d4cc06374k |
| This journal is © The Royal Society of Chemistry 2025 |