
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssembly of a trapped valent CeIII/IV–TCNQ complex through metal–ligand redox cooperativity†
Himanshu
Gupta
 ,
Brett D.
Vincenzini
,
Alexandra M.
Bacon
and
Eric J.
Schelter
*
,
Brett D.
Vincenzini
,
Alexandra M.
Bacon
and
Eric J.
Schelter
*
P. Roy and Diana T. Vagelos Laboratories, Department of Chemistry, University of Pennsylvania, 231 South 34th Street, Philadelphia, Pennsylvania 19104, USA. E-mail: schelter@sas.upenn.edu
First published on 7th June 2024
Abstract
Complex (Cp3CeIV)2(TCNQ)(CeIIICp3)2 (1) was prepared by reducing neutral TCNQ0 (tetracyanoquinodimethane) with Cp3Ce(THF). Two types of cerium centres with a dianionic TCNQ2− moiety are present in 1, wherein each of the four cyano-groups are bound by a cation. Formation of this trapped-valent organocerium compound 1 is facilitated by metal–ligand redox cooperativity. Characterization of 1 was carried out using structural-, magnetometry-, and IR-spectroscopic analyses. Photophysical studies on this compound reveal CeIII luminescence, and opens up avenues for promising multifunctional, mixed-valent lanthanide materials.
Metal mixed valency is of interest in reticular chemistry to achieve long-range electronic coupling and associated physical properties.1,2 Fine-tuning of metal frontier orbital overlap and associated redox potentials can afford discovery of emergent phenomena in molecule-based materials, tuning that is typically not possible in intermetallic nor other solid-state materials.3–5 For example, redox active organics such as pyrazine have been shown to induce variable magnetic and conducting properties in materials comprising transition metal salts.6–8 Lanthanide compounds that are otherwise dominated by the trivalent oxidation state can exhibit notable behaviours in formally divalent or tetravalent materials upon combining with suitable electron donors/acceptors.9–11
Electron acceptors such as fullerenes, TCNQ (tetracyanoquinodimethane), TCNE (tetracyanoethylene) and others have been shown to assemble notable structures and associated, collective properties through charge transfer processes.12–14 TCNQ and TCNE offer multiple accessible redox states and four terminal cyano-functionalities that bind metal coordination sites to assemble magnetic, conducting and multifunctional materials.5,15,16 Mixed valent ferrocene-TCNQ materials comprising π–π interactions have also been assembled by charge transfer processes.17 On the other hand, redox assembly of lanthanide based TCNQ materials remain unexplored with the exception of ytterbocene, (C5Me5)2YbII(THF)2, which undergoes an unusual oxidative reorganization with TCNQ to form the YbIII dimer, [(C5Me5)2YbIII(CH3CN){(μ-CN)2(C6H4)(CN)2}]2.18 A previous report of a ribbon-like coordination polymer of CeIII and TCNQX2 X = –Cl, –Br showed notable magnetic coercivity, but was not formed through redox processes.13
Among organometallic lanthanide complexes, cerium offers relatively mild and tunable CeIII/IV oxidation potentials,19,20 and isolable tetravalent compounds.21–26 Tris(cyclopentadienyl) cerium(III) complexes have previously been shown to be a useful platform to access organocerium(IV) compounds: Cp3Ce–X, X = –halides, –alkoxides, and –aryloxides.27–29 As such, we expected that this platform could be used for molecule-based materials with multifunctional charge transfer properties if assembled with suitable organic acceptors. Our group recently reported that (C5Me4H)3Ce reacts with C60 to form [(C5Me4H)3Ce]2·C60 where intermolecular interactions promoted the emergence of Mott insulator behaviour reminiscent of Ce2O3.30 In the current work, we provide an initial demonstration that high valent, organocerium-based materials can be achieved with metal–ligand redox cooperativity using the organic acceptor TCNQ.
The first reduction of TCNQ0 reportedly occurs at E1/2 = –0.34 V versus Fc in THF while the second reduction occurs at E1/2 = –1.01 V.31 We set out to explore organocerium-based material by reducing TCNQ with Cp3Ce(THF), the latter of which shows an oxidation potential of E1/2 = –0.26 V versus Fc in THF.29 Reaction of Cp3Ce(THF) with TCNQ in toluene results in immediate precipitation of a brown solid. Using the tetramethylated precursor: (C5Me4H)3Ce towards the goal of preparing a compound with increased solubility, does not show any color change or precipitation upon reaction with TCNQ0 in toluene. We postulate that the larger steric demand offered by three C5Me4H− ligands prevent interaction with TCNQ0 to initiate the redox reaction. An optimized synthesis of the former reaction with Cp3Ce(THF) produced a crystalline, brown-black material, suitable for single crystal X-ray diffraction analysis, in 71% yield (see ESI† for details). The isolated material was insoluble in toluene, DCM, and Et2O at RT. Addition of THF and acetonitrile to the solid 1 produces yellow coloration, likely due to reductive decomposition. Notably, single crystal X-ray diffraction shows a neutral tetrameric complex (Cp3CeIV)2(TCNQ)(CeIIICp3)2 (1) with the TCNQ2− ligand in the doubly reduced state (Scheme 1).
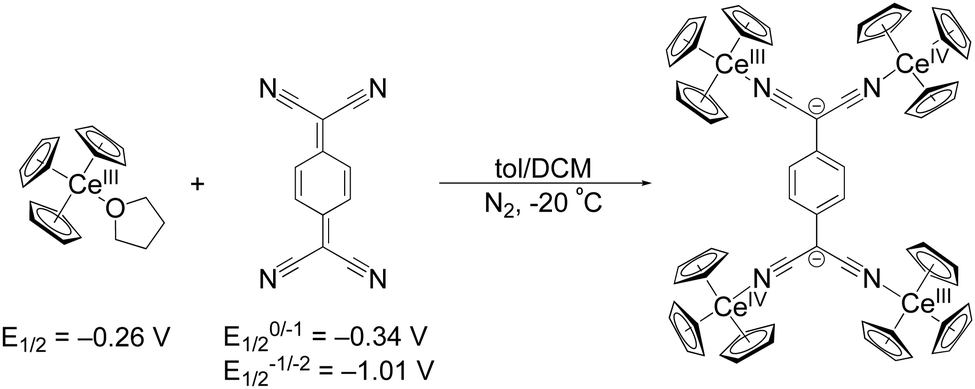 | ||
| Scheme 1 Synthetic conditions for 1 from Cp3Ce(THF) and TCNQ along with their respective redox potentials versus Fc in THF. | ||
The X-ray diffraction data for 1 are shown in the Table 1. The tetrameric TCNQ2− complex consists of two, structurally distinct, types of cerium centres. The Ce–N and Ce–Cp(centroid) bond distances at Ce(1) and Ce(3)* are 2.558(3) Å and 2.558(avg.) Å respectively, the latter of which is consistent with the structure of the previously reported Cp3CeIII–OC(NMe2)2, 2.576(avg.) Å.32 These same bond distances at Ce(2) and Ce(4)* are 2.414(3) Å and 2.458(avg.) Å, respectively, distances that more closely resemble those of Cp3CeIVCl with Ce–Cp(centroid) distance 2.460(avg.) Å.27 The redox state of the TCNQ2− ligand in 1 was evaluated by examining the bond length c (Fig. 1), where a change to the benzenoid form upon reduction is expressed through an elongation of that bond compared to neutral TCNQ. In 1, this bond length is 1.468(3) Å which is longer than the 1.374(3) Å observed in the structure of neutral TCNQ.33 The bond length c is also used in the Kistenmacher relationship calculation to estimate the charge distribution on TCNQ.34 (see ESI†). For 1, the net calculated charge on TCNQ is −2.19.
| Bond ‘c’ (Å) | M–N (Å) | ν CN (cm−1) | M–N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C (°) C (°) |
|
|---|---|---|---|---|
| 1 (M = Ce) | 1.469 (5) | 2.414(3), 2.558(3) | 2098, 2177 | 168.5(3), 159.9(3) |
| TCNQ0 (ref. 35) | 1.374(3) | n/a | 2223 | n/a |
| TCNQ˙− (ref. 17, M = Ce) | 1.400–1.432 | 2.654–2.666 | 2165, 2198 | 168.7(3), 149.0(3), 171.4(3) |
| TCNQ2− (ref. 36, M = Zn) | 1.472(5) | 2.160(3)–2.167(3) | 2121, 2194 | 159.26(3), 158.21(3) |
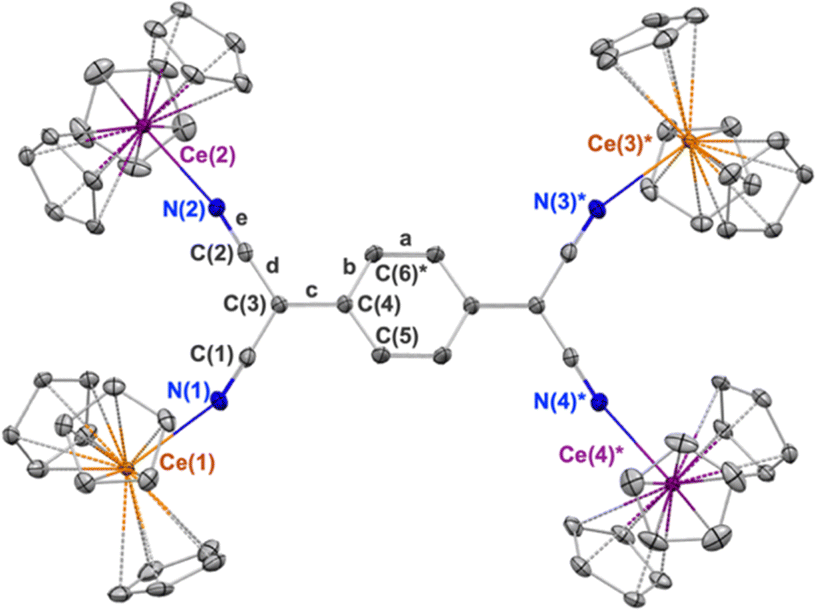 | ||
| Fig. 1 Thermal ellipsoid plot of 1 shown at the 30% probability level. CeIII centers are shown in orange and CeIV in purple color. The * indicates atoms at equivalent position (−x,1−y,1−z). Hydrogen atoms and co-crystalized toluene molecules are omitted for clarity. | ||
A notable feature of 1 is the bond angles described by the atoms: Ce–NC. For the Ce(2) and Ce(4)* sites comprising the tetravalent oxidation state, more linear Ce(IV)–NC bond angles of 168.5(3)° are observed. The Ce(1) and Ce(3)* sites exhibit more bent Ce(III)–NC bond angles of 159.9(3)°, presumably due to the weaker association of the cyano groups with the cerium(III) cations. Overall, based on the structural investigation, 1 comprises two cerium(III) cations and two cerium(IV) cations and is best described as a trapped-valent complex, class I under the Robin–Day classification.37
TCNQ vibrational signatures are sensitive to the extent of reduction of this moiety in molecules and materials. IR spectra were collected and compared with neutral TCNQ crystals and 1 in a KBr matrix. Compound 1 shows characteristic stretching frequencies associated with the cyano groups at 2098 and 2177 cm−1 (Table 1) whereas neutral TCNQ shows a single feature at 2223 cm−1. This evident decrease in frequency is attributed to 2e− reduction of TCNQ moiety in 1 as reported previously.38 Attempts to perform Raman measurements of 1 were unsuccessful due to a consistently poor signal-to-noise ratio.
Malischewski and co-workers recently reported that coordination of B(C6F5)3 to TCNQ shifted the first reduction potential of the latter by 1.2 V to 0.9 V versus Fc in CH2Cl2.39 We and others have previously shown such Lewis acid promoted potential shift (LAPPS) of organic acceptors using trivalent lanthanides.35,40 For example, we demonstrated LAPPS by cerium coordination and concomitant reduction of para-benzoquinone using [{Li3(thf)4}{(BINOLate)3Ce(thf)}].41 Similarly, 1 evidently assembles from a Lewis acid effect of CeIII bonding that shifts the redox potentials of the Cp3Ce donor and TCNQ acceptor, and relatively covalent bonding of CeIV coordination. The operative Lewis acid-assisted coordination significantly lowers the reduction potentials of TCNQ and concurrently reduces it to assemble the trapped valent cerium complex 1. This chemistry is reminiscent of transition metals based TCNQ reduction that has not been previously reported for lanthanides.42
Variable temperature magnetic susceptibility data performed at 2 T and variable field magnetization saturation experiments performed at 2 K were recorded for 1 to further verify the redox assignment of the cerium cations and TCNQ (Fig. 2). For Cp3Ce(THF), the χMT value is 0.68 emu K mol−1 at 300 K and slowly decreases to 0.25 emu K mol−1 at 2 K, similar to the reported value for Cp3CeOP(NMe2)3.36 For 1, the χMT value is 1.56 emu K mol−1 at 300 K and slowly decreases to 0.49 emu K mol−1 at 2 K. This data falls in the expected range for two non-interacting CeIII ions (1.60 emu K mol−1) and helps to confirm the presence of two CeIII cations in 1. With decreasing temperature, the χMT value decreases slowly and drops to 0.50 emu K mol−1 at 2 K. This decrease in the χT product is likely due to thermal depopulation of the cerium(III) Kramers doublets or weak antiferromagnetic coupling. A field dependent magnetization saturation experiment showed a value of M = 1.59μB at 2 K at an applied field of 7 T, which further supports the assignment of two cerium(III) centres in 1.43
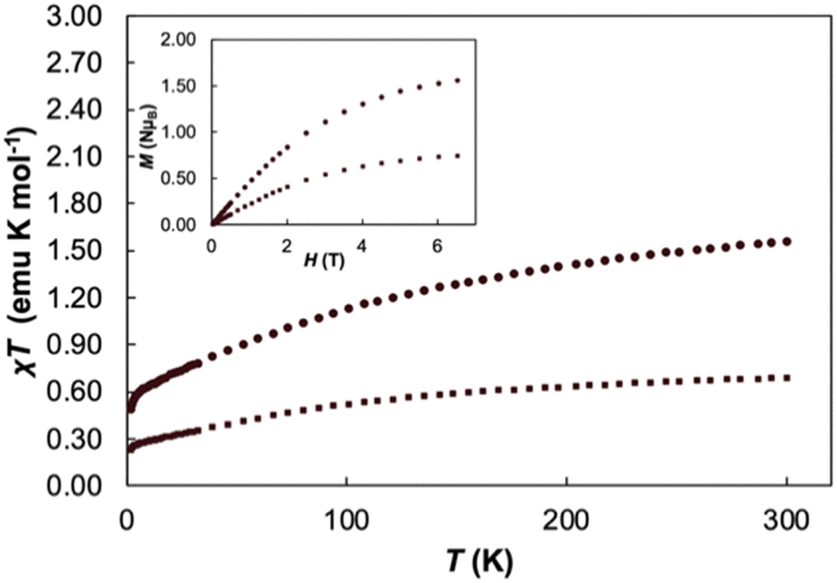 | ||
| Fig. 2 Variable temperature χT measurements of Cp3Ce(THF) (square markers) and 1 (circle markers) at 2T, variable field magnetization experiments (inset) conducted at T = 2 K. | ||
To further interrogate the photophysical properties of 1, photoluminescence spectra were collected on (only poorly soluble) suspensions of the compound in toluene (Fig. 3). With excitation at 340 nm, a broad emission band was observed with λmax = 414 nm. The excitation spectrum of 1, collected between 230 and 380 nm, contains a broad excitation band with λmax = 343 nm and a prominent shoulder located at 360 nm. The separation of λmax and the lower energy shoulder is 1377 cm−1 which falls in the range of peak separations commonly observed for cerium(III) 5d → 4f transitions between the 2D excited state and 2F5/2 and 2F7/2 ground state manifold.44 The Stokes shift for 1 was determined to be 54 nm (0.45 eV) which is comparable to those shifts reported for other luminescent cerium(III) compounds.45 Given that the luminescence profile of 1 resembles those observed for reported Ce(III) 5d → 4f emissions, we tentatively attribute the observed luminescence features to electronic transitions at the cerium(III) sites of 1.
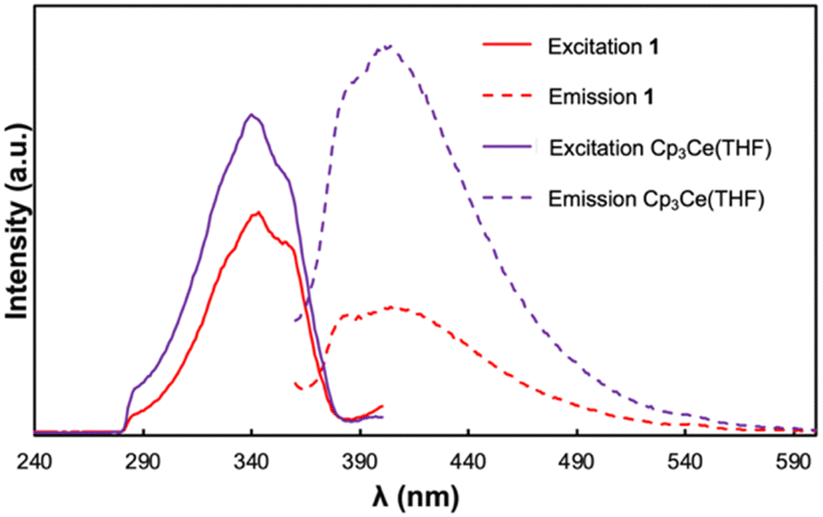 | ||
| Fig. 3 Photoluminescence spectra of 1 (red) and Cp3Ce(THF) (purple) as a suspension in toluene and a 0.005 mM solution respectively. The excitation spectra at 420 nm emission are shown as solid traces. The emission spectra at 340 nm excitation are shown as dashed traces. | ||
Previous studies of the photophysical properties of neutral TCNQ revealed that it exhibits solvent dependent photoluminescent behaviour.46 While TCNQ was not luminescent in toluene, the starting Cp3Ce(THF) complex exhibited a similar luminescence profile to 1 and resembled the spectra previously reported in THF.47 The excitation spectrum was essentially indistinguishable from that of 1 with an identical λmax value and prominent shoulder at 343 and 360 nm respectively. Notwithstanding, the emission band of Cp3Ce(THF) obtained upon excitation at 340 nm occurred at slightly higher energy with the λmax = 403 nm. As such, Cp3Ce(THF) exhibits a smaller Stokes shift than that of 1 (43 nm, 0.36 eV). Our group has previously demonstrated that the Stokes shift in cerium(III) emitters is correlated with the degree of nonradiative vibrational losses of energy in the emissive state(s).48 The results therefore indicate that 1 undergoes greater vibrational relaxation in the 2D excited state compared with the mononuclear Cp3Ce(THF) complex.
A synthetic pathway for cerium coordination-induced double reduction of TCNQ has been described. This strategy can be used to assemble molecule-based materials comprising reduced organics with otherwise poorly matched donor–acceptor redox potentials. Currently, studies of a range of these materials with various metals and Cp-substituents, and studies of their electronic structures towards multi-functional materials are underway in our laboratory.
We gratefully acknowledge the U.S. Department of Energy under Award DE-SC0020169 for support of this work. H. G. also acknowledges the Vagelos Institute for Energy Science and Technology for a graduate fellowship. And E. J. S. also thanks the University of Pennsylvania for financial support. We also thank the JASCO facility for Spectroscopic Excellence in the Chemistry Department at the University of Pennsylvania for access to the Raman spectrometer used in this work.
Data availability statement
Data supporting this article have been included as part of the ESI.† Crystallographic data for 1 have been deposited at the CCDC under accession number 2343799 and can be obtained from https://www.ccdc.cam.ac.uk/structures.Conflicts of interest
There are no conflicts to declare.Notes and references
- R. Murase, C. F. Leong and D. M. D’Alessandro, Inorg. Chem., 2017, 56, 14373–14382 CrossRef CAS PubMed.
- L. E. Darago, M. L. Aubrey, C. J. Yu, M. I. Gonzalez and J. R. Long, J. Am. Chem. Soc., 2015, 137, 15703–15711 CrossRef CAS PubMed.
- J. Xie, S. Ewing, J.-N. Boyn, A. S. Filatov, B. Cheng, T. Ma, G. L. Grocke, N. Zhao, R. Itani, X. Sun, H. Cho, Z. Chen, K. W. Chapman, S. N. Patel, D. V. Talapin, J. Park, D. A. Mazziotti and J. S. Anderson, Nature, 2022, 611, 479–484 CrossRef CAS PubMed.
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- A. E. Thorarinsdottir and T. D. Harris, Chem. Rev., 2020, 120, 8716–8789 CrossRef CAS PubMed.
- K. S. Pedersen, P. Perlepe, M. L. Aubrey, D. N. Woodruff, S. E. Reyes-Lillo, A. Reinholdt, L. Voigt, Z. Li, K. Borup, M. Rouzières, D. Samohvalov, F. Wilhelm, A. Rogalev, J. B. Neaton, J. R. Long and R. Clérac, Nat. Chem., 2018, 10, 1056–1061 CrossRef CAS PubMed.
- P. Perlepe, I. Oyarzabal, A. Mailman, M. Yquel, M. Platunov, I. Dovgaliuk, M. Rouzières, P. Négrier, D. Mondieig, E. A. Suturina, M.-A. Dourges, S. Bonhommeau, R. A. Musgrave, K. S. Pedersen, D. Chernyshov, F. Wilhelm, A. Rogalev, C. Mathonière and R. Clérac, Science, 2020, 370, 587–592 CrossRef CAS PubMed.
- P. Perlepe, I. Oyarzabal, L. Voigt, M. Kubus, D. N. Woodruff, S. E. Reyes-Lillo, M. L. Aubrey, P. Négrier, M. Rouzières, F. Wilhelm, A. Rogalev, J. B. Neaton, J. R. Long, C. Mathonière, B. Vignolle, K. S. Pedersen and R. Clérac, Nat. Commun., 2022, 13, 5766 CrossRef CAS PubMed.
- M. A. Dunstan, A. S. Manvell, N. J. Yutronkie, F. Aribot, J. Bendix, A. Rogalev and K. S. Pedersen, Nat. Chem., 2024, 16, 735–740 CrossRef CAS PubMed.
- L. Münzfeld, S. Gillhuber, A. Hauser, S. Lebedkin, P. Hädinger, N. D. Knöfel, C. Zovko, M. T. Gamer, F. Weigend, M. M. Kappes and P. W. Roesky, Nature, 2023, 620, 92–96 CrossRef PubMed.
- C. H. Booth, M. D. Walter, M. Daniel, W. W. Lukens and R. A. Andersen, Phys. Rev. Lett., 2005, 95, 267202 CrossRef CAS PubMed.
- J. Arvanitidis, K. Papagelis, S. Margadonna, K. Prassides and A. N. Fitch, Nature, 2003, 425, 599–602 CrossRef CAS PubMed.
- M. Ballesteros-Rivas, H. Zhao, A. Prosvirin, E. W. Reinheimer, R. A. Toscano, J. Valdés-Martínez and K. R. Dunbar, Angew. Chem., Int. Ed., 2012, 51, 5124–5128 CrossRef CAS PubMed.
- J. G. Park, D. E. Jaramillo, Y. Shi, H. Z. H. Jiang, H. Yusuf, H. Furukawa, E. D. Bloch, D. S. Cormode, J. S. Miller, T. D. Harris, E. Johnston-Halperin, M. E. Flatté and J. R. Long, ACS Cent. Sci., 2023, 9, 777–786 CrossRef CAS PubMed.
- B. F. Abrahams, R. W. Elliott, T. A. Hudson, R. Robson and A. L. Sutton, CrystEngComm, 2018, 20, 3131–3152 RSC.
- Ö. Üngör and M. Shatruk, Polyhedron, 2020, 177, 114254 CrossRef.
- T. Mochida, Y. Funasako, S. Yamazaki and H. Mori, Eur. J. Inorg. Chem., 2014, 3920–3926 CrossRef CAS.
- A. A. Trifonov, I. D. Gudilenkov, G. K. Fukin, A. V. Cherkasov and J. Larionova, Organometallics, 2009, 28, 3421–3425 CrossRef CAS.
- N. A. Piro, J. R. Robinson, P. J. Walsh and E. J. Schelter, Coord. Chem. Rev., 2014, 260, 21–36 CrossRef CAS.
- C. Uruburo, D. M. R. Y. P. Rupasinghe, H. Gupta, R. M. Knieser, L. M. Lopez, M. H. Furigay, R. F. Higgins, P. Pandey, M. R. Baxter, P. J. Carroll, M. Zeller, S. C. Bart and E. J. Schelter, Inorg. Chem., 2024, 63(21), 9418–9426 CrossRef CAS PubMed.
- G. B. Panetti, D.-C. Sergentu, M. R. Gau, P. J. Carroll, J. Autschbach, P. J. Walsh and E. J. Schelter, Nat. Commun., 2021, 12, 1713 CrossRef CAS PubMed.
- Y. Qiao, H. Yin, L. M. Moreau, R. Feng, R. F. Higgins, B. C. Manor, P. J. Carroll, C. H. Booth, J. Autschbach and E. J. Schelter, Chem. Sci., 2021, 12, 3558–3567 RSC.
- J. A. Bogart, C. A. Lippincott, P. J. Carroll, C. H. Booth and E. J. Schelter, Chem. - Eur. J., 2015, 21, 17850–17859 CrossRef CAS PubMed.
- N. T. Rice, I. A. Popov, D. R. Russo, T. P. Gompa, A. Ramanathan, J. Bacsa, E. R. Batista, P. Yang and H. S. La Pierre, Chem. Sci., 2020, 11, 6149–6159 RSC.
- I. J. Casely, S. T. Liddle, A. J. Blake, C. Wilson and P. L. Arnold, Chem. Commun., 2007, 5037–5039 RSC.
- A. J. Gremillion, J. Ross, X. Yu, P. Ishtaweera, R. Anwander, J. Autschbach, G. A. Baker, S. P. Kelley and J. R. Walensky, Inorg. Chem., 2024, 63(21), 9602–9609 CrossRef CAS PubMed.
- P. Dröse, A. R. Crozier, S. Lashkari, J. Gottfriedsen, S. Blaurock, C. G. Hrib, C. Maichle-Mössmer, C. Schädle, R. Anwander and F. T. Edelmann, J. Am. Chem. Soc., 2010, 132, 14046–14047 CrossRef PubMed.
- D. Schneider, N. Harmgarth, F. T. Edelmann and R. Anwander, Chem. - Eur. J., 2017, 23, 12243–12252 CrossRef CAS PubMed.
- L. Hirneise, J. Langmann, G. Zitzer, L. Ude, C. Maichle-Mössmer, W. Scherer, B. Speiser and R. Anwander, Organometallics, 2021, 40, 1786–1800 CrossRef CAS.
- P. Pandey, X. Wang, H. Gupta, P. W. Smith, E. Lapsheva, P. J. Carroll, A. M. Bacon, C. H. Booth, S. G. Minasian, J. Autschbach, E. Zurek and E. J. Schelter, ACS Appl. Mater. Interfaces, 2024, 16(14), 17857–17869 CrossRef CAS PubMed.
- E. J. Schelter, D. E. Morris, B. L. Scott, J. D. Thompson and J. L. Kiplinger, Inorg. Chem., 2007, 46, 5528–5536 CrossRef CAS PubMed.
- Â. Domingos, N. Marques, A. Pires de Matos, M. G. Silva-Valenzuela and L. B. Zinner, Polyhedron, 1993, 12, 2545–2549 CrossRef.
- R. E. Long, R. A. Sparks and K. N. Trueblood, Acta Crystallogr., 1965, 18, 932–939 CrossRef CAS.
- A. L. Sutton, B. F. Abrahams, D. M. D'Alessandro, R. W. Elliott, T. A. Hudson, R. Robson and P. M. Usov, CrystEngComm, 2014, 16, 5234–5243 RSC.
- S. Fukuzumi and K. Ohkubo, Chem. - Eur. J., 2000, 6, 4532–4535 CrossRef CAS PubMed.
- E. M. Aricó, B. Kanellakopulos, C. Apostolidis and L. B. Zinner, J. Alloys Compd., 1998, 275–277, 798–800 CrossRef.
- M. B. Robin and P. Day, in Mixed Valence Chemistry-A Survey and Classification, Advances in Inorganic Chemistry and Radiochemistry, ed. Emeléus, H. J., Sharpe, A. G., Academic Press, 1968, vol. 10, pp. 247–422 Search PubMed.
- S. Shimomura, R. Matsuda, T. Tsujino, T. Kawamura and S. Kitagawa, J. Am. Chem. Soc., 2006, 128, 16416–16417 CrossRef CAS PubMed.
- P. A. Albrecht, S. M. Rupf, M. Sellin, J. Schlögl, S. Riedel and M. Malischewski, Chem. Commun., 2022, 58, 4958–4961 RSC.
- S. Fukuzumi, Org. Biomol. Chem., 2003, 1, 609–620 RSC.
- J. R. Robinson, C. H. Booth, P. J. Carroll, P. J. Walsh and E. J. Schelter, Chem. - Eur. J., 2013, 19, 5996–6004 CrossRef CAS PubMed.
- R. Gross and W. Kaim, Angew. Chem., Int. Ed. Engl., 1987, 26, 251–253 CrossRef.
- O. Kahn, Molecular Magnetism, Wiley, 1993 Search PubMed.
- P. Pandey, Q. Yang, M. R. Gau and E. J. Schelter, Dalton Trans., 2023, 52, 5909–5917 RSC.
- Y. Qiao and E. J. Schelter, Acc. Chem. Res., 2018, 51, 2926–2936 CrossRef CAS PubMed.
- H. Tamaya, H. Nakano and T. Iimori, J. Lumin., 2017, 192, 203–207 CrossRef CAS.
- P. N. Hazin, J. W. Bruno and H. G. Brittain, Organometallics, 1987, 6, 913–918 CrossRef CAS.
- Y. Qiao, D.-C. Sergentu, H. Yin, A. V. Zabula, T. Cheisson, A. McSkimming, B. C. Manor, P. J. Carroll, J. M. Anna, J. Autschbach and E. J. Schelter, J. Am. Chem. Soc., 2018, 140, 4588–4595 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic Details, characterization, additional magnetometry, vibrational spectroscopy, and X-ray crystallographic data. CCDC 2343799. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4cc01478b |
| This journal is © The Royal Society of Chemistry 2024 |