
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStimuli responsive multicolour fluorescence emission in carbon nanodots and application in metal free hydrogen evolution from water†
Subir
Paul
and
Arindam
Banerjee
 *
*
School of Biological Sciences, Indian Association for the Cultivation of Science, Jadavpur, Kolkata, 700032, India. E-mail: bcab@iacs.res.in; Fax: +91-33-2473-2805
First published on 8th December 2020
Abstract
The present study convincingly demonstrates visible light induced continuous tuning of the fluorescence of carbon nanodots from yellow to cyan within two hours. These carbon dots are well characterized using field emission gun transmission electron microscopy (FEG-TEM), X-ray photoelectron spectroscopy (XPS), Fourier-transform infrared spectroscopy (FT-IR), fluorescence spectroscopy and UV-visible spectroscopy. Moreover, solid state fluorescence of different wavelengths has also been obtained from these colour tunable carbon dots by circumventing the common problem of aggregation induced quenching of fluorescence in the solid state. These carbon dots have also shown a very low band gap and the yellow emissive C-dots have been successfully utilized as a photocathode in metal free photo-electrochemical hydrogen evolution from water.
Introduction
The discovery and development of new fluorescent materials have attracted research interest from materials scientists over the years due to their applications in innumerable fields and also owing to the curiosity for understanding the fundamental principles behind this phenomenon. Fluorescent materials that show stimuli responsive fluorescence are of special importance as they have the advantage of on demand tuning of fluorescence.1–3 Fluorescent materials responsive to temperature variation, mechanical force, chemical stimuli and light irradiation have been reported in the literature.4–7 Photoresponsive fluorescent materials, whose fluorescence output can be tuned with light irradiation, have an edge over the materials with static output when it comes to biological applications, printing technology and others.8 Carbon dots, the newest entry into the carbon nanomaterial's family,9 have attracted tremendous research interest since its serendipitous discovery in 2004.10 Their fluorescence properties with very high quantum yield accompanied by their low intrinsic toxicity, cheap cost of production, photo-stability, and robust nature11–13 have already made them promising candidates in the field of photoluminescent nanomaterials. By virtue of these attractive properties, they have found widespread application in various fields including bioimaging, drug delivery, photoelectronics, photocatalysis and others.14–17 Several synthetic strategies have been employed by different research groups for the synthesis of carbon dots with different colour emission.18–20 Lei and coworkers reported an external heating free synthetic procedure for the fabrication of carbon dots using citric acid as the carbon source and 1-ethyl-3-(3-dimethylaminopropyl)carbodiimidehydrochloride (EDC) as the cross linking agent.21 Control of the reaction conditions and passivation of the surface of the carbon dots are the most frequently adopted strategies for the tuning of the fluorescence of carbon dots.22–24 In other cases, the solvatochromic properties of carbon dots have been utilized to gain access to multicolour emission from the carbon dots.25 However, using a simple stimulus like visible light is more desirable than using any other stimulus to change the emission of C-dots. To the best of our knowledge, there is no report on tuning the fluorescence of carbon dots by using visible light. One of the serious shortcomings for the majority of the carbon dots reported in the literature has been their inability to exhibit fluorescence properties in the solid state. Confinement of carbon dots in an external matrix is one of the adopted strategies to overcome this limitation.26,27 Generally aggregation induced quenching leads to the disappearance of the fluorescence in the solid state and this has been the mostly encountered observation in the case of carbon dots.28,29 However, carbon dots showing inherent solid state fluorescence have also been reported only in a few cases.30–32The demand for renewable energy resources is increasing by leaps and bounds as humanity faces the inevitable threat of complete consumption of non-renewable energy sources like coal, oil, natural gas and others.33 Hydrogen energy is one of those renewable resources which have the potential to fulfill the demands and can solve the energy crisis in the future.34 Transition metal based hybrid materials have been efficiently used as electrocatalysts in the hydrogen evolution reaction from water.35–37 Photobiocatalysis techniques which combine the photocatalytic process and photo-biological technologies have also been found to be effective for the hydrogen evolution reaction with high efficiency.38 Photo-electrochemical (PEC) hydrogen evolution through water splitting provides a useful way to obtain renewable hydrogen and already several successful attempts have been made using this approach.39–42 Evolution of hydrogen from water using hybrid materials in combination with carbon dots43–45 has been reported. However, metal free hydrogen evolution by water splitting using carbon dots has been scarcely reported.46,47 So, there is a genuine need to develop economically viable, new smart metal free nanomaterials for renewable energy sources.
In this study, a new class of smart fluorescent carbon nanodots has been synthesized and the emission color is continuously tunable from yellow to cyan by visible light irradiation within 2 hours. Solid state fluorescence with different emissive colours (namely yellow, green and cyan) has also been achieved by freeze drying the solutions of each of these different carbon dots. The yellow fluorescent carbon dot with a low band gap has been effectively utilized as a cathode for photoelectrochemical hydrogen evolution from water.
Experimental section
Chemicals
2,3-Dichloro-5,6-dicyano-benzoquinone (DDQ), ethylene glycol, benzyl alcohol and NaBH4 were purchased from SRL, India.UV-vis spectroscopic analysis
A Cary Varian 50 scan UV-vis optical spectrophotometer equipped with ‘Cary Win’ UV software was used to elucidate the optical properties of carbon dots in different solvents.Fluorescence spectroscopy
Fluorescence studies of carbon dots in a sealed cuvette were carried out in a PerkinElmer LS55 Fluorescence Spectrometer instrument. All experiments were carried out with an excitation slit width of 5 nm and emission slit width of 5 nm.FEG-TEM study
TEM studies of carbon dots in different solvents were carried out on a JEOL 2100 keV Ultra High Resolution Field Emission Gun (UHR-FEG) TEM instrument with a voltage of 200 keV using carbon coated copper grids.X-ray photoelectron spectroscopic (XPS) study
XPS analysis of carbon dots was carried out by using the X-ray photoelectron spectroscopy (XPS, Omicron, model: 1712-62-11) method. Measurement was carried out by using an Al-Kα radiation source under 15 kV voltages and 5 mA current.Fourier transform infrared (FTIR) study
FT-IR spectra were recorded by using the KBr pellet technique in a Nicolet 380 FT-IR spectrometer (Thermo Scientific).Mass spectrometry
The mass spectrum was recorded on a Q-Tofmicro™ (Waters Corporation) mass spectrometer by the positive mode electrospray ionization process.Synthesis of carbon dots
Carbon dots were synthesized from DDQ via a solvo-thermal route (Fig. S1†) using ethylene glycol as the solvent. In a typical procedure, 300 mg DDQ was dissolved in 20 ml of ethylene glycol through sonication. The solution was then heated at 220 °C for 3 hours using a Teflon lined autoclave without any stirring. After 3 hours the red coloured solution turned dark brown indicating the possible formation of carbon dots. Ethylene glycol was removed by heating at 115 °C for 4 hours in a vacuum oven. The carbon dots were re-dispersed in water, centrifuged and purified through dialysis to obtain the carbon C-dots1. In order to reduce C-dots1, 15 mg of C-dots1 was dissolved in water in a round-bottom flask (100 ml size) and 3 mg of NaBH4 was added to this solution. The mixture was then stirred at room temperature for five minutes. The reaction mixture was then centrifuged and subjected to dialysis to obtain C-dots2.Oxidation of benzyl alcohol by C-dots2 in the presence of light
50 μl benzyl alcohol was introduced into a 1 ml solution containing 5 mg of C-dots2 in a reaction tube. The reaction mixture was then stirred for 2 hours in the presence of a light source. The reaction mixture was found to be slightly basic and was neutralized with HCl solution and extracted with ethyl acetate.Photoelectrochemical measurements
Electrochemical measurements were performed on a CHI 700 electrochemical analyzer with a standard three-electrode configuration. The C-dots2 were dispersed in ethanol and this dispersion was spin coated on ITO-coated S5 glass. The C-dots2 coated ITO substrate was used as the working electrode for the electrochemical measurements after drying (Fig. S1†). A Pt wire acted as the counter electrode and a saturated calomel electrode (SCE) was used as the reference electrode. The conversion of potential from SCE to RHE was computed using eqn (1)| E vs. RHE = E vs. SCE + 0.2416 + 0.059 × pH | (1) |
An aqueous solution of Na2SO4 with 0.5 M strength was used as the electrolyte with pH ∼ 7. The linear sweep voltammograms (LSVs) were recorded at a 10 mV s−1 scan rate within a potential window of +0.2 V to −0.6 V (vs. RHE). Photocurrent density vs. time response of the sample-coated ITO electrode was recorded at −0.5 V vs. SCE in the same electrolyte and electrode configuration. The half-cell solar-to-hydrogen efficiency (HC-STH) was calculated from the current density–potential response of the photocathodes by using the following equation:
| HC-STH (%) = J × V × 100%/P |
Results and discussion
Carbon dots (C-dots1) were synthesized from 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) via a solvo-thermal route (Fig. S2†).They are treated with ethylene glycol as the solvent. The detailed preparation procedure is discussed in the Experimental section. C-dots1 show cyan green fluorescence when exposed to UV light from a hand held UV lamp at 365 nm. When C-dots1 are reduced with NaBH4, the solution emits a yellow colour (C-dots2) (Fig. S3†). Interestingly, C-dots2 show visible light tunable fluorescence. When a solution of C-dots2 in water is kept under continuous exposure to sunlight, the fluorescence of the solution changes from yellow to green to cyan (Fig. 1). The entire fluorescence shift is observed within 2 hours and the cyan fluorescent carbon dots are denoted as C-dots3. This spontaneous change in the fluorescence of C-dots2 does not take place when the solution is kept in a dark environment or in an inert argon atmosphere. To reveal the role of the oxidizing environment in this spontaneous fluorescence shift, the C-dots2 solution was taken in a vial and air was removed from the vial by using a vacuum. Then it was filled with argon gas. The C-dots2 solution was exposed to continuous sunlight for 5 hours. It was observed that, under these inert conditions, no change occurred in the fluorescence of the C-dots2 solution in the presence of visible sunlight. This observation indicates the importance of the presence of an oxygenated environment for this visible light induced fluorescence tuning of C-dots2.
 | ||
| Fig. 1 C-dots2 (a) initially and after (b) 30 minutes, (c) 1 hour, (d) 1.5 hours and (e) 2 hours of sunlight irradiation. (f) Cyan, yellow and green fluorescent solid carbon dots. | ||
A water dispersion of C-dots1 shows a strong cyan green fluorescence when held under the irradiation from a hand held UV lamp at 365 nm. From the emission spectrum of C-dots1 (Fig. S4†) a typical excitation dependent emission behavior of these carbon dots is observed with two major peaks centered at 465 nm and 522 nm. With an increase in the excitation wavelength, the peak at 522 nm becomes the major peak and the intensity of the peak at 465 nm starts decreasing to the minimum and then it vanishes for excitation at 425 nm. For C-dots2, the emission peak is centered at 567 nm (Fig. 2a) and the peak position does not change considerably when the excitation wavelength is altered. So, these carbon dots exhibit excitation independent emission behavior. After irradiating the C-dots2 solution with sunlight for 30 minutes, it shows a greenish yellow fluorescence (Fig. 1b) under UV light irradiation. The emission spectra show that the emission wavelength is blue shifted (Fig. 2b) compared to that for C-dots2 and the emission peak is centered at 556 nm with an excitation independent nature of fluorescence. After 1 hour of visible light irradiation, the carbon dot solution shows green emission under UV light (excitation at 365 nm) (Fig. 1c). The emission spectra of these carbon dots show a main peak centered at 550 nm with a weak shoulder at 502 nm (Fig. 2c). The nature of the fluorescence remains excitation independent. Irradiation of the carbon dot solution for 1.5 hours results in further blue shift of fluorescence compared to that for these carbon dots. This carbon dot solution shows a bluish green emission when it is held under a UV lamp (at 365 nm) (Fig. 1d). The emission spectra show a main peak around 534 nm and a shoulder around 478 nm (Fig. 2d). The emission peaks show a slight dependence on the excitation wavelength. However, the shift in the position of the emission is very low in magnitude. After 2 hours of irradiation, C-dots3 are obtained. This is the final fluorescent material that is obtained by the light induced tuning of C-dots2. Light irradiation of the C-dots3 solution causes no further changes to the fluorescence emission. In the fluorescence spectra of C-dots3, the main emission peak is centered at 482 nm. The peak position does not change with the altering of excitation wavelength. So the emission nature is excitation independent (Fig. 2e). The quantum yields of the cyan and green fluorescent carbon dots were found to be 26% and 34%, respectively (using quinine sulphate as a reference) while that of yellow carbon dots was found to be 16% (using Rhodamine 6G as a reference). Yellow, green and cyan fluorescence emission in the solid state (Fig. 1f) was also obtained by freeze drying of each of these different color emitting carbon dot solutions. The emission peak for the cyan emitting C-dots in the solid state is centered at 489 nm (Fig. S5†) and that of the green emitting C-dots is at 538 nm (Fig. S6†). The emission nature of the solid state cyan and green emission was found to be excitation independent. Yellow emissive solid state C-dots have 2 emission peaks, one at 582 nm and the other at 551 nm. The intensities of these two peaks are almost the same and the intensity ratio does not alter much upon varying the excitation wavelength (Fig. S7†).
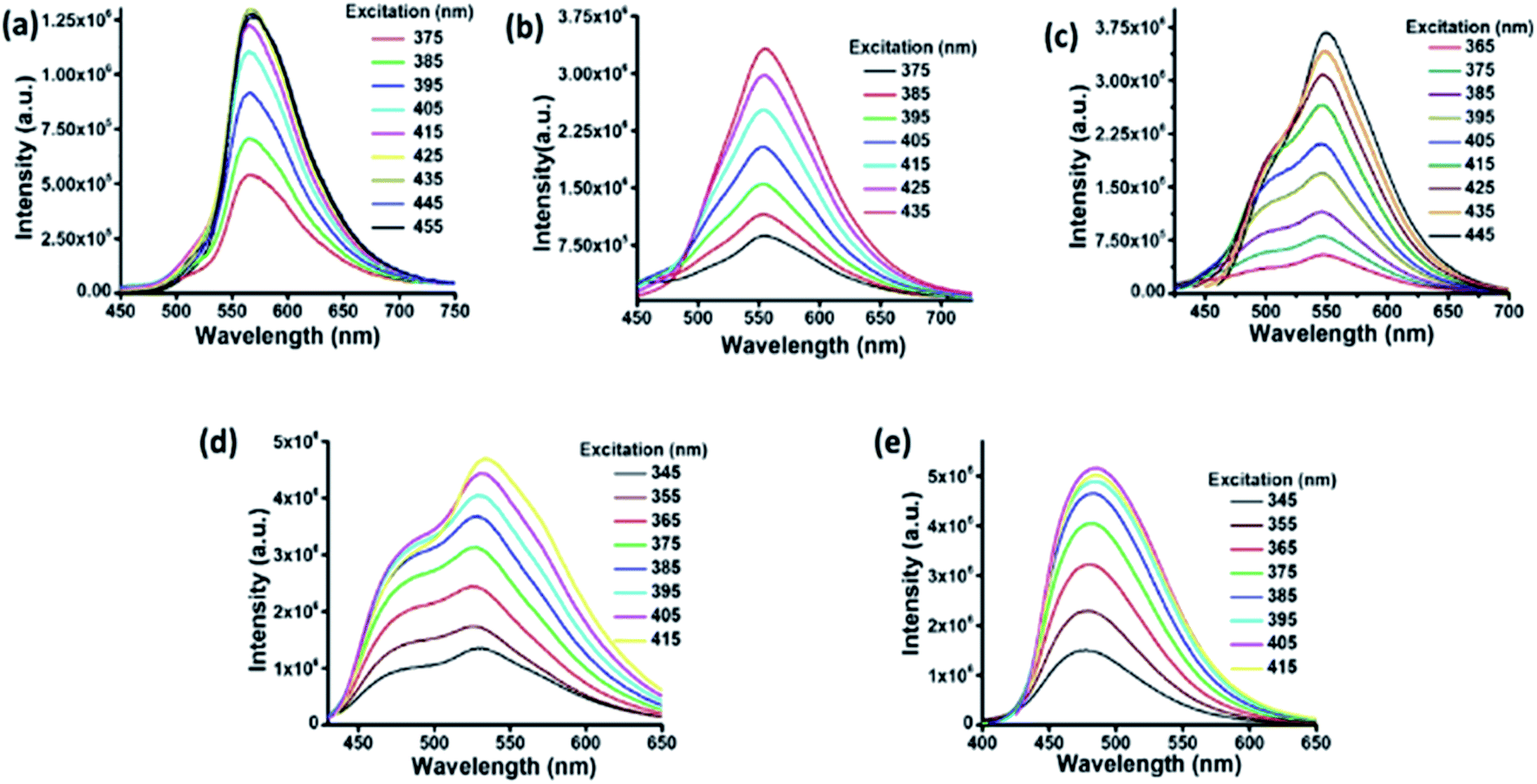 | ||
| Fig. 2 Fluorescence spectra of C-dots2 (a) initially and after (b) 30 minutes, (c) 1 hour, (d) 1.5 hours and (e) 2 hours. | ||
In the UV-visible spectrum of C-dots1 (Fig. 3a), the peak at 316 nm arises due to the π–π* transition in the carbogenic core. The peak at 349 nm represents the n–π* transition of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and CN bonds. In the UV visible spectrum of C-dots2 (Fig. 3b), the peak due to the π–π* transition does not shift its position and it is located at 320 nm. The peak due to n–π* transition of CO and CN bonds is no longer present in the case of C-dots2. However, a strong absorption in the visible region is observed in the case of C-dots2. Two peaks at 425 nm and 468 nm emerge in the UV-visible spectrum of C-dots2. Absorption at such higher wavelengths is rarely found in the case of carbon dots. These absorption peaks originate due to surface defects of the carbon dots.48 Since these peaks are not present in the case of C-dots1, incorporation of new surface states upon the reduction of C-dots1 with NaBH4 is indicated. The UV-visible spectrum of C-dots3 (Fig. 3c) does not show such long wavelength absorptions. The peak due to the π–π* transition appears at 322 nm and another peak is observed around 415 nm corresponding to surface state absorptions.
O and CN bonds. In the UV visible spectrum of C-dots2 (Fig. 3b), the peak due to the π–π* transition does not shift its position and it is located at 320 nm. The peak due to n–π* transition of CO and CN bonds is no longer present in the case of C-dots2. However, a strong absorption in the visible region is observed in the case of C-dots2. Two peaks at 425 nm and 468 nm emerge in the UV-visible spectrum of C-dots2. Absorption at such higher wavelengths is rarely found in the case of carbon dots. These absorption peaks originate due to surface defects of the carbon dots.48 Since these peaks are not present in the case of C-dots1, incorporation of new surface states upon the reduction of C-dots1 with NaBH4 is indicated. The UV-visible spectrum of C-dots3 (Fig. 3c) does not show such long wavelength absorptions. The peak due to the π–π* transition appears at 322 nm and another peak is observed around 415 nm corresponding to surface state absorptions.
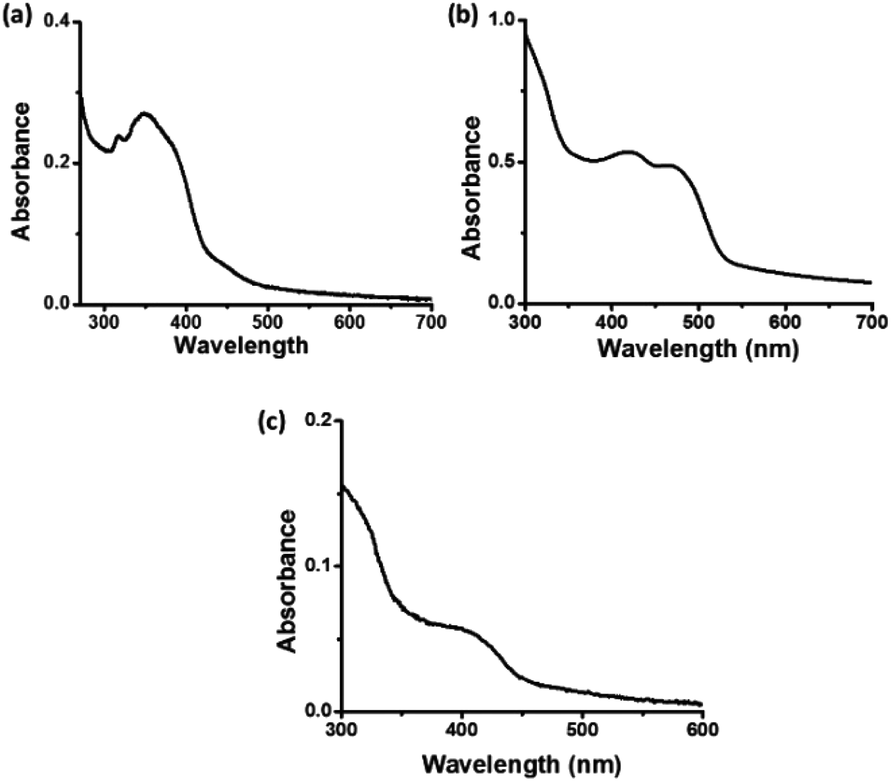 | ||
| Fig. 3 UV-visible spectra of (a) C-dots1, (b) C-dots2 and (c) C-dots3. | ||
Fig. 4a and b show the FEG-TEM images of C-dots1 and C-dots2. Since the C-dots2 are prepared by surface reduction of C-dots1, the size and morphological nature of these two types of carbon dots are expected to remain the same. It is evident from their FEG-TEM images that both C-dots1 and C-dots2 have similar sizes within a range of 4–7 nm, with an average size of 5 nm. The C-dots1 are crystalline in nature and this crystallinity is retained in C-dots2. The C-dots3, which are the final fluorescent species formed when C-dots2 are irradiated with visible light, also possess the same size and crystalline nature (Fig. 4c). The lattice spacings of C-dots1, C-dots2, and C-dots3 are also similar and are found to be around 0.21 nm. This indicates that the C-dots2 solution does not undergo any degradation or aggregation upon light irradiation. They are converted to a new type of carbon dot with similar sizes. Therefore the changes in the fluorescence output are not a result of the lack of stability of C-dots1 in the presence of light, but are due to the transformation of the materials into a new kind of carbon dot.
 | ||
| Fig. 4 FEG-TEM images of (a) C-dots1, (b) C-dots2 and (c) C-dots3. | ||
The FT-IR spectra of these three types of carbon dots were recorded in order to follow the changes in the functional groups present in these carbon dots. In the FT-IR spectrum of C-dots1 (Fig. 5a), the peaks at 3414 cm−1 and 3241 cm−1 arise due to the stretching vibrations of O–H and N–H bonds.49 The peaks at 3071 cm−1 and 2960 cm−1 are attributed to C–H and –C–H stretching vibrations. There is a sharp peak at 2228 cm−1 which arises due to the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching vibration. The peaks at 1719 cm−1 and 1627 cm−1 appear due to CO and CC stretching vibrations50 and the peaks at 1404 cm−1, 1219 cm−1 and 1062 cm−1 are assigned to the C–OH stretching peak and C–O stretching peaks, respectively.51 In the FTIR spectrum of C-dots2 (Fig. 5b), the changes expected upon reduction with NaBH4 are clearly reflected. The peak at 2228 cm−1 present in the FTIR spectrum of C-dots1 vanishes indicating the reduction of the –CN group to –CH2NH2. The CO stretching vibration at 1719 cm−1 is also absent in the FTIR spectrum of C-dots2. These observations indicate that upon reduction with NaBH4, the –CN and –CO groups present at the surface of the carbon dots are reduced to –CH2NH2 and –CH2OH, respectively. Upon comparison of the FTIR spectra of C-dots2 and C-dots3, a new peak can be observed in the FTIR spectrum of C-dots3 (Fig. 5c) at 1420 cm−1. This peak arises due to the stretching of the COO– group.52 The appearance of this peak in the C-dots3 spectrum after light irradiation of C-dots2 points towards the possible oxidation of the –CH2OH groups on the surface of the carbon dots upon light irradiation.
N stretching vibration. The peaks at 1719 cm−1 and 1627 cm−1 appear due to CO and CC stretching vibrations50 and the peaks at 1404 cm−1, 1219 cm−1 and 1062 cm−1 are assigned to the C–OH stretching peak and C–O stretching peaks, respectively.51 In the FTIR spectrum of C-dots2 (Fig. 5b), the changes expected upon reduction with NaBH4 are clearly reflected. The peak at 2228 cm−1 present in the FTIR spectrum of C-dots1 vanishes indicating the reduction of the –CN group to –CH2NH2. The CO stretching vibration at 1719 cm−1 is also absent in the FTIR spectrum of C-dots2. These observations indicate that upon reduction with NaBH4, the –CN and –CO groups present at the surface of the carbon dots are reduced to –CH2NH2 and –CH2OH, respectively. Upon comparison of the FTIR spectra of C-dots2 and C-dots3, a new peak can be observed in the FTIR spectrum of C-dots3 (Fig. 5c) at 1420 cm−1. This peak arises due to the stretching of the COO– group.52 The appearance of this peak in the C-dots3 spectrum after light irradiation of C-dots2 points towards the possible oxidation of the –CH2OH groups on the surface of the carbon dots upon light irradiation.
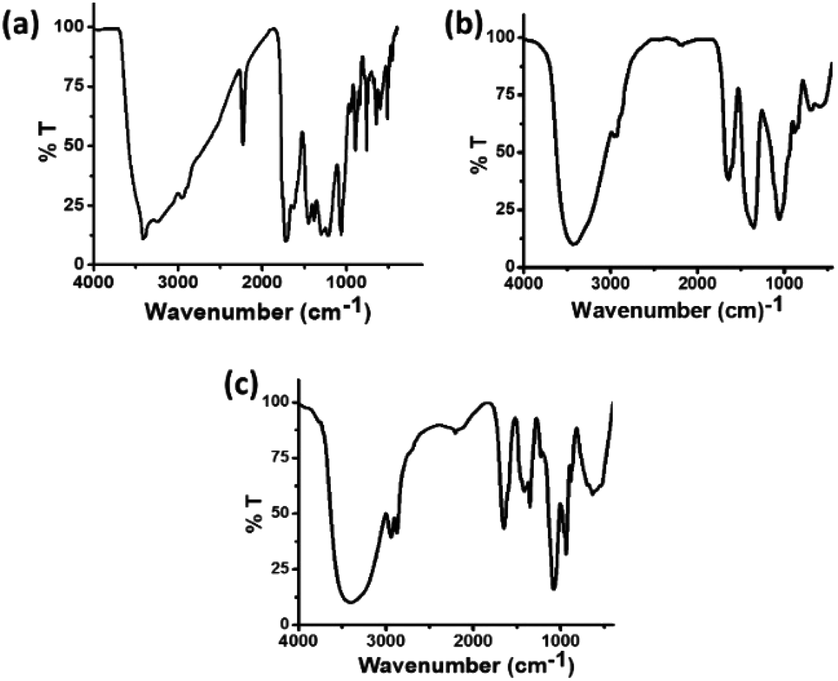 | ||
| Fig. 5 FT-IR spectra of (a) C-dots1, (b) C-dots2 and (c) C-dots3. | ||
In the high resolution C 1s XPS spectra of C-dots1 (Fig. 6a), the three peaks at 284.9 eV, 286.4 eV and 288.3 eV indicate the presence of CC, CO and COOH groups, respectively. In the case of C-dots2 (Fig. 6b), the peaks due to CC and –COOH are unaltered, but the peak due to the CO is no longer present. Instead, a new peak appears at 285.7 eV, which can be assigned to the –C–OH group.53 So, from the C 1s spectra of C-dots1 and C-dots2, the conversion of carbonyl groups on the surface of carbon dots to C–OH functionalities is indicated. The C 1s spectrum of C-dots3 (Fig. 6c) is similar to that of C-dots2.
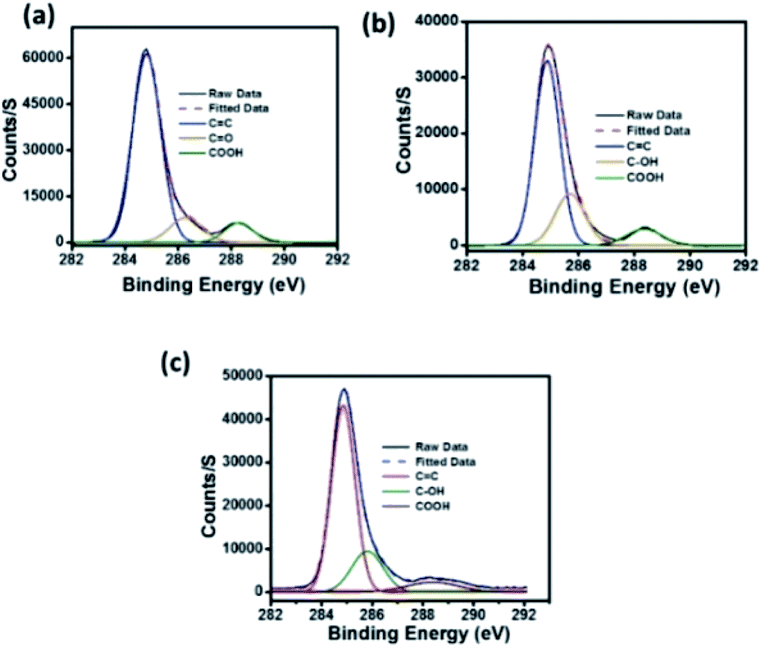 | ||
| Fig. 6 High resolution C 1s spectra of (a) C-dots1, (b) C-dots2 and (c) C-dots3. | ||
The changes in the fluorescence output of the carbon dots throughout the study can be explained on the basis of the changes in the functional groups present on the surface of the carbon dots. Functional groups with an electron donating nature reduce the energy gap between the HOMO and LUMO for the surface states in the carbon dots and thus shift the fluorescence emission to higher wavelengths, while electron withdrawing functional groups are expected to result in a blue shift of the fluorescence.54 In this study, when C-dots1 are reduced with NaBH4, the –CN groups on the carbon dot surface are transformed to –CH2NH2 groups. Thus removal of the electron withdrawing effect from the surface through the transformation of –CN groups to –CH2NH2 is the probable reason for the red shift in the fluorescence observed after the reduction of C-dots1 with NaBH4. The C-dots2 solution, when irradiated with sunlight, exhibits a spontaneous blue shift of fluorescence from yellow to cyan to give C-dots3. The FT-IR spectrum of C-dots3 suggests that C-dots3 bear –COOH groups that are not present in C-dots2. Since the colour change is only observed in the presence of an oxidizing environment, oxidation of –CH2OH groups of the carbon dots to –COOH groups is suggested. To further validate our hypothesis, we tested the possible catalytic activity of C-dots2 in the oxidation of benzyl alcohol to benzoic acid under similar conditions (details are given in the Experimental section). It was observed that C-dots2 could oxidize benzyl alcohol to benzoic acid in the presence of visible light (Fig. S8 and S9†). The transformation of the –CH2OH group to the electronegative –COOH group through catalysis by the carbon dots in the presence of visible light is the driving force for the light induced blue shift in the fluorescence of carbon dots in C-dots3.
Most of the carbon dots reported in the literature absorb light in the UV region of the spectrum and to some extent in the blue region.55–57 Carbon dots with significant absorption in the visible region of the spectrum have seldom been reported.58 This in turn limits the implementation of these nanomaterials in light harvesting applications. As discussed earlier, C-dot2 shows strong absorption in the visible region (Fig. 3b) and from Tauc's plot (Fig. 7a), their band gap was found to be around 2.6 eV. These observations prompted us to examine the ability of C-dots2 in the hydrogen evolution reaction from water. Reduction of water to produce hydrogen was carried out in a photoelectrochemical setup (the detailed procedure is provided in the Experimental section). Fig. 7b shows the linear sweep voltammogram of C-dots2. The onset of hydrogen evolution is at about 0.3 (V) vs. the reference hydrogen electrode (RHE) under visible light irradiation, while under dark conditions cathodic current is not obtained showing that the material does not show any activity in the absence of light irradiation. Fig. 7c provides the on-off data which further establish the photo responsive nature of the hydrogen evolution reaction in the presence of C-dots2. The half-cell solar to hydrogen efficiency (Fig. 7d) was calculated to be 7% for C-dots. The details of the calculation are mentioned in the ESI.†
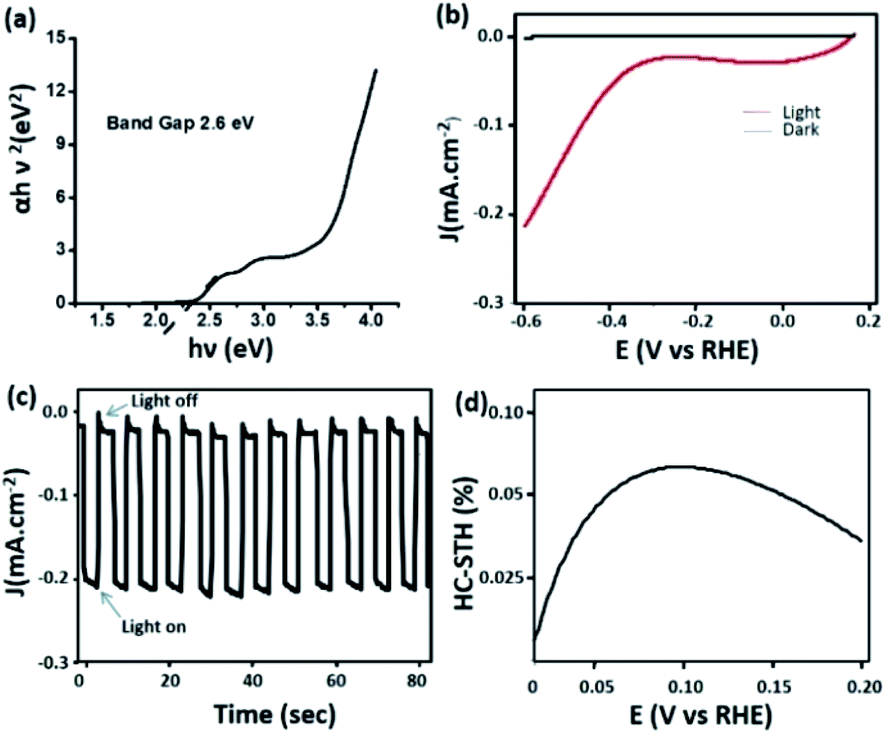 | ||
| Fig. 7 (a) Tauc's plot of C-dots2 and (b) the linear sweep voltammogram of C-dots2; (c) light on–off cycle and (d) efficiency curve of C-dots2. | ||
Conclusions
In this study, we report the continuous tuning of the carbon dot fluorescence using visible light as an external stimulus. The fluorescence colour changes from yellow to cyan within a short time frame upon irradiation with visible light. The presence of an oxygen environment was found to be necessary for the colour tuning phenomenon. The changes in the nature of the functional groups on the surface of the carbon dots are the driving force for light responsive fluorescence changes of carbon dots. Solid state fluorescence of carbon dots has also been achieved with cyan, green and yellow colour emission. Moreover, yellow emitting carbon dots (C-dots2) have also been employed as a photocathode for the hydrogen evolution reaction through water splitting in a photo-electrochemical set up. This work, thus, is a promising step towards the development of smart fluorescent materials with on demand tunability of emission output and metal free hydrogen evolution from water.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
S. P. has gratefully acknowledged the CSIR New Delhi, India for the fellowship.Notes and references
- X. Hou, C. Ke, C. J. Bruns, P. R. McGonigal, R. B. Pettman and J. F. Stoddart, Nat. Commun., 2015, 6, 6884 CrossRef.
- J. Xiong, K. Wang, Z. Yao, B. Zou, J. Xu and X.-H. Bu, ACS Appl. Mater. Interfaces, 2018, 10, 5819–5827 CrossRef CAS.
- R. Gao, X. Fang and D. Yan, J. Mater. Chem. C, 2019, 7, 3399–3412 RSC.
- J.-H. Tang, Y. Sun, Z.-L. Gong, Z.-Y. Li, Z. Zhou, H. Wang, X. Li, M. L. Saha, Y.-W. Zhong and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 7723–7729 CrossRef CAS.
- L. Wilbraham, M. Louis, D. Alberga, A. Brosseau, R. Guillot, F. Ito, F. Labat, R. Metivier, C. Allain and I. Ciofini, Adv. Mater., 2018, 30, 1800817 CrossRef.
- D. Zhang, Y. Zhang, W. Lu, X. Le, P. Li, L. Huang, J. Zhang, J. Yang, M. J. Serpe, D. Chen and T. Chen, Adv. Mater. Technol., 2019, 4, 1800201 CrossRef.
- W. Zhou, Y. Chen, Q. Yu, P. Li, X. Chen and Y. Liu, Chem. Sci., 2019, 10, 3346–3352 RSC.
- T. Fukaminato, S. Ishida and R. Métivier, NPG Asia Mater., 2018, 10, 859–881 CrossRef CAS.
- A. L. Himaja, P. S. Karthik and S. P. Singh, Chem. Rec., 2015, 15, 595–615 CrossRef CAS.
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS.
- H. Li, Z. Kang, Y. Liu and S.-T. Lee, J. Mater. Chem., 2012, 22, 24230–24253 RSC.
- K. J. Mintz, Y. Zhou and R. M. Leblanc, Nanoscale, 2019, 11, 4634–4652 RSC.
- R. Wang, K.-Q. Lu, Z.-R. Tang and Y.-J. Xu, J. Mater. Chem. A, 2017, 5, 3717–3734 RSC.
- M. Semeniuk, Z. Yi, V. Poursorkhabi, J. Tjong, S. Jaffer, Z.-H. Lu and M. Sain, ACS Nano, 2019, 13, 6224–6255 CrossRef CAS.
- H. Qi, M. Teng, M. Liu, S. Liu, J. Li, H. Yu, C. Teng, Z. Huang, H. Liu, Q. Shao, A. Umar, T. Ding, Q. Gao and Z. Guo, J. Colloid Interface Sci., 2019, 539, 332–341 CrossRef CAS.
- C. Shi, H. Qi, Z. Sun, K. Qu, Z. Huang, J. Li, M. Dong and Z. Guo, J. Mater. Chem. C, 2020, 8, 2238–2247 RSC.
- S. Singh, N. Shauloff and R. Jelinek, ACS Sustainable Chem. Eng., 2019, 7, 13186–13194 CrossRef CAS.
- F. Arcudi, L. Đorđević and M. Prato, Acc. Chem. Res., 2019, 52, 2070–2079 CrossRef CAS.
- J. Bai, Y. Ma, G. Yuan, X. Chen, J. Mei, L. Zhang and L. Ren, J. Mater. Chem. C, 2019, 7, 9709–9718 RSC.
- R. Ye, C. Xiang, J. Lin, Z. Peng, K. Huang, Z. Yan, N. P. Cook, E. L. G. Samuel, C. -C. Hwang, G. Ruan, G. Ceriotti, A.-R. O. Raji, A. A. Martí and J. M. Tour, Nat. Commun., 2013, 4, 2943 CrossRef.
- X. Ma, W. Zhong, J. Zhao, S. L. Suib and Y. Lei, Eng. Sci., 2019, 9, 44–49 Search PubMed.
- L. Li and T. Dong, J. Mater. Chem. C, 2018, 6, 7944–7970 RSC.
- L. Bao, C. Liu, Z.-L. Zhang and D.-W. Pang, Adv. Mater., 2015, 27, 1663–1667 CrossRef CAS.
- X. Miao, D. Qu, D. Yang, B. Nie, Y. Zhao, H. Fan and Z. Sun, Adv. Mater., 2018, 30, 1704740 CrossRef.
- H. Wang, C. Sun, X. Chen, Y. Zhang, V. L. Colvin, Q. Rice, J. Seo, S. Feng, S. Wang and W. W. Yu, Nanoscale, 2017, 9, 1909–1915 RSC.
- Q. Chang, S. Yang, C. Xue, N. Li, Y. Wang, Y. Li, H. Wang, J. Yanga and S. Hu, Nanoscale, 2019, 11, 7247–7255 RSC.
- D. Zhou, D. Li, P. Jing, Y. Zhai, D. Shen, S. Qu and A. L. Rogach, Chem. Mater., 2017, 29, 1779–1787 CrossRef CAS.
- Y. Chen, M. Zheng, Y. Xiao, H. Dong, H. Zhang, J. Zhuang, H. Hu, B. Lei and Y. Liu, Adv. Mater., 2016, 28, 312–318 CrossRef CAS.
- S. Qu, X. Wang, Q. Lu, X. Liu and L. Wang, Angew. Chem., Int. Ed., 2012, 51, 12215–12218 CrossRef CAS.
- D. Xu, F. Lei, H. Chen, L. Yin, Y. Shi and J. Xie, RSC Adv., 2019, 9, 8290–8299 RSC.
- J. He, Y. He, Y. Chen, B. Lei, J. Zhuang, Y. Xiao, Y. Liang, M. Zheng, H. Zhang and Y. Liu, Small, 2017, 13, 1700075 CrossRef.
- J. Yang, Y. Liu, J. Wang, S. Wang, X. Zhoua and H. Li, J. Mater. Chem. C, 2019, 7, 7806–7811 RSC.
- S. Chu and A. Majumdar, Nature (London, U. K.), 2012, 488, 294–303 CrossRef CAS.
- O. V. Marchenko and S. V. Solomin, Int. J. Hydrogen Energy, 2015, 40, 3801–3805 CrossRef CAS.
- L. Wei, K. Lozano and Y. Mao, Eng. Sci., 2018, 3, 62–66 Search PubMed.
- Q. Yuan, R. Wang, Q. Wang, P. Sun, R. Nie and X. Wang, ES Mater. Manuf., 2018, 2, 9–15 Search PubMed.
- P. Yang, H. Zhao, Y. Yang, P. Zhao, X. Zhao and L. Yang, ES Mater. Manuf., 2020, 7, 34–39 Search PubMed.
- B. Liu, Y. Jin, G. Xie, Z. Wang, H. Wen, N. Ren and D. Xing, ES Energy Environ., 2018, 1, 56–66 Search PubMed.
- D. Li, J. Shi and C. Li, Small, 2018, 14, 1704179 CrossRef.
- J.-Y. Jung, J.-Y. Yu, R. B. Wehrspohn and J.-H. Lee, J. Phys. Chem. C, 2019, 123, 1660–1668 CrossRef CAS.
- M. Barawi, F. Fresno, R. Pérez-Ruiz and V. A. de la Peña O'Shea, ACS Appl. Energy Mater., 2019, 2, 207–211 CrossRef CAS.
- H. Zhang, Z. Yang, W. Yu, H. Wang, W. Ma, X. Zong and C. Li, Adv. Energy Mater., 2018, 8, 1800795 CrossRef.
- B. C. M. Martindale, G. A. M. Hutton, C. A. Caputo and E. Reisner, J. Am. Chem. Soc., 2015, 137, 6018–6025 CrossRef CAS.
- C. Hu, M. Li, J. Qiu and Y.-P. Sun, Chem. Soc. Rev., 2019, 48, 2315–2337 RSC.
- L. Zhang, Y. Yang, M. A. Ziaee, K. Lu and R. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 9460–9467 CrossRef CAS.
- T.-F. Yeh, C.-Y. Teng, S.-J. Chen and H. Teng, Adv. Mater., 2014, 26, 3297–3303 CrossRef CAS.
- Y. Yan, J. Chen, N. Li, J. Tian, K. Li, J. Jiang, J. Liu, Qi. Tian and P. Chen, ACS Nano, 2018, 12, 3523–3532 CrossRef CAS.
- H. Ding, S.-B. Yu, J.-S. Wei and H.-M. Xiong, ACS Nano, 2016, 10, 484–491 CrossRef CAS.
- Y. Chen, Y. Hao, T. Kou, Q. Li and Q. Gao, Food Chem., 2019, 286, 467–474 CrossRef CAS.
- Y. Chen, G. Dai and Q. Gao, ACS Sustainable Chem. Eng., 2019, 7, 14064–14073 CrossRef CAS.
- S. Paul, K. Gayen, N. Nandi and A. Banerjee, Chem. Commun., 2018, 54, 4341–4344 RSC.
- M. Luo, G. K. Olivier and J. Frechette, Soft Matter, 2012, 8, 11923–11932 RSC.
- D. Shen, Y. Long, J. Wang, Y. Yu, J. Pi, L. Yang and H. Zheng, Nanoscale, 2019, 11, 5998–6003 RSC.
- N. Dhenadhayalan, K.-C. Lin, R. Suresh and P. Ramamurthy, J. Phys. Chem. C, 2016, 120, 1252–1261 CrossRef CAS.
- K. J. Mintz, Y. Zhou and R. M. Leblanc, Nanoscale, 2019, 11, 4634–4652 RSC.
- H. Luo, N. Papaioannou, E. Salvadori, M. M. Roessler, G. Ploenes, E. R. H. van Eck, L. C. Tanase, J. Feng, Y. Sun, Y. Yang, M. Danaie, A. B. Jorge, A. Sapelkin, J. Durrant, S. D. Dimitrov and M. M. Titirici, ChemSusChem, 2019, 12, 4432–4441 CrossRef CAS.
- N. Papaioannou, A. Marinovic, N. Yoshizawa, A. E. Goode, M. Fay, A. Khlobystov, M. Titirici and A. Sapelkin, Sci. Rep., 2018, 8, 6559 CrossRef.
- H. Nie, M. Li, Q. Li, S. Liang, Y. Tan, L. Sheng, W. Shi and S. X.-A. Zhang, Chem. Mater., 2014, 26, 3104–3112 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0na00799d |
| This journal is © The Royal Society of Chemistry 2021 |