DOI:
10.1039/D0OB01931C
(Review Article)
Org. Biomol. Chem., 2021,
19, 101-122
Synthesis and derivatization of hetera-buckybowls†
Received
19th September 2020
, Accepted 28th October 2020
First published on 30th October 2020
Abstract
Buckybowls have concave and convex surfaces with distinct π-electron cloud distribution, and consequently they show unique structural and electronic features as compared to planar aromatic polycycles. Doping the π-framework of buckybowls with heteroatoms is an efficient scheme to tailor inherent properties, because the nature of heteroatoms plays a pivotal role in the structural and electronic characteristics of the resulting hetera-buckybowls. The design, synthesis, and derivatization of hetera-buckybowls open an avenue for obtaining fascinating organic entities not only of fundamental importance but also of promising applications in optoelectronics. In this review, we summarize the advances in hetera-buckybowl chemistry, particularly the synthetic strategies toward these scaffolds.
 Wenbo Wang | Wenbo Wang was born in Shanxi Province, China. He received his B.S. and M.S. degrees in 2014 and 2017, respectively, from Hunan Institute of Technology and Henan University. He subsequently enrolled as a Ph.D. student in the group of Prof. Xiangfeng Shao at Lanzhou University working on the design and synthesis of buckybowls and nonplanar π-aromatic compounds. |
 Xiangfeng Shao | Prof. Xiangfeng Shao was born in Gansu Province, China. He received his B.S. and M.S. degrees from Lanzhou University in 1998 and 2001, respectively. He then joined Professor Daoben Zhu's group at ICCAS and received his Ph.D. degree in 2004. Thereafter, he worked as a post-doc in JST, Kyoto University, and Osaka Prefecture University until 2009. In October 2009, he became a Professor of Chemistry at Lanzhou University. His current research areas include synthesis, chemical reactivity, and optoelectronic properties of non-conventional π-systems. He received the Excellent Youth Fund from NSFC in 2015 and was appointed as vice-director of SKLAOC in 2018. |
1. Introduction
Inspired by the discovery by Kallmann and Pope that organic materials could be semiconducting upon injecting charge carriers,1 organic electronics has become a fast-growing research field and the requirement of organic compounds with optoelectronic properties adapted to various purposes has been largely increasing since then. Consequently, the synergetic interplay between organic chemistry and materials science has become essential. Taking advantage of the structural tunability of organic compounds, scientists are able to control specific properties of compounds to develop specialized materials.2
The last few decades have witnessed π-conjugated molecules showing multidisciplinary applications, for instance, in optoelectronics,3 porous materials,4 molecular sensing,5 and supramolecular chemistry.6 The most fundamental yet promising class of such compounds are polycyclic aromatic hydrocarbons (PAHs)7 which have been widely employed to fabricate organic light-emitting diodes (OLEDs),8 organic field-effect transistors (OFETs),9 organic photovoltaics (OPVs),10 and spintronics.11 It has been illustrated that the modifications in the size,12 shape,13 and periphery14 of PAHs present an opportunity to control their optoelectronic properties and assembly features in the solid state.6,15 Another promising strategy to tune the inherent properties of PAHs is embedding of main-group elements onto their π-framework, where the nature of the heteroatom plays a pivotal role in their structural and electronic characteristics.16
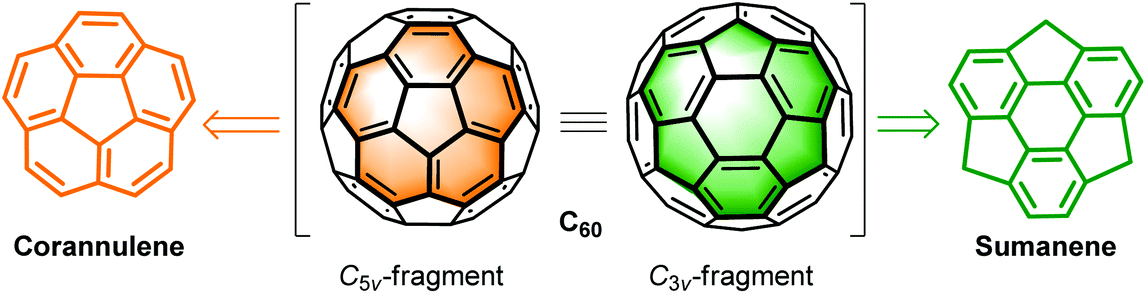
Most of the studies focus on planar PAHs, whereas investigations on non-planar PAHs are limited owing to the challenges in their synthesis. Thanks to the great progress in synthetic methodology in recent decades, scientists are able to synthesize non-planar PAHs with structural diversity such as bowl-shaped molecules (also denoted as buckybowls or π-bowls),13i,j,17 carbon nanorings,18 carbon nanobelts,19 carbon nanotubes,20 saddles,21 and helicenes.22 Compared to planar PAHs, non-planar PAHs show unique features in physical, chemical, and assembly aspects.12–21,23 Among non-planar PAHs, buckybowls have drawn intensive interest.13i,j,17 Buckybowls are mainly derived from the C5v- and C3v-symmetric fragments of fullerene C60, i.e., corannulenes24 and sumanenes,25 respectively (Scheme 1). From the viewpoint of synthetic chemistry, buckybowls can serve as synthons to produce fullerene C60,26 end-caps of carbon nanotubes,27 warped nanographene,13a carbon nanocones,28 and chiral PAHs.29
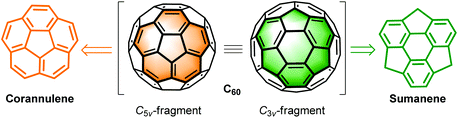 |
| | Scheme 1 Chemical structures of the corannulene and sumanene. | |
To gain value-added functionalities for buckybowls, several approaches have been unveiled: harnessing the periphery with an additional functional unit,30 expanding π-conjugated frameworks (fragments of higher fullerenes),31 embedding heteroatoms into carbonaceous skeletons (hetera-buckybowls),17i,32 coordinating with metal ions,33 and host–guest assembly.34 While the research on carbonaceous buckybowls has made substantial progress in the past decades, the potential of hetera-buckybowls has not been fully exploited to date. Herein, we provide an overview on the synthesis and derivatization of hetera-buckybowls published up to July 2020.
Heteroatom-centered triangulenes,35 subporphyrins,36 and subphthalocyanines37 may take bowl-shaped conformations. Therefore, they can also be regarded as hetera π-bowls. Meanwhile, there are “bottom-open”-type π-bowls generated via a “fold-in” strategy as reported by Stępień,38 Osuka,39 Isobe,40 and Wu.13i These exciting works are not included in this synopsis.
2. Synthesis and derivatization of hetera-corannulenes
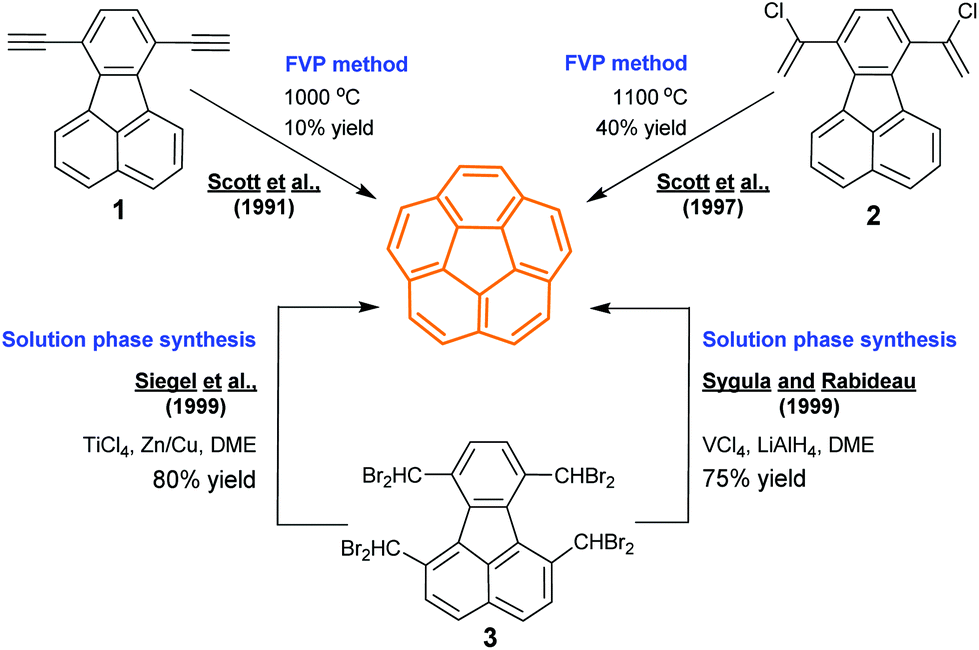
Corannulene is a C5v-symmetric fragment of fullerene C60. While the research on buckybowls is stimulated by the extraordinary properties of fullerenes, corannulene was found much earlier than the discovery of fullerenes in 1985.41 In 1966, Barth and Lawton synthesized corannulenes in 17 steps with a total yield less than 1%.24a,b The synthetic obstacles were surmounted by Scott's, Siegel's, and Rabideau's groups in 1990s (Scheme 2). Scott and co-workers took flash vacuum pyrolysis (FVP) as a key technique. In 1991, they synthesized corannulene from 1 (10% yield) via FVP at 1000 °C,24c and further improved the yield up to 40% by using 2 as a precursor.24d While FVP is widely used to create various buckybowls,17c it is not easy to perform this process. Therefore, non-pyrolytic synthesis of corannulenes has become an important goal. In 1996, Siegel's group reported non-pyrolytic synthesis of corannulene cyclophane.24g In 1999, Sygula and Rabideau24e and Siegel's group24f independently synthesized pristine corannulene in the solution phase. Later, the gram- and kilogram-scale synthesis of corannulenes was achieved by Sygula and Rabideau24h and Siegel's group.24i The progress in synthesis promotes the application of this peculiar scaffold in multiple terminals.17a–c
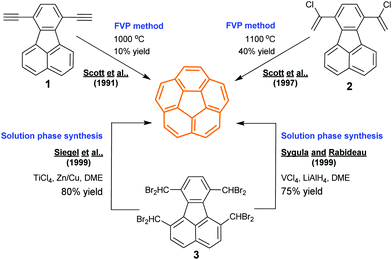 |
| | Scheme 2 Synthesis of the corannulene through FVP or in the solution phase (see ref. 24). | |
Replacing the carbon atoms on corannulene with heteroatoms is an efficient way to tailor its structural and electronic characteristics. To date, three kinds of molecular engineering have been performed to gain hetera-corannulenes (Scheme 3): embedding a nitrogen atom at the hub (blue circle) and rim (red circle) positions, and replacing two spoke C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds (green ellipses) with B–N bonds.42 In the following, we introduce several representative examples.
C bonds (green ellipses) with B–N bonds.42 In the following, we introduce several representative examples.
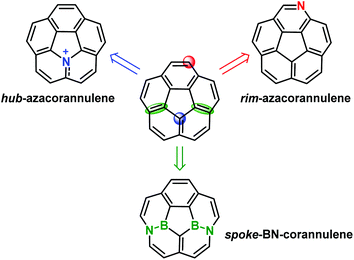 |
| | Scheme 3 Schematic depiction of the derivatization of a hetera-corannulene from a corannulene. | |
2.1.
hub-Azacorannulenes
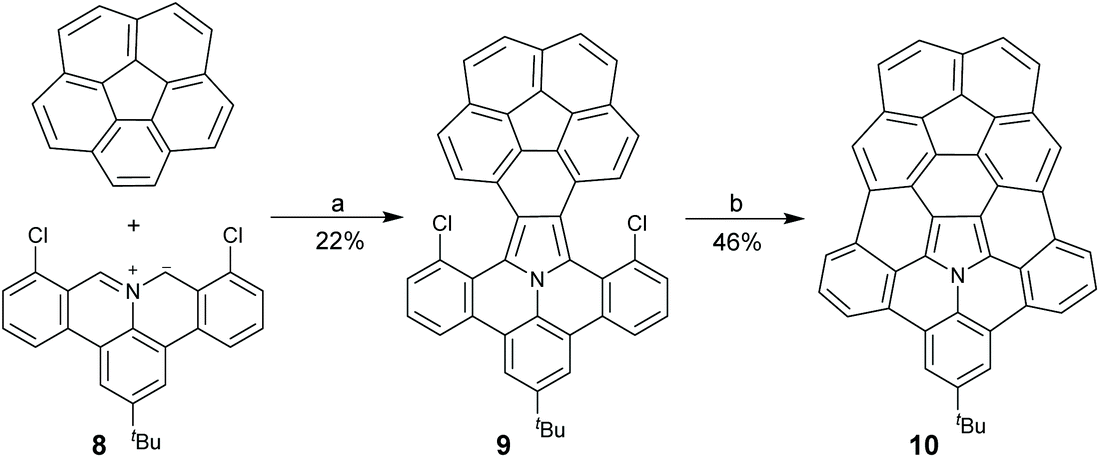
Chemically speaking, hub-azacorannulenes are derived from corannulenes by replacing the central carbon pentagon with pyrrole. Because the valences of carbon and nitrogen are different, the simple substitution of the carbon atom at the hub position of the corannulene with a nitrogen atom results in an unstable cationic species (Scheme 3). Fusing benzene rings at the flank positions is an efficient method to solve this problem. With this in mind, Ito and co-workers designed and synthesized hub-azacorannulene 7 as shown in Scheme 4.43 The key strategy for this synthesis is 1,3-dipolar44 cycloaddition of aromatic azomethine ylide 4 and diarylethyne 5, which affords 6. Compound 6 then undergoes Pd-catalyzed intramolecular cyclization to give 7. In 2017, Ito et al. found that 1,3-dipolar cycloaddition occurred between azomethine ylides and rim CC bonds of corannulenes to give pyrrole-fused corannulenes.45 Employing this reaction as a key step, they prepared a hub-nitrogen-doped C80 fragment 10 through 1,3-dipolar cycloaddition of the corannulene and 8, and the successive Pd-catalyzed intramolecular cyclization (Scheme 5).46
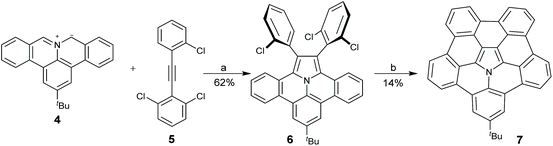 |
| | Scheme 4 Synthesis of hub-azacorannulene 7 (see ref. 43). Reagents and conditions: (a) DMSO, reflux; and (b) [Pd(PCy3)2Cl2], Cs2CO3, N,N-dimethylacetamide (DMA). | |
 |
| | Scheme 5 Synthesis of hub-nitrogen-doped C80 fragment 10 (see ref. 46). Reagents and conditions: (a) (1) DMSO, 140 °C, 20 h; (2) DDQ (1.8 eq.), r.t., 2 h; and (b) Pd(OAc)2 (1 eq.), [HPtBu2Me][BF4] (3 eq.), DMA/DBU, 150 °C, 24 h. DBU = 1,8-diazabicyclo[5,4,0]undec-7-ene. | |
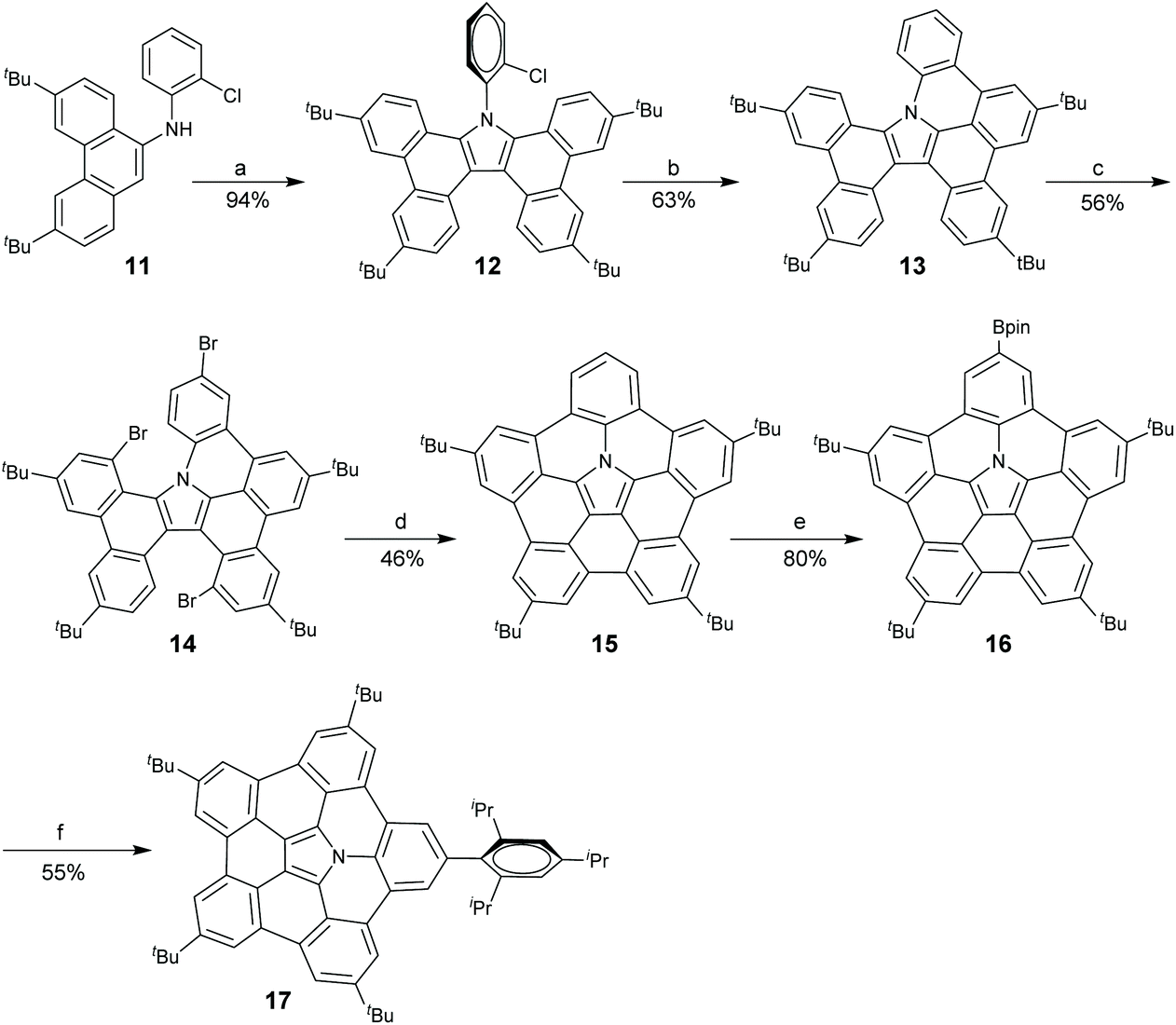
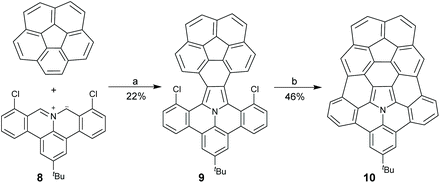
In 2015, Shinokubo and co-workers disclosed a different approach toward the hub-azacorannulene (Scheme 6).47 They have reported intermolecular oxidative annulation of 2-aminoanthracene derivatives in the presence of DDQ and trifluoroacetic acid (TFA) to form pyrrole-fused bisanthracenes.48 Under similar conditions, the oxidation of 9-aminophenanthrene (11) by DDQ gave tetrabenzocarbazole (12) which was converted to 13via Pd-catalyzed intramolecular coupling.47 Compound 13 was brominated to give 14, and successive Pd-catalyzed intramolecular coupling afforded hub-azacorannulene 15. It should be noted that two equivalents of Pd-catalyst was used in this reaction. Compound 15 was further transformed into 17via Ir-catalyzed C–H borylation.
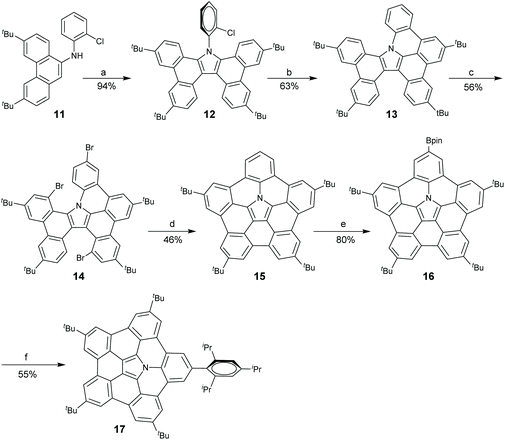 |
| | Scheme 6 Synthesis of hub-azacorannulenes 15–17 (see ref. 47). Reagents and conditions: (a) DDQ (2 eq.), TFA, toluene, r.t., 1 h; (b) Pd(OAc)2 (1 eq.), PCy3·HBF4 (2 eq.), K2CO3 (8 eq.), DMA, 130 °C, 43 h; (c) Br2 (30 eq.), CCl4, 70 °C, 12.5 h; (d) Pd(OAc)2 (2 eq.), PCy3·HBF4 (4 eq.), K2CO3 (8 eq.), DMA, 130 °C, 16 h; (e) bis(pinacolato)diboron (10 eq.), [Ir(OMe)cod]2, 4,4′-di-tert-butyl-2,2′-bipyridyl (1 eq.), octane, 110 °C, 10 h; and (f) 2-bromo-1,3,5-triisopropylbenzene, PdCl2(dppf)·CH2Cl2 (0.5 eq.), Cs2CO3 (2.5 eq.), 1,4-dioxane, 100 °C, 13 h. | |
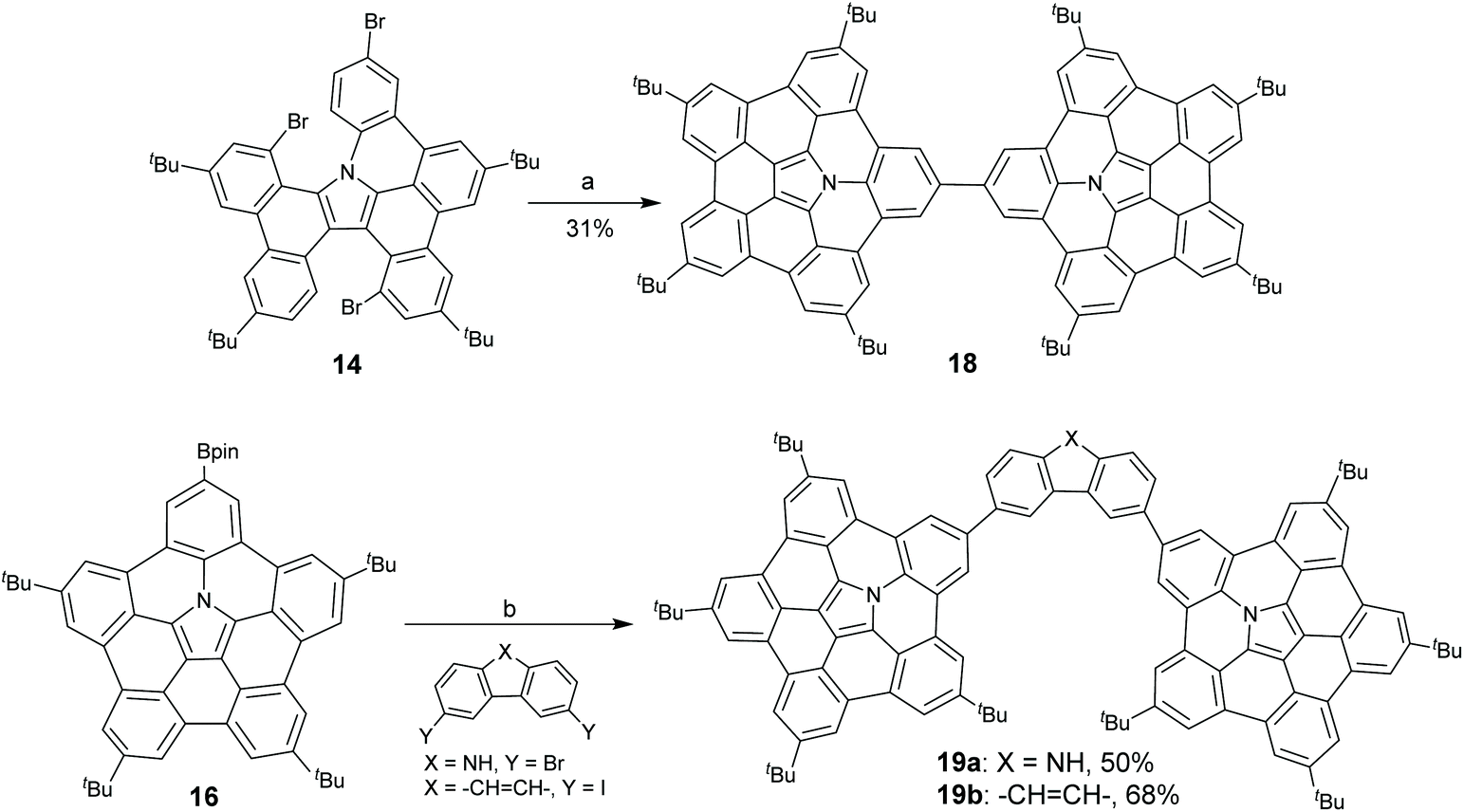
Interestingly, Shinokubo and co-workers found that a hub-azacorannulene dimer (18) was formed when catalytic amounts of Pd-catalyst (0.2 mol%) and PCy3·HBF4 (0.4 mol%) were used in the coupling reaction of 14 as shown in Scheme 7.49 Recently, they synthesized two hub-azacorannulene-based molecular tweezers 19a and 19b through Pd-catalyzed intermolecular coupling reaction of 16 with 3,6-dibromo-carbazole and 3,6-diiodophenanthrene, respectively.50
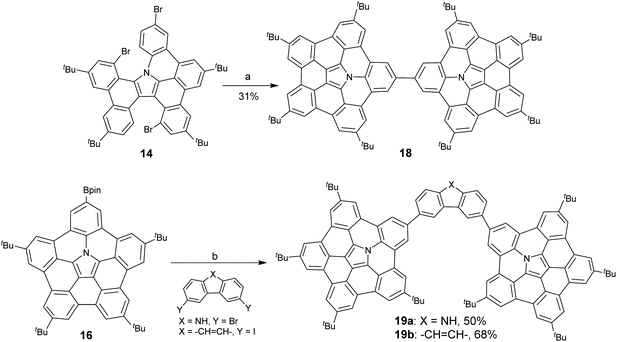 |
| | Scheme 7 Synthesis of hub-azacorannulene dimer 18 (see ref. 49) and hub-azacorannulene based molecular tweezer 19 (see ref. 50). Reagents and conditions: (a) Pd(OAc)2 (0.2 mol%), PCy3·HBF4 (0.4 mol%), K2CO3 (8 eq.), DMA, 130 °C, 43 h; and (b) for 19a, Pd(PPh3)4 (14 mol%), Cs2CO3 (7.7 eq.), THF/H2O (v/v = 4/1), reflux, 4 h; for 19b, Pd(PPh3)4 (13 mol%), Cs2CO3 (6.5 eq.), 1,4-dioxane/H2O (v/v = 20/3), 80 °C, 1 h. | |
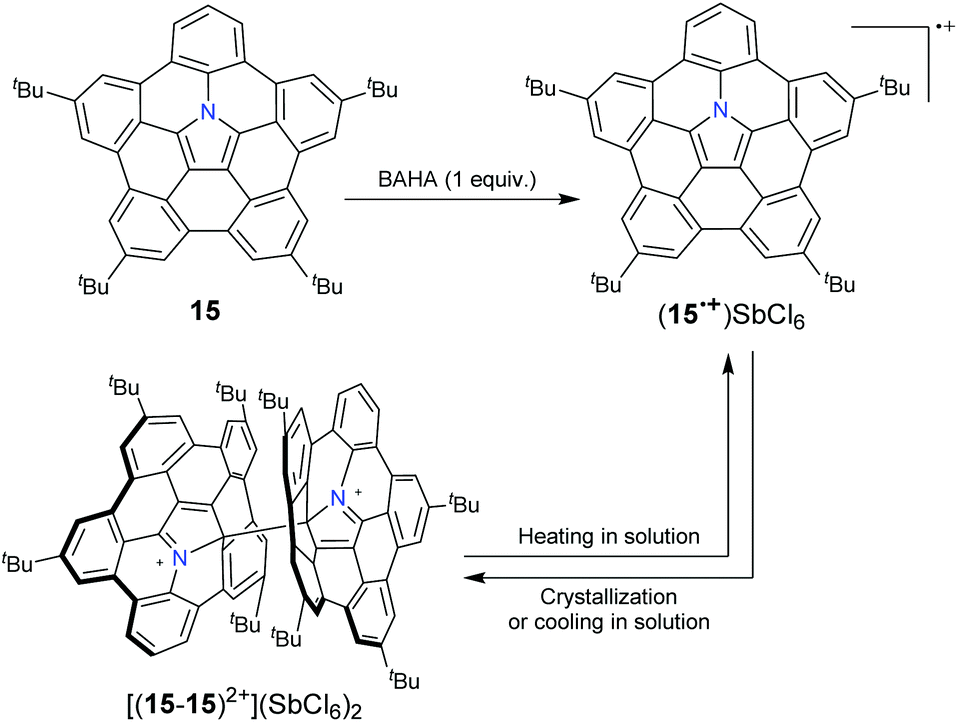
Compared to pristine corannulenes (bowl-depth: 0.87 Å), hub-azacorannulenes 7, 15, and 17 are deep π-bowls with bowl-depths of ca. 1.70 Å.43,47 As an N-doped C80 fragment, 10 has a bowl-depth of 4.19 Å.46 Due to the incorporation of a nitrogen atom into the π-skeleton and extension of the π-framework by fusing benzene at rim positions, compounds 7, 15, and 17 show broad absorption at 300–500 nm,43,47 clearly red-shifted as compared to that of the corannulene.51 They display green fluorescence with quantum yields (ΦF) over 17%,43,47 much higher than that of the corannulene.52 The π-system of 10 is further extended, and consequently it exhibits absorption at 600 nm and red fluorescence centered at 605 nm (ΦF = 17%).46 Upon protonation, the absorption of hub-azacorannulenes shows distinct changes. For example, 15 displays two new bands centered at 520 nm and 670 nm, and a broad band at 800–1150 nm in the presence of TFA. This is assigned to the formation of radical cations (15)˙+.47 A similar phenomenon is observed for 10.46
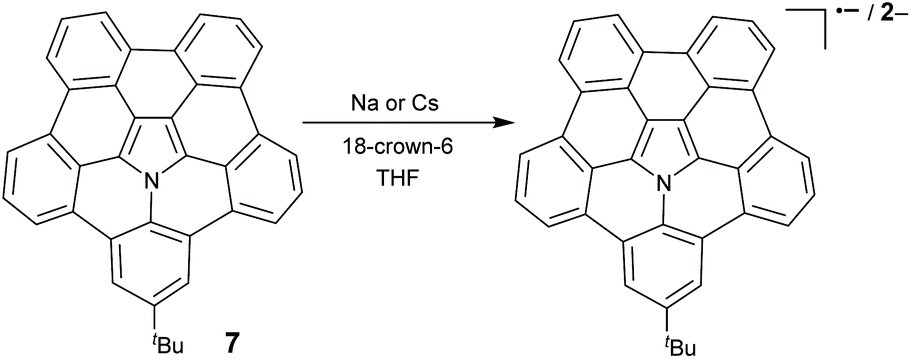
10 and 15 can be transformed into radical cation states ((10)˙+ and (15)˙+, respectively) upon oxidation with a one-electron oxidant tris(4-bromophenyl)aminium hexachloroantimonate (BAHA).46,47 (15)˙+ forms a σ-dimeric dication [(15–15)2+] in the crystalline phase, whereas the formation and dissociation of [(15–15)2+] are reversible in solution on varying the temperature (Scheme 8).53 Recently, the stepwise reduction of 7 has been reported (Scheme 9).54 Crystallographic analysis indicated that the chemical reduction of 7 with the Na metal afforded a dianion complex Na2[(7)2−]. However, the reduction of 7 with Cs gave rise to a radical anion complex Cs[(7)˙−] and a dianion complex Cs2[(7)2−]. The Cs+ ion selectively locates at the concave surface of 7˙− of Cs[(7)˙−], whereas two Cs+ ions locate at both the concave and convex surfaces of Cs2[(7)2−]. Owing to the electron-rich feature, hub-azacorannulenes can serve as fullerene hosts in solution and in the solid state.47,49,50 The co-crystal of 15 with C60 shows a high charge mobility of 0.17 cm2 V−1 s−1.47
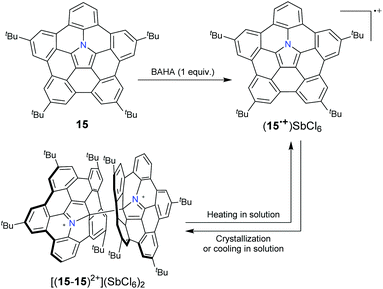 |
| | Scheme 8 Formation of hub-azacorannulene 15 radical cations along with their dimerization and dissociation behavior (see ref. 53). | |
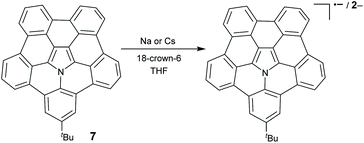 |
| | Scheme 9 Chemical reduction of hub-azacorannulene 7 with Na and Cs metals (see ref. 54). | |
2.2.
rim-Azacorannulenes
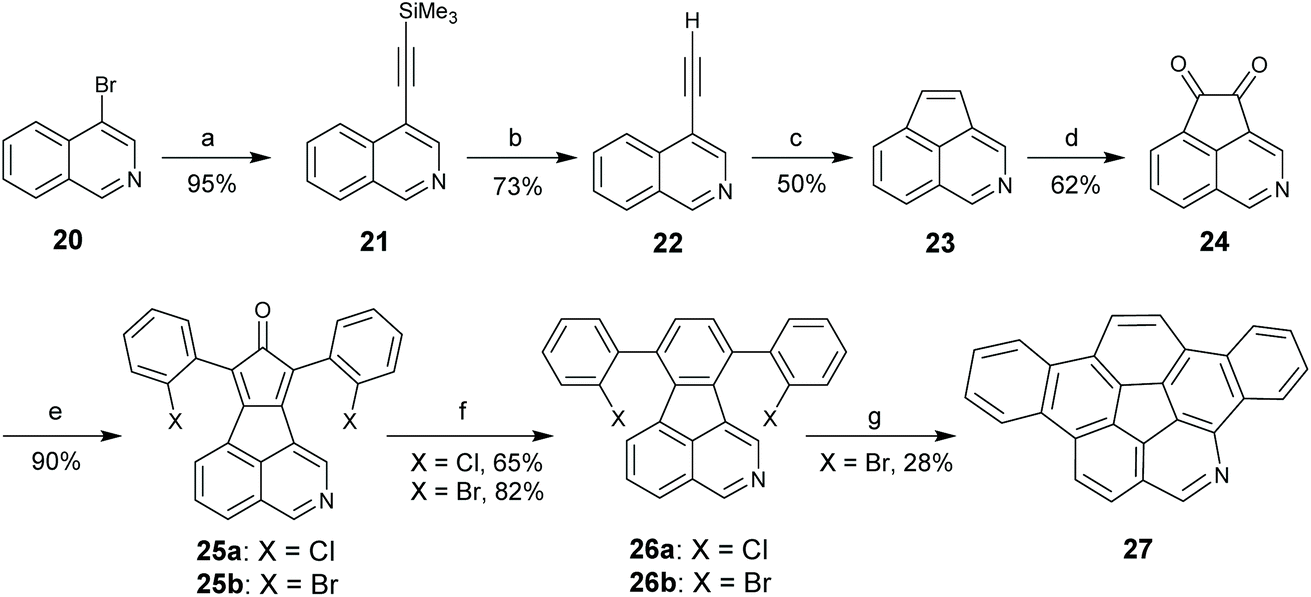
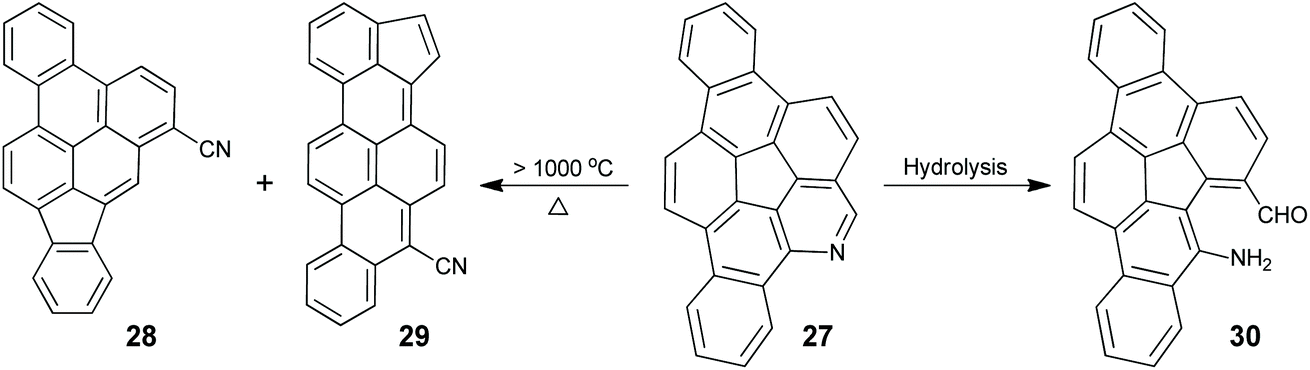
rim-Azacorannulenes are derived from corannulenes by replacing the rim benzenes with pyridines. Inspired by the FVP synthesis of corannulenes, Bratcher tried to synthesize rim-azacorannulenes from fluoranthene-7,10-dicarbonitrile.55 However, it failed due to several factors, i.e., stronger π-bonds in C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N than in CC,56 weaker aromatic stabilization energy of pyridine than that of benzene,57 and the shorter bond length of CN than that of CC.58 The successful synthesis of rim-azacorannulenes was achieved by Scott's group in 2017.59 By employing 4-bromoisoquinoline as a starting material, they synthesized a rim-azacorannulene 27 in seven steps (Scheme 10). In the final FVP step, ring closure only occurs for 26b. An attempt to prepare 27 from 26avia Pd-catalyzed intramolecular cyclization failed.5927 is not stable owing to the cleavage of its pyridine moiety. The pyridine ring on 27 is opened to give 28 and 29 at a temperature higher than 1000 °C (Scheme 11), assigned to the reversal of the ring-closure process for FVP synthesis of 27.59 The opening of the pyridine ring is also observed during purification of 27via chromatography on silica gel, affording amino aldehyde 30. The formation of 30 is attributed to the hydrolysis of the CN bond.
N than in CC,56 weaker aromatic stabilization energy of pyridine than that of benzene,57 and the shorter bond length of CN than that of CC.58 The successful synthesis of rim-azacorannulenes was achieved by Scott's group in 2017.59 By employing 4-bromoisoquinoline as a starting material, they synthesized a rim-azacorannulene 27 in seven steps (Scheme 10). In the final FVP step, ring closure only occurs for 26b. An attempt to prepare 27 from 26avia Pd-catalyzed intramolecular cyclization failed.5927 is not stable owing to the cleavage of its pyridine moiety. The pyridine ring on 27 is opened to give 28 and 29 at a temperature higher than 1000 °C (Scheme 11), assigned to the reversal of the ring-closure process for FVP synthesis of 27.59 The opening of the pyridine ring is also observed during purification of 27via chromatography on silica gel, affording amino aldehyde 30. The formation of 30 is attributed to the hydrolysis of the CN bond.
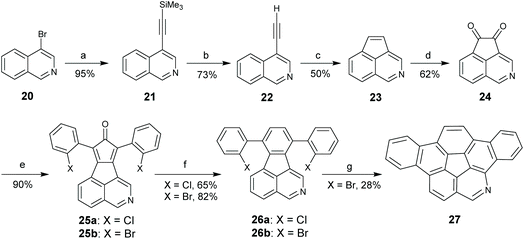 |
| | Scheme 10 Synthesis of rim-azacorannulene 27 (see ref. 59). Reagents and conditions: (a) Me3SiCCH, (Ph3P)2PdCl2, CuI, Et3N, 70 °C; (b) KOH, CH3OH; (c) FVP, 950 °C; (d) (PhSeO)2O, PhCl; (e) 1,3-bis(2-C6H4X)acetone, KOH, CH3OH (X = Br, Cl); (f) Norbornadiene, reflux; and (g) FVP, 1000 °C. | |
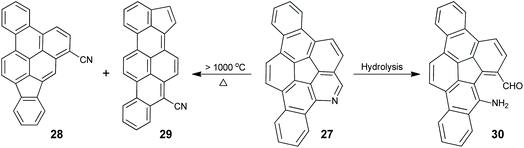 |
| | Scheme 11 Opening of the pyridine ring in rim-azacorannulene 27 under different conditions (see ref. 59). | |
2.3.
spoke-BN-corannulenes
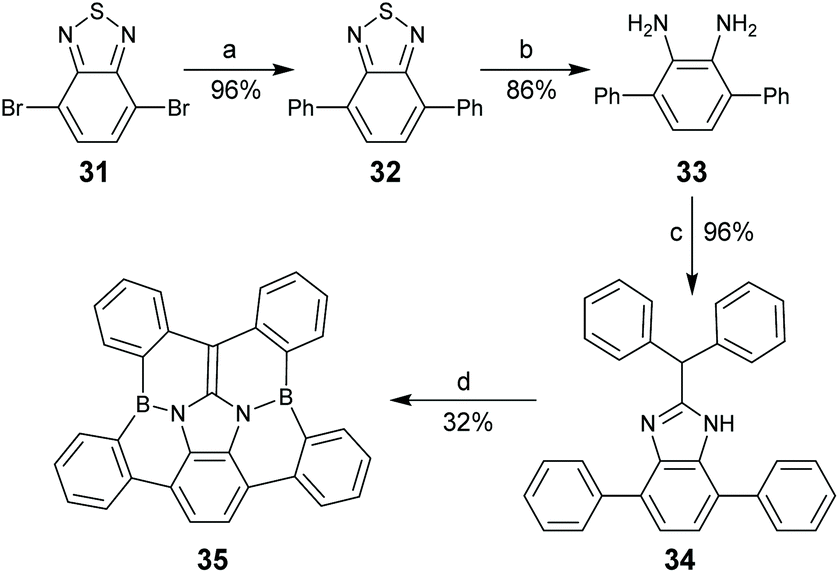
Replacing CC bonds in PAHs with isoelectronic B–N bonds gives rise to dramatic alteration in optoelectronic properties because the resulting molecules have local dipole moments and/or polarized frontier orbitals. The BN-doped arenes have drawn intensive attention due to their promising applications in biomedical and optoelectronic materials.60 In 2018, Hatakeyama and co-workers reported spoke-BN-corannulene 35.61 By employing 4,7-dibromobenzo[c][1,2,5]thiadiazole 31 as a starting material, compound 35 was synthesized in four steps, where the final step was electrophilic C–H borylation of 34 in the presence of 4.0 equiv. of BBr3 (Scheme 12).
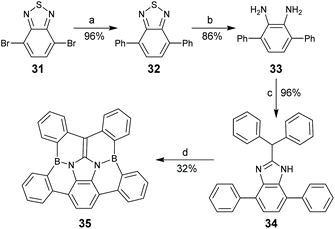 |
| | Scheme 12 Synthesis of spoke-BN-corannulene 35 (see ref. 61). Reagents and conditions: (a) phenylboronic acid (2.2 eq.), Pd(PPh3)4 (1 mol%), K2CO3 (3 eq.), THF/H2O (1/1), reflux, 3 days; (b) Zn (10 eq.), AcOH, 40 °C, 22 h; (c) 2,2-diphenylacetic acid (2 eq.), neat, 180 °C, 62 h; and (d) BBr3 (4 eq.), o-dichlorobenzene, reflux, 16 h. | |
While 35 is derived from the corannulene by replacing spoke CC bonds with B–N bonds, four rim C–C bonds on the corannulene are also replaced by C–B bonds. Crystallographic analysis shows that the bond length of C–B in 35 is 1.55–1.58 Å,61 which is longer than the corresponding C–C bond (1.44–1.45 Å) in the corannulene. Accordingly, the bowl-depth of 35 (0.15 Å) is much shallower than that of the corannulene (0.87 Å). Compound 35 shows strong blue emission (λem = 424 nm) with a ΦF of 69%, much higher than that of the corannulene (7%).5235 was further used as an emitter to fabricate an OLED, and the resulting device displayed sky-blue electroluminescence with the maximum luminance of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 cd m−2.
400 cd m−2.
3. Synthesis and derivatization of hetera-sumanenes
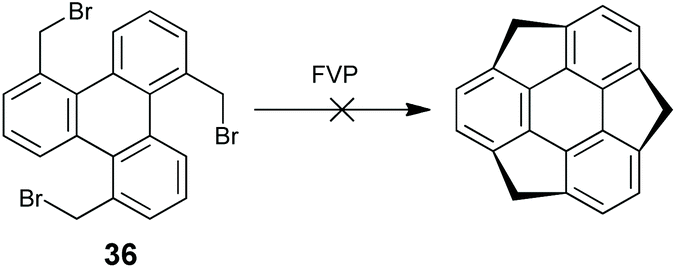
The C3v-symmetric sumanene is a more strained buckybowl than corannulenes; thus its synthesis is more challenging. Inspired by the great success of the FVP synthesis of corannulene, Mehta and co-workers attempted to synthesize sumanene through FVP (Scheme 13); however it failed since the sp3 benzylic carbons on the sumanene might not tolerate FVP conditions.62 Thereafter, several groups tried to construct sumanenes from planar aromatics, yet the goal was not achieved.63 These results imply that it would be impossible to prepare sumanenes from a planar π-skeleton.
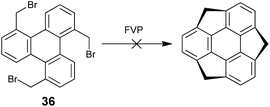 |
| | Scheme 13 An attempt to synthesize sumanenes through FVP (see ref. 62). | |
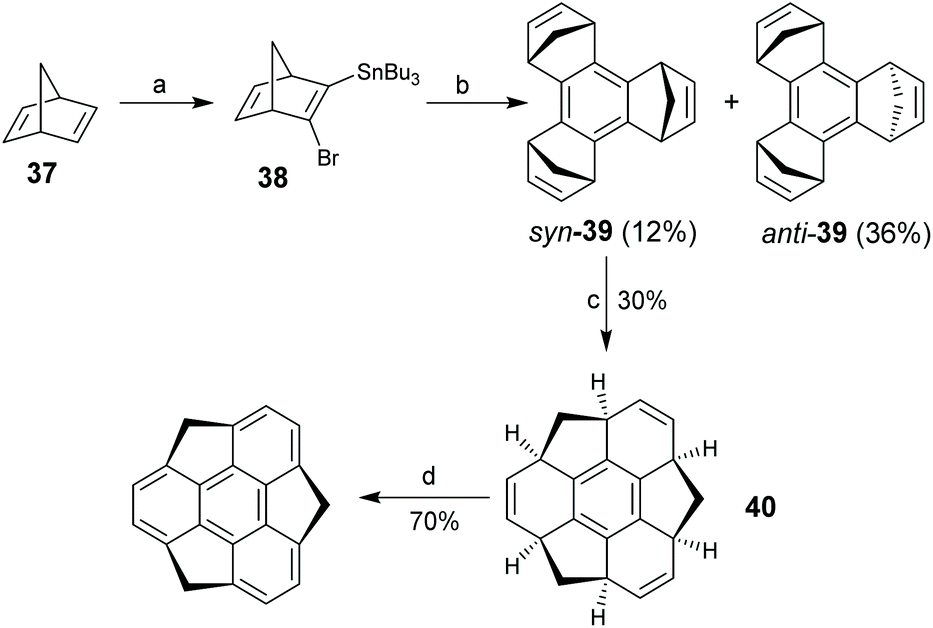
With this background, Sakurai, Daiko and Hirao proposed a decisive tandem ring-opening/ring-closure approach toward sumanenes, and for the first time they successfully synthesized sumanenes in four steps as shown in Scheme 14.25asyn- and anti-tris(norbornadieno)benzene syn-39 and anti-39, respectively, were prepared from 37 in two steps. Then, Ru-catalyzed tandem ring-opening metathesis (ROM) and ring-closure metathesis (RCM) reactions of syn-39 under an ethylene atmosphere afforded 40. It is worth noting that the cascade ROM/RCM reaction did not proceed for anti-39. Finally, dehydrogenation of 40 upon oxidation with DDQ gave the sumanene. This protocol was further used to enantioselectively synthesize chiral buckybowls, i.e., C3-symmetric trisubstituted sumanenes.29a,b Sumanene has a bowl-depth of 1.11 Å and shows bowl-to-bowl inversion with an energy barrier of 20.3 kcal mol−1.25b,64
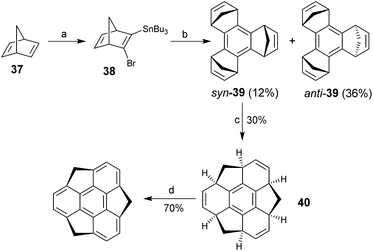 |
| | Scheme 14 Synthesis of sumanenes (see ref. 25). Reagents and conditions: (a) BuLi (0.5 eq.), tBuOK (0.5 eq.), BrCH2CH2Br (0.25 eq.), THF, −78 °C to −45 °C, then Bu3SnCl (0.3 eq.), r.t.; (b) Cu(2-C4H3SCO2) (1 eq.), NMP, −20 °C to r.t.; (c) [P(C6H11)3]2RuCl2CHPh (10 mol%), ethylene, toluene, −78 °C to r.t., 24 h; and (d) DDQ (6 eq.), toluene, 110 °C, 3h. | |
The synthetic breakthrough sparks further investigations in sumanene chemistry as well as the application of sumanenes in materials science.17d–f,h,i The needle-like crystals of sumanenes show a carrier mobility of 0.75 cm2 V−1 s−1 along the π-bowl stacking axis.29b,65 Recently, Sakurai and co-workers reported a hydrogen-bonded hexagonal framework30c and porous coordination networks30d based on sumanenes.
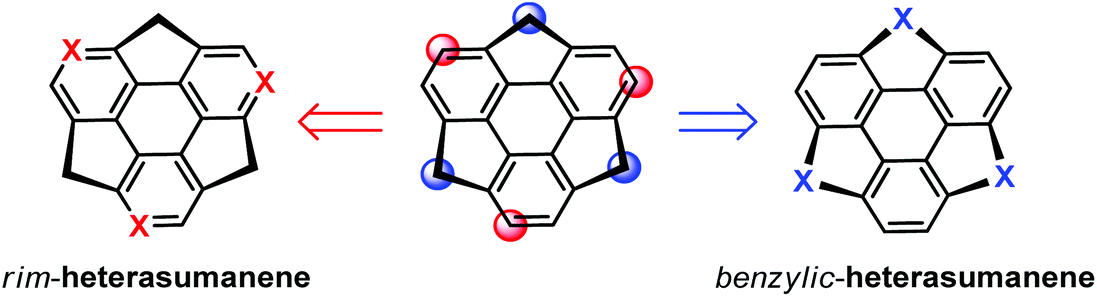
Hetera-sumanenes are of growing interest, because the nature of the heteroatom causes significant modulation in their physical and chemical properties.17i,32 Tracing the timeline of sumanene chemistry, one can find that hetera-sumanenes actually appeared earlier than sumanenes, i.e., Otsubo's group reported the synthesis of trithiasumanenes in 1999.66 Hetera-sumanenes are derived from sumanenes by replacing the rim and benzylic carbons with heteroatoms (Scheme 15). In the following, we introduce the synthesis and derivatization of hetera-sumanenes.
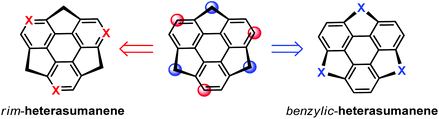 |
| | Scheme 15 Schematic depiction of the derivatization of heterasumanenes from sumanenes. | |
3.1.
rim-Triazasumanenes
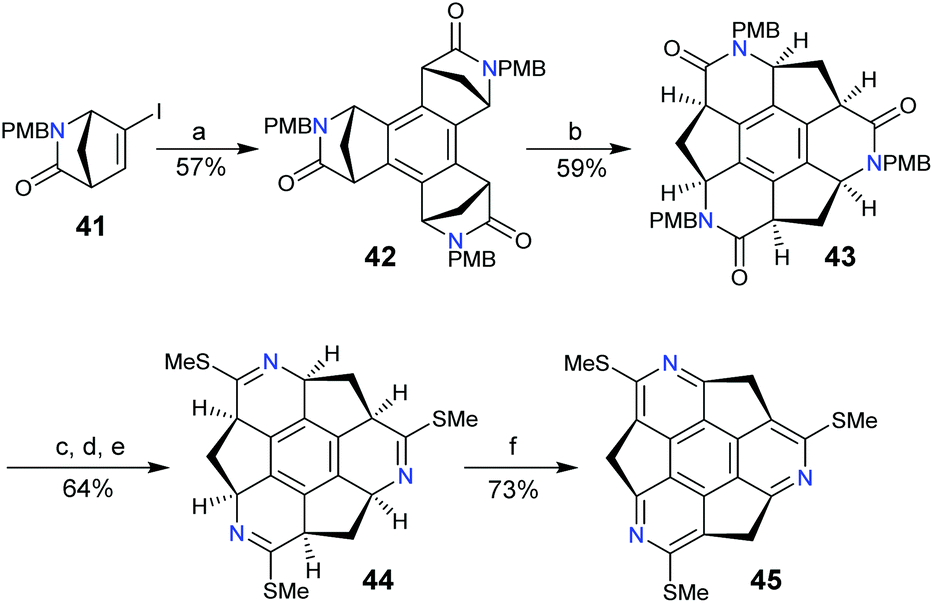
To date, rim-hetera-sumanenes have been only obtained by decorating nitrogen atoms at the rim-positions of sumanenes.67–69 Employing a similar synthetic strategy for sumanenes, Sakurai's group developed an enantioselective synthesis of the rim-triazasumanene 45 which has three thiomethyl groups at the rim positions (Scheme 16).67 The Pd-catalyzed cyclotrimerization of enantiopure 41 afforded 42, which was then hydrolysed, followed by condensation to give lactam 43. The direct aromatization of 43 failed to afford rim-triazasumanene; therefore 43 was converted to 44via three steps including microwave-assisted thiation, deprotection, and thiomethylation. Then 44 was dehydrogenated to give the desired 45 using Ph3CBF4 as an oxidant and DTBMP as an acid scavenger. 45 shows stable chirality as it has a bowl-to-bowl inversion energy (42.2 kcal mol−1) much higher than that of sumanenes. Recently, 45 was used as a synthon to prepare the pristine rim-triazasumanene68 and a rim-arylated one.69
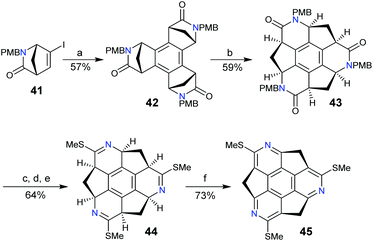 |
| | Scheme 16 Enantioselective synthesis of rim-triazasumanene 45 (see ref. 67). Reagents and conditions: (a) Pd(OAc)2 (5 mol%), PPh3 (0.1 eq.), Bu4NOAc (10 eq.), Na2CO3 (10 eq.), 4 Å molecular sieve, 1,4-dioxane, 100 °C, 2 h; (b) (i) HCl (12 M), AcOH, 60 °C, 3 h; (ii) C6F5OP(O)Ph2 (9 eq.), N,N-diisopropylethylamine (20 eq.), DMF, 0–60 °C; (c) Lawesson's reagent (0.5 eq.), ClCH2CH2Cl, microwave, 160 °C, 40 min; (d) TFA, microwave, 100 °C, 2 h; (e) MeI (30 eq.), K2CO3 (10 eq.), DMF, 30 °C, 3 h; and (f) Ph3CBF4 (6 eq.), DTBMP (8 eq.), CH2Cl2, 25 °C, 8 h. | |
3.2.
benzylic-Heterasumanenes
Most of the heterasumanenes obtained so far are benzylic-heterasumanenes, derived from sumanenes by substituting the benzylic carbons with group 14, 15, 16, and 17 elements.17i,32 According to the chemical structures of benzylic-hetera-sumanenes, a rational approach to synthesize them is to build heteroatom bridges at the bay regions of triphenylene. Theoretical studies indicate that it is challenging to synthesize curved hetera-sumanenes from planar triphenylene due to the large strain energy for ring cyclization.70 To achieve this purpose, many synthetic strategies have been exploited, i.e., FVP, sila-Friedel–Crafts reaction, C–H activation, and consecutive ring-cyclization/ring-contraction. In the following, we will introduce several representative examples.
3.2.1.
benzylic-Heterasumanenes involving group 14 elements.
Among this class of heterasumanenes, elements Si and Ge have been successfully introduced into the benzylic positions of sumanenes. In 2009, Furukawa, Kobayashi, and Kawashima developed an intramolecular sila-Friedel–Crafts reaction, which was then used to prepare trisilasumanene 50.71HBT was brominated to give a mixture of 46 and 47 (Scheme 17). The consecutive sila-Friedel–Crafts reaction of the 46/47 mixture afforded doubly cyclized monobromide 48, which was then converted to 49. Finally, intramolecular sila-Friedel–Crafts reaction of 49 afforded 50. Crystallographic analysis reveals that 50 adopts a planar conformation. 50 displays an intense absorption band at 299 nm and a weak band at 350–390 nm which are not observed for sumanenes.72 The latter is due to the σ*–π* conjugation on Si atoms. This compound exhibits strong blue emission both in solution (λmax = 427 nm) and in the solid state (λmax = 447 nm), plausibly ascribable to the unique photophysical nature of silole rings.73
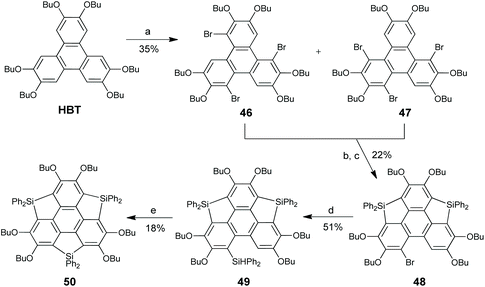 |
| | Scheme 17 Synthesis of trisilasumanene 50via sila-Friedel–Crafts reaction (see ref. 71). Reagents and conditions: (a) Br2, CH2Cl2, 0.5 h; (b) (i) n-BuLi (2 eq.), THF, −78 °C; (ii) Ph2SiCl2 (5 eq.), r.t.; (iii) LiAlH4 (3.6 eq.), reflux; (c) Ph3CB(C6F5)4 (2 eq.), 2,6-lutidine (2 eq.), CH2Cl2, r.t.; (d) (i) tBuLi (2 eq.), Et2O, −78 °C; (ii) Ph2SiH2 (10 eq.), reflux; and (e) Ph3CB(C6F5)4 (1 eq.), 2,6-lutidine (1 eq.), CH2Cl2, r.t. | |
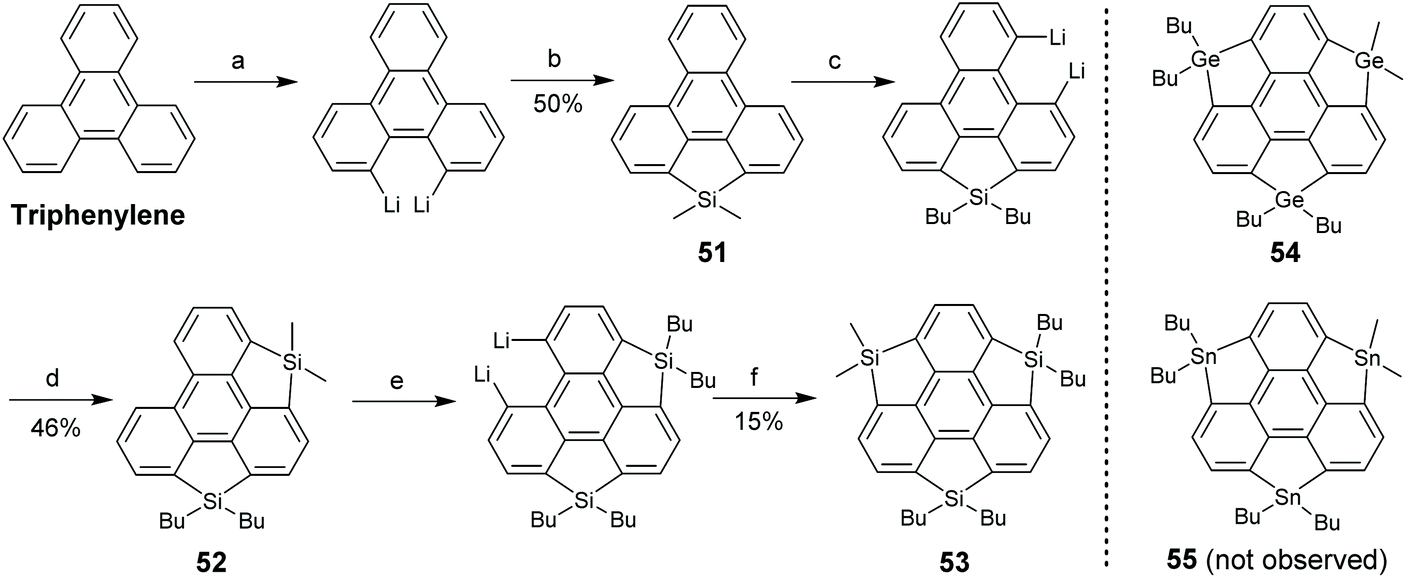
In 2011, Saito's group synthesized the pristine trisilasumanene 53via stepwise ring-cyclization (Scheme 18).74 Triphenylene was lithiated and then dichlorodimethylsilane was introduced to give 51. By repeating this operation, 52 and the ultimate product 53 were prepared. The methyl groups on silole moieties were replaced by butyl groups during this synthesis. Employing the same strategy, Saito's group synthesized trigermasumanene 54, but an attempt to create tristannosumanene 55 failed.75 Compared to 50 which has six butoxy groups at the rim positions, both absorption (λmax = 267 nm) and emission (λmax = 380 nm) of 53 are blue-shifted.74 This means that the rim-substituents play a pivotal role in the photophysical properties. The absorption spectrum of 54 is similar to that of 53, whereas the emission of 54 is further blue-shifted to 367 nm.75
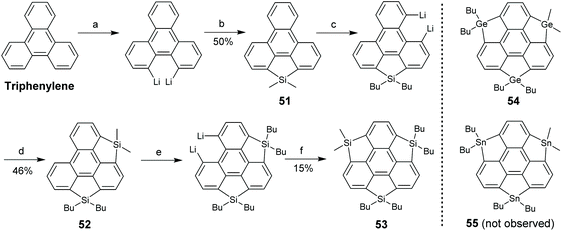 |
| | Scheme 18 Synthesis of trisilasumanene 53 (see ref. 74). Reagents and conditions: (a) BuLi (4 eq.), TMEDA (4 eq.), hexane, 60 °C, 3 h; (b) Me2SiCl2 (2 eq.), CuCN (0.1 eq.), THF, r.t., 14 h; (c) BuLi (6 eq.), TMEDA (6 eq.), hexane, 60 °C, 3 h; (d) Me3SiCl (6 eq.), THF, −78 °C to r.t., 13 h; (e) BuLi (10 eq.), TMEDA (10 eq.), hexane, 60 °C, 3 h; and (f) Me2SiCl2 (10 eq.), CuCN (0.6 eq.), THF, −88 °C to r.t., 13 h. | |
Transition-metal-catalyzed functionalization of aromatic C–H bonds has been widely used to synthesize π-conjugated systems with excellent optoelectronic properties.76 Tan and co-workers disclosed a Rh-catalyzed intramolecular cyclodehydrogenation of Si/Ge–H and C–H.77 Employing this reaction as a key step, they synthesized trisilasumanene 59 and trigermasumanene 61 as shown in Scheme 19.77 This synthesis is highly effective as compared to that in Kawashima's71 and Saito's74,75 work. Crystal structure analysis of 61 proves that this compound has a planar conformation.
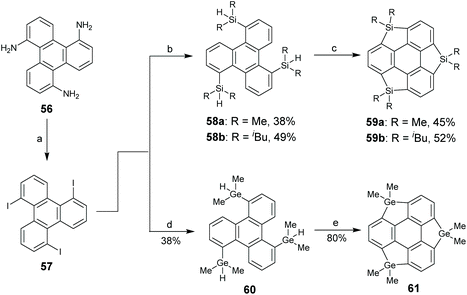 |
| | Scheme 19 Synthesis of trisilasumanene 59 and trigermasumanene 61via C–H activation (see ref. 77). Reagents and conditions: (a) NaNO2 (4 eq.), KI (16 eq.), H2SO4–H2O, 0 °C–65 °C; (b) n-BuLi (6.6 eq.), TMEDA (6.6 eq.), R2SiHCl (7.5 eq.), Et2O, −78 °C to r.t.; (c) 3,3-dimethyl-1-butene (30 eq.), RhCl(PPh3)3 (30 mol%), dioxane, 135 °C; (d) (i) n-BuLi (6.6 eq.), Me2GeCl2 (7.5 eq.), Et2O, −78 °C to r.t.; (ii) LiAlH4 (10 eq.), Et2O; and (e) 3,3-dimethyl-1-butene (30 eq.), RhCl(PPh3)3 (30 mol%), dioxane, 100 °C. | |
3.2.2.
benzylic-Heterasumanenes involving group 15 elements.
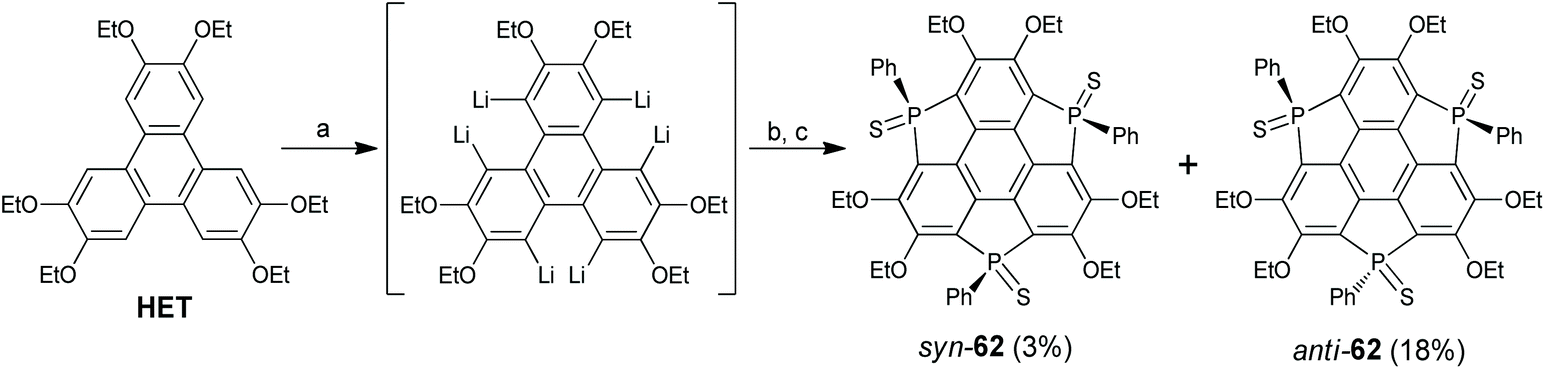
Among the group 15 elements, only phosphorus is introduced into the main skeleton of the sumanene, and these studies have been performed by Furukawa, Saito, and co-workers.78,79 In 2017, they reported a three-step synthesis of triphosphasumanene trisulfide as shown in Scheme 20.78HET was hexalithiated, followed by adding PhPCl2 and S8 in sequence to give triphosphasumanene trisulfide as a mixture of syn- and anti-isomers (syn-62 and anti-62). Crystallographic analyses reveal that syn-62 adopts a bowl-shaped skeleton (bowl-depth: 0.45 Å) and anti-62 is flat. A theoretical study shows that syn-62 has a large out-of-plane dipole moment of 13.7 D. Moreover, the PS of syn-62 can adsorb onto a Au(111) surface to form a molecular junction.78
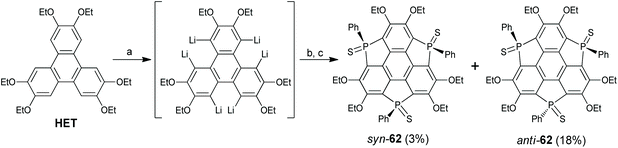 |
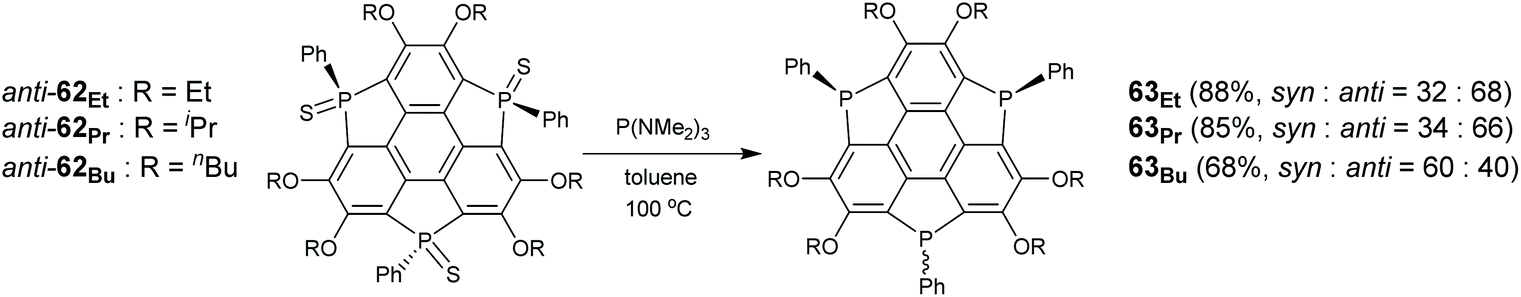
| | Scheme 20 Synthesis of triphosphasumanene trisulfide 3 (see ref. 78). Reagents and conditions: (a) n-BuLi (10 eq.), hexane, reflux, 4 h; (b) PhPCl2 (7.3 eq.), THF, −70 °C to r.t., 3 h; and (c) S8 (10.3 eq.), r.t. | |
Suda, Furukawa, and Saito synthesized triphosphasumanenes (Scheme 21).79 A series of anti-62 analogues were transformed into the corresponding triphosphasumanenes (63Et, 63Pr, and 63Bu) upon reduction with P(NMe2)3.79 Triphosphasumanenes were obtained as a mixture of syn- and anti-isomers. Due to a small energy difference between the syn- and anti-isomers, they show isomerization in solution. For example, 1H NMR analysis reveals that pure anti-63Et converts to a mixture of syn- and anti-isomers with an equilibrium ratio of syn-63Et:anti-63Et = 27:73. On the other hand, the syn- and anti-isomer mixture will transform to the single isomer on heating in the solid state.79
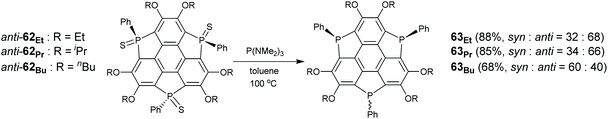 |
| | Scheme 21 Synthesis of triphosphasumanene 63via reduction of their sulfide forms (see ref. 79). | |
3.2.3.
benzylic-Heterasumanenes involving group 16 elements.
This type of heterasumanene is called trichalcogenasumanenes (TCSs), where three benzylic carbons are replaced with chalcogen atoms (S, Se, and Te). Compared to sumanenes, the pentagonal rings on TCSs are aromatic, which extends the π-framework. The valence state of chalcogen atoms can be chemically manipulated, leading to fine modulations of the structural and optoelectronic features. Moreover, chalcogen atoms can form various intermolecular interactions in the solid state, which are beneficial for electrical transport. Therefore, the chemistry of TCSs has attracted growing interest recently.
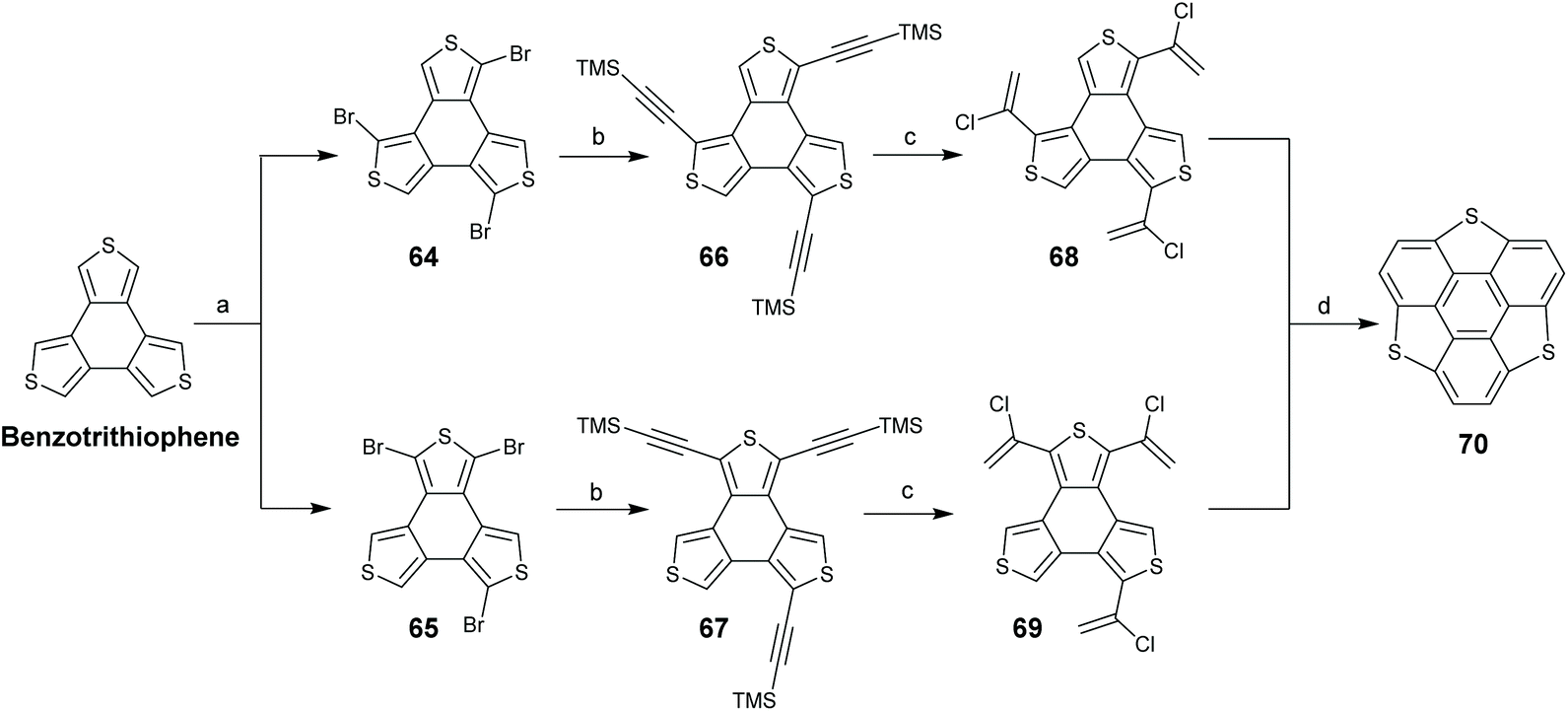
TCSs have two subunits, benzotrichalcogenole and triphenylene. With regard to their chemical structures, two synthetic routes are proposed: routes A and B start from benzotrichalcogenole and triphenylene, respectively. In 1999, Otsubo's group reported the first synthesis of trithiasumanenes through route A.66 Employing benzotrithiophene as a starting material, a four-step transformation afforded trithiasumanene 70 (Scheme 22). The key step is the ring-closure of 68 and 69 under FVP conditions (1000 °C, 0.005 Torr). Crystal structure analysis indicates that 70 takes a bowl-shaped conformation with a bowl-depth of 0.79 Å and forms convex-to-concave type stacks.
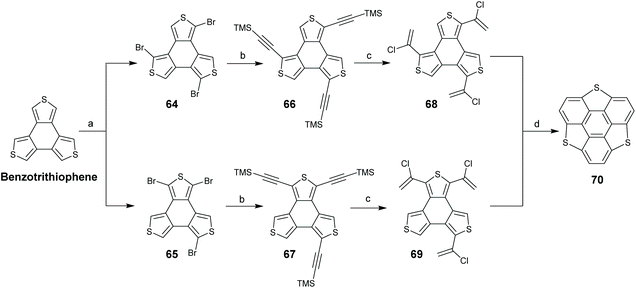 |
| | Scheme 22 Synthesis of pristine trithiasumanene 6via FVP (see ref. 66). Reagents and conditions: (a) NBS (3.0 eq.), DMF, r.t.; (b) TMSCHCH, Pd(PPh3)4, CuI, Et3N, reflux, 53% yield (two steps); (c) HCl (conc.), AcOH, 80 °C, 33% yield; and (d) 1000 °C, 0.005 Torr, N2 flow, 35% yield. | |
While a breakthrough in the synthesis of 70 was achieved by Otsubo's group,66 their method suffers from harsh reaction conditions for FVP, low yields (6% overall yield), and milligram-scale production. Needless to say, benzotrithiophene is more expensive than triphenylene. Theoretical study shows that it is thermodynamically unfavourable to prepare 70 through route B,70 say, with triphenylene as a starting material. Klemm and co-workers tried to synthesize 70 from triphenylene, but only mono- and di-bridged species were formed in trace amounts.80 These results imply that simultaneously forming three sulfur-bridges at the bay regions of triphenylene would be impossible.
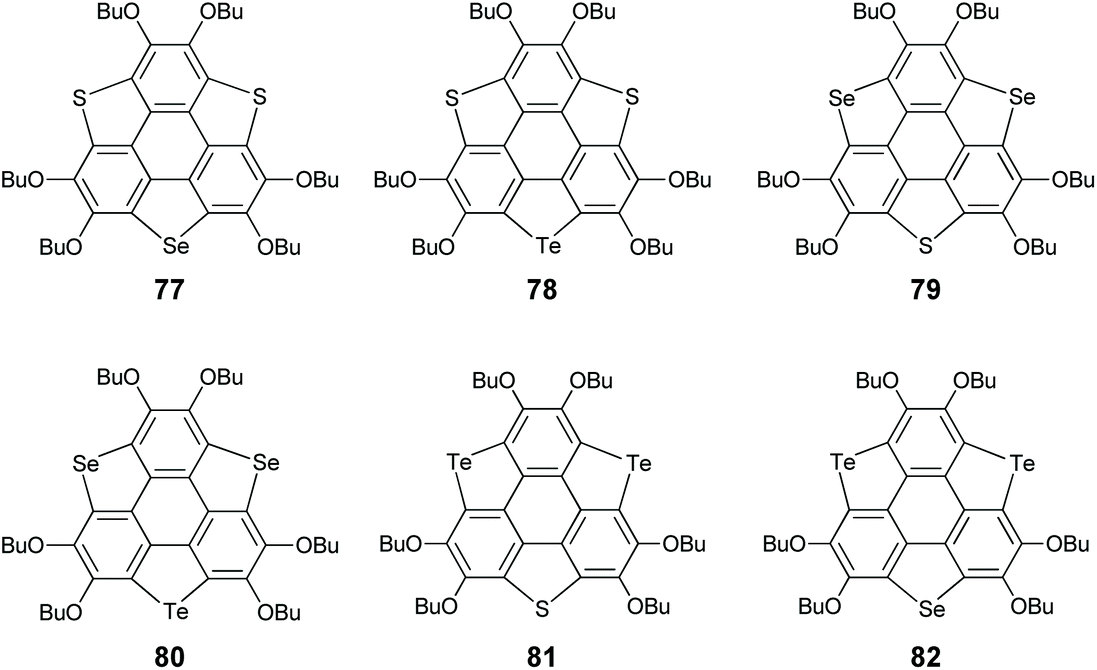
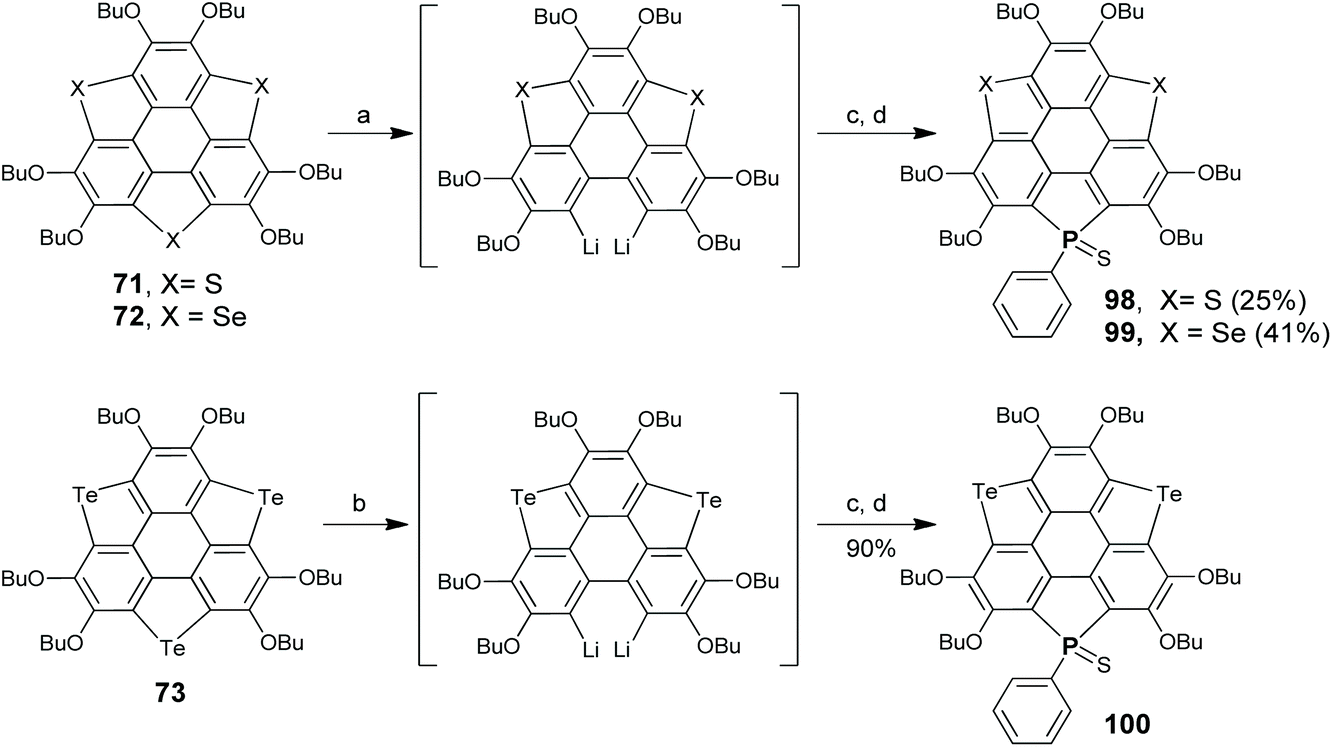
Aiming at this challenge, Shao's group proposed a ring-cyclization/ring-contraction two-step strategy and successfully prepared trithiasumanenes through route B under non-pyrolytic conditions (Scheme 23).81 In the ring-cyclization step, HBT was hexalithiated, followed by inserting S8 to form two 1,2-dithiin rings and one thiophene ring at the bay regions of HBT, i.e., compound 74. In the ring-contraction step, the dithiin rings on 74 were desulfurized by reacting with copper nanopowder in the solid state to give trithiasumanene 71, along with 75 which can be converted to 71. This progress led to two-step synthesis of triselenasumanene 7281 and one-pot synthesis of tritellurasumanene 73.82 A series of TCSs (77–82 in Scheme 24) containing different chalcogen atoms also have been created via this strategy.83 Compared to Otsubo's method, this approach shows advantages of cheaper starting materials, mild conditions, and large-scale production (ten gram scale). Moreover, it is a general method to create TCSs. It is worth noting that dechalcogenization of 74, 75, and 76 can be achieved via CuI-catalysis as reported by Wang's group in 2019.84
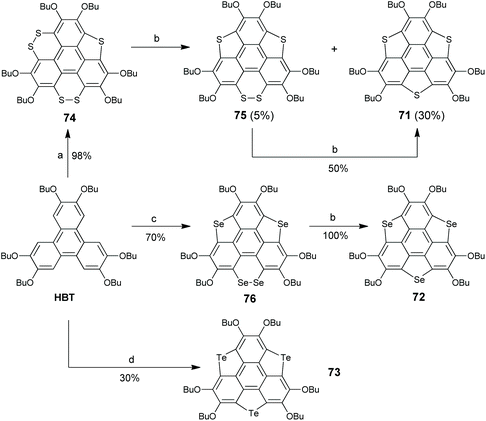 |
| | Scheme 23 Non-pyrolytic synthesis of TCSs 71–73 (see ref. 81 and 82). Reagents and conditions: (a) (i) TMEDA (10 eq.), n-BuLi (10 eq.), hexane, 60 °C, 3 h; (ii) S8 (15 eq.), −78 °C to r.t.; (b) Cu powder (80–100 nm, 10 eq.), 200 °C, 2 h; (c) (i) TMEDA (10 eq.), n-BuLi (10 eq.), hexane, 60 °C, 3 h; (ii) Se powder (10 eq.), −78 °C to r.t.; and (d) (i) n-BuLi (15 eq.), TMEDA (15 eq.), hexane, 60 °C, 3 h; (ii) Te powder (15 eq.), ultrasound, r.t., 12 h. | |
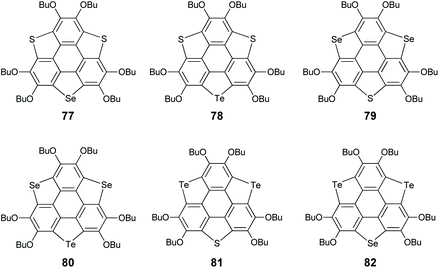 |
| | Scheme 24
TCSs containing different chalcogen atoms (see ref. 83). | |
In 2018, Tan and co-workers disclosed an approach toward trithiasumanenes via Pd-catalyzed intramolecular cyclodehydrogenation of S–H and C–H (Scheme 25).85 A two-step transformation of 56 afforded 83, which was converted to trithiasumanene 70 along with 84 under the catalysis of PdCl2/DMSO. Although this method works, it has drawbacks of low yields (2%) and utilization of a quantitative amount of Pd-catalyst.
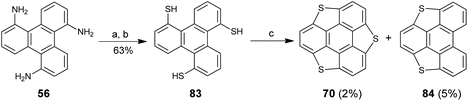 |
| | Scheme 25 Synthesis of trithiasumanenes via C–H activation (see ref. 85). Reagents and conditions: (a) NaNO2 (4.5 eq.), HCl–H2O, then KSCN (10 eq.), FeCl3 (1.5 eq.); (b) Na2S·9H2O (6 eq.), EtOH–H2O; and (c) PdCl2 (90 mol%), DMSO, 120 °C. | |
3.2.4.
benzylic-Heterasumanenes involving group 17 elements.
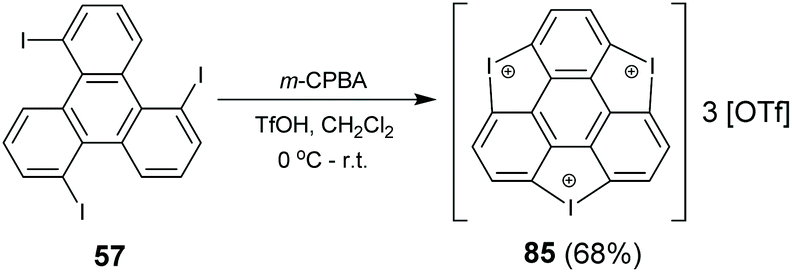
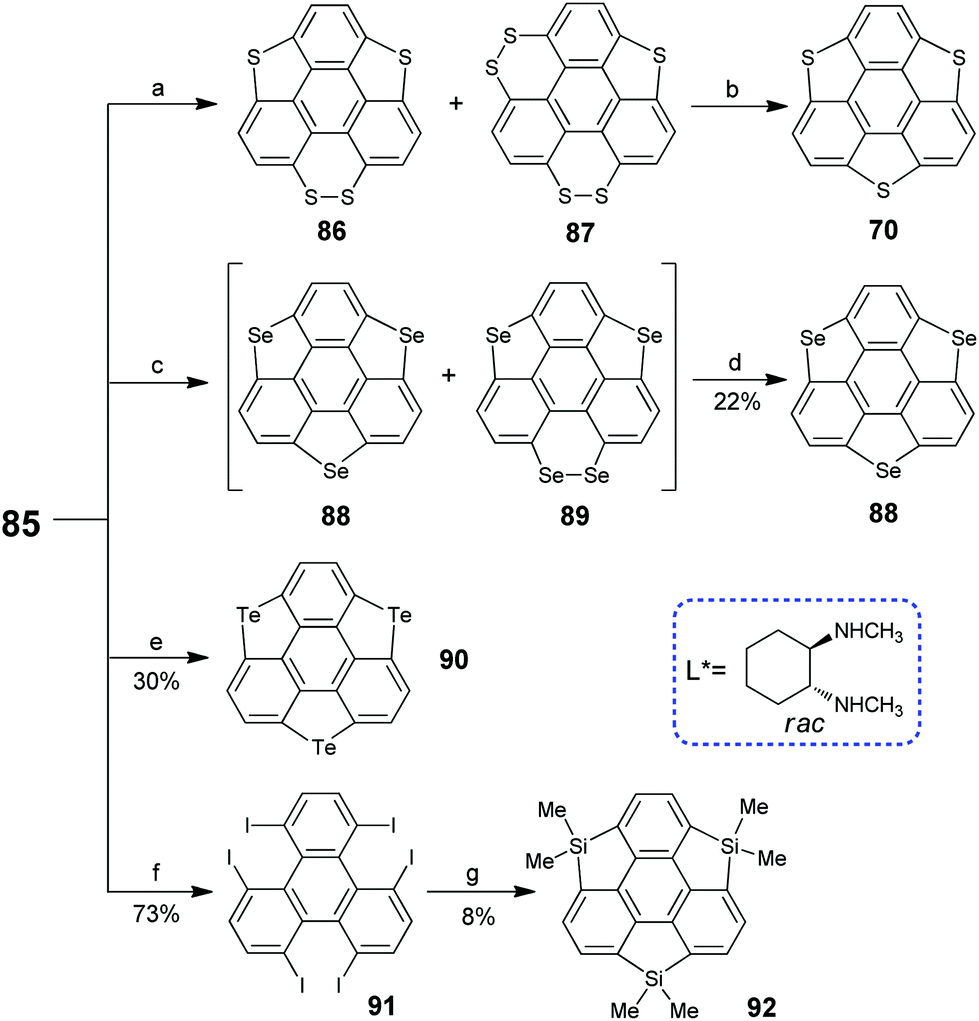
In 2017, Tan and co-workers synthesized triiodosumanene, the first halo-doped sumanene (Scheme 26).86 Treating 1,5,9-tribromotriphenylene 57 with m-CPBA in the presence of TfOH leads to the construction of three iodine bridges at the bay regions of triphenylene, i.e., triiodosumanene 85. In fact, 85 is an organohypervalent iodine compound. Organohypervalent iodine compounds are versatile synthons in organic chemistry87 and have been used to construct C–C, C–N, C–O, C–S, N–N and other bonds via “atom-transfer” oxidative coupling reactions.88 Therefore, 85 was used as a synthon to prepare the pristine trithia- (70), triselena- (88), tritellura- (90), and trisila-sumanenes (92) as shown in Scheme 27. This is an appealing approach to synthesize hetera-sumanenes. Moreover, 90 exhibits room-temperature phosphorescence in the solid state.89
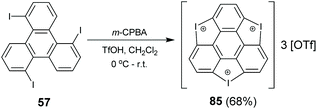 |
| | Scheme 26 Synthesis of triiodosumanene 85 (see ref. 86). | |
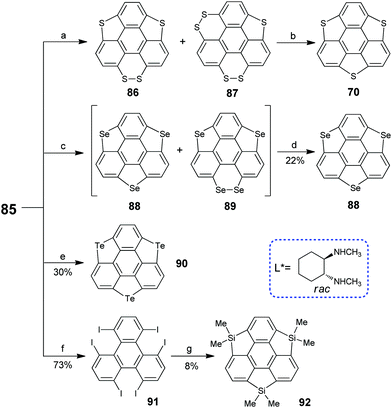 |
| | Scheme 27 Transformation of triiodosumanene into various hetera-sumanenes (see ref. 86 and 89). Reagents and conditions: (a) (i) AcSK (15 eq.), CuCl2 (0.12 eq.), DMSO, 110 °C, 86/87 > 20:1, 42% yield; or (ii) S8 (1.5 eq.), CsCO3 (15 eq.), DMSO, 12h, if 80 °C, 86/87 > 1:5.4, if 140 °C 86/87 > 14:1, 47% yield; (b) Cu (0.1 eq.), tetralin, 200 °C, 25% from 86, 58% from 87; (c) Se (9.0 eq.), KOtBu (15 eq.), DMSO, 70 °C; (d) Cu, tetralin, 200 °C; (e) Te (2 eq.), TBAB (2 eq.), 2-picoline-DMSO, 125 °C, 24 h, 30% yield; (f) KI (9 eq.), CuI (1.2 eq.), L*(2.4 eq.), DMSO, 70 °C, 20 h; and (g) n-BuLi (13.2 eq.), Me2SiCl2 (15 eq.), THF/toluene (1:1), −78 °C to r.t. | |
3.2.5.
benzylic-Heterasumanenes having heteroatoms from different groups.
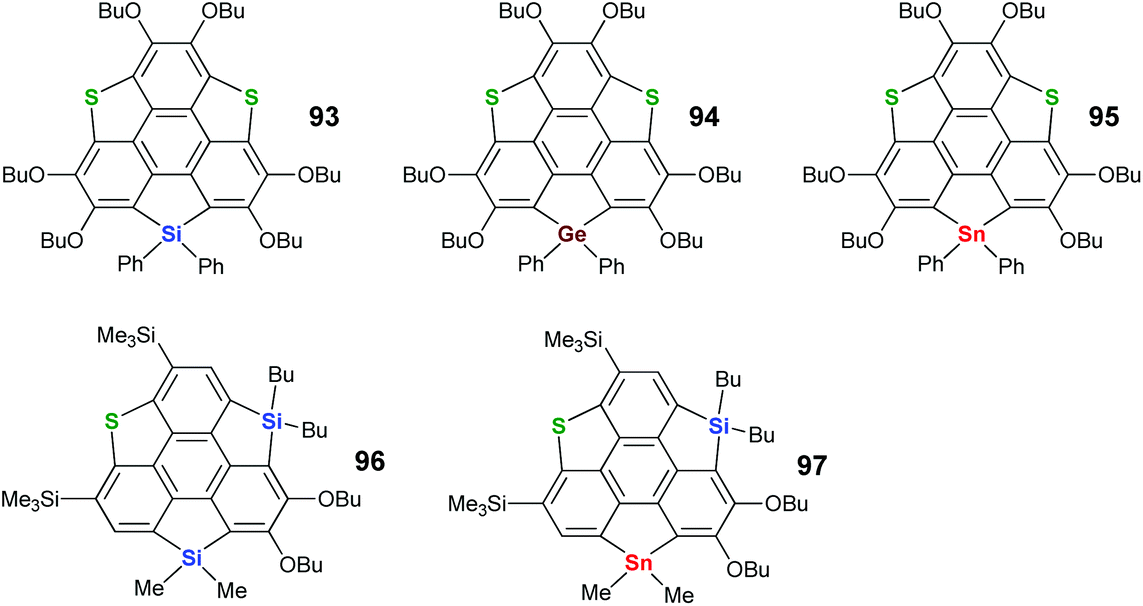
To create heterasumanenes with value-added functionalities, doping sumanenes with heteroatoms from different groups seems promising. With this intension, Shao's and Saito's groups exploited benzylic-heterasumanenes possessing heteroatoms from different groups. In 2010, Saito and co-workers developed a stepwise ring-cyclization approach to synthesize such hetera-sumanenes, and a series of hetera-sumanenes containing sulfur and group 14 elements (93–97 in Scheme 28) were successfully synthesized.90,91
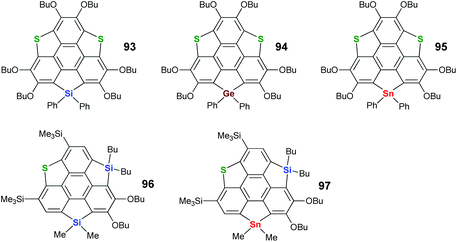 |
| | Scheme 28 Heterasumanenes co-doped with sulfur and group 14 elements (see ref. 90 and 91). | |
Recently, Shao's group produced a new type of hetera-sumanene co-doped with phosphorus and group 16 elements (98–100 in Scheme 29), which were derived from 71–73 by replacing one chalcogen atom with phosphorus.92 Upon treatment with n-BuLi, one of the chalcogenole rings on 71–73 opened to give dilithiated intermediates, which then reacted with PhPCl2, and then sulfur powder was added to obtain 98–100.92 This transformation has a large influence on the structural and electronic features, as well as chemical behaviors. The PS unit on 98–100 can coordinate with Ag+, leading to the first observation of a Ag⋯Te dative bond between Ag+ and a Te atom on tellurophene.92 Compound 98 is a sensitive fluorescent sensor for Ag+ with a limit of detection (0.21 μM) better than the WHO standard (0.50 μM) for drinking water.
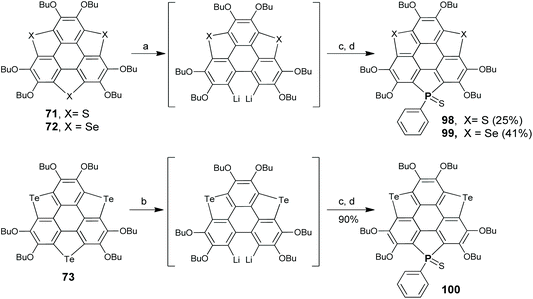 |
| | Scheme 29 Synthesis of heterasumanenes 98–100 (see ref. 92). Reagents and conditions: (a) n-BuLi (2.2 eq.), TMEDA, hexane, 60 °C, 3 h; (b) n-BuLi (2.2 eq.), TMEDA, hexane, −30 °C, 30 min; (c) PhPCl2 (5 eq.), THF, −78 °C to r.t., 4 h; and (d) S8 (10 eq.), r.t., 14 h. | |
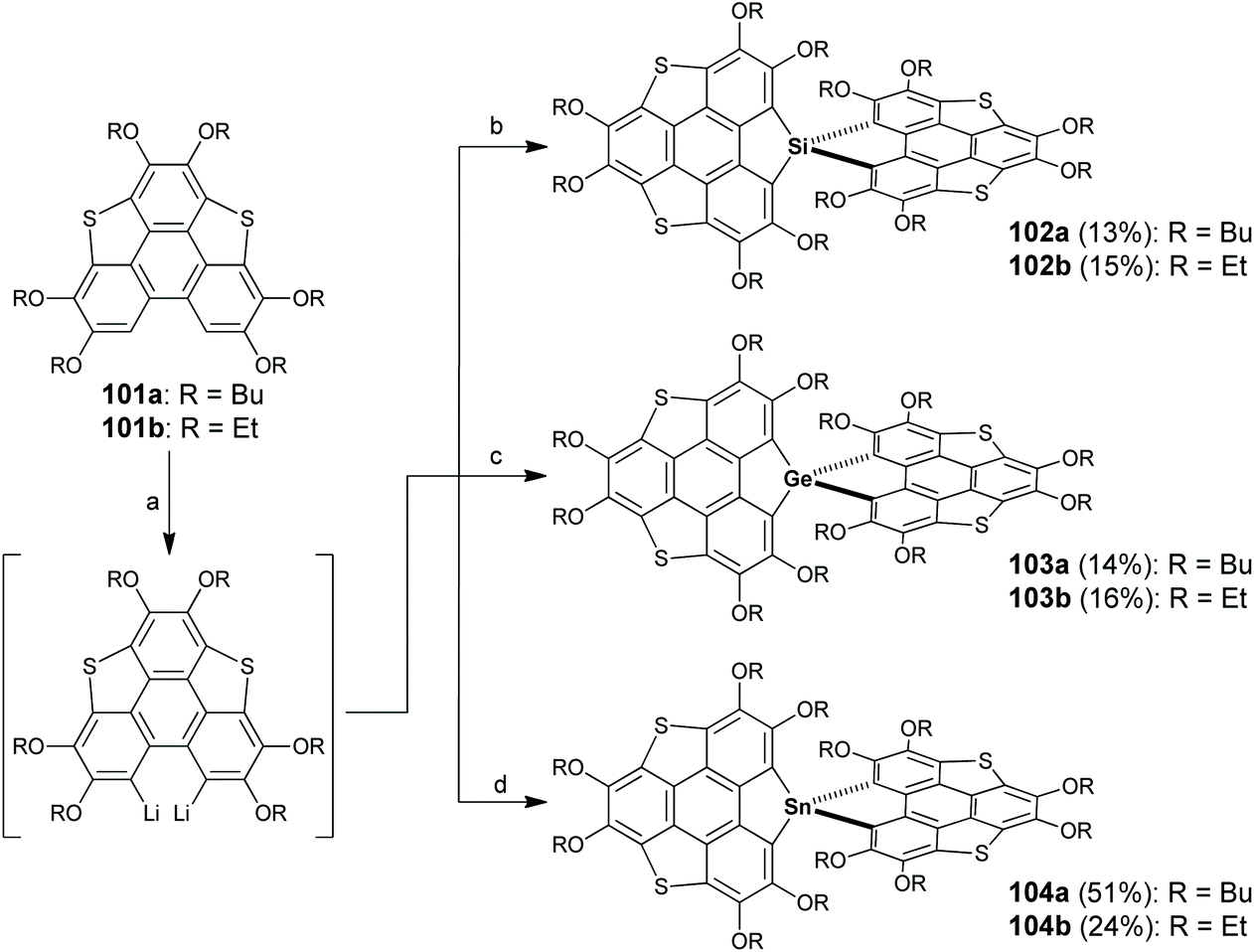
In 2018, Furukawa and co-workers reported spiro-type heterasumanenes which have sulfur and group 14 elements at benzylic positions (102–104 in Scheme 30).91 Compound 101 was dilithiated with two equivalents of n-BuLi, followed by the addition of heteroatom reagents (XCl4, X = Si, Ge, and Sn) to give 102–104. Crystallographic analyses revealed that two π-frameworks on 102–104 are almost orthogonally arrayed. Due to the existence of spiro-conjugation, the two π-frameworks show electronic communication in the oxidized state as proved by electrochemical, ESR, and theoretical investigations.91
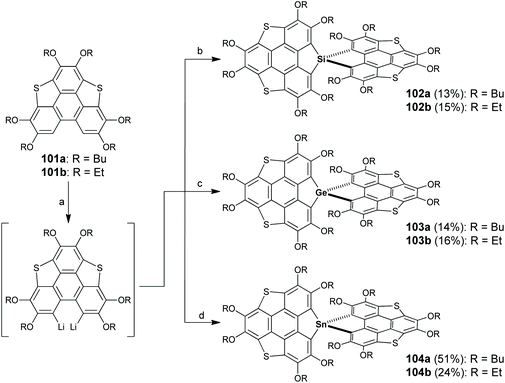 |
| | Scheme 30 Synthesis of spiro-type heterasumanenes 102–104 (see ref. 91). Reagents and conditions: (a) n-BuLi (2 eq.), TMEDA (2 eq.) hexane, reflux, 3 h; (b) for 102a, SiHCl3 (0.5 eq.), THF, −78 °C to r.t., then reflux, 12 h; for 102b, SiCl4 (0.5 eq.) THF, −78 °C to r.t., then reflux, 12 h; (c) GeCl4 (0.5 eq.), THF, −78 °C to r.t., then reflux, 12 h; and (d) SnCl4 (0.5 eq.), THF, −78 °C to r.t., then reflux, 12 h. | |
3.2.6. Reaction and transformation of TCSs.
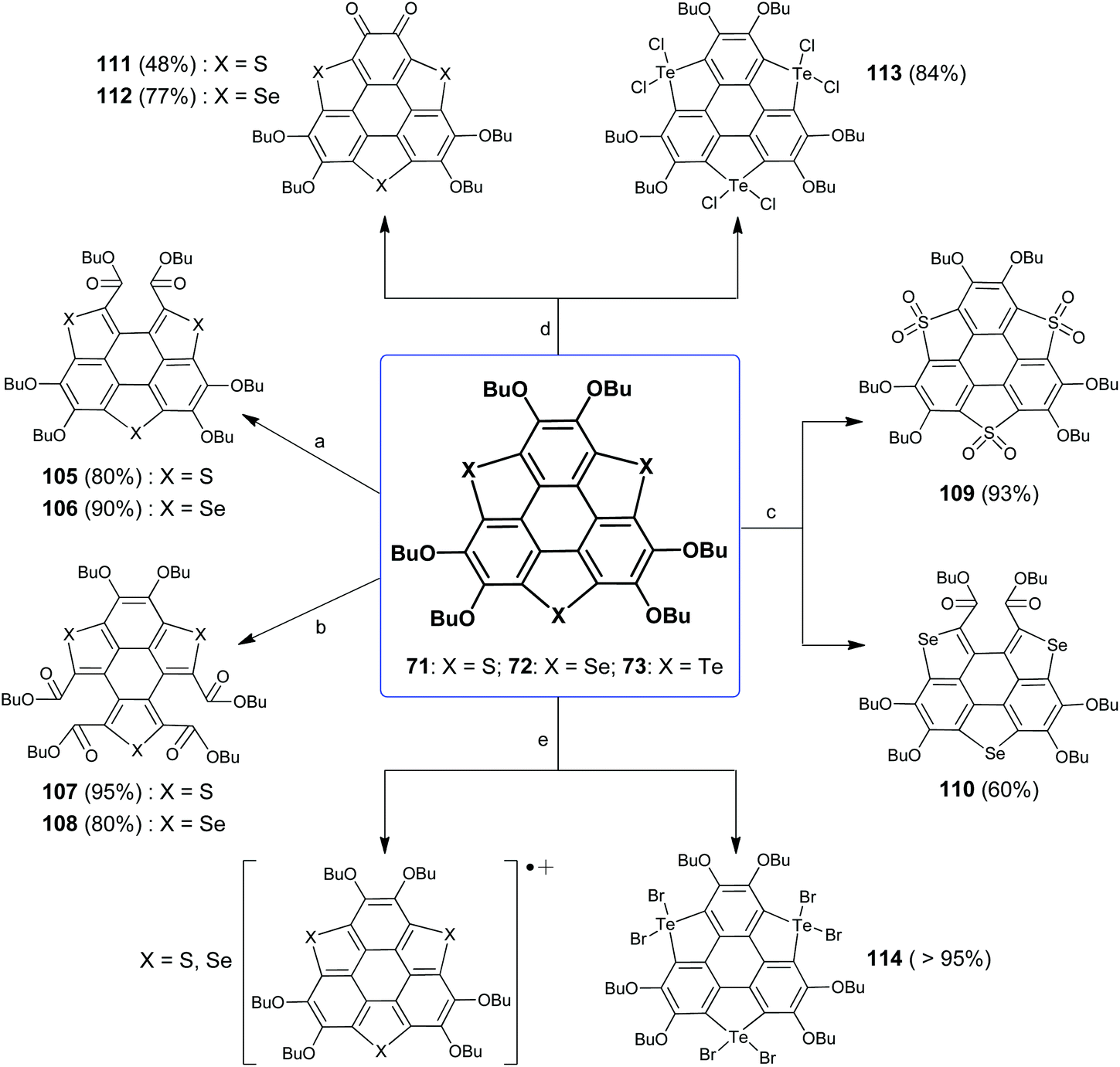
Among hetera-sumanenes so far obtained, the chemical behaviors of TCSs bearing six butoxy groups (71–73 and 77–82) have been thoroughly investigated.32d,82,93 Shao's group has evaluated the structure–property relationship of these TCSs and figured out the chemically active sites. Taking compound 71 for instance, it has three chemically active sites, the electron-rich rim CC bond, sulfur atom, and butoxy groups.
In connection with active sites, Shao's group has disclosed five kinds of reactions for 71. These reactions can be selectively performed (Scheme 31). Upon oxidation with oxone (condition a), one flank benzene ring on 71 is opened to form diester 105.93a Employing tert-butyl hydroperoxide (TBHP) as an oxidant (condition b), two flank benzene rings on 71 are broken to give tetraester 107.93d The thiophene rings on 71 are converted to thiophene-S,S-dioxides (109) under oxidation with H2O2 (condition c).93b Two butoxy groups on the same rim benzene ring of 71 are converted to ortho-quinone (111) in the presence of FeCl3 (condition d).93c71 is electron-rich owing to butoxy groups and thiophene moieties; thus charge-transfer occurs between 71 and Br2 to form the radical cation (71)˙+ as proved by ESR and absorption spectra (condition e).82
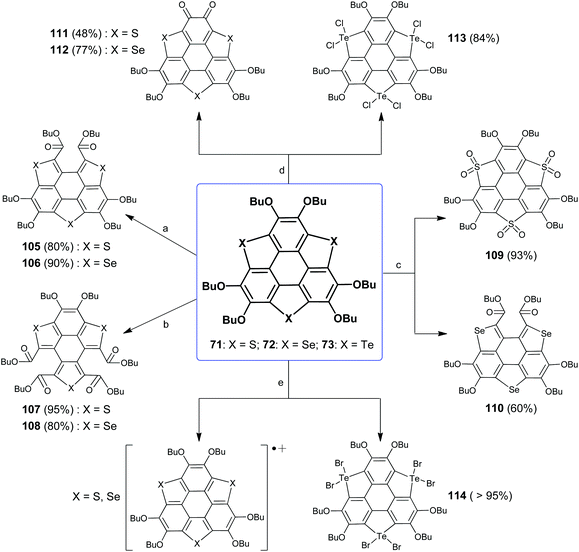 |
| | Scheme 31 Chemical reactions of TCSs 71–73 (see ref. 82 and 93). Reagents and conditions: (a) oxone (2 eq.), THF–H2O (v/v = 4:1), r.t., 2 h; (b) TBHP (6 eq.), CH2Cl2, 40 °C, 5 h; (c) H2O2 (30% aq.), CH2Cl2–AcOH (v/v = 1:2), 60 °C, 24 h; (d) FeCl3 (10 eq.), CH2Cl2–CH3NO2 (v/v = 1:1), r.t., 2 h; and (e) Br2 (10 eq.), CH2Cl2, r.t. | |
It is found that chalcogen atoms play an important role in the chemical reactivities of these TCSs. The reactions of 72 are same as those of 71 under conditions a, b, d, and e, which result in 106, 108, 112, and (72)˙+, respectively. An exceptional case is that one flank benzene ring on 72 is broken under condition c.93b73 shows different behaviors, and all the reactions occur at Te atoms. Particularly, six halogen atoms attach onto three Te atoms under conditions d and e, respectively, affording hypervalent adducts 113 and 114.82,93c On the other hand, the chemical reactivities of TCSs are dominated by the heavier chalcogen atom when the TCSs contain different chalcogen atoms.83
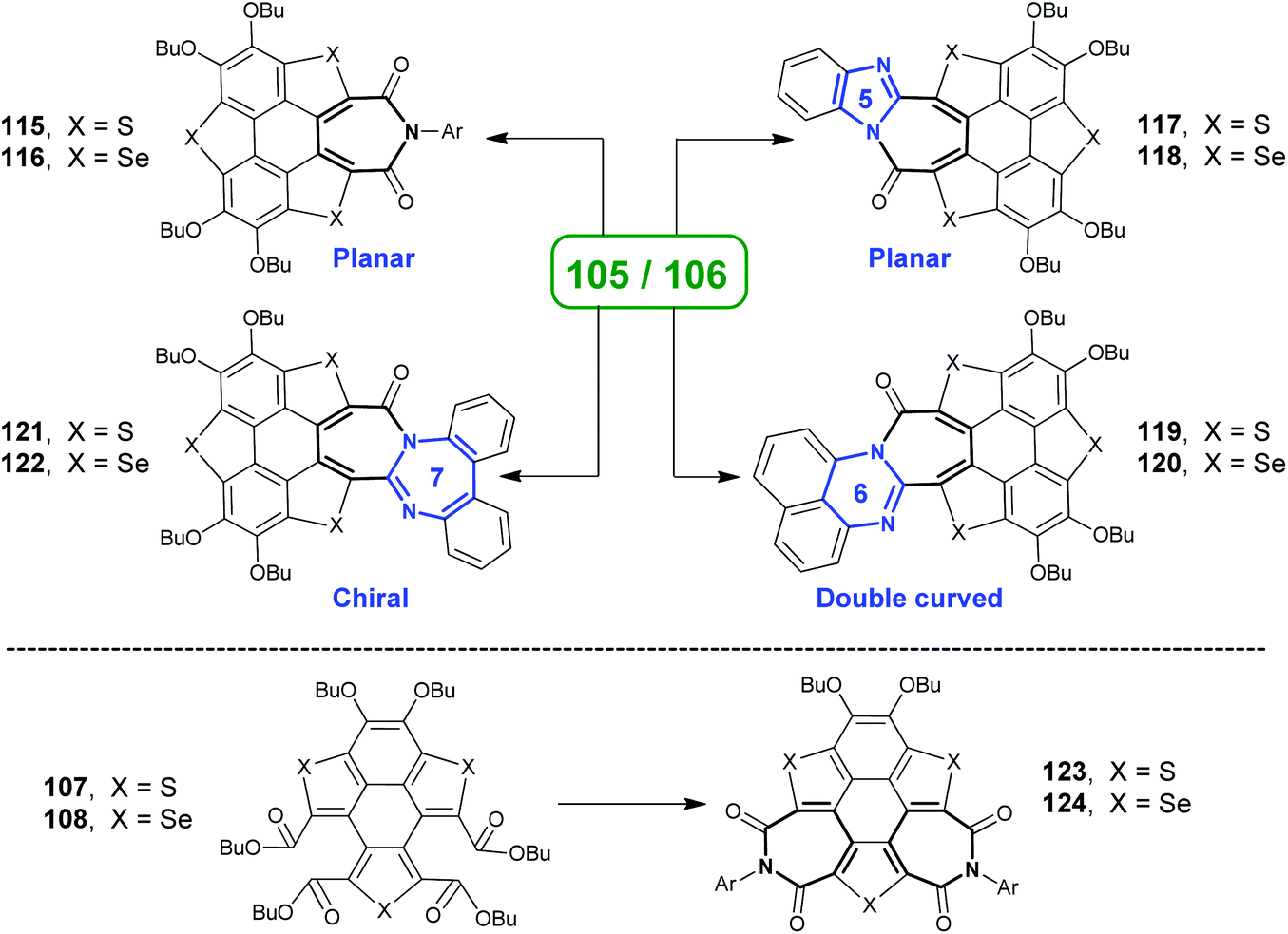
Employing these reactions as key step, TCSs 71–72 were transformed into various heteropolycycles.93–95 The ring-opening products 105/106 were converted into 115/116, 117/118, 119/120, and 121/122 as shown in Scheme 32.93a,94115/116 adopt a flat π-skeleton and show red emission (ΦF = 40%).93a As the size of amidine rings (painted in blue) increases from 117/118 to 119/120 to 121/122, the molecular geometries show distinct variation. 117/118 show a planar skeleton and exhibit strong red emission (ΦF = 52%) and two-photo absorption. 119/120 adopt a double curved geometry that is expected to show chirality, but the quick geometry inversion results in racemization. 121/122 are highly twisted and show stable chirality.94 Moreover, two-benzene-ring-opening products 107/108 were transformed into diimides 123/124, which show excellent optical-limiting (OL) behavior superior to that of C60.93d
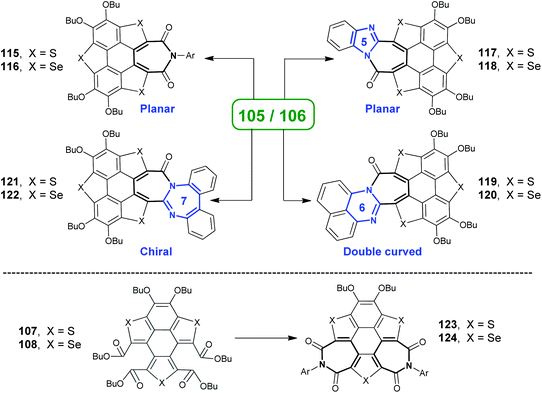 |
| | Scheme 32 Transformation of ring-opening products of TCSs into heteropolycycles (see ref. 93 and 94). | |
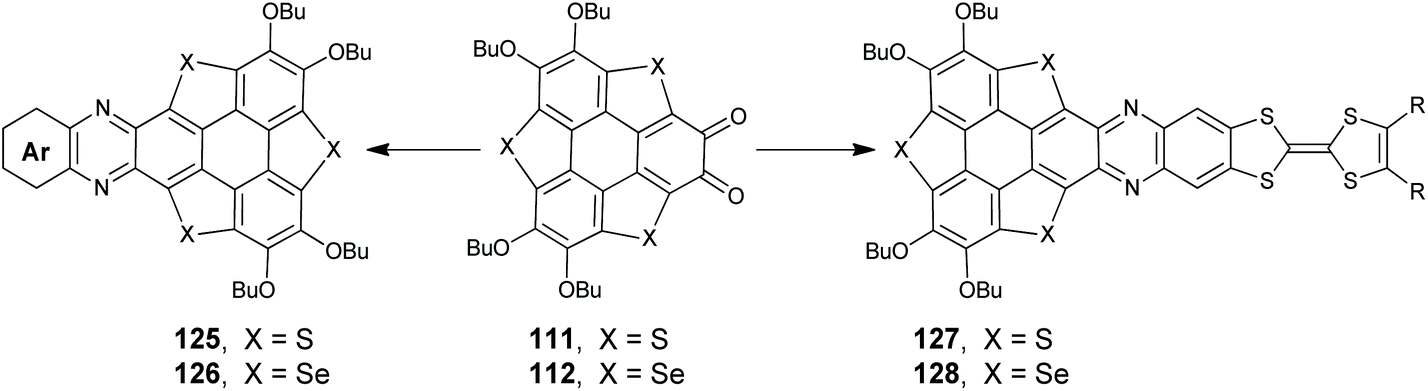
Incorporating pyridine and/or pyrazine units onto PAHs is an efficient method to tailor their optoelectronic properties.95 The construction of a pyrazine moiety can be conveniently achieved through the condensation of ortho-quinone and 1,2-diamine species. With the intention to gain pyrazine-fused TCSs, Shao's group set out to condense the ortho-quinones 111/112 and a variety of 1,2-diamino arenes (Scheme 33). In 2017, they synthesized 125/126 with a donor–acceptor (D–A) structure.93c125 adopts a spoon-like skeleton, and 126 takes a planar shape. The fusion of a pyrazine unit onto TCSs has a large influence on their optoelectronic properties. The D–A type structure will result in an intramolecular charge-transfer (ICT) transition, and accordingly 125/126 display distinctly red-shifted absorption and emission as compared to 71/72.93c Moreover, 125/126 exhibit OL behavior due to the large transition dipole moment arising from the D–A structure.9672 shows p-type semiconducting behavior with a hole mobility of 0.32 cm2 V−1 s−1,97 whereas the transport properties of 126 exhibit a high photosensitivity up to 2.58 × 107.98
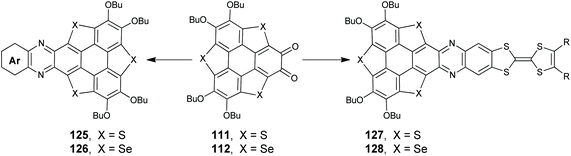 |
| | Scheme 33 Transformation of TCSortho-quinones into pyrazine-fused TCSs (see ref. 93 and 95). | |
Recently, electron-donor tetrathiafulvalene (TTF) was harnessed onto 71/72.95 The resulting 127/128 showed a donor–acceptor–donor (D1–A–D2) structure, where TTF, TCS, and pyrazine moieties acted as D1, D2, and A, respectively. Compared to D–A type 125/126, the absorptions of 127/128 shift bathochromically because both D1 and D2 moieties contribute to the ICT transition of 127/128. A femtosecond transient spectroscopic study reveals that 127/128 have two charge-separated transient states due to the ICT between (D1)+˙ and (D2)0 in the excited state. Moreover, 127/128 show protonation-induced intramolecular electron-transfer and self-sensitized photooxidation.95
4. Hetera π-bowls other than hetera-buckybowls
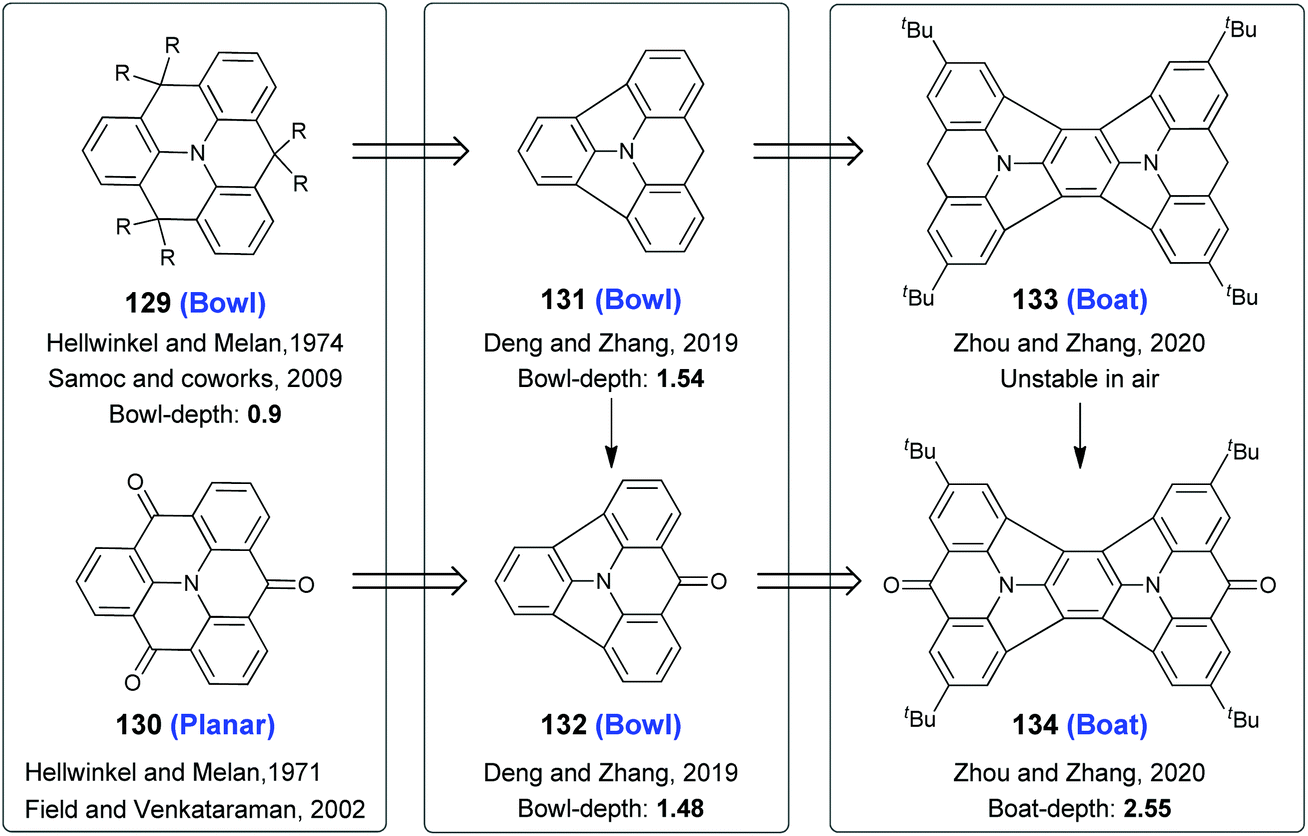
Among heteroatom-centered triangulenes,35 the boron-centered ones are planar,99,100 and the phosphorus-101 and silicon-centered100 ones take bowl-shaped conformations. On the other hand, the molecular geometries of N-centered triangulenes depend on ortho-bridging atoms.102 For example, 129102b,f and 130102a,c adopt bowl-shaped and planar skeletons (Scheme 34), respectively. To gain deep π-bowls based on N-centered triangulenes, two carbon-bridges on 129 and 130 were removed. The resulting 131 and 132 were all bowl-shaped with bowl-depths of 1.54 Å and 1.48 Å respectively, as reported by Deng and Zhang in 2019.103 This strategy was further extended by Zhou and Zhang, leading to the creation of molecular boats 133 and 134.104
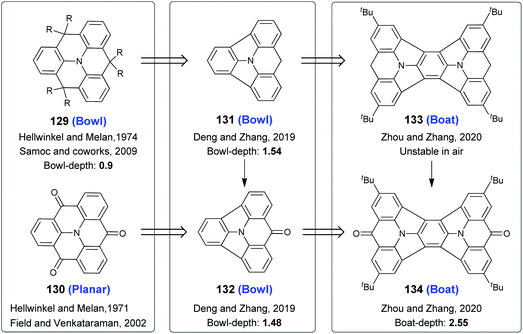 |
| | Scheme 34 Generation of π-bowls and molecular boats from nitrogen-centered triangulenes (see ref. 102–104). | |
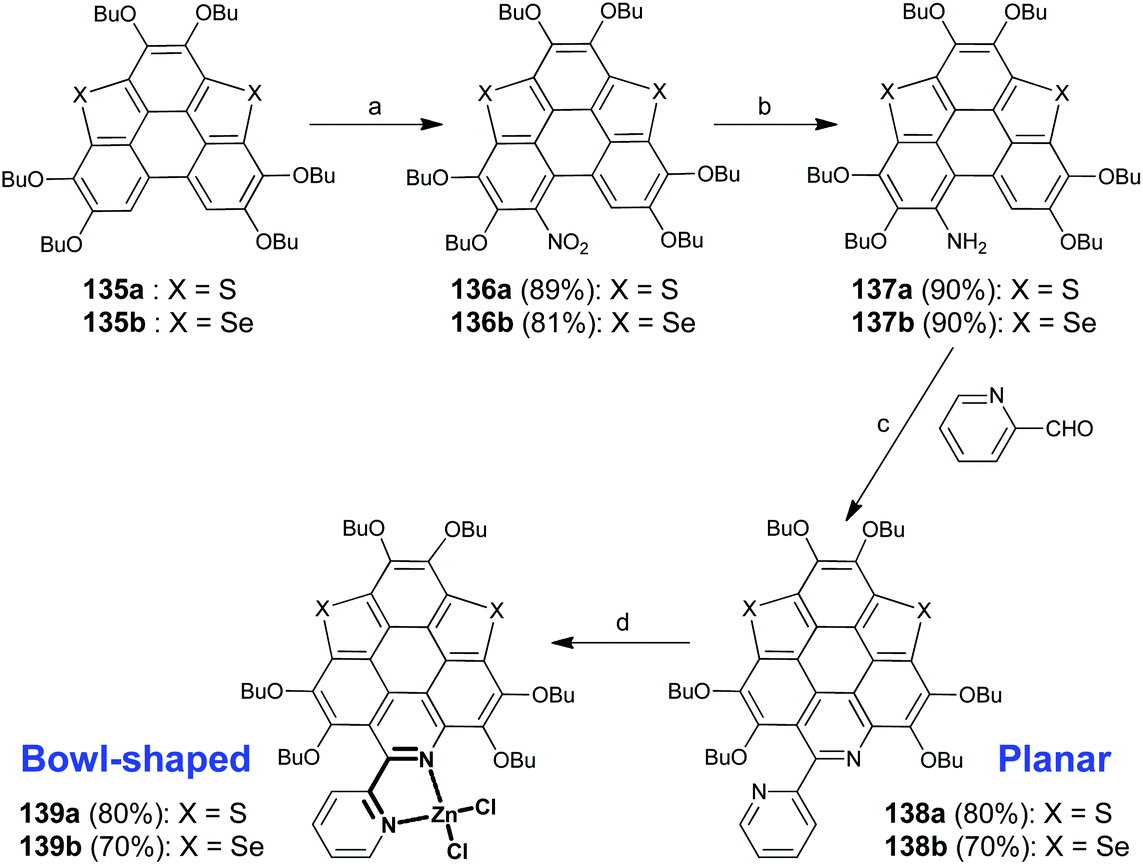
Apart from introducing pentagonal rings into the hexagonal sheets of PAHs, the π-bowls can also be generated via a lateral coordination. Recently, Shao's group disclosed a coordination-induced π-plane to π-bowl transformation.105 As shown in Scheme 35, the planar heteropolycycles bearing 2,2′-bipyridine (bpy) units (compounds 138a and 138b) were prepared from 135a/135b in three steps including nitration by tert-butyl nitrite (TBN), reduction by the zinc metal, and Pictet–Spengler reaction106 with picolinaldehyde. The bpy units on 138a/138b then chelate Zn2+ to form 139a/139b, whose π-frameworks adopt a bowl-shaped conformation. This transformation leads to not only a significant variation in the molecular geometry but also a distinct change in fluorescence, i.e., green and red emissions for 138a/138b and 139a/139b, respectively.
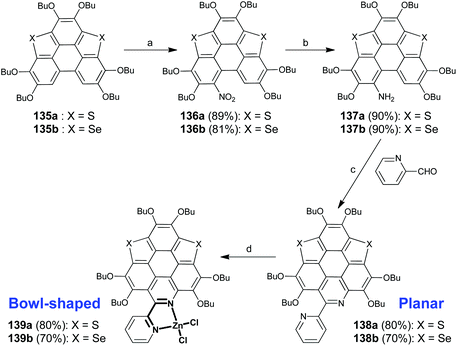 |
| | Scheme 35 Synthesis of hetera π-bowls via lateral coordination (see ref. 105). Reagents and conditions: (a) TBN (3 eq.), CH2Cl2, RT, 4h; (b) Zn (5 eq.), AcOH–EtOH (v/v = 1:70), 0 °C to r.t., 3 h; (c) picolinaldehyde (2 eq.), TFA, sealed Schlenk tube, 120 °C, 12 h; and (d) ZnCl2, dry CH2Cl2, r.t. | |
5. Physicochemical properties and applications
The research interest on buckybowls was initially inspired by the curiosity to create these unique π-conjugated systems. Recently, the development of buckybowls has been further stimulated by their prospects in multiple applications.30–34 Doping the π-scaffolds of buckybowls with heteroatoms is an efficient way to manipulate electronic structures, and consequently brings new functionalities and/or improved performance strongly related to the heteroatoms. To date, hetera-buckybowls have been successfully applied in coordination chemistry, supramolecular chemistry, and optoelectronic materials with excellent performance. Several representative examples are shown as follows.
5.1. Coordination chemistry
syn-62, 98, 99, and 100 possess PS units, which can coordinate with noble metals through S⋯M (M, noble metal) dative bonds. For example, the sulfur atoms of syn-62 bind to the Au(111) surface with an adsorption energy of 93.5 kcal mol−1.7898, 99, and 100 form complexes with Ag+via PS⋯Ag+ dative bonds. Particularly, the Ag⋯Te dative bond is observed between Ag+ and a Te atom on tellurophene of 100.92syn-63Pr has three phosphine groups which coordinate with Au+via P⋯Au+ bonds to form a complex (AuCl)3·(syn-63Pr).79
5.2. Supramolecular chemistry
Compound 109 possesses an electropositive center, and it forms a 1:1 cocrystal with HBTvia electrostatic interactions.93b The resulting cocrystal, (109)·HBT, has a much narrower HOMO–LUMO gap than HBT and 109, and consequently it displays yellow emission to be distinct from the violet emission of HBT and the indigo emission of 109. The hub-azacorannulene 15 is electron-rich and shows a shape complementary with C60. As a result, it shows tight binding with electron-deficient C60 in solution and forms 1:1 cocrystals with C60.47 The hub-azacorannulene dimer 18 also exhibits strong complexation with C60, whereas the stoichiometries of the complexes depend on the reaction conditions. The molecular ratios of 18 and C60 are 1:1 and 1:2 in solution and in the cocrystal, respectively.49 The hub-azacorannulene based molecular tweezers 19a and 19b are promising hosts for fullerene molecules.50 The former prefers to bind with C70, and the latter preferentially interacts with C60.
5.3. Optoelectronic materials
Compound 35 is used as an emitter to fabricate an OLED, and the resulting device shows high performance with the maximum luminance of 10400 cd m−2.61 Compound 90 exhibits room-temperature phosphorescence in the solid state.89 Due to the large transition dipole moment arising from the D–A structure, 123–126 show excellent OL performance superior to that of C60.93d,96 It has been proved that 98 is a sensitive fluorescent sensor for Ag+ with a limit of detection (0.21 μM) better than the WHO standard for drinking water.92syn-62 is employed to fabricate a molecular junction via adsorption onto the Au(111) surface.7872 shows p-type semiconducting behavior with a hole mobility of 0.32 cm2 V−1 s−1,97 and the transport properties of 126 exhibit a high photosensitivity up to 2.58 × 107.98
6. Conclusions and prospective
As discussed in the previous17 and present reviews, buckybowls have attracted intensive attention from the fundamental standpoint and also their application in various fields. While buckybowl chemistry goes back to 1970s, until recently great progress has been made in hetera-buckybowls. Hetera-buckybowls are derived from buckybowls by replacing their carbon atoms with heteroatoms (Si, Sn, Ge, N, P, S, Se, Te, and I) and/or replacing the C–C bond with a B–N bond. The main challenge with hetera-buckybowls is finding ways to create these unique entities. Aiming at this purpose, a variety of synthetic strategies have been developed such as FVP, tandem ring-opening and ring-closure metathesis reactions, 1,3-dipolar cycloaddition of azomethine ylides and CC/CC, intramolecular cyclodehydrogenation of X–H and C–H, successive ring-cyclization and ring-contraction, and “atom-transfer” reaction of organohypervalent iodine compounds. The synthetic progress could furnish multifarious hetera-buckybowls, yet the drawback lies in the lack of atom- and/or step-efficiency. The improvement of the synthetic strategy for hetera-buckybowls that meets the environmental and economical requirements is still in great demand.
Owing to the influence of heteroatoms on structural and electronic characteristics, hetera-buckybowls show different chemical and optoelectronic properties as compared to their parents. For example, the hub-azacorannulene can be controllably transformed into radical cation, radical anion, and dianion species, and its radical cation shows reversible dimerization behavior in solution. The TCSs bearing six butoxy groups show regio-selective chemical reactions. Meanwhile, hetera-buckybowls have found their way to applications in probing metal ions, hosting fullerenes, and displaying room-temperature phosphorescence. They can also act as active materials to fabricate OFETs, OLEDs, phototransistors, single-molecular junctions, and optical limiting devices with excellent performance.
While significant progress has been achieved in the synthesis and application of hetera-buckybowls, the current research has not been developed sufficiently to explore the irreplaceable properties of these peculiar entities. We are confident that the continuing endeavor to evaluate the structure–property relationship will give deeper insights into the inherent nature of hetera-buckybowls and put forward exciting discoveries.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the financial support from the National Natural Science Foundation of China (21871119 and 21522203) and the National Key R&D Program of China (2017YFA0204903).
References
- H. Kallmann and M. Pope, Nature, 1960, 186, 31 CrossRef.
-
(a)
Functional Organic Materials, ed. T. J. Muller and U. H. F. Bunz, Wiley-VCH, Weinheim, Germany, 2006 Search PubMed;
(b) Y. Shirota, J. Mater. Chem., 2000, 10, 1 RSC;
(c) M. Bendikov, F. Wudl and D. F. Perepichka, Chem. Rev., 2004, 104, 4891 CrossRef CAS;
(d) T. W. Kelley, P. F. Baude, C. Gerlach, D. E. Ender, D. Muyres, M. A. Haase, D. E. Vogel and S. D. Theiss, Chem. Mater., 2004, 16, 4413 CrossRef CAS;
(e) J. E. Anthony, Chem. Rev., 2006, 106, 5028 CrossRef CAS;
(f) Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742 CrossRef CAS.
-
(a) Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef CAS;
(b) A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897 CrossRef CAS;
(c) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS.
-
(a) J. J. Perry IV, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400 RSC;
(b) Y. He, B. Li, M. O'Keeffec and B. Chen, Chem. Soc. Rev., 2014, 43, 5618 RSC.
-
(a) L. Zang, Y. Che and J. S. Moore, Acc. Chem. Res., 2008, 41, 1596 CrossRef CAS;
(b) L. Basabe-Desmonts, D. N. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 993 RSC.
- M. D. Watson, A. Fechtenkötter and K. Müllen, Chem. Rev., 2001, 101, 1267 CrossRef CAS.
-
(a)
E. Clar, Polycyclic Hydrocarbons, Springer, Berlin, 1964, vol. 2 Search PubMed;
(b)
R. G. Harvey, Polycyclic Aromatic Hydrocarbons, Wiley-VCH, New York, 1997 Search PubMed.
- T. Yu, L. Liu, Z. Xie and Y. Ma, Sci. China: Chem., 2015, 58, 907 CrossRef CAS.
-
(a) Y. Zheng, J. Wang and J. Pei, Sci. China: Chem., 2015, 58, 937 CrossRef CAS;
(b) X. Gao and Z. Zhao, Sci. China: Chem., 2015, 58, 947 CrossRef CAS.
-
(a) O. Inganäs, Adv. Mater., 2018, 30, 1800388 CrossRef;
(b) P. Cheng, G. Li, X.-W. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131 CrossRef CAS;
(c) J. Urieta-Mora, I. García-Benito, A. Molina-Ontoria and N. Martín, Chem. Soc. Rev., 2018, 47, 8541 RSC.
-
(a) Y. Son, M. L. Cohen and S. G. Louie, Nature, 2006, 444, 347 CrossRef CAS;
(b) V. A. Dediu, L. E. Hueso, I. Bergenti and C. Taliani, Nat. Mater., 2009, 8, 707 CrossRef CAS.
-
(a) M. Müller, C. Kübel and K. Müllen, Chem. – Eur. J., 1998, 4, 2099 CrossRef;
(b) J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452 CrossRef CAS;
(c) X. Feng, W. Pisula and K. Müllen, Pure Appl. Chem., 2009, 81, 2203 CAS;
(d) W. Jiang, Y. Li and Z. Wang, Acc. Chem. Res., 2014, 47, 3135 CrossRef CAS;
(e) A. Narita, X.-Y. Wang, X. Feng and K. Müllen, Chem. Soc. Rev., 2015, 44, 6616 RSC;
(f) U. Beser, M. Kastler, A. Maghsoumi, M. Wagner, C. Castiglioni, M. Tommasini, A. Narita, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 4322 CrossRef CAS;
(g) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479 CrossRef;
(h) M. Stępień and D. Myśliwiec, Angew. Chem., Int. Ed., 2019, 58, 86 CrossRef;
(i)
Polycyclic Arenes and Heteroarenes: Synthesis, Properties, and Applications, ed. Q. Miao, John Wiley and Sons, 2015 Search PubMed.
- For selected examples of unconventional PAHs published recently:
(a) K. Kawasumi, Q. Zhang, Y. Segawa, L. T. Scott and K. Itami, Nat. Chem., 2013, 5, 739 CrossRef CAS;
(b) N. Nishina, M. Makino and J.-I. Aihara, J. Phys. Chem. A, 2016, 120, 2431 CrossRef CAS;
(c) J. Liu, S. Osella, J. Ma, R. Berger, D. Beljonne, D. Schollmeyer, X. Feng and K. Müllen, J. Am. Chem. Soc., 2016, 138, 8364 CrossRef CAS;
(d) G. Dai, J. Chang, J. Luo, S. Dong, N. Aratani, B. Zheng, K.-W. Huang, H. Yamada and C. Chi, Angew. Chem., Int. Ed., 2016, 55, 2693 CrossRef CAS;
(e) Y. Li, Z. Jia, S. Xiao, H. Liu and Y. Li, Nat. Commun., 2016, 7, 11637 CrossRef;
(f) G. E. Rudebusch, J. L. Zafra, K. Jorner, K. Fukuda, J. L. Marshall, I. Arrechea-Marcos, G. L. Espejo, R. P. Ortiz, C. J. Gómez-García, L. N. Zakharov, M. Nakano, H. Ottosson, J. Casado and M. M. Haley, Nat. Chem., 2016, 8, 753 CrossRef CAS;
(g) C. Liu, M. E. Sandoval-Salinas, Y. Hong, T. Y. Gopalakrishna, H. Phan, N. Aratani, T. S. Herng, J. Ding, H. Yamada, D. Kim, D. Casanova and J. Wu, Chem, 2018, 4, 1586 CrossRef CAS;
(h) X. Lu, T. Y. Gopalakrishna, H. Phan, T. S. Herng, Q. Jiang, C. Liu, G. Li, J. Ding and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 13052 CrossRef CAS;
(i) X. Lu, T. Y. Gopalakrishna, Y. Han, Y. Ni, Y. Zou and J. Wu, J. Am. Chem. Soc., 2019, 141, 5934 CrossRef CAS;
(j) I. C.-Y. Hou, Q. Sun, K. Eimre, M. D. Giovannantonio, J. I. Urgel, P. Ruffieux, A. Narita, R. Fasel and K. Müllen, J. Am. Chem. Soc., 2020, 142, 10291 CrossRef CAS;
(k) H. Xin, J. Li, X. Yang and X. Gao, J. Org. Chem., 2020, 85, 70 CrossRef CAS;
(l) H. Xin, J. Li, R.-Q. Lu, X. Gao and T. M. Swager, J. Am. Chem. Soc., 2020, 142, 13598 CrossRef CAS;
(m) Y. Han, Z. Xue, G. Li, Y. Gu, Y. Ni, S. Dong and C. Chi, Angew. Chem., 2020, 59, 9026 CrossRef CAS.
-
(a) X. Feng, V. Marcon, W. Pisula, M. R. Hansen, J. Kirkpatrick, F. Grozema, D. Andrienko, K. Kremer and K. Müllen, Nat. Mater., 2009, 8, 421 CrossRef CAS;
(b) J. Liu, B.-W. Li, Y.-Z. Tan, A. Giannakopoulos, C. Sanchez-Sanchez, D. Beljonne, P. Ruffieux, R. Fasel, X. Feng and K. Müllen, J. Am. Chem. Soc., 2015, 137, 6097 CrossRef CAS;
(c) K. Baumgartner, A. L. M. Chincha, A. Dreuw, F. Rominger and M. Mastalerz, Angew. Chem., Int. Ed., 2016, 55, 15594 CrossRef;
(d) H. Hopf, Chem. Rec., 2014, 14, 979 CrossRef CAS;
(e) Y.-Z. Tan, S. Osella, Y. Liu, B. Yang, D. Beljonne, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2015, 54, 2927 CrossRef CAS.
- W. Pisula, X. Feng and K. Müllen, Adv. Mater., 2010, 22, 3634 CrossRef CAS.
-
(a) S. Yamaguchi and K. Tamao, Chem. Lett., 2005, 34, 2 CrossRef CAS;
(b) H. Dong, H. Zhu, Q. Meng, X. Gong and W. Hu, Chem. Soc. Rev., 2012, 41, 1754 RSC;
(c) C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208 CrossRef CAS;
(d) J. Mei, Y. Diao, A. L. Appleton, L. Fang and Z. Bao, J. Am. Chem. Soc., 2013, 135, 6724 CrossRef CAS;
(e) W. Jiang, Y. Li and Z. Wang, Chem. Soc. Rev., 2013, 42, 6113 RSC;
(f) S. M. Parke, M. P. Boone and E. Rivard, Chem. Commun., 2016, 52, 9485 RSC.
- For reviews, see:
(a) P. W. Rabideau and A. Sygula, Acc. Chem. Res., 1996, 29, 235 CrossRef CAS;
(b) Y.-T. Wu and J. S. Siegel, Chem. Rev., 2006, 106, 4843 CrossRef CAS;
(c) V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868 CrossRef CAS;
(d) T. Amaya and H. Hirao, Chem. Commun., 2011, 47, 10524 RSC;
(e) A. Sygula, Eur. J. Org. Chem., 2011, 1611 CrossRef CAS;
(f) S. Higashibayashi and H. Sakurai, Chem. Lett., 2011, 40, 122 CrossRef CAS;
(g) Y.-T. Wu and J. S. Siegel, Top. Curr. Chem., 2014, 349, 63 CrossRef CAS;
(h) T. Amaya and T. Hirao, Chem. Rec., 2015, 15, 310 CrossRef CAS;
(i) M. Saito, H. Shinokubo and H. Sakurai, Mater. Chem. Front., 2018, 2, 635 RSC.
-
(a) K. Tahara and Y. Tobe, Chem. Rev., 2006, 106, 5274 CrossRef CAS;
(b) H. Omachi, Y. Segawa and K. Itami, Acc. Chem. Res., 2012, 45, 1378 CrossRef CAS;
(c) S. E. Lewis, Chem. Soc. Rev., 2015, 44, 2221 RSC;
(d) M. R. Golder and R. Jasti, Acc. Chem. Res., 2015, 48, 557 CrossRef CAS;
(e) Y. Segawa, A. Yagi, K. Matsui and K. Itami, Angew. Chem., Int. Ed., 2016, 55, 5136 CrossRef CAS.
-
(a) E. Nakamura, K. Tahara, Y. Matsuo and M. Sawamura, J. Am. Chem. Soc., 2003, 125, 2834 CrossRef CAS;
(b) D. Eisenberg, R. Shenhar and M. Rabinovitz, Chem. Soc. Rev., 2010, 39, 2879 RSC;
(c) G. Povie, Y. Segawa, T. Nishihara, Y. Miyauchi and K. Itami, Science, 2017, 356, 172 CrossRef CAS;
(d) G. Povie, Y. Segawa, T. Nishihara, Y. Miyauchi and K. Itami, J. Am. Chem. Soc., 2018, 140, 10054 CrossRef CAS;
(e) T.-H. Shi, Q.-H. Guo, S. Tong and M.-X. Wang, J. Am. Chem. Soc., 2020, 142, 4576 CrossRef CAS;
(f) Q. Zhang, Y.-E. Zhang, S. Tong and M.-X. Wang, J. Am. Chem. Soc., 2020, 142, 1196 CrossRef CAS.
- H. Omachi, T. Nakayama, E. Takahashi, Y. Segawa and K. Itami, Nat. Chem., 2013, 5, 572 CrossRef CAS.
- See representative examples:
(a) C.-N. Feng, M.-Y. Kuo and Y.-T. Wu, Angew. Chem., Int. Ed., 2013, 52, 7791 CrossRef CAS;
(b) Y. Sakamoto and T. Suzuki, J. Am. Chem. Soc., 2013, 135, 14074 CrossRef CAS;
(c) M. A. Majewski, Y. Hong, T. Lis, J. Gregoliński, P. J. Chmielewski, J. Cybińska, D. Kim and M. Stępień, Angew. Chem., Int. Ed., 2016, 55, 14072 CrossRef CAS;
(d) S. H. Pun, C. K. Chan, J. Luo, Z. Liu and Q. Miao, Angew. Chem., Int. Ed., 2018, 57, 1581 CrossRef CAS.
-
(a) Y. Shen and C. F. Chen, Chem. Rev., 2012, 112, 1463 CrossRef CAS;
(b) T. Hosokawa, Y. Takahashi, T. Matsushima, S. Watanabe, S. Kikkawa, I. Azumaya, A. Tsurusaki and K. Kamikawa, J. Am. Chem. Soc., 2017, 139, 18512 CrossRef CAS;
(c) V. Berezhnaia, M. Roy, N. Vanthuyne, M. Villa, J.-V. Naubron, J. Rodriguez, Y. Coquerel and M. Gingras, J. Am. Chem. Soc., 2017, 139, 18508 CrossRef CAS.
- N. Martín and L. T. Scott, Chem. Soc. Rev., 2015, 44, 6397 RSC.
-
(a) W. E. Barth and R. G. Lawton, J. Am. Chem. Soc., 1966, 88, 380 CrossRef CAS;
(b) R. G. Lawton and W. E. Barth, J. Am. Chem. Soc., 1971, 93, 1730 CrossRef CAS;
(c) L. T. Scott, M. M. Hashemi, D. T. Meyer and H. B. Warren, J. Am. Chem. Soc., 1991, 113, 7082 CrossRef CAS;
(d) L. T. Scott, P. Cheng, M. M. Hashemi, M. B. Bratcher, D. T. Meyer and H. B. Warren, J. Am. Chem. Soc., 1997, 119, 10963 CrossRef CAS;
(e) A. Sygula and P. W. Rabideau, J. Am. Chem. Soc., 1999, 121, 7800 CrossRef CAS;
(f) T. J. Seiders, E. L. Elliott, G. H. Grube and J. S. Siegel, J. Am. Chem. Soc., 1999, 121, 7804 CrossRef CAS;
(g) T. J. Seiders, K. K. Baldridge and J. S. Siegel, J. Am. Chem. Soc., 1996, 118, 2754 CrossRef CAS;
(h) A. Sygula and P. W. Rabideau, J. Am. Chem. Soc., 2000, 122, 6323 CrossRef CAS;
(i) A. M. Butterfield, B. Gilomen and J. S. Siegel, Org. Process Res. Dev., 2012, 16, 664 CrossRef CAS.
-
(a) H. Sakurai, T. Daiko and T. Hirao, Science, 2003, 301, 1878 CrossRef CAS;
(b) H. Sakurai, T. Daiko, H. Sakane, T. Amaya and T. Hirao, J. Am. Chem. Soc., 2005, 127, 11580 CrossRef CAS.
- L. T. Scott, M. M. Boorum, B. J. McMahon, S. Hagen, J. Mack, J. Blank, H. Wegner and A. de Meijere, Science, 2002, 295, 1500 CrossRef CAS.
- L. T. Scott, E. A. Jackson, Q. Y. Zhang, B. D. Steinberg, M. Bancu and B. Li, J. Am. Chem. Soc., 2012, 134, 107 CrossRef CAS.
-
(a) Z.-Z. Zhu, Z.-C. Chen, Y.-R. Yao, C.-H. Cui, S.-H. Li, X.-J. Zhao, Q. Zhang, H.-R. Tian, P.-Y. Xu, F.-F. Xie, X.-M. Xie, Y.-Z. Tan, S.-L. Deng, J. M. Quimby, L. T. Scott, S.-Y. Xie, R.-B. Huang and L.-S. Zheng, Sci. Adv., 2019, 5, eaaw0982 CrossRef CAS;
(b) K. Shoyama and F. Würthner, J. Am. Chem. Soc., 2019, 141, 13008 CrossRef CAS.
-
(a) S. Higashibayashi and H. Sakurai, J. Am. Chem. Soc., 2008, 130, 8592 CrossRef CAS;
(b) S. Higashibayashi, R. Tsuruoka, Y. Soujanya, U. Purushotham, G. N. Sastry, S. Seki, T. Ishikawa, S. Toyota and H. Sakurai, Bull. Chem. Soc. Jpn., 2012, 85, 450 CrossRef CAS;
(c) T. Fujikawa, D. V. Preda, Y. Segawa, K. Itami and L. T. Scott, Org. Lett., 2016, 18, 3992 CrossRef CAS;
(d) N. Ngamsomprasert, J.-S. Dang, S. Higashibayashi, Y. Yakiyama and H. Sakurai, Chem. Commun., 2017, 53, 697 RSC;
(e) D. Meng, G. Liu, C. Xiao, Y. Shi, L. Zhang, L. Jiang, K. K. Baldridge, Y. Li, J. S. Siegel and Z. Wang, J. Am. Chem. Soc., 2019, 141, 5402 CrossRef CAS.
-
(a) K. Shi, T. Lei, X.-Y. Wang, J.-Y. Wang and J. Pei, Chem. Sci., 2014, 5, 1041 RSC;
(b) R. Chen, R.-Q. Lu, K. Shi, F. Wu, H.-X. Fang, Z.-X. Niu, X.-Y. Yan, M. Luo, X.-C. Wang, C.-Y. Yang, X.-Y. Wang, B. Xu, H. Xia, J. Pei and X.-Y. Cao, Chem. Commun., 2015, 51, 13768 RSC;
(c) I. Hisaki, H. Toda, H. Sato, N. Tohnai and H. Sakurai, Angew. Chem., Int. Ed., 2017, 56, 15294 CrossRef CAS;
(d) Y. Yakiyama, T. Hasegawa and H. Sakurai, J. Am. Chem. Soc., 2019, 141, 18099 CrossRef CAS;
(e) T. K. Ronson, Y. Wang, K. Baldridge, J. S. Siegel and J. R. Nitschke, J. Am. Chem. Soc., 2020, 142, 10267 CrossRef CAS;
(f) X. Fu, Y. Zhen, Z. Ni, Y. Li, H. Dong, J. S. Siegel and W. Hu, Angew. Chem., Int. Ed., 2020, 59, 14024 CrossRef CAS.
-
(a) T.-C. Wu, H.-J. Hsin, M.-Y. Kuo, C.-H. Li and Y.-T. Wu, J. Am. Chem. Soc., 2011, 133, 16319 CrossRef CAS;
(b) T.-C. Wu, M.-K. Chen, Y.-W. Lee, M.-Y. Kuo and Y.-T. Wu, Angew. Chem., Int. Ed., 2013, 52, 1289 CrossRef CAS;
(c) G.-P. Gao, M. Chen, J. Roberts, M. Feng, C.-Y. Xiao, G.-W. Zhang, S. Parkin, C. Risko and L. Zhang, J. Am. Chem. Soc., 2020, 142, 2460 CrossRef CAS;
(d) M.-K. Chen, H.-J. Hsin, T.-C. Wu, B.-Y. Kang, Y.-W. Lee, M.-Y. Kuo and Y.-T. Wu, Chem. – Eur. J., 2014, 20, 598 CrossRef CAS.
-
(a) X. Li and X. Shao, Synlett, 2014, 25, 1795 CrossRef CAS;
(b) M. Saito, S. Furukawa, J. Kobayashi and T. Kawashima, Chem. Rec., 2016, 16, 64 CrossRef CAS;
(c) X. Hou, Y. Sun, L. Liu, S. Wang, R. Geng and X. Shao, Chin. Chem. Lett., 2016, 27, 1166 CrossRef CAS;
(d) X. Shao, Chin. Sci. Bull., 2019, 64, 3747 Search PubMed;
(e) D. Li and X. Shao, Synlett, 2020, 31, 1050 CrossRef CAS.
-
(a) M. A. Petrukhina, Coord. Chem. Rev., 2007, 251, 1690 CrossRef CAS;
(b) A. S. Filatov and M. A. Petrukhina, Coord. Chem. Rev., 2010, 254, 2234 CrossRef CAS;
(c) T. J. Seiders, K. K. Baldridge, J. M. O'Connor and J. S. Siegel, J. Am. Chem. Soc., 1997, 119, 4781 CrossRef CAS;
(d) M. A. Petrukhina, K. W. Andreini, J. Mack and L. T. Scott, Angew. Chem., Int. Ed., 2003, 42, 3375 CrossRef CAS;
(e) T. Amaya, H. Sakane and T. Hirao, Angew. Chem., Int. Ed., 2007, 46, 8376 CrossRef CAS;
(f) M. A. Petrukhina, Angew. Chem., Int. Ed., 2008, 47, 1150 CrossRef;
(g) A. V. Zabula, A. S. Filatov, S. N. Spisak, A. Y. Rogachev and M. A. Petrukhina, Science, 2011, 333, 1008 CrossRef CAS;
(h) T. Amaya, Y. Takahashi, T. Moriuchi and T. Hirao, J. Am. Chem. Soc., 2014, 136, 12794 CrossRef CAS.
-
(a) S. Mizyed, P. E. Georghiou, M. Bancu, B. Cuadra, A. K. Rai, P. Cheng and L. T. Scott, J. Am. Chem. Soc., 2001, 123, 12770 CrossRef CAS;
(b) A. Sygula, F. R. Fronczek, R. Sygula, P. W. Rabideau and M. M. Olmstead, J. Am. Chem. Soc., 2007, 129, 3842 CrossRef CAS;
(c) M. Gallego, J. Calbo, J. Aragó, R. M. K. Calderon, F. H. Liquido, T. Iwamoto, A. K. Greene, E. A. Jackson, E. M. Pérez, E. Ortí, D. M. Guldi, L. T. Scott and N. Martín, Angew. Chem., Int. Ed., 2014, 53, 2170 CrossRef CAS;
(d) Y.-M. Liu, D. Xia, B.-W. Li, Q.-Y. Zhang, T. Sakurai, Y.-Z. Tan, S. Seki, S.-Y. Xie and L.-S. Zheng, Angew. Chem., Int. Ed., 2016, 55, 13047 CrossRef CAS;
(e) Y. Shoji, T. Kajitani, F. Ishiwari, Q. Ding, H. Sato, H. Anetai, T. Akutagawa, H. Sakurai and T. Fukushima, Chem. Sci., 2017, 8, 8405 RSC;
(f) Y.-M. Liu, Y.-Q. Huang, S.-H. Liu, D.-D. Chen, C. Tang, Z.-L. Qiu, J. Zhu and Y.-Z. Tan, Angew. Chem., Int. Ed., 2019, 58, 13276 CrossRef CAS;
(g) Z.-L. Qiu, C. Tang, X.-R. Wang, Y.-Y. Ju, K.-S. Chu, Z.-Y. Deng, H. Hou, Y.-M. Liu and Y.-Z. Tan, Angew. Chem., Int. Ed., 2020, 59, 20868 CrossRef CAS.
-
(a) T. A. Schaub, K. Padberg and M. Kivala, J. Phys. Org. Chem., 2020, 33, e4022 CrossRef CAS;
(b) M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291 CrossRef CAS.
-
(a) S. Shimizu, Chem. Rev., 2017, 117, 2730 CrossRef CAS;
(b) P. J. Brothers, Chem. Commun., 2008, 2090 RSC;
(c) A. Osuka, E. Tsurumaki and T. Tanaka, Bull. Chem. Soc. Jpn., 2011, 84, 679 CrossRef CAS.
-
(a) A. Szumna, Chem. Soc. Rev., 2010, 39, 4274 RSC;
(b) G. d. l. Torre, G. Bottari, M. Sekita, A. Hausmann, D. M. Guldi and T. Torres, Chem. Soc. Rev., 2013, 42, 8049 RSC;
(c) S. Mori and N. Shibata, Beilstein J. Org. Chem., 2017, 13, 2273 CrossRef CAS.
-
(a) D. Myśliwiec and M. Stępień, Angew. Chem., Int. Ed., 2013, 52, 1713 CrossRef;
(b) M. A. Majewski, T. Lis, J. Cybińska and M. Stępień, Chem. Commun., 2015, 51, 15094 RSC.
- F. Chen, T. Tanaka and A. Osuka, Chem. Commun., 2017, 53, 2705 RSC.
- K. Ikemoto, R. Kobayashi, S. Sato and H. Isobe, Angew. Chem., Int. Ed., 2017, 56, 6511 CrossRef CAS.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162 CrossRef CAS.
- A. Borchardt, A. Fuchicello, K. V. Kilway, K. K. Baldridge and J. S. Siegel, J. Am. Chem. Soc., 1992, 114, 1921 CrossRef CAS.
- S. Ito, Y. Tokimaru and K. Nozaki, Angew. Chem., Int. Ed., 2015, 54, 7256 CrossRef CAS.
-
(a) R. Berger, A. Giannakopoulos, P. Ravat, M. Wagner, D. Beljonne, X. Feng and K. Müllen, Angew. Chem., Int. Ed., 2014, 53, 10520 CrossRef CAS;
(b) R. Berger, M. Wagner, X. Feng and K. Müllen, Chem. Sci., 2015, 6, 436 RSC.
- S. Ito, Y. Tokimaru and K. Nozaki, Angew. Chem., Int. Ed., 2017, 56, 15560 CrossRef.
- Y. Tokimaru, S. Ito and K. Nozaki, Angew. Chem., Int. Ed., 2018, 57, 9818 CrossRef CAS.
- H. Yokoi, Y. Hiraoka, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Nat. Commun., 2015, 6, 8215 CrossRef.
- K. Goto, R. Yamaguchi, S. Hiroto, H. Ueno, T. Kawai and H. Shinokubo, Angew. Chem., Int. Ed., 2012, 51, 10333 CrossRef CAS.
- H. Yokoi, S. Hiroto, D. Sakamaki, S. Seki and H. Shinokubo, Chem. Sci., 2018, 9, 819 RSC.
- M. Takeda, S. Hiroto, H. Yokoi, S. Lee, D. Kim and H. Shinokubo, J. Am. Chem. Soc., 2018, 140, 6336 CrossRef CAS.
- A. L. Lafleur, J. B. Howard, J. A. Marr and T. Yadav, J. Phys. Chem., 1993, 97, 13539 CrossRef CAS.
- J. Dey, A. Y. Will, R. A. Agbaria, P. W. Rabideau, A. H. Abdourazak, R. Sygula and I. M. Warner, J. Fluoresc., 1997, 7, 231 CrossRef CAS.
- H. Yokoi, S. Hiroto and H. Shinokubo, J. Am. Chem. Soc., 2018, 140, 4649 CrossRef CAS.
- Z. Zhou, Z. Wei, Y. Tokimaru, S. Ito, K. Nozaki and M. A. Petrukhina, Angew. Chem., Int. Ed., 2019, 58, 12107 CrossRef CAS.
-
M. S. Bratcher, Ph.D. dissertation, Boston College, Chestnut Hill, Massachusetts, 1996.
-
M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 6th edn, 2007, p. 29 Search PubMed.
-
P. J. Garratt, Aromaticity, John Wiley Sons, New York, N.Y., 1986 Search PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1 RSC.
- V. M. Tsefrikas, A. K. Greene and L. T. Scott, Org. Chem. Front., 2017, 4, 688 RSC.
-
(a) Z. Q. Liu and T. B. Marder, Angew. Chem., Int. Ed., 2008, 47, 242 CrossRef CAS;
(b) M. J. D. Bosdet and W. E. Piers, Can. J. Chem., 2009, 87, 8 CrossRef;
(c) P. G. Campbell, A. J. V. Marwitz and S.-Y. Liu, Angew. Chem., Int. Ed., 2012, 51, 6074 CrossRef CAS;
(d) X.-Y. Wang, J.-Y. Wang and J. Pei, Chem. – Eur. J., 2015, 21, 3528 CrossRef CAS;
(e) H. Helten, Chem. – Eur. J., 2016, 22, 12972 CrossRef CAS;
(f) M. M. Morgan and W. E. Piers, Dalton Trans., 2016, 45, 5920 RSC;
(g) J. Huang and Y. Li, Front. Chem., 2018, 6, 1 CrossRef;
(h) Z. X. Giustra and S.-Y. Liu, J. Am. Chem. Soc., 2018, 140, 1184 CrossRef CAS.
- S. Nakatsuka, N. Yasuda and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 13562 CrossRef CAS.
- G. Mehta, S. R. Shahk and K. Ravikumarc, J. Chem. Soc., Chem. Commun., 1993, 1006 RSC.
- H. K. Gupta, P. E. Lock and M. J. Mcglinchey, Organometallics, 1997, 16, 3628 CrossRef CAS.
-
(a) B. B. Shrestha, S. Karanjit, S. Higashibayashi, T. Amaya, T. Hirao and H. Sakurai, Asian J. Org. Chem., 2014, 4, 62 CrossRef;
(b) S. Higashibayashi, S. Onogi, H. K. Srivastava, G. N. Sastry, Y. Wu and H. Sakurai, Angew. Chem., Int. Ed., 2013, 52, 7314 CrossRef CAS.
- T. Amaya, S. Seki, T. Moriuchi, K. Nakamoto, T. Nakata, H. Sakane, A. Saeki, S. Tagawa and T. Hirao, J. Am. Chem. Soc., 2009, 131, 408 CrossRef CAS.
- K. Imamura, K. Takimiya, T. Otsubo and Y. Aso, Chem. Commun., 1999, 1859 RSC.
- Q. Tan, S. Higashibayashi, S. Karanjit and H. Sakurai, Nat. Commun., 2012, 3, 891 CrossRef.
- Q. Tan, P. Kaewmati, S. Higashihbayashi, M. Kawano, Y. Yakiyama and H. Sakurai, Bull. Chem. Soc. Jpn., 2018, 91, 531 CrossRef CAS.
- P. Kaewmati, Q. Tan, S. Higashibayashi, Y. Yakiyama and H. Sakurai, Chem. Lett., 2016, 46, 146 CrossRef.
-
(a) U. D. Priyakumar and G. N. Sastry, Tetrahedron Lett., 2001, 42, 1379 CrossRef CAS;
(b) G. N. Sastry and U. D. Priyakumar, J. Org. Chem., 2001, 66, 6523 CrossRef;
(c) G. N. Sastry and U. D. Priyakumar, J. Chem. Soc., Perkin Tran. 2, 2001, 30 RSC.
- S. Furukawa, J. Kobayashi and T. Kawashima, J. Am. Chem. Soc., 2009, 131, 14192 CrossRef CAS.
- T. Amaya, K. Mori, H.-L. Wu, S. Ishida, J. Nakamura, K. Murata and T. Hirao, Chem. Commun., 2007, 1902 RSC.
-
(a) J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC;
(b) M. Wang, G. Zhang, D. Zhang, D. Zhu and B. Z. Tang, J. Mater. Chem., 2010, 20, 1858 RSC.
- T. Tanikawa, M. Saito, J. Guo and S. Nagase, Org. Biomol. Chem., 2011, 9, 1731 RSC.
- T. Tanikawa, M. Saito, J. Guo, S. Nagase and M. Minoura, Eur. J. Org. Chem., 2012, 7135 CrossRef CAS.
-
(a) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2 CrossRef CAS;
(b) N. Kuhl, N. Schroder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443 CrossRef CAS;
(c) S. Tani, T. N. Uehara, J. Yamaguchi and K. Itami, Chem. Sci., 2014, 5, 123 RSC;
(d) S. A. Girard, T. Knauber and C. Li, Angew. Chem., Int. Ed., 2014, 53, 74 CrossRef CAS;
(e) V. S. Thirunavukkarasu, S. I. Kozhushkov and L. Ackermann, Chem. Commun., 2014, 50, 29 RSC.
- D. Zhou, Y. Gao, B. Liu, Q. Tan and B. Xu, Org. Lett., 2017, 19, 4628 CrossRef CAS.
- S. Furukawa, Y. Suda, J. Kobayashi, T. Kawashima, T. Tada, S. Fujii, M. Kiguchi and M. Saito, J. Am. Chem. Soc., 2017, 139, 5787 CrossRef CAS.
- Y. Suda, S. Furukawa and M. Saito, Chem. Lett., 2020, 49, 419 CrossRef CAS.
-
(a) L. H. Klemm, E. Hall, L. Cousins and C. E. Klopfensitein, J. Heterocycl. Chem., 1987, 24, 1749 CrossRef CAS;
(b) L. H. Klemm, E. Hall, L. Cousins and C. E. Klopfensitein, J. Heterocycl.
Chem., 1989, 26, 345 CrossRef CAS.
- X. Li, Y. Zhu, J. Shao, B. Wang, S. Zhang, Y. Shao, X. Jin, X. Yao, R. Fang and X. Shao, Angew. Chem., Int. Ed., 2014, 53, 535 CrossRef CAS.
- S. Wang, X. Li, X. Hou, Y. Sun and X. Shao, Chem. Commun., 2016, 52, 14486 RSC.
- S. Wang, J. Shang, C. Yan, W. Wang, C. Yuan, H. Zhang and X. Shao, Org. Chem. Front., 2019, 6, 263 RSC.
- Y. Wang, J. Deng, J. Chen, F. Cao, Y. Hou, Y. Yang, X. Deng, J. Yang, L. Wu, X. Shao, T. Shi and Z. Wang, ACS Catal., 2020, 10, 2707 CrossRef CAS.
- T. Zhang, G. Deng, H. Li, B. Liu, Q. Tan and B. Xu, Org. Lett., 2018, 20, 5439 CrossRef CAS.
- Q. Tan, D. Zhou, T. Zhang, B. Liu and B. Xu, Chem. Commun., 2017, 53, 10279 RSC.
-
(a) A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328 CrossRef CAS;
(b) A. Yoshimura, M. S. Yusubov and V. V. Zhdankin, Org. Biomol. Chem., 2016, 14, 4771 RSC;
(c) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052 CrossRef CAS.
-
(a) J. Charpentier, N. Fruh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef CAS;
(b) J. P. Brand, D. F. Gonzalez, S. Nicolai and J. Waser, Chem. Commun., 2011, 47, 102 RSC;
(c) Y. Li, D. P. Hari, M. V. Vita and J. Waser, Angew. Chem., Int. Ed., 2016, 55, 4436 CrossRef CAS.
- M. Jiang, J. Guo, B. Liu, Q. Tan and B. Xu, Org. Lett., 2019, 21, 8328 CrossRef CAS.
- M. Saito, T. Tanikawa, T. Tajima, J. Guo and S. Nagase, Tetrahedron Lett., 2010, 51, 672 CrossRef CAS.
- S. Furukawa, K. Hayashi, K. Yamagishi and M. Saito, Mater. Chem. Front., 2018, 2, 929 RSC.
- S. Wang, C. Yan, J. Shang, W. Wang, C. Yuan, H. Zhang and X. Shao, Angew. Chem., Int. Ed., 2019, 58, 3819 CrossRef CAS.
-
(a) X. Li, Y. Zhu, J. Shao, L. Chen, S. Zhao, B. Wang, S. Zhang, Y. Shao, H. Zhang and X. Shao, Angew. Chem., Int. Ed., 2015, 54, 267 CrossRef CAS;
(b) X. Hou, Y. Zhu, Y. Qin, L. Chen, X. Li, H. Zhang, W. Xu, D. Zhu and X. Shao, Chem. Commun., 2017, 53, 1546 RSC;
(c) Y. Sun, X. Li, C. Sun, H. Shen, X. Hou, D. Lin, H. Zhang, C. Di, D. Zhu and X. Shao, Angew. Chem., Int. Ed., 2017, 56, 13470 CrossRef CAS;
(d) X. Hou, J. Sun, Z. Liu, C. Yan, W. Song, H. Zhang and X. Shao, Chem. Commun., 2018, 54, 10981 RSC.
- X. Hou, X. Li, C. Sun, L. Chen, Y. Sun, Z. Liu, H. Zhang and X. Shao, Chem. – Eur. J., 2017, 23, 14375 CrossRef CAS.
- L. Liu, C. Yan, Y. Li, Z. Liu, C. Yuan, H.-L. Zhang and X. Shao, Chem. – Eur. J., 2020, 26, 7083 CrossRef CAS.
- J. Sun, Y. Sun, C. Yan, D. Lin, Z. Xie, S. Zhou, C. Yuan, H. Zhang and X. Shao, J. Mater. Chem. C, 2018, 6, 13114 RSC.
- B. Fu, X. Hou, C. Wang, Y. Wang, X. Zhang, R. Li, X. Shao and W. Hu, Chem. Commun., 2017, 53, 11407 RSC.
- B. Fu, C. Wang, Y. Sun, J. Yao, Y. Wang, F. Ge, F. Yang, Z. Liu, Y. Dang, X. Zhang, X. Shao, R. Li and W. Hu, Adv. Mater., 2019, 31, 1901437 CrossRef.
-
(a) Z. Zhou, A. Wakamiya, T. Kushida and S. Yamaguchi, J. Am. Chem. Soc., 2012, 134, 4529 CrossRef CAS;
(b) Y. Kitamoto, T. Suzuki, Y. Miyata, H. Kita, K. Funaki and S. Oi, Chem. Commun., 2016, 52, 7098 RSC.
- S. Nakatsuka, H. Gotoh, K. Kinoshita, N. Yasuda and T. Hatakeyama, Angew. Chem., Int. Ed., 2017, 56, 5087 CrossRef CAS.
-
(a) D. Hellwinkel, A. Wiel, G. Sattler and B. Nuber, Angew. Chem., Int. Ed. Engl., 1990, 29, 689 CrossRef;
(b) F. C. Krebs, P. S. Larsen, J. Larsen, C. S. Jacobsen, C. Boutton and N. Thorup, J. Am. Chem. Soc., 1997, 119, 1208 CrossRef CAS;
(c) M. Yamamura, T. Saito and T. Nabeshima, J. Am. Chem. Soc., 2014, 136, 14299 CrossRef CAS;
(d) M. Yamamura, K. Sukegawa and T. Nabeshima, Chem. Commun., 2015, 51, 12080 RSC;
(e) M. Yamamura, T. Hasegawa and T. Nabeshima, Org. Lett., 2016, 18, 816 CrossRef CAS;
(f) A. Heskia, T. Maris, P. M. Aguiar and J. D. Wuest, J. Am. Chem. Soc., 2019, 141, 18740 CrossRef CAS.
-
(a) D. Hellwinkel and M. Melan, Chem. Ber., 1971, 104, 1001 CrossRef CAS;
(b) D. Hellwinkel and M. Melan, Chem. Ber., 1974, 107, 616 CrossRef CAS;
(c) J. E. Field and D. Venkataraman, Chem. Mater., 2002, 14, 962 CrossRef CAS;
(d) M. Kuratsu, M. Kozaki and K. Okada, Angew. Chem., Int. Ed., 2005, 44, 4056 CrossRef CAS;
(e) Z. Fang, T.-L. Teo, L. Cai, Y.-H. Lai, A. Samoc and M. Samoc, Org. Lett., 2009, 11, 1 CrossRef CAS;
(f) Z. Jiang, Y. Chen, C. Yang, Y. Cao, Y. Tao, J. Qin and D. Ma, Org. Lett., 2009, 11, 1503 CrossRef CAS;
(g) Z. Fang, X. Zhang, Y. H. Lai and B. Liu, Chem. Commun., 2009, 920 RSC;
(h) C.-M. Chou, S. Saito and S. Yamaguchi, Org. Lett., 2014, 16, 2868 CrossRef CAS;
(i) K. Cui, F. Schlütter, O. Ivasenko, M. Kivala, M. G. Schwab, S.-L. Lee, S. F. L. Mertens, K. Tahara, Y. Tobe, K. Müllen, K. S. Mali and S. de Feyter, Chem. – Eur. J., 2015, 21, 1652 CrossRef CAS;
(j) S. Nakatsuka, Y. Watanabe, Y. Kamakura, S. Horike, D. Tanaka and T. Hatakeyama, Angew. Chem., 2020, 59, 1435 CrossRef CAS.
- N. Deng and G. Zhang, Org. Lett., 2019, 21, 5248 CrossRef CAS.
- L. Zhou and G. Zhang, Angew. Chem., Int. Ed., 2020, 59, 8963 CrossRef CAS.
- R. Geng, X. Hou, Y. Sun, C. Yan, Y. Wu, H. Zhang and X. Shao, Mater. Chem. Front., 2018, 2, 1456 RSC.
-
(a) E. D. Cox and J. M. Cook, Chem. Rev., 1995, 95, 1797 CrossRef CAS;
(b) J. Royer, M. Bonin and L. Micouin, Chem. Rev., 2004, 104, 2311 CrossRef CAS;
(c) A. K. Mandadapu, M. Saifuddin, P. K. Agarwal and B. Kundu, Org. Biomol. Chem., 2009, 7, 2796 RSC;
(d) J. Wei, B. Han, Q. Guo, X. Shi, W. Wang and N. Wei, Angew. Chem., Int. Ed., 2010, 49, 8209 CrossRef CAS;
(e) B. Liu, D. Shi, Y. Yang, D. Liu, M. Li, E. Liu, X. Wang, Q. Zhang, M. Yang, J. Li, X. Shi, W. Wang and J. Wei, Eur. J. Org. Chem., 2018, 869 CrossRef.
Footnote |
| † Dedicated to Prof. Zhongli Liu on the occasion of his 80th birthday. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*