
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent highlights of biosynthetic studies on marine natural products†
Jamshid
Amiri Moghaddam
 *a,
Theresa
Jautzus
a,
Mohammad
Alanjary
b and
Christine
Beemelmanns
*a
*a,
Theresa
Jautzus
a,
Mohammad
Alanjary
b and
Christine
Beemelmanns
*a
aJunior Research Group Chemical Biology of Microbe-Host Interactions, Leibniz Institute for Natural Product Research and Infection Biology (HKI), Beutenbergstr. 11a, 07745 Jena, Germany. E-mail: christine.beemelmanns@hki-jena.de
bBioinformatics Group, Wageningen University, Wageningen, The Netherlands
First published on 20th November 2020
Abstract
Marine bacteria are excellent yet often underexplored sources of structurally unique bioactive natural products. In this review we cover the diversity of marine bacterial biomolecules and highlight recent studies on structurally novel natural products. We include different compound classes and discuss the latest progress related to their biosynthetic pathway analysis and engineering: examples range from fatty acids over terpenes to PKS, NRPS and hybrid PKS–NRPS biomolecules.
 Jamshid Amiri Moghaddam | Jamshid Amiri Moghaddam obtained his PhD under the supervision of Prof. Gabriele M. König at the University of Bonn (Germany) with the focus on genome mining of marine bacteria for bioactive metabolites. He also, performed a research stay at the Scripps Institution of Oceanography in La Jolla/CA, USA with Prof. Bradley Moore. He is currently working at the Leibniz Institute for Natural Product Research and Infection Biology (Jena, Germany) as a postdoctoral scientist working on the biosynthetic pathways and enzymes involved in the biosynthesis of secondary metabolites in marine bacteria. |
 Theresa Jautzus | After obtaining a Master of Science degree in Microbiology at the Friedrich-Schiller-University Jena, Theresa Jautzus joined the Terrestrial Biofilms group to perform her PhD under supervision of Prof. Ákos T. Kovács at FSU Jena. Since end of 2018, she is a postdoctoral researcher in the lab of Christine Beemelmanns at the Leibniz Institute for Natural Product Research and Infection Biology in Jena. Currently, she is working on natural metabolite biosynthesis and marine invertebrate–microbe interactions. |
 Mohammad Alanjary | Dr Mohammad Alanjary is a post-doctoral researcher focused on developing computational platforms to accelerate drug discovery. In 2018 he received his PhD in bioinformatics at the University of Tübingen, Germany where he developed genome-mining tools for gene cluster prioritization and antibiotic resistance detection. Previously he aided in the launch of the first commercial semi-conductor gene sequencing system and developed several procedures for performance optimization at IonTorrent. Since then he is focused on leveraging comparative genomics for natural product discovery and engineering at the University of Wageningen, Netherlands. |
 Christine Beemelmanns | Christine Beemelmanns performed her PhD under the supervision of Prof. Hans-Ulrich Reissig at the FU Berlin. After two postdoctoral stays in the group of Prof. Keisuke Suzuki at the Tokyo Institute of Technology (Tokyo, Japan) and Prof. Jon Clardy at Harvard Medical School (USA), she joined the Leibniz Institute for Natural Product Research and Infection Biology (Jena, Germany) as junior research group leader in the end of 2013. Her research focuses of the isolation, characterization and total synthesis of microbial signalling molecules. |
1. Introduction
The marine environment is unique in terms of pH, temperature, pressure, oxygen, light, and salinity. It also contains most of the Earth's surface and nearly 87% of the world's biosphere (fauna and flora),1 and thus represents an exceptional reservoir of microbial diversity and microbial-derived bioactive compounds, nutraceuticals and other potential compounds of commercial value.2,3Marine microorganisms with unique biochemical capabilities have been found within all areas of the marine environment, including different water columns of open waters, the bottom of the Mariana Trench, known as the deepest point of the world's oceans, and many other extreme environments including deep-sea cold-water habitats and deep-sea vents.4
In addition, marine macro-organisms mostly belonging to the phyla Porifera (sponges) and Coelenterata (corals),5 have been found to contain a large portion of host-adapted microorganisms with up to 30% of the hosts biomass.6 Subsequent genomic and metagenomics analyses clearly showed that host-specific and co-evolved symbiotic microbial communities, often non-culturable outside the host environment, are responsible for most of the isolated natural products isolated from marine macro-organisms.7
A comprehensive study aiming at describing the bacterial diversity within different marine environments identified more than 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecular operational taxonomic units with 3% sequence dissimilarity threshold (OTU0.03) from 509 global marine samples.8 From the identified taxonomic units, Gammaproteobacteria and Alphaproteobacteria were dominant in ocean waters, followed by a high proportion of Flavobacteria and Cyanobacteria. The dominant groups in benthic communities were Gammaproteobacteria, Deltaproteobacteria, Planctomycetes, Actinobacteria, and Acidobacteria. The microbial community composition, however, differed greatly depending on their sampling origin.
000 molecular operational taxonomic units with 3% sequence dissimilarity threshold (OTU0.03) from 509 global marine samples.8 From the identified taxonomic units, Gammaproteobacteria and Alphaproteobacteria were dominant in ocean waters, followed by a high proportion of Flavobacteria and Cyanobacteria. The dominant groups in benthic communities were Gammaproteobacteria, Deltaproteobacteria, Planctomycetes, Actinobacteria, and Acidobacteria. The microbial community composition, however, differed greatly depending on their sampling origin.
Research in marine ecology and natural product chemistry has also shown that bacterial taxonomy, ecological role and metabolic diversity are likely fundamentally interrelated,9–12 with low nutrient concentrations, high dilution rates, and/or highly competitive environments within host and surface-associated microbial communities as major driving forces for the evolution of regulated biosynthetic traits.13–15 Many of the produced marine natural products are assumed to serve as defensive metabolites or siderophores, but also as chemical language allowing communication among species and with other organisms and thereby conferring a significant survival advantage. Bacterial lineages specifically adapted to a host-dependent life style, are assumed to secrete metabolites that might even influence physiological processes of the host in favor of the associated microorganisms.16
Overall, the analyses of bacterial symbionts and/or microbial communities in general are likely to reveal unique biosynthetic traits and new natural products, which can be exploited for human welfare.14,15,17
Biosynthesis of secondary metabolites
Secondary metabolites are biosynthesized by dedicated enzymatic machineries, which are often encoded within in biosynthetic gene cluster regions (BGCs) within the genome of the producing organisms. To date, more than 1500 BGCs have been experimentally characterized and their products been investigated.18 But it is estimated that many more gene cluster families still await characterization and even with conservative assumptions, the total number of bacterial BGC families present in the biosphere may be estimated to be ∼6000.19–21 AntiSMASH,22 the most versatile platform to predict BGCs, lists 46 different families of BGCs in its database (until 1st August, 2020), which contain 172395 BGCs from 24776 unique species/strains. The most abundant BGCs encode for nonribosomal peptide-synthetases (NRPS) (23%), polyketide synthases (PKS) (17%), ribosomally synthesized and post-translationally modified peptides (RiPPs) (9%) and/or bacteriocin biosynthesis (13%) as well as terpene synthases (12%).
Catalyzed by the availability genomic dereplication tools such as antiSMASH22 or Antibiotic Resistant Target Seeker (ARTS),23 many emerging (meta)genomic studies are now directed at exploring the biosynthetic capacities of rare or non-culturable microbes and/or microbial communities for yet undescribed transformations and potentially novel natural products.24,25 Today, low genome sequencing costs, the availability of easy-to-use web-based genome mining and metabolomic analysis tools26 allow for the efficient dereplication of known metabolites from complex metabolite mixtures resulting in increasing discovery rates of novel metabolites and biosynthetic traits.27–29 Many research groups have also picked up the challenge to fully characterize and manipulate the underlying biosynthetic transformations thereby applying or developing state-of-the art biotechnological approaches.24 Although many approaches were of mixed success, steadily improving computational power and methods are beginning to provide solutions to leverage these complex systems making semi-automated and design-based engineering of novel peptide and polyketide products more reachable than ever.30
Aim of the review
This review aims to illustrate the enormous biosynthetic potential of marine bacteria to produce structurally diverse small secondary metabolites, which includes fatty acids, terpenes, PKS, NRPS, hybrid PKS/NRPS, and alkaloids. The review commences with a global analysis of BGCs in marine bacteria, which is followed by chapters sorted according to the described biosynthetic origin. Each chapter features selected examples of small secondary metabolites accompanied by biosynthetic studies, which have been reported between 2014 and 2020. Due to the eminent lack of ecological studies related to many of the isolated compounds, speculation about their role in nature will be omitted. We also apologize in advance to those authors whose work could not be cited within the frame of this perspective.
The perspective builds on and complements several excellent preceding reviews. In 2010, McClintock et al. discussed the ecology and structures of biomolecules derived from phylogenetically diverse marine invertebrates of diverse geographic origin.31
Later, Skropeta and coworkers made a remarkable effort to review the literature of deep-sea natural products up to 2013.32 The diversity of marine natural products of 2014 and 2015 were covered by Blunt et al.33 The metabolic and biosynthetic diversity of secondary metabolites in marine Myxobacteria were covered in 2018.34 Also, the recent molecular tools and strategies applied in marine proteobacteria were reviewed in 2020 and demonstrated them as new platform for natural product research.35
Global analysis of BGCs in marine bacteria
A global analysis of 2734 genomes of marine bacterial strains retrieved from the JGI genome portal revealed that the dominant portion belonged to the widely distributed Gammaproteobacteria (27%) and Alphaproteobacteria (23%) followed by unclassified bacteria (17%), flavobacteria (6%), Deltaproteobacteria (4%), Actinobacteria (3%), and other classes of bacteria, respectively (Fig. 1B).
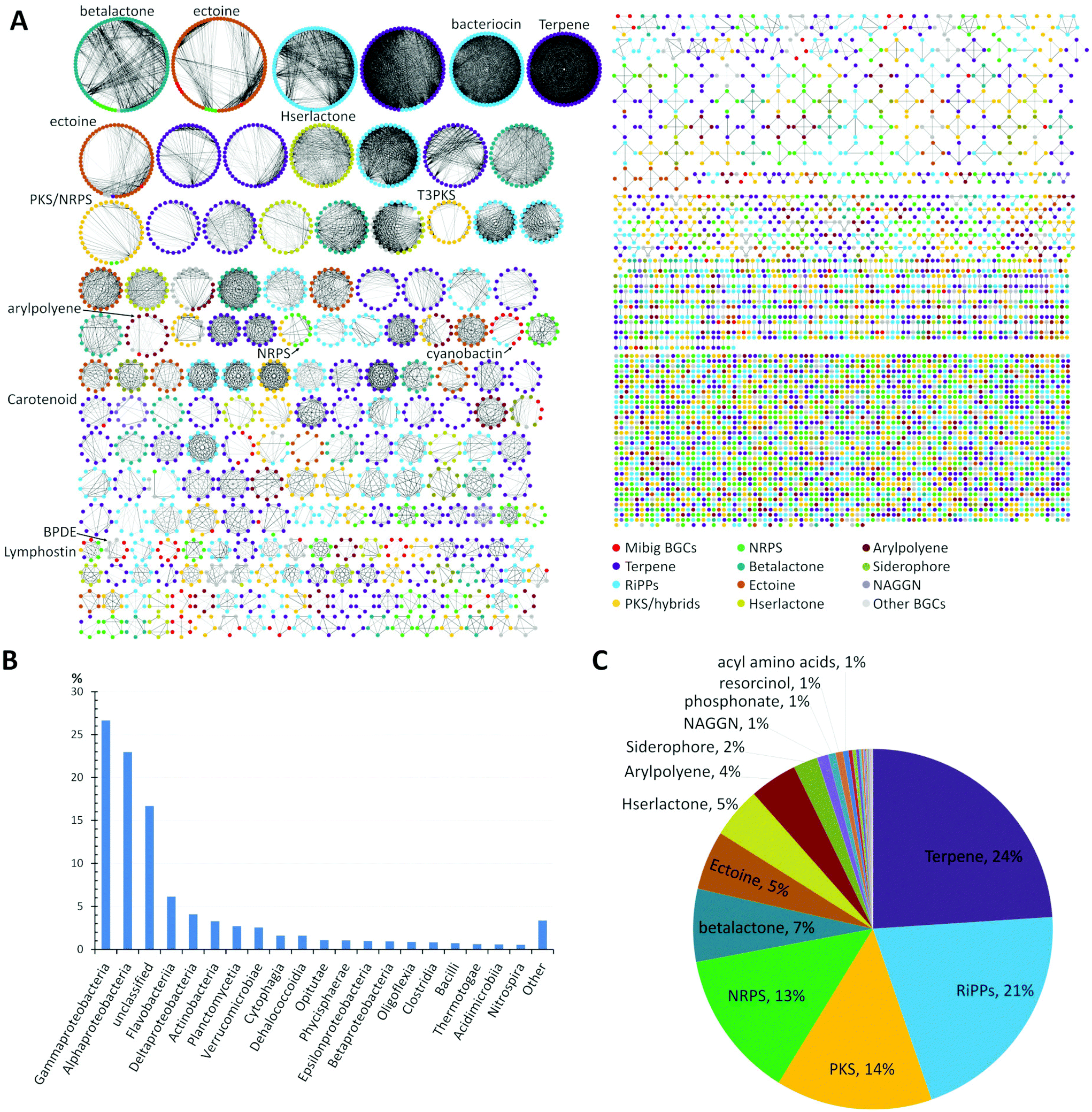 | ||
| Fig. 1 (A) Similarity network of BGCs in marine bacteria, detected by antiSMASH v.5 from a set of 2734 genome sequences acquired from JGI genome portal. Similarity network of the BGCs among different genomes was obtained using BiG-SCAPE.39 Each node represents a BGC and those with similar Pfam domain metrics are connected by edges. A cut-off of 0.75 was used for the analysis and final similarity network was visualized using Cytoscape 3.6.1. The similarity network file is available at NDEx36 (https://doi.org/10.18119/N9V31Z). (B) Taxonomic distribution (class) of the bacterial genomes used in the network. (C) Distribution of different families of BGCs in the genomes of marine bacteria. BGCs present in less than 1%: e.g. ladderane, butyrolactone, indole, phenazine, nucleosides, tropodithietic acid. | ||
A detailed comparative analysis of the collected genomes by antiSMASH v.5 (Fig. 1A) revealed the presence of more than 7480 BGCs and similarity network analysis revealed that BGCs related to terpene (24%) and RiPPs (21%) biosynthesis were amongst the most common BGCs within marine bacteria followed by PKS, NRPS and betalactone-related pathways, as well as the biosynthesis of the global osmolyte ectoine (Fig. 1C).
2. Fatty acids
Fatty acids (FAs) are ubiquitous in nature and are mostly known as primary metabolites important for nutrition and bacterial physiology. In addition to their physiological functions, several modified FAs are also known for their antifungal, antibacterial, and antimalarial activity.36 Bacterial FAs are mostly biosynthesized by discrete enzymes belonging to Type II fatty acid synthases (FASs).37,38 In brief, the biosynthesis is initiated by the formation of acetyl-ACP, which is catalyzed by a 3-ketoacyl-ACP synthase. Then, an iterative elongation sequence takes place, where malonyl-ACP is condensed with the acyl chain in a Claisen-type condensation reaction, which is followed by a reduction, dehydration and a final reduction reaction yielding an acyl-ACP that could undergo another round of elongation. In some bacterial species, the iterative elongation is interrupted after five cycles (ten carbon chain length) by an isomerase activity, which causes the isomerization of the 2-trans-decenoyl intermediate into the 3-cis-isomer. The latter is subsequently extended to long-chain monounsaturated fatty acids instead of being reduced.Medium-chain cyclopropane-containing fatty acids
Marine bacteria are known to biosynthesize many structurally-modified FAs, including cyclopropane-containing fatty acids (CFAs), which have been associated with bacterial adaptation to environmental stress factors such as cryodesiccation or low pH.40,41 The antimicrobial activities of CFAs are likely a result of to their potential to inhibit the synthesis of structurally-related fatty acids in other microorganisms or to influence cell membrane integrity.42,43Bacteria of the genus Labrenzia are known as surface colonizers in marine environments and are suggested to act as protective symbionts of oysters.44 In a recent study, Labrenzia sp. strain 011, isolated from Baltic Sea coastal sediment was found to produce two medium-chain fatty acids containing a cyclopropane moiety: cis-4-(2-hexylcyclopropyl)-butanoic acid (1) and cis-2-(2-hexylcyclopropyl)-acetic acid (2).44,45 Both compounds were isolated using absorber resin Sepabeads from 20 L culture and the structures were established by NMR analyses. Activity studies revealed their inhibition activity against a range of bacteria and fungi, including a causative agent of Roseovarius oyster disease (ROD), Pseudoroseovarius crassostreae DSM 16950. Comparative genome mining of members of the genus Labrenzia revealed several BGCs with conserved gene sequences encoding cyclopropane fatty acid synthases (CFASs). In E. coli, it was shown that CFAs catalyze not only the transfer of a methyl group from S-adenosyl L-methionine (SAM) to a cis double bond of an unsaturated fatty acid, but also the deprotonation of the newly attached methyl group and subsequent ring closure to form a cyclopropane ring (Fig. 2).46,47
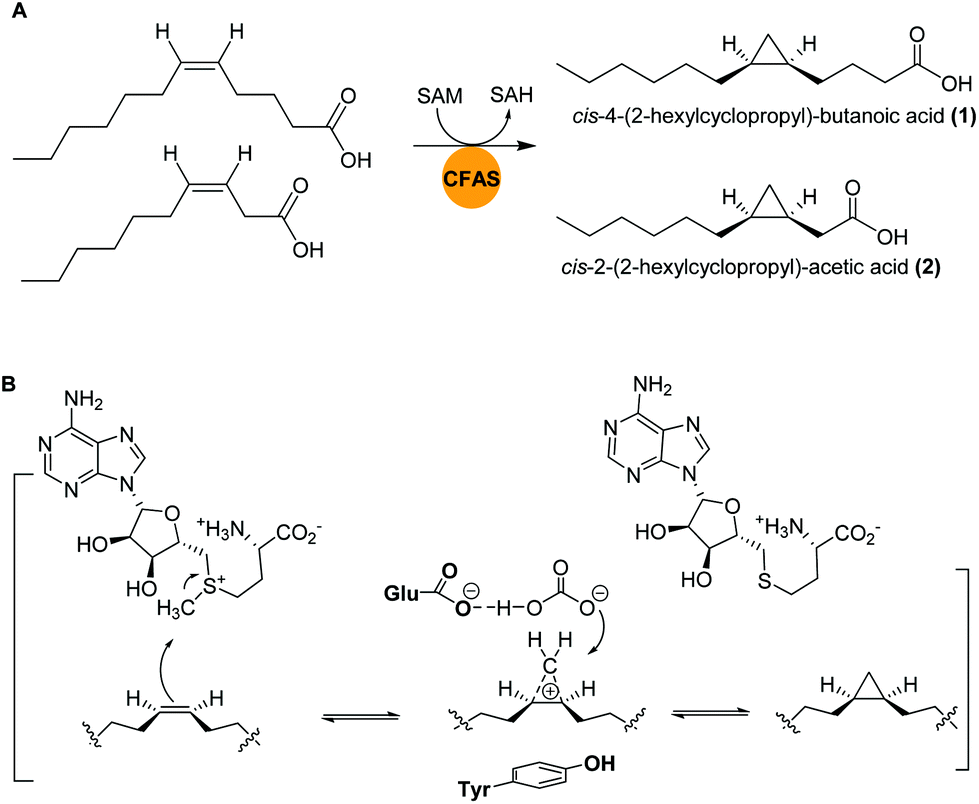 | ||
| Fig. 2 (A) Cyclopropanation of two medium-chain fatty acids by CFASs in Labrenzia sp. 011. (B) Generalized SAM-dependent cyclopropane biosynthesis catalyzed by CFA synthases. | ||
3. Terpenoids
More than 50000 terpenoids (also known as isoprenoids) grouped in numerous distinct structural families have been isolated so far.48 Although mostly isolated from marine and terrestrial eukaryotes, research within the last two decades has increasingly uncovered new terpenoids of prokaryotic origin, most notably from Gram positive Actinomycetes.49
In fact, the majority of bacterial-derived cyclic terpenes have only recently been uncovered due to targeted global genomic and metabolomic mining approaches. As highlighted in Fig. 1, in particular marine bacteria hold a yet untapped biosynthetic potential to produce terpenoids.50–53
The biosynthesis of terpenes requires the action of specific terpene synthases (TSs), which convert isoprene derived building blocks ((oligo)prenyl diphosphates) into linear and cyclic core structures, that are categorized into hemiterpenes (C5), monoterpenes (C10), sesquiterpenes (C15), diterpenes (C20), sesterterpenes (C25) and triterpenes (C30). While hemi-(C5) to di-(C20) terpenes are biosynthesized from prenyl-pyrophosphate units via the action of terpene synthases belonging to class I and class II, most of the tri-(C30) and tetra-(C40) terpenes are derived from isoprene precursors via a “head-to-head” condensation catalysed by class II enzymes.
Most bacteria produce the required C-5 building blocks for oligoprenyl diphosphates via the MEP (methylerythritol 4-phosphate) pathway; while others use the MVA (mevalonate) pathway instead of the MEP pathway, or possess both pathways.54 Despite the broad abundance of these biosynthetic traits in bacteria, and marine species in particular, only few studies in the past were directed at understanding the structural diversity, the biosynthetic origin and ecological functions of terpenes in the marine environment. Amongst increasing numbers of emerging reports, we highlight here three recent examples including a terpene-based toxin isolated from a marine diatom (eukaryote) and apologize to those authors whose work has been omitted.
Recently, a new TS (spata-13,17-diene synthase, SpS) from Streptomyces xinghaiensis, a marine actinomycete isolated from sediments near Dalian (China), was characterized and found to have unique sesqui-, di- and sesterterpene synthase activity with a broad substrate spectrum and Mg2+ as well as Mn2+ cofactor dependency. Amongst the many identified products (known and novel derivatives) were the terpenes prenylkelsoene (3) and spata-13,17-diene (4) (Fig. 3A).55
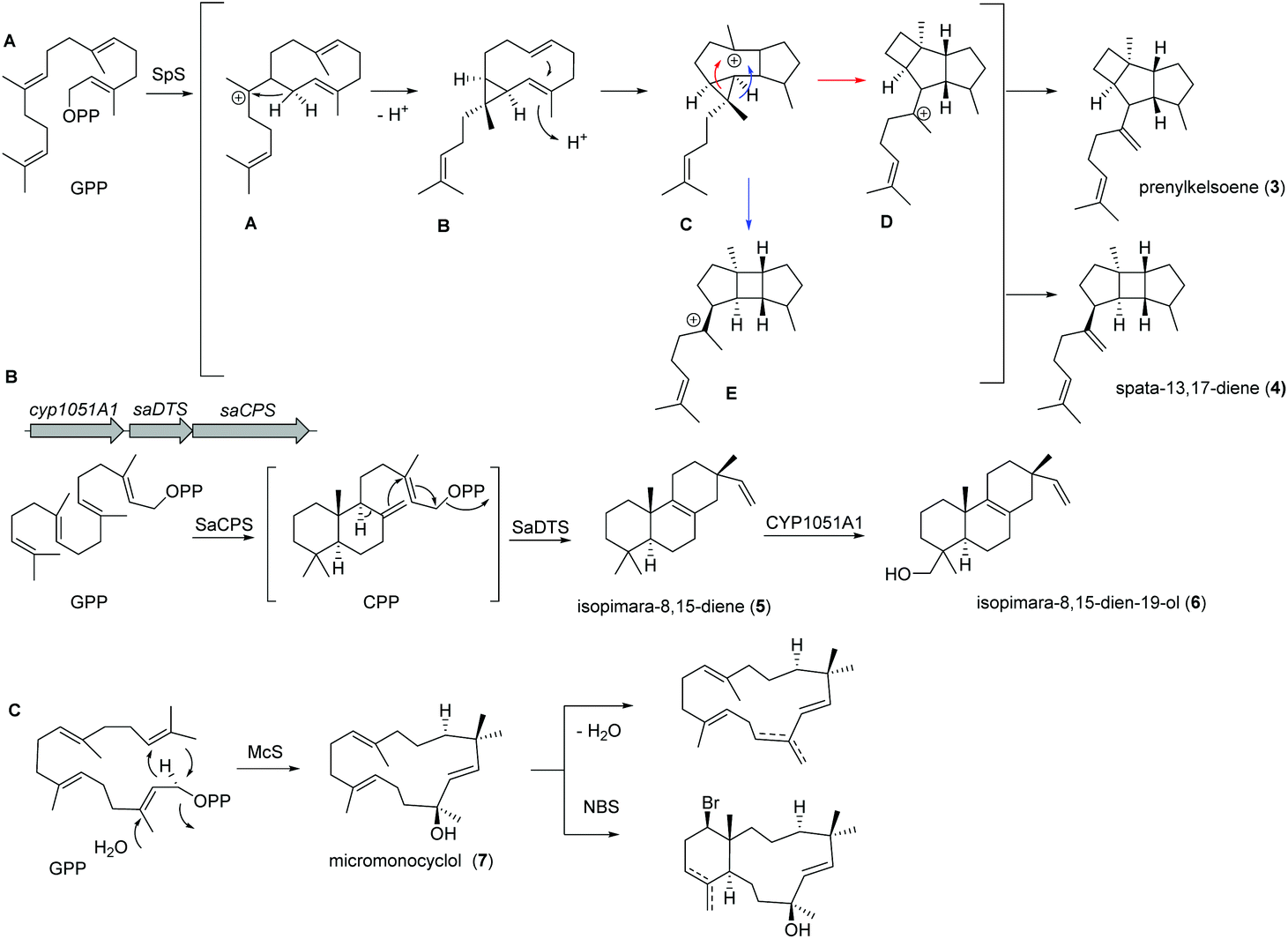 | ||
| Fig. 3 (A) Biosynthesis of prenylkelsoene (3) and spata-13,17-diene (4) by spata-13,17-diene synthase (SpS) from Streptomyces xinghaiensis. (B) Biosynthetic pathway of isopimara-8,15-diene (5) and isopimara-8,15-dien-19-ol (6) in Salinispora arenicola; (C) studies on the micromonocyclol synthase (McS) yielding micromonocyclol (7). | ||
In case of isopimara-8,15-dien-19-ol (6), the responsible BGC was uncovered by genome mining of the producing marine actinomycete Salinispora arenicola, and was found to consist of three genes encoding a type II diterpene synthase (DTS), a type I DTS and a cytochrome P450 monooxygenase (CYP; Fig. 3B).56 The type II DTS was then identified as a copalyl diphosphate synthase (SaCPS) catalyzing the first cyclization step. The resulting product CPP is further cyclized by the type I enzyme SaDTS into isopimara-8,15-diene (5) followed by oxidation to isopimara-8,15-dien-19-ol (6) by CYP1051A1.
Recent genome sequencing efforts unraveled the presence of phylogenetically distinct class I TS gene sequences that differentiated from previously reported sequences within all sequenced Micromonospora isolates.57 Subsequent functional studies on one of those class I TS from Micromonospora marina resulted in the production of the 15-membered micromonocyclol (7), which allowed the absolute structure determination by biomimetic chemical transformations and studies on the stereochemical cyclization model (Fig. 3C).
Domoic acid
Domoic acid (DA, 8) is a terpene-derived toxin, which is produced by Pseudo-nitzschia diatoms during harmful algal blooms thereby causing a serious danger to oceanic and human life.58 Although not of bacterial origin, we have included a short summary due to the highly intriguing biosynthetic pathway and structural features. Transcriptomic studies led to the identification of a conserved gene cluster (dabA-dabD) encoding a terpene cyclase, a hypothetical protein, a dioxygenase, and a CYP450 (Fig. 4).59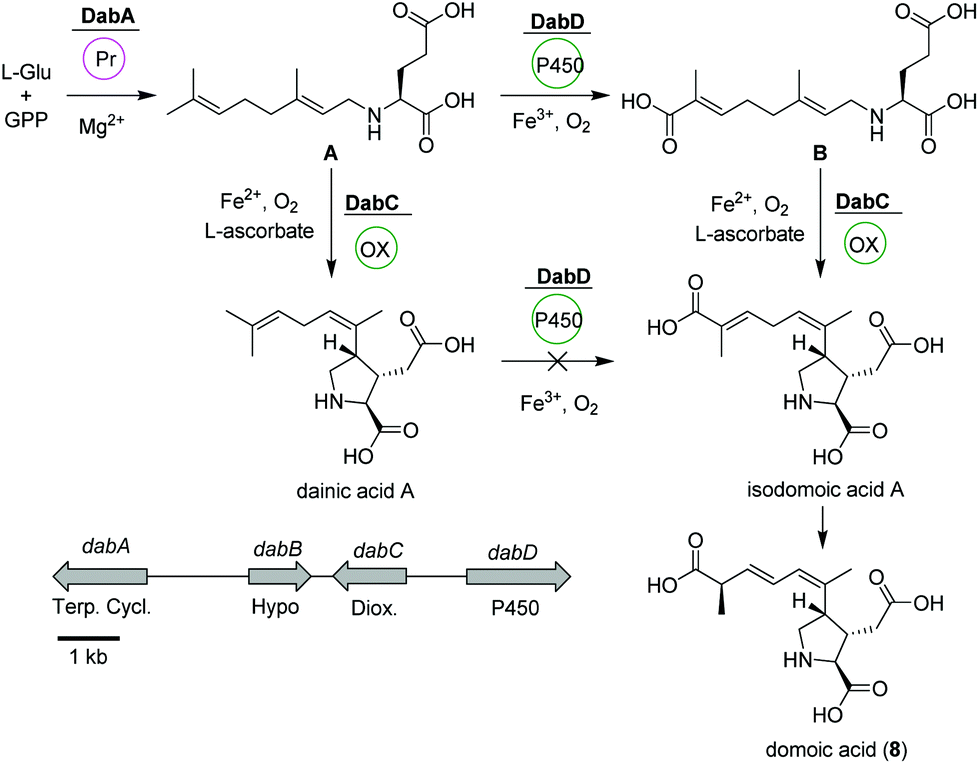 | ||
| Fig. 4 Domoic acid (8) biosynthetic pathway based on dab gene cluster in Pseudo-nitzschia multiseries. | ||
In vitro assays using recombinant DabA revealed a Mg2+-dependent N-geranylation activity toward L-glutamate yielding N-geranyl-L-glutamic acid (L-NGG, A). Next, DabD oxidizes the L-NGG (A) on the 7′-methyl end in three successive oxidation reactions yielding 7′-carboxy-L-NGG (B), which is then cyclized to isodomoic acid A by DabC. In a final step, it is speculated that a yet unknown isomerase converts isodomoic acid A to domoic acid (8).60 Interestingly, DabC is abale to catalyze the subsequent oxidative cyclization of L-NGG (A) and formation of pyrrolidine ring resulting in the formation of dainic acids A and derivatives (not shown). However, dainic acid isomers were not accepted as substrates by DabD CYP.
4. Polyketide synthases (PKS)
Polyketides are a large class of structurally diverse secondary metabolites, which are biosynthesized via the decarboxylative condensation of malonyl-CoA derived extender units catalyzed by dedicated polyketide synthases (PKSs).61,62 PKSs are characterized as mega-enzymes and are classified into different types (types I–III) based on their assembly line architecture and mode of action.63 PKSs are comprised of different modules, which can be further subdivided into domains, i.e. enzymatic units responsible for loading, condensation and further modification of the extender units.64 The vast diversity of polyketides is mainly derived from their substrate building blocks as well as module/domain organization of PKSs. The diversity and intricacy of PKS-derived natural products is enormously impacted by the various substrates that can serve as starter and extender units.65 Amongst the many reported PKS-derived natural products isolated from marine bacteria, we want to highlight the following compounds due to recent breakthroughs in the elucidation of their biosynthetic pathways.Nocapyrones
Actinomycetes of the genus Nocardiopsis have been repeatedly reported to be associated with marine invertebrates like sponges or mollusks. As an example, N. alba CR167 was isolated from the shell- and venom-protected cone snail Conus rolani and was identified as a nocapyrone (9) producer.66 Bioinformatic and chemical evidence could tie the production of these heterocyclic natural products of the γ-pyrone class to the bacteria in contrast to many previous studies that isolated these metabolites from marine invertebrates without further elucidation of their true biosynthetic origin. Sequencing and analysis of the N. alba CR167 genome as well as comparison with other pyrone biosynthetic genes led to the identification of the putative type I PKS BGC ncp, which is composed of four genes: the oxidoreductase ncpA, the methyltransferase ncpB, the PKS ncpC and the putative single acyltransferase ncpD.Based on the similarity to other pyrone biosynthetic gene clusters, the nocapyrone biosynthetic pathway presumably starts with the incorporation of a fatty acyl CoA via NcpD to NcpC (Fig. 5). Then, iterative elongation with two methylmalonyl-CoA units yields the C-core skeleton, which is subjected to O-methylation catalyzed by NcpB; final C-oxidation is performed by NcpA.66 To provide further confirmation of the proposed enzyme activity, the recombinant methyl transferase NcpB was tested using different substrates, which resulted in a narrow specificity for the demethylated metabolites e.g. demethylated nocapyrone P.65
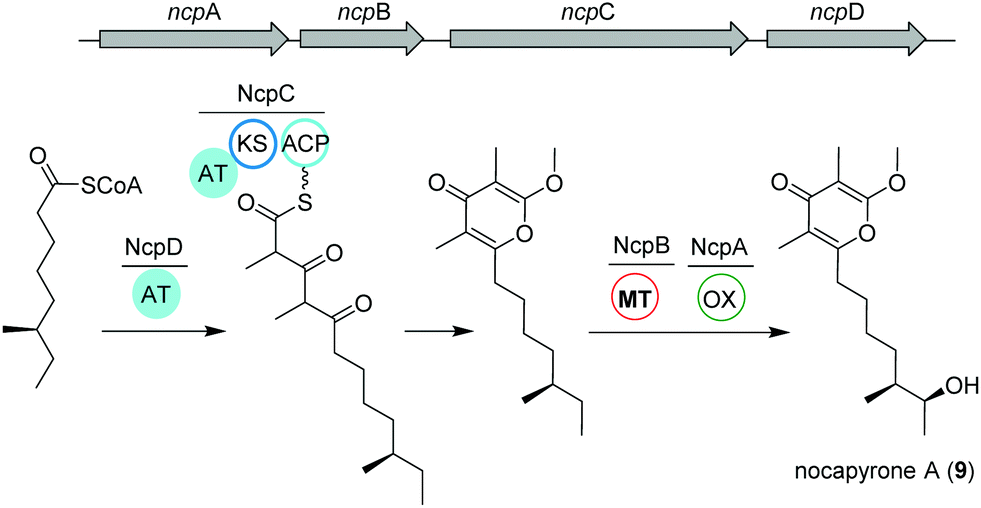 | ||
| Fig. 5 Organization of the ncp BGC and key biosynthetic transformations leading to the production of nocapyrone A (9). | ||
Anthracimycin and chlorotonil A
The unique macrolide antibiotic anthracimycin (10) is a 14-membered macrocyclic lactone ring with a decalin core structure. Anthracimycin was first isolated from a marine actinobacterium Streptomyces sp. T676 in 1995 and recently reported again from another marine Streptomyces sp. CNH36567 using a directed bioassays fractionation. Anthracimycin (10) has excellent inhibitory activities against Gram-positive pathogens including Bacillus anthracis and methicillin- and vancomycin-resistant Staphylococcus aureus (MRSA and VRSA).68Interestingly, a structurally related compound, chlorotonil A (11), was also isolated from a myxobacterial strain Sorangium cellulosum sp. So ce1525. The structure of chlorotonil A differs from anthracimycin at position C-8 by an additional methyl group, and at C-4 by a gem-dichloro moiety and a different stereochemistry. Similar to anthracimycin, chlorotonil A also has strong inhibitory activity against Plasmodium falciparum, the malaria pathogen and strong antibacterial and moderate antifungal activity.69
Both compounds (10) and (11) were assumed to be derived from a trans-AT PKS type I based biosynthetic pathway due to structural similarities, e.g. several β,γ-shifted double bonds, with other trans-AT PKS products such as bacillaene70 and rhizoxin.71 Genome mining studies of the producing strains revealed two highly homologous trans-AT PKS gene clusters, namely atc and cto, respectively (Fig. 6A and B).
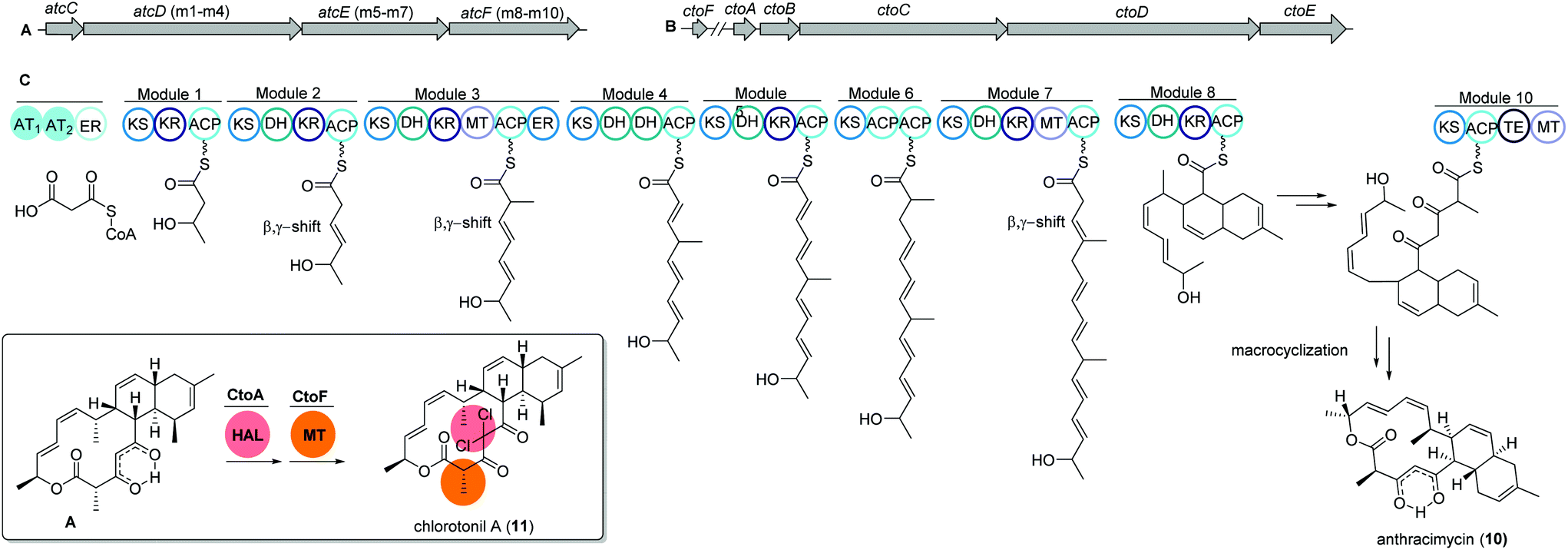 | ||
| Fig. 6 (A) Gene map of the trans-AT PKS gene cluster atc from Streptomyces sp. CNH365 and (B) cto BGC from Sorangium cellulosum So ce1525 responsible for the production of anthracimycin 10 and chlorotonil 11, respectively. (C) Architecture of the modules and domains in atc PKS AtcC-AtcF together with proposed reactions likely happening in each module. | ||
The atc BGC consists of 53 kbp and nine genes (atcA to atcI) where atcC encodes a protein with trans AT activity using malonyl-CoA as the substrate. The following atcDEF contains ten modules, which are responsible for the elongation of the polyketide chain. Similarly, the cto BGC contains a homologous trans-AT domain encoded by ctoB and ten KS domains encoded by ctoCDE, which are responsible for the necessary incorporation of acetate-derived extender units. In addition, an extra flavin dependent halogenase ctoA is held accountable for the chlorination reaction to yield chlorotonil.72 The putative biosynthetic gene clusters were both confirmed by gene inactivation and isotope labeling studies.
Misakinolides
Similar to nocapyrones, natural products isolated from the marine sponge Theonella swinhoei WA from the isle Hachijo-Jima in Japan have been shown to originate from associated bacteria.73 The metabolic profile of this specific sponge revealed the presence of only one polyketide, misakinolide A (12). This cytotoxic lactone macrolide acts as an actin inhibitor and is produced by filamentous bacterial symbionts “Candidatus entotheonella” variants as demonstrated by Ueoka and colleagues.74 Using bioinformatic analysis of a sponge DNA clone library, they discovered a ca. 90 kb long trans-AT PKS BGC, termed mis (Fig. 7), which includes the PKS genes misC-F as well as the AT misG. Predictions based on phylogeny of the ketosynthase domains and the BGC architecture strongly pointed toward a misakinolide-like product. Briefly, the biosynthesis was proposed to start with the incorporation of a malonyl-CoA unit followed by elongation, O-methylation and cyclization catalyzed by MisC. Consecutive biosynthetic steps include several elongation and methylation steps catalyzed by MisD and MisE, whereas MisF is responsible for dihydropyran ring formation, dimerization and release of the final product.74,75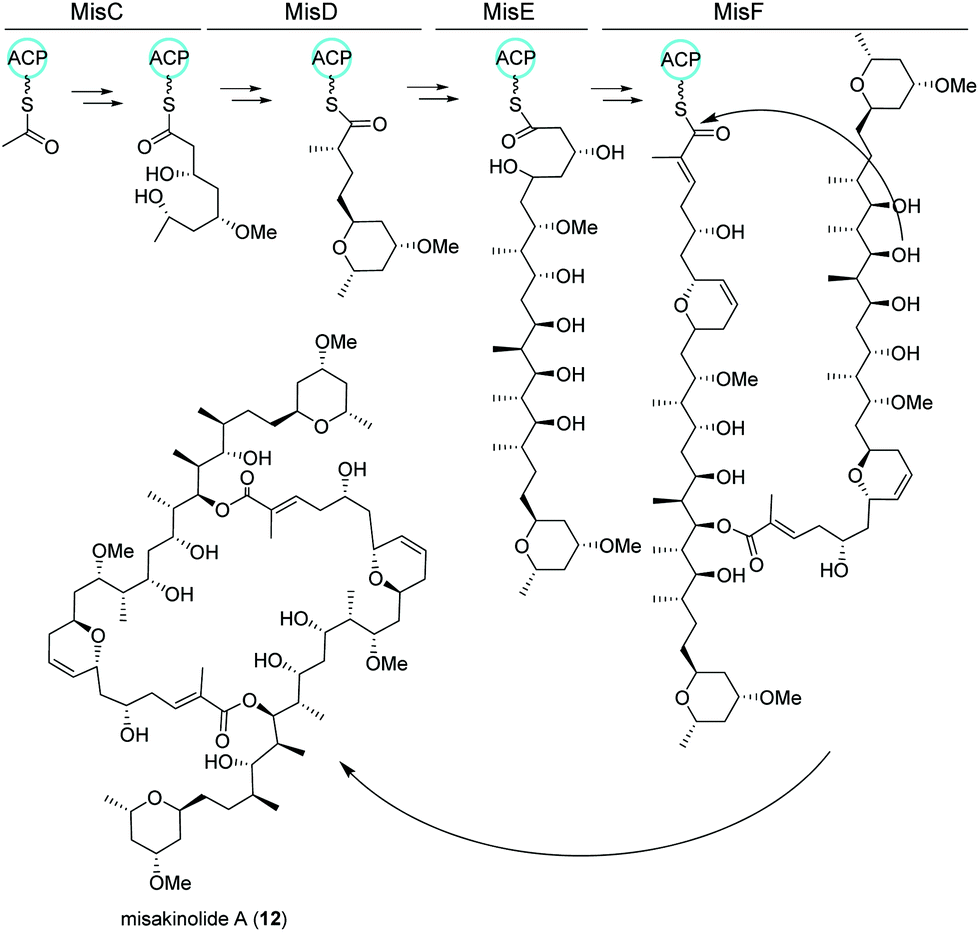 | ||
| Fig. 7 trans-AT PKS biosynthetic pathway of misakinolide A (12) isolated from the filamentous bacterial sponge symbiont “Candidatus entotheonella”. | ||
5. Non-ribosomal peptide synthetases (NRPS)
NRPS-derived secondary metabolites are of great pharmacological and industrial significance.76 Similar to the PKS, the NRPS is organized in an assembly-line fashion and dedicated protein modules are responsible for incorporating of the amino acid building blocks into the growing chain. Each module consists of several domains with defined functions.77 Unlike ribosomes, NRPSs can accept numerous possible monomers in their assembly line (>520 monomers including rare nonproteinogenic amino acids), which makes them highly diverse. Additional product diversifications arise during the chain assembly, chain termination and post assembly-line tailoring reactions.78 Most interestingly, hybrid PKS–NRPS assembly lines can incorporate both acyl and aminoacyl building blocks into structurally complex polyketide–peptide hybrid molecules. Their combined functionalities expand the biological activities of these molecules by mixing their chemical properties.79Endolides
Tetrapeptides endolide A (13) and B (14) are unusual N-methylated peptides, which were produced by the marine-derived fungus Stachylidium sp. isolated from the sponge Callyspongia sp. cf. C. flammea.80,81 Both were non-cytotoxic but endolide A exhibited affinity to the vasopressin receptor while endolide B interacted with the serotonin receptor 5-HT2b. Due to their cyclic nature as well as the presence of N-methylated amide bonds, a NRPS-based biosynthesis was proposed. Both peptides contain the very rare amino acid 3-(3-furyl)-alanine, which is most likely biosynthesized via the shikimate pathway from phosphoenolpyruvate (PEP) and erythrose-4-phosphate (E-4-P) (Fig. 8). Although isolated from a marine fungus, it is speculated that bacterial endosymbionts of the fungus might be the primary producer as 3-(3-furyl)alanine containing peptides, i.e., rhizonins A and B, were isolated from the fungal endosymbiont Burkholderia endofungorum82 and bingchamide B from Streptomyces bingchenggensis.83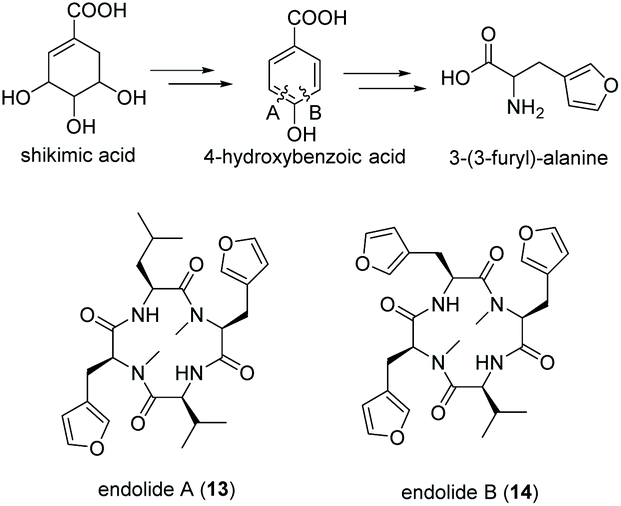 | ||
| Fig. 8 Biosynthesis of 3-(3-furyl)-alanine via the shikimate pathway and structures of endolide A (13) and B (14) produced by the marine fungus Stachylidium sp. | ||
Surugamides
Streptomycetes isolated from deep-sea sediment originating from Suruga or Sagami bay in Japan produce the surugamide peptides that show inhibitory activity against cathepsin B.84 The surugamides can be structurally divided in two classes: cyclic octapeptide surugamides A–E (surugamide A (15)), whereas surugamide F (16) and cyclosurugamide F are decapaptides with a linear and cyclic structure, respectively.85 They are all produced by homologs of the four-gene NRPS biosynthetic cluster surABCD, first identified in Streptomyces sp. JAMM992 (Fig. 9).81,86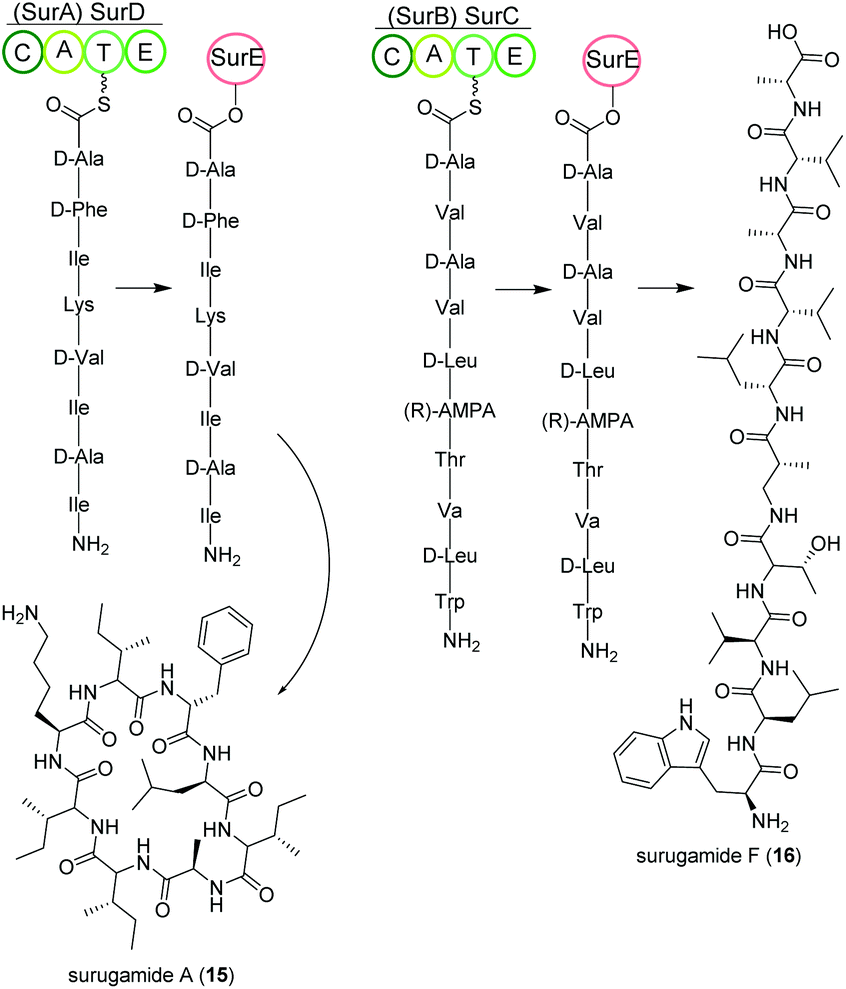 | ||
| Fig. 9 Key steps of the NRPS-based biosynthesis (surABCD) of octa- and decapeptidic surugamides. | ||
This cluster exhibits unique features: (i) Two genes are responsible for the production of one class of surugamides, respectively. While surA and surD are responsible for the biosynthesis of surugamides A–E, surB and surC encode for the biosynthesis of cyclosurugamide F with surugamide F probably being a degradation product of cyclosurugamide F. The organization of each of the gene cluster is noteworthy: as the names suggest, surA and surD are separated by surB and surC but work together as a distinct natural product assembly-line, differing from the usual linear setup of NRPS gene clusters.86 (ii) The gene cluster lacks a thioesterase (TE) domain, condensation (C) domain or reductase (R) domain, which usually catalyze the release of the natural product from the final enzyme of the production line.86 Instead, the macrocyclization and subsequent release of the surugamide products is catalyzed by the penicillin-binding-protein (PBP) SurE, acting in trans. Its gene, surE, is located directly in front of the surABCD NRPS gene cluster.87,88
SurE is a special case not only because it represents a stand-alone TE domain, but it was also shown to possess broad substrate specificity. On the one hand, it catalyzes the release of surugamides from both the SurBC and SurAD assembly line.87 On the other hand, SurE is also able to accept other synthetic substrates indicating a possible application in novel natural product diversification. Additionally, the crucial function of the amino acid residues S63, K66 and N156 for SurE activity was determined in Streptomyces albidoflavus LHW3101.89,90
Marformycins
The deep-sea sediment-derived isolate Streptomyces drozdowiczii SCSIO 10141 was analyzed for the biosynthesis of the natural product class marformycins (17).91,92 Marformycin A and its derivatives are N-formylated cyclodepsipeptides consisting of five non-proteinogenic amino acids. They were shown to selectively inhibit Micrococcus luteus as well as two Propionibacterium species. Genome analysis of the producing organism revealed a large operon with 20 open reading frames (ORFs), mfn, including six NRPS genes (Fig. 10). NRPS genes mfnC, mfnD and mfnE were identified as core biosynthetic genes for marformycin production using knock-out studies. Additionally, three stand-alone enzymes a stand-alone adenylase (MfnK), a free PCP (MfnL) and an MbtH-like protein (MfnF) were identified.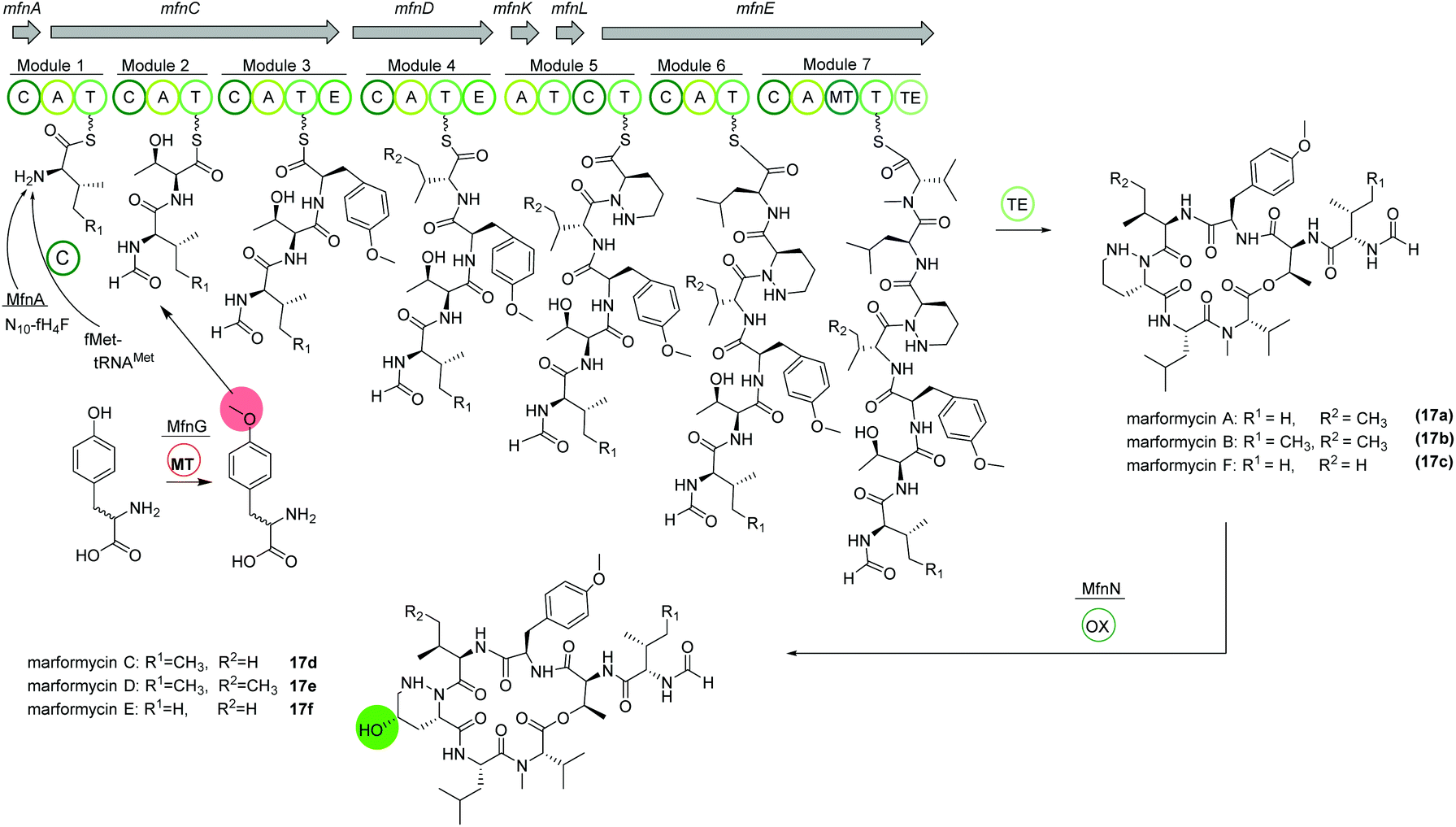 | ||
| Fig. 10 Organization of the mfn NRPS BGC in Streptomyces drozdowiczii SCSIO 10141 responsible for the biosynthesis of the marformycins (17). | ||
The biosynthesis is proposed to commence with the transfer of a formyl group through MfnA, either via fMet-tRNAMet (from primary metabolism) and subsequent condensation by MfnC or via direct transfer of the formyl group from the cofactor N10-fH4F to the valine residue on the PCP of MfnC. After incorporation of threonine, O-methylated tyrosine is integrated. Biochemical characterization revealed the activity of MfnG as methyltransferase, methylating L- or D-tyrosine prior to its incorporation in the assembly line by the A3 domain of MfnC. Next, L-valine or L-allo-isoleucine is included by MfnD, followed by incorporation of L-piperazic acid via MfnK. The presence of D-tyrosine, D-valine or D-allo-isoleucine in marformycin derivatives can be explained by two epimerization domains in MfnC and MfnD, respectively. Here, it is noteworthy that only O-methylated D-tyrosine is found in marformycins. Two final amino acids, L-leucine and L-valine, are integrated by MfnE, followed by cyclization and release of the cyclic marformycin via the TE domain of MfnE. Finally, analysis of the products of a mutant with inactivated cytochrome P450 monooxygenase MfnN indicated its function in regio- and stereoselective hydroxylation of the marformycin piperazic acid moiety after release of the product.91
Due to its ubiquitous presence in nature the biosynthesis of the non-proteinogenic amino acid L-allo-isoleucine was examined in S. drozdowiczii SCSIO 10141.93 A stereochemical comparison suggested a potential origination of L-allo-isoleucine from L-isoleucine via a deamination reaction catalyzed by an aminotransferase and an epimerization reaction by an isomerase. Interestingly, the mfn gene cluster itself encoded for homologous gene sequences, the aminotransferase gene mfnO and the isomerase gene mfnH. Gene inactivation and biochemical characterization revealed a reversible conversion of L-isoleucine to L-allo-isoleucine with a pyridoxal 5′-phosphate (PLP) linked intermediate and an in nature rarely observed epimerization at the Cβ-position, synergistically performed by MfnO and MfnH.93
Thiocoraline
The bisintercalator and anti-tumor active peptide thiocoraline (18) was discovered in 1997 from the marine actinomycete Micromonospora sp. L-13-ACM2-092,94,95 but its biosynthesis remained elusive at that time. Recent endeavors have deciphered details of the thiocoraline biosynthesis.The biosynthesis is initiated by the conversion of L-tryptophan to 3-hydroxyquinaldic acid (3HQA) as shown in previous studies.96 More recently, the incorporation of 3HQA was examined closely by Mori et al. (Fig. 11).97 Here, in a first step 3HQA is activated via adenylation and transported via a carrier protein to TioR to be incorporated as starter unit of the NRPS assembly line. By comparison of the amino acid sequence of other aryl substrate activating A-domains with enzymes from the thiocoraline biosynthetic gene cluster, TioJ was identified as a candidate for activation of 3HQA. Knockout experiments and substrate-specificity analyses confirmed that TioJ is essential for thiocoraline biosynthesis and that its natural substrate is 3HQA. In addition, TioJs substrate seems to be determined also by the overall enzyme structure and not only by the active site as revealed by the substrate specificity of various mutants. Despite being the only free T domain in the gene cluster and therefore a likely candidate as partner of TioJ, TioO failed to accept 3HQA. Successively, the acyl carrier protein FabC from the fatty acid biosynthesis was examined as it was found to be involved in the biosynthesis of other bisintercalator natural products. FabC could accept 3HQA verifying its function as carrier protein of the latter. By interaction studies the role of FabC as ACP acting in concert with TioJ as A-domain was further established.97
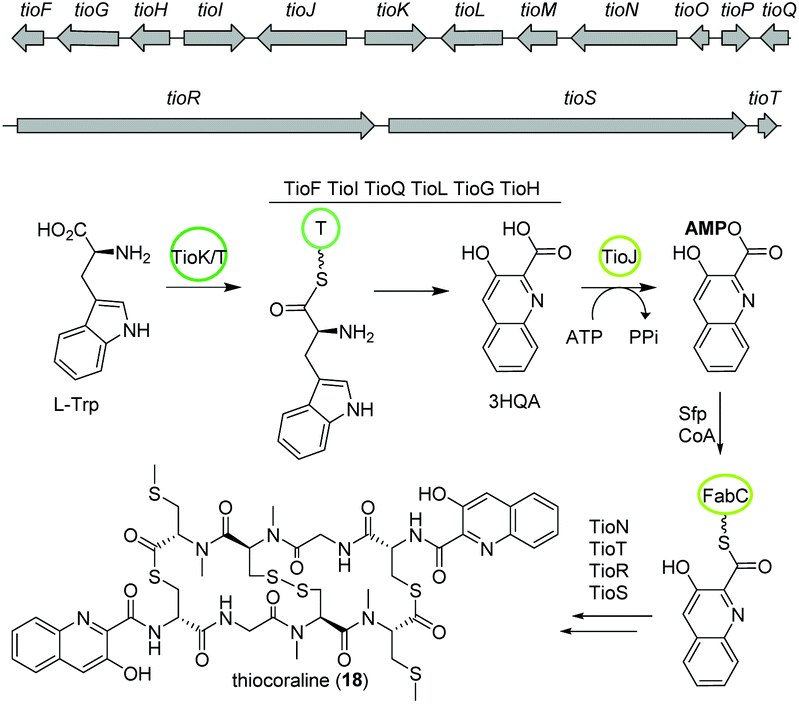 | ||
| Fig. 11 Partial gene map and key biosynthetic steps of thiocoraline (18). | ||
Thiocoraline consists of a bicyclic octathiodepsipeptide core structure with two S-methylated L-cysteine moieties and two 3-hydroxyquinaldic acid moieties that facilitate binding to DNA and its production is governed by the tio (A–Z) gene cluster (Fig. 11). Previous studies focused on the formation of the 3-hydroxyquinaldic acid moiety involving TioF, TioQ and TioK and are reviewed elsewhere.98 Recent work on the enzymes TioN, TioT by Al-Mestarihi and colleagues showed that the S-methylated L-cysteine moiety is generated by TioN and TioT.99 Knockout and co-expression studies confirmed that the MbtH-like protein TioT is necessary for activity of TioN and that both are crucial for thiocoraline production. The structure of TioN is unique since it represents a free A-domain that is interrupted by a methylation domain in an unprecedented position of the core signature sequence. Thereby, TioN possesses the rare dual function of adenylation and S-methylation. The formation of the S-methylated L-cysteine moiety by TioN could start via 2 pathways: (i) methylation of L-cysteine followed by adenylation or (ii) adenylation of L-cysteine followed by either direct methylation or attachment to TioS followed by methylation, both generating S-Me-L-Cys-S-TioS. Finally, TioS incorporates the moiety into the growing molecule. The investigation of the substrate specificity of TioN using radioactive adenylation assays revealed poor acceptance of S-Me-L-cysteine in contrast to L-cysteine eliminating pathway (i). In addition, it was shown that TioN can methylate both L-cysteine-S-TioS and L-cysteine-AMP and both, L-cysteine-AMP and S-Me-L-cysteine, are accepted by TioS.99 In light of the dependence of TioN on TioT and with respect to the before discussed marformycin biosynthesis, a role of the MbtH-like protein MfnF in aiding a free adenylation domain, e.g. MfnK, seems possible.
6. PKS–NRPS hybrids
Haliamide
Haliangium ochraceum SMP-2 (DSM 14365T) was the first marine myxobacterium isolated from a seaweed sample (Miura, Japan) and produced a new hybrid PKS–NRPS compound haliamide (19).100 Haliamide showed cytotoxicity activity against HeLa-S3 cells with IC50 of 12 μM. In silico analysis of H. ochraceum SMP-2 revealed a 21.7 kbp biosynthetic gene cluster (hla) consisting of one NRPS module followed by four PKS modules (Fig. 12). The biosynthesis of haliamide was proposed to commence with benzoyl CoA and its condensation to alanine via the NRPS module. Loading mechanism of benzoyl CoA is unknown due to the absence of the loading module. Nevertheless, the incorporation of benzoic acid was confirmed via isotope labeling study. Next, PKS modules incorporate two malonyl-CoAs and two methylmalonyl-CoAs following decarboxylative chain termination by the sulfotransferase (ST)-thioesterase (TE) domains. The assembly line contains some unusual features such as the absence of AT domains in modules 2 and 5, and a missing KR domain in module 5 and it was speculated that some trans-acting enzymes may perform the function of the missing domains.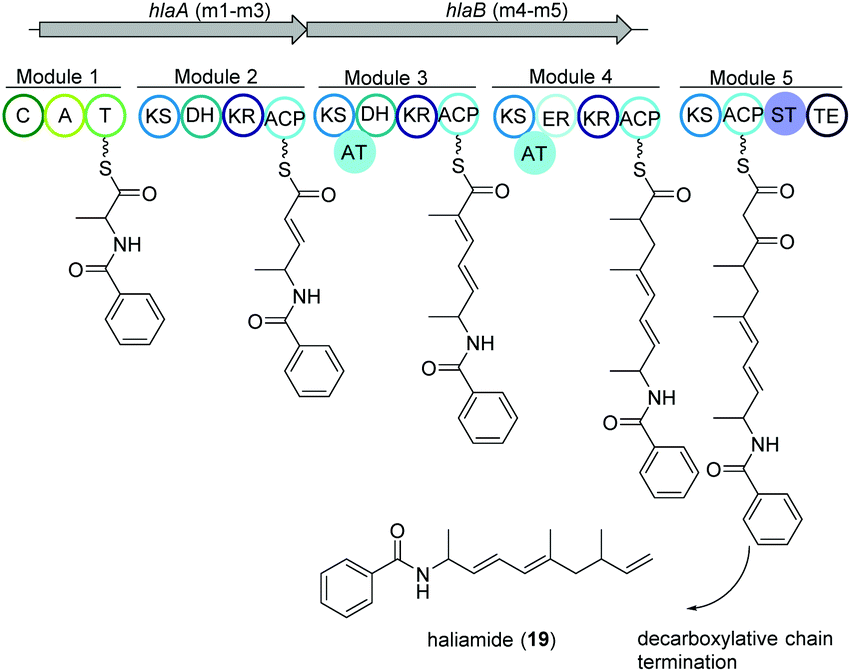 | ||
| Fig. 12 Organization of the hla BGC of haliamide consisting one NRPS module followed by four PKS modules in H. ochraceum SMP-2 (DSM 14365T). | ||
Thiolactomycin and thiotetromycins
Thiolactomycin A (20a) belongs to the group of thiotetronic acid natural products and was discovered from the Nocardia sp. no. 2-200 in 1982.101 This compound class contains a unique thiolactone moiety, exhibits a broad antibacterial activity and is known amongst other bioactivities to act as FASII inhibitors.First, isotope labeling experiments suggested a polyketide pathway involving acetate and three propionate building blocks and cysteine as origin of the sulfur atom.102 Later, Leadlay and co-workers were the first to uncover the underlying biosynthetic pathway using a comparative genomics approach.103 Genome analysis of Streptomyces olivaceus Tü 3010 indicated that such thiotetronates are assembled by a novel iterative PKS–NRPS. Subsequently, three other thiotetronate-producing actinomycetes were found to have near-identical clusters. In one example, the identity of the cluster from Streptomyces thiolactonus NRRL 15439 was validated by loss of thiotetronate production in in-frame deletion mutants and heterologous expression. In a later study, Moore and coworkers used target-directed genome mining on the pan-genome of 86 Salinispora bacterial genomes to identify a hybrid PKS/NRPS gene cluster (tlm) putatively responsible for the production (Fig. 13).104,105 The tlm gene cluster was then heterologously expressed in Streptomyces coelicolor using a TAR cloning strategy, which resulted in the identification of thiolactomycin and three new analogues. Later, mutagenesis and comparative metabolic analyses revealed the involvement of tlmF-tlmI genes in the γ-thiolactone skeleton construction of this class of antibiotics.106 The tlmG gene harbors a double KS-ACP domain, which is independent from the AT domain and responsible for initiation of the polyketide chain using acetate. Then, TlmH, an iterative PKS module (KS-AT-DH-KR-ACP-C) incorporates three methylmalonate units to form the backbone of thiolactomycin. The NRPS TlmI (A-PCP-TE) integrates sulfur derived from cysteine by an unknown mechanism and the cytochrome P450 TlmF forms the 5-membered thiolactone ring (Fig. 13). Further, heterologous expression of a closely related gene cluster from Streptomyces afghaniensis with some additional biosynthetic enzymes resulted in a series of new thiolactomycin analogues (20b–20d).
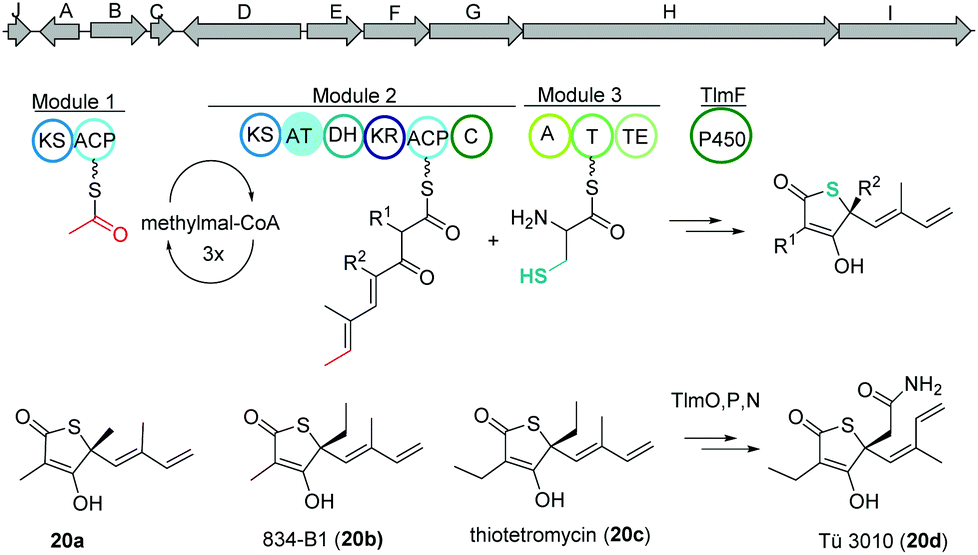 | ||
| Fig. 13 Gene map of the thiolactomycin hybrid PKS/NRPS gene cluster (tlm), key biosynthetic steps and structures of derivatives. | ||
Thiomarinol
Amongst other bioactive molecules, members of the genus Pseudoalteromonas have been shown to produce the marinolic acid-dithiopyrrolone hybrid thiomarinol (21) (Fig. 14).107 The hybrid molecule exhibits antimicrobial activity against various Gram positive and -negative bacteria, and has caught attention because of its potency against methicillin-resistant Staphylococcus aureus. Thiomarinol and its derivatives are produced by a combinatorial pathway involving PKS, NRPS and FASs in Pseudoalteromonas sp. SANK73390.108 In general, the PKS moiety is synthesized by a trans-AT modular PKS (TmpA, C, D) where the AT domain is separated from the PKS and requires the collaboration of a FAS (probably TmpB) to produce marinolic acid. The FAS starter units are generated by the class I adenylate forming enzyme TmlQ, the reductase TmlS and the ACP TacpD.109 The NRPS produces holothin, which is incorporated into marinolic acid by the ligase TmlU and the acetyltransferase HolE, yielding thiomarinol A.110 Work by Murphy and colleagues could finally show that the 8-hydroxyoctanoic acid side-chain originates from 4-hydroxybutyrate, which is elongated stepwise by the FAS.111 In addition, 13C and pyrrothine feeding experiments to a ΔNRPS mutant indicated that the pyrrothine moiety is produced by the NRPS and integrated into the assembly line as intact unit.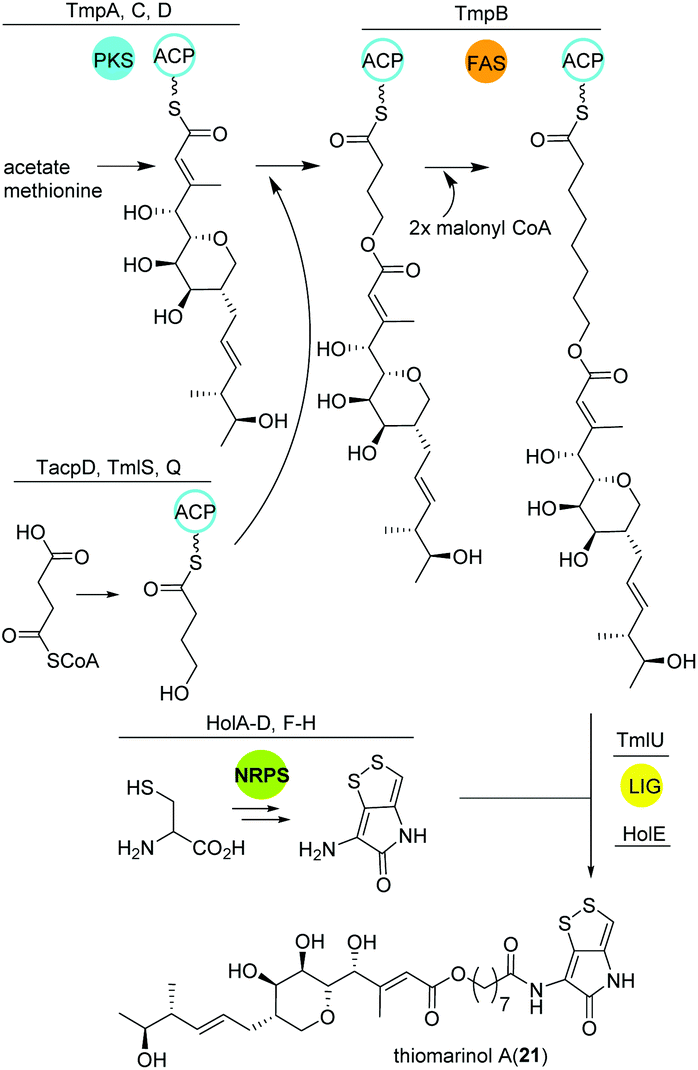 | ||
| Fig. 14 Key biosynthetic steps leading to thiomarinol A (21) and related metabolites in Pseudoalteromonas. | ||
Alterochromides
Marine species of the genus Pseudoalteromonas (γ-proteobacteria) have been identified as a very promising source of biologically important natural products and their genomes were found to encode for many different classes of BGCs. Amongst several other identified natural products, a group of lipopeptides called alterochromides (22, Fig. 15B) were found to be produced by multiple species of Pseudoalteromonas strains. Alterochromides possess unusual chemical structural features, such as a brominated lipid moiety, and show strong antibacterial and cytotoxic activities.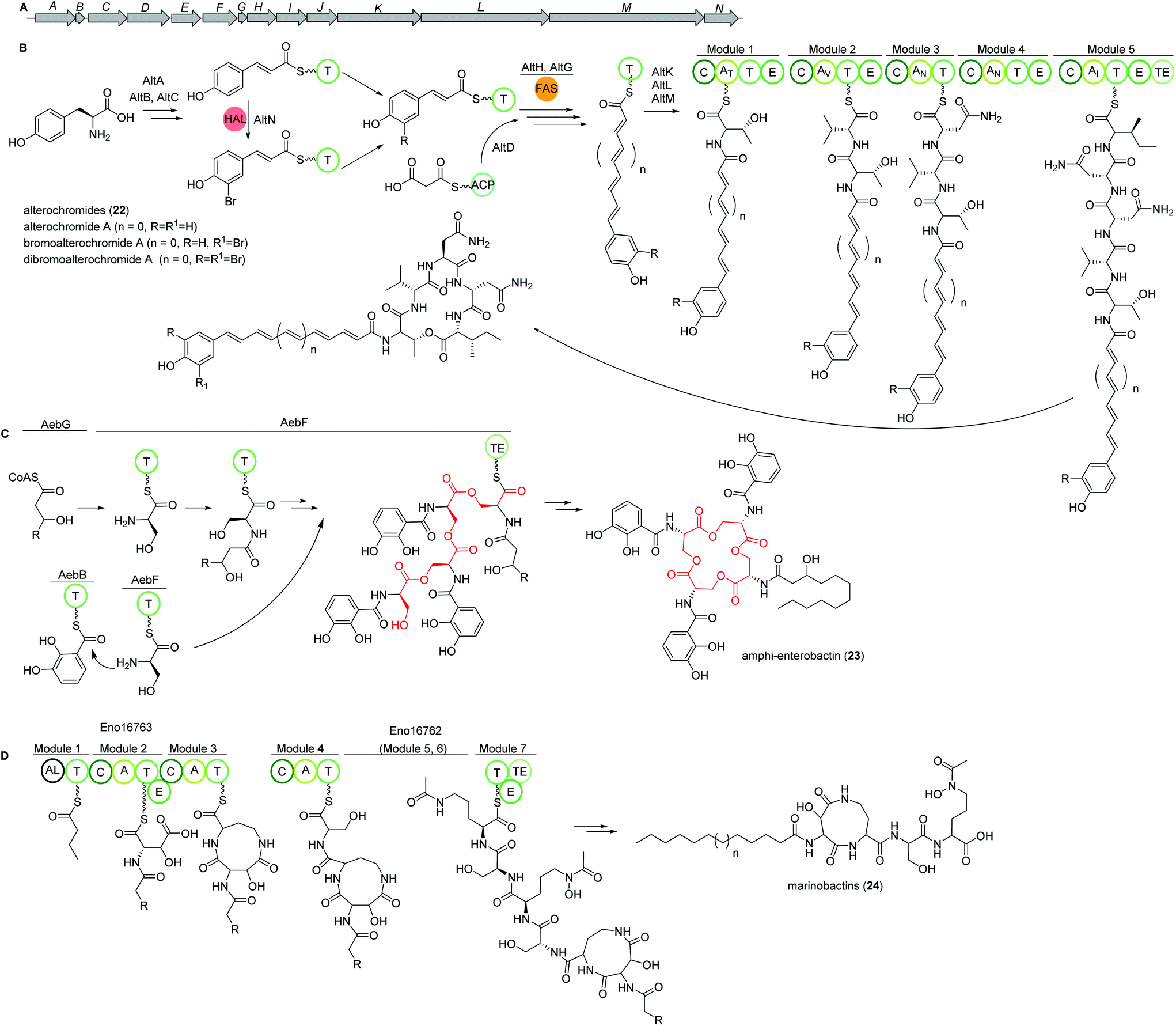 | ||
| Fig. 15 (A) Gene map of the alterochomide natural products in Pseudoalteromonas piscicida JCM 20779. Key biosynthetic steps of (B) alterochromide BGC in Pseudoalteromonas piscicida JCM 20779, (C) amphi-enterobactin siderophore biosynthesis based on the iterative enterobactin “waiting room” model, and (D) marinobactins in Marinobacter nanhaiticus D15-8W (n = 5–7). | ||
Genome mining studies unraveled a putative BGC (alt), which was proposed to be responsible for their biosynthesis. The alt gene cluster consists of a unique combination of NRPS, FAS, flavin-dependent halogenase, and transporter proteins. Presumably, the formation of the alterochromide unsaturated fatty acid chain is catalyzed by the combined action of a tyrosine ammonia lyase (AltA) and halogenase (AltN) on the phenolic residue via the formation of a coumaric acid residue derived from tyrosine. Subsequent elongation by a FAS-like enzyme should generate the unsaturated lipid chain with varying length. Next, five NRPS modules (AltK, L, M) incorporate the necessary amino acids to yield the structure of alterochromides.
Using TAR cloning strategy, the putative 34 kb hybrid NRPS biosynthetic gene cluster (alt) from P. piscicida JCM 20779 was captured in E. coli BL21 (DE3) (Fig. 15A).112 Based on GNPS molecular networking analysis and co-expression of the alt gene cluster and a PPTase from the related strain Pseudoalteromonas luteoviolacea 2ta16 under direct control of an upstream T7 promoter resulted in the detection of a large number of alterochromide molecules.
Amphi-enterobactins
Amphi-enterobactin siderophores (23) are triscatechol macrolactone iron-scavengers containing an N-acyl fatty acid moiety with varying hydroxylation status, chain length and saturation degree (Fig. 15C).114 They are produced by Vibrio harveyi BAA-1116 V, a marine bacterium that has been reclassified as V. campbellii, and has been subject to intense studies on quorum sensing dependent bioluminescence.113The biosynthesis of amphi-enterobactin siderophores (23) was elucidated in 2014 based on the similarity of its gene cluster (aeb) to known enterobactin BGCs, and was found to involve a condensation domain with a unique activity and a long-chain fatty acid CoA ligase (FACL).114 This finding was in accordance with the presence of an FACL encoding gene, aebG, upstream of the six-gene aeb NRPS BGC. The abolished amphi-enterobactin production in a ΔaebG mutant and function of similar enzymes led the authors to propose the activation of the fatty acid as fatty acid CoA thioester by AebG as initial biosynthesis step. Due to its high similarity to the enterobactin EntF NRPS and via mutant analysis, AebF was identified as the main NRPS enzyme of the assembly line (Fig. 15C). Interestingly, in vitro activity assays of the cloned and recombinantly purified AebF revealed two functions in contrast to the single function of EntF: (i) like EntF, AebF catalyzes L-serine-DHBA formation; (ii) in addition, AebF activates L-serine by fatty acid acylation using the fatty acid CoA thioester formed by AebG. Both condensation reactions contribute to amphi-enterobactin biosynthesis since derivatives with two different amide bonds, either L-serine-2,3 dihydroxybenzoic acid (DHBA) or L-serine-fatty, are produced. The rest of the biosynthetic pathway is proposed to be similar to the enterobactin biosynthesis with AebE and AebB catalyzing the formation of DHBA-AMP and functioning as a carrier, respectively.114
Another group was able to show that the amphi-enterobactin BGC is repressed by iron and quorum sensing, i.e. the presence of autoinducers.115 In addition, they revealed the production of the soluble siderophores dihydroxybenzoyl-serine and DHBA produced by the same gene cluster by release of biosynthetic intermediates or degradation of the amphi-enterobactins. Although they were present in a 100× higher concentration than amphi-enterobactins, the latter probably represent the main contributors to iron uptake in planktonic conditions as the soluble siderophores diffuse away. However, under e.g. biofilm conditions, V. harveiy could use dihydroxybenzoyl-serine and DHBA since their concentration in the cell's vicinity increases and the amphi-enterobactins could act as shuttles on the cell surface.115
Marinobactins
Other siderophores produced by marine bacteria, namely some Marinobacter species, are the amphiphilic marinobactins (24) which are composed of a headgroup of six amino acids and an attached fatty acid tail of varying length (Fig. 15D).116–118 In Marinobacter nanhaiticus D15-8W, two putative NRPS genes ENO16763 and ENO16762 are proposed to govern the biosynthesis of marinobactins (Fig. 15D).117 The biosynthesis pathway is predicted to follow the linear NRPS logic: the gene product of ENO16763 as putative acyl-CoA ligase initiates the pathway by activation of a fatty acid (module 1) and incorporation of the starter unit L-aspartic acid which is epimerized by ENO16763 module 2. The A domain of module 3 is predicted to incorporate L-diaminobutyric acid, but the domain/enzyme catalyzing the cyclization reaction is still elusive. The biosynthesis continues with incorporation of L-serine, which is epimerized to its D form, incorporation of L-N5-hydroxyornithine, again L-serine and L-N5-hydroxyornithine, which is also epimerized. These reactions are catalyzed by ENO16762 gene product.117 Similar to other siderophores, like pyoverdine from Pseudomonas fluorescens, the fatty acid can be cleaved off by an acylase, yielding a more hydrophilic molecule (marinobactin head group, MHG). By search for similar genes to the P. fluorescens acylase gene pvdQ, bntA was found in Marinobacter sp. DS40M6 and the lack of MHG production in a ΔbntA mutant indicated its function in marinobactin acylation. In Marinobacter nanhaiticus D15-8W, the two PvdQ-like enzymes MhtA and MhtB were identified and the activity of recombinant MhtA in MHG production was verified in vitro. In addition, MhtA was able to hydrolyze acyl homoserine lactones, which act as quorum sensing molecules, indicating a putative role as quorum quenchers.116,117Polybrominated diphenyl ethers (PBDEs) and polybrominated bipyrroles
PBDEs are ubiquitous in all trophic level of marine life and are likely to be toxic to humans at higher concentrations. Many derivatives are known to be produced by Pseudoalteromonas strains often associated with eukaryotic marine hosts. Interestingly, one of their building blocks, tetrabromopyrrole, is known as an inducer for coral larval settlement and metamorphosis which is also produced by Pseudoalteromonas strains.119Biosynthesis of polybrominated diphenyl ethers (PBDEs) and polybrominated bipyrroles was recently described by Moore and coworkers.120 Using genome mining approach, a conserved gene cluster (bmp) was detected from marine Pseudoalteromonas strains consist of ten genes bmp1 to bmp10 (Fig. 16). Heterologous expression of the bmp gene cluster revealed that bromopyrroles are derived from L-proline which initiates by activation (bmp4) and thiolation reactions (bmp1) following the oxidation by the flavin-dependent dehydrogenase (bmp3) and tribromination by the flavin-dependent halogenase (bmp2). After offloading the tribromopyrrole (25A) from the ACP domain, cytochrome P450 (bmp7) dimerizes two molecules to hexabromo-2,2′-bipyrrole (25C). Bromophenoles are produced by conversion of chorismate to 4-HBA using a chorismate lyase (bmp6) following bromination by a flavin-dependent halogenase (bmp5) yielding 2,4-dibromophenol (25D). Next, cytochrome P450 (bmp7) couples the bromophenol and bromopyrrole monomers and generates a suite of diverse homo/heterodimeric molecules (including 25E, 25F, and 25G).
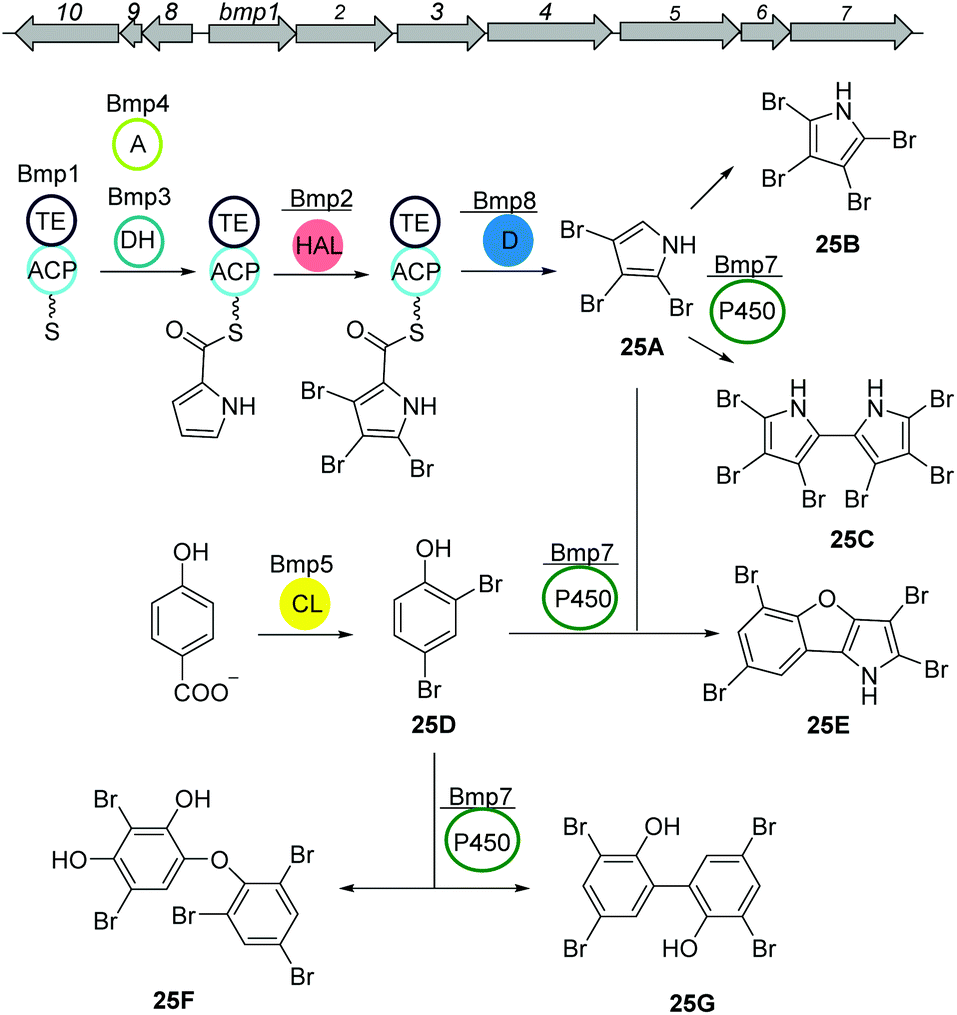 | ||
| Fig. 16 Biosynthetic pathway of polybrominated diphenyl ethers (PBDEs) and polybrominated bipyrroles from Pseudoalteromonas luteoviolacea. | ||
7. Alkaloids
Alkaloids are a group of natural products containing one or more basic nitrogen atoms and are produced by a large variety of organisms including many of marine origin.121 Alkaloids are most often biosynthesized from amino acids such as tryptophan or aspartic acid. Here, we will discuss the group of 3-(2-aminophenyl)pyrroles (APP) isolated from Rapidithrix thailandica122 and N-methylated marinoquinolines detected using molecular networking.123Aminophenylpyrrole-derived alkaloids
Aminophenylpyrrole-derived alkaloids (APPAs) are highly important compounds such as widely used fungicide pyrrolnitrin produced by several proteobacterial strains such as Pseudomonas pyrrocinia.124 APP was reported as a common APPA precursor candidate and a dead-end product in pyrrolnitrin biosynthesis which is catalyzed from tryptophan by a PrnB-like enzyme.125 Using bioinformatics and phylogenetic analysis of Rapidithrix thailandica, the biosynthetic gene cluster of APPAs was identified (Fig. 17) and heterologously verified in E. coli. Tryptophan undergoes an oxidation step by the indolamine 2,3-dioxygenase enzyme MarC (homologous to PrnB) yielding APP. Next, a spontaneous nonenzymatic Pictet–Spengler-like coupling reaction creates a library of tricyclic alkaloid marinoquinolines (26). Furthermore, a SAM-dependent methyltransferase (MarB) catalyzes the methylation reaction before the nonenzymatic reaction to create N-methylated marinoquinolines (27).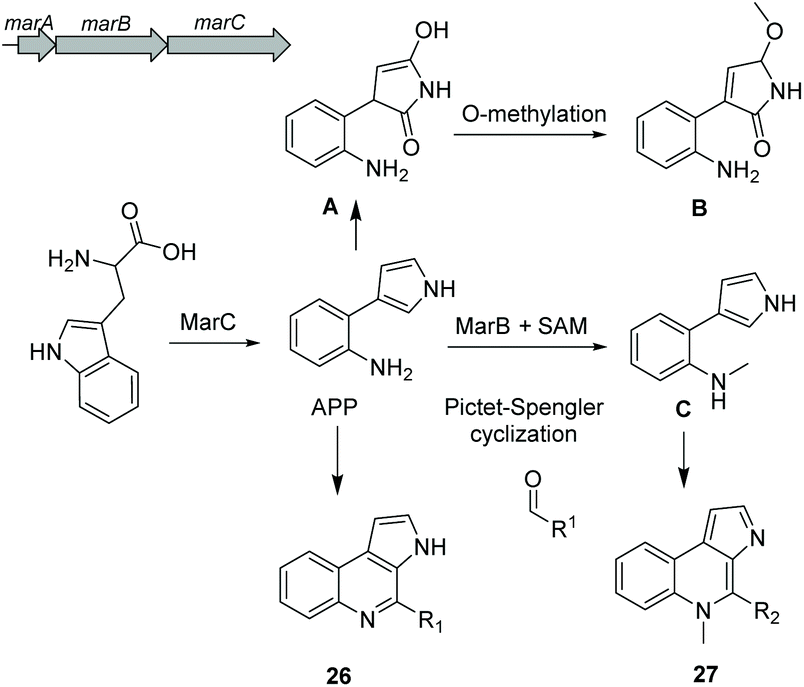 | ||
| Fig. 17 Biosynthesis of alkaloid marinoquinolines. | ||
8. Conclusions
Marine organisms such as bacteria, fungi, sponges and algae represent an enormous and still mostly unexplored source of novel natural products and biosynthetic traits. In addition to the structural identification of novel metabolites, the elucidating of their biosynthesis is a key prerequisite step for understanding and ultimately exploiting the biosynthetic pathway. In particular advances in genomics, molecular biology and directed evolution provide us with a unique opportunity to engineer and optimize these pathways resulting often in new bioactive small metabolites with altered bioactvities.30 Despite the current wealth of chemical knowledge and biotechnological methodologies, examples of fully chemically and biosynthetically described examples from the marine environment remain comparably low and much has to be done to explore the given diversity more in detail.Abbreviations
| KS | β-Keto acyl synthase |
| AT | Acyl transferase |
| ACP | Acyl carrier protein |
| mMCoA | Methyl malonyl CoA |
| MT | Methyltransferase |
| Ox | Oxidoreductase |
| T | Thiolation domain |
| C | Condensation domain |
| A | Adenylation domain |
| E | Epimerase domain |
| TE | Thioesterase domain |
| NRPS | Nonribosomal peptide-synthetase, ribosomally synthesized and post-translationally modified peptides, RIPPs. |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
JAM and CB have received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (Project: 802736 MORPHEUS). TJ and CB greatly acknowledge funding provided by the Boehringer Ingelheim Stiftung (Exploration Grant Engina).References
- E. DeLong and D. Karl, Nature, 2005, 437, 336–342 CrossRef CAS.
- D. M. Karl, Nat. Rev. Microbiol., 2007, 5, 759–769 CrossRef CAS.
- K. Venkateskumar, S. Parasuraman, L. Yu Chuen, V. Ravichandran and S. Balamurgan, Curr. Drug Discovery Technol., 2020, 17, 507–514 CrossRef CAS.
- A. Poli, I. Finore, I. Romano, A. Gioiello, L. Lama and B. Nicolaus, Microorganisms, 2017, 5, 25 CrossRef.
- G.-P. Hu, J. Yuan, L. Sun, Z.-G. She, J.-H. Wu, X.-J. Lan, X. Zhu, Y.-C. Lin and S.-P. Chen, Mar. Drugs, 2011, 9, 514–525 CrossRef.
- T. Wichard and C. Beemelmanns, J. Chem. Ecol., 2018, 44, 1–14 CrossRef.
- M. Rust, E. J. N. Helfrich, M. F. Freeman, P. Nanudorn, C. M. Field, C. Rückert, T. Kündig, M. J. Page, V. L. Webb, J. Kalinowski, S. Sunagawa and J. Piel, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 9508–9518 CrossRef CAS.
- L. L. Zinger, L. A. Amaral-Zettler, J. A. Fuhrman, M. C. Horner-Devine, S. M. Huse, D. B. M. Welch, J. B. H. Martiny, M. Sogin, A. Boetius and A. Ramette, PLoS One, 2011, 6, e24570 CrossRef CAS.
- P. R. Jensen and W. Fenical, Annu. Rev. Microbiol., 1994, 48, 559–584 CrossRef CAS.
- S. Sunagawa, L. P. Coelho, S. Chaffron, J. R. Kultima, K. Labadie, G. Salazar, B. Djahanschiri and G. Zeller, et al. , Science, 2015, 348, 6237 CrossRef.
- P. Yilmaz, P. Yarza, J. Z. Rapp and F. O. Glöckner, Front. Microbiol., 2016, 6, 1524 Search PubMed.
- R. Subramani and D. Sipkema, Mar. Drugs, 2019, 17, 249 CrossRef CAS.
- D. Giordano, D. Coppola, R. Russo, R. Denaro, L. Giuliano, F. M. Lauro, G. Di Prisco and C. Verde, Adv. Microb. Physiol., 2015, 66, 357–428 CrossRef CAS.
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2016, 33, 382–431 RSC.
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2017, 34, 235–294 RSC.
- J. C. Venter, K. Remington, J. F. Heidelberg, A. L. Halpern, D. Rusch, J. A. Eisen, D. Wu, I. Paulsen, K. E. Nelson, W. Nelson, D. E. Fouts, S. Levy, A. H. Knap, M. W. Lomas, K. Nealson, O. White, J. Peterson, J. Hoffman, R. Parsons, H. Baden-Tillson, C. Pfannkoch, Y.-H. Rogers and H. O. Smith, Science, 2004, 304, 66–74 CrossRef CAS.
- D. A. Dias, S. Urban and U. Roessner, Metabolites, 2012, 2, 303–336 CrossRef CAS.
- M. H. Medema, mSystems, 2018, 3, e00182–e00117 CrossRef CAS.
- T. A. Scott and J. Piel, Nat. Rev. Chem., 2019, 3, 404–425 CrossRef.
- P. Cimermancic, M. H. Medema, J. Claesen, K. Kurita, L. C. Wieland Brown, K. Mavrommatis, A. Pati, P. A. Godfrey, M. Koehrsen, J. Clardy, B. W. Birren, E. Takano, A. Sali, R. G. Linington and M. A. Fischbach, Cell, 2014, 158, 412–421 CrossRef CAS.
- H. Wang, D. P. Fewer, L. Holm, L. Rouhiainen and K. Sivonen, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 9259–9264 CrossRef CAS.
- K. Blin, M. H. Medema, R. Kottmann, S. Y. Lee and T. Weber, Nucleic Acids Res., 2017, 45, D555–D559 CrossRef CAS.
- M. Alanjary, B. Kronmiller, M. Adamek, K. Blin, T. Weber, D. Huson, B. Philmus and N. Ziemert, Nucleic Acids Res., 2017, 45, W42–W48 CrossRef CAS.
- M. A. Schorn, M. M. Alanjary, K. Aguinaldo, A. Korobeynikov, S. Podell, N. Patin, T. Lincecum, P. R. Jensen, N. Ziemert and B. S. Moore, Microbiology, 2016, 162, 2075–2086 CrossRef CAS.
- J. Piel and J. Cahn, Angew. Chem., Int. Ed., 2019 DOI:10.1002/anie.201900532.
- M. Wang, J. J. Carver, V. V. Phelan, L. M. Sanchez, N. Garg, Y. Peng and D. Duy Nguyen, et al. , Nat. Biotechnol., 2016, 34, 828 CrossRef CAS.
- J. Y. Yang, L. M. Sanchez, C. M. Rath, X. Liu, P. D. Boudreau, N. Bruns, E. Glukhov, A. Wodtke, R. de Felicio, A. Fenner, W. R. Wong, R. G. Linington, L. Zhang, H. M. Debonsi, W. H. Gerwick and P. C. Dorrestein, J. Nat. Prod., 2013, 76, 1686–1699 CrossRef CAS.
- M. Maansson, N. G. Vynne, A. Klitgaard, J. L. Nybo, J. Melchiorsen, D. D. Nguyen, L. M. Sanchez, N. Ziemert, P. C. Dorrestein, M. R. Andersen and L. Gram, mSystems, 2020, e00028–e00015 Search PubMed.
- L. M. Naughton, S. Romano, F. O'Gara and A. D. W. Dobson, Front. Microbiol., 2017, 8, 1494 CrossRef.
- M. Alanjary, C. Cano-Prieto, H. Gross and M. H. Medema, Nat. Prod. Rep., 2019, 36, 1249–1261 RSC.
- J. B. McClintock, C. D. Amsler and B. J. Baker, Integr. Comp. Biol., 2010, 50, 967–980 CrossRef.
- D. Skropeta and L. Wei, Nat. Prod. Rep., 2014, 31, 999–1025 RSC.
- J. W. Blunt, A. R. Carroll, B. R. Copp, R. A. Davis, R. A. Keyzers and M. R. Prinsep, Nat. Prod. Rep., 2018, 35, 8–53 RSC.
- K. Gemperlein, N. Zaburannyi, R. Garcia, J. La Clair and R. Müller, Mar. Drugs, 2018, 16, 314 CrossRef.
- Y. Buijs, P. K. Bech, D. Vazquez-Albacete, M. Bentzon-Tilia, E. C. Sonnenschein, L. Gram and S.-D. Zhang, Nat. Prod. Rep., 2020, 1333–1350 Search PubMed.
- N. M. Carballeira, Prog. Lipid Res., 2008, 47, 50–61 CrossRef CAS.
- E. Schweizer and J. Hofmann, Microbiol. Mol. Biol. Rev., 2004, 68, 501–517 CrossRef CAS.
- J. E. Cronan and J. Thomas, Methods Enzymol., 2009, 459, 395–433 CAS.
- J. C. Navarro-Muñoz, N. Selem-Mojica, M. W. Mullowney, S. A. Kautsar, J. H. Tryon, E. I. Parkinson, E. L. C. De Los Santos, M. Yeong, P. Cruz-Morales, S. Abubucker, A. Roeters, W. Lokhorst, A. Fernandez-Guerra, L. T. D. Cappelini, A. W. Goering, R. J. Thomson, W. W. Metcalf, N. L. Kelleher, F. Barona-Gomez and M. H. Medema, Nat. Chem. Biol., 2019, 16, 60–68 CrossRef.
- T. M. H. To, C. Grandvalet and R. Tourdot-Maréchal, Appl. Environ. Microbiol., 2011, 77, 3327–3334 CrossRef CAS.
- H. Velly, M. Bouix, S. Passot, C. Penicaud, H. Beinsteiner, S. Ghorbal, P. Lieben and F. Fonseca, Appl. Microbiol. Biotechnol., 2015, 99, 907–918 CrossRef CAS.
- Antifungal free fatty acids: A review. Science Against Microbial Pathogens: Communicating Current Research and Technological Advances, ed. C. H. Pohl, J. L. F. Kock and V. S. Thibane, Badajoz, Spain, 2011 Search PubMed.
- N. M. Carballeira, Prog. Lipid Res., 2008, 47, 50–61 CrossRef CAS.
- J. Amiri Moghaddam, A. Dávila-Céspedes, M. Alanjary, J. Blom, G. König and T. Schäberle, Data, 2019, 4, 33 CrossRef.
- J. Amiri Moghaddam, A. Dávila-Céspedes, S. Kehraus, M. Crüsemann, M. Köse, C. Müller and G. M. König, Mar. Drugs, 2018, 16, 369 CrossRef.
- E. Guangqi, D. Lesage and O. Ploux, Biochimie, 2010, 92, 1454–1457 CrossRef CAS.
- C. J. Thibodeaux, W.-C. Chang and H.-W. Liu, Chem. Rev., 2012, 112, 1681–1709 CrossRef CAS.
- D. E. Cane and H. Ikeda, Acc. Chem. Res., 2012, 45, 463–472 CrossRef CAS.
- E. J. N. Helfrich, G.-M. Lin, C. A. Voigt and J. Clardy, Beilstein J. Org. Chem., 2019, 15, 2889–2906 CrossRef CAS.
- J. D. Rudolf and C. Y. Chang, Nat. Prod. Rep., 2020, 37, 425–463 RSC.
- M. Baunach, J. Franke and C. Hertweck, Angew. Chem., Int. Ed., 2015, 54, 2604–2626 CrossRef CAS.
- J. S. Dickschat, Nat. Prod. Rep., 2016, 33, 87–110 RSC.
- J. S. Dickschat, Angew. Chem., Int. Ed., 2019, 45, 15964–15976 CrossRef.
- J. Pérez-Gil and M. Rodríguez-Concepción, Biochem. J., 2013, 452, 19–25 CrossRef.
- J. Rinkel, L. Lauterbach and J. S. Dickschat, Angew. Chem., Int. Ed., 2017, 56, 16385–16389 CrossRef CAS.
- M. Xu, M. L. Hillwig, A. L. Lane, M. S. Tiernan, B. S. Moore and R. J. Peters, J. Nat. Prod., 2014, 77, 2144–2147 CrossRef CAS.
- J. Rinkel and J. S. Dickschat, Org. Lett., 2019, 21, 9442–9445 CrossRef CAS.
- P. F. Cook, C. Reichmuth, A. A. Rouse, L. A. Libby, S. E. Dennison, O. T. Carmichael, K. T. Kruse-Elliott, J. Bloom, B. Singh, V. A. Fravel, L. Barbosa, J. J. Stuppino, W. G. Van Bonn, F. M. D. Gulland and C. Ranganath, Science, 2015, 350, 1545–1547 CrossRef CAS.
- J. K. Brunson, S. M. K. McKinnie, J. R. Chekan, J. P. McCrow, Z. D. Miles, E. M. Bertrand, V. A. Bielinski, H. Luhavaya, M. Oborník, G. J. Smith, D. A. Hutchins, A. E. Allen and B. S. Moore, Science, 2018, 361, 1356–1358 CrossRef CAS.
- Y. Maeno, Y. Kotaki, R. Terada, Y. Cho, K. Konoki and M. Yotsu-Yamashita, Sci. Rep., 2018, 8, 356 CrossRef.
- Y. A. Chan, A. M. Podevels, B. M. Kevany and M. G. Thomas, Nat. Prod. Rep., 2009, 26, 90–114 RSC.
- J. A. Robinson, Philos. Trans. R. Soc., B, 1991, 332, 107–114 CrossRef CAS.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS.
- J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380–416 RSC.
- L. Ray and B. S. Moore, Nat. Prod. Rep., 2016, 33, 150–161 RSC.
- Z. Lin, J. P. Torres, M. A. Ammon, L. Marett, R. W. Teichert, C. A. Reilly, J. C. Kwan, R. W. Hughen, M. Flores, M. D. Tianero, O. Peraud, J. E. Cox, A. R. Light, A. J. L. Villaraza, M. G. Haygood, G. P. Concepcion, B. M. Olivera and E. W. Schmidt, Chem. Biol., 2013, 20, 73–81 CrossRef CAS.
- K. H. Jang, S.-J. Nam, J. B. Locke, C. A. Kauffman, D. S. Beatty, L. A. Paul and W. Fenical, Angew. Chem., Int. Ed., 2013, 52, 7822–7824 CrossRef CAS.
- S. Alt and B. Wilkinson, ACS Chem. Biol., 2015, 10, 2468–2479 CrossRef CAS.
- K. Jungmann, R. Jansen, K. Gerth, V. Huch, D. Krug, W. Fenical and R. Müller, ACS Chem. Biol., 2015, 10, 2480–2490 CrossRef CAS.
- C. T. Calderone, W. E. Kowtoniuk, N. L. Kelleher, C. T. Walsh and P. C. Dorrestein, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8977–8982 CrossRef CAS.
- T. Bretschneider, J. B. Heim, D. Heine, R. Winkler, B. Busch, B. Kusebauch, T. Stehle, G. Zocher and C. Hertweck, Nature, 2013, 502, 124–128 CrossRef CAS.
- K. Jungmann, R. Jansen, K. Gerth, V. Huch, D. Krug, W. Fenical and R. Müller, ACS Chem. Biol., 2015, 10, 2480–2490 CrossRef CAS.
- A. Bhushan, E. E. Peters and J. Piel, in Blue Biotechnology: From Gene to Bioactive Product, ed. W. E. G. Müller, H. C. Schröder and X. Wang, Springer International Publishing, Cham, 2017, pp. 291–314 Search PubMed.
- R. Ueoka, A. R. Uria, S. Reiter, T. Mori, P. Karbaum, E. E. Peters, E. J. N. Helfrich, B. I. Morinaka, M. Gugger, H. Takeyama, S. Matsunaga and J. Piel, Nat. Chem. Biol., 2015, 11, 705–712 CrossRef CAS.
- R. Uria, J. Piel and T. Wakimoto, in Methods in Enzymology, ed. B. S. Moore, Elsevier Inc., 1st edn, 2018, vol. 604, pp. 287–330 Search PubMed.
- M. A. Martínez-Núñez and V. E. López y López, Sustainable Chem. Processes, 2016, 4, 13 CrossRef.
- D. L. Niquille, D. A. Hansen, T. Mori, D. Fercher, H. Kries and D. Hilvert, Nat. Chem., 2018, 10, 282–287 CrossRef CAS.
- J. Grünewald and M. A. Marahiel, in Handbook of biologically active peptides, ed. A. J. Kastin, AP, Amsterdam, 2013, pp. 138–149 Search PubMed.
- A. Miyanaga, F. Kudo and T. Eguchi, Nat. Prod. Rep., 2018, 35, 1185–1209 RSC.
- F. El Maddah, S. Kehraus, M. Nazir, C. Almeida and G. M. König, J. Nat. Prod., 2016, 79, 2838–2845 CrossRef CAS.
- C. Almeida, F. El Maddah, S. Kehraus, G. Schnakenburg and G. M. König, Org. Lett., 2016, 18, 528–531 CrossRef CAS.
- L. P. Partida-Martinez, C. F. de Looß, K. Ishida, M. Ishida, M. Roth, K. Buder and C. Hertweck, Appl. Environ. Microbiol., 2007, 73, 793–797 CrossRef CAS.
- W.-S. Xiang, J.-D. Wang, X.-J. Wang and J. Zhang, J. Antibiot., 2009, 62, 501–505 CrossRef CAS.
- K. Takada, A. Ninomiya, M. Naruse, Y. Sun, M. Miyazaki, Y. Nogi, S. Okada and S. Matsunaga, J. Org. Chem., 2013, 78, 6746–6750 CrossRef CAS.
- K. Matsuda, M. Kobayashi, T. Kuranaga, K. Takada, H. Ikeda, S. Matsunaga and T. Wakimoto, Org. Biomol. Chem., 2019, 17, 1058–1061 RSC.
- A. Ninomiya, Y. Katsuyama, T. Kuranaga, M. Miyazaki, Y. Nogi, S. Okada, T. Wakimoto, Y. Ohnishi, S. Matsunaga and K. Takada, ChemBioChem, 2016, 17, 1709–1712 CrossRef CAS.
- T. Kuranaga, K. Matsuda, A. Sano, M. Kobayashi, A. Ninomiya, K. Takada, S. Matsunaga and T. Wakimoto, Angew. Chem., Int. Ed., 2018, 57, 9447–9451 CrossRef CAS.
- K. Matsuda, M. Kobayashi, T. Kuranaga, K. Takada, H. Ikeda, S. Matsunaga and T. Wakimoto, Org. Biomol. Chem., 2019, 17, 1058–1061 RSC.
- X. Zhou, H. Huang, J. Li, Y. Song, R. Jiang, J. Liu, S. Zhang, Y. Hua and J. Ju, Tetrahedron, 2014, 70, 7795–7801 CrossRef CAS.
- Y. Zhou, X. Lin, C. Xu, Y. Shen, S. P. Wang, H. Liao, L. Li, H. Deng and H. W. Lin, Cell Chem. Biol., 2019, 26, 737–744 CrossRef CAS.
- J. Liu, B. Wang, H. Li, Y. Xie, Q. Li, X. Qin, X. Zhang and J. Ju, Org. Lett., 2015, 17, 1509–1512 CrossRef CAS.
- X. Zhou, H. Huang, J. Li, Y. Song, R. Jiang, J. Liu, S. Zhang, Y. Hua and J. Ju, Tetrahedron, 2014, 70, 7795–7801 CrossRef CAS.
- Q. Li, X. Qin, J. Liu, C. Gui, B. Wang, J. Li and J. Ju, J. Am. Chem. Soc., 2016, 138, 408–415 CrossRef CAS.
- F. Romero, F. Espliego, J. P. Baz, T. G. De Quesada, D. Gravalos, F. De Calle and J. L. Fernandez-puentes, J. Antibiot., 1997, 50, 734–737 CrossRef CAS.
- J. PérezBaz, L. M. Canedo and J. L. F. Puentes, J. Antibiot., 1997, 50, 738–741 CrossRef.
- O. E. Zolova and S. Garneau-Tsodikova, Med. Chem. Commun., 2012, 3, 950–955 RSC.
- S. Mori, S. K. Shrestha, J. Fernández, M. Álvarez San Millán, A. Garzan, A. H. Al-Mestarihi, F. Lombó and S. Garneau-Tsodikova, Biochemistry, 2017, 56, 4457–4467 CrossRef CAS.
- J. Fernández, L. Marín, R. Álvarez-Alonso, S. Redondo, J. Carvajal, G. Villamizar, C. J. Villar and F. Lombó, Mar. Drugs, 2014, 12, 2668–2699 CrossRef.
- A. H. Al-Mestarihi, G. Villamizar, J. Fernández, O. E. Zolova, F. Lombó and S. Garneau-Tsodikova, J. Am. Chem. Soc., 2014, 136, 17350–17354 CrossRef CAS.
- Y. Sun, T. Tomura, J. Sato, T. Iizuka, R. Fudoi and M. Ojika, Molecules, 2016, 21, 59 CrossRef.
- H. Oishi, T. Noto, H. Sasaki, K. Suzuki, T. Hayashi, H. Okazaki, K. Ando and M. Sawada, J. Antibiot., 1980, 35, 391–395 CrossRef.
- M. S. Brown, K. Akopiants, D. M. Resceck, H. A. I. McArthur, E. McCormick and K. A. Reynolds, J. Am. Chem. Soc., 2003, 125, 10166 CrossRef CAS.
- W. Tao, M. E. Yurkovich, S. Wen, K. E. Lebe, M. Samborskyy, Y. Liu, A. Yang, Y. Liu, Y. Ju, Z. Deng, M. Tosin, Y. Sun and P. F. Leadlay, Chem. Sci., 2016, 7, 376–385 RSC.
- X. Tang, J. Li, N. Millán-Aguiñaga, J. J. Zhang, E. C. O'Neill, J. A. Ugalde, P. R. Jensen, S. M. Mantovani and B. S. Moore, ACS Chem. Biol., 2015, 10, 2841–2849 CrossRef CAS.
- M. L. Rothe, J. Li, E. Garibay, B. S. Moore and S. M. K. McKinnie, Org. Biomol. Chem., 2019, 17, 3416–3423 RSC.
- X. Tang, J. Li and B. S. Moore, ChemBioChem, 2017, 18, 1072–1076 CrossRef CAS.
- H. Shiozawa, T. Kagasaki, T. Kinoshita, H. Haruyama, H. Domon, Y. Utsui, K. Kodama and S. Takahashi, J. Antibiot., 1993, 46, 1834–1842 CrossRef CAS.
- A. C. Murphy, S. S. Gao, L. C. Han, S. Carobene, D. Fukuda, Z. Song, J. Hothersall, R. J. Cox, J. Crosby, M. P. Crump, C. M. Thomas, C. L. Willis and T. J. Simpson, Chem. Sci., 2014, 5, 397–402 RSC.
- P. D. Walker, M. T. Rowe, A. J. Winter, A. N. M. Weir, N. Akter, L. Wang, P. R. Race, C. Williams, Z. Song, T. J. Simpson, C. L. Willis and M. P. Crump, ACS Chem. Biol., 2020, 15, 494–503 CrossRef CAS.
- Z. D. Dunn, W. J. Wever, N. J. Economou, A. A. Bowers and B. Li, Angew. Chem., Int. Ed., 2015, 54, 5137–5141 CrossRef CAS.
- A. C. Murphy, S.-S. Gao, L.-C. Han, S. Carobene, D. Fukuda, Z. Song, J. Hothersall, R. J. Cox, J. Crosby, M. P. Crump, C. M. Thomas, C. L. Willis and T. J. Simpson, Chem. Sci., 2014, 5, 397–402 RSC.
- A. C. Ross, L. E. S. Gulland, P. C. Dorrestein and B. S. Moore, ACS Synth. Biol., 2015, 4, 414–420 CrossRef CAS.
- A. S. Ball, R. R. Chaparian and J. C. van Kessel, J. Bacteriol., 2017, 199, e00105–e00117 CrossRef CAS.
- H. K. Zane, H. Naka, F. Rosconi, M. Sandy, M. G. Haygood and A. Butler, J. Am. Chem. Soc., 2014, 136, 5615–5618 CrossRef CAS.
- D. L. McRose, O. Baars, M. R. Seyedsayamdost and F. M. M. Morel, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 7581–7586 CrossRef CAS.
- J. S. Martinez and A. Butler, J. Inorg. Biochem., 2007, 101, 1692–1698 CrossRef CAS.
- M. P. Kem and A. Butler, BioMetals, 2015, 28, 445–459 CrossRef CAS.
- M. P. Kem, H. Naka, A. Iinishi, M. G. Haygood and A. Butler, Biochemistry, 2015, 54, 744–752 CrossRef CAS.
- M. J. H. van Oppen and L. L. Blackall, Nat. Rev. Microbiol., 2019, 17, 557–567 CrossRef CAS.
- V. Agarwal, A. A. El Gamal, K. Yamanaka, D. Poth, R. D. Kersten, M. Schorn, E. E. Allen and B. S. Moore, Nat. Chem. Biol., 2014, 10, 640–647 CrossRef CAS.
- Modern Alkaloids, ed. E. Fattorusso and O. Taglialatela-Scafati, Wiley, 2007 Search PubMed.
- A. Kanjana-opas, S. Panphon, H.-K. Fun and S. Chantrapromma, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o2728–o2730 CrossRef CAS.
- L. Linares-Otoya, Y. Liu, V. Linares-Otoya, L. Armas-Mantilla, M. Crüsemann, M. L. Ganoza-Yupanqui, J. Campos-Florian, G. M. König and T. F. Schäberle, ACS Chem. Biol., 2019, 14, 176–181 CrossRef CAS.
- K. H. van Pée and J. M. Ligon, Nat. Prod. Rep., 2000, 17, 157–164 RSC.
- A. R. Carroll, S. Duffy and V. M. Avery, J. Org. Chem., 2010, 75, 8291–8294 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The similarity network file is available at NDEx36 (https://doi.org/10.18119/N9V31Z). See DOI: 10.1039/d0ob01677b |
| This journal is © The Royal Society of Chemistry 2021 |