
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular hydrogels enable co-delivery of chemotherapeutics with synergistic efficacy against patient-derived glioblastoma cells and spheroids†
Robert J.
Cavanagh
*ab,
Saif
Baquain
c,
Cameron
Alexander
 a,
Oren A.
Scherman
c and
Ruman
Rahman
*b
a,
Oren A.
Scherman
c and
Ruman
Rahman
*b
aSchool of Pharmacy, University of Nottingham, NG7 2RD, UK. E-mail: pazrjc@exmail.nottingham.ac.uk
bSchool of Medicine, Biodiscovery Institute, University of Nottingham, NG7 2RD, UK. E-mail: ruman.rahman@nottingham.ac.uk
cMelville Laboratory for Polymer Synthesis, Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
First published on 1st August 2024
Abstract
Drug combinations have been shown to be highly effective in many cancer therapies but the ratios of the individual drugs must be adjusted carefully and formulated appropriately to ensure synergistic action. Here we assessed combinations of doxorubicin and gemcitabine for post-surgical treatment of IDH1 wild-type glioblastoma (GBM). 2D and 3D spheroid in vitro models of GBM were generated from patient-derived glioblastoma cells resected from brain tumour cores and invasive margins. Drug combinations were screened for synergy using the Chou–Talalay method and mechanisms of action investigated using measures of caspase 3/7-mediated apoptosis and γH2AX-mediated DNA damage. Single drug and drug combinations were formulated in a supramolecular hydrogel based on a peptide-functionalised hyaluronic acid backbone dynamically linked by cucurbit[8]uril-mediated host–guest interactions as an implantable drug-delivery vehicle. Drug efficacy data from in vitro assays demonstrated synergistic activity with doxorubicin and gemcitabine combinations in a molar ratio-dependent manner. These compounds were included in the drug screen as exemplars of DNA intercalators and nucleoside analogue respectively. Consistent with this, enhanced apoptosis and DNA damage were also observed in a synergistic manner. Overall, these drug-loaded hydrogels demonstrated potency and maintenance of synergy with drug-combination hydrogels, in an easy-to-administer in situ gelling formulation suitable for post-resection delivery to prevent GBM recurrence.
1. Introduction
Isocitrate dehydrogenase wild-type Glioblastoma (GBM) remains one of the most hard-to-treat cancers, with an invariable median overall survival of 14–16 months from diagnosis over the past two decades.1,2 As gross total resection fails to safely remove all tumour cells, and adjuvant standard-of-care of temozolomide and radiation therapy confers only a ∼2-month survival benefit, there is an urgent clinical need to identify more effective chemotherapeutics including synergistic combinations. A significant impediment for most chemotherapeutic agents is poor penetration of the blood–brain barrier when delivered systemically, thus limiting the effective dose which reaches brain parenchyma and causing dose-limiting systemic toxicity.3 A further challenge for GBM therapy is cellular and genetic inter- and intra-tumour heterogeneity which manifests from variable microenvironmental selection pressures. This results in rapid sub-clonal divergence which confers therapeutic resistance.4 This is confounded by the capability of GBM cells to rapidly infiltrate the healthy brain parenchyma adjacent to the primary tumour mass, forming new tumour micro-deposits which are responsible for tumour recurrence.5 As surgically resected primary tumour is not reflective of infiltrative residual disease, repurposed or experimental therapies must aim to prevent or delay local tumour recurrence, which typically arises 2 cm from the resection margin. One means by which this may be achieved is to deliver anti-cancer drugs directly into the resection cavity during surgery.6 This mode of interstitial delivery has several advantages over systemic routes of administration, as higher drug concentrations can in principle, be achieved at the tumour site, whilst systemic toxicity can be avoided or reduced as the blood–brain barrier prevents escape of the drugs into the main circulation. Although several post-surgical resection drug delivery systems have been considered preclinically,7–13 only the Gliadel® formulation, which is a biodegradable polymer wafer for local release of carmustine, has been approved for clinical use.14 However, despite the success of Gliadel®, there can be difficulties in ensuring wafers remain proximal to surgical resection margin, and the formulation is restricted as a monotherapy. Moreover, the mechanical mismatch between the hard Gliadel® wafer and soft brain tissue have been shown to result in unwanted side effects including seizures, wound infections, headaches and lethargy.15 Recently, a number of studies have detailed the use of drug-containing hydrogels as potential strategies to improve post-surgical resection delivery.16–23 The advantages of hydrogel-based delivery systems include the ability to conform fully to the resection cavity, and for in situ gelling materials, the possibility to be injected as a free-flowing solution with subsequent setting in place to ensure persistent access to the non-resected tissue. In addition, a variety of drugs (and distinct drug chemistries) can be incorporated into gel-forming systems, potentially allowing for therapies to be stratified for different patients and cancer types. These delivery systems thus allow for combination drug treatments predicated against GBM heterogeneity, which can simultaneously target multiple oncoproteins or pathways associated with GBM survival and growth.24,25 Rational choice of drug combinations can potentially achieve synergistic effects, where the combined action of the drugs is greater than the sum of their individual potencies. In addition, by targeting more than one pathway it is possible to reduce the risk of drug resistance, as multiple mutations are required for GBM cells to adapt to the therapeutic challenge. Currently, several drug combinations are being evaluated for GBM treatment, such as veliparib with adjuvant temozolomide,26,27 and lomustine with bevacizumab.28 However, to date there have been rather fewer reports of hydrogel-mediated delivery of drug combinations.29–31 Here, we describe the evaluation of a new drug combination treatment, using the topoisomerase II inhibitor doxorubicin (DOX) combined with the nucleoside analogue gemcitabine (GEM), and their incorporation in a hydrogel to mimic their application in post-resection delivery. The hydrogel used for delivery is a hyaluronic acid-based system, in which some of the hydroxyl side-chains have been functionalised via a linker with a dipeptide, cysteine-phenylalanine (HA-CF), enabling assembly into a non-covalently cross-linked network via interactions of the Phe residues with the macrocyclic host molecule cucurbit[8]uril (CB[8]).32,33 These hydrogels have been previously demonstrated to be biocompatible and facilitate drug delivery in vitro, in vivo and ex vivo. Moreover, the mechanical properties of the HA-CF/CB[8] hydrogels can be tuned to match the stiffness of the native tissue to improve tolerability and enhance drug delivery.34,35 To determine an efficacious hydrogel cargo, we evaluated the potencies of drug combinations at various ratios in 2D culture and 3D cell spheroids, using GBM patient-derived cells from the core and invasive margins, the latter representing tissue isolated from the tumour edge which blends into brain parenchyma and is a more accurate proxy of residual disease spared by surgery. We investigated the mechanisms of action of these combination therapies and showed that synergistic effects could be achieved in the patient-derived cells in both 2D and 3D cultures, including from drugs released from the hydrogel formulations.2. Materials and methods
2.1. Materials
Standard reagents, solvents, monomers and other materials for chemical synthesis were purchased from Sigma-Aldrich, ThermoFisher or Cayman High glucose DMEM (phenol red free, cat. # 31053-028) was acquired from Gibco (Life technologies). High glucose DMEM (cat. # D6546), Trypsin–EDTA (cat. # T3924), foetal bovine serum (FBS, cat. # F7524), penicillin–streptomycin antibiotic solution (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U penicillin and 10 mg mL−1 streptomycin, cat. # P0781), L-glutamine (200 mm, cat. # G7513) and DMSO (cat. # D2438) were purchased from Sigma-Aldrich. Hoechst 33342 (cat.# H1399) and Presto Blue 10× solution (cat. # A13262 100 mL) were acquired from Invitrogen. Gemcitabine and Doxorubicin were purchased from Cayman Chemical Company. Float-A-lyzer® cellulose ester (CE) dialysis tubes with (molecular weight cut-off) MWCO of 50 kDa were purchased from Spectrum Laboratories. Cucurbit[8]uril was synthesised and separated according to literature procedures.36,37
000 U penicillin and 10 mg mL−1 streptomycin, cat. # P0781), L-glutamine (200 mm, cat. # G7513) and DMSO (cat. # D2438) were purchased from Sigma-Aldrich. Hoechst 33342 (cat.# H1399) and Presto Blue 10× solution (cat. # A13262 100 mL) were acquired from Invitrogen. Gemcitabine and Doxorubicin were purchased from Cayman Chemical Company. Float-A-lyzer® cellulose ester (CE) dialysis tubes with (molecular weight cut-off) MWCO of 50 kDa were purchased from Spectrum Laboratories. Cucurbit[8]uril was synthesised and separated according to literature procedures.36,37
2.2. Formation and characterisation of drug-loaded hydrogels
2.3. Rheology
Rheological analysis was conducted on a TA Discovery DH-2 Rheometer equipped with a 20 mm parallel plate geometry at 20 °C. Hydrogel samples were loaded onto the rheometer with approximately a 500 μm loading gap. Dynamic oscillatory strain amplitude sweep measurements were conducted at a frequency of 10 rad s−1. Dynamic oscillatory frequency sweep measurements were conducted at a 1% strain amplitude. All data was analysed using TRIOS software.2.4. Drug release from hydrogels
Hydrogels loaded with GEM and/or DOX (1 mg ml−1, 0.1 wt%) were placed in float-a-lysers (50 kDa CE membrane). The dialysis devices were then immersed in 15 ml pre-warmed buffer and incubated at 37 °C. At pre-determined time points the entire release media was removed and replaced with fresh pre-warmed buffer. Aliquots of the collected release media were then analysed by HPLC-UV/vis. The concentration of drug released was calculated using a standard calibration curve.:acetonitrile (90:10). For DOX and a combination of GEM:DOX detection the mobile phase consisted of water:acetonitirile (30:70) acidified to pH 3.0 using orthophosphoric acid. Data acquisition was carried out using Chemstation software.
2.5. Cell culture
Culture conditions were maintained at 37 °C with 5% CO2 and 90% relative humidity. GIN8 (Glioma INvasive margin cells) were isolated from medial front invasive margin (54 years female, wild-type IDH (primary GBM), intact ATRX, 0% MGMT promoter methylation, 90% resection plus Gliadel wafers; treatment 60 Gy radiotherapy, concurrent and adjuvant temozolomide; patient died 5 months after surgery). GIN28 were isolated from the 5-aminolevulinic acid (5-ALA) fluorescence-positive invasive margin, and GCE28 (Glioma Contrast Enhanced core cells) from the core region of the same tumour (71 years male, wild-type IDH (primary GBM), intact ATRX, 0% MGMT promoter methylation 99% resection; no adjuvant therapy (patient choice); died 3 months after surgery). GIN31 were isolated from the 5-ALA fluorescence-positive invasive margin, and GCE31 from the core region of the same tumour (wild-type IDH (primary GBM), intact ATRX, 0% MGMT promoter methylation, 100% resection; treatment 60 Gy radiotherapy, concurrent and adjuvant temozolomide; patient died 16.1 months after surgery). Cell lines were cultured in Dulbecco's Modified Eagle Medium (DMEM; Sigma-Aldrich, St Louis, MO, USA) supplemented with 10% FBS, 1 g L−1 glucose and 2 mM L-glutamine (Sigma-Aldrich) at 37 °C with 5% CO2. Cells were used within a passage window of 10.2.6. Cytotoxicity
Screening of chemotherapeutic drug potency was performed on GBM 2D monolayers and 3D spheroids using the PrestoBlue™ metabolic assay and CellTiter Glo® 3D assay, respectively. Drugs were applied to cells in 100 μL per well in phenol red free DMEM containing 10% (v/v) FBS for 72 hours. In vitro testing of blank and drug-loaded hydrogels, alongside single drug and combination drug controls were conducted using the PrestoBlue™ assay and live/dead staining (Invitrogen™ LIVE/DEAD™ Viability/Cytotoxicity Kit; L3224). Hydrogels were applied at 100 μl per well in 96-well plates followed by the addition of 100 μl phenol red free DMEM containing 10% (v/v) FBS. Prior to assaying hydrogel treated cells, hydrogel was removed via pipetting, washed with PBS and cells were assessed for metabolic activity or underwent Live/Dead staining (as described below).2.7. Immunofluorescence microscopy of γH2AX
GIN28 cells were seeded on LabTek chamber slides (Thermo Fisher Scientific) with 3 × 104 cells per chamber well and cultured for 24 hours. Cells were then treated with doxorubicin, gemcitabine or combinations for 24 hours. Cells were fixed with 4% PFA for 15 minutes at room temperature, permeabilised with 0.1% (v/v) TX100 for 15 minutes at room temperature and then blocked with 1.5% (v/v) bovine serum albumin (BSA) in PBS for 60 minutes at room temperature. Cells were then incubated for 1 hour at room temperature with (1:1000) mouse monoclonal anti-gH2AX/ser139, washed thrice with PBS and then incubated with (1:500) goat anti-mouse IgG AlexaFluor 488 for 1 hour at room temperature. The chamber slides were also counterstained with 0.5 μg mL−1 DAPI to visualise nuclei. Slides were washed twice with PBS and then mounted with coverslips and ProLong™ Gold Antifade Mountant. Microscopy was performed using an EVOS M5000 fluorescent microscope, with subsequent image analysis and counting of foci conducted using ImageJ software (v1.5).
2.8. Detection of activated caspase-3/7
The CellEvent® caspase-3/7 green detection reagent (Thermo Fisher Scientific) was used to assess levels of activated caspase-3 and 7 as an indicator of apoptosis. GIN28 cells were cultured as 2D monolayers and treated as described above in 96-well plates. After treatment, 100 μl 2% (v/v) CellEvent® probe in PBS was applied per well for 30 minutes at 37 °C. Fluorescent intensity was measured at 502/530 nm (λex/λem) and normalized to the untreated control (set as a value of 1).2.9. Statistical analysis
All values expressed as mean ± standard deviation (S.D.) unless otherwise stated (n ≥ 3 biological replicates). Dose response curves fitted using nonlinear fits. Statistical analysis and modelling performed using GraphPad Prism 7. One-way Analysis of Variance (ANOVA) or two-way ANOVA followed by Dunnett's post hoc test, as indicated, was used to test statistical difference between measurements; p-values less than 0.05 were considered significant (*p < 0.05; **p < 0.005; ***p < 0.0005; ****p < 0.0001).2.10. Ethical statement
Experiments using primary cell lines derived from human brain tumour tissue were approved by the National Health Service Research and Ethics Committee East Midlands (Ref: 11/EM/0076), and such research is subject to audits by the Human Tissue Authority. Informed consents were obtained from human participants. All samples were handled and monitored in accordance with national guidelines of the UK Human Tissue Act 2004, specifically ‘Part 2 – Regulation of activities involving human tissue’.3. Results
3.1. Drug screening in GBM spheroids
Initial potency screening was performed to identify lead drug compounds for future loading into hydrogels. The use of a hydrogel matrix for post-surgical-resection delivery enabled chemotherapeutics to be chosen without the constraint of blood brain barrier transport. Drug compounds were prepared and applied in culture media and potency was tested in patient-derived core and margin GBM cell lines cultured both as 2D monolayers (ESI Fig. 2†) and as 3D spheroids (Table 1). IC50 data presented in Table 1 indicates the topoisomerase II inhibitor DOX demonstrated the highest potency across the panel of GBM spheroids tested. Furthermore, the first-in-line GBM drug treatment temozolomide was observed as the least potent across the spheroids tested. In terms of drug potency ranking, similar trends were observed in drug treatment on 2D cultured GBM lines, albeit with increased drug potency, as expected when comparing 2D and 3D culture models for drug testing.38:DMEM for a further 72 hours. Potency was assessed via spheroid ATP levels measured using the CellTiter Glo 3D assay. IC50 values calculated via GraphPad prism. Data is presented as mean ± S.D
| Patient derived GBM cell line | |||||
|---|---|---|---|---|---|
| Drug | GIN8 (invasive margin) | GIN28 (invasive margin) | GCE28 (tumour core) | GIN31 (invasive margin) | GCE31 (tumour core) |
| Temozolomide | 965 ± 85 μM | 623 ± 70 μM | 309 ± 33 μM | >1000 μM | >1000 μM |
| Etoposide | 205 ± 51 μM | 221 ± 65 μM | 162 ± 35 μM | 148 ± 37 μm | 162 ± 50 μm |
| Irinotecan | 190 ± 43 μM | 162 ± 35 μM | 221 ± 60 μM | 190 ± 63 μM | 163 ± 42 μM |
| Olaparib | 113 ± 35 μM | 60.2 ± 21 μM | 56.7 ± 9.2 μM | 422 ± 54 μM | 135 ± 25 μM |
| Gemcitabine | 55.3 ± 15 μM | 20.8 ± 3.4 μM | 39.4 ± 2.5 μM | 93.5 ± 5.1 μM | 230 ± 19.8 μM |
| Doxorubicin | 0.67 ± 0.18 μM | 1.42 ± 0.41 μM | 3.22 ± 0.38 μM | 0.85 ± 0.2 μM | 1.90 ± 0.20 μM |
Drug potency comparisons between invasive margin (GIN) and tumour core (GCE) isolated GBM cells can be made from counterpart patient samples, i.e., GIN28 vs. GCE28, and GIN31 vs. GCE31; drug sensitivity in invasive margin cells was of particular interest owning to its representation of a post-surgical GBM cell population compared to core cells which are predominantly removed. Of note, DOX was significantly more potent (2-fold) in invasive cells compared to non-invasive core cells in GIN28/GCE28 spheroids (p value = 0.005) and in GIN31/GCE31 (p value = 0.003). A similar significant 2-fold increase in potency in invasive cells was observed for GEM, a nucleoside analogue, in GIN28/GCE28 (p value = 0.0015) and GIN31/GCE31 (p value = 0.0003). Temozolomide demonstrated 2-fold increased sensitivity to GCE28 tumour cells than GIN28 invasive margin cells. Etoposide and irinotecan, topoisomerase I and II inhibitors, respectively, demonstrated similar potency levels across all GBM cell lines tested. The toxic effects of olaparib, an inhibitor of poly ADP ribose polymerase (PARP), were consistent across the majority of GBM cell lines, with the exception of GIN31 whereby an increase in IC50 value of approximately 4-fold is observed relative to other lines.
3.2. Identification of synergistic drug combinations
Next, drug combinations were studied owing to their potential to overcome drug resistance mechanisms and increase clinical therapy.39 GEM and DOX were selected as the lead drug compounds for combination testing due to the high potency observed in monotherapy, relative to all other compounds tested (Table 1). In vitro calculation of DOX:GEM combination synergy was performed using the established Chou and Talalay method40,41 and resulting combination index (CI) values are shown in Fig. 1. Additionally, the effect of varying the molar ratios of the DOX:GEM combination was investigated to inform future hydrogel drug loading to the optimal combination formulation. Synergy screening was initially undertaken in 2D monolayer cultured GBM cells (Fig. 1A) and the data indicated that synergistic efficacy (CI < 1) was induced at 10:1 DOX:GEM molar ratio in all cell lines with the exception of GCE31, and at 1:1 DOX:GEM ratio with invasive cell lines (GIN8, GIN28 and GIN31) but not the counterpart tumour core lines (GCE28 and GCE31). Antagonism (CI > 1) was observed at 1:10 DOX:GEM molar ratio for 4 out of 5 cell lines.
 | ||
| Fig. 1 (A) Combination index (CI) values (mean ± S.D) for DOX and GEM at different molar ratios using Tou and Chalay method for quantification of synergy. Data generated from PrestoBlue™ metabolic assays following drug combination treatment for 48 hours. (B) Micrographs of GBM spheroid morphologies pre- and post-treatment with DOX and GEM 1:1 combination. Scale bar, 250 μm. (C) CI values of DOX and GEM combinations in 3D GBM spheroid models. CI values generated following treatment and assaying with CellTiter Glo® 3D probing cellular ATP levels. | ||
In order to confirm the observed synergy in more physiologically relevant models, 3D spheroids of the invasive GBM cell lines were employed (Fig. 2B and C). Data indicate that synergy is maintained in spheroidal models at DOX:GEM combination molar ratios of 1:1 and 10:1, and antagonistic or additive (CI ≈ 1) effects are observed at 1:10 ratio. Moreover, the synergy of 1:1 DOX:GEM combination can be observed via morphological changes in spheroids, as shown in Fig. 1B, supporting the CI values calculated from dose response curves of intracellular ATP levels (CellTiter Glo® 3D assay).
 | ||
| Fig. 2 Assessment of dsDNA breaks using γH2AX immunostaining upon GBM exposure to DOX and GEM. (A–C) GIN28 cells exposed to single dosed DOX and GEM and DOX:GEM combinations for 24 hours. DOX concentration was kept constant at 1 μM and GEM concentration ranged from (A) 0.1 μM, (B) 1 μM, and (C) 10 μM as either single drug or in combination with DOX, to represent DOX:GEM combinations of 10:1, 1:1 and 1:10, respectively. Cells were fixed and stained for γH2AX (green) and nuclei (DAPI, blue) and imaged on an EVOS M5000 fluorescent microscope. Scale bar, 10 μm. (D) Foci quantification was performed using ImageJ and data presented in bar charts (mean ± S.D). Symbol identification, +, 0.1 μM; ++, 1 μM; +++, 10 μM. (E) Induced foci per cell data presented for response additivity analysis. Statistical significance tested using one-way ANOVA and Dunnett's multiple comparisons tests (**p < 0.005; ***p < 0.0005). | ||
3.3. Synergistic induction of dsDNA breaks and caspase activation
In order to ascertain the mechanism of synergy of DOX and GEM combinations, the levels of double stranded breaks (DSBs) in DNA were investigated using the γH2AX assay (Fig. 2). There is evidence that monotherapy with DOX or GEM induces DSBs, albeit via distinct mechanisms.42,43 Therefore, it was of interest to explore if DSBs also resulted following mono-treatment in patient-derived GBM cells, and if the observed synergy, and antagonism, with DOX and GEM combinations is related to DNA damage.These data demonstrate that DOX mono-treatment induces significant increases in DSBs, as assessed by γH2AX foci at concentrations ≥0.1 μM, and GEM mono-treatment at concentrations ≥10 μM (Fig. 2 and ESI Fig. 3†). Fig. 2 further illustrates the effects of applying DOX and GEM combinations whereby DOX concentration is maintained at 1 μM and varying GEM concentrations of 0.1, 1 and 10 μM are co-applied to represent molar ratios of 10:1, 1:1 and 1:10 DOX:GEM, respectively. At these drug concentrations, all combinations were observed to induce significant induction in DSBs relative to the vehicle control (Fig. 2D). Despite the lack of significant DSB induction in monotherapy (Fig. 2D), combinations containing 0.1 and 1 μM GEM resulted in significant synergistic efficacy with 1 μM DOX, as determined by a responsive additivity-based model,44i.e., the sum of the drug combination elicits a greater effect than the sum of the individual drugs (Fig. 2E). These observations are consistent with the previously noted synergistic nature of the 10:1 and 1:1 DOX:GEM molar ratios (Fig. 1). Interestingly, for the 1:10 DOX:GEM combination, in which a GEM concentration of 10 μM is applied in combination with 1 μM DOX, the response additivity model indicates only additive effects, as no significant difference is calculated between the drug combination and the sum of the individual drugs (Fig. 2E). This observation is again consistent with previous CI data (Fig. 2).
To further investigate combination mechanisms of cytotoxicity, activation of caspase-3/7 was assessed as an indicator of apoptosis (Fig. 3). The data indicate significant caspase activation upon exposure of GBM cells to DOX or GEM as single drugs at concentrations ≥0.1 μM. Application of all combinations resulted in significant caspase activation relative to the untreated control; however relatively lower caspase activation is observed with a 1:10 DOX:GEM ratio (0.1 μM DOX + 1.0 μM GEM) (Fig. 3A). Further analyses using response additivity modelling (Fig. 3B), illustrates significant increases in combination caspase activation, relative to the sum of single DOX and GEM treatment, with DOX:GEM combination ratios of 10:1 and 1:1. No significant difference to single drug summations is evident with 1:10 DOX:GEM combination; indeed a slight decrease in caspase activation is noted, indicating additive to antagonistic effects.
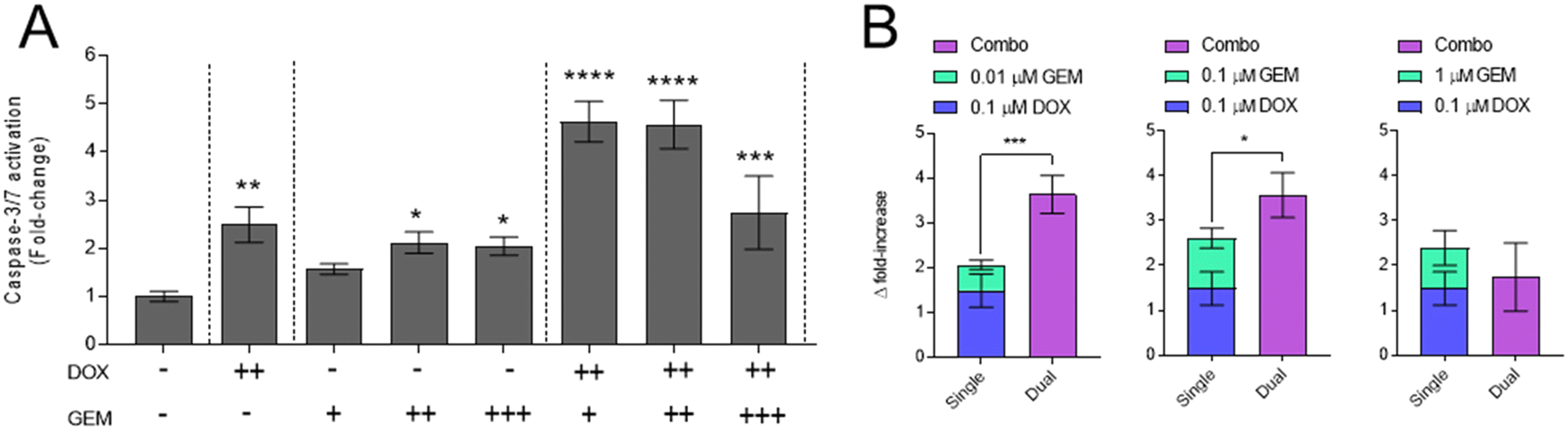 | ||
| Fig. 3 Detection of Caspase-3/7 activation upon GBM exposure to DOX and GEM. (A) GIN28 cells cultured in 2D and incubated with single, combination or the vehicle control for 24 hours. Values normalised to vehicle control. Symbol identification, +, 0.01 μM; ++, 0.1 μM; +++, 1 μM. (B) Response additivity graphs of combinations showing caspase level fold-increase. Data presented as mean ± S.D. Statistical significance tested using one-way ANOVA and Dunnett's multiple comparisons tests. (*p < 0.05; **p < 0.005; ***p < 0.0005; ****p < 0.0001). | ||
3.4. Formation and characterisation of drug-loaded hydrogels
In order to form a drug delivery system suitable for use following surgical removal of a tumour, we generated HA-CF/CB[8] hydrogels, which have already been shown to have advantageous properties for post-resection local drug delivery to the brain.34,35 These gels were synthesised by reacting hyaluronic acid with methacrylic anhydride, such that a proportion of the pendant 6-OH groups on the hyaluronate backbone were functionalised with Michael acceptor methacrylate groups, and then adding 3,3′-disulfanediylbis(2-(2-amino-3-phenylpropanamido)propanoic acid) dihydrochloride, in the presence of dithiothreitol. In this way, in situ generated thiol functionality from the peptide reacted with the methacrylate-functional side-chains to form the HA-CF gel precursor, with ∼10% of the 6-OH groups being methacrylated and bearing these peptide pendent groups to form gels in the presence of CB[8]. Loading of DOX and GEM was possible via simple mixing of components with the functionalised HA-CF backbone (Fig. 4A), prior to adding CB[8] and forming drug-loaded HA-CF/CB[8] hydrogels. This facile formulation approach enabled a range of drug-loaded hydrogel materials to be prepared. | ||
| Fig. 4 Formation of drug-loaded hydrogels, rheological analysis of blank and drug-loaded (0.1% w/w) gels at 20 °C and drug release. In (A) is a schematic of hydrogel formation from peptide-modified hyaluronic acid (HA-CF) and CB[8] in the presence of candidate drugs. (B) Oscillatory rheological frequency sweep at an oscillation strain of 1% sweep comparing blank, GEM, DOX and Combo hydrogels. (C) Oscillatory rheological amplitude sweep at an angular frequency of 1 rad s−1 comparing blank, GEM, DOX and Combo hydrogels. (D, E and F) Therapeutic cargo release from hydrogels at 37 °C. Release of GEM (D) and DOX (E) as individual components, and as dual components in hydrogel (F): n = 3, error bars represent the range. | ||
Initial studies showed that the presence of drugs in the supramolecular hydrogels was not deleterious to their rheological properties. The blank (empty) and the three drug-loaded hydrogels (GEM, DOX, 1:1 GEM:DOX) displayed frequency dependant behaviour, common for hydrogels mediated by dynamic host–guest interactions (Fig. 4B) and broad linear viscoelastic (LVE) regions were observed in all oscillatory strain amplitude sweeps (Fig. 4C). The mechanical properties of both the empty and drug-loaded gels were well matched to the properties of brain tissue noted in human GBM and “normal” tissue,35 indicating that the presence of the drug molecules, alone or in combination with one and other, does not impact the properties of the hydrogels.
 | ||
| Fig. 5 Cytotoxicity of blank and drug loaded hydrogels. (A) Micrographs of live dead staining of GIN28 cells following 72-hour hydrogel incubation. (B) Quantification of live dead images and (C) metabolic activity assessment of hydrogel cytotoxicity using PrestoBlue™ assay at 24, 48 and 72 hours. Scale bar is 250 μm. Additional images of live/dead staining at 24 and 48 hours are presented in ESI Fig. 6.† Statistical analysis performed using two-way ANOVA with comparison to relevant vehicle control, unless indicated to demonstrate free drug versus hydrogel comparison. | ||
Neither free drug alone, at the concentrations tested (1 μM), nor hydrogel treatment was observed to be toxic to cells following 24 hours incubation via live dead staining or metabolic activity assessment (Fig. 5). Treatment with GEM and GEM hydrogels induced cell killing after 72 hours incubation as indicated by live/dead staining and significant loss in metabolic activity relative to controls (Fig. 5). No significant difference was calculated between free drug and GEM-hydrogel and no loss in cell viability was noted at 24 or 48 hours with free GEM or GEM hydrogels. Cell death was observed with DOX and DOX hydrogels at 48 and 72 hours of treatment (Fig. 5). At 48 hours of treatment, a decrease in metabolic activity was induced by DOX hydrogels (85.6 ± 6.3%); however, this was not significant compared to the unloaded hydrogel and was significantly less relative to the free DOX at this time point (70.3 ± 5.4%). Cell death subsequently increased following 72 hours incubation with DOX and DOX hydrogels and no significant difference was noted between these treatments, an observation supported by both live dead staining and metabolic screening.
DOX and GEM combinations, either as free drugs or loaded into hydrogels, demonstrated higher cytotoxicity (Fig. 5), as expected from the prior drug synergy studies. Increased numbers of dead cells and fewer live cells as denoted from staining assays and significant loss in metabolic activity were observed from 48 hours of incubation with both free and hydrogel loaded combinations. At 48 hours incubation it can be noted that the combination treatments were comparable in efficacy, and not significantly different from the level of cell killing induced by the DOX counterpart treatments. The combination treatments however, induced substantial GBM cell death following 72 hours incubation and in a manner that was significantly higher than single DOX treatment. Moreover, at 72 hours incubation, compared to the GEM-hydrogel and DOX-hydrogel treatments, there was a higher level of cell killing with the combination-hydrogel, indicating retention of synergistic drug efficacy upon release from the hydrogels (ESI Fig. 5†).
4. Discussion
Patient-derived GBM cells isolated from the infiltrative margin (GIN lines) and tumour core (GCE lines) of adult patients have been used throughout this study.45 Typically, commercial GBM cell lines are derived from the MRI contrast-enhanced tumour core region, which do not enable a phenotypically accurate model of the post-surgical microenvironment whereby remaining infiltrative cells provide a basis of GBM recurrence.46 Thus, the use of infiltrative GBM cells provides a better proxy of residual disease spared by surgery, represents a more clinically relevant model for testing new therapies, and provides an interesting comparison to the more commonly investigated tumour core derived cells. Drug testing demonstrated substantial differences in drug potencies between cell lines derived from separate patients (Table 1), reflecting intertumoural heterogeneity.47,48Interestingly, of the two anthracycline topoisomerase II inhibitors tested, only DOX and not etoposide, demonstrated significantly increased anti-cancer activity against invasive margin GBM cells relative to tumour core cells (Table 1). Moreover, the topoisomerase I inhibitor irinotecan induced no significant invasive margin or tumour core specificity in potency. Therefore, the increased invasive margin cell potency exhibited by DOX is likely not related to its topoisomerase inhibition or DNA intercalation, which is a conserved mechanism across anthracycline drugs.49 Additionally, both DOX and etoposide are known to generate reactive oxygen species (ROS) intracellularly which can mediate increased cell apoptosis.50,51 Nevertheless, due to enhanced cancer cell redox buffering systems, differences in the magnitude of ROS generation of the drugs may play a role;52 however, this requires further investigation of the specific GBM cells tested here.
GEM was also observed to demonstrate increased potency in invasive margin GBM cells (Table 1), highlighting its potential in preventing GBM recurrence post-surgery. Further work is required to determine the specific GEM-mediated anticancer mechanism that is responsible for the observed increase in invasive margin GBM cell potency. Potential mechanisms include GEM-mediated DNA chain termination,53 inhibition of ribonucleotide reductase53,54 or nucleotide excision repair (NER) inhibition.55,56 Identification of the specific mechanism(s) will aid the selection of additional compounds for post-surgical GBM treatment.
Temozolomide is the standard-of-care chemotherapeutic for post-surgical treatment of GBM;57 however, the results generated here demonstrate it is the least potent drug across all patient derived cell lines tested (Table 1). Moreover, temozolomide presents as more potent on tumour core cells as opposed to invasive margin derived cells, thus indicating that studies testing temozolomide on standard commercially available GBM cell lines that are typically derived from the tumour core (i.e., U87 cells), may overestimate the effectiveness of the drug for GBM treatment post-surgery. Potency aside, temozolomide does however present certain pharmacological advantages that are exploited for clinical use; it has good oral bioavailability and due to its lipophilic properties and small size is able to cross the blood brain barrier effectively.58,59 The use of local delivery therapies in the post-surgical cavity, such as the hydrogel investigated here, mitigate the necessity of crossing the blood brain barrier. As such, the more potent chemotherapeutics identified, DOX or GEM, present as promising candidates for local drug delivery over the selection of temozolomide, for post-surgical GBM treatment. Moreover, local delivery may confer additional benefits such as reducing systemic drug toxicity associated with nonspecific biodistribution. For example, treatment with intravenous DOX is associated with serious dose-limiting cardiotoxicity,60–62 however a number of in vivo studies have demonstrated reduced systemic DOX toxicity following local delivery mediated via gels of various materials in gastric,63 breast64 and sarcoma65 tumour bearing mice.
The emergence of drug resistance to monotherapy is prevalent in GBM tumours owing to their highly heterogenous nature, and therefore the use of multi-drug therapy represents a means to overcome this.66–68 Moreover, in addition to decreased chemoresistance, drug combinations that perform in a synergistic manner may achieve greater therapeutic value, enabling lower doses of individual drugs to be administered and thus offering the potential to reduce off site toxicity.39 Here we investigated the synergy of combinations of the most potent single agents identified in our drug screening, DOX and GEM, as potential combination drug cargo for hydrogel loading.
Moreover, varying molar ratios of the drugs in combination were studied, as combination synergy and effects are known to be dependent on molar ratios in addition to distinct drug mechanism of actions.69–71 Combinations of DOX and GEM applied to 2D and 3D spheroid models of GBM demonstrated molar ratio-dependent cell killing synergy (Fig. 1). Synergy was induced at 1:1 and 10:1 DOX:GEM ratios, and additive/antagonistic effects presenting at 1:10 ratio. Moreover, this ratio profile was consistent between 2D and 3D models, broadly consistent between cell lines tested, and more uniform cell line response observed in 3D relative to 2D in vitro models. Further study revealed that ratios 10:1 and 1:1 DOX:GEM presented with synergistic DNA damage, as demonstrated with γH2AX immunostaining (Fig. 2) and effector caspase-3/7 activation (Fig. 3); responses that correlate with enhanced cell killing observed in Fig. 1. Collectively, these observations indicate a pro-apoptotic mechanism driven via the synergistic induction of DNA damage.
There are several proposed mechanisms of DOX mediated DNA damage, including topoisomerase II poisoning,42,72 intercalation and formulation of DOX-DNA adducts,73,74 and oxidative stress resulting from DOX induced release of free radicals.42,75 In contrast, GEM-induced DNA breaks are reported to result via the stalling of replication forks following partial chain termination.43,76 These distinct mechanisms are likely responsible for the DNA damage observed with single drug treatments and potentially the additive response observed with 1:10 DOX:GEM combination; however, the synergistic mechanism resulting in DNA damage at 10:1 and 1:1 combination ratios remains unclear. To the best of our knowledge, this drug combination has not been investigated previously for GBM and only sparse evidence is reported from other cancer conditions. For example, DOX and GEM combinations have been reported as synergistic in models of triple negative breast cancer; however the exact mechanism of synergy remained unknown.77,78 One potential insight into the synergistic mechanism may be provided from studies on GEM and platinum-based drug combinations which have been investigated clinically for pancreatic and non-small cell lung cancers.79,80 Other in vitro studies have reported that GEM synergises with cisplatin via GEM-mediated inhibition of DNA repair mechanisms including nucleotide excision repair (NER), homologous recombination (HR) and nonhomologous end-joining (NHEJ).55,56 HR and NHEJ are major repair pathways for double stranded DNA breaks and inhibitors of such pathways have been shown to increase sensitivity to DOX treatment.81–83 Therefore, it is hypothesised that GEM-mediated inhibition of HR and NHEJ may be associative mechanism(s) responsible for the synergy with DOX that we have observed in GBM cell lines; however, further work is required to confirm this. Interestingly, the 1:10 DOX:GEM combination demonstrated additive/antagonistic effects represented by additive levels of DNA damage (Fig. 2) and reduced levels of caspase-3/7 activation (Fig. 3). Further work is required to elucidate these responses which may be related to the opposing stages of cell cycle arrest induced by DOX and GEM. Prevailing GEM-mediated S phase arrest may subsequently hinder DOX-mediated DNA damage which is cell cycle dependent (G2/M phase).84,85
Following the identification of a synergistic DOX and GEM combination we next investigated loading of the drugs into the hyaluronic acid peptide conjugate/CB[8] hydrogel and established that the key rheological properties of the supramolecular network were not significantly altered. This established the suitability of the hydrogel as a reservoir for combination therapeutics, and drug release studies indicated complete release of GEM and no further release of DOX after 24 h. Drug-free hydrogels were found to be non-toxic in vitro to patient-derived GBM cell lines in agreement with data previously obtained in patient-derived GBM xenograft hydrogel implant models.35 Following 24 hours incubation, no loss in viability was observed with free drugs or hydrogel-loaded drugs. This is likely a result of this 24 hours timepoint being too short to observe any substantial loss in cell number due to anti-proliferative or apoptotic effects. For example, at this time point significant DNA damage (Fig. 2) and caspase activation (Fig. 3) is noted with free drugs. Therefore, it can be concluded that the cells are indeed undergoing cell death related mechanisms however their influence is yet to be lethal at this time point. Regarding drug loaded hydrogels at 48 hours the DOX-loaded hydrogel was significantly less potent than free DOX (Fig. 5), an effect that likely reflects the sub-maximal DOX release from the hydrogel (<50%) (Fig. 4). On the contrary, rapid (>10 h) and near maximal (>95%) GEM release was observed from the hydrogel, and subsequently no significant differences in cytotoxicity were observed between free GEM and GEM-hydrogels. Testing of DOX and GEM (1:1) combination hydrogels revealed comparable cytotoxicity to the free drug combination. Moreover, comparison to single drug loaded hydrogels indicated that the GBM cell killing synergy of the drug combination was maintained by GEM and DOX together when released from hydrogels, suggesting promise for future in vivo efficacy studies. The HA-CF/CB[8] hydrogels themselves have been shown to be cytocompatible, and to have no deleterious immune-activating properties,34 and are expected to degrade to simple, excretable, saccharides and amino acids. It is not known as yet the rate of degradation of these gels in patient-relevant in vivo settings, but it is unlikely to affect the therapeutic properties of these drug combinations, thus we believe these materials can progress rapidly to more detailed pre-clinical studies.
5. Conclusions
The work undertaken here has identified a highly potent novel drug combination of DOX and GEM that demonstrates synergistic cytotoxic efficacy in patient derived GBM cells. The focus was on in vitro testing of invasive margin GBM cells that commonly remain post-surgery and form the basis of disease recurrence. Subsequent drug combination loading into a hydrogel-based drug delivery reservoir was achieved and maintenance of in vitro synergy demonstrated. Collectively, the data demonstrate that the reported DOX:GEM combination hydrogels hold promise for post-surgical treatment of GBM and warrant further investigation in in vivo surgical resection models amenable for interstitial delivery.
Data availability
The data supporting this article have been included as part of the ESI.† All other relevant data are available on request via the authors at pazrjc@exmail.nottingham.ac.uk, oas23@cam.ac.uk and ruman.rahman@nottingham.ac.uk.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC)-UKRI [Grant Numbers EP/S009000/1, EP/V049291/1]. This work was also funded by the Royal Society [Wolfson Research Merit Award WM150086] to C. A., and a philanthropic donation by Michael Hodgkinson to R. R. The authors thank the Nanoscale and Microscale Research Centre (nmRC) for providing access to instrumentation, Paul Cooling, Tom Hyde and Esme Ireson for expert technical help, and Sudha Cotterill and Carol Turrill for excellent administrative support.References
- Y. Zhou, W. J. Wu, H. Y. Bi, D. Y. Yang and C. Z. Zhang, Cancer Lett., 2020, 475, 79–91 CrossRef CAS.
- M. E. Davis, Clin. J. Oncol. Nurs., 2016, 20, S2–S8 CrossRef PubMed.
- W. M. Pardridge, Drug Discovery Today, 2007, 12, 54–61 CrossRef CAS PubMed.
- S. J. Smith, M. Diksin, S. Chhaya, S. Sairam, M. A. Estevez-Cebrero and R. Rahman, Int. J. Mol. Sci., 2017, 18, 2452–2452 CrossRef PubMed.
- A. Salmaggi, A. Boiardi, M. Gelati, A. Russo, C. Calatozzolo, E. Ciusani, F. L. Sciacca, A. Ottolina, E. A. Parati, C. La Porta, G. Alessandri, C. Marras, D. Croci and M. De Rossi, Glia, 2006, 54, 850–860 CrossRef PubMed.
- G. D. Cha, T. Kang, S. Baik, D. Kim, S. H. Choi, T. Hyeon and D. H. Kim, J. Controlled Release, 2020, 328, 350–367 CrossRef CAS.
- C. Bastiancich, A. Malfanti, V. Preat and R. Rahman, Adv. Drug Delivery Rev., 2021, 177, 113951 CrossRef CAS PubMed.
- P. McCrorie, C. E. Vasey, S. J. Smith, M. Marlow, C. Alexander and R. Rahman, J. Controlled Release, 2020, 328, 917–931 CrossRef CAS PubMed.
- M. Alghamdi, M. Gumbleton and B. Newland, Biomater. Sci., 2021, 9, 6037–6051 RSC.
- C. Bastiancich, E. Bozzato, I. Henley and B. Newland, J. Controlled Release, 2021, 337, 296–305 CrossRef.
- A. R. Anderson and T. Segura, Adv. Mater. Interfaces, 2020, 7, 2001055 CrossRef.
- A. Turek, K. Stoklosa, A. Borecka, M. Paul-Samojedny, B. Kaczmarczyk, A. Marcinkowski and J. Kasperczyk, Pharm. Res., 2020, 37, 90 CrossRef.
- D. Han, R. Serra, N. Gorelick, U. Fatima, C. G. Eberhart, H. Brem, B. Tyler and A. J. Steckl, Sci. Rep., 2019, 9, 17936 CrossRef PubMed.
- L. S. Ashby, K. A. Smith and B. Stea, World J. Surg. Oncol., 2016, 14, 225 CrossRef.
- H. C. Lawson, P. Sampath, E. Bohan, M. C. Park, N. Hussain, A. Olivi, J. Weingart, L. Kleinberg and H. Brem, J. Neuro-Oncol., 2007, 83, 61–70 CrossRef PubMed.
- E. Bozzato, N. Tsakiris, A. Paquot, G. G. Muccioli, C. Bastiancich and V. Preat, Int. J. Pharm., 2022, 628, 122341 CrossRef PubMed.
- S. H. Chen, Q. J. Qiu, D. D. Wang, D. J. She, B. Yin, G. L. Gu, M. H. Chai, D. N. Heo, H. N. He and J. X. Wang, J. Controlled Release, 2022, 349, 565–579 CrossRef CAS.
- M. Bouche, Y. C. Dong, S. Sheikh, K. Taing, D. Saxena, J. C. Hsu, M. H. Chen, R. D. Salinas, H. J. Song, J. A. Burdick, J. Dorsey and D. P. Cormode, ACS Biomater. Sci. Eng., 2021, 7, 3209–3220 CrossRef CAS PubMed.
- M. Norouzi, J. Firouzi, N. Sodeifi, M. Ebrahimi and D. W. Miller, Int. J. Pharm., 2021, 598, 120316 CrossRef CAS.
- G. Brachi, J. Ruiz-Ramirez, P. Dogra, Z. H. Wang, V. Cristini, G. Ciardelli, R. C. Rostomily, M. Ferrari, A. M. Mikheev, E. Blanco and C. Mattu, Nanoscale, 2020, 12, 23838–23850 RSC.
- M. C. Garrett, T. M. O'Shea, A. L. Wollenberg, A. M. Bernstein, D. Hung, B. Staarman, H. Soto, T. J. Deming, M. V. Sofroniew and H. I. Kornblum, PLoS One, 2020, 15, e0219632 CrossRef CAS.
- P. Schiapparelli, P. C. Zhang, M. Lara-Velazquez, H. Guerrero-Cazares, R. Lin, H. Su, R. W. Chakroun, M. Tusa, A. Quinones-Hinojosa and H. G. Cui, J. Controlled Release, 2020, 319, 311–321 CrossRef CAS PubMed.
- M. Zhao, E. Bozzato, N. Joudiou, S. Ghiassinejad, F. Danhier, B. Gallez and V. Préat, J. Controlled Release, 2019, 309, 72–81 CrossRef CAS PubMed.
- T. Johanssen, L. McVeigh, S. Erridge, G. Higgins, J. Straehla, M. Frame, T. Aittokallio, N. O. Carragher and D. Ebner, Front. Oncol., 2023, 12, 1075559 CrossRef.
- M. N. Zhao, D. van Straten, M. L. D. Broekman, V. Preat and R. M. Schiffelers, Theranostics, 2020, 10, 1355–1372 CrossRef CAS.
- S. K. Gupta, S. H. Kizilbash, B. L. Carlson, A. C. Mladek, F. Boakye-Agyeman, K. K. Bakken, J. L. Pokorny, M. A. Schroeder, P. A. Decker, L. Cen, J. E. Eckel-Passow, G. Sarkar, K. V. Ballman, J. M. Reid, R. B. Jenkins, R. G. Verhaak, E. P. Sulman, G. J. Kitange and J. N. Sarkaria, J. Natl. Cancer Inst., 2016, 108, djv369 CrossRef PubMed.
- ClinicalTrials.gov identifier: NCT02152982, Updated May 17, 2024, https://clinicaltrials.gov/study/NCT02152982.
- X. Ren, D. Ai, T. Li, L. Xia and L. Sun, Front. Neurol., 2020, 11, 603947 CrossRef.
- C. C. Parkins, J. H. McAbee, L. Ruff, A. Wendler, R. Mair, R. J. Gilbertson, C. Watts and O. A. Scherman, Biomaterials, 2021, 276, 120919 CrossRef CAS.
- E. A. Appel, M. J. Rowland, X. J. Loh, R. M. Heywood, C. Watts and O. A. Scherman, Chem. Commun., 2012, 48, 9843–9845 RSC.
- A. Tabet, M. P. Jensen, C. C. Parkins, P. G. Patil, C. Watts and O. A. Scherman, Adv. Healthcare Mater., 2019, 8, e1801391 CrossRef.
- M. J. Rowland, M. Atgie, D. Hoogland and O. A. Scherman, Biomacromolecules, 2015, 16, 2436–2443 CrossRef CAS PubMed.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS.
- M. J. Rowland, C. C. Parkins, J. H. McAbee, A. K. Kolb, R. Hein, X. J. Loh, C. Watts and O. A. Scherman, Biomaterials, 2018, 179, 199–208 CrossRef CAS.
- C. C. Parkins, J. H. McAbee, L. Ruff, A. Wendler, R. Mair, R. J. Gilbertson, C. Watts and O. A. Scherman, Biomaterials, 2021, 276, 120919 CrossRef CAS.
- J. Kim, I. S. Jung, S. Y. Kim, E. Lee, J. K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS.
- A. Day, A. P. Arnold, R. J. Blanch and B. Snushall, J. Org. Chem., 2001, 66, 8094–8100 CrossRef CAS PubMed.
- A. S. Mikhail, S. Eetezadi and C. Allen, PLoS One, 2013, 8, e62630 CrossRef CAS.
- R. Bayat Mokhtari, T. S. Homayouni, N. Baluch, E. Morgatskaya, S. Kumar, B. Das and H. Yeger, Oncotarget, 2017, 8, 38022–38043 CrossRef.
- T. C. Chou, Cancer Res., 2010, 70, 440–446 CrossRef CAS.
- T. C. Chou, Pharmacol. Rev., 2006, 58, 621–681 CrossRef CAS.
- C. F. Thorn, C. Oshiro, S. Marsh, T. Hernandez-Boussard, H. McLeod, T. E. Klein and R. B. Altman, Pharmacogenet. Genomics, 2011, 21, 440–446 CrossRef CAS PubMed.
- R. M. Jones, P. Kotsantis, G. S. Stewart, P. Groth and E. Petermann, Mol. Cancer Ther., 2014, 13, 2412–2421 CrossRef CAS PubMed.
- B. K. Slinker, J. Mol. Cell Cardiol., 1998, 30, 723–731 CrossRef CAS PubMed.
- S. J. Smith, M. Diksin, S. Chhaya, S. Sairam, M. A. Estevez-Cebrero and R. Rahman, Int. J. Mol. Sci., 2017, 18, 2452 CrossRef PubMed.
- S. L. Perrin, M. S. Samuel, B. Koszyca, M. P. Brown, L. M. Ebert, M. Oksdath and G. A. Gomez, Biochem. Soc. Trans., 2019, 47, 625–638 CrossRef CAS PubMed.
- E. Skaga, E. Kulesskiy, A. Fayzullin, C. J. Sandberg, S. Potdar, A. Kyttala, I. A. Langmoen, A. Laakso, E. Gaal-Paavola, M. Perola, K. Wennerberg and E. O. Vik-Mo, BMC Cancer, 2019, 19, 628 CrossRef CAS PubMed.
- D. Nehama, A. S. Woodell, S. M. Maingi, S. D. Hingtgen and G. Dotti, Neuro-Oncol., 2023, 25, 1551–1562 CrossRef CAS.
- L. A. Lipscomb, M. E. Peek, F. X. Zhou, J. A. Bertrand, D. VanDerveer and L. D. Williams, Biochemistry, 1994, 33, 3649–3659 CrossRef CAS.
- M. Songbo, H. Lang, C. Xinyong, X. Bin, Z. Ping and S. Liang, Toxicol. Lett., 2019, 307, 41–48 CrossRef CAS PubMed.
- H. J. Shin, H. K. Kwon, J. H. Lee, M. A. Anwar and S. Choi, Sci. Rep., 2016, 6, 34064 CrossRef CAS PubMed.
- Y. Li, X. Zhang, Z. Wang, B. Li and H. Zhu, Front. Pharmacol., 2023, 14, 1156538 CrossRef CAS PubMed.
- P. Huang, S. Chubb, L. W. Hertel, G. B. Grindey and W. Plunkett, Cancer Res., 1991, 51, 6110–6117 CAS.
- V. Heinemann, Y. Z. Xu, S. Chubb, A. Sen, L. W. Hertel, G. B. Grindey and W. Plunkett, Mol. Pharmacol., 1990, 38, 567–572 CAS.
- M. A. Moufarij, D. R. Phillips and C. Cullinane, Mol. Pharmacol., 2003, 63, 862–869 CrossRef CAS PubMed.
- M. Crul, R. C. van Waardenburg, S. Bocxe, M. A. van Eijndhoven, D. Pluim, J. H. Beijnen and J. H. Schellens, Biochem. Pharmacol., 2003, 65, 275–282 CrossRef CAS PubMed.
- K. Petrecca, M. C. Guiot, V. Panet-Raymond and L. Souhami, J. Neurooncol., 2013, 111, 19–23 CrossRef PubMed.
- E. S. Newlands, M. F. Stevens, S. R. Wedge, R. T. Wheelhouse and C. Brock, Cancer Treat. Rev., 1997, 23, 35–61 CrossRef CAS PubMed.
- S. Ostermann, C. Csajka, T. Buclin, S. Leyvraz, F. Lejeune, L. A. Decosterd and R. Stupp, Clin. Cancer Res., 2004, 10, 3728–3736 CrossRef CAS.
- T. Simunek, M. Sterba, O. Popelova, M. Adamcova, R. Hrdina and V. Gersl, Pharmacol. Rep., 2009, 61, 154–171 CrossRef CAS PubMed.
- D. B. Sawyer, N. Engl. J. Med., 2013, 368, 1154–1156 CrossRef CAS PubMed.
- P. K. Singal and N. Iliskovic, N. Engl. J. Med., 1998, 339, 900–905 CrossRef CAS PubMed.
- A. M. Al-Abd, K. Y. Hong, S. C. Song and H. J. Kuh, J. Controlled Release, 2010, 142, 101–107 CrossRef CAS.
- G. Cinar, A. Ozdemir, S. Hamsici, G. Gunay, A. Dana, A. B. Tekinay and M. O. Guler, Biomater. Sci., 2016, 5, 67–76 RSC.
- W. Wu, H. Chen, F. Shan, J. Zhou, X. Sun, L. Zhang and T. Gong, Mol. Pharm., 2014, 11, 3378–3385 CrossRef CAS.
- D. Ghosh, S. Nandi and S. Bhattacharjee, Clin. Transl. Med., 2018, 7, 33 CrossRef PubMed.
- M. A. Qazi, P. Vora, C. Venugopal, S. S. Sidhu, J. Moffat, C. Swanton and S. K. Singh, Ann. Oncol., 2017, 28, 1448–1456 CrossRef CAS.
- A. Sottoriva, I. Spiteri, S. G. Piccirillo, A. Touloumis, V. P. Collins, J. C. Marioni, C. Curtis, C. Watts and S. Tavare, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4009–4014 CrossRef CAS.
- A. Dicko, L. D. Mayer and P. G. Tardi, Expert Opin. Drug Delivery, 2010, 7, 1329–1341 CrossRef CAS.
- S. Eetezadi, J. C. Evans, Y. T. Shen, R. De Souza, M. Piquette-Miller and C. Allen, Mol. Pharm., 2018, 15, 472–485 CrossRef CAS.
- R. J. Cavanagh, P. F. Monteiro, C. Moloney, A. Travanut, F. Mehradnia, V. Taresco, R. Rahman, S. G. Martin, A. M. Grabowska, M. B. Ashford and C. Alexander, Biomater. Sci., 2024, 12, 1822–1840 RSC.
- D. A. Gewirtz, Biochem. Pharmacol., 1999, 57, 727–741 CrossRef CAS PubMed.
- L. P. Swift, A. Rephaeli, A. Nudelman, D. R. Phillips and S. M. Cutts, Cancer Res., 2006, 66, 4863–4871 CrossRef CAS.
- R. A. Forrest, L. P. Swift, A. Rephaeli, A. Nudelman, K. Kimura, D. R. Phillips and S. M. Cutts, Biochem. Pharmacol., 2012, 83, 1602–1612 CrossRef CAS PubMed.
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef CAS PubMed.
- B. Ewald, D. Sampath and W. Plunkett, Mol. Cancer Ther., 2007, 6, 1239–1248 CrossRef CAS PubMed.
- D. R. Vogus, A. Pusuluri, R. Chen and S. Mitragotri, Bioeng. Transl. Med., 2018, 3, 49–57 CrossRef CAS PubMed.
- D. R. Vogus, M. A. Evans, A. Pusuluri, A. Barajas, M. Zhang, V. Krishnan, M. Nowak, S. Menegatti, M. E. Helgeson, T. M. Squires and S. Mitragotri, J. Controlled Release, 2017, 267, 191–202 CrossRef CAS.
- L. Crino, G. Scagliotti, M. Marangolo, F. Figoli, M. Clerici, F. De Marinis, F. Salvati, G. Cruciani, L. Dogliotti, F. Pucci, A. Paccagnella, V. Adamo, G. Altavilla, P. Incoronato, M. Trippetti, A. M. Mosconi, A. Santucci, S. Sorbolini, C. Oliva and M. Tonato, J. Clin. Oncol., 1997, 15, 297–303 CrossRef CAS PubMed.
- C. Louvet, T. Andre, G. Lledo, P. Hammel, H. Bleiberg, C. Bouleuc, E. Gamelin, M. Flesch, E. Cvitkovic and A. de Gramont, J. Clin. Oncol., 2002, 20, 1512–1518 CrossRef CAS PubMed.
- W. M. Ciszewski, M. Tavecchio, J. Dastych and N. J. Curtin, Breast Cancer Res. Treat., 2014, 143, 47–55 CrossRef CAS PubMed.
- Y. Zhao, H. D. Thomas, M. A. Batey, I. G. Cowell, C. J. Richardson, R. J. Griffin, A. H. Calvert, D. R. Newell, G. C. Smith and N. J. Curtin, Cancer Res., 2006, 66, 5354–5362 CrossRef CAS PubMed.
- J. Lai, H. Yang, Y. Zhu, M. Ruan, Y. Huang and Q. Zhang, BMC Cancer, 2019, 19, 602 CrossRef.
- M. Zimmermann, A. S. Arachchige-Don, M. S. Donaldson, R. F. Dallapiazza, C. E. Cowan and M. C. Horne, J. Biol. Chem., 2012, 287, 22838–22853 CrossRef CAS PubMed.
- Y. H. Ling, A. K. el-Naggar, W. Priebe and R. Perez-Soler, Mol. Pharmacol., 1996, 49, 832–841 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4pm00177j |
| This journal is © The Royal Society of Chemistry 2024 |