
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComparison of emulsion and spray methods for fabrication of rapamycin-loaded acetalated dextran microparticles†
Stephen A.
Ehrenzeller
a,
Nicole Rose
Lukesh
 a,
Rebeca T.
Stiepel
a,
Denzel D.
Middleton
a,
Steven M.
Nuzzolo
a,
Aliyah J.
Tate
a,
Cole J.
Batty
a,
Eric M.
Bachelder
a and
Kristy M.
Ainslie
*abc
a,
Rebeca T.
Stiepel
a,
Denzel D.
Middleton
a,
Steven M.
Nuzzolo
a,
Aliyah J.
Tate
a,
Cole J.
Batty
a,
Eric M.
Bachelder
a and
Kristy M.
Ainslie
*abc
aDivision of Pharmacoengineering and Molecular Pharmaceutics, Eshelman School of Pharmacy, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, USA. E-mail: ainsliek@email.unc.edu
bJoint Department of Biomedical Engineering, University of North Carolina at Chapel Hill and North Carolina State University, Chapel Hill, North Carolina, USA
cDepartment of Microbiology & Immunology, UNC School of Medicine, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina, USA
First published on 8th July 2024
Abstract
Rapamycin (rapa), an immunosuppressive medication, has demonstrated considerable effectiveness in reducing organ transplant rejection and treating select autoimmune diseases. However, the standard oral administration of rapa results in poor bioavailability, broad biodistribution, and harmful off-target effects, necessitating improved drug delivery formulations. Polymeric microparticles (MPs) are one such solution and have demonstrated promise in pre-clinical studies to improve the therapeutic efficacy of rapa. Nevertheless, MP formulations are highly diverse, and fabrication method selection is a critical consideration in formulation design. Herein, we compared common fabrication processes for the development of rapa-loaded MPs. Using the biopolymer acetalated dextran (Ace-DEX), rapa-loaded MPs were fabricated by both emulsion (homogenization and sonication) and spray (electrospray and spray drying) methods, and resultant MPs were characterized for size, morphology, surface charge, and drug release kinetics. MPs were then screened in LPS-stimulated macrophages to gauge immunosuppressive efficacy relative to soluble drug. We determined that homogenized MPs possessed the most optimal combination of sizing, tunable drug release kinetics, and immunosuppressive efficacy, and we subsequently demonstrated that these characteristics were maintained across a range of potential rapa loadings. Further, we performed in vivo trafficking studies to evaluate depot kinetics and cellular uptake at the injection site after subcutaneous injection of homogenized MPs. We observed preferential MP uptake by dendritic cells at the depot, highlighting the potential for MPs to direct more targeted drug delivery. Our results emphasize the significance of fabrication method in modulating the efficacy of MP systems and inform improved formulation design for the delivery of rapa.
Introduction
Rapamycin (Rapa) is a macrolide-class inhibitor of the mammalian target of rapamycin (mTOR), a critical regulator of cell growth and metabolism.1 It has been extensively studied as an immunosuppressant for renal transplant patients2 and as an anti-proliferative in drug-eluting stents for coronary interventions.3,4 Furthermore, rapa has recently been employed as a therapeutic for autoimmune cytopenia in several clinical trials5,6 and has shown substantial promise in pre-clinical work to expand regulatory T cell (Treg) populations in patients with autoimmune disease.7,8 However, despite its potential to treat various inflammatory conditions, rapa suffers from poor bioavailability, broad biodistribution, and a relatively narrow therapeutic window under current modes of administration (e.g., oral suspensions and tablets) – significantly impairing therapeutic efficacy.9 Moreover, given the rather ubiquitous role of mTOR in mammalian cell metabolism, substantial off-target effects are associated with prolonged systemic rapa treatment, including new-onset metabolic dysfunction and increased susceptibility to infection.10 Thus, innovative formulation strategies are required to maximize the therapeutic potential of rapa.Polymeric microparticles (MPs) are well-reported drug delivery systems that can ameliorate off-target effects of systemic drug administration, enhance drug efficacy, and improve drug delivery to target tissues and cells via several mechanisms. Namely, by encapsulation of drug cargo in a polymeric matrix, polymeric MPs shield encapsulated drug from premature metabolism and allow for controlled release of drug via diffusion and polymer degradation, limiting systemic drug release and often improving pharmacokinetics.11 Furthermore, MPs can passively target phagocytic cell populations via sizing, as non-phagocytic cells cannot engulf particulates greater than 0.2 μm in diameter and phagocytic cells exhibit preferential uptake of particulates ranging from 0.5 to >3 μm in diameter.12–14 For immunosuppressive agents such as rapa, this permits localized drug release to phagocytic antigen presenting cells (APCs) such as dendritic cells (DCs) and macrophages, which directly regulate both local and global immune responses. Indeed, previous research has demonstrated improved immunosuppressive efficacy of rapa-loaded poly(lactic-co-glycolic acid) (PLGA) MPs compared to soluble drug in DCs in vitro.15,16 Additionally, in vivo administration of MPs co-encapsulating rapa and disease relevant antigens has shown significant therapeutic efficacy in several murine models of autoimmunity, including experimental autoimmune encephalomyelitis (EAE),17,18 vitiligo,19 and type 1 diabetes.20,21
Despite recent advances, polymeric MP systems are relatively understudied with respect to comparative analysis of fabrication methods and parameters. Considerations of size, morphology, surface charge, and encapsulate are critical for developing polymeric MP formulations, and selection of fabrication process directly determines these parameters. Perhaps the most widely utilized polymeric MP fabrication methods include bulk emulsification methods such as homogenization and sonication. In these fabrication protocols, shear (i.e., homogenization) or pulsatile (i.e., sonication) forces are used to emulsify drug–polymer droplets into a continuous aqueous phase, and subsequent solvent evaporation facilitates droplet hardening into MPs (Fig. 1A). Although versatile with respect to potential encapsulates and MP architectures, bulk emulsion-based approaches are limited by scalability and batch-to-batch variability.22 Additional common fabrication methods include spray-based approaches, such as electrohydrodynamic atomization (i.e., electrospraying) and spray drying, which create MPs via aerosolization of drug–polymer solutions and co-current solvent evaporation to facilitate MP hardening. In electrospraying, aerosolization of the drug-polymer solution is performed via ionization (Fig. 1B). At the nozzle tip a Taylor cone is formed, and electrostatic repulsion results in small, charged droplets, and an oppositely charged collection plate serves as an attachment substrate for newly formed MPs following solvent evaporation.23 In spray drying, aerosolization is achieved through a pressure nozzle, and drug–polymer solutions are sprayed into a heated chamber to accelerate solvent evaporation and MP formation (Fig. 1C). Compared to bulk-emulsion approaches, spray-based approaches are highly scalable yet are rather limited by accessibility and solvent compatibilities.24,25 Furthermore, cargo denaturation or inactivation is a significant concern for spray drying, as the drug-polymer solution is sprayed into a heated chamber to facilitate MP hardening. Given the range of potential fabrication methods of polymeric MPs and their potential differential efficacies in drug delivery, a comprehensive comparison among both spray and emulsion-based techniques for MP fabrication is necessary.
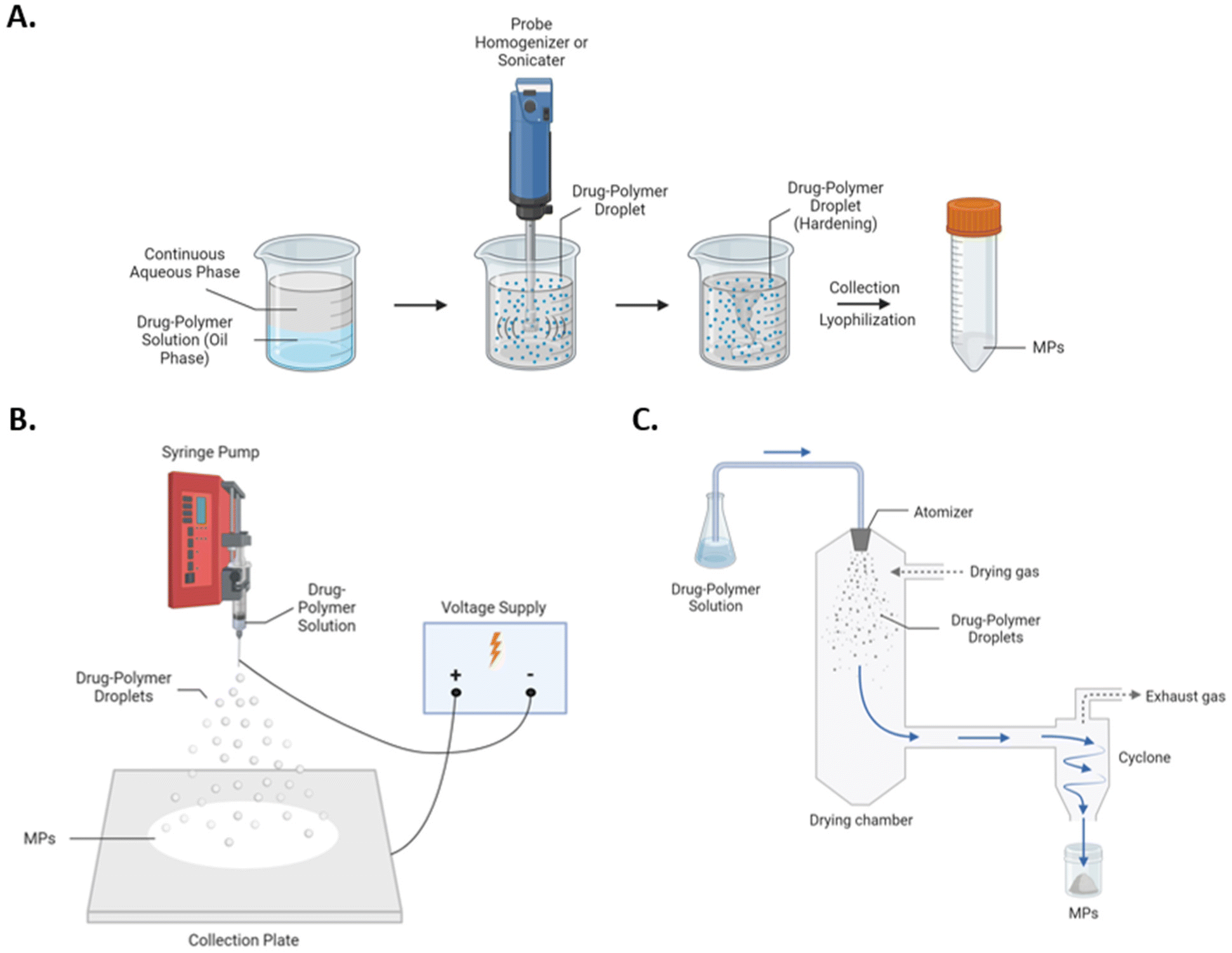 | ||
| Fig. 1 Schematics of (A) homogenization and sonication, (B) electrospray, and (C) spray drying for the fabrication of polymeric MPs. | ||
Herein, emulsion and spray-based approaches to MP fabrication are compared for the delivery of rapa using acetalated dextran (Ace-DEX), a biopolymer synthesized from the FDA-approved polysaccharide dextran. Compared to more traditional polymers used in MP fabrication such as PLGA and other polyesters, Ace-DEX is advantageous with respect to biocompatibility and degradation kinetics. More specifically, the degradation of PLGA generates acidic byproducts, which can lower local pH, result in local toxicities, and contribute to inflammation.26–28 Moreover, although PLGA possesses tunable degradation rates depending on the molar ratios of lactic and glycolic acids within the polymer chain, PLGA polymers largely degrade on the order of weeks to months, limiting possible therapeutic applications.29 In comparison, Ace-DEX degrades into well-tolerated, pH-neutral byproducts, including dextran and trace quantities of acetone and ethanol. Furthermore, Ace-DEX possesses tunable degradation rates ranging from hours to months, enabling a broad range of drug release kinetics and potential therapeutic applications. Degradation rates are tightly controlled by synthesis reaction time, wherein continued reaction favors increased coverage of stable cyclic acetals (i.e., cyclic acetal coverage, or CAC) and, in turn, a slower degrading polymer. Ace-DEX is also acid-sensitive. In the context of MP platforms to passively target phagocytic immune cells, Ace-DEX will rapidly degrade in the acidic endosomal environment, resulting in more targeted drug delivery.30 Given these characteristics, Ace-DEX is an ideal platform for the delivery of immunosuppressive agents such as rapa. Indeed, Ace-DEX MPs co-encapsulating rapa with disease relevant antigens have shown significant efficacy in murine models of multiple sclerosis17 and type 1 diabetes.20
In the present study, rapa-loaded Ace-DEX MPs were fabricated by both emulsion (homogenization and sonication) and spray-based (spray drying and electrospray) methods and evaluated for physiochemical characteristics, rapa encapsulation efficiency (EE), rapa release kinetics, and immunosuppressive efficacy in macrophages to inform how fabrication method impacts relevant drug delivery parameters. Trafficking studies were performed to understand depot trafficking of subcutaneously administered Ace-DEX MPs for the optimized formulation. Overall, this work sought to determine the optimal fabrication method and loading parameters for rapa-loaded Ace-DEX MPs, thereby informing future formulation design.
Methods
All chemicals were acquired from Sigma Aldrich (St Louis, MO) unless otherwise specified. All consumables (e.g., plastics), biologics, and assays were acquired from Thermo Fisher Scientific (Waltham, MA) unless otherwise specified. Rapamycin was purchased from LC Laboratories (Woburn, MA).Ace-DEX synthesis
Acetalated dextran (Ace-DEX) was synthesized as previously described.31 Dextran (71 kDa) was first dissolved in dimethyl sulfoxide (DMSO) with the acid-catalyst pyridinium p-toluenesulfonate (PPTS). The acetalation reaction was initiated via addition of 2-ethoxypropene (Matrix Scientific; Columbia, SC) under anhydrous conditions. Reactions were quenched at 4, 10, and 135 minutes via addition of triethylamine (TEA) to yield 20%, 40%, and 60% CAC Ace-DEX, respectively. Following reaction quenching, Ace-DEX was precipitated in basic water (0.04% v/v TEA in water), washed, and placed on a rotary evaporator to remove residual organic solvent. The polymer was then frozen and lyophilized overnight. The next day, the Ace-DEX was dissolved in ethanol, centrifuged to remove impurities, and precipitated in basic water. The Ace-DEX was then rotovapped, frozen, lyophilized, and stored at −20 °C until use. CAC was estimated using Inova 400 MHz NMR, as previously reported.31 All glassware was soaked in a 0.1 M sodium hydroxide solution prior to use to remove endotoxin.Alexa-DEX synthesis
Dextran was dissolved in borate buffer (pH = 8.5) and heated to 70 °C. Diaminobutane was added at 7.5 wt%, sodium sulfate was added at 30 wt%, and sodium cyanoborohydride was added at 1.2 wt%. The resulting solution was reacted for 5 h in a reaction microwave at 70 °C (delta = 5) using SPS mode at 100 W (CEM Discover Microwave Synthesizer; CEM Corporation; Mattews, NC),32 resulting in dextran with a terminal amine, which we termed “DAB-DEX” (Fig. S1A†). DAB-DEX was frozen and lyophilized for 2 days. The extent of reaction is measured using a bicinchoninic acid assay (BCA) assay. Using EDC-NHS chemistry,33 DAB-DEX was then reacted with Alexa Fluor 647-NHS-ester (Thermo Fisher) for 2 h in 0.1 M MES (4-(20sulfonatoehyl)morpholin-4-ium) buffer (pH = 6) to form “Alexa-DEX”, dextran with a fluorescent terminus (Fig. S1B†). Alexa-DEX was frozen then lyophilized for 2 days and stored at −20 °C until use. We then acetalated Alexa-DEX with a 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 wt mixture of Alexa-DEX:dextran using the same conditions and characterization for 60 CAC above to form Alexa-DEX polymer.
:80 wt mixture of Alexa-DEX:dextran using the same conditions and characterization for 60 CAC above to form Alexa-DEX polymer.
Homogenized particle fabrication
Blank and rapa-loaded homogenized MPs were prepared from 20, 40, and 60 CAC Ace-DEX via a water-in-oil-in-water (W/O/W) emulsion method adapted from a previously reported method (Fig. 1A).34 Blank Alexa-DEX MPs were fabricated from 60 CAC Alexa-DEX by the same method. In short, Ace-DEX or Alexa-DEX was first dissolved in dichloromethane (DCM), with or without rapa (0.5%, 1%, 3%, and 5% wt/wt). A small volume of phosphate buffered saline (PBS) was added to form the primary emulsion, which was probe-homogenized at 18000 rpm for 30 seconds (IKA T25 Digital Ultra-Turrax; Cole Parmer; Vernon Hills, IL). Then, 3% polyvinyl alcohol (PVA) in PBS was added to the primary emulsion to create a secondary emulsion, which was subsequently probe-homogenized at 18000 rpm for 30 seconds. The secondary emulsion was then stirred in 0.3% PVA in PBS solution for 2 hours to facilitate evaporation of the DCM and hardening of the MPs. The MPs were then collected, washed with basic water, frozen, lyophilized, and stored at −20 °C prior to use. All glassware was soaked in a 0.1 M sodium hydroxide solution prior to use to remove endotoxin.
Sonicated particle fabrication
Blank and rapa-loaded sonicated MPs were prepared from 20, 40, and 60 CAC Ace-DEX via an oil-in-water (O/W) emulsion method similar to the homogenized MPs (Fig. 1A). Ace-DEX was first dissolved in DCM, with or without rapa (1% wt/wt). To form the primary emulsion, 3% PVA in PBS solution was added to the drug–polymer solution, and the mixture was then probe-sonicated at 40% Amps for 30 seconds (1 second on, 1 second off) (Q500 Sonicator; QSonica Sonicators; Newtown, CT). The emulsion was then stirred in 0.3% PVA in PBS solution for 2 hours to facilitate evaporation of the DCM and hardening of the MPs. The resulting MPs were collected, washed with basic water, frozen, lyophilized, and stored at −20 °C prior to use. All glassware was soaked in a 0.1 M sodium hydroxide solution prior to use to remove endotoxin.Electrosprayed particle fabrication
Blank and rapa-loaded electrosprayed MPs were prepared from 20, 40, and 60 CAC Ace-DEX by a monoaxial electrospray method (Fig. 1B). Ace-DEX was first dissolved in absolute ethanol to a final concentration of 20 mg mL−1, with or without rapa (2% wt/wt). The drug-polymer solution was loaded into a 2.5 mL glass syringe equipped with a 20-gauge stainless-steel blunt needle (Hamilton Company; Reno, NV). The syringe was then mounted on a syringe pump (Harvard Apparatus; Holliston, MA) and positioned vertically above a stainless-steel collection plate. The pump sprayed at a constant rate of 0.2 mL h−1, with a −5 kV potential applied to the needle and a +2.5 kV potential applied to the plate. The resulting MPs were collected, frozen, lyophilized, and stored at −20 °C until use. To ensure the removal of endotoxin prior to spraying, all glassware was soaked in a 0.1 M sodium hydroxide solution and the collection plate was heat-treated overnight.Spray dried particle fabrication
Blank and rapa-loaded spray dried MPs were prepared from 20, 40, and 60 CAC Ace-DEX using a B290 Mini Spray Dryer (BUCHI Corporation; New Castle, DE) equipped with a collection cyclone (Fig. 1C). Ace-DEX was first dissolved in absolute ethanol to a final concentration of 2.5 mg mL−1, with or without rapa (1% wt/wt). The drug-polymer solution was then fed into a pressure nozzle at 10 mL min−1 and an inlet temperature of 75 °C. The Q-flow was maintained at 60 mm. Following completion of the spray, the resultant MPs were collected and stored at −20 °C until use. To ensure removal of endotoxin prior to spraying, the collection cyclone was soaked in a 0.1 M sodium hydroxide solution overnight and the drying chamber was UV-sterilized.Characterization of MPs
Assessment of MP degradation
To measure blank MP degradation, MP suspensions were prepared in triplicate at 1 mg mL−1 in PBS (pH 7.4) and placed on a shaker plate set to 37 °C. Time points were collected at 0, 0.5, 2, 4, 8, 24, 48, 72, 96, 168, and 336 hours. At each time point, the MP suspensions were briefly vortexed, and an aliquot was taken and centrifuged at 21000g for 10 minutes. The supernatant was collected and stored at −80 °C prior to analysis. The dextran content of the supernatant was measured by a microplate BCA assay, following the manufacturer's protocol. BCA data was normalized to a fully degraded sample of each MP group, which was prepared by treating an MP suspension with high heat (80 °C) overnight.
Assessment of rapa release from MPs
To measure rapa release from the MPs, MP suspensions were prepared in triplicate at 1 mg mL−1 in PBS (pH 7.4) and placed on a shaker plate set to 37 °C. Time points were collected at 0, 0.5, 2, 4, 8, 24, 48, 72, 96, 168, and 336 hours. At each time point, the MP suspensions were briefly vortexed, and an aliquot was taken and centrifuged at 21000g for 10 minutes. The supernatant was separated from the pellet, and the pellet was frozen and lyophilized. Pellets were stored at −80 °C prior to analysis. Rapa content in the pellet was quantified by the HPLC method described above. Percent release was quantified by the decline in the rapa content of the pellet relative to the known rapa content of the MPs.
Cell culture and treatments
RAW 264.7 murine macrophages (ATCC; Manassas, VA) were cultured in Dulbecco's modified Eagle's medium (DMEM, Corning; Corning, NY) supplemented with 10% fetal bovine serum (FBS, Corning) and 1% penicillin–streptomycin solution (VWR; Radnor, PA). DC2.4 cells (ATCC; Manassas, VA) were cultured in RPMI 1640 Media (Corning) supplemented with 10% FBS and 1% penicillin–streptomycin solution. All cells were kept at 37 °C, 5% CO2, and 100% humidity for the duration of the experiments.To test for anti-inflammatory activity in macrophages, RAWs were seeded overnight at 2.5 × 104 cells per well in a tissue culture-treated 96-well plate. For DCs, DC2.4 cells were seeded overnight at 1.0 × 104 cells per well in a TC-treated 96-well plate. The next day, the cells were stimulated with 20 ng mL−1 lipopolysaccharide (LPS, Escherichia coli O111:B4) unless otherwise indicated. One hour after LPS treatment, the cells were treated with the indicated concentrations of soluble or encapsulated rapa and incubated for 24 hours. The cells were then centrifuged at 500g for 5 minutes, and supernatants were collected for lactose dehydrogenase assays (LDH, Invivogen; San Diego, CA) to measure cytotoxicity and TNF-α ELISA (Biolegend; San Diego, CA) to gauge inflammation. LDH assays were run immediately, but cell supernatants were stored at −80 °C until use in the ELISA.
In vivo trafficking study
The use of mice in the following study was conducted with the approval of the Institutional Animal Care and Use Committee (IACUC) at the University of North Carolina at Chapel Hill. 10–12-week-old Balb/c mice (Jackson Laboratory) were administered 2mgs of Alexa-DEX MPs in each subcutaneous flank. On days 0, 1, 2, 3, 4, 7, 10, 14, 17, 21, animals were imaged on an In Vivo Imaging System (IVIS Lumina, PerkinElmer Inc.; Walthman, MA) to track the fluorescent intensity (excitation 600 nm, emission 640 nm) of the subcutaneous depots formed by the MPs. On days 2, 7, 14, and 21, a cohort of animals were sacrificed, and their depots (n = 4 Alexa-DEX MP treated, n = 2 PBS) were harvested. Depots were processed into single-cell suspension using a previously reported method with some modifications.35 Briefly, approximately a 2 cm × 2 cm piece of skin with the attached depot is harvested upon animal euthanasia. The skin is washed in naked RPMI for 30 minutes at 37 °C on a shaker plate to remove loosely adhered debris. The skin is then minced using surgical scissors to about 2 mm × 2 mm pieces. These refined pieces are placed in a collagenase D digest containing 1 mg mL−1 collagenase D and 50 μg mL−1 DNase I in RPMI media with 10% heat-inactivated fetal bovine serum and 1% pen strep for 30 minutes at 37 °C on a shaker plate. After the digest, the skin pieces are passed through a 70 μm filter and mashed through the filter with the back of a 5 mL syringe. Once in single-cell suspension, depots were plated and stained for flow cytometry using a surface staining innate cell panel (Table S1†).Statistical analysis
Figures were prepared with GraphPad Prism 10. Statistics were generated via an ordinary two-way ANOVA with Dunnett's multiple comparisons test unless otherwise specified. A P-value of <0.05 was considered significant between groups.Results & discussion
Physiochemical characterization of Ace-DEX MPs
Blank and rapa-loaded Ace-DEX MPs were successfully fabricated via homogenization, sonication, electrospray, and spray drying. For each fabrication method, MPs were fabricated with 20, 40, and 60 CAC Ace-DEX to mimic fast, moderate, and slow-degrading MPs, respectively.36–38 In keeping with these different fabrication processes, each method yielded MPs with distinct physiochemical characteristics (Fig. 2 and Table 1). | ||
| Fig. 2 Representative scanning electron micrographs of (A) homogenized (60CAC 1% rapa MPs), (B) sonicated (20CAC blank MPs), (C) electrosprayed (60CAC 1% rapa MPs) and (D) spray dried (60CAC 1% rapa MPs) Ace-DEX MPs. | ||
| Fabrication method | Theoretical loading | CAC (%) | Diameter (nm) | Zeta potential (mV) |
|---|---|---|---|---|
| Homogenization | Blank | 20 | 981 ± 415 | −10.3 ± 1.9 |
| 40 | 543 ± 221 | −13.5 ± 0.5 | ||
| 60 | 385 ± 175 | −5.5 ± 3.6 | ||
| 1% w/w | 20 | 654 ± 221 | −7.3 ± 5.3 | |
| 40 | 353 ± 216 | −7.1 ± 3.3 | ||
| 60 | 318 ± 155 | −6.0 ± 4.3 | ||
| Sonication | Blank | 20 | 93 ± 22 | — |
| 40 | 64 ± 16 | — | ||
| 60 | 64 ± 15 | — | ||
| 1% w/w | 20 | 90 ± 17 | — | |
| 40 | 67 ± 17 | — | ||
| 60 | 61 ± 16 | — | ||
| Electrospray | Blank | 20 | 1150 ± 206 | −6.3 ± 0.5 |
| 40 | 802 ± 114 | −12.0 ± 0.2 | ||
| 60 | 1136 ± 178 | −10.6 ± 0.9 | ||
| 2% w/w | 20 | 1157 ± 191 | −11.1 ± 2.5 | |
| 40 | 1120 ± 209 | −11.8 ± 0.6 | ||
| 60 | 1216 ± 140 | −15.1 ± 1.4 | ||
| Spray Dry | Blank | 20 | 914 ± 316 | −15.4 ± 3.6 |
| 40 | 1085 ± 382 | −14.7 ± 4.2 | ||
| 60 | 1183 ± 287 | −17.9 ± 0.7 | ||
| 1% w/w | 20 | 753 ± 257 | −15.7 ± 2.6 | |
| 40 | 918 ± 246 | −17.9 ± 0.7 | ||
| 60 | 1255 ± 490 | −10.6 ± 0.9 |
MP diameter and morphology were first evaluated via SEM followed by ImageJ analysis (Fig. S2†), as non-spherical MP morphologies and MP aggregation complicated dynamic light scattering analysis. Thus, the measured diameters are representative of dehydrated MPs, which may be slightly lower than diameters observed in solution.39 Emulsion-based methods – homogenization (Fig. 2A) and sonication (Fig. 2B) – resulted in spherical MPs. Homogenized MPs were fairly polydisperse, with batches ranging in diameter from 318 ± 155 nm to 981 ± 415 nm (Table 1). In contrast, sonicated MPs exhibited a higher degree of monodispersity at a much smaller scale, with all batches possessing average diameters below 100 nm (Table 1). Notably, however, sonicated MPs were prone to aggregation, often forming micron-scale clusters in suspension despite extensive measures to generate a single-MP suspension (data not shown). Spray-based methods – electrospraying (Fig. 2C) and spray drying (Fig. 2D) – yielded MPs with a collapsed morphology, consistent with rapid solvent evaporation during the spray process (i.e., deflation).23 Both fabrication methods yielded MPs approximately 1 μm in diameter (Table 1), but spray dried MPs were more polydisperse and amorphic compared to electrosprayed MPs. Broadly, these data suggest that homogenized, electrosprayed, and spray dried MPs are most optimal for passive targeting of phagocytic cells, given that phagocytic cells preferentially phagocytose materials ranging from roughly 0.5 μm to >3 μm in diameter.12,14 In contrast, sonicated MPs may traffic in a more non-specific manner, as non-phagocytic cells are capable of engulfing materials up to 200 nm in diameter via endocytosis.13,14 However, it is important to note that these data are representative of a single fabrication protocol, and optimization of fabrication parameters might yield more favorable MP sizes and morphologies. For each fabrication method, no dramatic morphological or sizing differences were observed between blank and rapa-loaded MPs or MPs of different CAC (Table 1). Considering the potential for Ace-DEX MPs to act as a tunable release platform, it is beneficial that similarly sized MP batches can be generated despite differing polymer characteristics.
In addition to sizing and morphology, MP surface charge (i.e., zeta potential) was measured via ELS, as MP surface charge is a key determinant of MP-cell and MP-environment interactions. For example, cationic MPs have been reported to activate phagocytic immune cells and are considered to traffic more non-specifically than anionic MPs, although these phenomena are heavily dependent on the magnitude of the charge.40 Furthermore, cationic MPs have exhibited accelerated clearance in vivo compared to neutral and anionic MPs.41 For these reasons, MPs with a slight negative charge are preferred for the delivery of rapa.
In accordance, all MP batches bore slight negative charges (Table 1), indicating that Ace-DEX MPs provide an ideal carrier system for rapa. Most notably, spray-based methods imparted slightly more negative surface charge on resultant MP batches compared to emulsion-based methods, likely due to rapid solvent evaporation concentrating charge at the MP surface. Nevertheless, the differences were fairly marginal: the MP surface charge of spray dried MP batches ranged from −10.6 ± 0.9 to −17.9 ± 0.7 mV; electrosprayed, −6.3 ± 0.5 to −15.1 ± 1.4 mV; and homogenized, −6.0 ± 4.3 to −13.5 ± 0.5 mV (Table 1). Sonicated MPs were excluded from ELS analysis as MP aggregation skewed instrument measurement of surface charge, but previous reports have indicated a comparable negative charge for sonicated Ace-DEX MPs.42 Similar to the size analysis, neither the presence of rapa nor CAC appeared to trend with MP surface charge. Once again, these results demonstrate that Ace-DEX itself is a fairly robust carrier, with fabrication method seeming to impart the most substantial differences in sizing and surface charge for Ace-DEX MPs.
Rapa encapsulation and release by Ace-DEX MPs
Rapa is a hydrophobic (logP ∼6.0) small molecule, lending itself useful for organic solvent systems and effective incorporation into a polymeric matrix. As such, rapa was fairly well-encapsulated in Ace-DEX MPs across the four encapsulation methods (Table 2). Spray drying resulted in the most EEs, reliably at or around 100% across all CACs. Some MP batches boasted EEs in excess of 100%, which may be attributable to polymer loss during the fabrication process. Homogenization and sonication also resulted in near full rapa encapsulation, although these methods demonstrated slightly higher batch-to-batch variability compared to spray drying. Surprisingly, electrosprayed MPs exhibited substantially weaker encapsulation of rapa, achieving only about 50% EE across CACs. Consequently, the theoretical loading of rapa was doubled to ensure comparable drug loading across fabrication methods. Such poor encapsulation was unable to be corrected by adjusting the spray solvent or flow rate, which may imply complications with the current fabrication parameters or incompatibility between Ace-DEX and rapa in electrospray systems.
| Fabrication method | Theoretical loading | CAC (%) | Encapsulation efficiency | Rapa loading (μg mg−1) |
|---|---|---|---|---|
| Homogenization | 1% w/w | 20 | 99.2 ± 12.7% | 9.92 ± 1.27 |
| 40 | 86.2 ± 4.7% | 8.62 ± 0.47 | ||
| 60 | 111.8 ± 8.5% | 11.17 ± 0.85 | ||
| Sonication | 1% w/w | 20 | 72.2 ± 3.2% | 7.22 ± 0.32 |
| 40 | 86.5 ± 6.7% | 8.65 ± 0.67 | ||
| 60 | 96.2 ± 3.5% | 9.62 ± 0.35 | ||
| Electrospray | 2% w/w | 20 | 50.5 ± 12.5% | 10.10 ± 2.50 |
| 40 | 45.7 ± 6.9% | 9.13 ± 1.39 | ||
| 60 | 61.9 ± 1.8% | 11.67 ± 1.11 | ||
| Spray Dry | 1% w/w | 20 | 101.8 ± 12.7% | 10.18 ± 1.27 |
| 40 | 95.2 ± 11.2% | 9.52 ± 1.12 | ||
| 60 | 123.8 ± 3.7% | 12.38 ± 0.37 |
Although rapa release from Ace-DEX MPs has been previously reported,34,43 no studies have sought direct comparison among common MP fabrication methods. Thus, Ace-DEX MPs of different CACs and fabrication methods were assessed for their ability to modulate rapa release over two weeks at physiological conditions (pH 7.4, 37 °C). Homogenized (Fig. 3A) and sonicated (Fig. 3B) MPs performed fairly similarly, with both MP groups exhibited strongly tunable release profiles. Following initial burst release, which is likely attributable to immediate drug diffusion from the polymeric matrix upon suspension in solution, 20 CAC MPs and 40 CAC MPs fully released their cargo by 48 hours and 1 week, respectively. Interestingly, 40 CAC homogenized MPs exhibited zero-order (i.e., linear) release kinetics (Fig. 3A, purple), which are relatively uncommon for polymeric MP platforms. These kinetics are possibly attributable to favorable MP degradation kinetics and uneven distribution of rapa within the MP. With linear MP degradation kinetics, an increasing concentration of rapa towards the core would compensate for decreased MP surface area, resulting in zero-like kinetics. In contrast, 60 CAC MPs released approximately 50% of their cargo by the end of the two-week study. Across both fabrication methods, these release profiles align strongly with the blank MP degradation curves obtained under the same conditions (Fig. S3A and B†), indicating that polymer degradation is a key determinant for rapa release. Indeed, previous work has shown that homogenized Ace-DEX MPs degrade by surface erosion, and drug-release can be mathematically modeled using an equation based on polymer degradation and drug diffusion rates.34
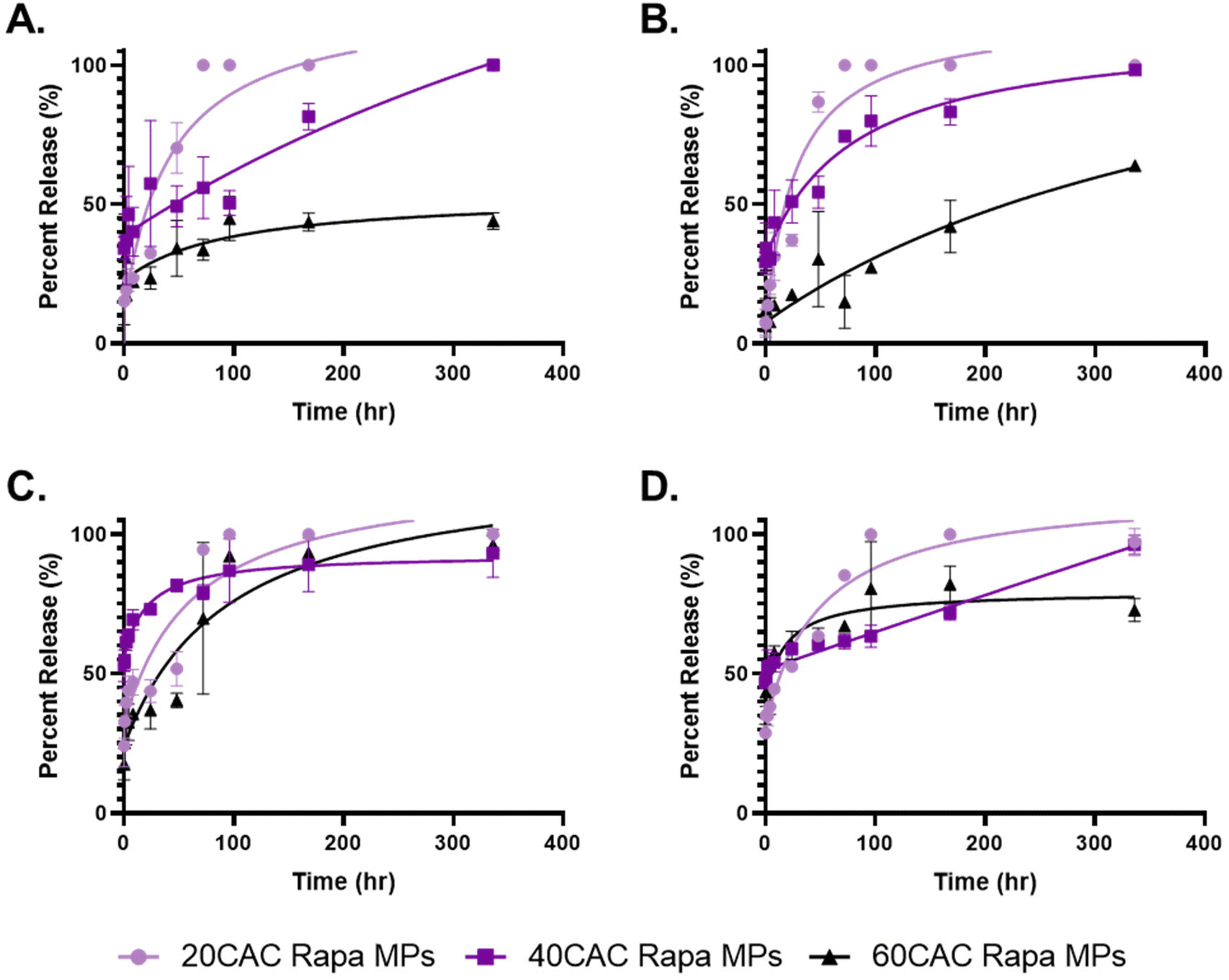 | ||
| Fig. 3 Rapa release profiles of (A) homogenized, (B) sonicated, (C) electrosprayed, and (D) spray dried Ace-DEX MPs by CAC. Release was measured over two weeks at pH 7.4 and 37 °C. Data are presented as average ± standard deviation (n = 3). Non-linear curves were interpolated using a Pade (1,1) approximate on GraphPad Prism. | ||
In contrast, electrosprayed (Fig. 3C) and spray dried (Fig. 3D) MPs exhibited much less tunable rapa release profiles. For electrosprayed MPs, all three CACs released greater than 80% of their cargo by two weeks, with the slowest degrading Ace-DEX (60 CAC) closely mirroring the fastest degrading Ace-DEX (20 CAC). However, the blank MP degradation profiles reflected much more typical CAC-dependent degradation kinetics (Fig. S3C†), which suggests that rapa release from electrosprayed MPs is additionally governed by alternative mechanisms. Namely, ionization of the drug–polymer solution might alter interactions between rapa and Ace-DEX, resulting in increased rapa density towards the periphery of the MP matrix or preferential surface adsorption of rapa to MPs. Alternatively, electrosprayed Ace-DEX MPs have previously been reported to possess a porous morphology,44,45 and perhaps the internal architecture of these MPs differs significantly from the other fabrication methods. However, further studies would be required to evaluate these hypotheses.
Spray dried MPs also performed similarly across CACs through the first 48 hours, with a considerably high burst release (∼50%) compared to the other fabrication methods (Fig. 3D). The burst is corroborated by the blank MP degradation curves (Fig. S3D†), which suggests poor stability upon resuspension. This may be attributable to the collapsed MP morphologies, which lends greater surface area for hydrolysis and weaker MP architectures (i.e., higher surface area-to-volume ratio). Nevertheless, subsequent timepoints revealed more substantial CAC-dependent release compared to electrosprayed MPs, with increasing CAC showing more sustained release profiles. Once again, 40 CAC MPs exhibited zero-order release kinetics (Fig. 3D, purple), indicating a more nuanced mechanism of release as seen with the 40 CAC homogenized MPs.
The release data indicates that emulsion-based methods are more efficacious in fabricating MP formulations with tunable rapa release kinetics. Although the blank MP degradation kinetics were similar across fabrication methods at each CAC (Fig. S4†), only homogenization and sonication demonstrated highly tunable rapa release kinetics, especially with respect to 60 CAC release (Fig. S5†). However, further optimization work may address the shortcomings of spray-based methods and their rapa release profiles, including the incorporation of cryoprotectant to MP batches for improved storage or the modification of spray solvents to control MP morphologies.
Inhibition of LPS-induced inflammation by Ace-DEX MPs
The efficacy of MP systems as drug delivery vehicles often hinges on delivery to and phagocytosis by target cell populations. Macrophages comprise a key immune cell compartment targeted by MP platforms and are integral regulators of inflammatory pathologies observed in transplant rejection and autoimmunity.46,47 Furthermore, macrophages are highly plastic, enabling facile induction and resolution of inflammation. Thus, we were interested as to whether fabrication methods altered phagocytic responses, and, in turn, delivery of rapa to macrophages in an in vitro setting.To this end, murine macrophages were first stimulated with LPS, a toll-like receptor 4 (TLR-4) agonist. TLR-4 signaling results in the activation of a number of downstream pro-inflammatory regulators, such as the transcription factor NF-κB, and is understood to be a contributing factor to several inflammatory disorders.48,49 After induction of inflammation, cells were then treated with varying concentrations of soluble or encapsulated rapa for 24 hours, and ELISAs were performed to measure pro-inflammatory cytokine secretion in the cell supernatant. Namely, we primarily focused on TNF-α production, as selective inhibition of TNF-α production and signaling has been a long-standing approach to treating inflammatory disorders.50,51 Other common inflammatory cytokines such as IL-1β, IL-6, IFN-γ, and IL-12p70 were measured via ELISA, but little signal was observed (data not shown).
After 24 hours, all rapa-containing treatments demonstrated marginal impacts on cell viability (Fig. 4A) and significant inhibition of TNF-α production relative to the LPS-alone control (LPS, Fig. 4B). Of note, spray-based MPs elicited slight reductions in cell viability at the highest MP doses (Fig. 4A), which may be attributable to aggregation of MPs at the cell surface. Rapa-loaded homogenized MPs performed most similarly to soluble rapa after 24 hours across all doses of rapa (Fig. 4B and S6A†). Sonicated (Fig. 4B and S6B†) and electrosprayed (Fig. 4B and S6C†) MPs exhibited reduced inhibitory effects at increased doses of rapa, suggesting that MP concentration may alter the immunosuppressive efficacy of rapa in these formulations. Finally, rapa-loaded spray dried MPs induced the weakest inhibition of TNF-α production compared to the other MP groups, especially at lower rapa doses (Fig. 4B and S6D†). Interestingly, spray dried MPs were the only MP batch shown to exacerbate LPS-induced TNF-α production in the absence of rapa (Fig. S6D†), indicating the selected spray drying parameters may not be the optimal fabrication process for rapa-loaded Ace-DEX MPs.
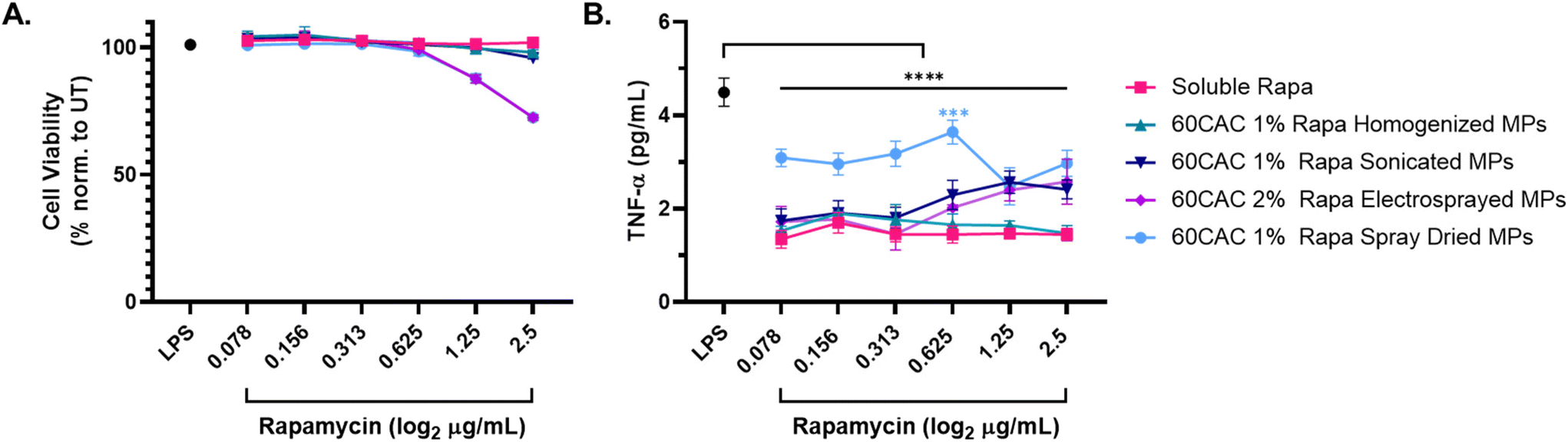 | ||
| Fig. 4 Inhibition of LPS-induced inflammation following treatment with rapa-loaded Ace-DEX MPs of different fabrication method. Macrophages were stimulated with 20 ng mL−1 LPS for 1 hour, followed by concurrent treatment with the indicated concentrations of soluble or encapsulated rapa for 24 hours. The “LPS” control (black) represents cells only receiving LPS stimulation. (A) Cell viability measures collected via lactose dehydrogenase (LDH) assay. Viability is quantified as 100% – LDH Cytotoxicity. (B) TNF-α content from cell supernatants, as measured by TNF-α ELISA. Data are presented as an average ± standard deviation (n = 3). Statistical significance is presented as ***p < 0.001, and ****p < 0.0001 for a two-way ANOVA test applied between MP groups and LPS alone (*). | ||
Evaluation of drug-to-polymer ratio for immunosuppressive efficacy
The inhibition of TNF-α production in LPS-stimulated macrophages validated the immunosuppressive efficacy of rapa-loaded Ace-DEX MPs. Moreover, given that homogenized MPs performed most similarly to soluble drug across all rapa doses, and homogenized MPs exhibited favorable sizing and tunable release profiles, homogenization was selected as the optimal fabrication method for rapa-loaded Ace-DEX MPs in subsequent studies.In particular, we were interested in understanding how the relative drug loading in MPs impacts the physiochemical properties and immunosuppressive efficacy of rapa-loaded Ace-DEX MPs. Exploring the limits of rapa loading in Ace-DEX MPs is a significant consideration for therapeutic design, as increased drug loading reduces required MP doses and limits potential vehicle-dependent effects. Indeed, high drug-loaded MPs overcome many administrations demands, such as limited local injection volumes and concerns of dosing frequencies.52 Thus, 60 CAC Ace-DEX MPs were fabricated at 0.5, 1, 3, and 5% w/w rapa using the previously described emulsion homogenization method. Consistent with previous data, homogenization yielded spherical MPs approximately 500 to 600 nm in diameter, with no significant sizing differences observed between rapa loadings (Fig. S7 and Table S2†). Likewise, all MPs possessed surface charges of roughly −10 mV (Table S2†), suggesting that rapa encapsulation does not dramatically alter size, morphology, or surface charge under the current fabrication parameters. Encapsulation efficiency of rapa was inversely related with the theoretical drug loading (Table 3), indicating further optimization in the fabrication protocol may be required to achieve full encapsulation at higher theoretical loadings. Of note, the encapsulation efficiency of the 1% rapa MPs (94.6 ± 1.8%, Table 2) was slightly lower than the previously reported batch (118 ± 8.5%, Table 1), highlighting a known limitation of homogenization as a batch-process. Nevertheless, the MPs displayed similar release kinetics to previous batches and across the different rapa loadings, exhibiting approximately 50% cargo release by 96 hours followed by minimal additional release through 2 weeks (Fig. S8†).
| Fabrication method | Theoretical loading | CAC (%) | Encapsulation efficiency | Rapa loading (μg mg−1) |
|---|---|---|---|---|
| Homogenization | 0.5% w/w | 60 | 115.6 ± 8.1% | 5.78 ± 0.40 |
| 1% w/w | 94.6 ± 1.8% | 9.46 ± 0.18 | ||
| 3% w/w | 75.1 ± 4.4% | 22.54 ± 1.33 | ||
| 5% w/w | 60.0 ± 2.7% | 30.02 ± 1.35 |
As before, immunosuppressive efficacy was measured via inhibition of TNF-α production in LPS-stimulated macrophages. After 24 hours, all rapa treatments exhibited minimal impact on cell viability (Fig. 5A), consistent with the previous homogenized MP data. Furthermore, all rapa treatments similarly inhibited TNF-α production, with no statistically significant differences observed between the different rapa-loaded MP batches nor between soluble or encapsulated rapa (Fig. 5B). These results suggest that increasing the rapa loading of MPs does not compromise the biocompatibility or immunosuppressive effects of rapa-loaded homogenized Ace-DEX MPs. In fact, fabricating MPs at a higher theoretical loading of rapa allows for the same immunosuppressive efficacy with a substantially lower MP dose – an attractive characteristic for high drug-loaded therapeutics.
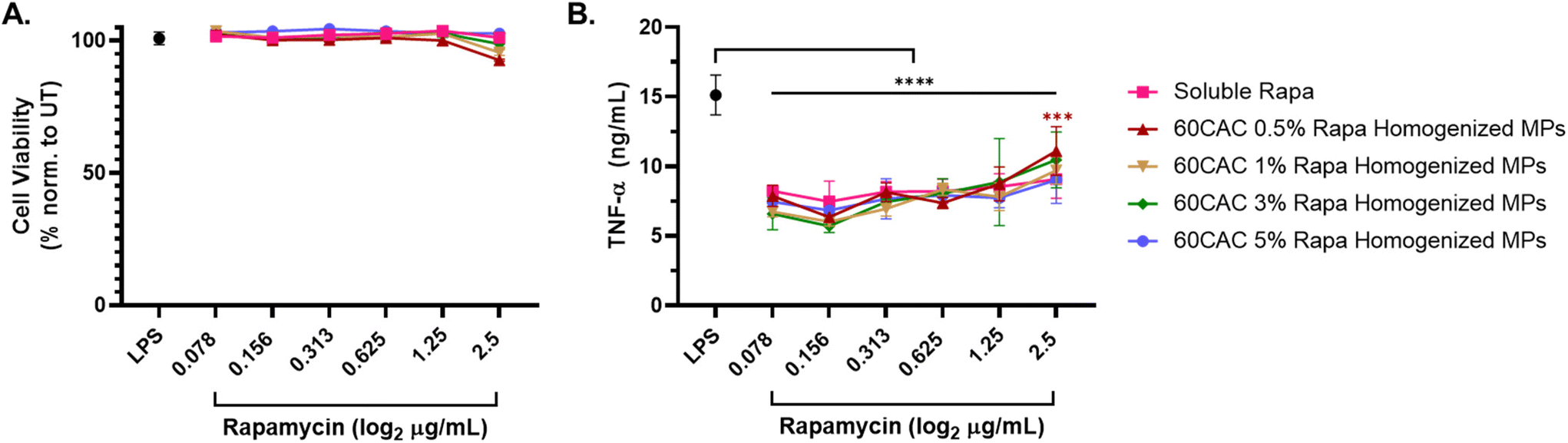 | ||
| Fig. 5 Inhibition of LPS-induced inflammation following treatment with rapa-loaded Ace-DEX MPs of different rapa-loading. Macrophages were stimulated with 20 ng mL−1 LPS for 1 hour, followed by concurrent treatment with the indicated concentrations of soluble or encapsulated rapa for 24 hours. The “LPS” control (black) represents cells only receiving LPS stimulation. (A) Cell viability measures collected via lactose dehydrogenase (LDH) assay. Viability is quantified as 100% – LDH Cytotoxicity. (B) TNF-α content from cell supernatants, as measured by TNF-α ELISA. Data are presented as an average ± standard deviation (n = 3). Statistical significance is presented as ***p < 0.001, and ****p < 0.0001 for a two-way ANOVA test applied between MP groups and LPS alone (*). | ||
In vivo depot trafficking of homogenized Ace-DEX MPs
Since the immunosuppressive efficacy of rapa-loaded Ace-DEX MPs in target immune cell populations was observed in vitro, we sought to understand if Ace-DEX MPs reach these target populations in vivo. Compared to the more static in vitro model, in vivo administration of MP platforms involves dynamic physiological processes that may challenge intended biodistributions (e.g., lymphatic or vascular circulation, competing target and non-target cell types). Thus, to track the local responses post-injection, fluorescent AF647-labelled blank homogenized Ace-DEX MPs (subsequently called Alexa-DEX MPs, Fig. S9†) were injected subcutaneously into BALB/c mice. Alexa-DEX MPs formed a depot in the subcutaneous space which was tracked over 21 days for residence time and cellular uptake.Live-animal IVIS imaging revealed that the depot fluorescence declined rapidly over the first 48 hours post-injection, indicating clearance of MPs from the injection site (Fig. 6A and B). Nevertheless, a sustained signal was observed through 21 days, as indicated by the approximately 2-fold greater mean fluorescent intensity (MFI) than the saline control (Fig. 6A and B). These results are consistent with previous reports of trafficking fluorescent indocyanine green-labeled homogenized Ace-DEX MPs following subcutaneous injection, which additionally identified notable MP trafficking to the draining lymph nodes, a critical site for antigen presentation and immune cell education.20
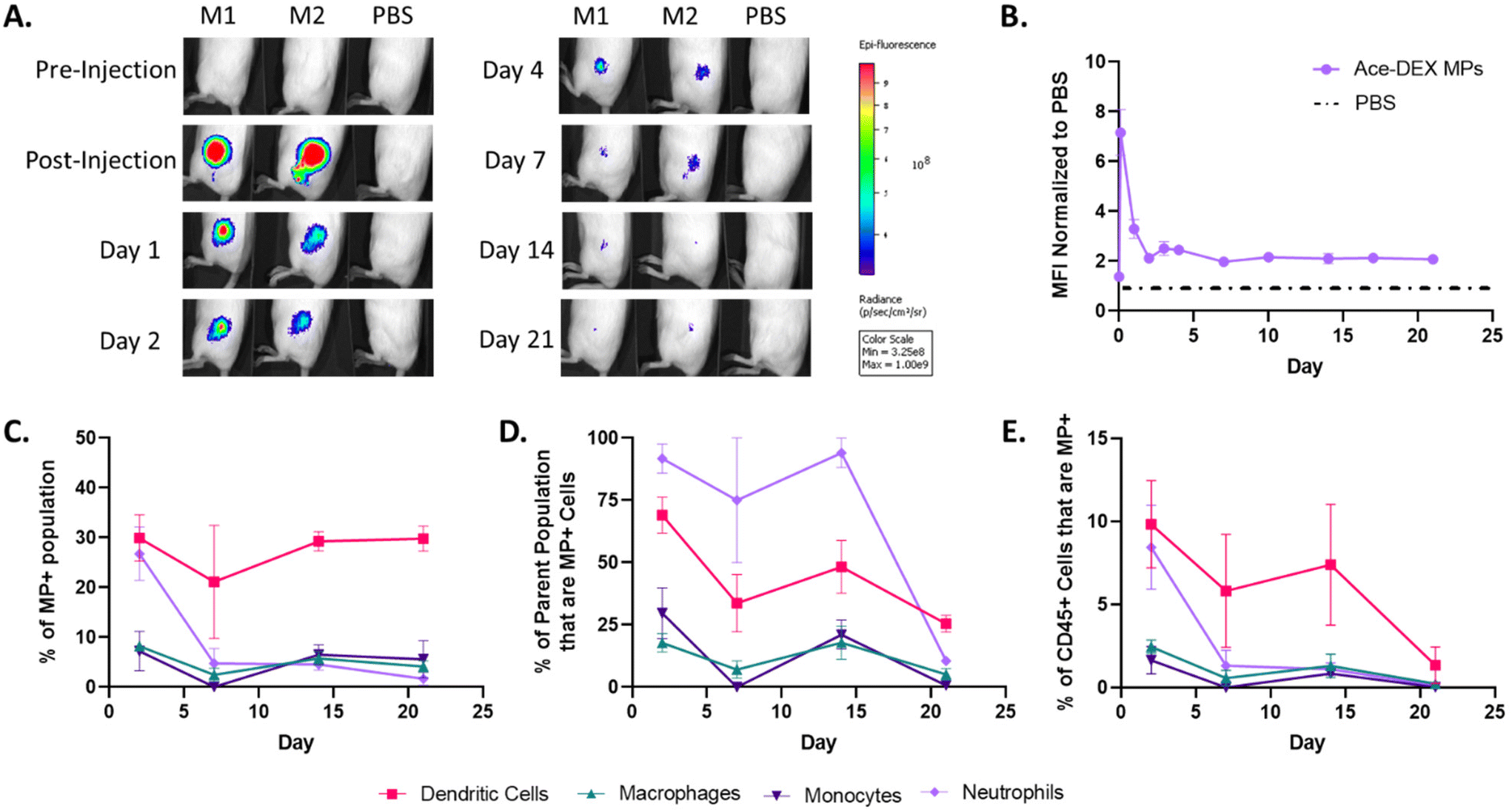 | ||
| Fig. 6 Blank homogenized Alexa-DEX MP trafficking in vivo. BALB/c mice were injected subcutaneously in each flank with 2 mgs of fluorescent Alexa-DEX MPs. (A) Representative IVIS images from Alexa-DEX MP-treated (M1, M2) and PBS control (PBS) mice tracked through day 21 and (B) the mean fluorescent intensity (MFI) of Alexa-DEX MP depots normalized to PBS mice. At each time point, the subcutaneous depot was harvested and processed for flow cytometry. (C) Percent of MP+ population by cell type. (D) Percent of CD45+ immune cell (immune cells) population that are MP+, by cell type (i.e., on Day 1, approximately 10% of all CD45+ immune cells at the depot are MP+ DCs). (E) Percent of parent population that are MP+ (i.e., on Day 1, approximately 75% of DCs at the depot are MP+). Data are presented as mean ± SEM. | ||
On days 2, 7, 14, and 21, animals were sacrificed, and the injection depots were harvested to characterize the Alexa-DEX MP+ cell populations at the depot. While many have reported on MP trafficking solely looking at APCs such as DCs and macrophages,53–57 we looked at a more extensive panel including markers for monocytes (CD45+CD11b+Ly6Ghi) and neutrophils (CD45+CD11b+Ly6G+) to give a broader view on innate responders to MPs (Fig. S10†).
DCs (CD45+CD11c+MHCII+) were the primary immune cell population targeted by homogenized MPs, comprising approximately 30% of the MP+ population at all time points (Fig. 6C). This supports previous reports that have indicated that MPs on the order of 0.5 to 2 μm are ideal for DC uptake.13 Additionally, Alexa-DEX MP+ DCs account for a significant portion of all immune cells present in the depot, ranging between 5–10% through day 14 (Fig. 6D). This is indicative of a sustained DC response in which DCs are continually sampling the MP depot compared to other cells. Given this and the role of DCs as critical modulators of local inflammation, rapa-loaded Ace-DEX MPs were additionally screened in LPS-stimulated DCs and shown to inhibit TNF-α production in vitro (Fig. S11†).
Neutrophils exhibited MP uptake comparable to DCs the Day 2 timepoint, followed by a dramatic decline by Day 7 (Fig. 6C). Although unexpected, neutrophils are phagocytic first responders that react to the tissue modulation caused by the injection. Neutrophils rapidly sample their environment, digesting foreign particles, mediating local inflammation, and recruiting other immune cell populations. However, because the neutrophil response is short-lived while the DC response is persistent, this suggests that MPs are both reaching target APCs and inducing minimal inflammation. In comparison, monocytes and macrophages represented only a marginal fraction of the MP+ population across all timepoints (Fig. 6C), contradicting our previous expectation of MP uptake by macrophages. Therefore, the current homogenized MP formulation may be better equipped for primarily DC-associated pathologies (e.g., systemic lupus erythematosus, multiple sclerosis) compared to macrophage-associated pathologies (e.g., rheumatoid arthritis, atherosclerosis). Nevertheless, route of administration and MP sizing have previously been shown to direct organ and cellular tropism, and these parameters could possibly be investigated to drive greater macrophage uptake.13,58
The uptake of Alexa-DEX MPs by DCs illustrates the potential of homogenized Ace-DEX MPs as a local therapeutic platform. Additionally, for each cell type, the frequencies of MP+ cells within both the total immune cell (Fig. 6D) and specific immune cell (Fig. 6E) populations at the depot decreased during the time course, which is consistent with cellular migration away from the depot. Future studies will focus on the analysis of relevant organs such as the draining lymph node to understand where MP+ innate cells traffic to and how MP+ innate cells interact with the adaptive immune system. Furthermore, future work will address how MP fabrication methods and cargo modulate trafficking.
The present study sought to elucidate the role of fabrication method in determining the drug delivery efficacy of rapa-loaded Ace-DEX MPs. As such, rapa-loaded Ace-DEX MPs were fabricated via emulsion (homogenization and sonication) and spray (spray drying and electrospraying) methods and first characterized for physiochemical parameters such as size, morphology, and surface charge. Emulsion-based MPs exhibited spherical morphologies ranging from the micro (homogenization) to nano (sonication) scale. Additionally, these MPs bore slightly negative surface charge. In contrast, spray-based MPs were approximately a micron in size, exhibited collapsed morphologies, and possessed slightly more negative surface charge. Rapa-loaded Ace-DEX MPs were then characterized for relevant drug-delivery parameters, including drug loading and drug release kinetics. With the exception of electrosprayed MPs, rapa was either fully or nearly fully encapsulated in Ace-DEX MPs. Highly tunable rapa release kinetics were achieved for emulsion-based MPs based on CAC, whereas spray-based MPs exhibited less tunable release profiles, more rapid release kinetics, and higher burst release. For further analysis, the immunosuppressive efficacy of rapa-loaded Ace-DEX MPs was compared to soluble rapa in vitro. All MPs demonstrated significant inhibition in TNF-α production in LPS-stimulated macrophages, but homogenized MPs performed most similarly to the soluble drug. Therefore, homogenized MPs were employed to explore potential dosing ranges of rapa-loaded Ace-DEX MPs, and increasing the rapa loading within homogenized Ace-DEX MPs did not significantly alter MP physiochemical parameters, drug release kinetics, or in vitro immunosuppressive efficacy. Finally, to understand MP trafficking in vivo, fluorescently labeled homogenized Ace-DEX MPs were injected into mice. The injection depot was rapidly cleared within the first 48 hours, and sustained, preferential MP uptake by DCs was observed. These results demonstrate the potential of rapa-loaded Ace-DEX MP formulations to improve the delivery of rapa. Future work may aim to further optimize the fabrication methods discussed herein, as the conclusions of the present study are limited to the select set of fabrication parameters used for each method. With more optimal protocols established, comparative analysis among fabrication methods may extend to mouse models of inflammatory disease and inform translatable formulations.
Ethical statement
All animal procedures were reviewed and approved by the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee (IACUC). All studies were ethically performed in compliance with the policies set forth by the University of North Carolina at Chapel Hill IACUC and the United States Public Health Service (PHS) Policy on Humane Care and Use of Laboratory Animals.Author contributions
K.M.A, E.M.B, and R.T.S. conceived and supervised the research. S.A.E., N.R.L., R.T.S., D.D.M., S.M.N, and A.J.T. performed and analyzed experiments. C.J.B. developed and validated Alexa-DEX synthesis. S.A.E. primarily prepared figures and wrote the manuscript. N.R.L. assisted with figure preparation and discussion for in vivo trafficking. N.R.L., R.T.S., D.D.M., S.M.N., A.J.T., E.M.B., and K.M.A. reviewed and edited the manuscript. K.M.A. secured the funding.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed in part at the Chapel Hill Analytical and Nanofabrication Laboratory, CHANL, a member of the North Carolina Research Triangle Nanotechnology Network, RTNN, which is supported by the National Science Foundation, Grant ECCS-2025064, as part of the National Nanotechnology Coordinated Infrastructure, NNCI. The UNC Flow Cytometry Core Facility (RRID: SCR_019170) is supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center, the North Carolina Biotech Center Institutional Support Grant 2017-IDG-1025, and by the National Institutes of Health 1UM2AI30836-01. This work was supported, in part, by National Institutes of Health (NIH) NIAID R01AI137525 (PI: Ainslie) and NIDDK R01DK130225 (PI: Ainslie). Graphics created with BioRender.com. Reaction schematics created with ChemDraw. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.References
- J. Li, S. G. Kim and J. Blenis, Rapamycin: One Drug, Many Effects, Cell Metab., 2014, 19(3), 373–379 CrossRef CAS PubMed.
- J. J. Augustine, K. A. Bodziak and D. E. Hricik, Use of Sirolimus in Solid Organ Transplantation, Drugs, 2007, 67(3), 369–391 CrossRef CAS.
- A. Lansky, W. Wijns, B. Xu, H. Kelbæk, N. Van Royen and M. Zheng, et al., Targeted therapy with a localised abluminal groove, low-dose sirolimus-eluting, biodegradable polymer coronary stent (TARGET All Comers): a multicentre, open-label, randomised non-inferiority trial, Lancet, 2018, 392(10153), 1117–1126 CrossRef CAS.
- B. Xu, Y. Saito, A. Baumbach, H. Kelbæk, N. Van Royen and M. Zheng, et al., 2-Year Clinical Outcomes of an Abluminal Groove–Filled Biodegradable-Polymer Sirolimus-Eluting Stent Compared With a Durable-Polymer Everolimus-Eluting Stent, JACC Cardiovasc. Interv., 2019, 12(17), 1679–1687 CrossRef PubMed.
- K. L. Bride, T. Vincent, K. Smith-Whitley, M. P. Lambert, J. J. Bleesing and A. E. Seif, et al., Sirolimus is effective in relapsed/refractory autoimmune cytopenias: results of a prospective multi-institutional trial, Blood, 2016, 127(1), 17–28 CrossRef CAS.
- D. T. Teachey, R. Greiner, A. Seif, E. Attiyeh, J. Bleesing and J. Choi, et al., Treatment with sirolimus results in complete responses in patients with autoimmune lymphoproliferative syndrome, Br. J. Haematol., 2009, 145(1), 101–106 CrossRef CAS.
- M. Battaglia, A. Stabilini, B. Migliavacca, J. Horejs-Hoeck, T. Kaupper and M. G. Roncarolo, Rapamycin Promotes Expansion of Functional CD4 + CD25 + FOXP3+ Regulatory T Cells of Both Healthy Subjects and Type 1 Diabetic Patients, J. Immunol., 2006, 177(12), 8338–8347 CrossRef CAS.
- L. Passerini, F. Barzaghi, R. Curto, C. Sartirana, G. Barera and F. Tucci, et al., Treatment with rapamycin can restore regulatory T-cell function in IPEX patients, J. Allergy Clin. Immunol., 2020, 145(4), 1262–1271 CrossRef CAS PubMed.
- D. J. Trepanier, H. Gallant, D. F. Legatt and R. W. Yatscoff, Rapamycin: distribution, pharmacokinetics and therapeutic range investigations: an update, Clin. Biochem., 1998, 31(5), 345–351 CrossRef CAS PubMed.
- D. W. Lamming, Inhibition of the Mechanistic Target of Rapamycin (mTOR)–Rapamycin and Beyond, Cold Spring Harbor Perspect. Med., 2016, 6(5), a025924 CrossRef.
- R. T. Stiepel, E. Duggan, C. J. Batty and K. M. Ainslie, Micro and nanotechnologies: The little formulations that could, Bioeng. Transl. Med., 2023, 8(2), e10421 CrossRef CAS PubMed.
- J. A. Champion, A. Walker and S. Mitragotri, Role of Particle Size in Phagocytosis of Polymeric Microspheres, Pharm. Res., 2008, 25(8), 1815–1821 CrossRef CAS.
- V. Manolova, A. Flace, M. Bauer, K. Schwarz, P. Saudan and M. F. Bachmann, Nanoparticles target distinct dendritic cell populations according to their size, Eur. J. Immunol., 2008, 38(5), 1404–1413 CrossRef CAS PubMed.
- M. V. Baranov, M. Kumar, S. Sacanna, S. Thutupalli and G. Van Den Bogaart, Modulation of Immune Responses by Particle Size and Shape, Front. Immunol., 2021, 11, 607945 CrossRef.
- A. Haddadi, P. Elamanchili, A. Lavasanifar, S. Das, J. Shapiro and J. Samuel, Delivery of rapamycin by PLGA nanoparticles enhances its suppressive activity on dendritic cells, J. Biomed. Mater. Res., Part A, 2008, 84A(4), 885–898 CrossRef CAS PubMed.
- T. K. Kishimoto, M. Fournier, A. Michaud, G. Rizzo, C. Roy and T. Capela, et al., Rapamycin nanoparticles increase the therapeutic window of engineered interleukin-2 and drive expansion of antigen-specific regulatory T cells for protection against autoimmune disease, J. Autoimmun., 2023, 140, 103125 CrossRef CAS.
- N. Chen, K. J. Peine, M. A. Collier, S. Gautam, K. A. Jablonski and M. Guerau-de-Arellano, et al., Co–Delivery of Disease Associated Peptide and Rapamycin via Acetalated Dextran Microparticles for Treatment of Multiple Sclerosis, Adv. Biosyst., 2017, 1(3), 1700022 CrossRef.
- R. A. LaMothe, P. N. Kolte, T. Vo, J. D. Ferrari, T. C. Gelsinger and J. Wong, et al., Tolerogenic Nanoparticles Induce Antigen-Specific Regulatory T Cells and Provide Therapeutic Efficacy and Transferrable Tolerance against Experimental Autoimmune Encephalomyelitis, Front. Immunol., 2018, 9, 281 CrossRef PubMed.
- X. Zhang, D. Liu, M. He, M. Lin, C. Tu and B. Zhang, Polymeric nanoparticles containing rapamycin and autoantigen induce antigen-specific immunological tolerance for preventing vitiligo in mice, Hum. Vaccines Immunother., 2021, 17(7), 1923–1929 CrossRef CAS PubMed.
- N. Chen, C. J. Kroger, R. M. Tisch, E. M. Bachelder and K. M. Ainslie, Prevention of Type 1 Diabetes with Acetalated Dextran Microparticles Containing Rapamycin and Pancreatic Peptide P31, Adv. Healthcare Mater., 2018, 7(18), 1800341 CrossRef.
- J. S. Lee, P. Han, R. Chaudhury, S. Khan, S. Bickerton and M. D. McHugh, et al., Metabolic and immunomodulatory control of type 1 diabetes via orally delivered bile-acid-polymer nanocarriers of insulin or rapamycin, Nat. Biomed. Eng., 2021, 5(9), 983–997 CrossRef CAS.
- E. Lagreca, V. Onesto, C. Di Natale, S. La Manna, P. A. Netti and R. Vecchione, Recent advances in the formulation of PLGA microparticles for controlled drug delivery, Prog. Biomater., 2020, 9(4), 153–174 CrossRef CAS.
- A. D. Duong, S. Sharma, K. J. Peine, G. Gupta, A. R. Satoskar and E. M. Bachelder, et al., Electrospray Encapsulation of Toll-Like Receptor Agonist Resiquimod in Polymer Microparticles for the Treatment of Visceral Leishmaniasis, Mol. Pharm., 2013, 10(3), 1045–1055 CrossRef CAS.
- T. Marante, C. Viegas, I. Duarte, A. S. Macedo and P. Fonte, An Overview on Spray-Drying of Protein-Loaded Polymeric Nanoparticles for Dry Powder Inhalation, Pharmaceutics, 2020, 12(11), 1032 CrossRef CAS PubMed.
- R. T. Steipel, M. D. Gallovic, C. J. Batty, E. M. Bachelder and K. M. Ainslie, Electrospray for generation of drug delivery and vaccine particles applied in vitro and in vivo, Mater. Sci. Eng., C, 2019, 105, 110070 CrossRef CAS PubMed.
- L. Lu, S. J. Peter, M. D. Lyman, H. L. Lai, S. M. Leite and J. A. Tamada, et al., In vitro and in vivo degradation of porous poly(dl-lactic-co-glycolic acid) foams, Biomaterials, 2000, 21(18), 1837–1845 CrossRef CAS PubMed.
- R. Nicolete, D. F. D. Santos and L. H. Faccioli, The uptake of PLGA micro or nanoparticles by macrophages provokes distinct in vitro inflammatory response, Int. Immunopharmacol., 2011, 11(10), 1557–1563 CrossRef CAS PubMed.
- Y. Ramot, M. Haim-Zada, A. J. Domb and A. Nyska, Biocompatibility and safety of PLA and its copolymers, Adv. Drug Delivery Rev., 2016, 107, 153–162 CrossRef CAS PubMed.
- H. K. Makadia and S. J. Siegel, Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier, Polymers, 2011, 3(3), 1377–1397 CrossRef CAS PubMed.
- E. M. Bachelder, T. T. Beaudette, K. E. Broaders, J. Dashe and J. M. J. Fréchet, Acetal-Derivatized Dextran: An Acid-Responsive Biodegradable Material for Therapeutic Applications, J. Am. Chem. Soc., 2008, 130(32), 10494–10495 CrossRef CAS PubMed.
- K. J. Kauffman, C. Do, S. Sharma, M. D. Gallovic, E. M. Bachelder and K. M. Ainslie, Synthesis and Characterization of Acetalated Dextran Polymer and Microparticles with Ethanol as a Degradation Product, ACS Appl. Mater. Interfaces, 2012, 4(8), 4149–4155 CrossRef CAS.
- M. S. Verma and F. X. Gu, Microwave-enhanced reductive amination via Schiff's base formation for block copolymer synthesis, Carbohydr. Polym., 2012, 87(4), 2740–2744 CrossRef CAS.
- D. Sehgal and I. K. Vijay, A Method for the High Efficiency of Water-Soluble Carbodiimide-Mediated Amidation, Anal. Biochem., 1994, 218(1), 87–91 CrossRef CAS.
- R. T. Stiepel, E. S. Pena, S. A. Ehrenzeller, M. D. Gallovic, L. M. Lifshits and C. J. Genito, et al., A predictive mechanistic model of drug release from surface eroding polymeric nanoparticles, J. Controlled Release, 2022, 351, 883–895 CrossRef CAS.
- C. J. Benck, T. Martinov, B. T. Fife and D. Chatterjea, Isolation of Infiltrating Leukocytes from Mouse Skin Using Enzymatic Digest and Gradient Separation, J. Visualized Exp., 2016,(107), 53638 Search PubMed.
- N. Chen, M. M. Johnson, M. A. Collier, M. D. Gallovic, E. M. Bachelder and K. M. Ainslie, Tunable degradation of acetalated dextran microparticles enables controlled vaccine adjuvant and antigen delivery to modulate adaptive immune responses, J. Controlled Release, 2018, 273, 147–159 CrossRef CAS.
- N. Chen, M. D. Gallovic, P. Tiet, J. P. Y. Ting, K. M. Ainslie and E. M. Bachelder, Investigation of tunable acetalated dextran microparticle platform to optimize M2e-based influenza vaccine efficacy, J. Controlled Release, 2018, 289, 114–124 CrossRef CAS.
- N. Chen, M. A. Collier, M. D. Gallovic, G. C. Collins, C. C. Sanchez and E. Q. Fernandes, et al., Degradation of acetalated dextran can be broadly tuned based on cyclic acetal coverage and molecular weight, Int. J. Pharm., 2016, 512(1), 147–157 CrossRef CAS.
- A. Bootz, V. Vogel, D. Schubert and J. Kreuter, Comparison of scanning electron microscopy, dynamic light scattering and analytical ultracentrifugation for the sizing of poly(butyl cyanoacrylate) nanoparticles, Eur. J. Pharm. Biopharm., 2004, 57(2), 369–375 CrossRef CAS PubMed.
- C. J. Genito, C. J. Batty, E. M. Bachelder and K. M. Ainslie, Considerations for Size, Surface Charge, Polymer Degradation, Co–Delivery, and Manufacturability in the Development of Polymeric Particle Vaccines for Infectious Diseases, Adv. NanoBiomed Res., 2021, 1(3), 2000041 CrossRef CAS PubMed.
- L. Chen, J. D. Simpson, A. V. Fuchs, B. E. Rolfe and K. J. Thurecht, Effects of Surface Charge of Hyperbranched Polymers on Cytotoxicity, Dynamic Cellular Uptake and Localization, Hemotoxicity, and Pharmacokinetics in Mice, Mol. Pharm., 2017, 14(12), 4485–4497 CrossRef CAS.
- C. J. Batty, E. S. Pena, E. A. Amouzougan, K. M. Moore, K. M. Ainslie and E. M. Bachelder, Humoral Response to the Acetalated Dextran M2e Vaccine is Enhanced by Antigen Surface Conjugation, Bioconjugate Chem., 2023, 34(8), 1447–1458 CrossRef CAS.
- K. J. Kauffman, N. Kanthamneni, S. A. Meenach, B. C. Pierson, E. M. Bachelder and K. M. Ainslie, Optimization of rapamycin-loaded acetalated dextran microparticles for immunosuppression, Int. J. Pharm., 2012, 422(1–2), 356–363 CrossRef CAS PubMed.
- C. J. Batty, M. D. Gallovic, J. Williams, T. M. Ross, E. M. Bachelder and K. M. Ainslie, Multiplexed electrospray enables high throughput production of cGAMP microparticles to serve as an adjuvant for a broadly acting influenza vaccine, Int. J. Pharm., 2022, 622, 121839 CrossRef CAS PubMed.
- D. A. Hendy, E. S. Pena, C. J. Batty, L. Ontiveros-Padilla, J. A. Roque III and T. A. Dixon, et al., Cobra Hemagglutinin and cGAMP Loaded Ace–Dex Microparticles Provide a Broadly Active and Shelf–Stable Influenza Vaccine Platform, Adv. Ther., 2024, 7(2), 2300273 CrossRef CAS.
- S. C. Funes, M. Rios, J. Escobar-Vera and A. M. Kalergis, Implications of macrophage polarization in autoimmunity, Immunology, 2018, 154(2), 186–195 CrossRef CAS.
- K. R. Wyburn, M. D. Jose, H. Wu, R. C. Atkins and S. J. Chadban, The Role of Macrophages in Allograft Rejection, Transplantation, 2005, 80(12), 1641–1647 CrossRef.
- F. Braza, S. Brouard, S. Chadban and D. R. Goldstein, Role of TLRs and DAMPs in allograft inflammation and transplant outcomes, Nat. Rev. Nephrol., 2016, 12(5), 281–290 CrossRef CAS.
- Y. Liu, H. Yin, M. Zhao and Q. Lu, TLR2 and TLR4 in Autoimmune Diseases: a Comprehensive Review, Clin. Rev. Allergy Immunol., 2014, 47(2), 136–147 CrossRef CAS.
- R. L. Ang and A. T. Ting, Tumor necrosis factor-driven cell death in donor organ as a barrier to immunological tolerance, Curr. Opin. Organ Transplant., 2019, 24(1), 12–19 CrossRef CAS PubMed.
- N. Parameswaran and S. Patial, Tumor Necrosis Factor-α Signaling in Macrophages, Crit. Rev. Eukaryotic Gene Expression, 2010, 20(2), 87–103 CrossRef CAS.
- M. W. Tibbitt, J. E. Dahlman and R. Langer, Emerging Frontiers in Drug Delivery, J. Am. Chem. Soc., 2016, 138(3), 704–717 CrossRef CAS.
- J. J. Cho, J. M. Stewart, T. T. Drashansky, M. A. Brusko, A. N. Zuniga and K. J. Lorentsen, et al., An antigen-specific semi-therapeutic treatment with local delivery of tolerogenic factors through a dual-sized microparticle system blocks experimental autoimmune encephalomyelitis, Biomaterials, 2017, 143, 79–92 CrossRef CAS.
- B. P. Gross, A. Wongrakpanich, M. B. Francis, A. K. Salem and L. A. Norian, A Therapeutic Microparticle-Based Tumor Lysate Vaccine Reduces Spontaneous Metastases in Murine Breast Cancer, AAPS J., 2014, 16(6), 1194–1203 CrossRef CAS PubMed.
- C. M. Jewell, S. C. Bustamante López and D. J. Irvine, In situ engineering of the lymph node microenvironment via intranodal injection of adjuvant-releasing polymer particles, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(38), 15745–15750 CrossRef CAS PubMed.
- J. S. Lewis, T. D. Zaveri, C. P. Crooks and B. G. Keselowsky, Microparticle surface modifications targeting dendritic cells for non-activating applications, Biomaterials, 2012, 33(29), 7221–7232 CrossRef CAS.
- B. Phillips, K. Nylander, J. Harnaha, J. Machen, R. Lakomy and A. Styche, , et al., A Microsphere-Based Vaccine Prevents and Reverses New-Onset Autoimmune Diabetes, Diabetes, 2008, 57(6), 1544–1555 CrossRef CAS PubMed.
- X. Huang, F. Zhang, L. Zhu, K. Y. Choi, N. Guo and J. Guo, et al., Effect of Injection Routes on the Biodistribution, Clearance, and Tumor Uptake of Carbon Dots, ACS Nano, 2013, 7(7), 5684–5693 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 contains the reaction schematics for DAB-DEX and Alexa-DEX. Table S1 contains the antibodies and staining parameters used in the in vivo trafficking studies. Fig. S2 contains representative ImageJ tracings of SEM images to evaluate MP diameter. Fig. S3 contains degradation curves of blank Ace-DEX MPs by fabrication method. Fig. S4 contains degradation curves of blank Ace-DEX MPs by CAC. Fig. S5 contains rapa release curves of rapa-loaded Ace-DEX MPs by CAC. Fig. S6 contains TNF-α ELISA data from LPS-stimulated macrophages treated with blank or rapa-loaded Ace-DEX MPs, organized by fabrication method. Fig. S7 contains representative SEM images of rapa-loaded Ace-DEX MPs of increasing rapa loading. Table S2 contains MP diameter and surface charge data of rapa-loaded Ace-DEX MPs of increasing rapa loading. Fig. S8 contains rapa release curves of rapa-loaded Ace-DEX MPs of increasing rapa loading. Fig. S9 contains a representative SEM image of fluorescent Alexa-DEX MPs. Fig. S10 contains the gating strategy used to phenotype cells in the in vivo trafficking studies. Fig. S11 contains TNF-α ELISA data from LPS-stimulated dendritic cells treated with soluble rapa or rapa-loaded Ace-DEX MPs. See DOI: https://doi.org/10.1039/d4pm00054d |
| This journal is © The Royal Society of Chemistry 2024 |