
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulating the thickness of nanofiltration membranes for efficient water purification†
Ke
Tang‡
a,
LinSheng
Zhu‡
a,
Piao
Lan
a,
YunQiang
Chen
b,
Zhou
Chen
 *a,
Yihong
Lan
b and
WeiGuang
Lan
*ab
*a,
Yihong
Lan
b and
WeiGuang
Lan
*ab
aXiamen University Center for Membrane Application and Advancement, College of Materials, Xiamen University, Xiamen 361005, Fujian, China. E-mail: zhouchen@xmu.edu.cn; wglan@xmu.edu.cn
bSuntar Membrane Technology (Xiamen) Co., Ltd., Xiamen 361022, Fujian, China
First published on 3rd August 2023
Abstract
Fabrication of an organic polymer nanofiltration membrane with both high water permeability and high salt rejection is still a big challenge. Herein, phytic acid (PhA)-modified graphene oxide (GO) was used as the membrane thickness modifier, which was introduced into the thin-film nanoparticle composite (TFN) membrane via in situ interfacial polymerization (IP) on a porous substrate. The water flux of the optimally tuned TFN-GP-0.2 composite membrane is 48.9 L m−2 h−1, which is 1.3 times that of the pristine thin-film composite (TFC) nanofiltration membrane (37.9 L m−2 h−1) (GP represents the PhA modified GO composite). The rejection rate of 2000 ppm MgSO4 for TFN-GP-0.2 membranes was maintained at 97.5%. The increased water flux of the TFN-GP composite membrane compared to that of the TFN nanofiltration membrane was mainly attributed to enhanced hydrophilicity and reduced thickness of the polyamide (PA) layer. Molecular dynamics (MD) simulations confirm that the diffusion rate of amine monomers is reduced by the presence of a GP complex in the IP process, which facilitates the formation of PA layer with thinner thickness. In addition, the TFN-GP-0.2 composite membrane also showed good long-term stability; after 12 h of continuous operation, the water flux only decreased by 0.1%. This study sheds new light on the development of GO-based nanofiltration for potential implementation, as well as a unique concept for manufacturing high-performance nanofiltration membranes.
1 Introduction
Efficient water treatment technologies have become urgent global demands due to industrial development and environmental pollution. Membrane separation technology features high separation efficiency, low energy demand, ease of operation, and low toxicity making it widely employed in wastewater purification and desalination.1–6 Nanofiltration is an alternative to conventional filtration technologies, and is very effective in removing organic molecules and multivalent salts at a relatively low operational pressure that is difficult to be achieved by conventional ultrafiltration.7–9 Nanofiltration membranes, such as polyamide (PA) nanofiltration membranes, are typically manufactured by interfacial polymerization (IP), a polymerization reaction that occurs at or near the interface boundary between two immiscible aqueous and organic solutions. However, conventional PA nanofiltration membranes usually suffer from poor anti-pollution, high energy consumption and “trade-off” limits between permeability and selectivity, which limit their lengthy recurring runs and applications.10–12 Thence, there is an increasing demand for constructing new nanofiltration materials with excellent separation factors and water permeability.It is well known that the performance and selectivity of thin-film nanoparticle composite (TFN) nanofiltration membranes mainly depend on the PA layer during the separation process. The structure and properties of the PA layer affect the comprehensive performance, such as permeability and selectivity of the nanofiltration membrane, far and even more than the impact of the substrate. It has been well investigated that the surface hydrophilic properties, internal transport channel and thickness of the PA layer are pivotal for membrane separation performance. The introduction of nanoparticles able to generate numerous low resistance channels in their intrinsic nanopores or interface voids for water transport, result in a significant increase in water flux. Wang et al. incorporated covalent organic frameworks (COFs) into the PA active layer by the IP method. Compared to the pristine nanofiltration membrane, the water flux of the as-prepared membrane can be increased twofold. The Na2SO4 rejection rate for a COF modified nanofiltration membrane is higher than 80%.13 Preparing a thinner PA layer is another practical approach for optimizing the PA layer for reducing water transport resistance. However, the thickness of most membranes is usually greater than 100 nm due to the difficulty in controlling the rapid reaction of IP, resulting in a relatively low water flux. A PA layer within 100 nm is only obtained in few studies. For example, Livingston et al. fabricated a polyamide layer with thickness below 10 nm on a cadmium hydroxide nanostrand support layer, which greatly improved the permeation performance.14 There are some other reports on optimizing the separation layer, Fan et al. used phenolic compounds instead of polyols to fabricate 10 nm ultrathin polyurethane membranes, which exhibited higher flux and rejection of dye.15 Zhang et al. used dopamine as the intermediate layer for interfacial polymerization, and successfully prepared a defect-free separation layer thinner than 44 nm, taking into account the high flux and excellent rejection.16 It can be seen that the modification of the separation layer is a good strategy to optimize the nanofiltration membrane. However, how to fabricate a PA membrane within 100 nm on a traditional porous support is still a big challenge.
Recently, various nanomaterials, such as graphene oxide (GO),17–19 boron nitride (BN),20 zeolites21,22 and metal–organic frameworks (MOFs)23,24 have been used to improve the separation performance of TFN membranes. Among these nanomaterials, GO has gained widespread attention, because of its two-dimensional (2D) layered structure as well as desired physical and mechanical properties.25 The presence of abundant functional groups on GO nanosheets, including hydroxyl, carboxyl, and epoxy groups, enables GO to easily interact with both piperazine (PIP) and organic trimesoyl chloride (TMC).26 These oxygen-containing functional groups on GO nanosheets assist in the dispersion of GO in water and provide suitable sites for interactions with transport components (water molecules and ions) during the water treatment process, which results in increased water flux and better anti-fouling performance. Meanwhile, the hydrophilic GO may allow water molecules to intercalate into its interlayer structure, providing water molecules with fast-transport channels through the PA layer. Bano et al.27 used an aqueous mixture of m-phenylenediamine (MPD)-GO and organic TMC solutions to fabricate a GO-modified PA layer on a polysulfone support. The permeate water flux of the resultant membrane was from 1.8 L m−2 h−1 to 22 L m−2 h−1, but salt rejection was only maintained at about 80%. Li et al.28 proved that TFC membranes containing a PA layer with 0.12 wt% GO can increase the water flux up to 80% while decreasing salt rejection from 96.8% to 87.5%. Hu et al.29 developed an ultrathin TFN membrane with a wrinkled sandwich structure by changing the lateral size and additive content of GO. The optimized composite membrane provides a high water flux, but only 90% rejection. We can see that the addition of pure GO nanosheets to the PA layer provides only limited improvement in water flux and usually results in reduced membrane selectivity.30 This is due to the fact that GO membranes are unstable and may disintegrate in an aqueous environment because of the strong electrostatic repulsion between GO nanosheets. Hence, it is difficult to fabricate a nanofiltration membrane based solely on the GO additive with a satisfactory trade-off between permeability and selectivity. To overcome these difficulties, proper modification or functionalization of GO nanosheets can improve the obtained TFN nanofiltration performance and also be expected to change the hydrophilic nature of the film layer surface, which would be very worthwhile research if the thickness of the PA layer could be further reduced.
Phytic acid (PhA), a natural organic compound that is water soluble, is easily produced from grains and beans.31,32 It consists of a cyclohexanehexol ring with six phosphate groups symmetrically connected to it. PhA has a specific molecular structure that allows for controlling the diffusion rate of amine monomers and acid chloride toobtain a thin PA layer because the high spatial charge density of PhA makes it capable of forming strong electrostatic interactions with amine monomers. PhA has strong hydrophilicity due to the presence of hydroxyl groups, and its phosphate groups can easily interact with the oxygen-containing functional groups of GO nanosheets by forming multiple hydrogen bonds. Hence, PhA can be applied to GO-based membranes as both a surface modifier and an additive. The addition of PhA to the GO membrane could enhance the membrane hydrophilicity. Therefore, the GP complex formed by GO and PA can be applied to the PA layer of TFC membranes (GP represents the PhA modified GO composite), and is expected to improve the hydrophilicity of TFC membranes and optimize the thickness of the PA layer.
In this work, we prepared a series of TFN-GP nanofiltration membranes by the IP method using the GP complex as an aqueous phase additive. The surface morphologies and chemical structures of the modified membranes were observed by scanning electron microscopy (SEM), transmission electron microscopy (TEM), atomic force microscopy (AFM), energy dispersive spectroscopy (EDS), X-ray photoelectron spectroscopy (XPS) and Fourier transform infrared (FT-IR) spectroscopy. The role of GP complexes in the IP process was investigated by molecular dynamics (MD) simulations and diffusion experiments, and the results confirmed that GP complexes can effectively reduce the thickness of PA layers. The optimized membrane maintained a high salt rejection rate of over 97.5%, and the water flux of the optimized composite membrane increased 1.3 times from 37.9 L m−2 h−1 for the TFC membrane to 48.9 L m−2 h−1 for the TFN-GP-0.2 membrane. The reduction of the polyamide layer thickness and the improvement of hydrophilicity are two main reasons for this enhancement in membrane performance. In addition, the TFN-GP-0.2 membrane also demonstrates long-term stability. The present work provides useful insights into fabricating next generation TFN nanofiltration membranes for better separation performance.
2 Experimental
2.1 Materials
Phytic acid (PhA, 70 wt%) was provided by Shanghai Dibo Chemicals Technology Co. Ltd. (China). Magnesium sulphate (MgSO4, 99%), sodium sulphate (Na2SO4, 99%), magnesium chloride (MgCl2, 95%), sodium chloride (NaCl, 99%), camphorsulfonic acid (C10H16O4S, 99%), triethylamine (C6H15N, 99%), Isopar G (CH3(CH2)4CH3, 99%), and trimesoyl chloride (C9H3Cl3O3, 99%) were supplied by XiLong Chemical Reagents Co. Ltd. (China). Piperazine (C4H10N2, 99%) and heptane (C7H16, 99%) were obtained from Sinopharm Chemical Reagents Co. Ltd. (China). A polysulfone (PSU) ultrafiltration membrane, and VNF1, DNF2 and VNFK commercial membranes were supplied by Suntar Membrane Technology (Xiamen). The reagents mentioned above are of analytical grade.2.2 Preparation of the GP complex
According to our earlier research, the GO solution was prepared using a modified Hummers' method.33 The GP complex was prepared as follows: 50 mL of GO dispersion (2 mg mL−1) was added to different amounts of PhA solution (0.143 g, 0.714 g, 1.429 g, and 2.143 g) and mixed for 2 min. Following this, the mixed solution was sonicated for 30 min to ensure a homogeneous GP suspension with different mass ratios of GO to PhA (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:5, 1:10, and 1:15). The mass ratio determined the component ratio of the obtained complex, expressed as GPY (GP1, GP5, GP10, and GP15), in which Y represents the mass ratio of PhA.
:1, 1:5, 1:10, and 1:15). The mass ratio determined the component ratio of the obtained complex, expressed as GPY (GP1, GP5, GP10, and GP15), in which Y represents the mass ratio of PhA.
2.3 Preparation and characterization of the TFN nanofiltration membrane
We used a porous PSU ultrafiltration membrane as a support and prepared a nanofiltration membrane by interfacial polymerization of the PIP aqueous phase and TMC organic phase monomer. After stirring 0.3 wt% PIP and 0.21 wt% camphorsulfonic acid in deionized water for 20 min, 0.16 wt% triethylamine was added, and stirred for 10 min, and then a certain amount of GP complex was added, subjected to ultrasonic dispersion for 10 min and then stirred for 20 min to prepare PIP-GP aqueous solution. 0.1 wt% of TMC was dissolved in Isopar G and stirred for 4 h to prepare the uniform organic phase solution. The porous PSU ultrafiltration membrane was mounted on the fixture and blown dry. The PIP-GP aqueous solution was poured on the surface of the support membrane and held for 2 min before it was blown dry again. Then, the TMC organic solution was also poured on the support membrane surface. The reaction was kept for 20 s, and then the organic solution was poured off. The freshly prepared membranes were immediately held at 90 °C for 2 min to complete the preparation, and were rinsed with deionized water and preserved in water before testing. For the preparation of TFN-GO and TFC-PhA membranes, the GP was replaced by the corresponding weight of GO and PhA in the PIP aqueous phase; and for the preparation of TFC, there are no additives of GO or GP nanomaterials in aqueous. The specific aqueous phase solution formulation is shown in Table S1.† The GP-modified TFN nanofiltration membrane was named TFN-GPY-Z, where Z represents the concentration of the GPY complex. Unless otherwise specified, TFN-GP refers to TFN-GP10 and Z refers to 0.2.2.4 Diffusion experiment of PIP from the aqueous phase to the organic phase
For the diffusion experiment, the composition of the reaction water phase is identical to that for membrane preparation, and in the solution of the organic phase TMC was replaced by n-heptane. In detail, the PIP solution was prepared by mixing 0.3 wt% PIP and a certain amount of GP complex in deionized water. For the simulation of TFN-GO and TFC-PhA membranes, the GP was replaced by the corresponding weights of GO and PhA in the PIP solution. For the simulation of the TFC membrane, there was no additive in PIP solution. First, 30 mL of homogeneous PIP solution containing different additives was poured into a 50 mL glass container, followed by 10 mL of n-heptane being injected above the aqueous phase. After 60 s, 3 mL of n-heptane with PIP was removed from the organic solution and added to the cuvettes. PIP absorbance in n-heptane was measured using an ultraviolet-visible (UV-vis) spectrophotometer (Persee Co, TU-1901, China).2.5 Separation performance test of the TFN nanofiltration membrane
The separation performances of TFC and TFN-GP composite nanofiltration membranes were evaluated using a cross-flow filtration membrane module with an effective test area of 42 cm2 at 25 °C. The feed solution is 2000 ppm MgSO4. Before the membrane performance test, all membranes were pre-pressurized for 25 min with an operation pressure of 6.9 bar. Water flux was measured by weighing the permeated water every 5 min. Furthermore, a conductivity meter was used to record the conductivities of the penetrated and feed solutions. The following calculations were made for salt rejection and the water flux: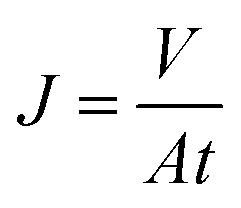 | (1) |
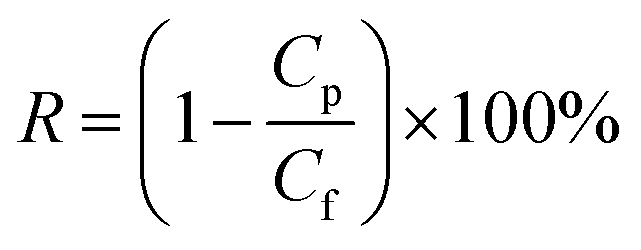 | (2) |
2.6 Membrane stability performance
To evaluate the stability of TFN-GP membranes, TFC membranes and TFN-GP-0.2 membranes were tested for stability using the following steps.MgSO4 solution with a concentration of 2000 ppm was used as the feed solution, and the membranes were pre-pressurized for 25 min with an operation pressure of 6.9 bar, and the filtrate was collected every 5 min for the determination of the permeate flux and rejection rate, and then the filtrate was collected and measured every 25 min for 12 h to determine the stability of the TFN nanofiltration membrane.
Detailed materials, membrane characterization methods, and other relevant data are available in the ESI.
3 Results and discussion
3.1 Membrane surface morphology and physicochemical properties
Fig. 1a depicts the synthesis process of the GP composite membrane. First, PhA was attached to GO by ultrasonic dispersion to form a GP complex through hydrogen bonding. The formation of a PA layer on the surface of the porous PSU ultrafiltration membrane happened immediately at the water–oil interface. The GP complex worked as an aqueous phase additive, which was introduced into the PA layer by interfacial polymerization. The transmission electron microscopy (TEM) image of the GP complex and the corresponding element mapping are shown in Fig. S1.† The morphology of GO nanosheets is well maintained, and the C, O, and P elements are uniformly distributed on the GP complex. These results indicate that PhA molecules successfully bonded onto the GO surface.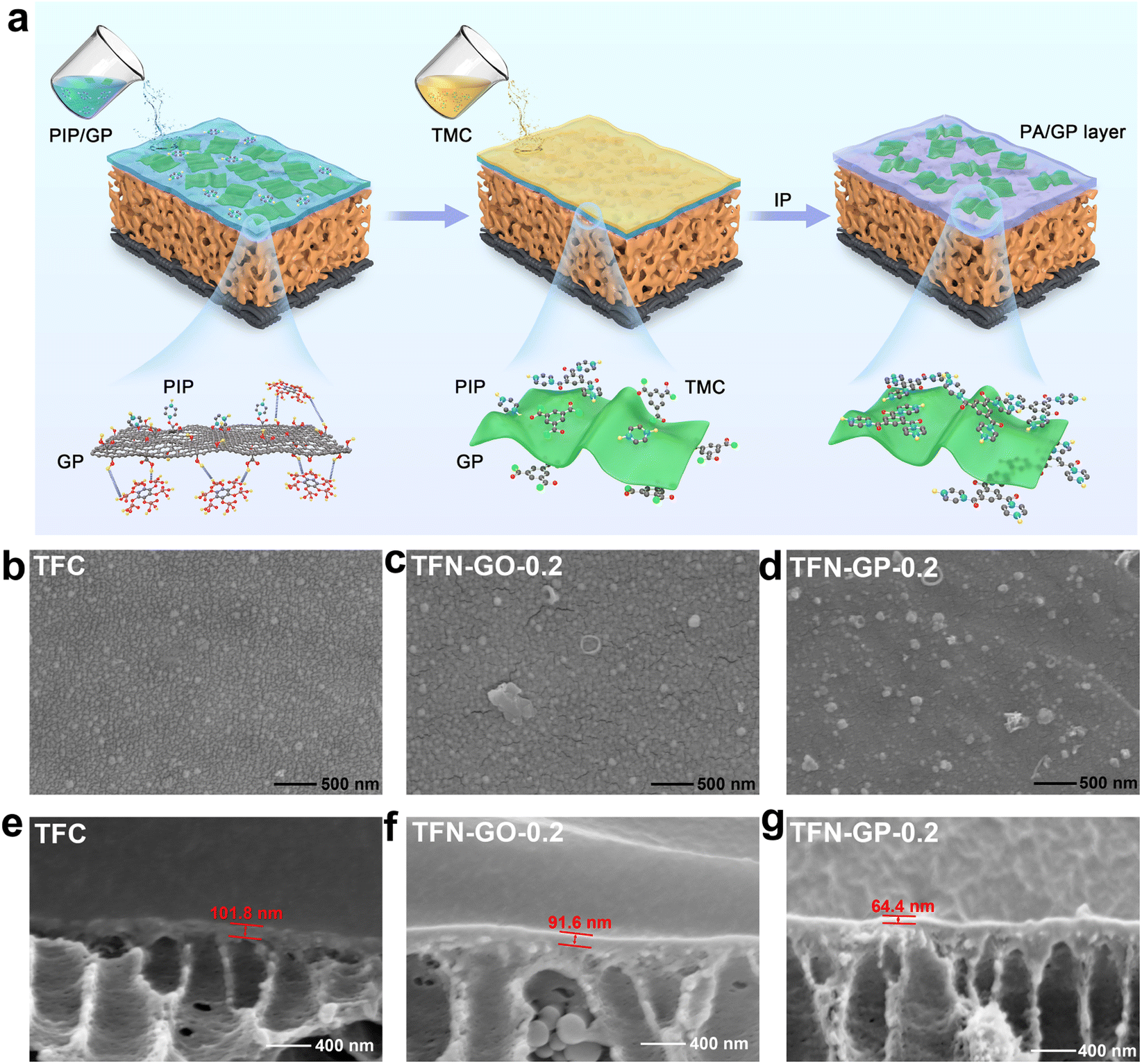 | ||
| Fig. 1 (a) Preparation process of the TFN-GP composite membrane; SEM surface images of (b) TFC, (c) TFN-GO-0.2 and (d) TFN-GP-0.2 membranes; SEM cross-section images of (e) TFC, (f) TFN-GO-0.2 and (g) TFN-GP-0.2 membranes. | ||
Fig. 1b–d show digital photographs and SEM images, and we can see that the surface of the TFC nanofiltration membrane is relatively dense and smooth, with no obvious nodules and protrusions. After the introduction of the GP complex, cracks started to appear on the membrane surface, and nodules and protrusions of smaller sizes were uniformly dispersed on the surface, mainly due to the synergistic effect of GO and PhA (Fig. S2†). The cross-sectional images in Fig. 1e–g and S3† show that the thickness of the PA layers reduces linearly as the GP complex dosage increases. The thickness of TFN-GP-0.2 (64.4 nm) is lower than that of TFC (101.8 nm) (Fig. 2g). This is mainly because the introduction of GO slows down the IP reaction rate to some extent.34 The synergistic effect of the PhA-modified GO nanosheets further slows down the rate of interfacial polymerization. Reducing the thickness of the PA separation layer lowers the water molecule transport resistance and thus enhances the membrane water flux more effectively.
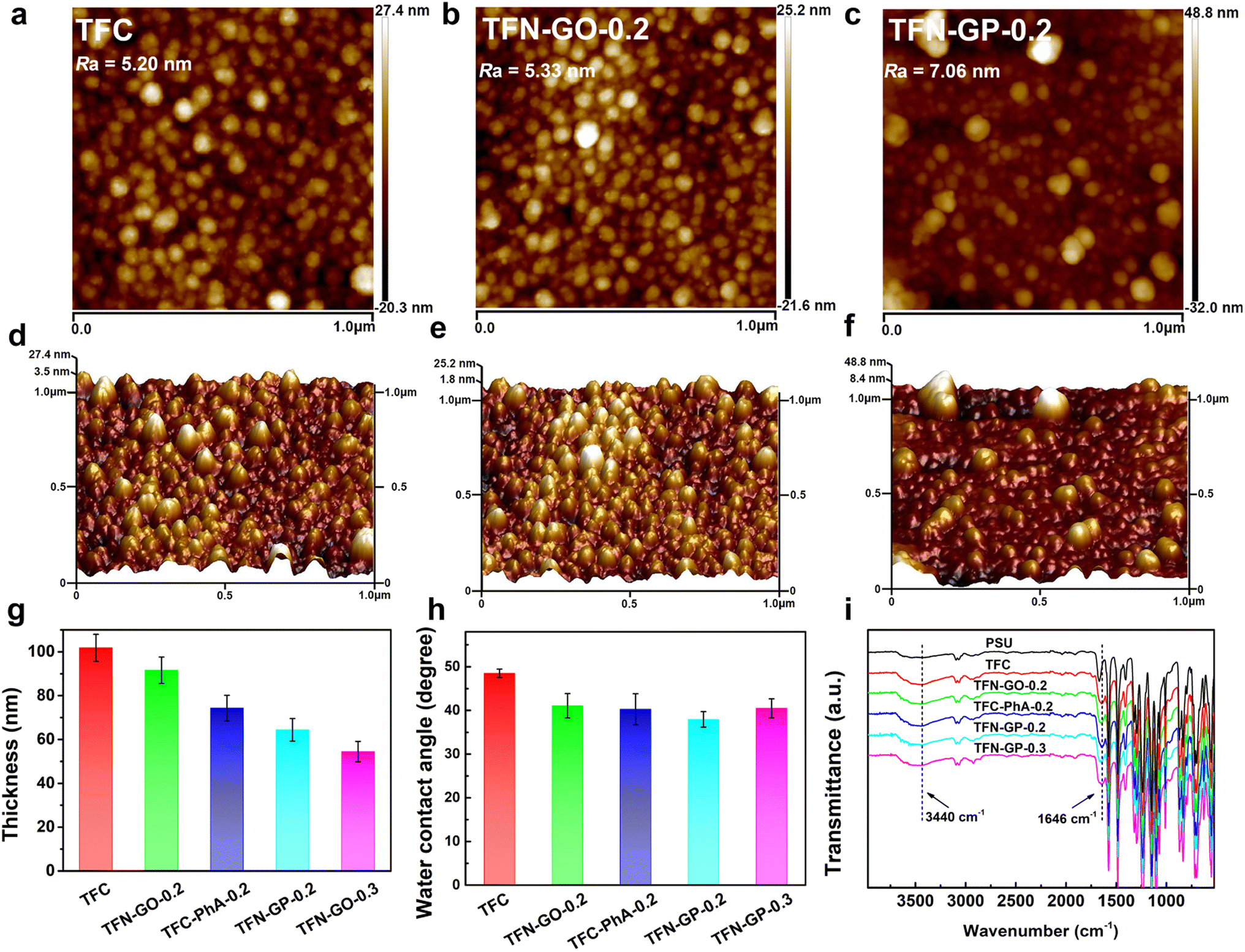 | ||
| Fig. 2 AFM surface images and the corresponding three-dimensional (3D) surface morphology images of (a), (d) TFC, (b), (d) TFN-GO-0.2 and (c), (f) TFN-GP-0.2 membranes; (g) thickness of TFC and TFN composite membranes; (h) water contact angles of TFC and TFN nanofiltration membranes; (i) FT-IR spectra of PSU, TFC and TFN composite membranes. | ||
The membrane's hydrophilicity and surface roughness are two main factors that could influence water permeability. Fig. 2a–f and S4 show the AFM surface images and corresponding 3D topographies of the TFC and TFN composite membranes, and the roughness values are listed in Table S2.† It is clear that the TFC nanofiltration membrane has low roughness and a relatively smooth surface. The roughness of TFN-GO nanofiltration membranes increased slightly after adding GO (Fig. 2b and e). However, when pure PhA molecules were introduced, the membrane surface roughness of the TFC-PhA-0.2 membrane was significantly increased, which was associated with the large size nodules and protrusions on the membrane surface (Fig. S4a†). When 0.2 wt% of the GP complex was added, the TFN-GP-0.2 membrane had significantly lower roughness than the TFC-PhA-0.2 and TFN-GP-0.3 membranes (Fig. S4b†), and there were no large size nodules, protrusions or clusters on the surface (Fig. 2c and f). These results are consistent with the SEM results. It can be concluded that TFN-GP-0.2 nanofiltration membranes have a rougher membrane surface than TFC membranes, and the increased roughness usually means an increase in the effective membrane filtration area, which is beneficial for further improving the membrane permeation performance.35,36
The water contact angles of TFC membranes were measured to determine their hydrophilicity. The average contact angles of TFC, TFN-GO-0.2, TFC-PhA-0.2, TFN-GP-0.2, and TFN-GP-0.3 are 48.5°, 41.1°, 40.3°, 37.9°, and 40.5°, respectively (Fig. 2h). The introduction of both pure GO or PhA molecules can effectively reduce the water contact angle and improve the hydrophilicity of the membrane, which is mainly due to the fact that they both contain abundant hydrophilic carboxyl or hydroxyl groups. Among them, TFN-GP-0.2 has the lowest water contact angle of 37.9°, and its hydrophilicity is the best, which is beneficial to accelerate the diffusion rate of water molecules and thus obtain better permeation performance. For the TFN-GP-0.3 nanofiltration membrane, although the addition of the GP complex increased, its contact angle is higher than that of TFN-GP-0.2. It can be speculated that the agglomeration of excess GP complex or PIP-PhA complex made them unevenly distributed on the membrane surface, which led to a large difference in hydrophilicity at various points on the membrane surface, which therefore lowered the hydrophilicity of the membrane.
Fig. 2i illustrates the FT-IR spectra of the PSU, TFC and TFN membranes. The spectra of the nanofiltration membrane show absorption peaks at 3440 cm−1, which are attributed to the hydroxyl group or the unreacted N–H bond in PIP.37 Compared with PSU, TFC and TFN membranes, a new absorption peak appears at 1646 cm−1, attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–N stretching vibrations in the amide bond (–CONH–),38 which indicates that the chloride group in TMC and the amino group in PIP successfully formed a PA separation layer through interfacial polymerization.
O and C–N stretching vibrations in the amide bond (–CONH–),38 which indicates that the chloride group in TMC and the amino group in PIP successfully formed a PA separation layer through interfacial polymerization.
3.2 Membrane surface chemical composition
XPS analysis was used to further characterize the surface element compositions of membranes, and these five membranes show three peaks at 285.0, 400.0, and 523.0 eV that are attributed to C 1s, N 1s, and O 1s, respectively (Fig. 3a). For TFN-GP-0.2 membranes, two new peaks appeared at 133.0 and 192.0 eV, attributed to the P 2s and P 2p derived from the PhA molecule,39 which indicates that the GP complex has been embedded in the TFN nanofiltration membrane. In addition, Fig. 3b shows that the C 1s spectrum of the TFN-GO-0.2 membrane has three divided peaks at binding energies of 248.8, 285.9, and 287.7 eV, which are assigned to C–C/CC, C–O, and O–CO bonds, respectively.40,41 The O 1s spectrum of TFN-GO-0.2 (Fig. 3c) also contains three main peaks at 531.0, 532.3, and 533.4 eV, corresponding to O–CO, OC, and O–C, respectively.42 After modification of the GP complex (Fig. 3d), the C 1s spectrum is divided into four peaks including C–C/CC (284.8 eV), C–P (285.4 eV), C–O (286.2 eV), and O–CO (288 eV).43,44 The O 1s component peaks (Fig. 3e) at 531.1, 532.0, 533.3, and 535.4 eV are identified as those of O–CO, OC/OP, HO–P, and O–C/P–O–C, respectively.42,45 Meanwhile, in the P 2p spectrum, two divided peaks located at 134.0 and 135.0 eV are assigned to P–C and P–O, respectively (Fig. 3f).44,46 The new additional peaks (C–P, PO, and P–OH) confirm that PhA successfully modified GO nanosheets on the TFN membrane.
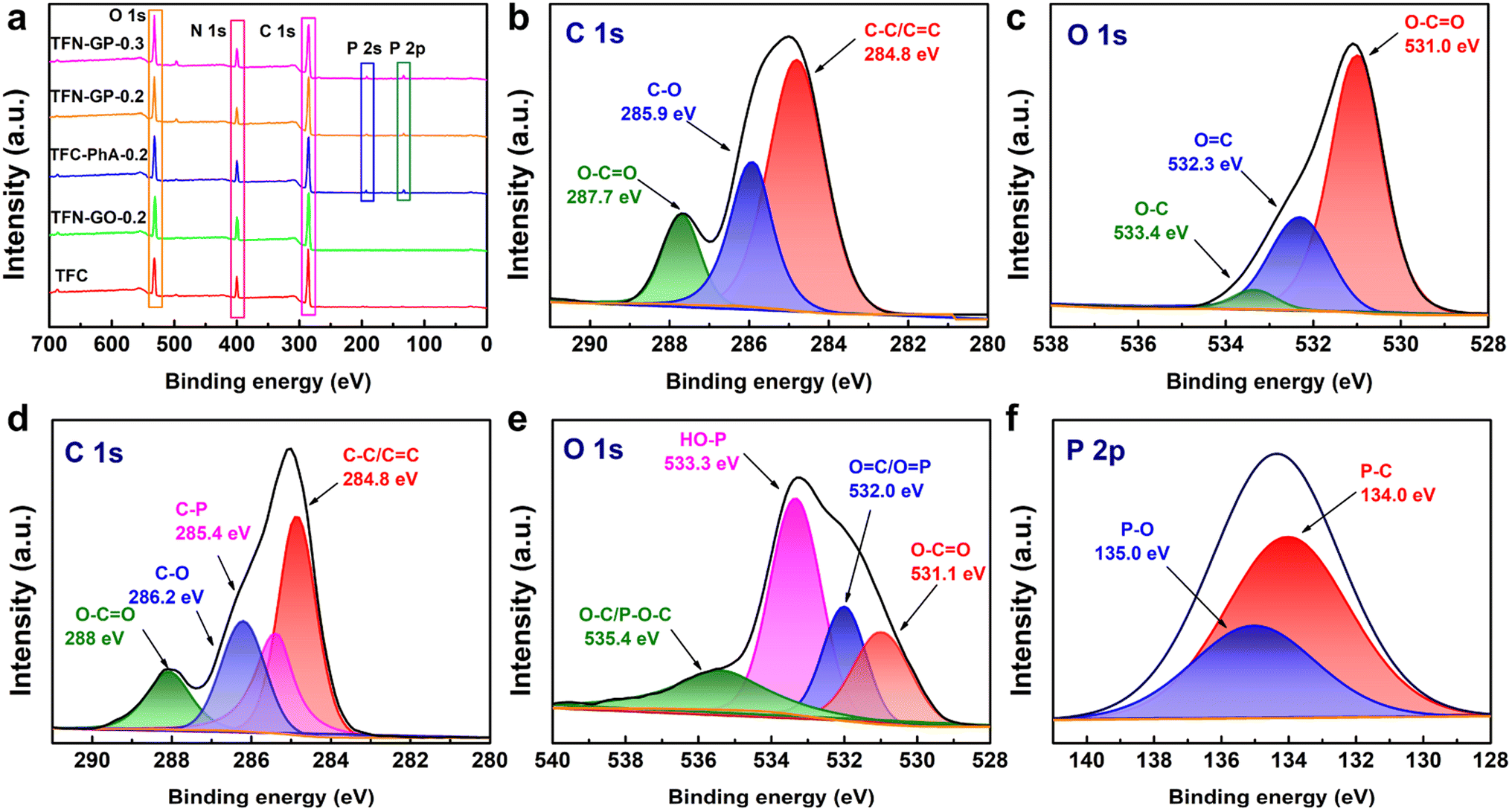 | ||
| Fig. 3 (a) XPS survey spectra of TFC and TFN composite membranes; (b) C 1s and (c) O 1s XPS spectra of the TFN-GO-0.2 membrane; (d) C 1s, (e) O 1s, and (f) P 2p XPS spectra of the TFN-GP-0.2 membrane. | ||
The specific elemental composition of the membrane surface of these membranes is summarized in Table S3.† As can be seen from Table S3,† the TFC nanofiltration membrane has a higher oxygen content after adding GO, PhA and the GP complex. In addition, the P-element content on the surface of TFC-PhA-0.2, TFN-GP-0.2 and TFN-GP-0.3 nanofiltration membranes increased with the increase in PhA molecule content. To further investigate the elemental composition of the TFN-GP-0.2 nanofiltration membrane at different depths, we further tested the XPS signals at different depths of 0, 20 nm, 40 nm and 60 nm (Table S4†). Obviously, the presence of the P element was detected in the depth range of 0–60 nm, indicating that our GP complex was successfully introduced not only to the membrane surface, but also to the interior of the PA layer. In addition, we also obtained the elemental distribution map of the surface of the TFN-GP-0.2 nanofiltration membrane with optimal performance by EDS. According to the elemental distribution (Fig. S5†), it can be seen that the elements C, O, N and P are uniformly distributed on the TFN-GP-0.2 nanofiltration membrane, indicating that the GP complex is uniformly distributed on the surface of the modified membrane.
3.3 Diffusion experiment and molecular dynamics simulation
The thickness of a polyamide dense layer is proportional to the one-third power of the diffusivity of amine monomers, according to Freger's kinetic model for the formation of polyamide nanofilms.47,48 Details are as follows: | (3) |
As shown in Fig. 4a, when the GP complex is added to the aqueous phase, PIP cannot freely diffuse due to interactions with the GP complex via hydrogen bonding, electrostatic attraction, and steric hindrance.49 To confirm this hypothesis, we used a UV-vis spectrophotometer to detect the absorbance variation of PIP in the organic phase after 60 s of diffusion to characterize the diffusion rate of PIP in n-heptane solution. A characteristic absorption peak at about 230 nm can be ascribed to the PIP molecule, and the peak intensity represents the concentration of PIP. The peak intensity reduces with increasing GP complex concentration, indicating that the GP complex can decelerate the rate of PIP diffusion and thus reduce the thickness of the PA layer.
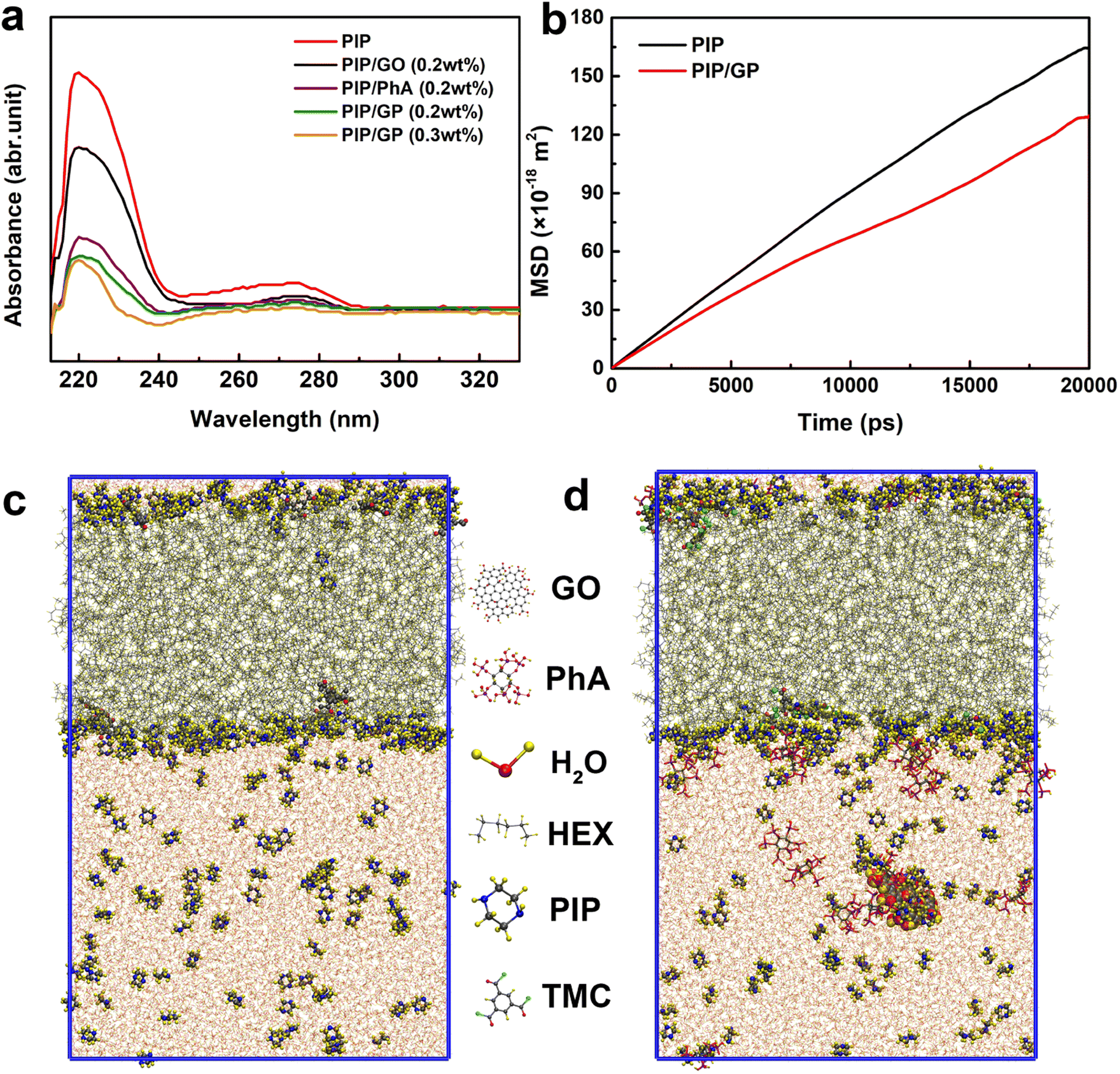 | ||
| Fig. 4 (a) Ultraviolet-visible (UV-vis) spectra of PIP in the organic phase after 60 s of diffusion; (b) the simulated diffusion coefficients of PIP toward the n-hexane phase with/without the GP complex; snapshots of the water/n-hexane interface in the MD simulations (c) without and (d) with the GP complex. | ||
To further validate our hypothesis, molecular dynamics simulation was conducted to study the diffusion of PIP monomers toward the organic phase across the aqueous phase with/without the GP complex, to effectively gain in-depth insights into how the GP complex modification influences the membrane's thickness for the transport of water molecules (Fig. 4c and d). The diffusivity of PIP was evaluated using the mean square displacement (MSD) and the diffusion coefficient, Dα, calculated from Einstein's equation, as follows:
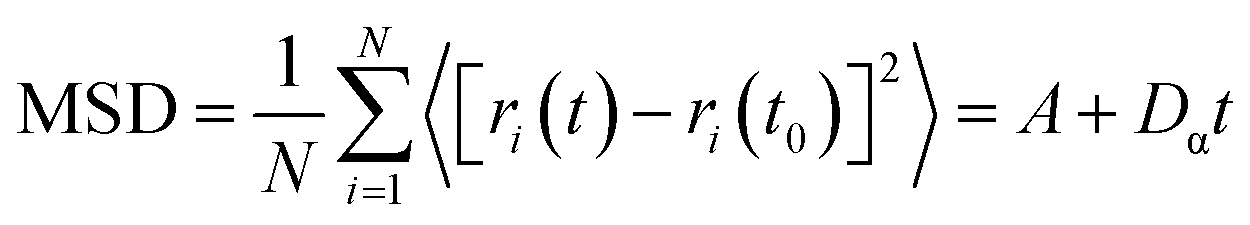 | (4) |
The MD results are shown in Fig. 4b (detailed computational simulation methods are shown in the ESI), and clearly, in the absence of the GP complex, the simulated diffusion coefficient of PIP monomers is 1.40 × 10−9 m2 s−1. In contrast to what happened in the aqueous phase, the simulated diffusion coefficients of PIP molecules is 40% lower (1.00 × 10−9 m2 s−1), supporting our hypothesis that the addition of GP allows us to control the diffusion rate of the amine monomer to obtain a thin PA layer.
Since the diffusion rate in the presence of the GP complex at the water/hexane interface is lower, it is possible that the binding energy at the water/hexane interface is higher after the incorporation of the GP complex. Therefore, the process of diffusion of PIP molecules from the aqueous solution to the interface results in an energy loss; the GP complex significantly increases the binding energy penalty for transporting PIP molecules from the aqueous to hexane phases. Finally, a thinner PA layer could be successfully synthesized by adding the GP complex.
3.4 Membrane permeability and separation performance
The TFC membrane's pure water flux was only 37.9 L m−2 h−1, as shown in Fig. 5a. Compared to TFC membranes, the TFN-GP composite membrane has a higher pure water flux due to the thinner PA layer and increased surface hydrophilicity. Meanwhile, it should be noted that compared to the TFN-GP-0.2 membrane, the enhancement of membrane flux by introducing GO nanosheets alone was limited, while for the TFC-PhA-0.2 nanofiltration membrane, the MgSO4 rejection rate decreased significantly to 94.7%, which was closely related to the more sparse structure and larger crack width of the separation layer surface as well as the large size of nodules and protrusions.60 To determine the optimal mass ratio of GO to PhA, we compared the nanofiltration performance of different mass ratios of GO to PhA (GP1, GP5, GP10, and GP15), and the total GP additive is controlled at 0.2 wt%. We can see that the water flux gradually increases as the proportion of PhA increases. However, a decrease in water flux occurs when the mass ratio of PhA exceeds 20. Therefore, GP10 is considered the best additive, and for the convenience of future naming, GP10 is written as GP.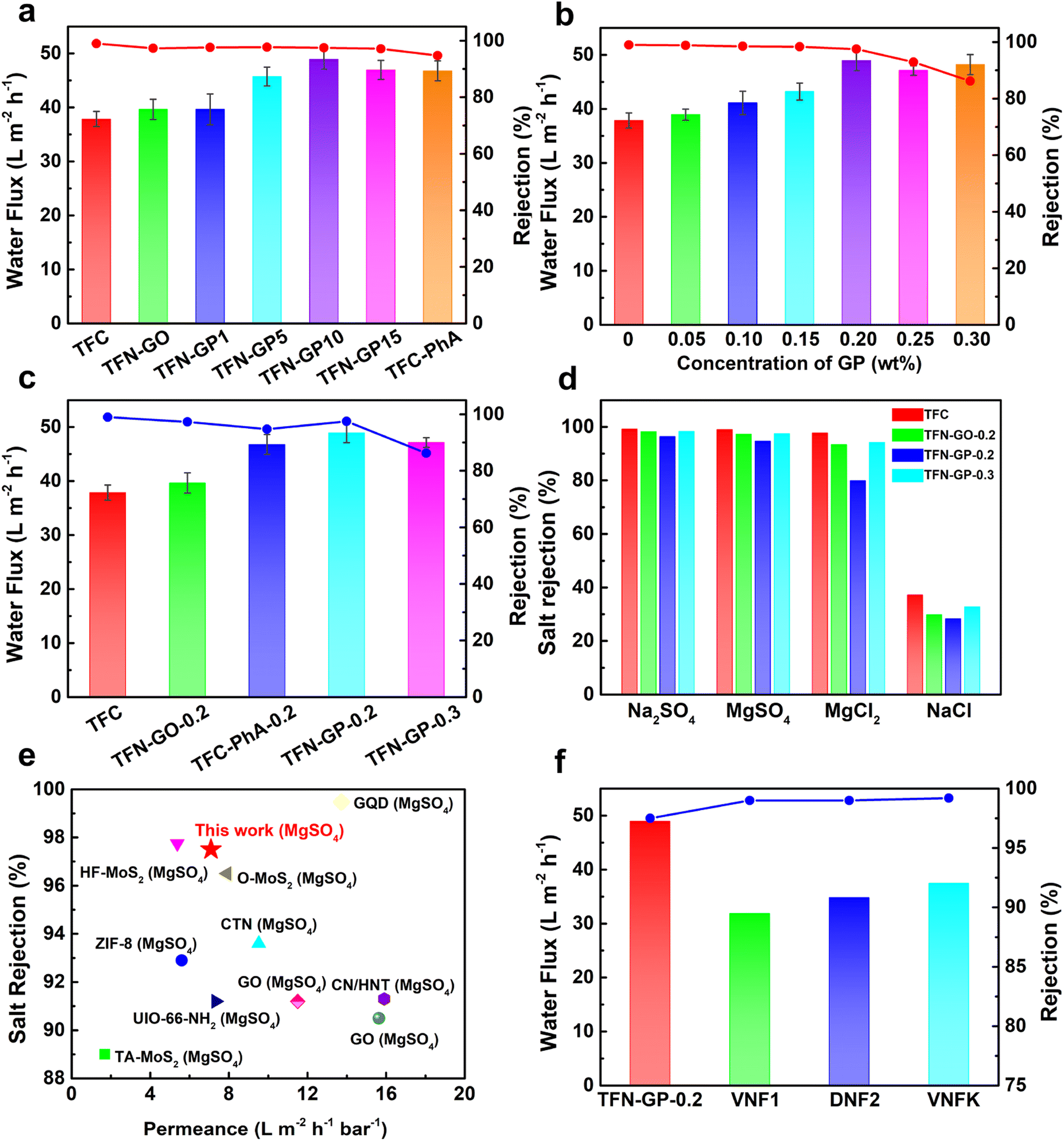 | ||
| Fig. 5 (a) Water flux and MgSO4 rejection of different mass ratio TFN-GP nanofiltration membranes; (b) effect of different concentrations of the GP complex on the separation performance of nanofiltration membranes; (c) separation performance of the TFC and TFN nanofiltration membranes; (d) salt rejection of 2000 ppm salt solution by the TFC and TFN composite membrane; (e) comparison of the flux and rejection of the TFN-GP-0.2 membrane prepared in our work with those of TFN nanofiltration membranes modified with nanomaterials, data from ref. 50–59; (f) comparison of the separation performance of the TFN-GP-0.2 membrane with that of commercial polymeric membranes. | ||
Subsequently, the separation performance of nanofiltration membranes was tested using different concentrations of the GP complex (Fig. 5b), and it can be seen that the water flux increases as the content of GP complexes increases. However, when the concentration of GP exceeds 0.2 wt%, the water flux and salt rejection show a significant decrease, which is attributed to the excess GP complex that will form large-sized clusters in the separation layer and lead to larger defects.
To further explore the reasons for the optimal separation performance of the TFN-GP-0.2 nanofiltration membrane, we summarized the separation performance of TFC, TFN-GO-0.2, TFC-PhA-0.2, TFN-GP-0.2 and TFN-GP-0.3 nanofiltration membranes for MgSO4 solutions as shown in Fig. 5c. It is worth noting that compared with the TFC membrane, although the TFN-GP-0.2 membrane has slightly decreased densification, it has a significantly higher pure water flux. This is mainly because of the synergistic effect of GO and PhA in the GP complex, which makes the TFN-GP-0.2 nanofiltration membrane have better hydrophilicity, greater roughness, and more water molecule channels.
The salt separation performance of the TFC, TFN-GO-0.2, TFC-PhA-0.2, and TFN-GP-0.2 nanofiltration membranes was then evaluated using four different salt ions (2000 ppm Na2SO4, MgSO4, MgCl2, and NaCl solutions) (Fig. 5d). As can be seen, the rejection of all four membranes for the four salt solutions is in the order of Na2SO4 > MgSO4 > MgCl2 > NaCl, which is consistent with the rejection characteristics of negatively charged PA nanofiltration membranes reported in the literature,61 and is the result of the synergistic effect of the spatial site resistance effect, the Donnan effect and the dielectric effect. The rejection rate of the TFN-GP-0.2 nanofiltration membrane for the four salt solutions was only slightly reduced compared with that of the TFC nanofiltration membrane, which was also mainly attributed to the synergistic effect of spatial site resistance and the Donnan effect. In summary, the introduction of the GP complex will cause the membrane surface to become relatively loose and create some cracks between the contact interface of the PA layer and the GP complex, resulting in a lower rejection rate. However, both the GO nanosheets and PhA in the GP complex have a strong negative charge, which makes the surface of the TFN-GP-0.2 nanofiltration membrane have enhanced electronegativity, which facilitates the enhancement of the Donnan effect, thus improving the membrane rejection rate. With the synergy of these two effects, the salt rejection rate of the TFN-GP-0.2 nanofiltration membrane remains high.
As shown in Fig. 5e and in Table S5,† we compared the membrane performance of TFN-GP-0.2 to earlier research. Different nanomaterials have been used as membrane additives in the PA active layer to improve the performance of TFC nanofiltration membranes, such as ZIF-8,50 covalent triazine framework nanosheets (CTNs),51 O–MoS2 (ref. 52) and other nanomaterials,53–59 which can improve the water flux of GO membranes, and the salt rejection rate is only slightly lower. The TFN-GP-0.2 composite membrane works efficiently, with superior salt rejection and high pure water flux. Significantly, the water flux and the salt rejection of the TFN-GP-0.2 membrane are significantly higher than those of the three commercial membranes (i.e., VNF1, DNF2, and DK) (Fig. 5f), which were studied in our previous work. As a result, our TFN-GP-0.2 composite membrane has promising applications in water purification.
3.5 Durability of the membrane
The membrane's durability is a crucial consideration when deciding whether it can be used in practical water treatment. The water flux and salt rejection of TFN-GP-0.2 for 2000 ppm MgSO4 solution in 12 h are shown in Fig. 6a and b. Compared with TFC nanofiltration membranes, the presence of strong GO nanosheets and PIP-PhA polymer complexes in the PA layer of TFN-GP-0.2 nanofiltration membranes has a strong supporting effect on the structure of the PA layer and gives higher mechanical strength to the PA layer.52 Therefore, the denseness of the TFN-GP-0.2 nanofiltration membrane did not change much under the extrusion of the salt solution, and the water flux only decreased by 0.1% after 12 h of long operation, which improves the TFN-GP-0.2 composite membrane's application prospects in the water treatment industry.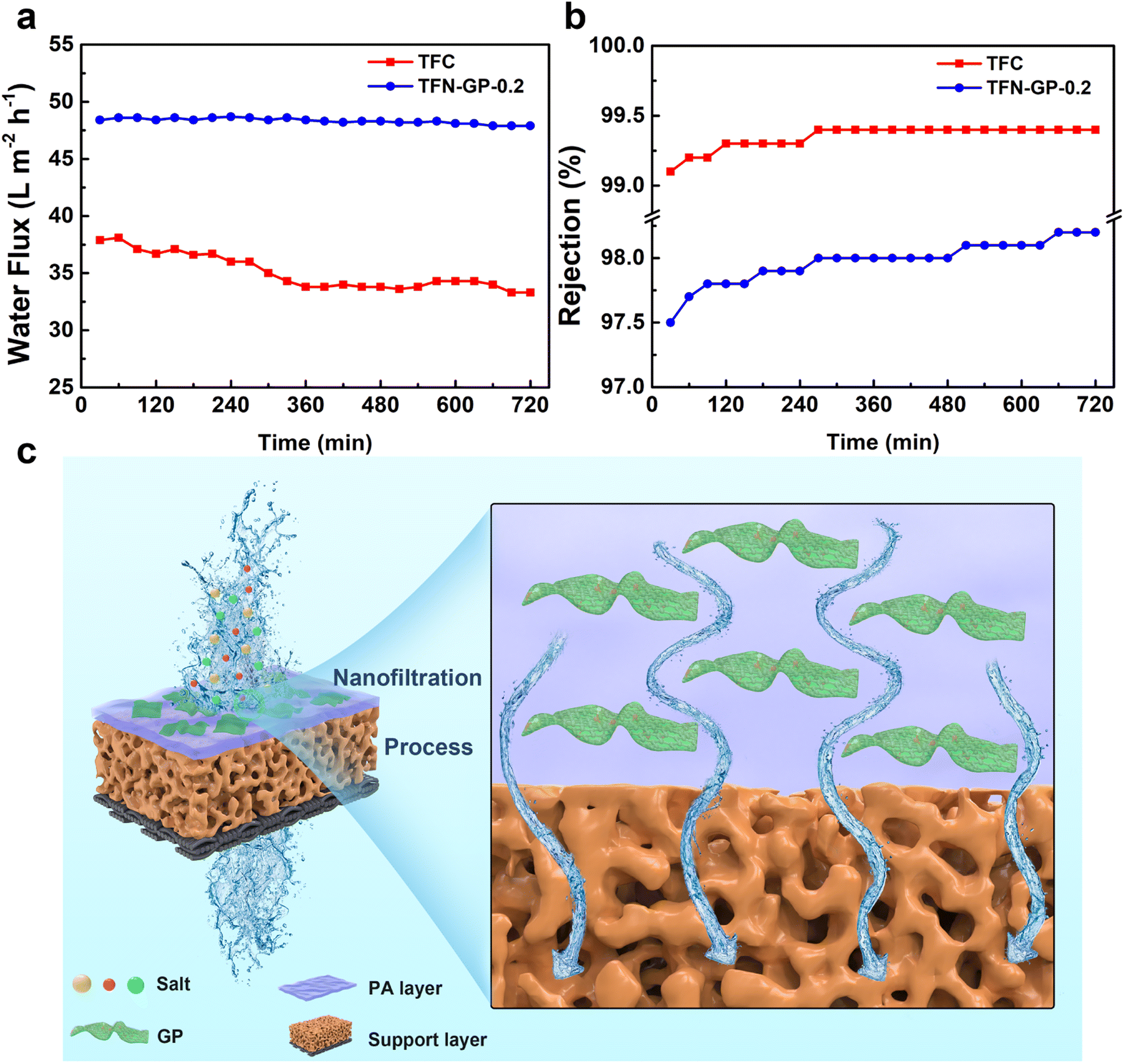 | ||
| Fig. 6 Stability of water flux (a) and rejection (b) of TFC and TFN nanofiltration membranes for 12 h of continuous operation; (c) separation mechanism of the TFN-GP composite membrane. | ||
Fig. 6c depicts the mechanism for separation by the TFN-GP membrane. Specific physical and structural properties lead to excellent water flux. Due to the highly hydrophilic surface, more water molecules cross the membrane, which is mainly attributed to the nodules and protrusions that make the surface rougher. The GP complex allows the formation of a thinner PA layer during the IP process, which can hasten their passage across the membrane, resulting in a significantly enhanced water flux. In addition, the high rejection of the TFN-GP membrane for salt is mainly attributed to the enhanced Donnan effect. Large SO42− ions are directly rejected and Mg2+ ions are held back due to the size exclusion and electrostatic repulsion.
4 Conclusions
In this paper, TFN-GP composite nanofiltration membranes were fabricated using the GP complex as an aqueous phase additive and by introducing it into the PA layer by interfacial polymerization. The optimal aqueous phase additive GP complex was obtained by regulating the ratio of GO nanosheets to PhA. Optimal performance of the TFN-GP-0.2 nanofiltration membrane was obtained when 0.2 wt% GP complex was added, and the water flux of 2000 ppm MgSO4 solution reached 48.9 L m−2 h−1 at an operating pressure of 6.9 bar, which was about 1.3 times higher than that of the original TFC nanofiltration membrane. The superior water flux is mainly attributed to the slow diffusion rate of PIP in the presence of the GP composite, which results in the generation of a thinner PA layer, confirmed by the MD simulations and diffusion experiment studies. Meanwhile, additional water transport channels are formed to accelerate the passage of water. In addition, the GP complex is rich in negatively charged groups, which makes the nanofiltration membrane electronegative and has an enhanced Donnan effect when rejecting salt solutions. Therefore, although the membrane surface structure becomes relatively sparse, the TFN-GP-0.2 composite nanofiltration membrane still maintains a high salt rejection rate of 98.3% for 2000 ppm Na2SO4 and the TFN-GP-0.2 nanofiltration membrane has excellent long-term cycle stability and comparable nanofiltration performance to the three leading commercial nanofiltration membranes. This work reports a novel design of high-performance GO-based nanofiltration membranes, as well as a comprehensive understanding of the permeation and rejection mechanisms of such membranes.Author contributions
Ke Tang: investigation, writing – original draft. LinSheng Zhu: investigation, data curation. Piao Lan: investigation. YunQiang Chen: conceptualization. Zhou Chen: conceptualization, methodology, writing-original draft. Yihong Lan: resources, supervision, writing – review & editing. WeiGuang Lan: resources, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Suntar Membrane Technology (Xiamen) Co., Ltd, Xiamen.References
- B. Lee, Y. Baek, M. Lee, D. H. Jeong, H. H. Lee, J. Yoon and Y. H. Kim, Nat. Commun., 2015, 6, 7109 CrossRef CAS PubMed.
- H. B. Park, J. Kamcev, L. M. Robeson, M. Elimelech and B. D. Freeman, Science, 2017, 356, 6343 CrossRef PubMed.
- W. Y. Zhang, H. Xu, F. Xie, X. H. Ma, B. Niu, M. Q. Chen, H. Y. Zhang, Y. Zhang and D. H. Long, Nat. Commun., 2022, 13, 471 CrossRef CAS PubMed.
- K. P. Wang, X. M. Wang, B. Januszewski, Y. L. Liu, D. Y. Li, R. Y. Fu, M. Elimelech and X. Huang, Chem. Soc. Rev., 2022, 51, 672–719 RSC.
- J. Wang, J. Y. Zhu, Y. T. Zhang, J. D. Liu and B. Van der Bruggen, Nanoscale, 2017, 9, 2942–2957 RSC.
- B. Li, C. G. Wang, N. E. Surat'man, X. J. Loh and Z. Li, Nanoscale Adv., 2021, 3, 5265–5276 RSC.
- J. Y. Zhu, A. Uliana, J. Wang, S. S. Yuan, J. Li, M. M. Tian, K. Simoens, A. Volodin, J. Y. Lin, K. Bernaerts, Y. T. Zhang and B. Van der Bruggen, J. Mater. Chem. A, 2016, 4, 13211–13222 RSC.
- D. Fritsch, P. Merten, K. Heinrich, M. Lazar and M. Priske, J. Membr. Sci., 2012, 401, 222–231 CrossRef.
- Z. Chen, Y. H. Lan, Y. B. Hong and W. G. Lan, ACS Appl. Nano Mater., 2022, 5, 12343–12365 CrossRef CAS.
- I. Levitsky, D. Tavor and V. Gitis, Sep. Purif. Technol., 2021, 268, 118710 CrossRef CAS.
- W. Y. Ye, H. W. Liu, M. Jiang, J. Y. Lin, K. F. Ye, S. Q. Fang, Y. D. Xu, S. F. Zhao, B. Van der Bruggen and Z. He, Water Res., 2019, 157, 555–563 CrossRef CAS PubMed.
- G. K. Zhao, K. Zhou, R. R. Hu and H. W. Zhu, Sep. Purif. Technol., 2023, 304(122321) CAS.
- C. B. Wang, Z. Y. Li, J. X. Chen, Z. Li, Y. H. Yin, L. Cao, Y. L. Zhong and H. Wu, J. Membr. Sci., 2017, 523, 273–281 CrossRef CAS.
- S. Karan, Z. W. Jiang and A. G. Livingston, Science, 2015, 348, 1347–1351 CrossRef CAS PubMed.
- F. Yang, Y. Q. Zhang, J. H. Huang, G. Gao, J. Q. Zhu, J. Ma and L. Shao, Sci. Bull., 2023, 68, 29–33 CrossRef CAS PubMed.
- Y. Q. Zhang, F. Yang, H. G. Sun, Y. P. Bai, S. W. Li and L. Shao, Engineering, 2022, 16, 247–255 CrossRef CAS.
- Z. Z. Wang, C. Ma, C. Y. Xu, S. A. Sinquefield, M. L. Shofner and S. Nair, Nat. Sustain., 2021, 4, 402–408 CrossRef.
- Y. J. Qian, J. Shang, D. Liu, G. L. Yang, X. G. Wang, C. Chen, L. Z. Kou and W. W. Lei, J. Am. Chem. Soc., 2021, 143, 5080–5090 CrossRef CAS PubMed.
- X. F. Chen, M. H. Qiu, H. Ding, K. Y. Fu and Y. Q. Fan, Nanoscale, 2016, 8, 5696–5705 RSC.
- N. G. Doménech, Á. Coogan, F. Purcell-Milton, M. L. Casasín García, A. S. Arjona, M. B. Cabré, A. Rafferty, K. McKelvey, P. Dunne and Y. K. Gun'ko, Nanoscale Adv., 2022, 4, 4895–4904 RSC.
- S. F. Anis, B. S. Lalia, A. Lesimple, R. Hashaikeh and N. Hilal, Chem. Eng. J., 2022, 428, 131184 CrossRef CAS.
- G. L. Zhang, K. J. Tang, X. Zhang, L. S. Xu, C. Shen and Q. Meng, J. Membr. Sci., 2021, 617, 118612 CrossRef CAS.
- S. Z. Cong, Y. Yuan, J. X. Wang, Z. Wang, F. Kapteijn and X. L. Liu, J. Am. Chem. Soc., 2021, 143, 20055–20058 CrossRef CAS PubMed.
- Y. L. Ji, B. X. Gu, S. J. Xie, M. J. Yin, W. J. Qian, Q. Zhao, W. S. Hung, K. R. Lee, Y. Zhou, Q. F. An and C. J. Gao, Adv. Mater., 2021, 33, 2102292 CrossRef CAS PubMed.
- A. Iakunkov and A. V. Talyzin, Nanoscale, 2020, 12, 21060–21093 RSC.
- S. Maiti and S. Bose, Nanoscale Adv., 2022, 4, 467–478 RSC.
- S. Bano, A. Mahmood, S. J. Kim and K. H. Lee, J. Mater. Chem. A, 2015, 3, 2065–2071 RSC.
- L. He, L. F. Dumee, C. F. Feng, L. Velleman, R. Reis, F. H. She, W. M. Gao and L. X. Kong, Desalination, 2015, 365, 126–135 CrossRef CAS.
- R. R. Hu, R. J. Zhang, Y. J. He, G. K. Zhao and H. W. Zhu, J. Membr. Sci., 2018, 564, 813–819 CrossRef CAS.
- Y. Jiang, Q. Q. Zeng, P. Biswas and J. D. Fortner, J. Membr. Sci., 2019, 581, 453–461 CrossRef CAS.
- S. M. Xue, C. H. Ji, Z. L. Xu, Y. J. Tang and R. H. Li, J. Membr. Sci., 2018, 545, 185–195 CrossRef CAS.
- L. S. Zhu, X. X. Guo, Y. Q. Chen, Z. Chen, Y. H. Lan, Y. B. Hong and W. G. Lan, ACS Appl. Nano Mater., 2022, 5, 3643–3653 CrossRef CAS.
- K. Q. Zheng, S. Q. Li, Z. Chen, Y. Q. Chen, Y. B. Hong and W. G. Lan, Nanoscale, 2021, 13, 10061–10066 RSC.
- H. R. Chae, J. Lee, C. H. Lee, I. C. Kim and P. K. Park, J. Membr. Sci., 2015, 483, 128–135 CrossRef CAS.
- R. J. Zhang, J. Y. Tian, S. S. Gao and B. van der Bruggen, J. Mater. Chem. A, 2020, 8, 8831–8847 RSC.
- W. G. Wang, G. H. Hong, Y. Q. Zhang, X. B. Yang, N. M. Hu, J. L. Zhang, P. Sorokin and L. Shao, J. Membr. Sci., 2023, 675, 121534 CrossRef CAS.
- Q. L. Xie, W. Y. Shao, S. S. Zhang, Z. Hong, Q. Q. Wang and B. R. Zeng, RSC Adv., 2017, 7, 54898–54910 RSC.
- M. Safarpour, V. Vatanpour, A. Khataee and M. Esmaeili, Sep. Purif. Technol., 2015, 154, 96–107 CrossRef CAS.
- H. J. Kim, S. Im, J. C. Kim, W. G. Hong, K. Shin, H. Y. Jeong and Y. J. Hong, ACS Sustainable Chem. Eng., 2017, 5, 6654–6664 CrossRef CAS.
- Y. P. Tang, D. R. Paul and T. S. Chung, J. Membr. Sci., 2014, 458, 199–208 CrossRef CAS.
- B. Liang, W. Zhan, G. G. Qi, S. S. Lin, Q. Nan, Y. X. Liu, B. Cao and K. Pan, J. Mater. Chem. A, 2015, 3, 5140–5147 RSC.
- J. H. Liu, Q. Yu, M. Yu, S. M. Li, K. Zhao, B. Xue and H. Zu, J. Alloys Compd., 2018, 744, 728–739 CrossRef CAS.
- B. H. Yuan, C. L. Bao, X. D. Qian, L. Song, Q. L. Tai, K. M. Liew and Y. Hu, Carbon, 2014, 75, 178–189 CrossRef CAS.
- J. Wang and F. Ciucci, Appl. Catal. B Environ., 2019, 254, 292–299 CrossRef CAS.
- W. Wang, X. Wang, Y. Pan, K. M. Liew, O. A. Mohamed, L. Song and Y. Hu, Ind. Eng. Chem. Res., 2017, 56, 6664–6670 CrossRef CAS.
- D. Q. Lin, C. G. Hu, H. Chen, J. Qu and L. M. Dai, Chem.–Eur. J., 2018, 24, 18487–18493 CrossRef CAS PubMed.
- V. Freger, Langmuir, 2005, 21, 1884–1894 CrossRef CAS PubMed.
- V. Freger, Langmuir, 2003, 19, 4791–4797 CrossRef CAS.
- Z. Tan, S. F. Chen, X. S. Peng, L. Zhang and C. J. Gao, Science, 2018, 360, 518–521 CrossRef CAS PubMed.
- J. Li, R. R. Liu, J. Y. Zhu, X. Li, S. S. Yuan, M. M. Tian, J. Wang, P. Luis, B. V. der Bruggen and J. Y. Lin, Desalination, 2021, 512, 115125 CrossRef CAS.
- N. A. Khan, H. Wu, J. Q. Yuan, M. Y. Wu, P. F. Yang, M. Y. Long, A. U. Rahman, N. M. Ahmad, R. N. Zhang and Z. Y. Jiang, Sep. Purif. Technol., 2021, 274, 119046 CrossRef CAS.
- S. S. Yang, Q. L. Jiang and K. S. Zhang, J. Membr. Sci., 2020, 604, 118052 CrossRef CAS.
- Y. Q. Gong, S. J. Gao, Y. Y. Tian, Y. Z. Zhu, W. X. Fang, Z. G. Wang and J. Jin, J. Membr. Sci., 2020, 600, 117874 CrossRef CAS.
- R. R. Hu, Y. J. He, C. M. Zhang, R. J. Zhang, J. Li and H. W. Zhu, J. Mater. Chem. A, 2017, 5, 25632–25640 RSC.
- W. Zhao, H. Y. Liu, N. Meng, M. P. Jian, H. T. Wang and X. W. Zhang, J. Membr. Sci., 2018, 565, 380–389 CrossRef CAS.
- Y. F. Li, X. D. You, Y. Li, J. Q. Yuan, J. L. Shen, R. N. Zhang, H. Wu, Y. L. Su and Z. Y. Jiang, J. Mater. Chem. A, 2020, 8, 23930–23938 RSC.
- Y. L. Liu, X. M. Wang, X. Q. Gao, J. F. Zheng, J. Wang, A. Volodin, Y. F. Xie, X. Huang, B. Van der Bruggen and J. Y. Zhu, J. Membr. Sci., 2020, 596, 117717 CrossRef CAS.
- H. Zhang, X. Y. Gong, W. X. Li, X. H. Ma, C. Y. Tang and Z. L. Xu, J. Membr. Sci., 2020, 616, 118605 CrossRef CAS.
- X. P. Wang, Q. Xiao, C. Wu, P. Li and S. J. Xia, Chem. Eng. J., 2021, 416, 129154 CrossRef CAS.
- L. L. Gui, Y. Q. Cui, Y. Z. Zhu, X. Q. An, H. C. Lan and J. Jin, Sep. Purif. Technol., 2022, 293(121125) CAS.
- J. Y. Zhu, J. W. Hou, S. S. Yuan, Y. Zhao, Y. Li, R. J. Zhang, M. M. Tian, J. Li, J. Wang and B. Van der Bruggen, J. Mater. Chem. A, 2019, 7, 16313–16322 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3na00110e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |