Multilayer composite beads constructed via layer-by-layer self-assembly for lysozyme controlled release
Jiemin Zhao†
ab,
Xiaoping Wang†c,
Yanshen Kuangd,
Yufeng Zhangb,
Xiaowen Shia,
Xingyun Liua and
Hongbing Deng*a
aDepartment of Environmental Science, School of Resource and Environmental Science, Wuhan University, Wuhan 430079, China. E-mail: hbdeng@whu.edu.cn; alphabeita@yahoo.com; Fax: +86-2768778501; Tel: +86-2768778501
bHubei-MOST KLOS & KLOBME, Wuhan University Stomatological Hospital, Wuhan University, Wuhan 430079, China
cDepartment of Thoracic Surgery, Tangdu Hospital, Fourth Military Medical University, Xi'an 710038, China
dZhongnan Hospital, Wuhan University, Wuhan 430079, China
First published on 23rd May 2014
Abstract
Alginate (ALG)–lysozyme (LZ) beads were fabricated by a cross-linking process. Negatively charged ALG and positively charged LZ were alternately deposited on the positively charged ALG–LZ beads via layer-by-layer (LBL) self-assembly technique. The mechanical properties and the enzymatic activity of these samples were studied by regulating the number of deposition bilayers and the composition of the outermost layer. The scanning electron microscopy images indicated that the resultant samples exhibited good sphericity and porosity. The Fourier transform infrared spectra results implied the presence of electrostatic interactions between ALG and LZ. The pore size distribution results revealed that the samples mainly possessed mesopores with radius in the range of 2–7 nm. In vitro LZ release test performed at different time intervals showed that LZ could be released from ALG–LZ beads and LBL film-coated beads. The amount of released LZ increased with extended time intervals.
1. Introduction
Recently, significant efforts have been made to develop different protein delivery systems.1–4 The design of novel protein delivery systems was required for the development of successful products, reduction of adverse reactions and side effects, convenient model of delivery and so on.5 Herein, various measures have been taken to facilitate the delivery of protein. The application of microspheres and beads was an useful method for protein delivery, which could protect the protein from their microenvironment and keep their long-term biological activity.6,7Based on the above considerations, the carriers for protein delivery should be critically evaluated by considering their toxicity, biological activity and biodegradability etc. Additionally, many research efforts were aimed towards choosing alginate (ALG) as an ideal candidate for protein delivery due to its nontoxicity, good biocompatibility and biodegradability etc. In detail, it was a family of linear anionic polysaccharide, which consisted of (1–4) linked β-D-mannuronic acid and α-L-guluronic acid units in various composition and sequence and existed widely in many species of brown seaweeds.8,9 ALG was studied extensively in drug delivery systems because its droplets could be transformed into rigid beads by gelation with the addition of divalent cations in aqueous solution, such as calcium or barium ions.10 The relatively mild gelation process enabled proteins to be incorporated into ALG beads with retention of full biological activity, so ALG beads were considered as a perfect carrier for protein delivery.
Owing to the high stability of lysozyme (LZ) within a wide range of pH and temperature, LZ was chosen as the model protein for drug delivery.11,12 LZ, the natural defense substance produced by living organisms with an isoelectric point value of 10.7,13 was selected as the target protein for its positive charge in aqueous solutions. Moreover, LZ has been extensively used for antibacterial agents,14,15 wound dressing16 and protein separation.17 Compared with the free LZ, immobilized LZ exhibited improved stability to environmental changes. There were many investigations focused on the immobilization of LZ.13,18,19 In our research, the major means of immobilizing LZ was encapsulation, which could fabricate rigid beads by dropping ALG into excessive LZ solutions containing calcium ions. Interestingly, after the encapsulation, the surface of beads was positively charged with LZ on the outmost layer. Although the significance of protein immobilization has been stressed, few researches have paid attention to the further immobilization of protein on the surface of protein loaded template. Here, the technique applied for further immobilization of much more LZ was electrostatic layer-by-layer self-assembly technique (LBL), which has rapidly spread within various researchers due to the simplicity of the procedure.19–21 Based on this technology, relatively high concentration of the solute in solution led to its excess adsorption where charge neutralization and resaturation resulted in charge reversal. Alternation of the surface charge resulted in a continuous assembly between negatively and positively charged materials affording great freedom in the number of layers.22–24
In this paper, ALG–LZ beads were firstly produced via cross-linking process. Encouraged by our recent progress on the deposition of LBL films on electrospun nanofibers,20,25 negatively charged ALG and positively charged LZ were alternately deposited on the surface of ALG–LZ beads through LBL self-assembly technique. The effect of the outermost layer variation and the number of coating bilayers on the formation of the LBL films deposited ALG–LZ beads were explored. Additionally, the catalytic activity of immobilized LZ was measured and the in vitro release experiments were carried out to determine the feasibility of the immobilized LZ release from ALG–LZ beads and LBL films coated beads.
2. Materials and methods
2.1. Materials
The starting materials included as follows: sodium alginate (ALG, Mw = 2.5 × 105 Da) was from Aladdin Chemical Reagent, China. Lysozyme (LZ, activity 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U mg−1) was purchased from Amresco Co., USA. Micrococcus lysodeikticus used for checking the catalytic activity of LZ was supplied by Nanjing Jiancheng, Bioengineering Institute, China. Coomassie Brilliant Blue (G250) was obtained from Amresco Co., USA. Other chemical reagents used in this experiment were analytical grade.
000 U mg−1) was purchased from Amresco Co., USA. Micrococcus lysodeikticus used for checking the catalytic activity of LZ was supplied by Nanjing Jiancheng, Bioengineering Institute, China. Coomassie Brilliant Blue (G250) was obtained from Amresco Co., USA. Other chemical reagents used in this experiment were analytical grade.
2.2. Fabrication of beads
According to the previous report,26 ALG–LZ beads were prepared by using cross-linking process. The schematic diagram of the beads formation was shown in Scheme 1. Briefly, ALG and LZ solutions were dissolved in purified water, and their concentrations were both fixed at 2%. Then, using a hypodermic syringe the prepared ALG solutions were dropped slowly into excessive LZ solutions containing Ca2+ (1%, w/w). The LZ solutions were still under gentle magnetic stirring at room temperature during the dripping process. The obtained beads were filtered and washed.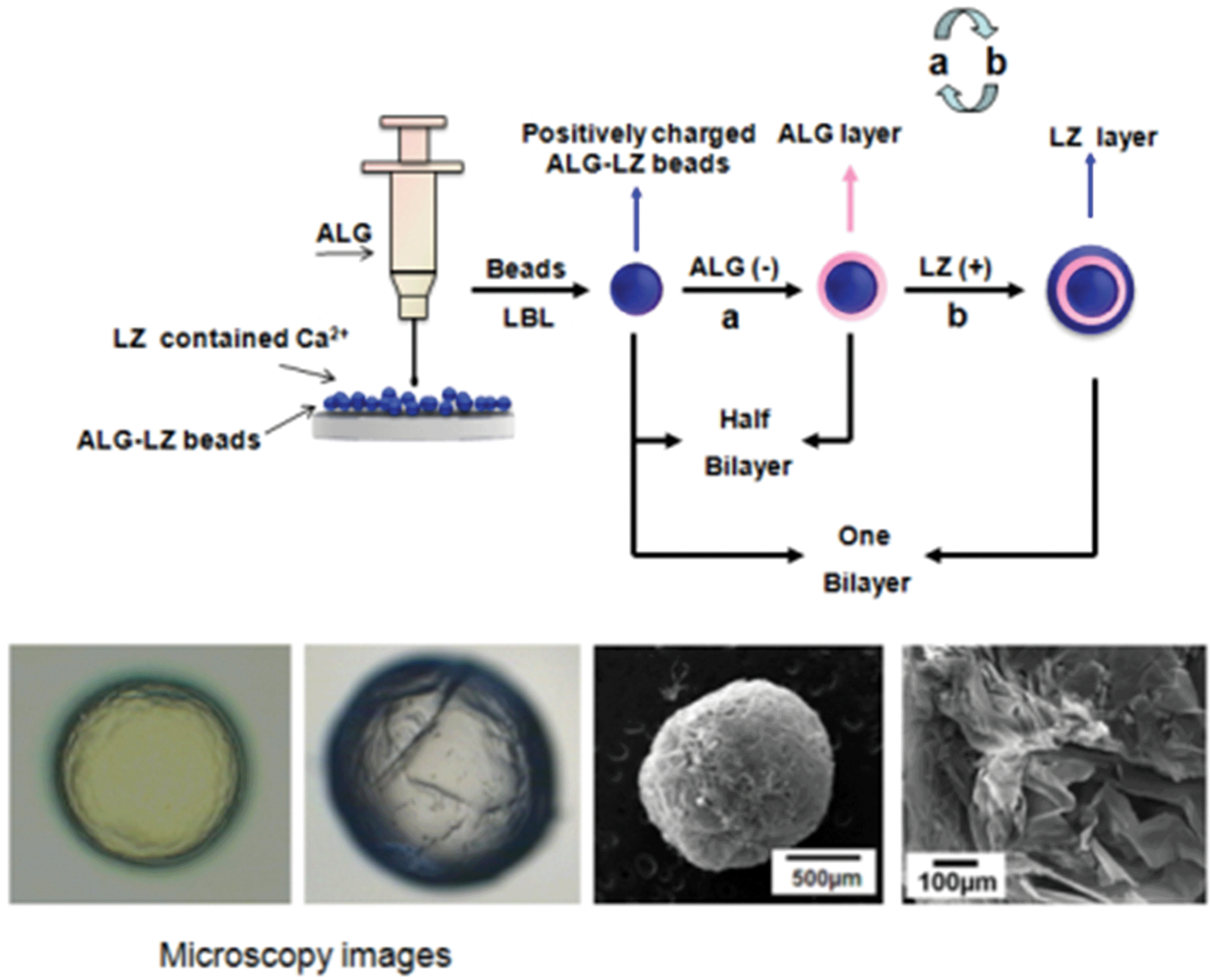 | ||
| Scheme 1 Schematic diagram illustrating the fabrication process of LBL films coated ALG–LZ beads. | ||
2.3. Preparation of dipping solutions for LBL process
The dipping solutions for LBL process including the negatively charged ALG solutions and the positively charged LZ solutions with the same concentration of 1 mg mL−1 by pouring them into purified water. The pH values of ALG and LZ solutions were adjusted at 4 and 6.5, respectively. The ionic strength of all the dipping solutions was regulated by the addition of sodium chloride with the concentration of 0.1 mol L−1.2.4. Formation of LBL structured composite beads
The fabrication process of the LBL structured beads was identical with that in our previous reports.14,20 Briefly, the LBL films coated beads were fabricated by adsorption of negatively charged ALG (−52.0 mV) and positively charged LZ (+25.0 mV) on the surface of positively charged ALG–LZ beads (+37.8 mV). First, ALG–LZ beads were soaked with ALG suspensions for 20 min, and then rinsed in pure water baths for 2 min and repeated three times. The beads were then immersed into the LZ solutions for 20 min followed by identical rinsing procedures. The adsorption and rinsing steps were repeated until the desired number of deposition bilayers obtained. Then the composite beads were filtered and freeze-dried for further characterizations. Herein, (ALG–LZ)n was used as a formula to label the LBL structured films, where n was the number of (ALG–LZ) bilayers. When n equaled to 5 or 10, the outermost layer on the composite beads was LZ. When n equaled to 5.5 or 10.5, the outermost layer was ALG.2.5. Characterizations
The scanning electron microscopy (SEM, JSM-6700F, JEOL Co., Ltd., Japan) was applied to observe the morphology of the beads. Fourier transform infrared (FT-IR) spectra were recorded by using a Nicolet 170-SX (Thermo Nicolet Ltd. USA). The N2 adsorption isotherm data collected at 77.3 K (Autosorb-1-MP, Quantachrome Co., USA) was applied for evaluating BET surface areas of the prepared samples. Prior to analysis, adsorbent samples were outgassed for 12 h at 313 K. Pore size distribution analysis was performed by conducting N2 adsorption experiments, and pore volume was calculated using the BJH method.27 The mechanical properties of the developed structures were examined by a texture analyzer TA.XT2i TA.XTplus (SMS) at a test speed of 2 mm s−1 with 90% strain.2.6. Measurement of LZ activity
The determination of LZ activity was using the M. lysodeikticus Fleming (turbidity) method. The activity measurement of free LZ was identical with our former report.14 The activity of immobilized LZ was evaluated according to the method of free LZ determination. 1 mg freeze-dried beads were added into the cuvette to conduct the test.2.7. In vitro LZ release profiles
The LZ release experiments were done in 10 mmol L−1 phosphate buffer with the pH value of 7.3.28 10 mg beads were put into a centrifuge tube containing 10 mL of the above solution, and then incubated on a constant temperature shaking bed with 100 rpm at 37 °C. With 4 h or 24 h intervals, 1 mL medium was withdrawn and immediately replaced with the same amount of fresh medium. The amounts of released LZ were determined using Coomassie Brilliant Blue (G250) method through UV-1800 spectrophotometry at 595 nm. All experiments were done in triplicate and mean values were reported. The above mentioned method, commonly referred as the Bradford assay, was based on the absorption shift from 470 to 595 nm when the brilliant blue G dye binds to protein. The brilliant blue G dye bound most readily to arginyl and lysyl residues in the protein, which could lead to variation in the response of the assay to different proteins. The preparation for the Coomassie Brilliant Blue solution was as follow: a total of 100 mg of Coomassie Brilliant Blue G-250 was dissolved in 50 mL of 95% ethanol solution. 100 mL 85% phosphoric acid (w/v) solution was added and then the blending solution was diluted to 1 L with distilled water.3. Results and discussion
3.1. Particle size and mechanical properties of beads
The particle size was measured with a micrometer caliper. The data was shown in Table 1. Obviously, the diameter of the wet beads ranged from 1.5 to 3.0 mm, and that of the freeze dried beads ranged from 1.3 to 2.7 mm. The average diameter of the beads slightly increased with the increasing number of coating films both in wet and dry state. The average thickness of each bilayer of the LBL films coated beads could be estimated to 225 (n = 5/5.5) and 127 (n = 10/10.5) nm, respectively.| Beads coated with LBL films | Average diameter (mm) | |
|---|---|---|
| Wet beads | Freeze dried beads | |
| a Data shown are the mean ± SD (n = 10), measured with a micrometer. | ||
| (ALG–LZ)0 | 1.50 ± 0.09 | 1.25 ± 0.03 |
| (ALG–LZ)5 | 2.62 ± 0.07 | 2.34 ± 0.03 |
| (ALG–LZ)5.5 | 2.74 ± 0.03 | 2.45 ± 0.03 |
| (ALG–LZ)10 | 2.76 ± 0.02 | 2.52 ± 0.05 |
| (ALG–LZ)10.5 | 2.83 ± 0.12 | 2.67 ± 0.14 |
Fig. 1 shows the mechanical properties including the hardness (Fig. 1A) and the resilience (Fig. 1B) of the beads. Both the hardness and the resilience of the ALG–LZ beads were higher than those of LBL film-coated beads, which were attributed to the sufficient soaking time and the increased amount of LZ. LZ was a kind of alkaline enzyme which had low hardness,29 thus the hardness of the beads with the outermost layer of ALG (Fig. 1A 5.5 and 10.5) was a little higher than that with LZ on the outermost layer (Fig. 1A 5 and 10).
 | ||
| Fig. 1 The mechanical properties including (A) hardness and (B) resilience of LBL structured beads coated with: (ALG–LZ)0, (ALG–LZ)5, (ALG–LZ)5.5, (ALG–LZ)10 and (ALG–LZ)10.5. Data shown are the mean ± standard deviations (n = 3). Significant difference: **p < 0.01. | ||
3.2. Morphology of the beads
Fig. 2 presents the SEM images of ALG–LZ beads and LBL films coated beads after freeze-drying treatment. Coincide with our previous report,30 the SEM images of the beads displayed good sphericity and porosity. In order to investigate the influences of the number of coating bilayers on the formation of LBL films coated beads, different number of LBL structured films were deposited on ALG–LZ beads. With the different number of coating bilayers, the morphology of the LBL structured beads was different from each other. The surface of the LBL films coated beads became coarse which was distinguished from that of the uncoated beads, verifying that the films were successfully assembled on the surface of the ALG–LZ beads.31 Interestingly, the surface roughness of LBL films coated beads was clearly observed. The figures show the cross-section of ALG–LZ beads and (ALG–LZ)10.5 films coated beads and high magnification image of the surface of the (ALG–LZ)10.5 films coated beads, respectively. Remarkably, the pores were both on the surface and internal section of beads. The reason for the presence of the small pores on the surface of LBL films coated beads was that the LBL films were split into webs during the drying process,20 the formation of the pores on the internal section was ascribed to the pressure developing inside the beads, and some of that pressure could be released from the pores. When more polymers were coated on the surface of beads, they could contribute to the structural support of beads during the solvent evaporation,32 so more polymers coating resulted in less pores inside beads. Because of the high treacliness of the ALG solution, the beads were produced with a tiny tail-like part (Fig. 2c). | ||
| Fig. 2 SEM morphology of LBL structured beads coated with: (a) (ALG–LZ)0, (b) (ALG–LZ) 5.5, (c) (ALG–LZ)10, (d) (ALG–LZ)10.5. Images (e) and (f) showed high magnification image and internal section of (ALG–LZ)10.5, respectively. | ||
The porous structure was assumed to affect drug release ability. The N2 adsorption and desorption isotherm, the pore size distribution were performed (Fig. 3). The cumulative surface area of ALG–LZ beads and (ALG–LZ)10.5 films coating was 28.2 and 13.076 m2 g−1, respectively. Obviously, the cumulative surface area of the LBL films coated beads was smaller than that of ALG–LZ beads, which further confirmed that the LBL structured films modification was effective. As mentioned above, the deposition space on the beads was limited and could be filled with the polymers via LBL deposition, so the surface area of the beads would become smaller with increasing the number of coating bilayers. On the basis of BJH results, the beads before and after LBL modification mainly possessed mesopores with radius in the 2–7 nm, and the LBL films coated beads had more mesopores with radius in the 4–7 nm than ALG–LZ beads. After LBL modification, pores with different size could be observed from BJH curves (Fig. 3b′ and c′), which presumably related to the freeze-drying treatment. According to the previous report,33 the porosity of ALG gel affected by drying the beads and complete dehydration of ALG beads could resulted in surface cracking.
 | ||
| Fig. 3 Nitrogen adsorption and desorption isotherms at 77.3 K of: (a) (ALG–LZ)0, (b) (ALG–LZ)5 and (c) (ALG–LZ)10.5 film-coated beads, respectively. BJH pore size distribution images derived from the adsorption isotherm were shown: (a′) (ALG–LZ)0, (b′) (ALG–LZ)5 and (c′) (ALG–LZ)10.5 film-coated beads, respectively. | ||
3.3. FT-IR Analysis
The FT-IR spectra of composite beads and raw materials were shown in Fig. 4. In the spectrum of ALG,34 the characteristic peaks at 3430, 1615 and 1417 cm−1 stood for the –OH groups vibration, asymmetric and symmetric –COO stretching vibrations, respectively. The band around the 1030 cm−1 (C–O stretching) was ascribed to its saccharine structure. As we know, the amide linkages between amino acid residues in polypeptides and proteins gave the well-known fingerprints in their FT-IR spectra, displaying the character of those substances. In the FT-IR spectra of proteins, the position of the amide I band acted as a sensitive indicator of conformation changes in the protein secondary structure,35 and the position of the amide I peak around 1650 cm−1 could be observed in LZ, ALG–LZ beads and LBL films coated beads, which indicated that the secondary structure of the protein was retained in the immobilized LZ molecules. The peak at 1450 cm−1, corresponded to the C–C stretching vibration of LZ molecules.36 Additionally, the peak of –COO– became widely at 1417 cm−1 and the peak at 1530 cm−1, corresponding to the amide II band even disappeared, which indicated that the carboxyl group of ALG interacted with the amino group of LZ. | ||
| Fig. 4 FT-IR Spectra of LBL structured beads coated with: (a) (ALG–LZ)0, (b) (ALG–LZ)5 and (c) (ALG–LZ)10.5. | ||
3.4. Enzymatic catalysis
The activity of immobilized LZ was listed in Fig. 5. The results were obtained from the freeze-dried samples and free LZ was employed as control. The enzymatic activity of LZ immobilized on ALG–LZ beads was only 18.78% of that of free LZ. The decrease in activity was likely due to the amount of LZ aggregates formed as the result of encapsulation procedure.37 The activity of immobilized LZ on (ALG–LZ)n films coating was higher than that of ALG–LZ beads. The ratio of the activity of immobilized LZ on (ALG–LZ)5 and (ALG–LZ)5.5 films coating and free LZ was 41.14% and 35.73%, respectively. It revealed that when the number of coating films reached 5 or 5.5, catalytic activity of the samples with LZ on the outermost layer was higher than that with ALG on the outermost layer, but when the number of coating films reached 10 or 10.5, the catalytic activity of the samples with ALG on the outermost layer was higher than that with LZ on the outermost layer. The reason for the above results was explained as follows: after the first step of LBL, the beads showed low catalytic activity because ALG was on the outermost layer of ALG–LZ beads. After LZ was assembled on the surface of the beads, the beads were covered with LZ, which could contact with M. lysodeikticus directly, resulting in high catalytic activity. However, when the number of coating films reached 10 or 10.5, with the thickness of each bilayer of the LBL films coated beads became thin, the hindered diffusion of LZ caused by ALG got weaken accordingly. Besides, as much more LZ deposited on the surface of film-coated beads, the catalytic activity of the samples with ALG on the outermost layer was higher than that with LZ on the outermost layer. The result was identical with that of the release behavior of LZ from the beads in the following release experiments. And the mechanism is not clear enough now, we will investigate it next. | ||
| Fig. 5 The enzymatic activity of immobilized LZ of LBL structured beads coated with: (ALG–LZ)0, (ALG–LZ)5, (ALG–LZ)5.5, (ALG–LZ)10 and (ALG–LZ)10.5. Significant difference: **p < 0.01. | ||
Herein, ALG and LZ were successfully assembled on the surface of ALG–LZ beads via LBL technique. After immobilization, the catalytic activity of LZ was still maintained, and with the different number of coating films, different parameters dominated the catalytic activity of the samples. According to previous literature, ALG could interact with various kinds of proteins in a protective or destructive manner. Obviously, it deduced that ALG had a protective effect on immobilized LZ.38
3.5. In vitro release profiles
In order to explore the controlled release properties of ALG–LZ and LBL films coated beads, in vitro release experiments were performed at different time intervals. Fig. 6 shows that LZ could be released from both ALG–LZ beads and LBL films coated beads. Obviously, the initial burst phenomenon was exhibited in all samples which could be attributed to the diffusion of water molecules into the polymeric beads structure, leading to the release of immobilized LZ into aqueous solutions from the beads.39 Actually, beads made from high α-L-guluronic acid would reswell only slightly upon rehydration, so all the beads immersed in phosphate buffer had similar swelling behavior which caused the previously mentioned diffusion.40 Besides, the equivalent release rates of immobilized LZ released from the LBL structured beads could be observed. Moreover, in the controlled release test, more LZ could be released from LBL films coated beads than that from uncoated beads, which confirmed that LBL self-assembly technique was effective for the immobilization of more LZ. | ||
| Fig. 6 Release profiles of LZ from LBL structured beads (a) every 4 h and (b) every 24 h. Data shown are the mean ± standard deviations (n = 3). | ||
Fig. 6a presents that the ALG–LZ beads had lower initial release quantity (2.88%) than that of LBL films coated beads (13.16%) during the period of 8 h, which resulted from the insufficient immersion time for the degradation of ALG and few LZ loading on the uncoated beads. Besides, LBL films had the lower densities that could promote materials diffusion,41 and the release of LZ could be related to a difference in the diffusion barrier at the surface of the beads.42
Obviously, in Fig. 6b, the amount of LZ released from ALG–LZ beads was twice as much as that from ALG–LZ beads in Fig. 6a, because of the long time immersing in medium containing phosphate ions which caused the degradation of Ca2+ crosslinked ALG gel by removal of the Ca2+ ions.43 Besides, the amount of LZ released from all the beads reached maximum after 24 h (Fig. 6b), which was presumably ascribed to the diffusion of immobilized LZ through the pores. It demonstrated that with growing number of coating films, both the porosity of the beads and the degradative phosphate ions in the release medium had the great influences on the LZ release profiles. Fig. 6b presents that more LZ could be released from the (ALG–LZ)10 or (ALG–LZ)10.5 films coated beads than that from (ALG–LZ)5 or (ALG–LZ)5.5 films coated beads, because total amount of LZ assembled on former beads was more than that on latter beads. Consequently, the amount of LZ released from the beads partially depended on the total amount of LZ in the beads when the release time was long enough. Besides, after long time immersion, more LZ could be released via the pores on the beads that suggests the open pore structure could be related the different LZ release behaviors from coated beads.44
On the contrary, during the short time immersion (Fig. 6a), the amount of released LZ was affected by the amount of LZ assembled on the outermost layer of the beads. Hence, more LZ could be released from (ALG–LZ)5 or (ALG–LZ)5.5 films coated beads than that from (ALG–LZ)10 or (ALG–LZ)10.5 films coating. The reason was as follows: ALG–LZ beads had higher positive charge and larger specific surface area than LBL structured beads, so the thick and large amount of ALG could be assembled on the surface of ALG–LZ beads in the first step of LBL process, which would adsorb more LZ in the next step. Notably, the LZ assembled on the surface of the LBL films coated beads was less than that in ALG–LZ beads. When the second layer (ALG) was deposited, its amount was less than that of ALG in the former layer. Therefore, the amount of LZ assembled on each bilayer decreased with increasing the number of coating bilayers.
In Fig. 6, especially 24 h later the amount of LZ released from LBL structured beads reduced more or less. The reason was that several free LZ in the solution could be reabsorbed onto the surface of the beads. The result was identical with the previous report.45
4. Conclusion
ALG–LZ beads were selected as the template and modified with negatively charged ALG and positively charged LZ through LBL self-assembly technology. The morphology of LBL films coated ALG–LZ beads was affected by the composition of the outermost layer of the beads. The BET surface area results proved that the ALG and LZ were successfully assembled on the surface of ALG–LZ beads. Surface porosity and phosphate ions had significant influences on the release of LZ. In vitro release assay indicated that immobilized LZ could be released into aqueous solutions from both ALG–LZ beads and LBL films coated beads, and the immobilized LZ still maintained its enzymatic activity, which could be used for the nutrition delivery, drug-loading, catalysis, antimicrobial, etc.Acknowledgements
This project was funded by the Open Research Fund Program of Hubei-MOST KLOS & KLOBME.References
- H. J. Lee, Y. H. Park and W. G. Koh, Adv. Funct. Mater., 2013, 23, 652 CrossRef.
- J. E. Galán and A. Collmer, Science, 1999, 284, 1322 CrossRef.
- Y. Li, J. Zheng, H. Xiao and D. J. McClements, Food Hydrocolloids, 2012, 27, 517 CrossRef CAS PubMed.
- T. Sessler, J. Weiss and Y. Vodovotz, Food Hydrocolloids, 2013, 32, 294 CrossRef CAS PubMed.
- V. R. Sinha and A. Trehan, J. Controlled Release, 2003, 90, 261 CrossRef CAS.
- M. Ye, S. Kim and K. Park, J. Controlled Release, 2010, 146, 241 CrossRef CAS PubMed.
- Y. Li and D. J. McClements, Food Hydrocolloids, 2013, 33, 368 CrossRef CAS PubMed.
- G. Ma, D. Fang, Y. Liu, X. Zhu and J. Nie, Carbohydr. Polym., 2012, 87, 737 CrossRef CAS PubMed.
- L. Salvia-Trujillo, M. A. Rojas-Graü, R. Soliva-Fortuny and O. Martín-Belloso, Food Hydrocolloids, 2013, 30, 401 CrossRef CAS PubMed.
- P. de Vos, M. M. Faas, B. Strand and R. Calafiore, Biomaterials, 2006, 27, 5603 CrossRef CAS PubMed.
- S. Sershen and J. West, Adv. Drug Delivery Rev., 2002, 54, 1225 CrossRef CAS.
- S. Singh and J. Singh, Int. J. Pharm., 2004, 271, 189 CrossRef CAS PubMed.
- K. Zhu, T. Ye, J. Liu, Z. Peng, S. Xu, J. Lei, H. Deng and B. Li, Int. J. Pharm., 2013, 441, 721 CrossRef CAS PubMed.
- W. Huang, X. Li, Y. Xue, R. Huang, H. Deng and Z. Ma, Int. J. Biol. Macromol., 2013, 53, 26 CrossRef CAS PubMed.
- Y. Li, S. Kadam, T. Abee, T. M. Slaghek, J. W. Timmermans, M. A. Cohen Stuart, W. Norde and M. J. Kleijn, Food Hydrocolloids, 2012, 28, 28 CrossRef CAS PubMed.
- R. Jayakumar, M. Prabaharan, P. Sudheesh Kumar, S. Nair and H. Tamura, Biotechnol. Adv., 2011, 29, 322 CrossRef CAS PubMed.
- Q.-Q. Gai, F. Qu, Z.-J. Liu, R.-J. Dai and Y.-K. Zhang, J. Chromatogr. A, 2010, 1217, 5035 CrossRef CAS PubMed.
- N. Charernsriwilaiwat, P. Opanasopit, T. Rojanarata and T. Ngawhirunpat, Int. J. Pharm., 2012, 427, 379 CrossRef CAS PubMed.
- W. Huang, H. Xu, Y. Xue, R. Huang, H. Deng and S. Pan, Food Res. Int., 2012, 48, 784 CrossRef CAS PubMed.
- H. Deng, X. Zhou, X. Wang, C. Zhang, B. Ding, Q. Zhang and Y. Du, Carbohydr. Polym., 2010, 80, 474 CrossRef CAS PubMed.
- W. Li, X. Li, T. Wang, X. Li, S. Pan and H. Deng, Eur. Polym. J., 2012, 48, 1846 CrossRef CAS PubMed.
- K. Ariga, Y. M. Lvov, K. Kawakami, Q. Ji and J. P. Hill, Adv. Drug Delivery Rev., 2011, 63, 762 CrossRef CAS PubMed.
- K. Ariga, J. P. Hill and Q. Ji, Macromol. Biosci., 2008, 8, 981 CrossRef CAS PubMed.
- K. Ariga, J. P. Hill and Q. Ji, Phys. Chem. Chem. Phys., 2007, 9, 2319 RSC.
- R. Huang, Y. Li, X. Zhou, Q. Zhang, H. Jin, J. Zhao, S. Pan and H. Deng, Carbohydr. Polym., 2012, 90, 957 CrossRef CAS PubMed.
- X. W. Shi, Y. M. Du, L. P. Sun, J. H. Yang, X. H. Wang and X. L. Su, Macromol. Biosci., 2005, 5, 881 CrossRef CAS PubMed.
- A. Gupta, C. D. Saquing, M. Afshari, A. E. Tonelli, S. A. Khan and R. Kotek, Macromolecules, 2009, 42, 709 CrossRef CAS.
- C. Pérez, P. De Jesús and K. Griebenow, Int. J. Pharm., 2002, 248, 193 CrossRef.
- M. Tachibana, Y. Kobayashi, T. Shimazu, M. Ataka and K. Kojima, J. Cryst. Growth, 1999, 198, 661 CrossRef.
- R. Xu, X. Feng, W. Li, S. Xin, X. Wang, H. Deng and L. Xu, J. Biomater. Sci., Polym. Ed., 2013, 24, 1 CrossRef CAS PubMed.
- S. Xin, X. Li, Z. Ma, Z. Lei, J. Zhao, S. Pan, X. Zhou and H. Deng, Carbohydr. Polym., 2013, 92, 1880 CrossRef CAS PubMed.
- A. Bohr, J. Kristensen, M. Dyas, M. Edirisinghe and E. Stride, J. R. Soc., Interface, 2012, 9, 2437 CrossRef CAS PubMed.
- W. R. Gombotz and S. F. Wee, Adv. Drug Delivery Rev., 2012, 64, 194 CrossRef PubMed.
- W. Li, X. Li, Y. Chen, X. Li, H. Deng, T. Wang, R. Huang and G. Fan, Carbohydr. Polym., 2013, 92, 2232 CrossRef CAS PubMed.
- A. Gole, J. Thakar and M. Sastry, Colloids Surf., B, 2003, 28, 209 CrossRef CAS.
- H.-M. Ding, L. Shao, R.-J. Liu, Q.-G. Xiao and J.-F. Chen, J. Colloid Interface Sci., 2005, 290, 102 CrossRef CAS PubMed.
- C. Pérez and K. Griebenow, J. Pharm. Pharmacol., 2001, 53, 1217 CrossRef.
- L. A. Wells and H. Sheardown, Eur. J. Pharm. Biopharm., 2007, 65, 329 CrossRef CAS PubMed.
- X. Huang and C. S. Brazel, J. Controlled Release, 2001, 73, 121 CrossRef CAS.
- J.-W. Wang and M.-H. Hon, J. Appl. Polym. Sci., 2005, 96, 1083 CrossRef CAS.
- M. Onda, Y. Lvov, K. Ariga and T. Kunitake, Biotechnol. Bioeng., 1996, 51, 163 CrossRef.
- W. R. Gombotz and S. Wee, Adv. Drug Delivery Rev., 1998, 31, 267 CrossRef CAS.
- K. Y. Lee and D. J. Mooney, Prog. Polym. Sci., 2012, 37, 106 CrossRef CAS PubMed.
- R. Censi, P. Di Martino, T. Vermonden and W. E. Hennink, J. Controlled Release, 2012, 161, 680 CrossRef CAS PubMed.
- R. Xu, S. Xin, X. Zhou, W. Li, F. Cao, X. Feng and H. Deng, Int. J. Pharm., 2012, 438, 258 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |