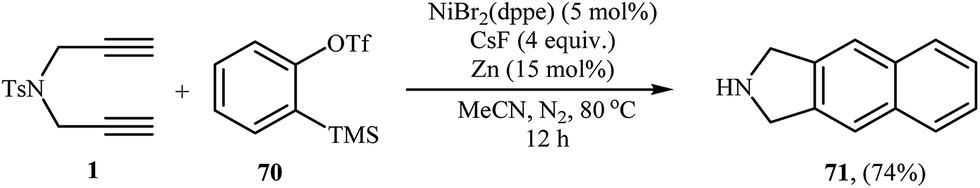DOI:
10.1039/C7RA05398C
(Review Article)
RSC Adv., 2017,
7, 43716-43736
Transition metal-catalyzed [2 + 2 + 2] cycloaddition of nitrogen-linked 1,6-diynes: a straightforward route to fused pyrrolidine systems
Received
12th May 2017
, Accepted 2nd August 2017
First published on 11th September 2017
Abstract
Transition metal-catalyzed [2 + 2 + 2] cycloadditions of nitrogen-linked 1,6-diynes with unsaturated motifs, such as alkynes, alkenes, nitriles, ketenes and isocyanates, have recently attracted more attention from synthetic organic chemists because of their high efficiency in the construction of numerous pyrrolidine based systems. Utilizing different chiral transition metal catalysts to access challenging chiral skeletons has been extensively explored in recent years. We hope to highlight the power of [2 + 2 + 2] cycloaddition chemistry to access a variety of fused pyrrolidine structures from inexpensive and easily available nitrogen-linked 1,6-diynes. The literature has been surveyed until the end of 2016.
 Mirzaagha Babazadeh | Mirzaagha Babazadeh is Associate Professor of Organic Chemistry in Department of Chemistry at Tabriz Branch, Islamic Azad University, Tabriz, Iran. He was born in Tabriz, Iran, in 1973. He received his B.S. degree in Applied Chemistry (1996) and his M.S. degree in Organic Chemistry from University of Tabriz, Tabriz, Iran (1999) under the supervision of Prof. A. A. Entezami and Prof. H. Namazi. He completed his Ph.D. degree under the supervision of Prof. K. D. Safa in University of Tabriz, Tabriz, Iran (2005). His research interests focus on organic synthesis, catalyst, polymer, green chemistry, drug delivery systems, organosilicon compounds and nanocomposites. |
 Somayeh Soleimani-Amiri | Somayeh Soleimani-Amiri was born in Tehran, Iran, in 1975. She received her B.S. degree in Pure Chemistry from Shahid Beheshti University, Tehran, Iran, and her M.S. degree in organic chemistry from Alzahra University, Tehran, Iran, in 2002 under the supervision of Prof. S. H. Abdi Oskooie and Prof. M. M. Heravi. She completed his Ph.D. degree in 2009 under the supervision of Prof. M. Z. Kassaee. Now she is working at Karaj Branch, Islamic Azad University as Assistant Professor. Her research interests include Computational Organic Chemistry, Nano Chemistry, Synthesis of Organic Compounds. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in Pure Chemistry from University of Tabriz, Tabriz, Iran, and his M.S. degree in organic chemistry from Tehran University, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his Ph.D. degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as full Professor of Organic Chemistry. His research interests include Theoretical Organic Chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
 Akram Hosseinian | Akram Hosseinian was born in Ahar, Iran, in 1973. She received her B.S. degree in Pure Chemistry from University of Tehran, Iran, and her M.S. degree in inorganic chemistry from Tarbiat Modares University, Tehran, Iran, in 2000 under the supervision of Prof. A. R. Mahjoub. She completed his Ph.D. degree in 2007 under the supervision of Prof. A. R. Mahjoub. Now she is working at University of Tehran as Asistant Professor. Her research interests include inorganic and organic synthesis, new methodologies in nano material synthesis. |
 Ladan Edjlali | Ladan Edjlali was born in Tabriz, Iran, in 1960. She received her B.S. degree in Applied Chemistry from University of Tabriz, Iran, and her M.S. degree in organic chemistry from University of Tabriz, Tabriz, Iran, in 1993 under the supervision of Prof. Y. Mirzaei. She completed his Ph.D. degree in 2000 under the supervision of Prof. Y. Mirzaei and Prof. S. M. Golabi. Now, she is working at Islamic Azad University, Tabriz Branch as Associate Professor. Her research interests include organic synthesis and new methodologies in organic synthesis. |
1. Introduction
The construction of carbo- and heterocyclic skeletons remains a fertile active area of research because of their ubiquitous occurrence in natural products1 and pharmaceutical agents.2 Transition metal-catalyzed [2 + 2 + 2] cycloadditions of unsaturated substrates, such as alkenes, alkynes, nitriles, imines, and isocyanates, provide a straightforward route to the synthesis of highly substituted carbo- and heterocyclic compounds in one step with ideal atom economy (all the atoms present in the starting materials are present in the product).3 In addition, enantioselective cycloaddition using chiral transition metal catalysts is a powerful synthetic tool for the construction of chiral cyclic frameworks.4 The recent growth in [2 + 2 + 2] cycloaddition reactions has led to a large number of excellent reviews on this chemistry.3,4 However, most of them focused on the general aspects of the field and organization of the reactions based on starting materials and desired products (to give the opportunity to compare the efficacy of procedures) are usually ignored. The literature review reveals that alkynes are the most popular substrates in this chemistry and 1,6-diynes are the most common types of them. Despite great popularity of these substrates in [2 + 2 + 2] cycloaddition reactions, no review in the literature is available covering different aspects (scope, limitations, advances, and so forth) of 1,6-diynes in [2 + 2 + 2] cycloaddition chemistry. Thus, considering the lack of such a review in the literature and in continuation of our interest on N-propargylamines chemistry,5 here we will focus exclusively on transition metal-catalyzed [2 + 2 + 2] cycloaddition of nitrogen-linked 1,6-diynes. The review is organized based on the desired products (e.g. isoindolines, 2,3-dihydro-1H-pyrrolo[3,4-c]pyridine, 2,3-dihydropyrano[4,3-c]pyrrol-6(1H)-ones, and 2,3-dihydro-pyrrolo[3,4-c]pyridin-6-imines), type of reactions (intermolecular and intramolecular), and metal catalysts (e.g. rhodium, ruthenium, cobalt, and nickel). With this review, we aim to inspire researchers to conduct more research in [2 + 2 + 2] cycloaddition of nitrogen-linked 1,6-diyne so that synthesis of important fused pyrrolidine-based natural products and pharmaceuticals through this reaction can become a reality.
2. Isoindolines
2.1. Isoindoline derivatives from nitrogen-linked 1,6-diynes and monoalkynes
2.1.1. Achiral isoindolines via intermolecular [2 + 2 + 2] cycloadditions.
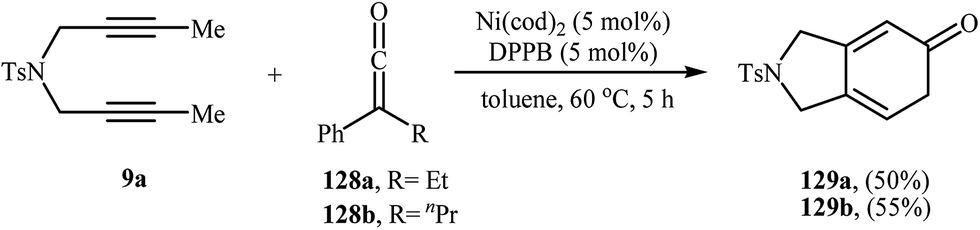
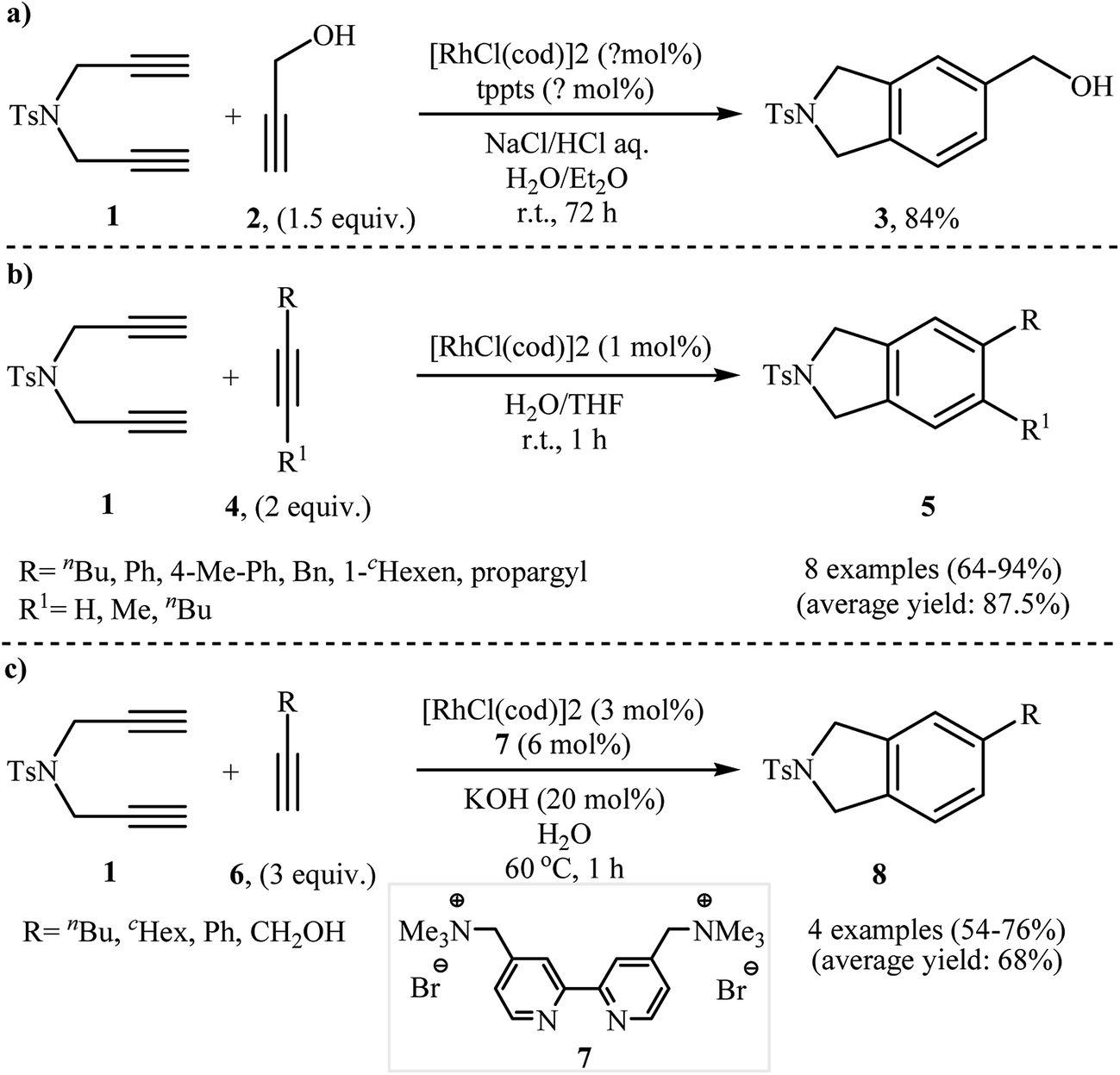
2.1.1.1. Rhodium. Among all the available catalysts, rhodium complexes have proved the most popular and efficient reagents for the [2 + 2 + 2] cycloaddition reaction of nitrogen-linked 1,6-diynes with alkynes. This ability of rhodium complexes discovered by T. Kim nearly 20 years ago.6 In general, Rh-catalyzed [2 + 2 + 2] cycloaddition reactions have been performed in common organic solvents, water, and biphasic systems at temperatures ranging from room temp. upwards. The first example of isoindoline synthesis through this strategy in biphasic system has been reported by Kinoshita, Shinokubo, and Oshima in 2003, when N-tosylated diyne 1 underwent a cycloaddition reaction with propargyl alcohol 2 in the presence of the [RhCl(cod)]2/tppts (tris(m-sulfonatophenyl)-phosphine)/NaCl/HCl/H2O/Et2O system (Scheme 1a).7 Later, Wu's group studied the scope of alkynes in this reaction. Thus, a variety of mono and disubstituted isoindolines were synthesized via selective [2 + 2 + 2] cyclization of 1 with both terminal and internal alkynes 4 using the [Rh(cod)Cl2]/H2O/THF system at room temperature. The results demonstrated that the substrates with terminal alkyne unite gave higher yields than those with internal unite, and arylacetylene derivatives gave higher yields than alkylacetylenes (Scheme 1b).8 With the objective of designing a greener procedure to isoindolines through [2 + 2 + 2] cycloaddition strategy, Tsai and co-workers were able to demonstrate that a series of 5-substituted isoindolines could be obtained from the reaction of 1 with terminal alkynes 6 employing [RhCl(cod)]2/cationic 2,2′-bipyridyl 7 combination as the catalytic system in the most environmentally benign solvent, water (Scheme 1c).9
 |
| | Scheme 1 Rh-catalyzed isoindoline synthesis from terminal dipropargyl amine and alkynes. | |
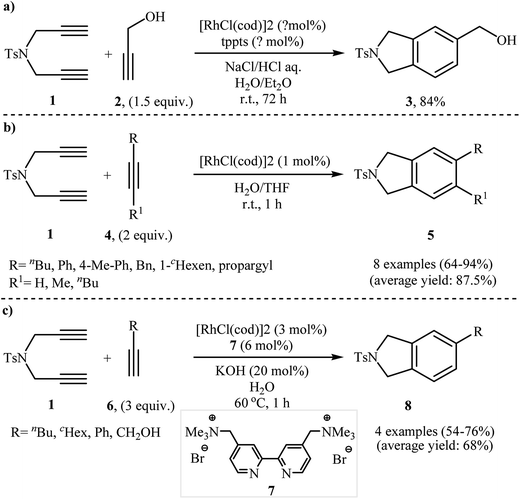
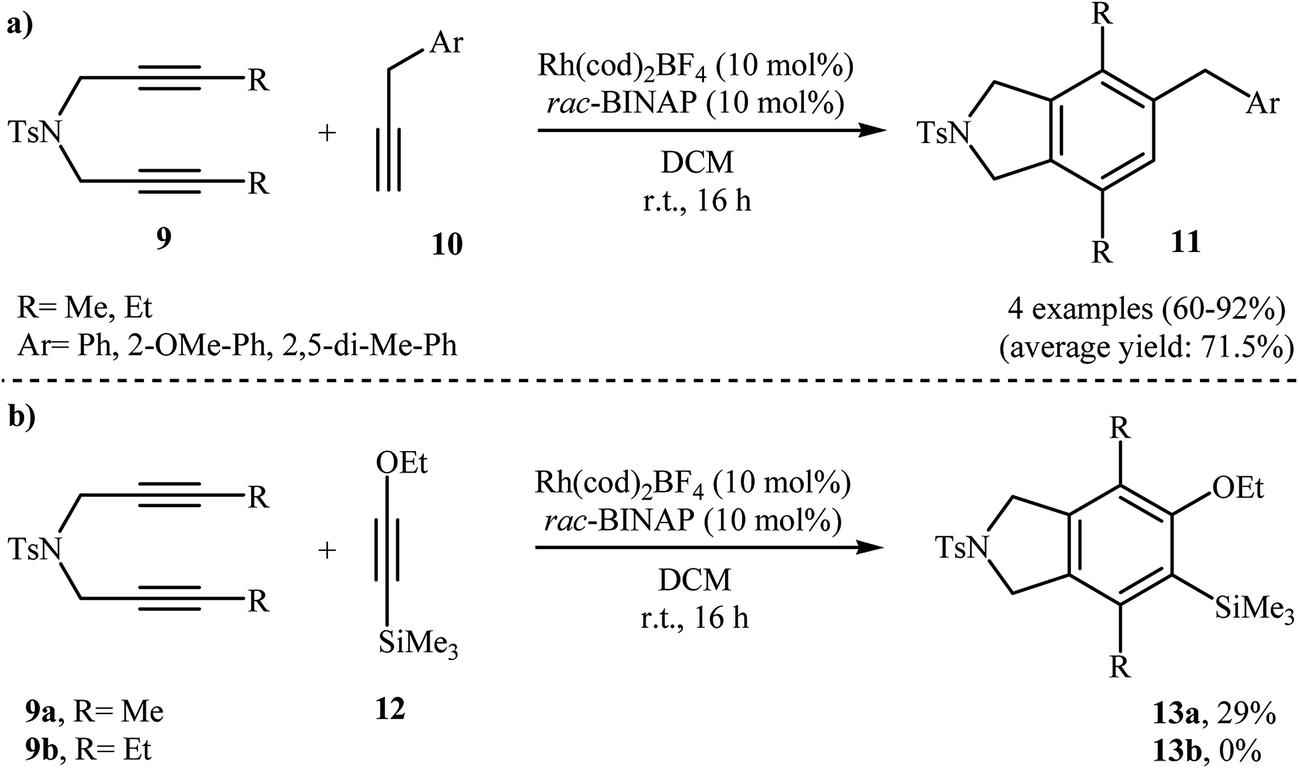
In addition to terminal diyne 1, internal diynes were also reacted with alkynes giving highly substituted isoindoline derivatives. In 2007, Clayden and Moran investigated the reaction of symmetric internal dipropargyl amines 9 with phenyl acetylenes 10 to 4,5,7-trisubstituted N-protected isoindolines 11. After studying a number of solvents, such as heptane, d-chloroform, dichloromethane, toluene, and THF, and catalyst, such as CpCo(CO)2, Rh(cod)2BF4, Pd(PPh3)4, and Pd/C, the system DCM/Rh(cod)2BF4/rac-BINAP at room temperature was found to be optimum with respect to the yield of product isolated (Scheme 2a). The authors also examined the cyclization of 9 with (2-ethoxyethynyl)trimethylsilane 12 under the standard reaction conditions. However, poor results were obtained (Scheme 2b).10
 |
| | Scheme 2 (a) Synthesis of three substituted isoindolines 11 described by Clayden; (b) synthesis of four substituted isoindolines 13 developed by Clayden. | |
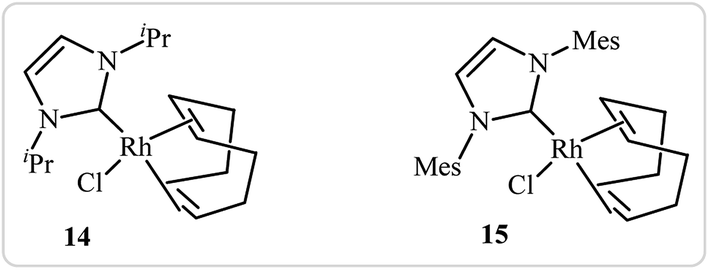
Roglans's group improved the efficiency of these reactions by using rhodium complexes of N-heterocyclic carbenes [RhCl(IPr)(cod)] 14 and [RhCl(IMes)(cod)] 15 as catalyst in ethanol or toluene (Fig. 1).11
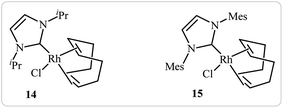 |
| | Fig. 1 Chemical structures of 14 and 15. | |
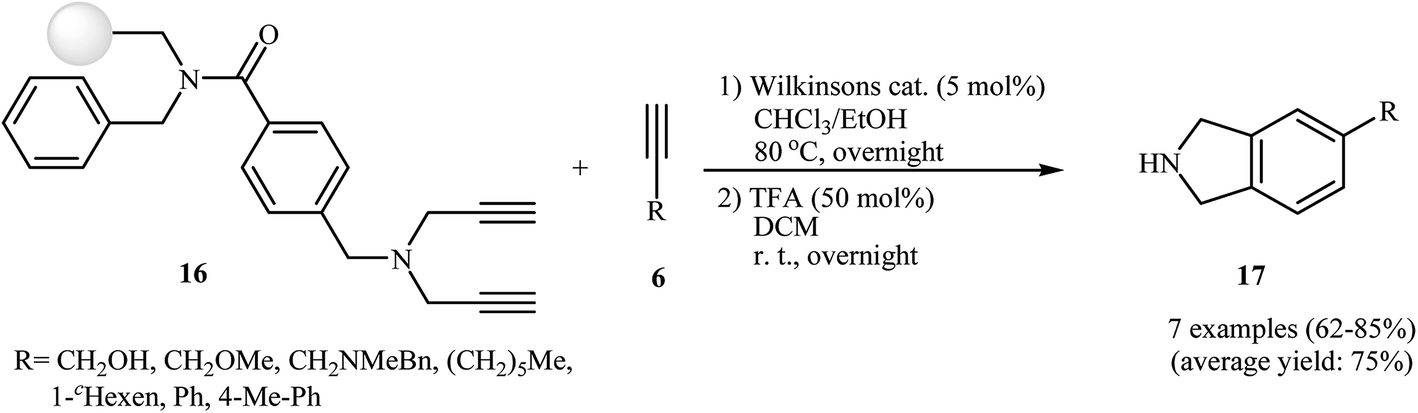
The possibility of solid-support [2 + 2 + 2] cycloaddition reaction of dipropargyl amines with alkynes in the presence of Wilkinson's catalyst was first realized by Sun and co-workers, who synthesized a series of NH-free isoindolines 17 from resin-bound dipropargyl amines 16 and terminal alkynes 6 in good yields (Scheme 3). It should be mentioned that the internal alkynes do not give good yields under this reaction conditions.12 Later, the group of Deiters improved the efficiency of this protocol using [RhCl(PPh3)3] as the catalyst.13
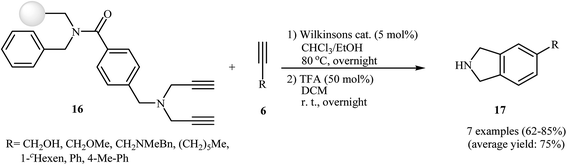 |
| | Scheme 3 Solid-phase synthesis of isoindolines 17. | |
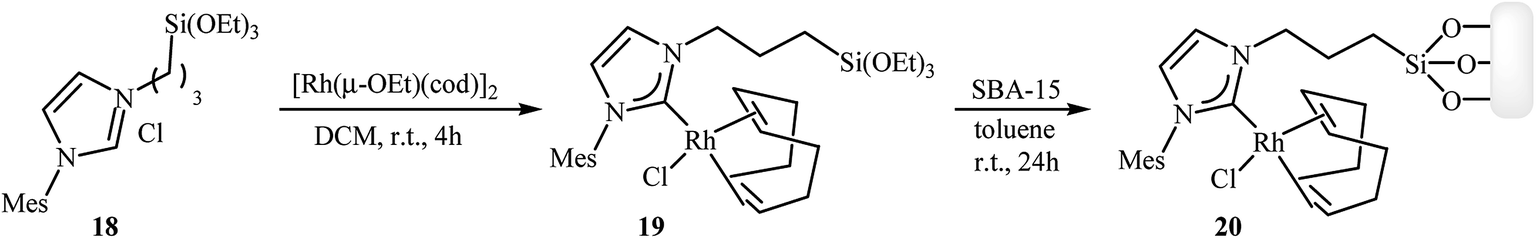
Along this line, recently Roglans and co-workers developed a beautiful rhodium–NHC hybrid silica material, which efficiency catalyzed both inter and intramolecular [2 + 2 + 2] cycloaddition of nitrogen-linked 1,6-diynes. This catalyst was prepared in two steps from silylated imidazolium salt 18: treatment of imidazolium salt 18 with [Rh(μ-OEt)(cod)]2 in anhydrous dichloromethane at room temperature for 4 hours led to the desired monosilylated rhodium complex 19 in a 91% isolated yield. The rhodium–NHC hybrid silica catalyst 20 was then obtained in high yield by grafting of 19 to the mesostructured silica SBA-15 in refluxing anhydrous toluene under N2 atmosphere over the course of 24 hours (Scheme 4).14
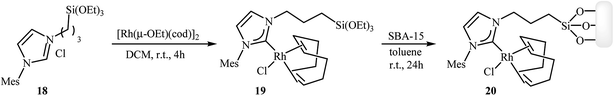 |
| | Scheme 4 Synthesis of rhodium–NHC hybrid silica material 20. | |
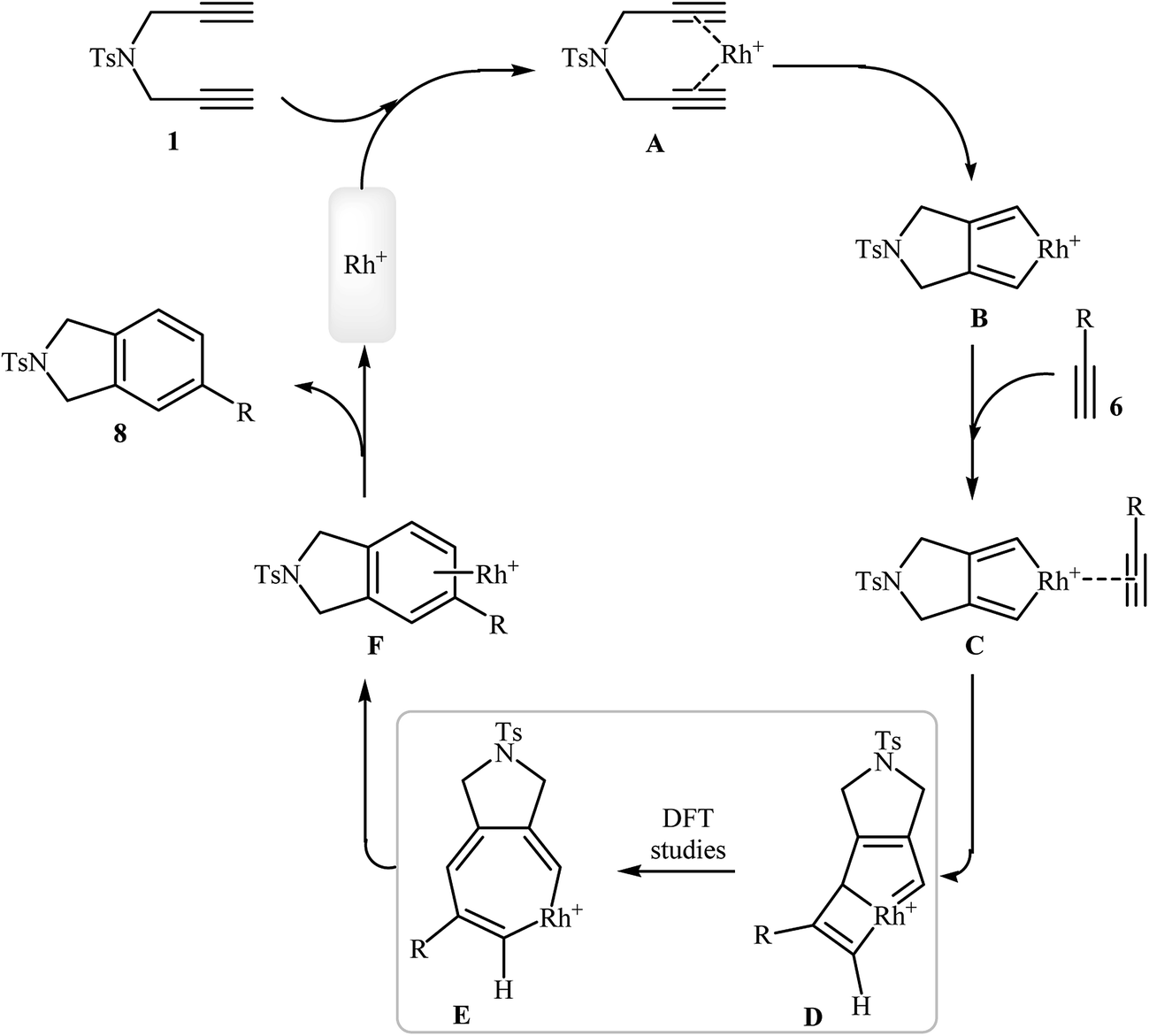
The documented mechanism for Rh-catalyzed [2 + 2 + 2] cycloaddition reaction of dipropargyl amines with alkynes in intermolecular as well as intramolecular fashion is rare, possibly due to the difficulty associated with the presence of paramagnetic species which made impossible the following by NMR. Recently, the groups of A. Roglans were able to detect of key intermediates in Rh(I)-catalyzed [2 + 2 + 2] cycloaddition of dipropargyl amines with alkynes by electrospray ionization mass spectrometry (ESI-MS) and DFT calculations. The authors proposed the mechanistic pathway shown in Scheme 5 that starts with the coordination of the rhodium(I) catalyst to the dipropargyl amine 1, leading to the formation of intermediate A, which undergoes oxidative addition–cyclization to furnish the rhodacyclopentadiene complex B. Its coordination to monoalkyne 6 yields complex C which undergoes monoyne insertion to give the rhodabicyclo[3.2.0]heptatriene intermediate D and then the cycloheptatriene intermediate E. Its reductive elimination yields the complex F, which finally furnishes the final product 8 and regenerates the Rh(I) catalytic species.15
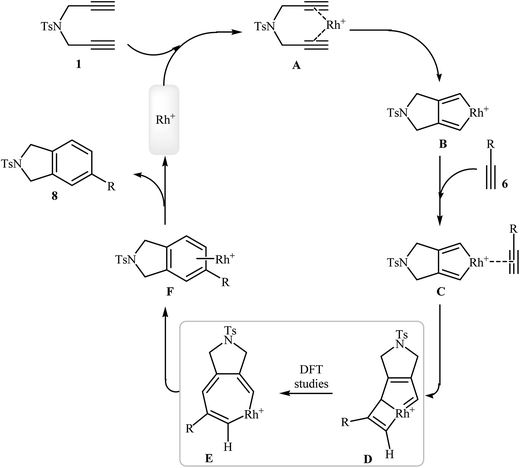 |
| | Scheme 5 Mechanism of the rhodium-catalyzed isoindoline synthesis from dipropargyl amines and alkynes. | |
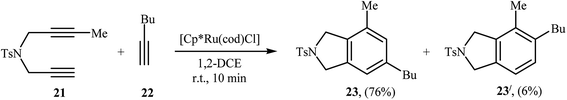 |
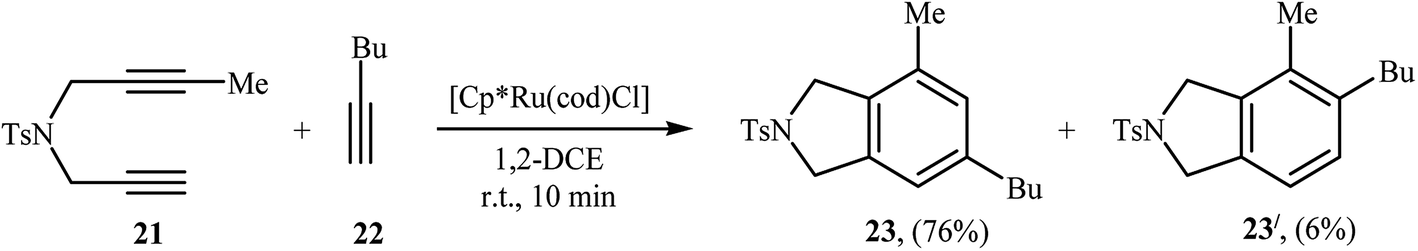
| | Scheme 6 Regioselective Ru(II)-catalyzed [2 + 2 + 2] cycloaddition of 1,6-diyene 21 with hex-1-yne 22. | |
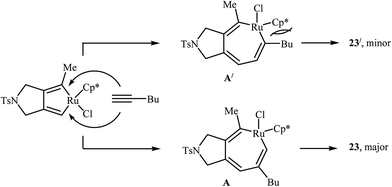 |
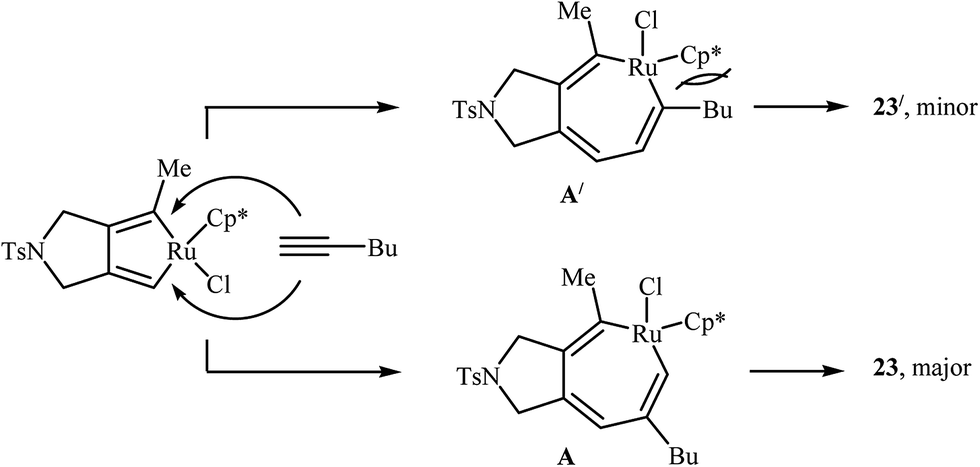
| | Scheme 7 Mechanistic proposal for the reaction in Scheme 6. | |
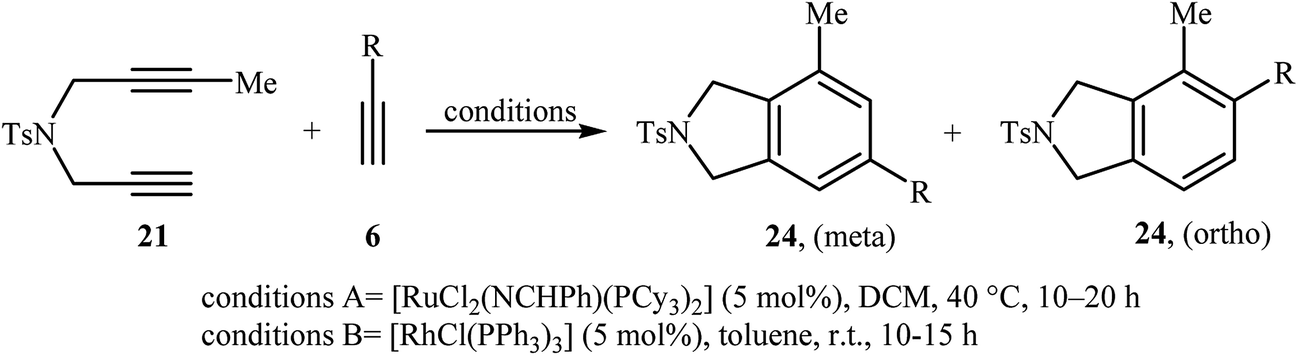
Inspired by this work, Witulski and co-workers compared the Ru-mediated [2 + 2 + 2] cycloaddition reaction of unsymmetrical dipropargyl amine 21 with a series of terminal alkynes 6 with Rh-mediated reaction of the same set of compounds. The results showed that the regioselectivity of the products were drastically changed by changing the catalysts. When [RuCl2(NCHPh)(PCy3)2] was applied as the catalyst, 4,6-disubstituted isoindolines (meta) were obtained as major product, whereas when [RhCl(PPh3)3] is used the yield of 4,6-disubstituted isoindolines are decreased in favor of the 4,5-disubstituted isoindolines (ortho) (Table 1).17
Table 1 Cycloaddition of 1,6-diyne 21 with alkynes 6 catalyzed by Grubbs'catalyst or Wilkinson's catalyst
Deiters groups have applied microwave heating for ruthenium-catalyzed [2 + 2 + 2] cycloaddition of polymer supported dipropargyl amines with both terminal and internal alkynes, generally reporting improved yields in shorter reaction times, with no effect on the regioselectivities.18 More recently it was found that the use of commercially available ruthenium trichloride allowed to catalyze this process at loadings as low as 2 mol% under solvent-free conditions. This system has several advantages such as environmentally friendly processes (Green Chemistry), good to high yields and number of functional groups tolerance (Scheme 8). This procedure was applied on the gram-scale without sacrificing the yield or outcome of the methodology.19 Other applications of Ru catalysts in [2 + 2 + 2] cycloaddition of di(3-iodopropargyl)amine with alkynes to afford 4,7-diiodoisoindoline derivatives are also known.20
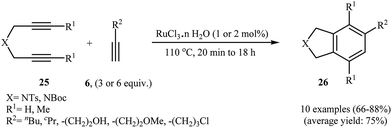 |
| | Scheme 8 Solvent-free Ru-catalyzed synthesis of isoindolines 26 reported by Ratovelomanana-Vidal. | |
Yamamoto and co-workers were able to demonstrate that a series of regioisomeric isoindolinones 28 and 29 be efficiently synthesized from the cyclization of amide-tethered diynes 27 with terminal alkynes 6 by treatment with [Cp*Ru(cod)Cl] in DCM. The results showed that the regiochemical effect of the reaction was strongly dependent on the substitution pattern of 27. Thus, when R1 = R2, the cyclization afford isomer 28 as major product, whereas when R1 ≠ R2, isomers 29 are regioselectively formed in higher yields than isomer 28 (Scheme 9).21 The regioselectivity of this process also depended on the solvent employed in the reaction. The use of cyclopentyl methyl ether (CPME) as the solvent afforded better regioselectivity and a higher yield than DCM for isomers 28.22
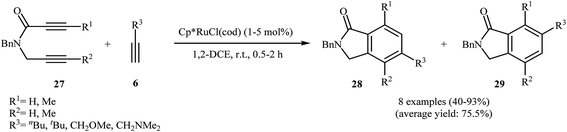 |
| | Scheme 9 Regioselective Ru(II)-catalyzed cycloaddition of 27 with terminal alkynes 6. | |
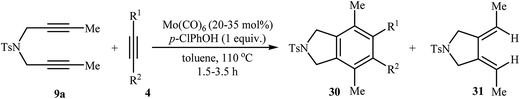
Table 2 [2 + 2 + 2] Cycloaddition of dipropargyl amine 9a with internal alkynes 4 using Mo(CO)6/p-ClPhOH
|
| Entry |
R1 |
R2 |
Equiv. |
30 (%) |
31 (%) |
| 1 |
Et |
Et |
2 |
10 |
8 |
| 2 |
Et |
Et |
5 |
25 |
6 |
| 3 |
Et |
Et |
8 |
34 |
3 |
| 4 |
Et |
Et |
15 |
44 |
5 |
| 5 |
Et |
Et |
15 |
43 |
7 |
| 6 |
Ph |
Ph |
15 |
43 |
5 |
| 7 |
Ph |
Me |
15 |
49 |
3 |
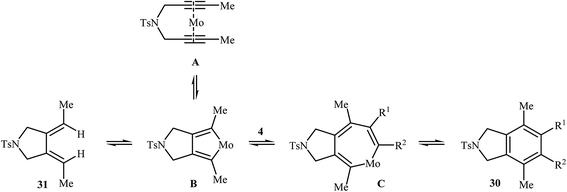 |
| | Scheme 10 Mechanism that accounts for the formation of 30 and 31. | |
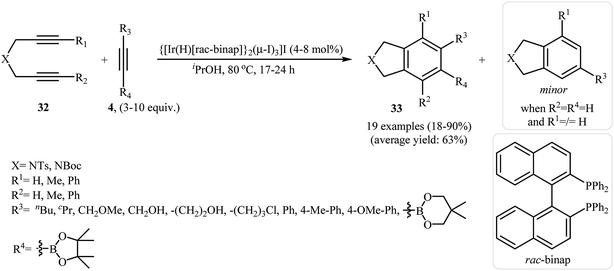 |
| | Scheme 11 Iridium(III)-catalyzed synthesis of highly substituted isoindolines 33. | |
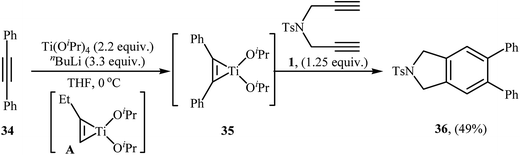 |
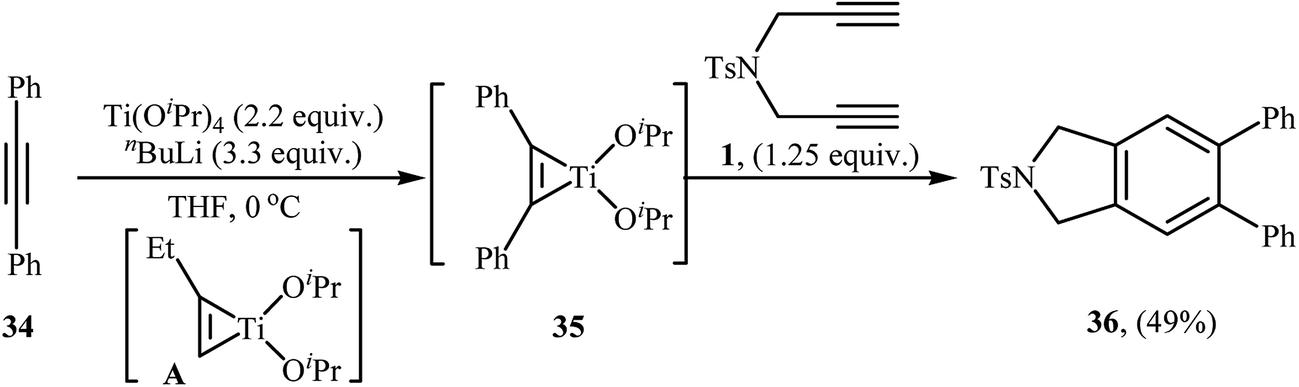
| | Scheme 12 Formation of isoindoline 36 by the Ti(OiPr)4/nBuLi-mediated cycloaddition of dipropargyl amine 1 with internal alkyne 34. | |
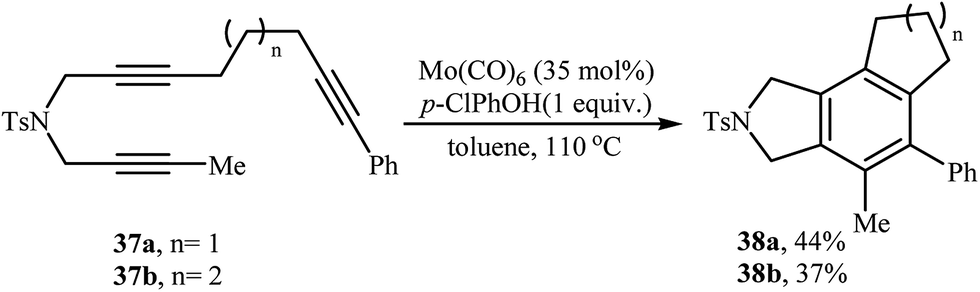
2.1.2. Achiral isoindolines via intramolecular [2 + 2 + 2] cycloadditions. Isoindoline derivatives can also be synthesized by completely intramolecular fashion of [2 + 2 + 2] cycloaddition reaction of three alkyne functionalities. The possibility of this reaction using triyne 37 in the presence of a Mo(CO)6/p-ClPhOH catalyst was first realized by Mori and co-workers, who synthesized highly substituted isoindolines 38 in moderate yields (Scheme 13).23
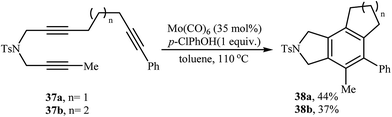 |
| | Scheme 13 Mo-catalyzed synthesis of isoindolines 38 through intramolecular cycloaddition reaction. | |
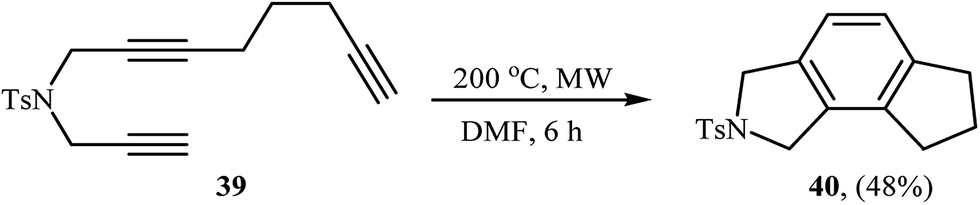
A method involving the microwave-assisted intramolecular cyclization of N-(prop-2-ynyl)octa-2,7-diyn-1-amine 39 was also developed, leading to the formation of 6,7,8-trihydrocyclopenta[e]isoindoline 40 in 48% yield (Scheme 14). The reaction was performed under metal-free conditions using focused microwave heating in DMF.27
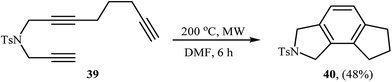 |
| | Scheme 14 Formation of 6,7,8-trihydrocyclopenta[e]isoindoline 40 through non-metal-catalyzed intramolecular alkyne cyclotrimerization reaction promoted by focussed microwave heating. | |
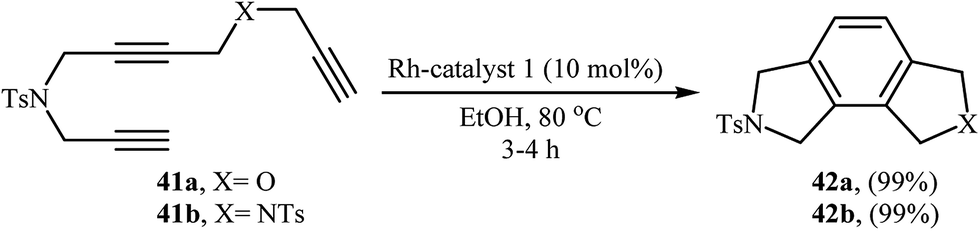
Recently, an excellent method for the generation of furo-isoindoline 42a and pyrrolo-isoindoline 51 from triynes 41a and 41b have been reported by Roglans and co-workers using hybrid silica material 20 as a catalyst in refluxing ethanol (Scheme 15).14 Previously, the group of Aubert applied this methodology to the preparation of 42b using [Co(H)(PMe3)4]/THF system.28 The same authors also synthesized 4,5-dichlorinated analogue of 42b form the corresponding triyne using Cp*Ru(cod)Cl/DCE and CpCo(CO)(dimethylfumarate)/toluene systems.29
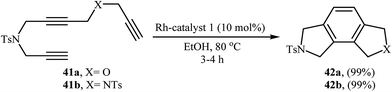 |
| | Scheme 15 Rh-catalyzed synthesis of furo-isoindoline 42a and pyrrolo-isoindoline 42b from triynes 41a and 41b, respectively. | |
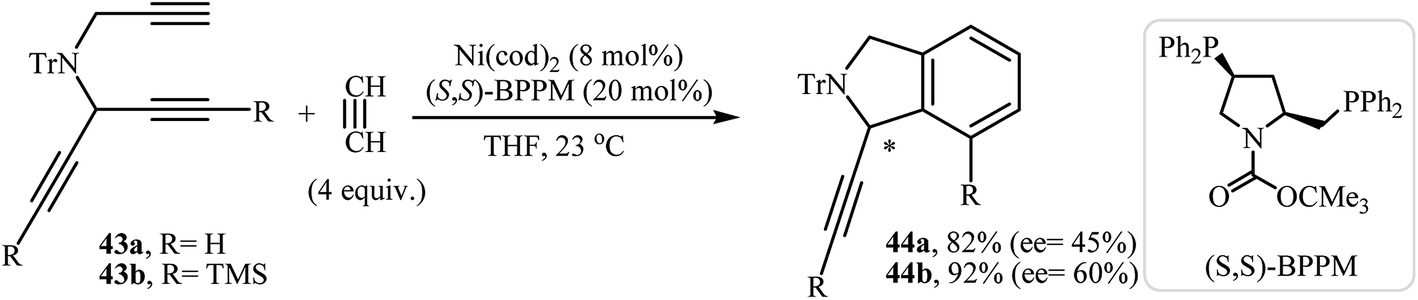
2.1.3. Chiral isoindolines. Mori and co-workers were the first to describe a Ni(0)-catalyzed enantioselective [2 + 2 + 2] cycloadditions of dipropargyl amines with alkynes for the synthesis of chiral isoindolines. They showed that the reaction of unsymmetrical N-(prop-2-ynyl)penta-1,4-diyn-3-amines 43 and acetylene in the presence of Ni(cod)2 as the catalyst and (S,S)-BPPM as the ligand furnishes 1,7-disubstituted isoindolines 44 in good yields with moderate enantioselectivity (Scheme 16).30
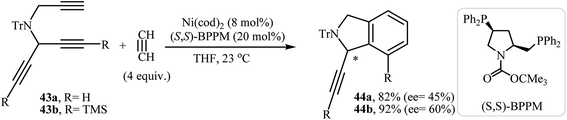 |
| | Scheme 16 Ni(0)-catalyzed enantioselective [2 + 2 + 2] cycloadditions of dipropargyl amines 43 with acetylene. | |
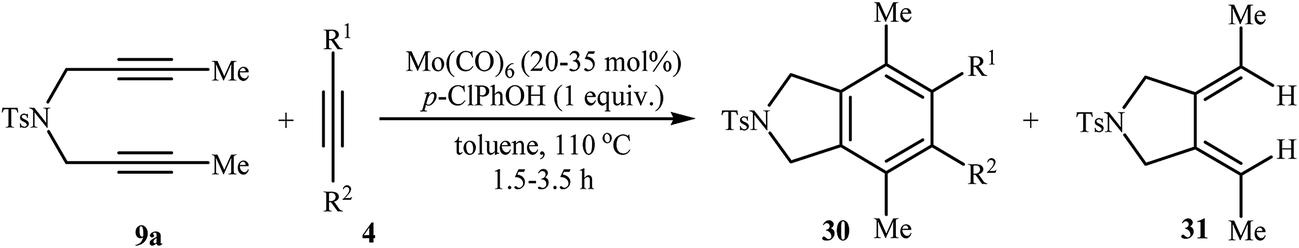
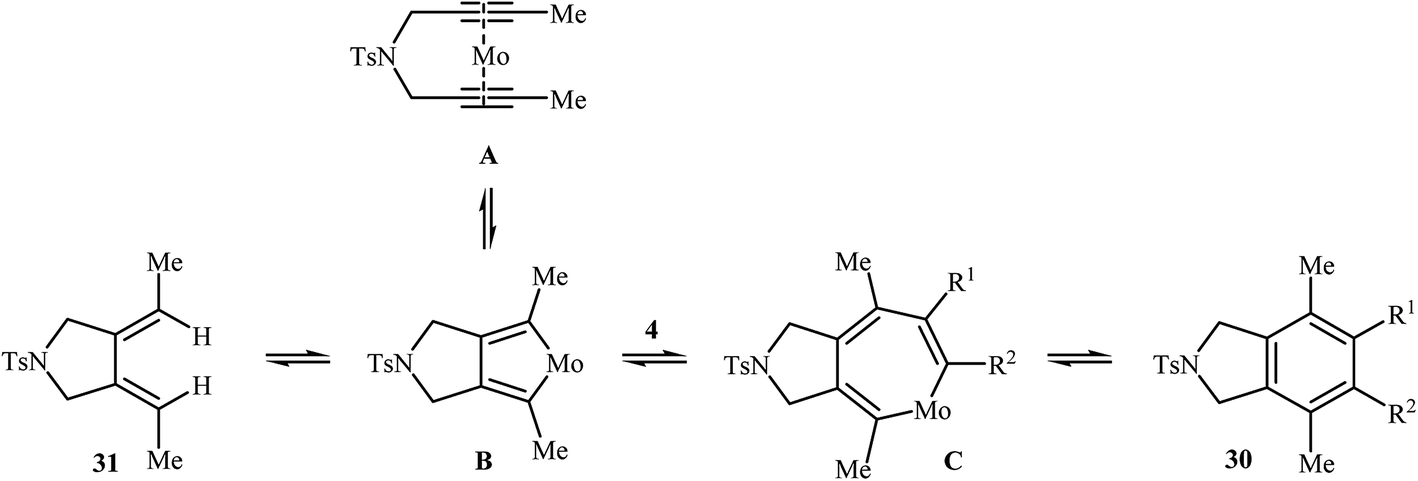
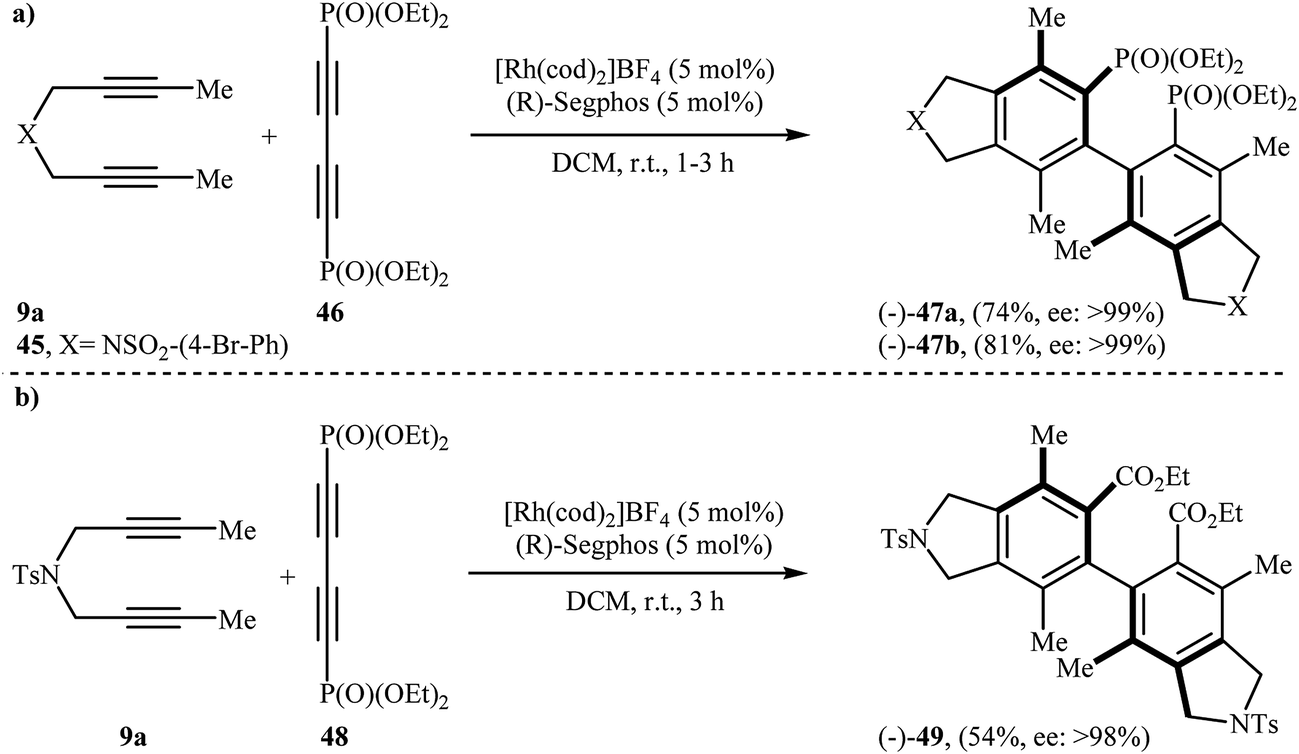
Along this line, Tanaka's group reported the enantioselective synthesis of axially chiral biaryl diphosphonates 47 and dicarboxylate 49 by cationic rhodium(I)/Segphos complex catalyzed [2 + 2 + 2] cycloaddition of internal di(but-2-ynyl)amines 9a (and 45) with a phosphonate- 46 and ester-substituted 1,3-butadiyne 48, respectively. The desired double-annulation products were obtained in moderate to good yields and outstanding enantiomeric excesses (Scheme 17). It is noted that the use of electron-deficient phosphonate- or ester-substituted 1,3-butadiyne as a cycloaddition partner is crucial for the success of this reaction.31
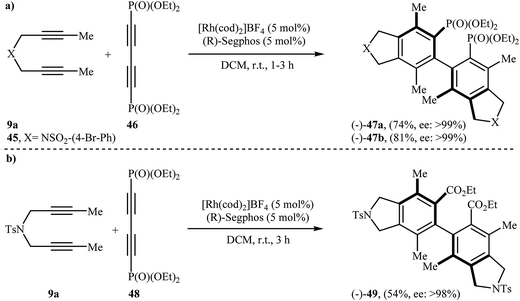 |
| | Scheme 17 (a) Formation of axially chiral biaryl diphosphonates 47 through Rh(I)-catalyzed [2 + 2 + 2] cycloaddition; (b) synthesis of axially chiral biaryl dicarboxylate 49 from 9a and ester-substituted 1,3-butadiyne 48. | |
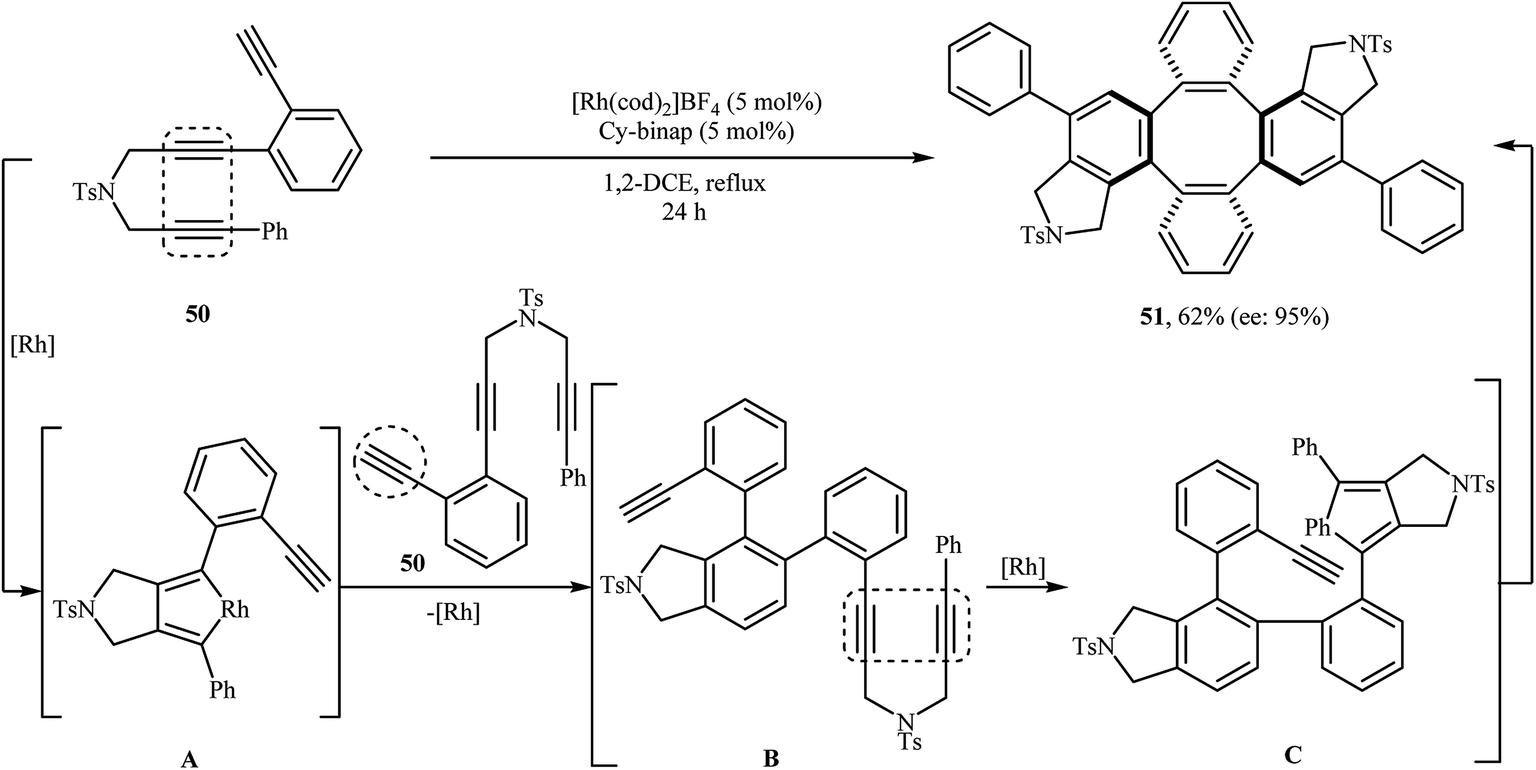
In 2009, Shibata and co-workers were able to synthesize the chiral tetraphenylenes 51 by the consecutive inter- and intramolecular cycloadditions of two N-(3-(2 ethynylphenyl)prop-2-ynyl)-3-phenylprop-2-yn-1-amines 50 using [Rh(cod)2]BF4 as the catalyst and Cy-BINAP as the chiral ligand (Scheme 18).32 Asymmetric Ir-catalyzed [2 + 2 + 2] cycloaddition of dipropargyl amines in both inter- and intramolecular fashions to isoindoline derivatives has also been well documented in the literature by the same research team.33–35
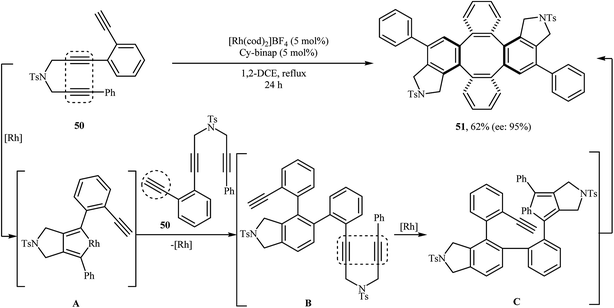 |
| | Scheme 18 Shibata's synthesis of chiral tetraphenylenes 51. | |
2.2. Isoindoline derivatives from nitrogen-linked 1,6-diynes and alkenes
2.2.1. Achiral isoindolines via intermolecular [2 + 2 + 2] cycloadditions.
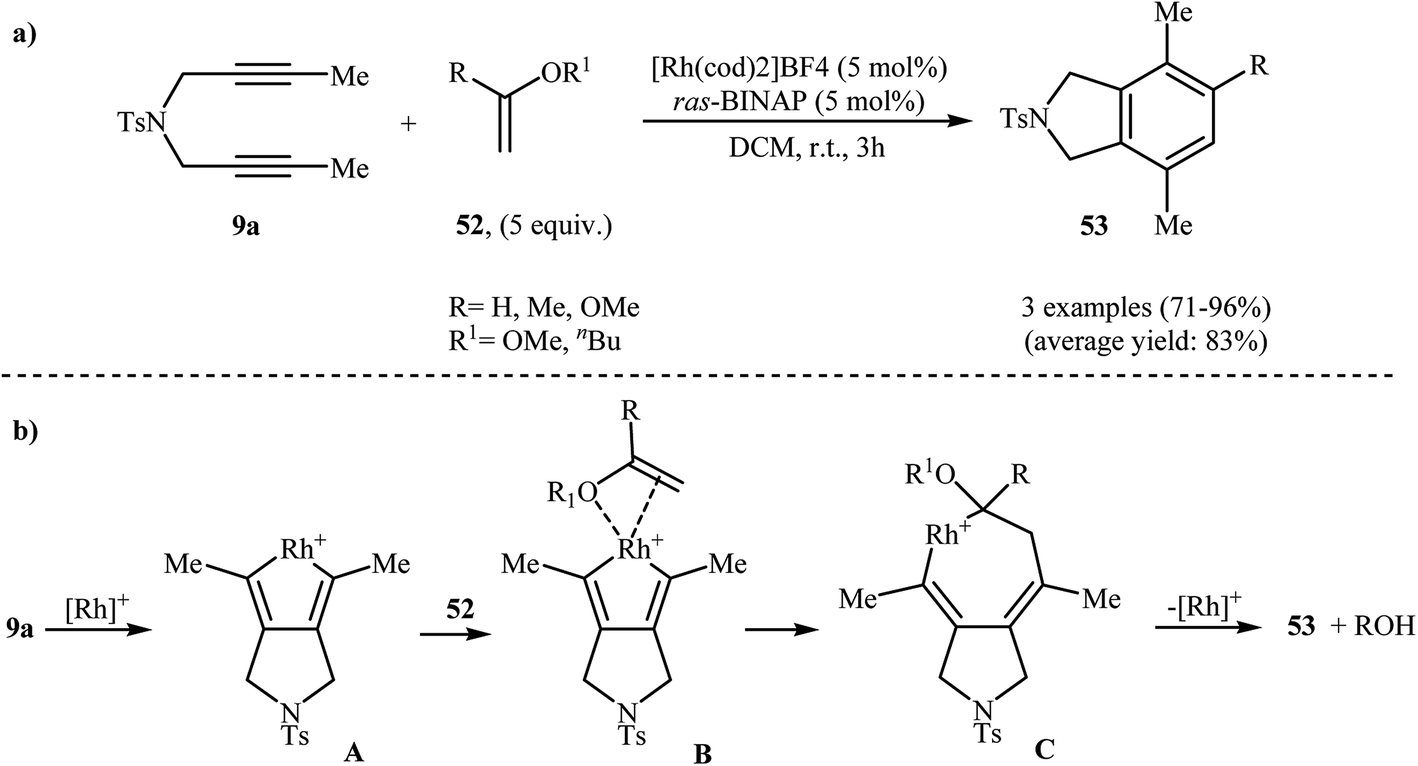
2.2.1.1. Rhodium. Rh-catalyzed intermolecular [2 + 2 + 2] cyclization of nitrogen-linked 1,6-diynes with olefins is a straightforward rout to the isoindoline cores. One of the earliest report of the applicability of these reactions, has been reported by Hara, Hirano, and Tanaka in 2008. They showed that intermolecular [2 + 2 + 2] cycloaddition of internal dipropargyl amine 9a with enol ethers 52 in the presence of [Rh(cod)2]BF4 as the catalyst and ras-BINAP as a biphosphine ligand in DCM led to the corresponding di- and trisubstituted isoindolines 53 in good to excellent yields (Scheme 19a). The mechanistic course of this reaction sequence is shown in Scheme 19b, and involves the initial formation of the rhodacyclopentadiene A from the reaction of diyne 9a with rhodium. Insertion of 52 into one of the Rh–C bonds gives intermediate C through intermediate D. Finally, reductive elimination of rhodium affords the observed isoindolines 53 (Scheme 19b).36,37
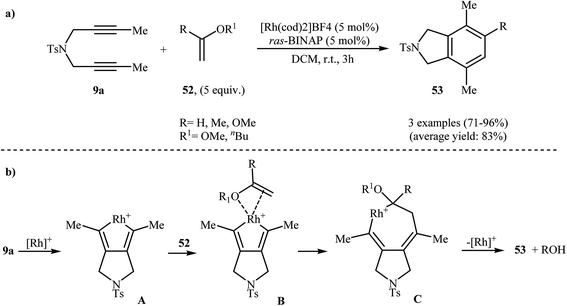 |
| | Scheme 19 (a) Rh(I)-catalyzed [2 + 2 + 2] cycloaddition of 9a with enol ethers 52 to isoindolines 53; (b) mechanism proposed to explain the isoindolines 53 synthesis developed by Tanaka. | |
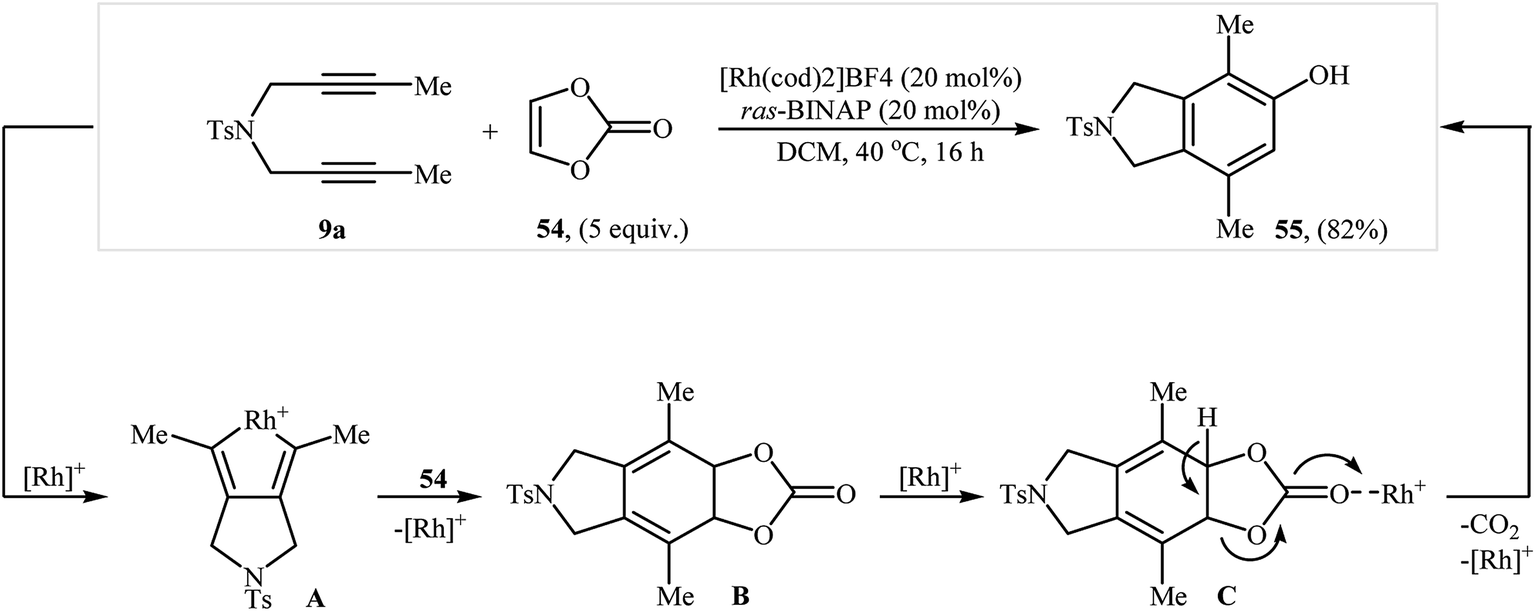
They also showed that treatment of diyne 9a with vinylene carbonate 54 under the aforementioned rhodium catalyst gave the corresponding 4,7-dimethyl-2-tosylisoindolin-5-ol 55 in yield of 82% (Scheme 20).38 More recently, the same authors extended their methodology to cyclization of dipropargyl amines with 2,3-dihydofuran to 2-(isoindolin-6-yl)ethanol derivatives.39
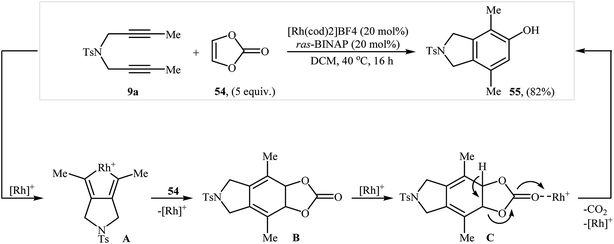 |
| | Scheme 20 Tanaka's synthesis of isoindoline 55 from 9a and vinylene carbonate 54 in the presence of Rh(I)/ras-BINAP. | |
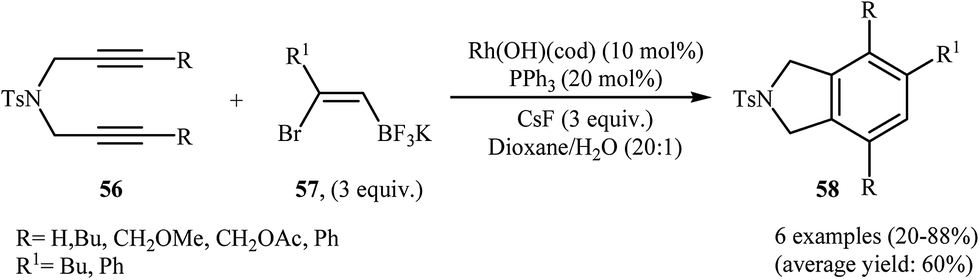
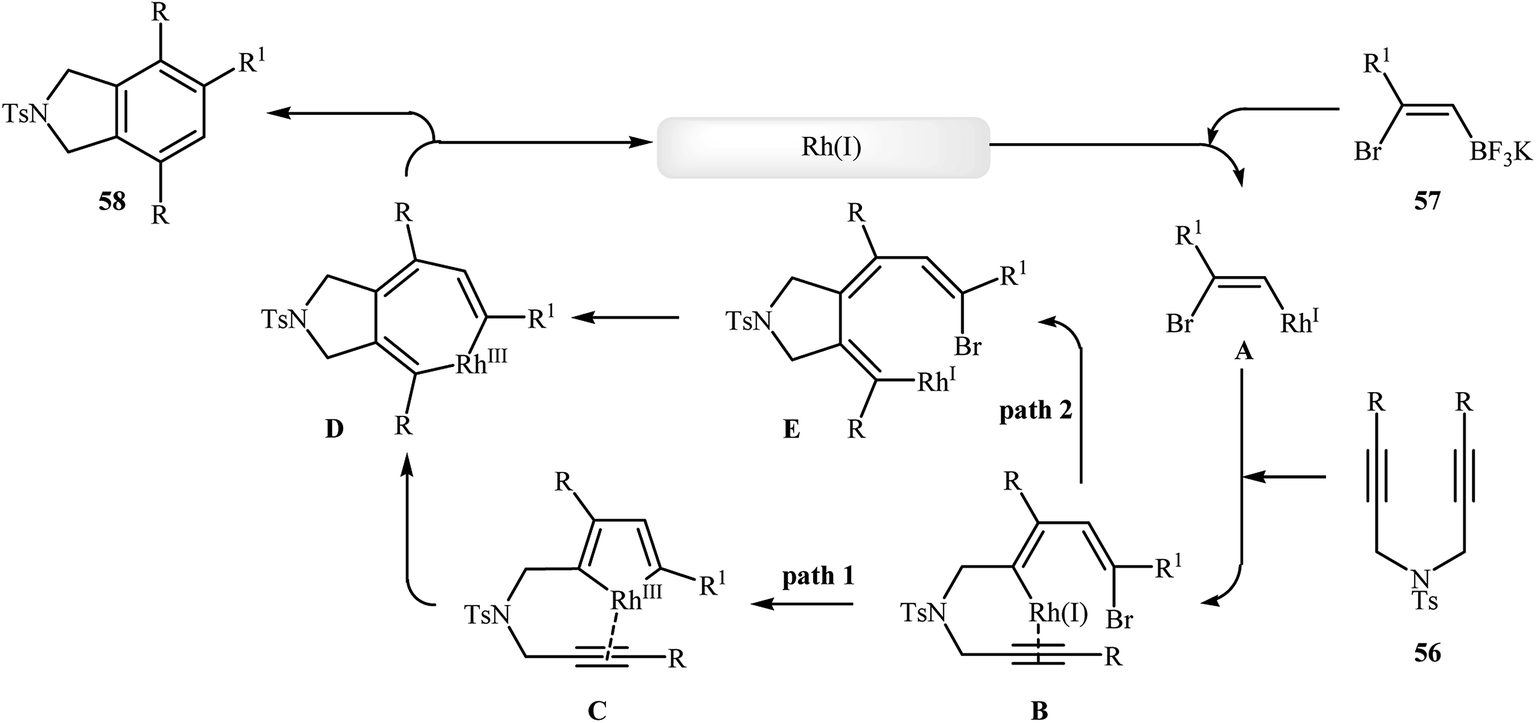
To develop an new protocol for the synthesis of isoindoline derivatives via [2 + 2 + 2] cycloaddition reaction, Tong and co-workers have investigated the cyclization of dipropargyl amines 56 with potassium (Z)-(2-bromovinyl)trifluoroborates 57 in the presence of Rh(OH)(cod)/PPh3/CsF system in dioxin/water, and moderate to good yields of desired products 58 were obtained (Scheme 21). The reaction scope appears that the diynes with internal alkene units are more reactive than those with terminal alkene units. A plausible catalytic cycle is depicted in Scheme 22.40
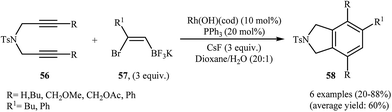 |
| | Scheme 21 Synthesis of highly substituted isoindolines 58 through Rh(I)-catalyzed [2 + 2 + 2] cycloadditions of 1,6-diynes 56 with potassium (Z)-(2-bromovinyl)trifluoroborates 57. | |
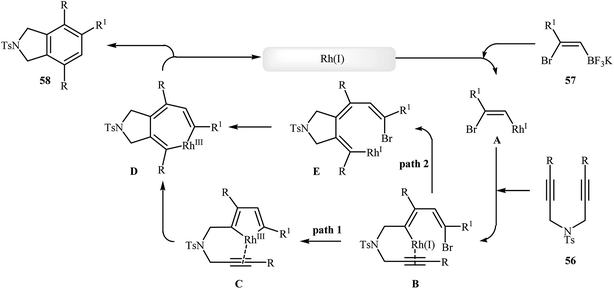 |
| | Scheme 22 Mechanism that accounts for the formation of 58. | |
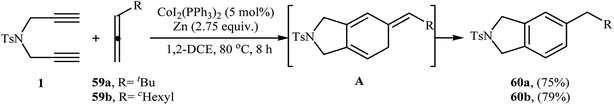 |
| | Scheme 23 Synthesis of isoindolines 60 described by Cheng. | |
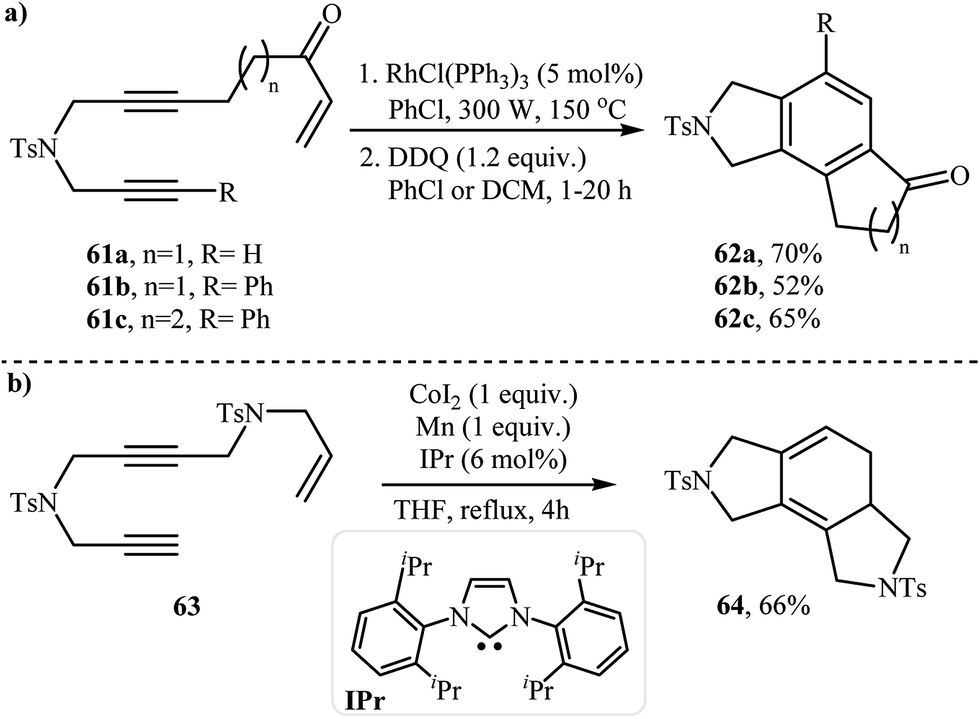
2.2.2. Achiral isoindolines via intramolecular [2 + 2 + 2] cycloadditions. The reported methods for the synthesis of isoindoline cores via intramolecular cyclization of dipropargyl amines with olefins are very scarce and to the best of our knowledge there is only one example for such reactions. A Rh-catalyzed polycyclization of liner diyne-enones 61 has been developed for construction of fused isoindolines 62 in two steps under microwave irradiation. The polycyclization takes place through Rh-catalyzed intramolecular [2 + 2 + 2] cycloaddition of vinyl ketone with the diyne followed by aromatization to give observed products in moderate yields (Scheme 24a).42 Co/Mn-catalyzed intramolecular [2 + 2 + 2] cycloaddition of enediynes 63, provided unsaturated isoindolines 64 instead (Scheme 24b).43
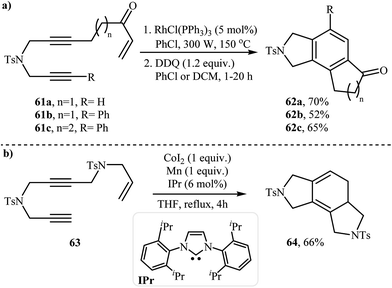 |
| | Scheme 24 (a) Microwave-assisted polycyclization of liner diyne-enones 61; (b) Co-catalyzed intramolecular [2 + 2 + 2] cycloaddition of enediynes 63. | |
2.2.3. Chiral isoindolines. Shibata and co-workers were the first to use a chiral rhodium complex-catalyzed intermolecular [2 + 2 + 2] cycloaddition of dipropargyl amines with olefins, to generate chiral isoindoline derivatives. In their investigations they reacted different unsymmetrical dipropargyl amines 65 with norbornene 66 in the presence of Rh–difluorphos complex, and obtained tetracyclic products 67 in moderate to high yields with good to excellent enantioselectivity. As shown in Table 3 the reaction was more efficient for diynes possessing an ester functionality at their alkyne terminus.44
Table 3 Rh-catalyzed asymmetric cycloaddition of nitrogen-linked 1,6-diyenes 65 with norbornene 66a

|
| Entry |
R1 |
R2 |
Yield (%) |
ee (%) |
| 5,5′-Bis(diphenylphosphino)-2,2,2′,2′-tetrafluoro-4,4′-bi-1,3-benzodioxole. |
| 1 |
Ph |
CO2Me |
83 |
96 |
| 2 |
Ph |
Me |
34 |
69 |
| 3 |
Ph |
COMe |
56 |
95 |
| 4 |
Ph |
CH2OMe |
70 |
69 |
| 5 |
4-OMe-Ph |
CO2Me |
90 |
95 |
| 6 |
4-(CO2Et)-Ph |
CO2Me |
93 |
96 |
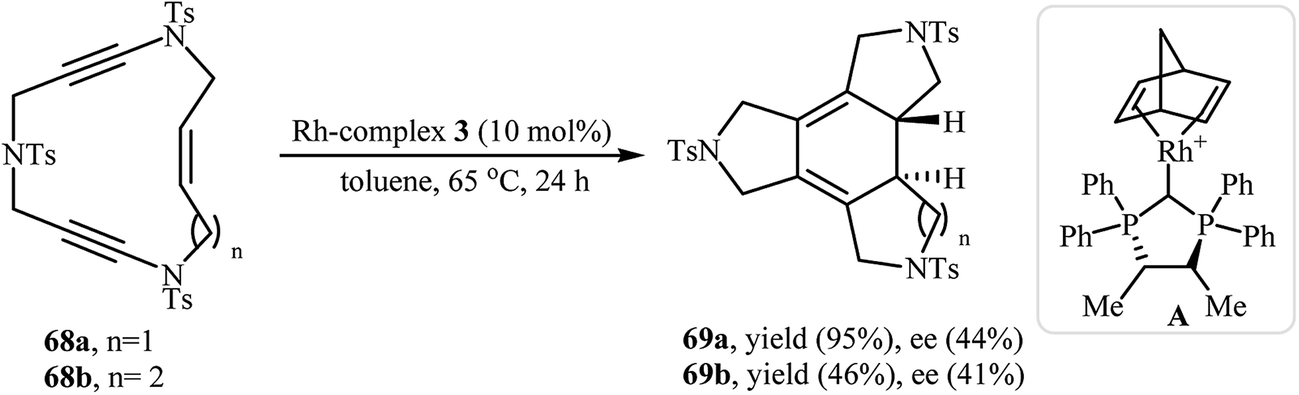
In another interesting report, Roglans and co-workers utilized rhodium complex A in the enantioselective intramolecular [2 + 2 + 2] cycloaddition of enediynic macrocycles 68 to prepare fused tetracycles 69 (Scheme 25).45 Interestingly, using PNSO (N-phosphino tert-butylsulfinamides)/Rh complex A combination as catalyst, the reaction afforded 69 in opposite enantioselectivity to that obtained with complex A.46
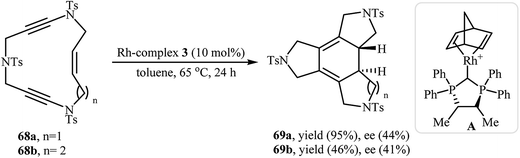 |
| | Scheme 25 Rh(I)-catalyzed [2 + 2 + 2] cycloisomerization of enediyne macrocycles 68. | |
2.3. Isoindoline derivatives from nitrogen-linked 1,6-diyenes and arynes
The intermolecular [2 + 2 + 2] cocyclotrimerization of arynes with diynes represents an interesting method to prepare naphthalene ring systems. The use of this protocol in the synthesis of benzene fused isoindolines has been scarcely studied; in fact, only one example of such a reaction was reported in the literature by Hsieh and Cheng. They observed that treatment of dipropargyl amine 1 with 2-(trimethylsilyl)phenyl trifluoromethanesulfonate 70 (as benzyne precursor) in the presence of NiBr2(dppe)/CsF/Zn system gave 2,3-dihydro-1H-benzo[f]isoindole 71 in yield of 74% (Scheme 26).47
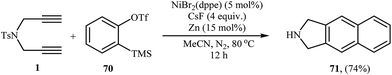 |
| | Scheme 26 Single example of a Ni-catalyzed [2 + 2 + 2] cycloaddition of dipropargyl amine 1 with 2-(trimethylsilyl)phenyl trifluoromethanesulfonate 70. | |
3. 2,3-Dihydro-1H-pyrrolo[3,4-c]pyridine derivatives from nitrogen-linked 1,6-diyenes and nitriles
3.1. Achiral 2,3-dihydro-1H-pyrrolo[3,4-c]pyridines via intermolecular [2 + 2+ 2] cycloadditions
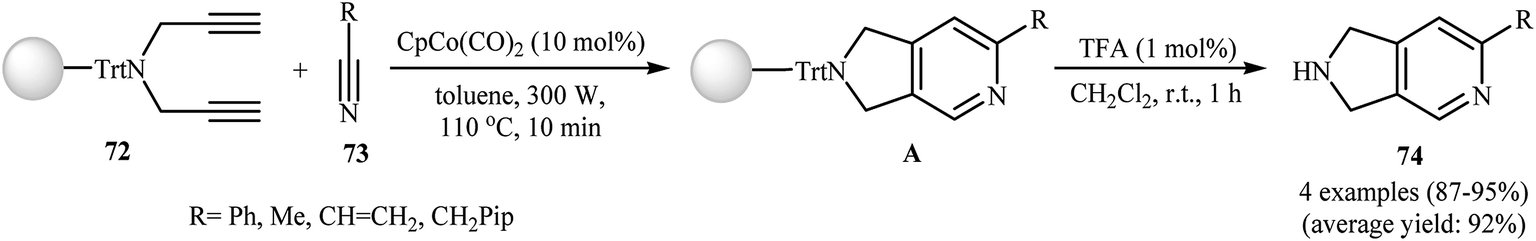
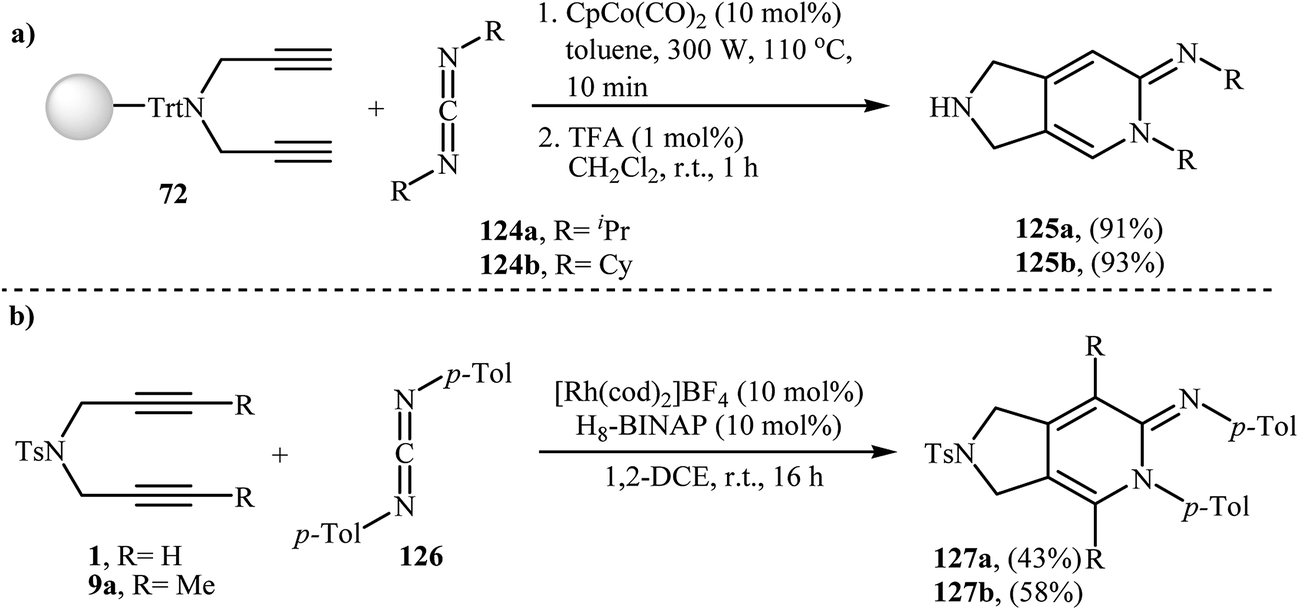
3.1.1. Cobalt. After the pioneering work of Chiusoli and Costa, in 1989,48,49 who showed that dicobalt octacarbonyl complex could catalyze intermolecular [2 + 2 + 2] cycloaddition of dipropargyl amines with nitriles to give 2,3-dihydro-1H-pyrrolo[3,4-c]pyridines, this methodology has developed into a general powerful tool for the synthesis of a wide range of 2,3-dihydro-1H-pyrrolo[3,4-c]pyridine derivatives. Thus, numerous cobalt-based catalytic system have been developed (Table 1). These include: CpCo(PPh3)2,50 dppe/CoCl2–6H2O/Zn,51 [CpCo(CO)(fumarate)],52 [CpCo(cod)],53 and (1-neomenthylindenyl)Co(cod).54 An important study on this interesting field was carried out by Young and Deiters, in which solid-supported N-trityl dipropargyl amine 72 were transformed into 2,3-dihydro-1H-pyrrolo[3,4-c]pyridine derivatives 74 by the treatment with nitriles 73 in the presence of CpCo(CO)2 under microwave irradiation (300 W) for 10 min, and then deprotection of trityl group with trifluoroacetic acid. The reaction tolerates both aromatic and aliphatic nitriles and gave final product in excellent yields (Scheme 27).55 DFT calculations suggest that this transformation takes place through cobaltacyclopentadiene intermediate.56
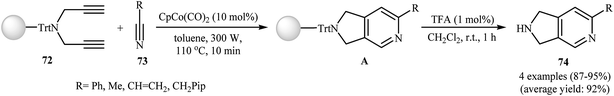 |
| | Scheme 27 Microwave-mediated solid-supported formation of 2,3-dihydro-1H-pyrrolo[3,4-c]pyridine derivatives 74. | |
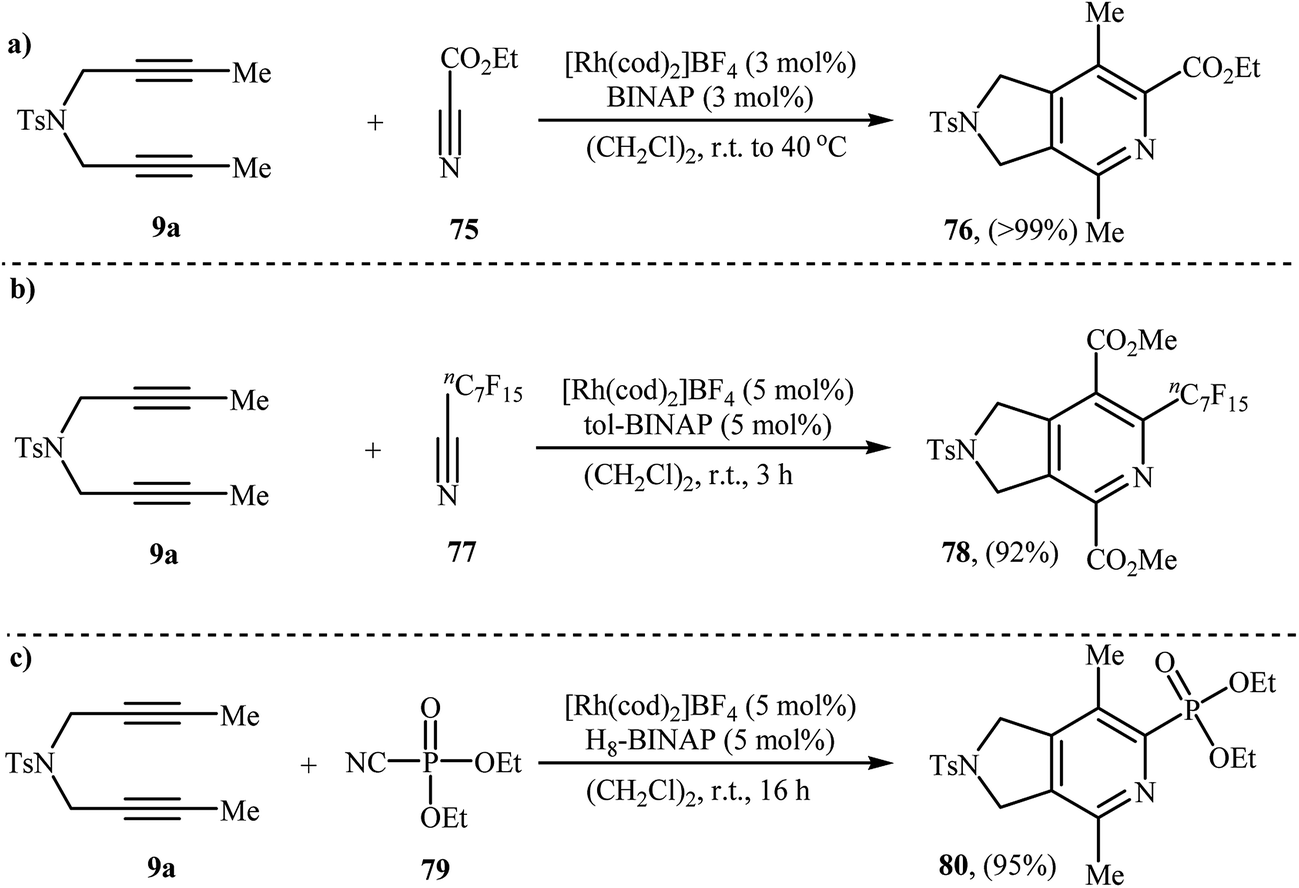
3.1.2. Rhodium. A preliminary example of Rh-catalyzed [2 + 2 + 2] cycloaddition of dipropargyl amines with nitriles has been reported by Tanaka et al., in 2006. They showed that treatment of diyne 9a with ethyl cyanoformate 75 in the presence of [Rh(cod)2]BF4/BINAP system afforded the corresponding 2,3-dihydro-1H-pyrrolo[3,4-c]pyridine 76 in a 99% isolated yield (Scheme 28a).57 Shortly afterwards, the same group has investigated the cycloaddition of 9a with perfluoroalkylacetylene 77 under the aforementioned rhodium catalyst but using a higher catalyst loading (Scheme 28b).58 The authors also reported an example of pyrrolidine-fused pyridine 80 preparation from 9a and diethyl phosphorocyanidate 79 (Scheme 28c).59
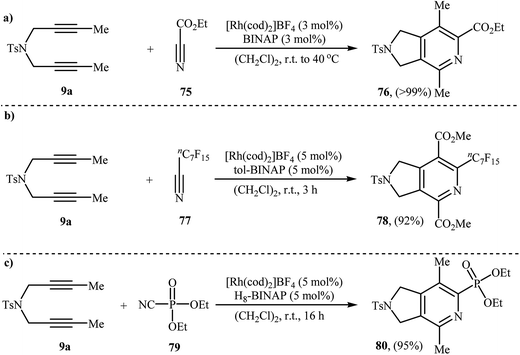 |
| | Scheme 28 Rh-catalyzed [2 + 2 + 2] cycloaddition of nitrogen-linked 1,6-diyene 9a with nitriles developed by Tanaka. | |
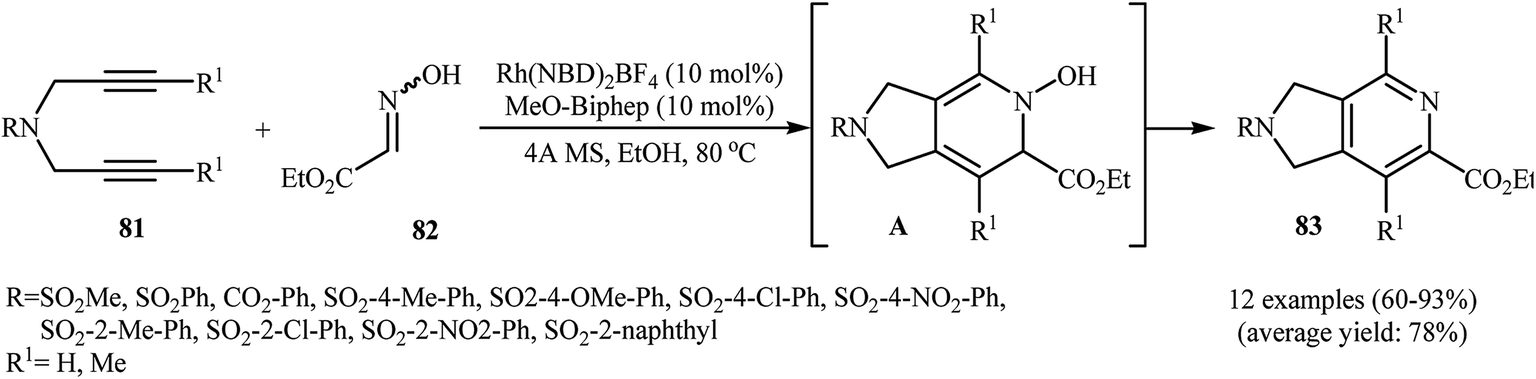
Recently, Wan and co-workers described a general and efficient synthesis of 4,6,7-trisubstituted 2,3-dihydro-1H-pyrrolo[3,4-c]pyridines 83 by the [2 + 2 + 2] cycloaddition of corresponding dipropargyl amines 81 with ethyl 2-(hydroxyimino)acetate 82 employing a Rh(NBD)2BF4/MeO-biphep combination as the catalytic system in refluxing ethanol. This process tolerated various sensitive functional groups and generally provided 83 in good to excellent yields (Scheme 29). However, dipropargyl amines derived from 4-bromobenzene-sulfonamide don't work well under this reaction conditions. The results also demonstrate that the reaction do not give good yields with aromatic aldoximes, and aliphatic aldoximes failed to participate in the reaction.60 The mechanism proposed by the authors to explain this reaction is based on the formation of hydroxylpyridine derivatives A as intermediates from the starting dipropargyl amines and oximes, followed by their spontaneous dehydration.61
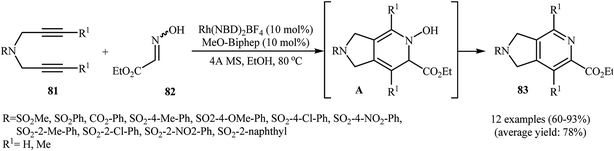 |
| | Scheme 29 Rhodium-catalyzed [2 + 2 + 2] cycloaddition of 1,6-diyenes 81 and ethyl 2-(hydroxyimino)acetate 82 developed by Wan. | |
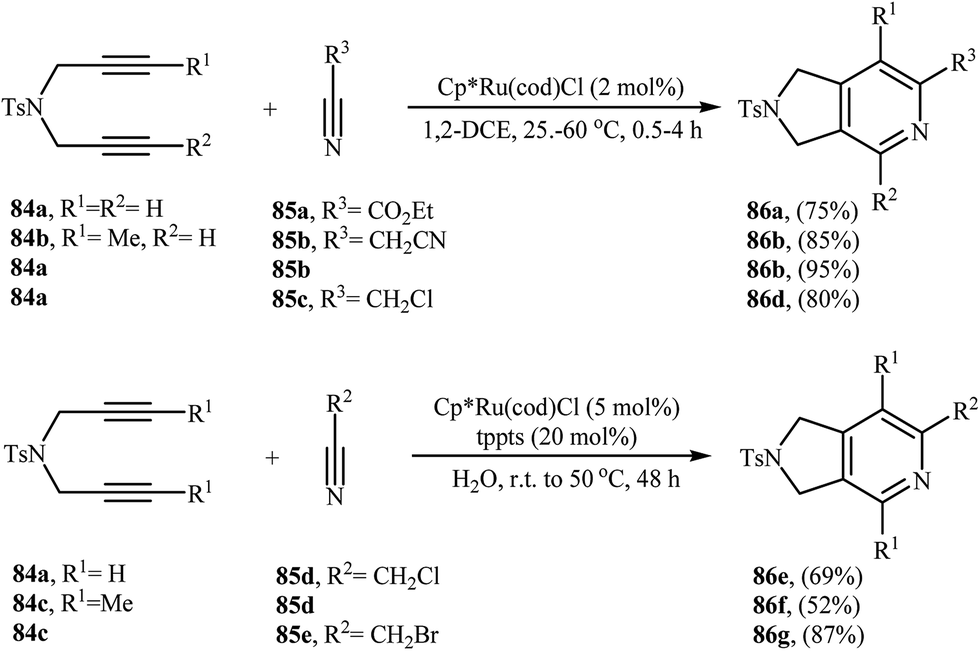
3.1.3. Ruthenium. Although, ruthenium catalyzed [2 + 2 + 2] cycloadditions of nitrogen-linked 1,6-diyne with nitriles are extremely efficient, they has received scarce attention. The most commonly used Ru-complex in general, and particularly in this chemistry, are commercially available Cp*Ru(cod)Cl, though only a few synthetic applications were reported (Scheme 30).62–67
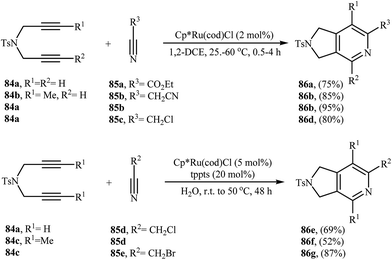 |
| | Scheme 30 Ruthenium catalyzed [2 + 2 + 2] cycloadditions of dipropargyl amines 84 with nitriles 85. | |
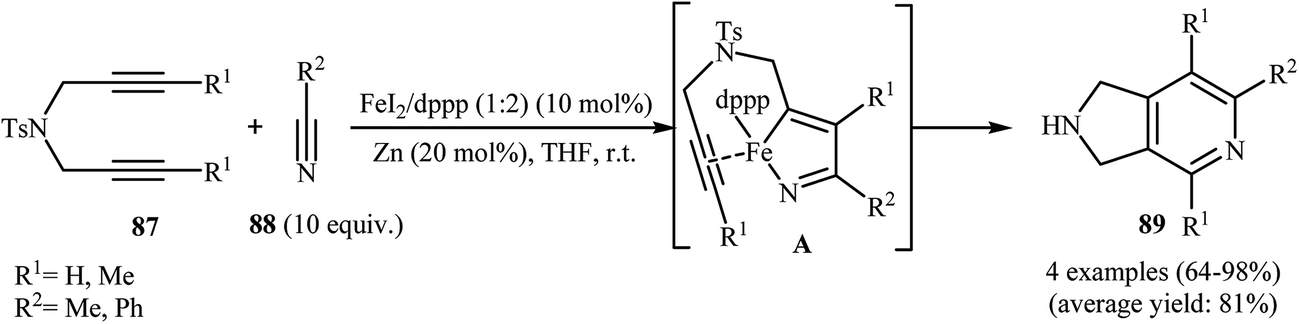
3.1.4. Iron. In 2011, B. Wan and co-workers demonstrated for the first time the usefulness of iron catalysts for the construction of 2,3-dihydro-1H-pyrrolo[3,4-c]pyridines. Thus, in the presence of FeI2/dppp/Zn as catalytic system in anhydrous tetrahydrofuran at room temperature, cycloaddition of dipropargyl amines 87 with nitriles 88 furnished corresponding pyrrolidine-fused pyridines 89 via azaferracyclopentadiene intermediates A (Scheme 31). The results showed that propargyl amines with internal alkyne unite gave higher yields than those with terminal unite, and aryl nitriles are slightly more reactive than alkyl nitriles.68 Shortly after, the same author showed that their catalytic system could effectively catalyze the cycloaddition of dipropargyl amines with cyanamides.69
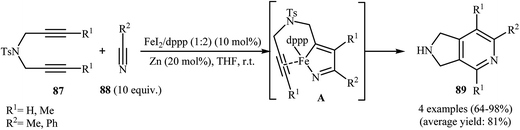 |
| | Scheme 31 Cycloaddition of 1,6-diyenes 87 with nitriles 88 catalyzed by FeI2/dppp/Zn system. | |
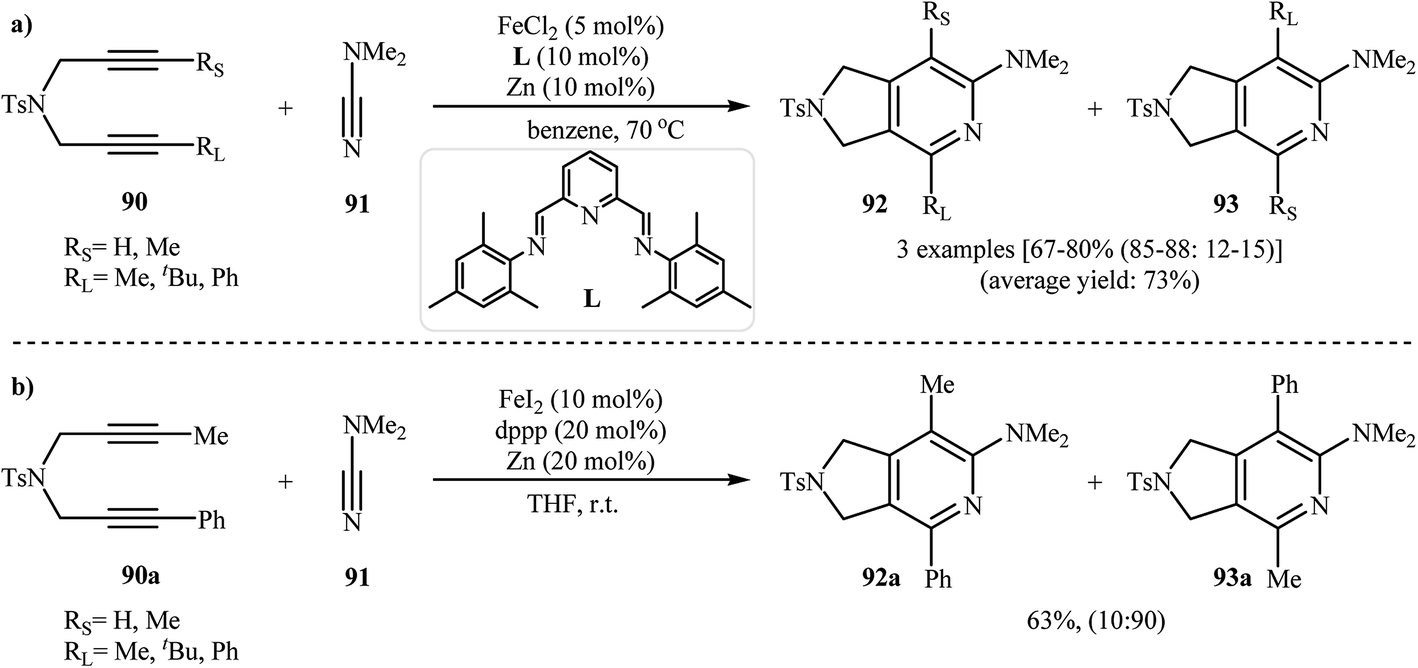
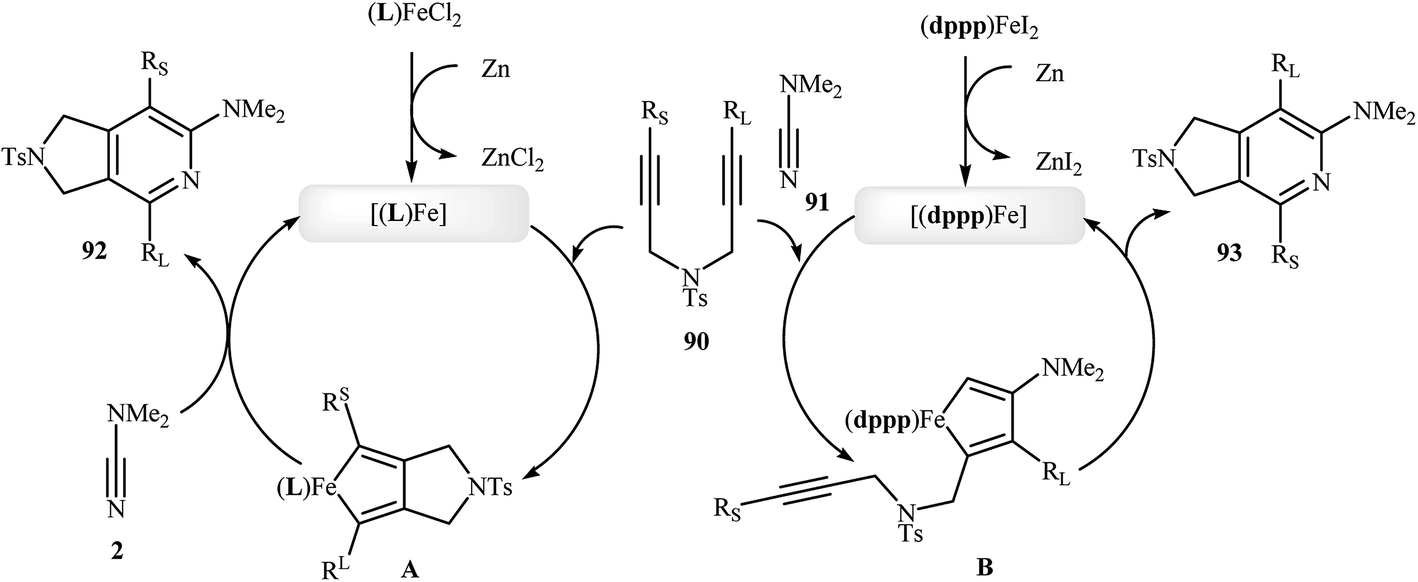
Along this line, Louie and co-workers reported an elegant regioselective iron-catalyzed [2 + 2 + 2] cycloaddition between unsymmetrical dipropargyl amines 90 and dimethylcyanamide 91 employing 5 mol% FeCl2 as catalyst, 10 mol% bis-(imino)pyridine as ligand, and 10 mol% Zn dust as reductant in benzene. Although both of possible regioisomers of the pyrrolidine-fused pyridines 92 and 93 are produced, their combined yields are good. Interestingly, in all cases the isomer that the larger alkyne substituent is placed next to the pyridine nitrogen has highest yield (Scheme 32a).70 In sharp contrast, the same reaction using Wan's catalytic system shows a strong preference to place the smaller substituent next to the pyridine nitrogen (Scheme 32b).69,70 The author proposed mechanism to explain this different reaction patterns is depicted in Fig. 2.
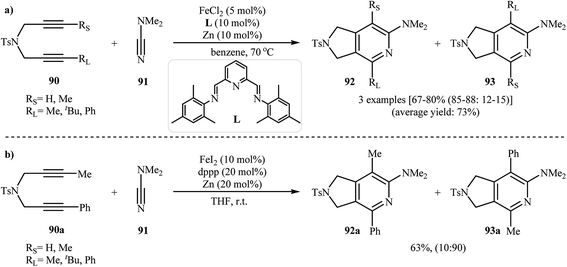 |
| | Scheme 32 (a) Iron-catalyzed cycloaddition reaction of diynes 90 and dimethylcyanamide 91 developed by Louie; (b) iron-catalyzed cycloaddition reaction of diyne 90a and dimethylcyanamide 91 developed by Wan. | |
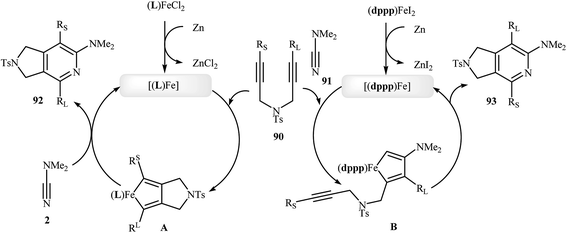 |
| | Fig. 2 Mechanistic proposal for the reactions in Scheme 32. | |
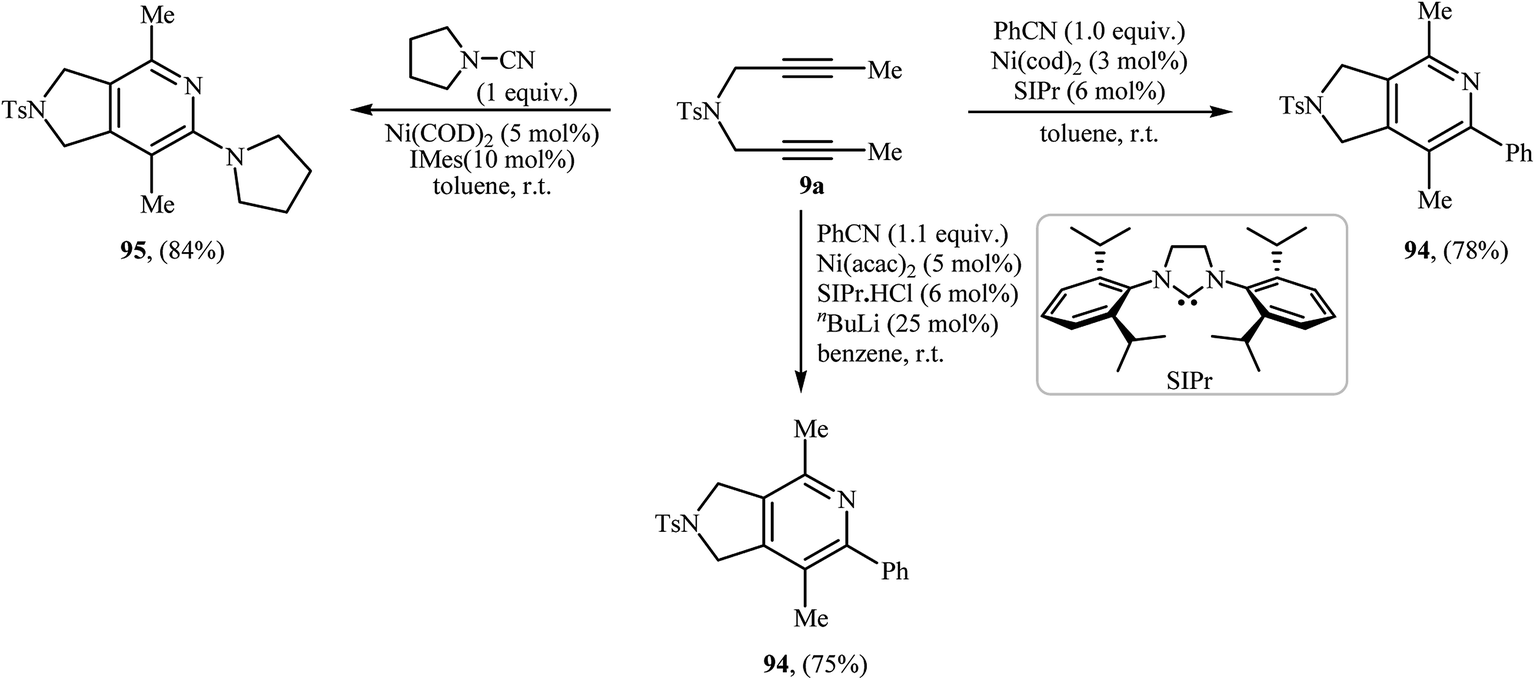
3.1.5. Nickel. Nickel catalyzed [2 + 2 + 2] cycloaddition of dipropargyl amines with nitriles has been scarcely studied; in fact, only three examples of such a reaction were reported in the literature (Scheme 33). In 2005, the group of J. Louie presented an example of Ni-catalyzed [2 + 2 + 2] cycloaddition between internal dipropargyl amine 9a and benzonitrile employing 3 mol% of Ni(cod)2 as catalyst and 6 mol% of SIPr as ligand in toluene at room temperature. The corresponding pyrrolidine-fused pyridines 94 was obtained in yield of 78%.71 Subsequently, the same group reinvestigated this reaction using Ni(acac)2/SIPr·HCl/nBuLi/benzene system but lower yield than previously reported was obtained.72 Ni-catalyzed cycloaddition of 9a and pyrrolidine-1-carbonitrile, was also reported by the same authors.73
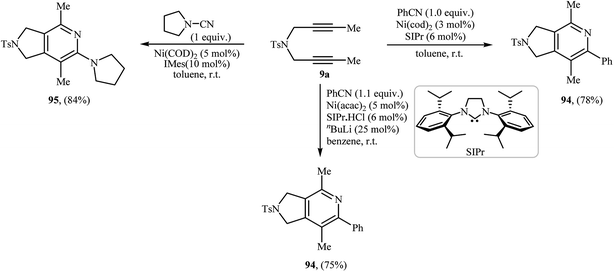 |
| | Scheme 33 Nickel catalyzed [2 + 2 + 2] cycloaddition reactions of dipropargyl amine 9a with nitriles. | |
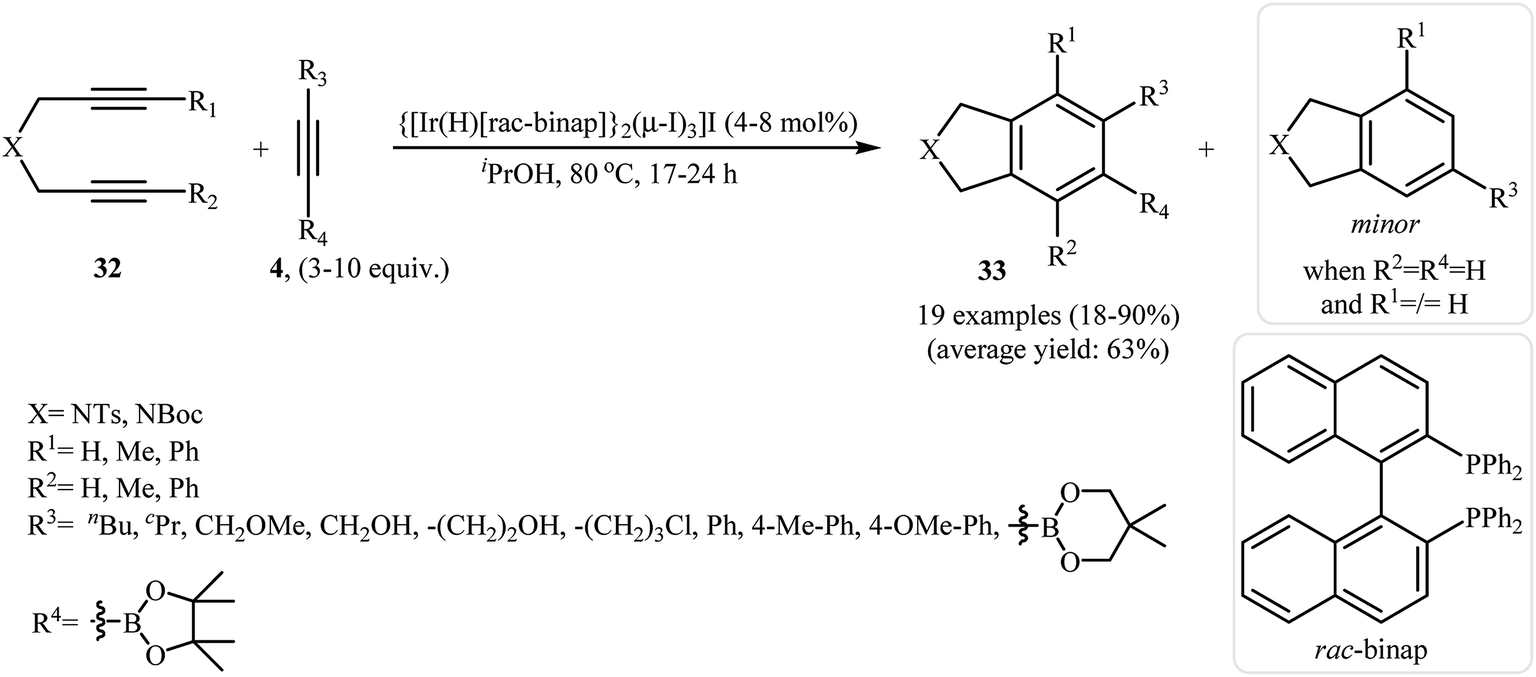
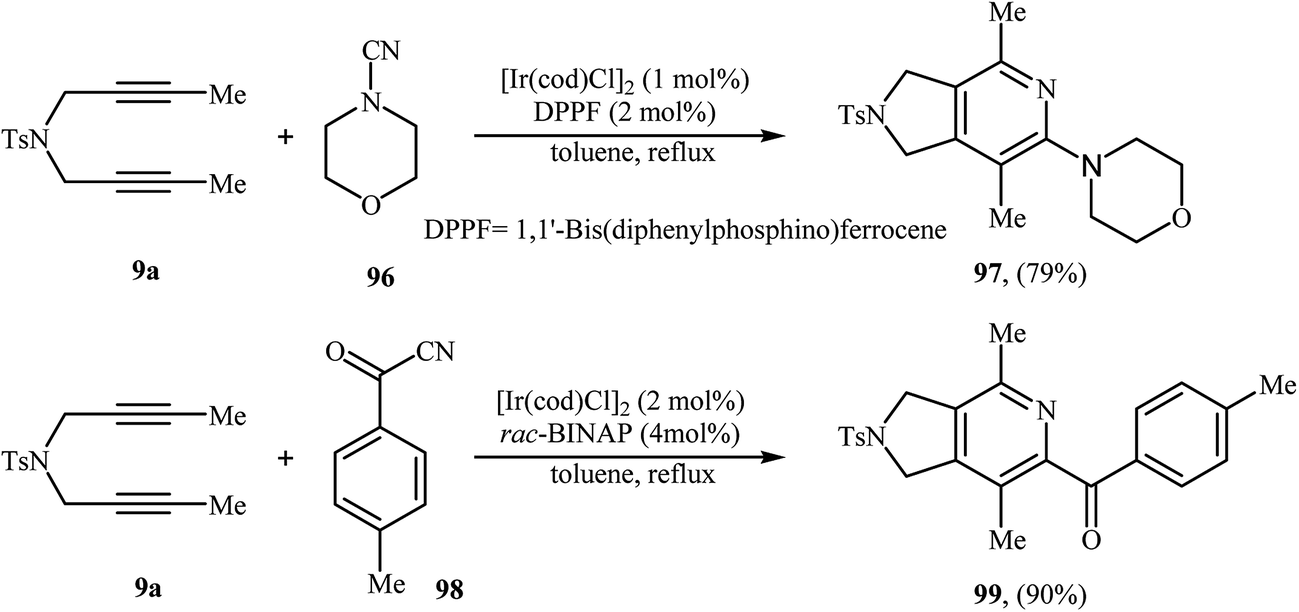
3.1.6. Iridium. The [2 + 2 + 2] cycloaddition of dipropargyl amines with nitriles can also be carried out efficiently by iridium(I) complex of [Ir(cod)Cl]2 (Scheme 34).74,75 However, the number of reported examples are very scarce and there is an urgent need to study the scope and limitations of this chemistry.
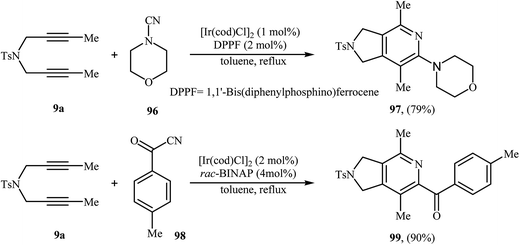 |
| | Scheme 34 Synthesis of pyrrolidine-fused pyridines 97 and 99 described by Takeuchi. | |
3.2. Achiral 2,3-dihydro-1H-pyrrolo[3,4-c]pyridines via intramolecular [2 + 2 + 2] cycloadditions
To develop a new protocol for the construction of 2,3-dihydro-1H-pyrrolo[3,4-c]pyridines via [2 + 2 + 2] cycloaddition reaction, Cheng and co-workers have investigated the purely intramolecular cycloaddition of dipropargyl amine-nitriles 101 in the presence of CoI2(dppe)/Zn system in refluxing acetonitrile, and good yields of polycyclic products 102 was obtained (Scheme 35a).76 The intramolecular cycloaddition of dipropargyl amine-nitriles was also performed under microwave-assisted conditions.77,78 For example, 2-(pyridin-2-yl)pyridine 103 was prepared in yield of 88% via Rh(I)-catalyzed double cocyclization of 102 under microwave irradiation at 90 °C (Scheme 35b).78
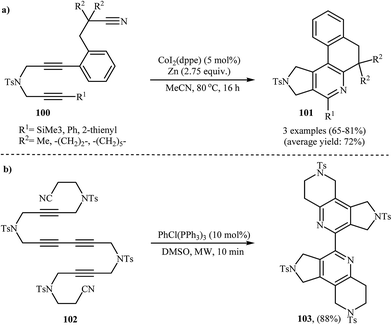 |
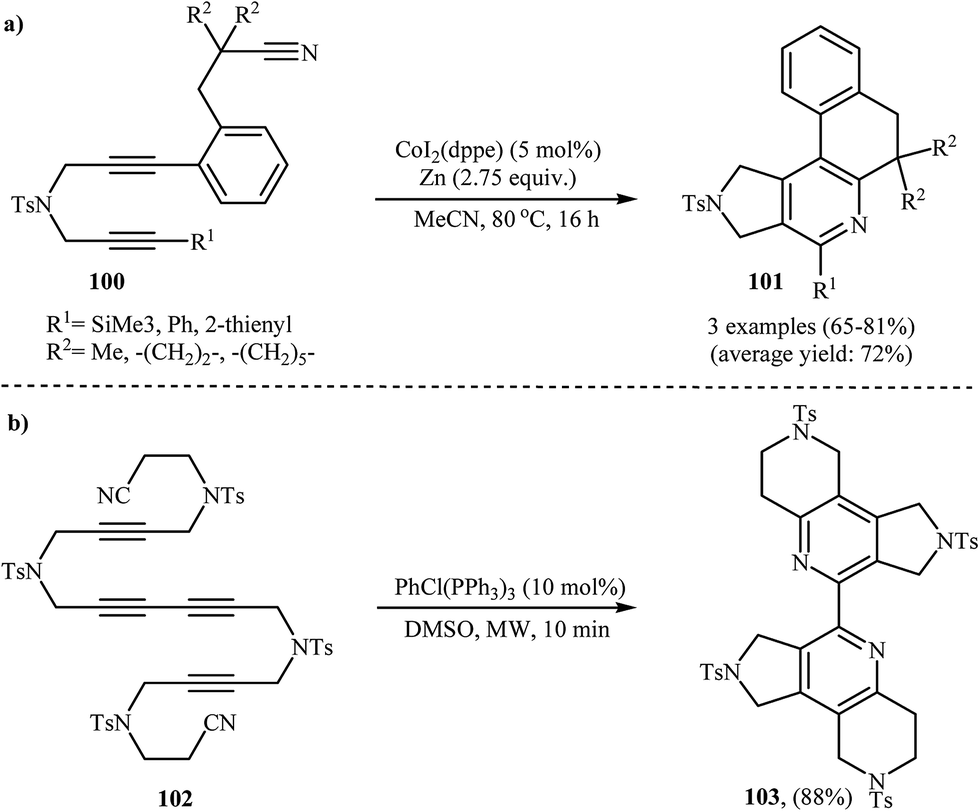
| | Scheme 35 (a) Synthesis of pyrrolidine-fused pyridine derivatives 101 through Co-catalyzed intramolecular cycloaddition of dipropargyl amine-nitriles 100; (b) microwave-assisted intramolecular cycloaddition of dicyanotetrayne 102. | |
3.3. Chiral 2,3-dihydro-1H-pyrrolo[3,4-c]pyridines
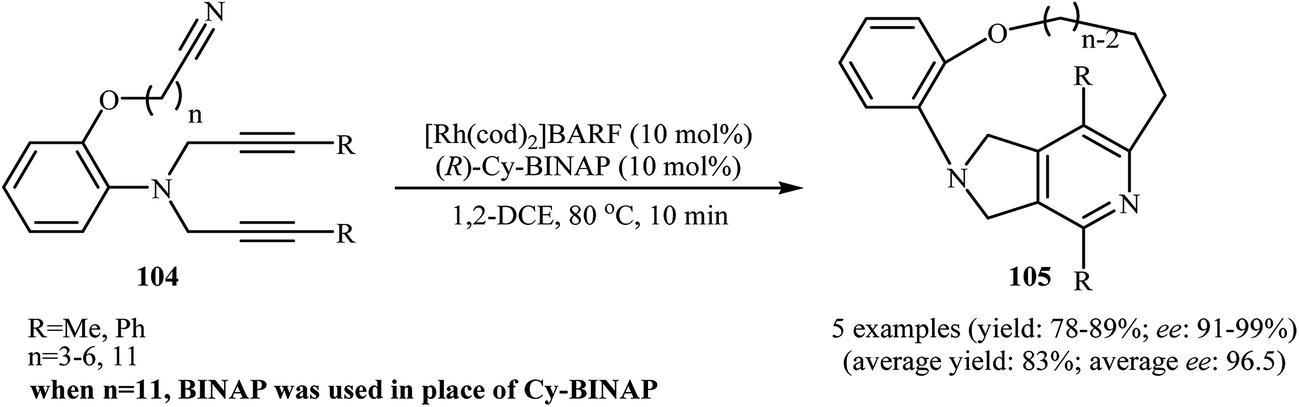
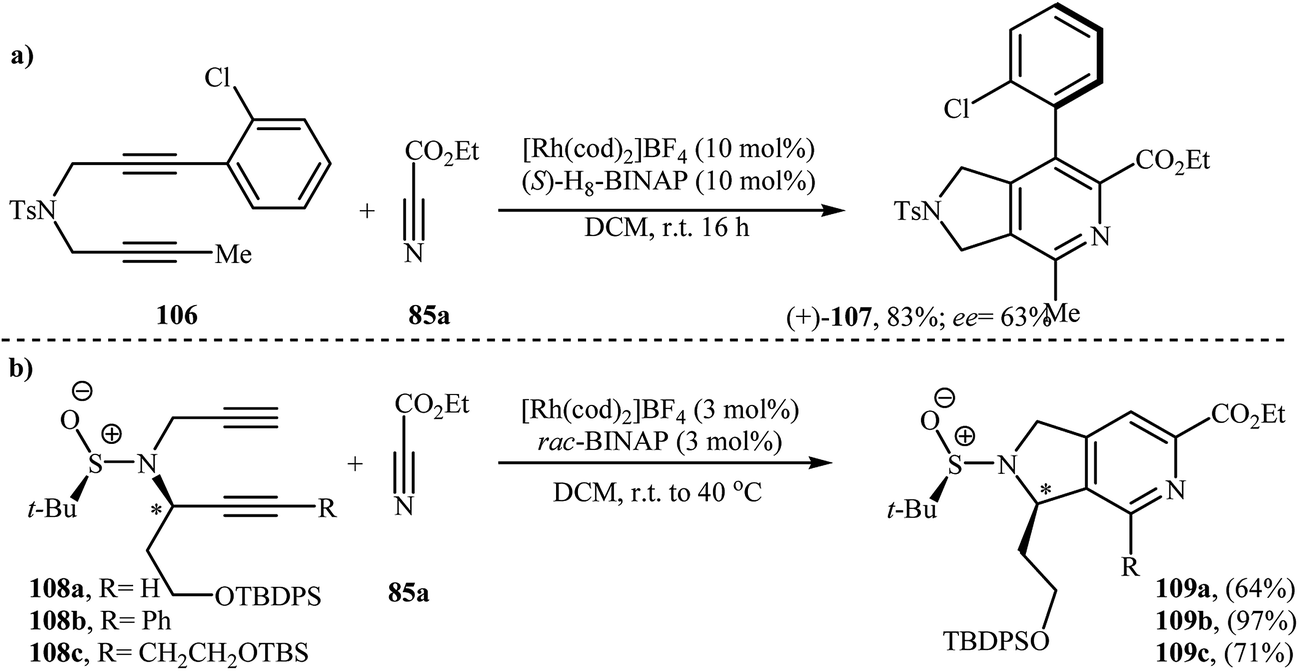
There has been a recent increase in the use of chiral substrates or ligands in the [2 + 2 + 2] cycloaddition of dipropargyl amines with nitriles for the construction of chiral cycloadducts. In an elegant study, shibata's group disclosed the catalytic enantioselective synthesis of planar-chiral macrocyclic tripodal cage compounds 105 via rhodium(I)/(R)-Cy-BINAP complex-catalyzed intramolecular cycloaddition of 2-aminophenol-tethered dipropargyl amine-nitriles 104 (Scheme 36).79,80 Under optimized conditions, cycloadducts 105 were obtained in high yields and excellent enantiomeric excess up to 99% ee. However, very poor enantioselectivities were achieved with diyne-nitriles possessing very long ansa chains (n = 11). To solve the problem, the authors replaced Cy-BINAP with BINAP. Along this line, Tanaka and co-workers reported an example of regio- and enantioselective synthesis of axially chiral 7-(2-chlorophenyl)-pyrrolidinopyridine 107 by cationic rhodium(I)/(S)-H8-BINAP complex catalyzed atroposelective [2 + 2 + 2] cycloaddition of unsymmetrical dipropargyl amine 106 with ethyl cyanoformate 85a (Scheme 37a)81 and Tan's group performed Rh-catalyzed [2 + 2 + 2] cycloaddition of chiral tert-butylsulfinamidated N-propargyl amines 108 with 85a to prepare chiral pyrrolidine-fused pyridine 109 (Scheme 37b).82
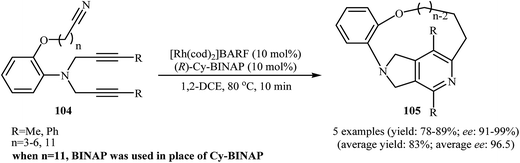 |
| | Scheme 36 Enantioselective synthesis of planar-chiral macrocyclic tripodal cage compounds 105 described by Shibata. | |
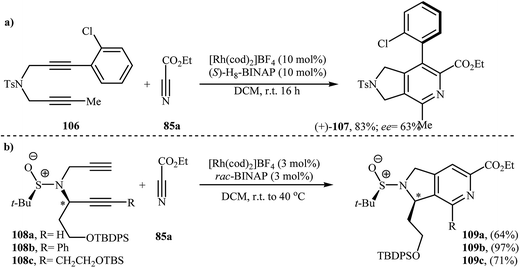 |
| | Scheme 37 (a) Rh-catalyzed atroposelective [2 + 2 + 2] cycloaddition of unsymmetrical diyene 106 with ethyl cyanoformate 85a described by Tanaka; (b) Rh-catalyzed cycloaddition of chiral tert-butylsulfinamidated N-propargylamines 108 with 85a reported by Tan. | |
4. 2,3-Dihydro-1H-pyrrolo[3,4-c]pyridin-6(5H)-one derivatives from nitrogen-linked 1,6-diyenes and isocyanates
4.1. Achiral 2,3-dihydro-1H-pyrrolo[3,4-c]pyridin-6(5H)-ones
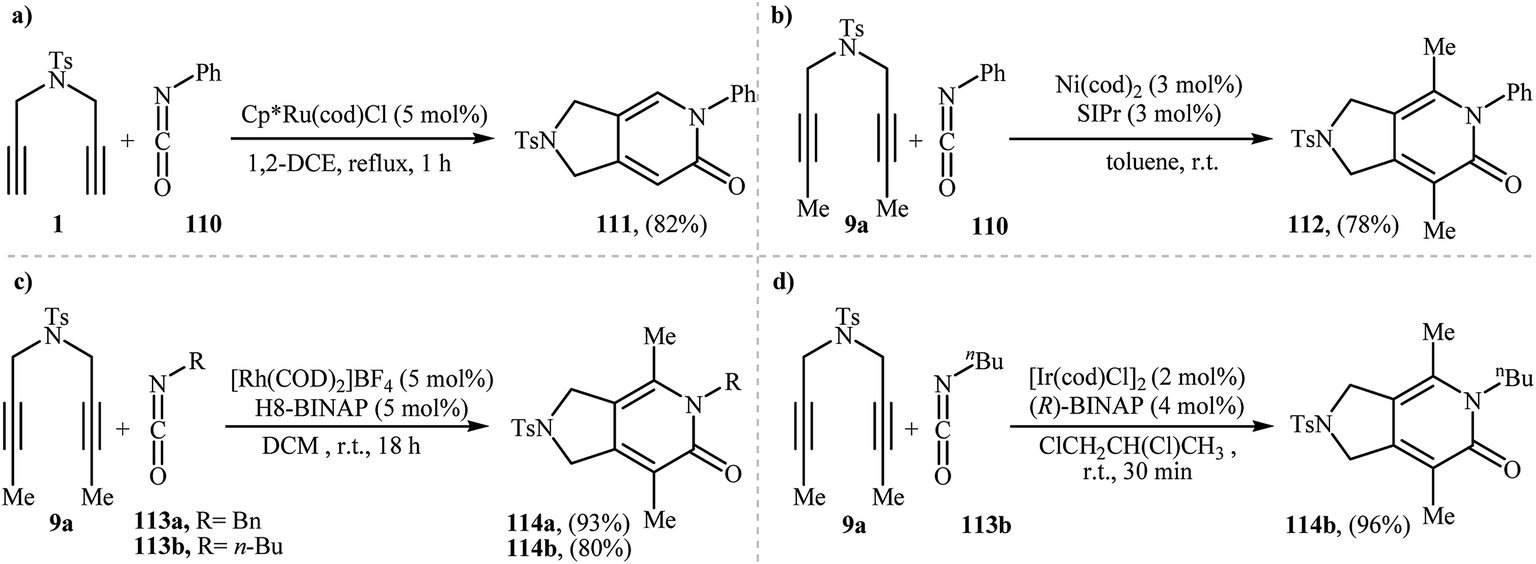
The first mention of the synthesis of 2,3-dihydro-1H-pyrrolo[3,4-c]pyridin-6(5H)-ones through a ruthenium(II)-catalyzed [2 + 2 + 2] cycloaddition between dipropargyl amines and isocyanates can be found in a 2001 paper by Itoh, although only one example was described (Scheme 38a).83 Louie and co-workers have subsequently shown that Ni(cod)2/SIPr system can also efficiency catalyze this reaction (Scheme 38b).84 Later, the group of Tanaka extended the scope of isocyanates to benzyl and alkyl isocyanates employing a cationic rhodium(I) catalytic system (Scheme 38c)85 and Takeuchi's group improved the efficiency of this reaction in terms of yield and reaction time by performing the process in refluxing 1,2-dichloropropane using [Ir(cod)Cl]2 as the catalyst (Scheme 38d).86
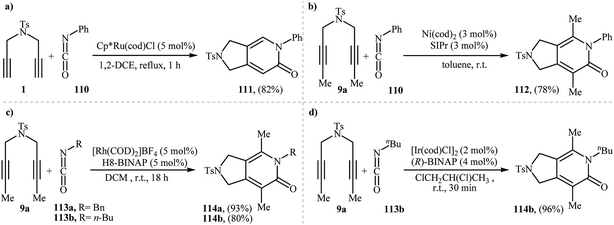 |
| | Scheme 38 Synthesis of pyrrolidine-fused pyridones via transition metal catalyzed [2 + 2 + 2] cycloaddition of nitrogen-linked 1,6-diyenes with isocyanates. | |
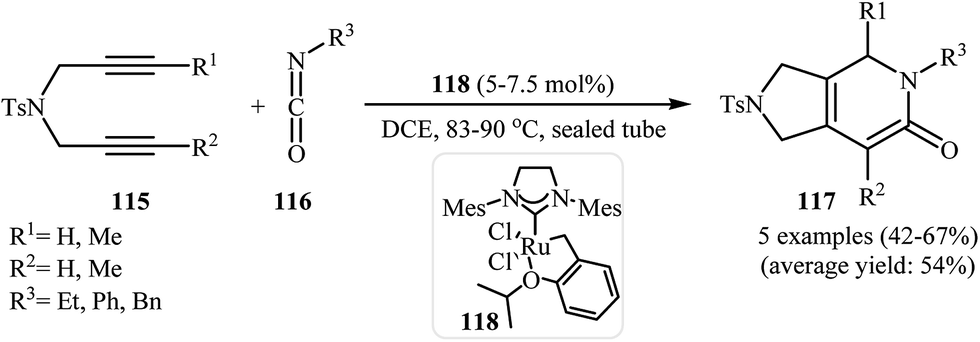
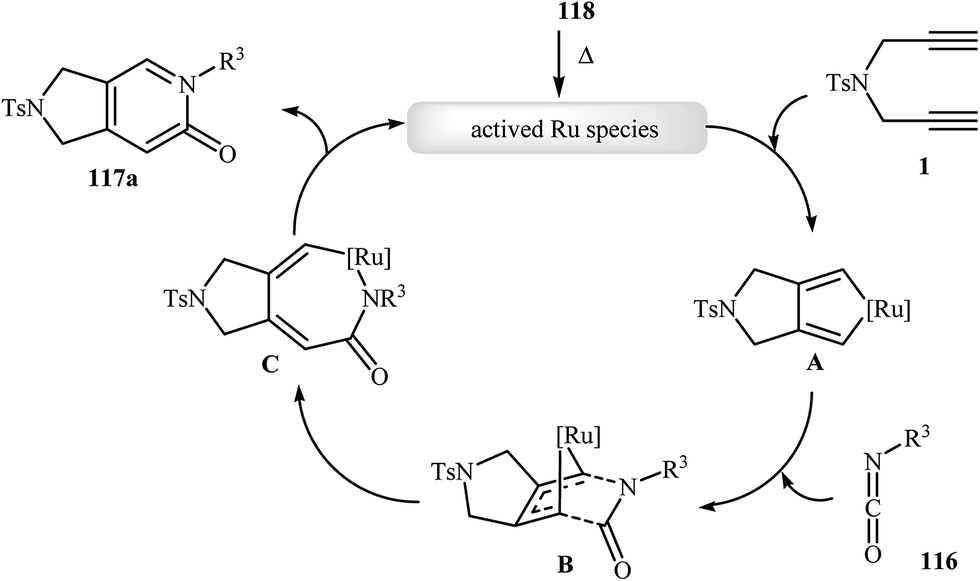
Recently, Pérez-Castells et al. described the preparation of 2,3-dihydro-1H-pyrrolo[3,4-c]pyridin-6(5H)-ones 117 in moderate yields via [2 + 2 + 2] cycloaddition of isocyanates 116 with both symmetrical and unsymmetrical dipropargyl amines 115 in the presence of catalytic amounts of ruthenium–alkylidene complexe 118 (Scheme 39). The reaction of unsymmetrical dipropargyl amines exhibits remarkable regioselectivity, and the substituent is located exclusively next to the 2-pyridone nitrogen. The proposed mechanism is outlined in Fig. 3.87
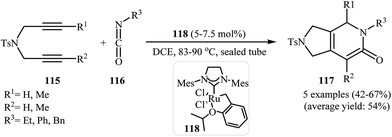 |
| | Scheme 39 Ruthenium–alkylidene complex 118 catalyzed cycloaddition of 1,6-diyenes 115 with isocyanates 116 described by Pérez-Castells. | |
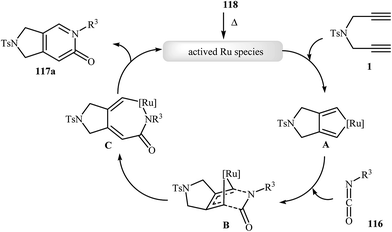 |
| | Fig. 3 Mechanism that accounts for the formation of 117a. | |
4.2. Chiral 2,3-dihydro-1H-pyrrolo[3,4-c]pyridin-6(5H)-ones
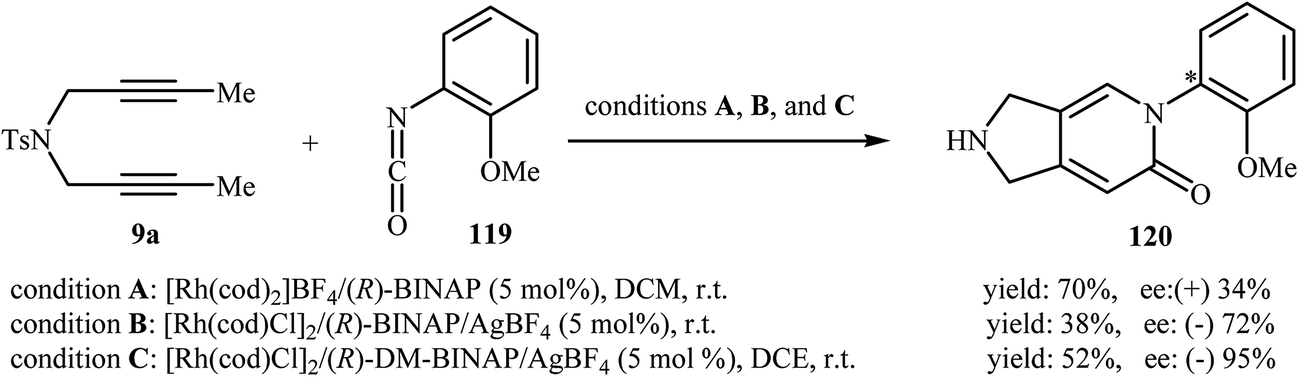
In 2008, Tanaka and co-workers reported an example of axially chiral 2,3-dihydro-1H-pyrrolo[3,4-c]pyridin-6(5H)-one preparation through a cationic rhodium(I)-catalyzed [2 + 2 + 2] cycloaddition strategy. They showed that dipropargyl amine 9a underwent cyclization with 2-methoxybenzene-isocyanate 119 in the presence of [Rh(cod)2]BF4/(R)-BINAP as catalytic system in DCM at room temperature and gave the corresponding pyrrolidine-fused pyridinone 120 in good yield (70%), although in low enantiomeric excess (34%) (Scheme 40, conditions A).88 five years later, the group of Aubert reinvestigated this reaction employing [Rh(cod)Cl]2/(R)-BINAP/AgBF4 combination as the catalytic system. Under these conditions, the cycloadduct 120 was obtained in good enantiomeric excess, although in low yield (Scheme 40, conditions B).89 This drawback was significantly improved by replacing of (R)-BINAP with (R)-DM-BINAP (Scheme 40, conditions C).90
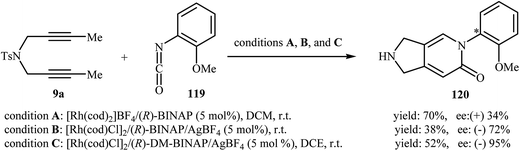 |
| | Scheme 40 Atroposelective [2 + 2 + 2] cycloaddition of dipropargyl amine 1 with 2-methoxybenzene-isocyanate 2. | |
5. 2,3-Dihydropyrano[4,3-c]pyrrol-6(1H)-ones from nitrogen-linked 1,6-diyenes and carbon dioxide
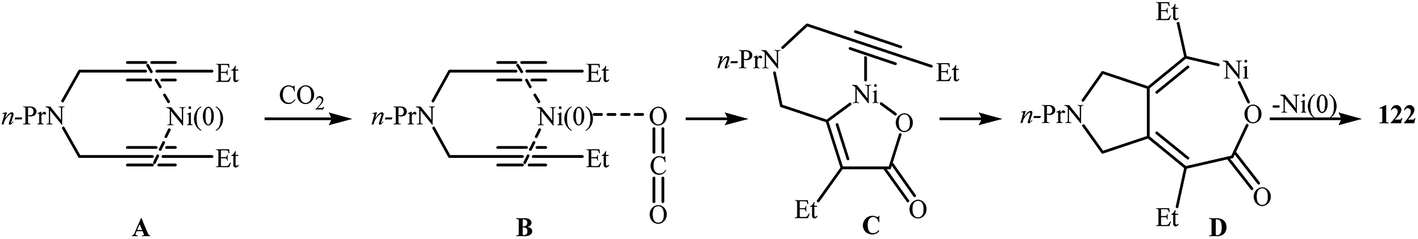
An interesting and rare example for the synthesis of 2,3-dihydropyrano[4,3-c]pyrrol-6(1H)-ones was reported by Saegusa et al. in 1988. Thus, in refluxing benzene, Ni(0)-catalyzed [2 + 2 + 2] cycloaddition between dipropargyl amine 121 and carbon dioxide, furnished -dihydropyrano[4,3-c]pyrrol-6(1H)-one 122 in yield of 78% (Scheme 41a). The mechanism proposed by the authors to explain this reaction is depicted in Scheme 42.91 26 years later, Tanaka and co-workers reinvestigated this reaction employing [Rh(cod)2]BF4/H8-BINAP/DCE system, but the corresponding 2-pyranone 123 was isolated in only 14% yield (Scheme 41b).92
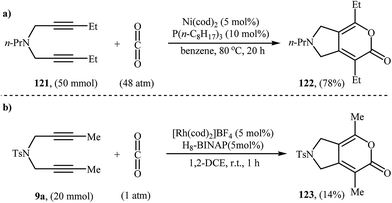 |
| | Scheme 41 (a) Synthesis of dihydropyrano[4,3-c]pyrrol-6(1H)-one 122 developed by Saegusa; (b) Tanaka's synthesis of dihydropyrano[4,3-c]pyrrol-6(1H)-one 123. | |
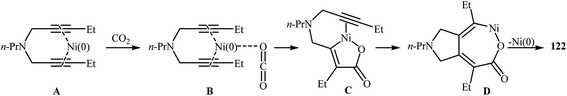 |
| | Scheme 42 Mechanistic proposal for the reaction in Scheme 41a. | |
6. 2,3-Dihydro-pyrrolo[3,4-c]pyridin-6-imines from nitrogen-linked 1,6-diyenes and carbodiimides
Young and Deiters have also developed a novel method for the preparation of 2,3-dihydro-pyrrolo[3,4-c]pyridin-6-imines 125 involving Co(II)-catalyzed [2 + 2 + 2] cycloaddition of resin-bound N-trityl dipropargyl amine 72 with carbodiimides 124 under microwave irradiation conditions, followed by deprotection of trityl group with trifluoroacetic acid (Scheme 43a). The authors found that microwave irradiation play a crucial role for the success of the reaction. When the same cyclotrimerization was conducted without microwave irradiation, no product formation was observed, even after a prolonged reaction time of 24 h at 110 °C.55 In a closely related investigation Tanaka and co-workers reported a cationic rhodium(I) catalytic system to get access to 2,3-dihydro-pyrrolo[3,4-c]pyridin-6-imines 127 (Scheme 43b).92
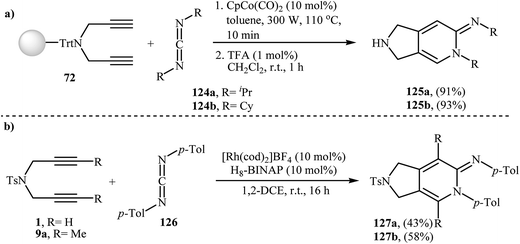 |
| | Scheme 43 (a) Synthesis of 2,3-dihydro-pyrrolo[3,4-c]pyridin-6-imines 125 developed by Deiters; (b) synthesis of 2,3-dihydro-pyrrolo[3,4-c]pyridin-6-imines 127 reported by Tanaka. | |
7. 2,3-Dihydro-1H-isoindol-5-ones from nitrogen-linked 1,6-diyenes and ketenes
Ketenes are one of the latest coupling partners to join the [2 + 2 + 2] cycloaddition story of dipropargyl amines. In 2011, Louie et al. successfully prepared 2,3-dihydro-1H-isoindol-5-ones 129 utilizing cycloaddition of dipropargyl amine 9a and 1.2 equivalents of ketenes 128 in the presence of Ni(cod)2/DPPB system in moderate yields (Scheme 44).93 Interestingly, under this reaction conditions no pyran product was observed, it means that exclusively the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds in the ketenes participated in the reactions.
C bonds in the ketenes participated in the reactions.
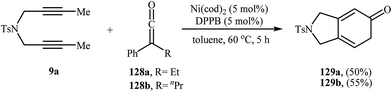 |
| | Scheme 44 Ni-catalyzed [2 + 2 + 2] cycloaddition of dipropargyl amine 9a with ketenes 128 to 2,3-dihydro-1H-isoindol-5-ones 129. | |
8. Outlook and conclusion
Transition metal catalyzed [2 + 2 + 2] cycloaddition of nitrogen-linked 1,6-diynes with unsaturated motifs, have drawn much interest from organic chemists because of their high efficiency in the construction of various pyrrolidine-fused systems. This method offers the opportunity to synthesis of complex frameworks, which may be difficult or impossible by other means. It also offers the possibility for the rapid synthesis of potential drugs from cheap and easily available dipropargyl amines in one step with ideal atom economy.
Despite the significant achievements during the past few years in this field, many challenges still remain to be overcome: (a) most of the [2 + 2 + 2] cycloadditions using nitrogen-linked 1,6-diyne reagents are limited to the use of expensive transition metal catalysts. Thus the exploration of cheaper transition metal catalysts (such as Fe and Cu catalysts) are highly desirable in terms of cost and availability; (b) generally, the number of reported examples are narrow and there is an urgent need to study the scope and limitations of reagents; (c) and finally, the mechanism of the reactions remains elusive, though some mechanistic courses were proposed based on DFT calculations.
Conflicts of interest
There are no conflicts to declare.
References
- Selected references:
(a) J. W. Blunt, B. R. Copp, M. H. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2005, 22, 15–61 RSC;
(b) J. P. Michael, Nat. Prod. Rep., 2008, 25, 166–187 RSC;
(c) D. O'Hagan, Nat. Prod. Rep., 2000, 17, 435–446 RSC;
(d) M. Ishikura, T. Abe, T. Choshi and S. Hibino, Nat. Prod. Rep., 2013, 30, 694–752 RSC;
(e) L. Le Bozec and C. J. Moody, Aust. J. Chem., 2009, 62, 639–647 CrossRef CAS;
(f) K. Speck and T. Magauer, Beilstein J. Org. Chem., 2013, 9, 2048–2078 CrossRef PubMed.
- Selected references:
(a) M. Baumann and I. R. Baxendale, Beilstein J. Org. Chem., 2013, 9, 2265–2319 CrossRef PubMed;
(b) A. Marella, O. P. Tanwar, R. Saha, M. R. Ali, S. Srivastava, M. Akhter, M. Shaquiquzzaman and M. M. Alam, Saudi Pharm. J., 2013, 21, 1–12 CrossRef PubMed;
(c) V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, RSC Adv., 2015, 5, 15233–15266 RSC;
(d) N. K. Kaushik, N. Kaushik, P. Attri, N. Kumar, C. H. Kim, A. K. Verma and E. H. Choi, Molecules, 2013, 18, 6620–6662 CrossRef CAS PubMed;
(e) Y. Bansal and O. Silakari, Bioorg. Med. Chem., 2012, 20, 6208–6236 CrossRef CAS PubMed;
(f) R. J. Nevagi, S. N. Dighe and S. N. Dighe, Eur. J. Med. Chem., 2015, 97, 561–581 CrossRef CAS PubMed;
(g) S. B. Ferreira and C. R. Kaiser, Expert Opin. Ther. Pat., 2012, 22, 1033–1051 CrossRef CAS PubMed.
- Selected references:
(a) J. A. Varela and C. Saá, Chem. Rev., 2003, 103, 3787–3802 CrossRef CAS PubMed;
(b) S. Kotha, E. Brahmachary and K. Lahiri, Eur. J. Org. Chem., 2005, 4741–4767 CrossRef CAS;
(c) Y. Yamamoto, Curr. Org. Chem., 2005, 9, 503–519 CrossRef CAS;
(d) V. Gandon, C. Aubert and M. Malacria, Chem. Commun., 2006, 2209–2217 RSC;
(e) P. R. Chopade and J. Louie, Adv. Synth. Catal., 2006, 348, 2307–2327 CrossRef CAS;
(f) B. Heller and M. Hapke, Chem. Soc. Rev., 2007, 36, 1085–1094 RSC;
(g) B. R. Galan and T. Rovis, Angew. Chem., Int. Ed., 2009, 48, 2830–2834 CrossRef CAS PubMed;
(h) M. R. Shaaban, R. El-Sayed and A. H. Elwahy, Tetrahedron, 2011, 67, 6095–6130 CrossRef CAS;
(i) G. Domínguez and J. Pérez-Castells, Chem. Soc. Rev., 2011, 40, 3430–3444 RSC;
(j) N. Weding and M. Hapke, Chem. Soc. Rev., 2011, 40, 4525–4538 RSC;
(k) K. Tanaka, Heterocycles, 2012, 85, 1017–1043 CrossRef CAS;
(l) S. Okamoto and Y.-k. Sugiyama, Synlett, 2013, 24, 1044–1060 CrossRef CAS.
- Selected references:
(a) T. Shibata and K. Tsuchikama, Org. Biomol. Chem., 2008, 6, 1317–1323 RSC;
(b) M. Amatore and C. Aubert, Eur. J. Org. Chem., 2015, 265–286 CrossRef CAS.
-
(a) E. Vessally, RSC Adv., 2016, 6, 18619–18631 RSC;
(b) E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 71662–71675 RSC;
(c) E. Vessally, L. Edjlali, A. Hosseinian, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 49730–49746 RSC;
(d) E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, RSC Adv., 2016, 6, 99781–99793 RSC;
(e) E. Vessally, A. Hosseinian, L. Edjlali, A. Bekhradnia and M. D. Esrafili, Curr. Org. Synth., 2017, 14, 557–567 CrossRef CAS;
(f) E. Vessally, S. Soleimani-Amiri, A. Hosseinian, L. Edjlali and A. Bekhradnia, RSC Adv., 2017, 7, 7079–7091 RSC;
(g) S. Arshadi, E. Vessally, L. Edjlali, E. Ghorbani-Kalhor and R. Hosseinzadeh-Khanmiri, RSC Adv., 2017, 7, 13198–13211 RSC;
(h) S. Arshadi, E. Vessally, M. Sobati, A. Hosseinian and A. Bekhradnia, J. CO2 Util., 2017, 19, 120–129 CrossRef CAS;
(i) S. Arshadi, E. Vessally, L. Edjlali, R. Hosseinzadeh-Khanmiri and E. Ghorbani-Kalhor, Beilstein J. Org. Chem., 2017, 13, 625–638 CrossRef CAS PubMed;
(j) E. Vessally, R. Hosseinzadeh-Khanmiri, E. Ghorbani-Kalhor, M. Es'haghi and A. Bekhradnia, RSC Adv., 2017, 7, 19061–19072 RSC;
(k) S. Arshadi, E. Vessally, A. Hosseinian, S. Soleimani-Amiri and L. Edjlali, J. CO2 Util., 2017, 21, 108–118 CrossRef CAS;
(l) S. Soleimani-Amiri, E. Vessally, M. Babazadeh, A. Hosseinian and L. Edjlali, RSC Adv., 2017, 7, 19061–19072 RSC;
(m) E. Vessally, M. Babazadeh, A. Hosseinian, L. Edjlali and L. Sreerama, Curr. Org. Chem., 2017 DOI:10.2174/1385272821666170519113904.
- T.-J. Kim, D.-H. Kim, S.-C. Shim and J.-H. Jeong, Bull. Korean Chem. Soc., 1995, 16, 1126–1127 CAS.
- H. Kinoshita, H. Shinokubo and K. Oshima, J. Am. Chem. Soc., 2003, 125, 7784–7785 CrossRef CAS PubMed.
- W. Wu, X. Y. Zhang and S. X. Kang, Chin. Chem. Lett., 2010, 21, 18–22 CrossRef CAS.
- Y.-H. Wang, S.-H. Huang, T.-C. Lin and F.-Y. Tsai, Tetrahedron, 2010, 66, 7136–7141 CrossRef CAS.
- J. Clayden and W. J. Moran, Org. Biomol. Chem., 2007, 5, 1028–1030 CAS.
- I. Gonzalez, A. Pla-Quintana and A. Roglans, Synlett, 2009, 2844–2848 CAS.
- Q. Sun, X. Zhou, K. Islam and D. J. Kyle, Tetrahedron Lett., 2001, 42, 6495–6497 CrossRef CAS.
- D. D. Young, R. S. Senaiar and A. Deiters, Chem.–Eur. J., 2006, 12, 5563–5568 CrossRef CAS PubMed.
- M. Fernández, M. Ferré, A. Pla-Quintana, T. Parella, R. Pleixats and A. Roglans, Eur. J. Org. Chem., 2014, 6242–6251 CrossRef.
- M. Parera, A. Dachs, M. Solà, A. Pla-Quintana and A. Roglans, Chem.–Eur. J., 2012, 18, 13097–13107 CrossRef CAS PubMed.
- Y. Yamamoto, R. Ogawa and K. Itoh, Chem. Commun., 2000, 549–550 RSC.
- B. Witulski, T. Stengel and J. M. Fernández-Hernández, Chem. Commun., 2000, 1965–1966 RSC.
- D. D. Young, L. Sripada and A. Deiters, J. Comb. Chem., 2007, 9, 735 CrossRef CAS PubMed.
- J. Jacquet, A. L. Auvinet, A. K. Mandadapu, M. Haddad, V. Ratovelomanana-Vidal and V. Michelet, Adv. Synth. Catal., 2015, 357, 1387–1392 CrossRef CAS.
- Y. Yamamoto, K. Hattori and H. Nishiyama, J. Am. Chem. Soc., 2006, 128, 8336–8340 CrossRef CAS PubMed.
- Y. Yamamoto, K. Kinpara, T. Saigoku, H. Nishiyama and K. Itoh, Org. Biomol. Chem., 2004, 2, 1287–1294 CAS.
- R. W. Foster, C. J. Tame, H. C. Hailes and T. D. Sheppard, Adv. Synth. Catal., 2013, 355, 2353–2360 CrossRef CAS PubMed.
- M. Nishida, H. Shiga and M. Mori, J. Org. Chem., 1998, 63, 8606–8608 CrossRef CAS.
- A. L. Auvinet, M. Ez-Zoubir, M. R. Vitale, J. A. Brown, V. Michelet and V. Ratovelomanana-Vidal, ChemSusChem, 2012, 5, 1888–1891 CrossRef CAS PubMed.
- A. L. Auvinet, M. Ez-Zoubir, S. Bompard, M. R. Vitale, J. A. Brown, V. Michelet and V. Ratovelomanana-Vidal, ChemCatChem, 2013, 5, 2389–2394 CrossRef CAS.
- V. Rassadin, E. Nicolas and Y. Six, Chem. Commun., 2014, 50, 7666–7669 RSC.
- S. Saaby, I. R. Baxendale and S. V. Ley, Org. Biomol. Chem., 2005, 3, 3365–3368 CAS.
- S. Ventre, C. Simon, F. Rekhroukh, M. Malacria, M. Amatore, C. Aubert and M. Petit, Chem.–Eur. J., 2013, 19, 5830–5835 CrossRef CAS PubMed.
- L. Iannazzo, N. Kotera, M. Malacria, C. Aubert and V. Gandon, J. Organomet. Chem., 2011, 696, 3906–3908 CrossRef CAS.
- Y. Sato, T. Nishimata and M. Mori, J. Org. Chem., 1994, 59, 6133–6135 CrossRef CAS.
- G. Nishida, S. Ogaki, Y. Yusa, T. Yokozawa, K. Noguchi and K. Tanaka, Org. Lett., 2008, 10, 2849–2852 CrossRef CAS PubMed.
- T. Shibata, T. Chiba, H. Hirashima, Y. Ueno and K. Endo, Angew. Chem., Int. Ed., 2009, 48, 8066–8069 CrossRef CAS PubMed.
- T. Shibata, T. Fujimoto, K. Yokota and K. Takagi, J. Am. Chem. Soc., 2004, 126, 8382–8383 CrossRef CAS PubMed.
- T. Shibata and K. Tsuchikama, Chem. Commun., 2005, 6017–6019 RSC.
- T. Shibata, Y. Arai, K. Takami, K. Tsuchikama, T. Fujimoto, S. Takebayashi and K. Takagi, Adv. Synth. Catal., 2006, 348, 2475–2483 CrossRef CAS.
- H. Hara, M. Hirano and K. Tanaka, Org. Lett., 2008, 10, 2537–2540 CrossRef CAS PubMed.
- H. Hara, M. Hirano and K. Tanaka, Tetrahedron, 2009, 65, 5093–5101 CrossRef CAS.
- H. Hara, M. Hirano and K. Tanaka, Org. Lett., 2009, 11, 1337–1340 CrossRef CAS PubMed.
- Y. Aida, S. Tooriyama, Y. Kimura, H. Hara, Y. Shibata and K. Tanaka, Eur. J. Org. Chem., 2016, 132–138 CrossRef CAS.
- X. Fang, J. Sun and X. Tong, Chem. Commun., 2010, 46, 3800–3802 RSC.
- M.-S. Wu, M. Shanmugasundaram and C.-H. Cheng, Chem. Commun., 2003, 718–719 RSC.
- A. L. Jones and J. K. Snyder, J. Org. Chem., 2009, 74, 2907–2910 CrossRef CAS PubMed.
- A. Geny, S. Gaudrel, F. Slowinski, M. Amatore, G. Chouraqui, M. Malacria, C. Aubert and V. Gandon, Adv. Synth. Catal., 2009, 351, 271–275 CrossRef CAS.
- T. Shibata, A. Kawachi, M. Ogawa, Y. Kuwata, K. Tsuchikama and K. Endo, Tetrahedron, 2007, 63, 12853–12859 CrossRef CAS.
- S. Brun, L. Garcia, I. Gonzalez, A. Torrent, A. Dachs, A. Pla-Quintana, T. Parella and A. Roglans, Chem. Commun., 2008, 4339–4341 RSC.
- S. Brun, M. Parera, A. Pla-Quintana, A. Roglans, T. León, T. Achard, J. Solà, X. Verdaguer and A. Riera, Tetrahedron, 2010, 66, 9032–9040 CrossRef CAS.
- J.-C. Hsieh and C.-H. Cheng, Chem. Commun., 2005, 2459–2461 RSC.
- L. P. Battaglia, D. Delledonne, M. Nardelli, G. Predieri, G. P. Chiusoli, M. Costa and C. Pelizzi, J. Organomet. Chem., 1989, 363, 209–222 CrossRef CAS.
- G. Predieri, A. Tiripicchio, M. T. Camellini, M. Costa and E. Sappa, J. Organomet. Chem., 1992, 423, 129–139 CrossRef CAS.
- Z. Zhou, L. P. Battaglia, G. P. Chiusoli, M. Costa, M. Nardelli, C. Pelizzi and G. Predieri, J. Organomet. Chem., 1991, 417, 51–63 CrossRef CAS.
- K. Kase, A. Goswami, K. Ohtaki, E. Tanabe, N. Saino and S. Okamoto, Org. Lett., 2007, 9, 931–934 CrossRef CAS PubMed.
- A. Geny, N. Agenet, L. Iannazzo, M. Malacria, C. Aubert and V. Gandon, Angew. Chem., Int. Ed., 2009, 48, 1810–1813 CrossRef CAS PubMed.
- F. Fischer, A. F. Siegle, M. Checinski, C. Fischer, K. Kral, R. Thede, O. Trapp and M. Hapke, J. Org. Chem., 2016, 81, 3087–3102 CrossRef CAS PubMed.
- M. Hapke, K. Kral, C. Fischer, A. Spannenberg, A. Gutnov, D. Redkin and B. Heller, J. Org. Chem., 2010, 75, 3993–4003 CrossRef CAS PubMed.
- D. D. Young and A. Deiters, Angew. Chem., 2007, 119, 5279–5282 CrossRef.
- A. M. Rodriguez, C. Cebrián, P. Prieto, J. I. García, A. de la Hoz and Á. Díaz-Ortiz, Chem.–Eur. J., 2012, 18, 6217–6224 CrossRef CAS PubMed.
- K. Tanaka, N. Suzuki and G. Nishida, Eur. J. Org. Chem., 2006, 3917–3922 CrossRef CAS.
- K. Tanaka, H. Hara, G. Nishida and M. Hirano, Org. Lett., 2007, 9, 1907–1910 CrossRef CAS PubMed.
- K. Kashima, M. Ishii and K. Tanaka, Eur. J. Org. Chem., 2015, 1092–1099 CrossRef CAS.
- F. Xu, C. Wang, H. Wang, X. Li and B. Wan, Green Chem., 2015, 17, 799–803 RSC.
- F. Xu, C. Wang, D. Wang, X. Li and B. Wan, Chem.–Eur. J., 2013, 19, 2252–2255 CrossRef CAS PubMed.
- Y. Yamamoto, S. Okuda and K. Itoh, Chem. Commun., 2001, 1102–1103 RSC.
- Y. Yamamoto, R. Ogawa and K. Itoh, J. Am. Chem. Soc., 2001, 123, 6189–6190 CrossRef CAS PubMed.
- Y. Yamamoto, K. Kinpara, T. Saigoku, H. Takagishi, S. Okuda, H. Nishiyama and K. Itoh, J. Am. Chem. Soc., 2005, 127, 605–613 CrossRef CAS PubMed.
- Y. Yamamoto, K. Kinpara, H. Nishiyama and K. Itoh, Adv. Synth. Catal., 2005, 347, 1913–1916 CrossRef CAS.
- Y. Yamamoto, K. Kinpara, R. Ogawa, H. Nishiyama and K. Itoh, Chem.–Eur. J., 2006, 12, 5618–5631 CrossRef CAS PubMed.
- F. Xu, C. Wang, X. Li and B. Wan, ChemSusChem, 2012, 5, 854–857 CrossRef CAS PubMed.
- C. Wang, X. Li, F. Wu and B. Wan, Angew. Chem., Int. Ed., 2011, 50, 7162–7166 CrossRef CAS PubMed.
- C. Wang, D. Wang, F. Xu, B. Pan and B. Wan, J. Org. Chem., 2013, 78, 3065–3072 CrossRef CAS PubMed.
- T. K. Lane, B. R. D'Souza and J. Louie, J. Org. Chem., 2012, 77, 7555–7563 CrossRef CAS PubMed.
- M. M. McCormick, H. A. Duong, G. Zuo and J. Louie, J. Am. Chem. Soc., 2005, 127, 5030–5031 CrossRef CAS PubMed.
- T. N. Tekavec, G. Zuo, K. Simon and J. Louie, J. Org. Chem., 2006, 71, 5834–5836 CrossRef CAS PubMed.
- R. M. Stolley, M. T. Maczka and J. Louie, Eur. J. Org. Chem., 2011, 3815–3824 CrossRef CAS PubMed.
- T. Hashimoto, S. Ishii, R. Yano, H. Miura, K. Sakata and R. Takeuchi, Adv. Synth. Catal., 2015, 357, 3901–3916 CrossRef CAS.
- T. Hashimoto, K. Kato, R. Yano, T. Natori, H. Miura and R. Takeuchi, J. Org. Chem., 2016, 81, 5393–5400 CrossRef CAS PubMed.
- H.-T. Chang, M. Jeganmohan and C.-H. Cheng, Org. Lett., 2007, 9, 505–508 CrossRef CAS PubMed.
- Y. Miclo, P. Garcia, Y. Evanno, P. George, M. Sevrin, M. Malacria, V. Gandon and C. Aubert, Synlett, 2010, 2314–2318 CAS.
- L. Garcia, A. Pla-Quintana, A. Roglans and T. Parella, Eur. J. Org. Chem., 2010, 3407–3415 CrossRef CAS.
- T. Shibata, T. Uchiyama and K. Endo, Org. Lett., 2009, 11, 3906–3908 CrossRef CAS PubMed.
- T. Shibata, M. Miyoshi, T. Uchiyama, K. Endo, N. Miura and K. Monde, Tetrahedron, 2012, 68, 2679–2686 CrossRef CAS.
- K. Kashima, K. Teraoka, H. Uekusa, Y. Shibata and K. Tanaka, Org. Lett., 2016, 18, 2170–2173 CrossRef CAS PubMed.
- R. A. Bauer, C. M. DiBlasi and D. S. Tan, Org. Lett., 2010, 12, 2084–2087 CrossRef CAS PubMed.
- Y. Yamamoto, H. Takagishi and K. Itoh, Org. Lett., 2001, 3, 2117–2119 CrossRef CAS PubMed.
- H. A. Duong, M. J. Cross and J. Louie, J. Am. Chem. Soc., 2004, 126, 11438–11439 CrossRef CAS PubMed.
- K. Tanaka, A. Wada and K. Noguchi, Org. Lett., 2005, 7, 4737–4739 CrossRef CAS PubMed.
- G. Onodera, M. Suto and R. Takeuchi, J. Org. Chem., 2012, 77, 908–920 CrossRef CAS PubMed.
- S. Alvarez, S. Medina, G. Dominguez and J. Perez-Castells, J. Org. Chem., 2013, 78, 9995–10001 CrossRef CAS PubMed.
- K. Tanaka, Y. Takahashi, T. Suda and M. Hirano, Synlett, 2008, 1724–1728 CrossRef CAS.
- M. Augé, M. Barbazanges, A. T. Tran, A. Simonneau, P. Elley, H. Amouri, C. Aubert, L. Fensterbank, V. Gandon and M. Malacria, Chem. Commun., 2013, 49, 7833–7835 RSC.
- M. n. Augé, A. Feraldi-Xypolia, M. Barbazanges, C. Aubert, L. Fensterbank, V. Gandon, E. Kolodziej and C. Ollivier, Org. Lett., 2015, 17, 3754–3757 CrossRef PubMed.
- T. Tsuda, S. Morikawa, R. Sumiya and T. Saegusa, J. Org. Chem., 1988, 53, 3140–3145 CrossRef CAS.
- M. Ishii, F. Mori and K. Tanaka, Chem.–Eur. J., 2014, 20, 2169–2174 CrossRef CAS PubMed.
- P. Kumar, D. M. Troast, R. Cella and J. Louie, J. Am. Chem. Soc., 2011, 133, 7719–7721 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
Somayeh Soleimani-Amiri
b,
Esmail Vessally
*a,
Somayeh Soleimani-Amiri
b,
Esmail Vessally





![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: