
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A sustainable and scalable multicomponent continuous flow process to access fused imidazoheterocycle pharmacophores†
Blake J. M.
Baker
ab,
William J.
Kerr
 *b,
David M.
Lindsay
b,
Vipulkumar K.
Patel
a and
Darren L.
Poole
*a
*b,
David M.
Lindsay
b,
Vipulkumar K.
Patel
a and
Darren L.
Poole
*a
aGlaxoSmithKline Medicines Research Centre, Gunnels Wood Road, Stevenage, Hertfordshire SG1 2NY, UK. E-mail: darren.l.poole@gsk.com
bDepartment of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: w.kerr@strath.ac.uk
First published on 4th January 2021
Abstract
Described herein is a green, continuous flow process for the synthesis of various aminoimidazoheterocycles, through the Gröebke–Blackburn–Bienaymé reaction (GBBR). This multicomponent procedure combines aminoazines, aldehydes and isocyanides to generate a wide variety of medicinally privileged, aminated imidazoheterocycle architectures. This method is performed in ethanol, using only mineral acid rather than the standard metal-based catalysts typical to the field. These sustainability benefits have been demonstrated even on multigram scale, exemplifying the facile scalability of the procedure. The process also boasts shorter reaction times, wider scope robustness, and improved yields compared to the currently available methods, with no requirement for an aqueous work-up procedure, affording resulting scaffolds of notable relevance, to a range of medicinal targets of academic and industrial interest.
Introduction
Continuous flow processes constitute a powerful and increasingly popular approach to chemical synthesis within the bulk and petrochemical industries. In comparison to batch methods, they offer benefits in terms of thermal transfer, superior mixing, safe handling of dangerous reagents, and the potential for in-line analyses and facile route telescoping.1 Moreover, the inherent safety of flow reactions, as well as their reduced requirement for reaction volume, and the quantities of reagents and solvents used, makes them ideally aligned to the 12 principles of green chemistry.2 Despite these advantages, the majority of chemical processes within the pharmaceutical and fine chemical industries remain batch in nature.3 Based on this, the benefits of applying flow chemistry methods for the sustainable synthesis of pharmaceutically relevant compounds are yet to be fully realised.Within medicinal chemistry, fused bicyclic heterocycles containing an imidazole motif, such as imidazopyridines, imidazopyrimidines, and imidazopyrazines are privileged pharmacophores, which are ubiquitous within a range of disease areas. For example, these scaffolds find application in health indications including cancer,4 inflammation,5 bacterial infection,6 HIV,7 cancer-induced osteoporosis,8 Alzheimer's disease,9 and diabetes.10 Moreover, several approved drug molecules and therapeutic probes, including olprinone11 (cardiotonic), and KA140712 (anti-malarial probe) also contain the fused imidazole functionality within their core heterocyclic framework. Furthermore, aminated derivatives of these imidazoheterocycles also possess elevated levels of medicinal importance, and include patented compounds such as the anti-inflammatory agent 1,13 and the GLP-1 (glucagon-like peptide-1) fusion peptide component 2, for diabetes and neurodegenerative disorders (Fig. 1).14 Given the prominent importance of this framework, an efficient, robust, sustainable, and scalable approach to aminated imidazole-containing heterocycles and their derivatives would be of appreciable value to the broad community of scientists aiming to access new therapeutic agents (Scheme 1).
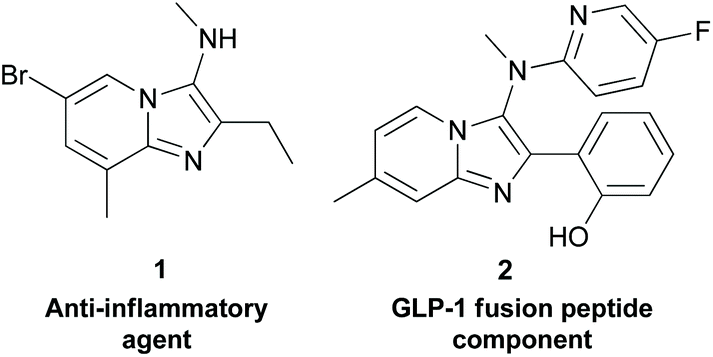 | ||
| Fig. 1 Examples of aminoimidazo[1,2-a]pyridines with therapeutic activity. | ||
 | ||
| Scheme 1 Summary of the sustainable, scalable, robust, and efficient flow-based GBBR process reported herein. | ||
Various methods have been reported for the synthesis of fused 3-aminoimidazoheterocycles (Scheme 2). For example, the triflic anhydride-mediated cyclisation of α-aminopyridinylamides delivers a variety of 3-aminoimidazo[1,2-a]heterocycles.15 Alternatively, aromatic ketones can be combined with 2 equivalents of a 2-aminopyridine, facilitated by iodine, to deliver 2-aryl-3-(pyridine-2-ylamino)imidazo[1,2-a]pyridines,16 and the synthesis of 3-amino[1,2-a]pyridines from α-ketovinyl azides and 2-aminopyridines has also been described.17 Finally, Katritzky and co-workers reported a synthesis of 3-aminoimidazoheterocycles bearing tertiary amine substituents.18 However, all of these methodologies possess distinct issues limiting their substrate applicability, scalability, versatility, efficiency, and preparative sustainability, as noted in Scheme 2, in addition to product work-up and purification demands.
 | ||
| Scheme 2 (Top) Reported synthetic methods to generate 3-aminoimidazoheterocycles. In all cases, a combination of several limitations is observed concerning preparative utility, including scalability, sustainability, substrate accessibility and desirability, scope robustness, and prolonged required reaction times. (Bottom) The Gröebke–Blackburn–Bienaymé reaction (GBBR) and the efficient, scalable, robust, and sustainable flow-based GBBR (this work). | ||
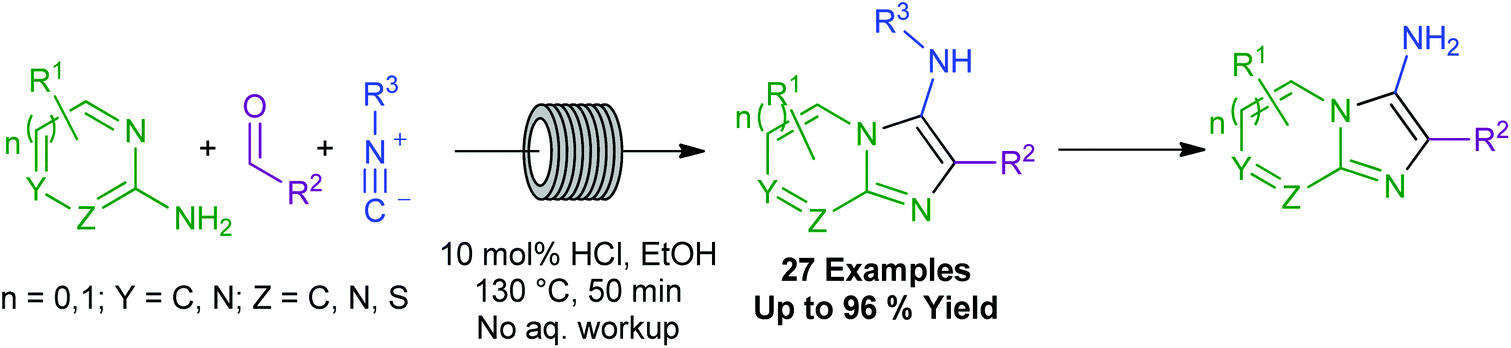
Based on these various drawbacks, the preferred synthetic route to 3-aminoimidazoheterocycles uses the Gröebke–Blackburn–Bienaymé reaction (GBBR, Scheme 2). The GBBR is a multicomponent reaction combining an aldehyde, an aminoazine, and an isocyanide to furnish the corresponding 3-aminoimidazoheterocycle in a one-pot procedure.19–22 The reaction involves an initial condensation of an aldehyde and an aminoazine, before a non-concerted [4 + 1] cycloaddition between the activated Schiff base intermediate and the isocyanide, to deliver the product (Scheme 2).23 The GBBR has been catalysed by a variety of Lewis and Brønsted acids,19–21,24–30 and more recent developments include the use of microwave heating,31 solvent- and acid-free conditions,32 biocatalysis,33 micellar reactions,34 ionic liquids as solvents and catalysts,35 and solid phase catalysis.36 Somewhat limited flow-based procedures have also been reported, with these methods typically providing the desired products in low to-moderate yields, and not tolerating aliphatic aldehydes.29,37
More broadly, all current GBBR methods suffer from a variety of drawbacks, most significantly including the use of unsustainable or expensive solvents and/or metal catalysts for long reaction times, as well as more general issues of variable yields and limited substrate scope or scalability. Moreover, the noxious nature of the isocyanide component limits the desirability of performing the GBBR under traditional batch conditions.
Herein, we present an easily-scalable, robust, environmentally benign, and efficient continuous flow-based GBBR process. The benefits of this method include the use of ethanol as a green solvent,38 and a dilute mineral acid catalyst in place of non-sustainable metal-based Lewis acids. The method requires no aqueous work-up procedure, and the effect of superheating within the flow setup results in a high-yielding, efficient reaction. The process is readily scalable and is demonstrated on a multigram scale. Additionally, the flexibility of the flow conditions allows control of reagent addition combinations to now bring sensitive and valuable functional groups into the scope of the GBBR.
Results and discussion
Initially, the model reaction of 2-aminopyridine 1a, benzaldehyde 2a, and tert-butyl isocyanide 3a was used for optimisation (Scheme 3 and Table 1). Consumption of the starting material was measured using HPLC concentration gradient measurements (see ESI†). Investigations began by evaluating reaction conversion at 50 °C over 24 h, before transfer of conditions to flow (entries 1–3), using conditions previously utilised within our laboratories. As expected, though inefficient, all reactions were fairly high yielding. The conditions in entry 1 were not pursued further due to the requirement for homogeneity within a flow procedure. The use of metal catalysts was also disfavoured when considering scale-up sustainability issues, and hydrochloric acid was preferred over perchloric acid, due to safety concerns related to superheating (entry 2 vs. entry 3). Ethanol (entry 4) was chosen as the reaction solvent owing to its beneficial green metrics and superior performance to acetonitrile (entry 3).38 In order to achieve high conversion on a timescale more amenable to flow, the reaction was heated to 130 °C and afforded a conversion of 93% by HPLC analysis, and an isolated yield of 84% in only 50 minutes (entry 5). The optimal loading of catalyst was found to be 10 mol%, as increasing the loading to 100 mol% (entry 6), or removing the catalyst completely (entry 7), both inhibited the reaction efficiency. From entry 8, an excess of aldehyde and isocyanide were employed to favour high conversion with potentially unreactive starting materials. Also, the less sustainable dioxane (entry 8) was replaced by ethanol (entry 9) as the mineral acid solvent; in both cases, quantitative conversion was observed. These optimised conditions were then transitioned into a continuous flow setup and, in this mode, delivered a 96% isolated yield of product 4a (entry 10), with the only purification required being the removal of volatile starting materials (see ESI† for diagrams and schematics of the flow equipment used).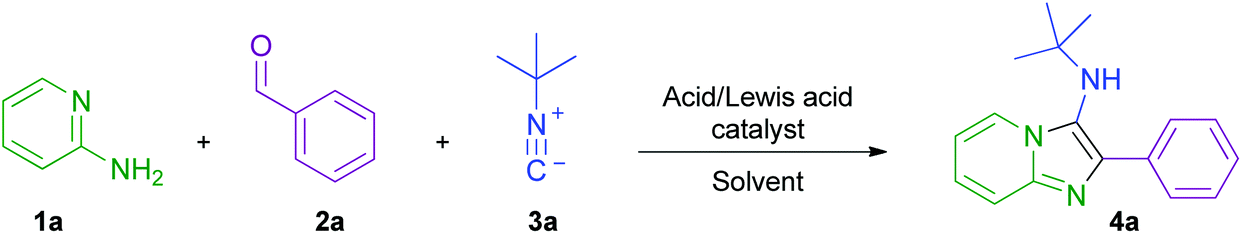 | ||
| Scheme 3 Model GBBR reaction between 2-aminopyridine 1a, benzaldehyde 2a, and tert-butyl isocyanide 3a, used for initial reaction optimisation. | ||
| Entry | Heating source | Temp (°C) | Solvent | Catalysta | Reaction/residence time | Conversionb (isolated yield) |
|---|---|---|---|---|---|---|
a All catalyst loadings at 10 mol % unless stated otherwise.
b Starting material conversion by HPLC analysis.
c 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 stoichiometry (1a :1:1 stoichiometry (1a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :2a:3a).
d Heterogenous mixture.
e 4 M solution in dioxane.
f 100 mol% catalyst.
g 1:2:2 stoichiometry (1a:2a:3a).
h 1.25 M in EtOH. :2a:3a).
d Heterogenous mixture.
e 4 M solution in dioxane.
f 100 mol% catalyst.
g 1:2:2 stoichiometry (1a:2a:3a).
h 1.25 M in EtOH.
|
||||||
| 1c | Thermal | 50 | Toluene | Sc(OTf)3d | 24 h | >99% |
| 2c | Thermal | 50 | MeOH | HClO4 | 24 h | 92% |
| 3c | Thermal | 50 | MeCN | HCle | 24 h | 77% |
| 4c | Thermal | 50 | EtOH | HCle | 24 h | 91% |
| 5c | MW | 130 | EtOH | HCle | 50 min | 93% (84%) |
| 6c | MW | 130 | EtOH | HCle,f | 50 min | 75% |
| 7c | MW | 130 | EtOH | — | 50 min | 36% |
| 8g | MW | 130 | EtOH | HCle | 50 min | >99% |
| 9g | MW | 130 | EtOH | HClh | 50 min | >99% |
| 10g | Flow | 130 | EtOH | HClh | 50 min | 97% (96%) |
With these optimised flow conditions in hand, the process was applied to a range of substrates. As shown in Scheme 4, the reaction is highly robust, allowing considerable variation of all starting materials. Solutions of the isocyanide component were present in an isolated input line in each case, meaning that exposure to their odour was eliminated. In terms of the amidine component, 2-aminopyridines (4b–k), 2-aminothiazole (4l, 4m), 2-aminopyrimidine (4n), 2-aminobenzothiazole (4o), and aminopyrazines (4p, 4q) were all tolerated well, giving good yields of the corresponding heterocycles. The reaction also allowed for various substitution patterns on the amidine component, allowing the use of methyl, hydroxymethyl, methoxy, ethyl ester, and bromine substituents on various positions of the aminated heterocycle. It has been observed that in the use of 2-aminopyrimidine in the GBBR, 2 regioisomers are possible, dependent on whether the initial imine condensation step occurs on the endocyclic or exocyclic nitrogen of the amidine.39 Whilst several reports have focused on the regioselective synthesis of aminoimidazopyrimidines,29,39–41 using our methodology an inherent selectivity of >10:1 for the 3-amino substituted product is seen in 4n (which has no substituents on the heteroaryl component that could be implicated in driving a regioisomeric bias).
 | ||
| Scheme 4 Substrate scope for the continuous flow GBBR process using aminopyridine, aminopyrimidine, aminopyrazine, aminothiazole, and aminobenzothiazole substrates with various aryl and heteroaryl aldehydes and a variety of commercial isocyanide reagents. Isolated yield is shown in parenthesis alongside starting material consumption (by HPLC analysis). a>10:1 regioselectivity by NOESY NMR spectroscopy. | ||
Focusing on the isocyanide component, aryl-, benzyl-, alkyl-, and other functionalised isocyanides are all tolerated well. The benzyl- and 1,1,3,3-tetramethylbutyl isocyanide-derived products are particularly useful, owing to the ready liberation of the latent primary amine product following debenzylation or dealkylation strategies (vide infra).42,43 Remarkably, under the mildly acidic conditions employed, (trimethylsilyl)methyl isocyanide can be used to generate products with the silylmethylamino functionality intact at the 3-position, for further manipulation (4e, 4h). Additionally, the use of an isocyanide reagent bearing ethyl ester functionality gave product 4i in good yield, and without significant by-product formation.
The aldehyde component can also be subject to considerable variation. Aryl aldehydes containing electron-donating and -withdrawing substituents performed well, affording a range of products in good yield, with methyl-, bromo-, hydroxyl-, and nitro-substituted aryl aldehydes all employed successfully to afford decorated bicyclic products 4b–d, 4h, 4m, 4n, and 4p. Heteroaryl aldehydes were also applied with good levels of effectiveness (4f, 4g, 4i, and 4j). Furthermore, example 4q illustrates the use of the GBBR with trans-cinnamaldehyde to deliver the styryl-substituted product.
A survey of the reported scope of the GBBR reveals limited and, moreover, generally low yielding incorporation of alkyl aldehydes and formaldehyde, likely owing to by product formation.4,21,29,45–47 Indeed, formaldehyde equivalents have been sought as a method to circumvent these issues.48–50 Accordingly, we next looked to challenge our protocol by the application of a range of alkyl aldehydes and formaldehyde (Scheme 5). Remarkably, using our method, and with the direct employment of aqueous (37% wt.) formaldehyde solution, reactions to form products 4r–4t proceeded cleanly and were uniformly high-yielding. Acetaldehyde could also be employed, using 3 equivalents to compensate for any acid-mediated acetaldehyde polymerisation.51 In this case, a good 65% yield of 4u was obtained, demonstrating another benefit of the flow methodology: the ability to modify the combination of starting materials in the input solutions, to bring highly reactive or sensitive reagents into the reaction scope. Primary, secondary, and tertiary alkyl aldehydes were all utilised to prepare a range of functionalised GBBR products 4u–4z. In all cases for the products shown in Schemes 4 and 5, no aqueous work-up procedure was required prior to purification. In the case of product 4y, our method was compared to a published batch GBBR procedure,44 where an extremely low conversion was observed, even after an appreciably extended reaction time (6 days). Additionally, in the batch reaction, multiple undesired by-products were observed, further highlighting the benefits of our developed flow procedure.
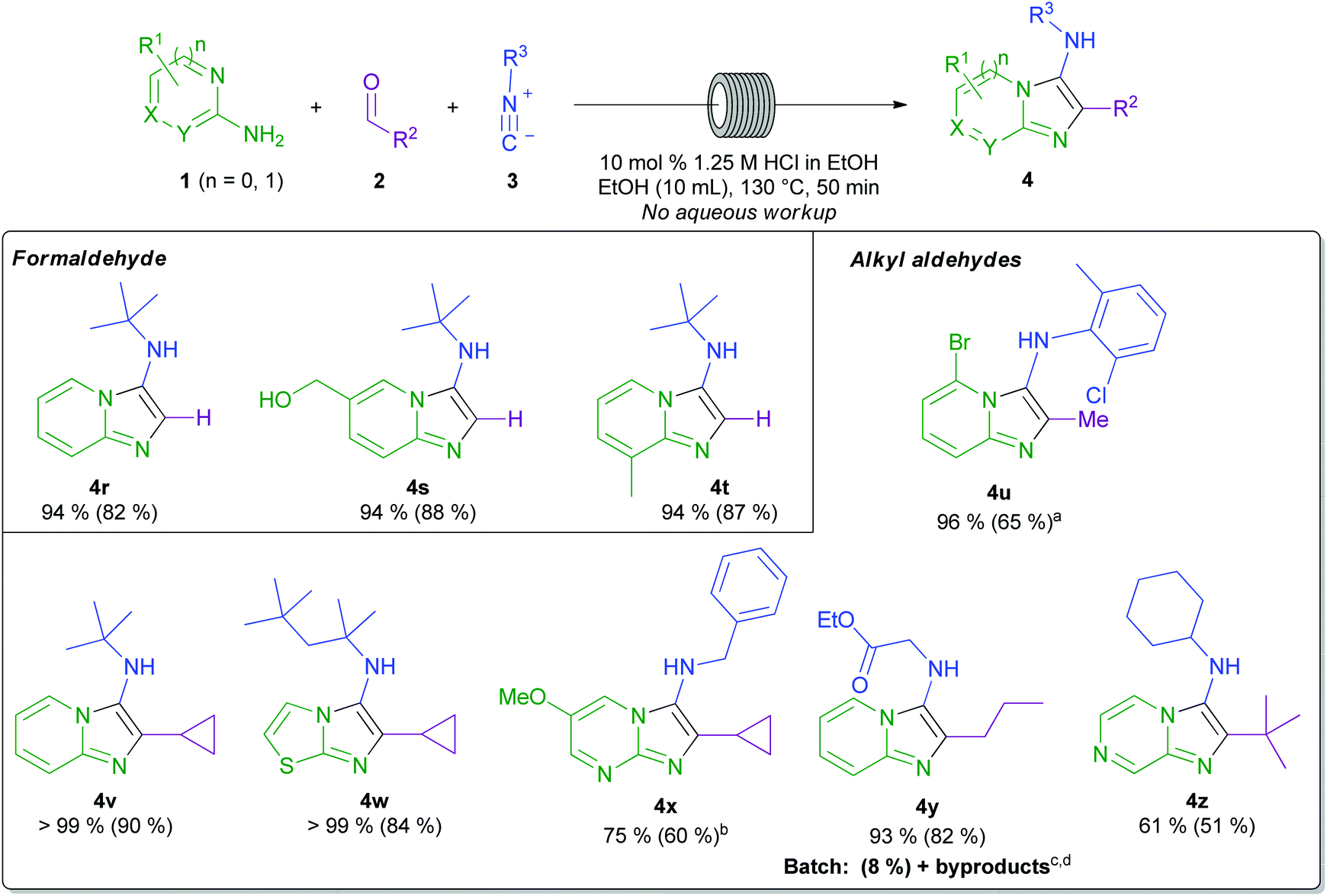 | ||
| Scheme 5 Substrate scope with formaldehyde and alkyl aldehydes. Typical conditions: 1 mmol amidine, 2 mmol isocyanide, 1.1 mmol 37% aq. formaldehyde or 2 mmol alkyl aldehyde, 10 mol% 1.25 M HCl in EtOH, EtOH, 50 min residence time. Isolated yield is shown in parenthesis alongside starting material consumption (by HPLC analysis). a3 equivalents of acetaldehyde was used, bonly observed regioisomer by 15N HMBC analysis, cliterature batch conditions: 1 mmol amidine, 1.1 mmol aldehyde, 1.2 mmol isocyanide, 0.05 mmol Sc(OTf)3 catalyst, 6 days, dvarious by products observed, see the ESI† for one example.44 | ||
Having demonstrated the effectiveness of this GBBR procedure on small scale, the ready scalability of the developed flow chemistry process was exploited in the multigram synthesis of 4aa in 70% isolated yield (Scheme 6). A facile deprotection of the 1,1,3,3-tetramethylbutyl group in 4aa then cleanly afforded the disubstituted imidazopyridine 4ab without any requirement for purification. To the best of our knowledge, this is the most efficient and sustainable example of a GBBR process using formaldehyde as the aldehyde component, and is the first scale-up GBBR procedure to be performed in flow.29,37,52 The reaction could be performed continuously for 8 h, and the starting materials and products were observed to be soluble up to a 0.5 M concentration, further enhancing the capability of the process for industrial application in order to generate medicinally relevant pharmacophores suitable for further modification. When reported batch conditions were again applied,44 a slightly lower yield was obtained, but only after a considerably extended reaction time, further highlighting the efficiency benefit gained from using the developed flow-based GBBR process.
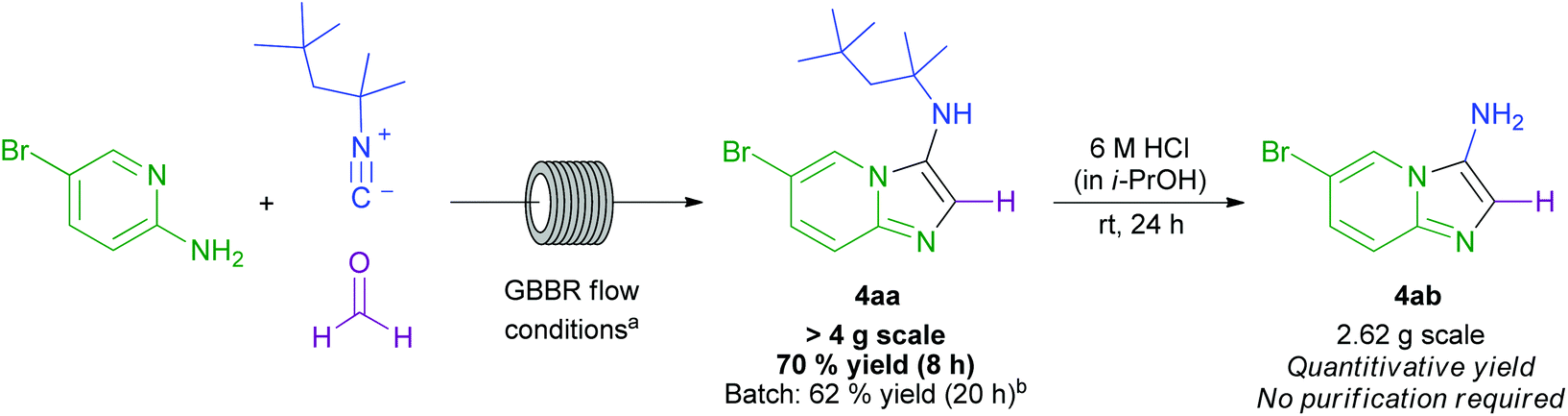 | ||
| Scheme 6 Scale up GBBR procedure and subsequent derivatisation to generate a difunctionalised imidazo[1,2-a]pyridine. aGBBR flow conditions: amidine (1.0 eq.); isocyanide (2.0 eq.), formaldehyde (37% wt. solution in H2O; 1.1 eq.), 10 mol% HCl (1.25 M in EtOH), 0.4 mL min−1 flow rate, 130 °C, ∼8 h total reaction time; bpatent literature batch conditions used: 18 mmol amidine, 19.8 mmol aldehyde, 21.6 mmol isocyanide, 0.900 mmol Sc(OTf)3 catalyst.44 | ||
Conclusions
In conclusion, we have developed a novel and sustainable continuous flow procedure for the synthesis of 3-aminoimidazoheterocycles, promoted by a simple dilute mineral acid in ethanol. The procedure is environmentally benign, robust, efficient, and scalable, requires no aqueous work-up procedure, and performs well even on larger scale. This multicomponent process displays an excellent substrate scope across all three reaction partners, enabling facile access to a range of privileged scaffolds relevant to a broad array of academically and industrially driven medicinal chemistry programmes.Synthetic methods
For a schematic and diagram of the specific flow system setup see the ESI.† The flow reactor setup consisted of a Syrris® Asia syringe module with Asia green syringes (250 μL/500 μL internal volume) joined via PFA tubing to a Vapourtec® RS-200 heating module with an attached 10 mL loop reactor. 2 × 10 mL loop reactors were used during the scale-up reaction. The overall internal volume of the apparatus was approximately 15.9 mL for the scope examples, as estimated using Rose Bengal solution in the system which had been flushed with toluene. The flow equipment was fitted with two back pressure regulators of 75 and 40 psi (8.0 bar total internal pressure).Representative flow procedure using aryl and heteroaryl aldehydes (Scheme 4, entry 4g)
Two stock 10 mL solutions were made up. The first solution contained 5-methanolyl-2-aminopyridine (248 mg, 2.00 mmol), picolinaldehyde (0.380 mL, 4.00 mmol), and HCl (0.160 mL, 0.200 mmol, 1.25 M in ethanol) in ethanol (10.0 mL total volume) as an amber coloured, clear solution. The second solution contained (isocyanomethyl)trimethylsilane (0.522 mL, 4.00 mmol) in ethanol (10.0 mL overall solution), as a colourless, clear solution. 5.00 mL from each solution was pumped into the flow reactor, heated to 130 °C, at a flow rate of 0.100 mL per minute per line for 50 min. Ethanol was then collected for 25 min to account for the 5 mL dead volume in the reactor, before the product solution was collected for 61 min 45 s (12.35 mL crude product mixture collected as a bronze clear solution). A 10 μL aliquot was removed from the solution and diluted with 1 mL methanol for quantitative HPLC analysis against a concentration gradient of the amidine starting material. 92% consumption of the starting material was observed, by quantitative HPLC analysis. The solvent was removed in vacuo, and the product was isolated by flash column chromatography, using a gradient elution of 5 to 30% ethyl acetate in cyclohexane. Following solvent removal under reduced pressure, (3-((2-morpholinoethyl) amino)-2-(pyridine-2-yl)imidazo[1,2-a]pyridin-6-yl)methanol 4g (309 mg, 0.874 mmol, 87% yield) was isolated as a beige powder.Representative flow procedure using formaldehyde (Scheme 5, entry 4s)
Two stock 10 mL solutions were prepared. The first solution contained 5-methanolyl-2-aminopyridine (248 mg, 2.00 mmol) and 2-isocyano-2-methylpropane (0.452 mL, 4.00 mmol) in ethanol (10.0 mL total volume) as a clear yellow solution. The second solution contained HCl (0.160 mL, 0.200 mmol, 1.25 M in ethanol) and aqueous 37% wt. formaldehyde (0.166 mL, 2.24 mmol) in ethanol (10.0 mL total volume) as a colourless, clear solution. 5.00 mL from each solution was pumped into the flow reactor, heated to 130 °C, at a flow rate of 0.100 mL per minute per line for 50 min. Ethanol was then collected for 25 min to account for the 5 mL dead volume in the reactor, before the product solution was collected for 61 min 45 s (12.35 mL crude product mixture collected as a golden clear solution). A 10 μL aliquot was removed from the solution and diluted with 1 mL methanol for quantitative HPLC analysis against a concentration gradient of the amidine starting material. 94% consumption of the starting material was observed, by quantitative HPLC analysis. The solvent was removed in vacuo, and the product was isolated by flash column chromatography, using a gradient elution of 5 to 10% ethanol in ethyl acetate. Following solvent removal under reduced pressure, (3-(tert-butylamino)imidazo[1,2-a]pyridin-6-yl) methanol 4s (192 mg, 0.876 mmol, 88% yield) was isolated as a white crystalline solid.Notes
The data that support the findings of this study are available within the article and its ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for funding via Prosperity Partnership EP/S035990/1. The authors would also like to thank Steve Richards (PDS-ASD-Global Spectroscopy-NMR UK, GSK) for his assistance with NMR analysis, and PDS API chemistry, GSK for consultation regarding the use of flow chemistry apparatus.References
- R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- B. Gutmann, D. Cantillo and C. O. Kappe, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS.
- A. T. Baviskar, C. Madaan, R. Preet, P. Mohapatra, V. Jain, A. Agarwal, S. K. Guchhait, C. N. Kundu, U. C. Banerjee and P. V. Bharatam, J. Med. Chem., 2011, 54, 5013–5030 CrossRef CAS.
- M. R. McKeown, D. L. Shaw, H. Fu, S. Liu, X. Xu, J. J. Marineau, Y. Huang, X. Zhang, D. L. Buckley, A. Kadam, Z. Zhang, S. C. Blacklow, J. Qi, W. Zhang and J. E. Bradner, J. Med. Chem., 2014, 57, 9019–9027 CrossRef CAS.
- N. M. Shukla, D. B. Salunke, E. Yoo, C. A. Mutz, R. Balakrishna and S. A. David, Bioorg. Med. Chem., 2012, 20, 5850–5863 CrossRef CAS.
- Tazeem, X. Han, Q. Zhou, J. Wei, P. Tien, G. Yang, S. Wu and C. Dong, Med. Chem. Commun., 2019, 10, 89–100 RSC.
- K. V. Sashidhara, L. R. Singh, D. Choudhary, A. Arun, S. Gupta, S. Adhikary, G. R. Palnati, R. Konwar and R. Trivedi, RSC Adv., 2016, 6, 80037–80048 RSC.
- S. Azimi, A. Zonouzi, O. Firuzi, A. Iraji, M. Saeedi, M. Mahdavi and N. Edraki, Eur. J. Med. Chem., 2017, 138, 729–737 CrossRef CAS.
- D. Flesch, S.-y. Cheung, J. Schmidt, M. Gabler, P. Heitel, J. Kramer, A. Kaiser, M. Hartmann, M. Lindner, K. Lüddens–Dämgen, J. Heering, C. Lamers, H. Lüddens, M. Wurglics, E. Proschak, M. Schubert–Zsilavecz and D. Merk, J. Med. Chem., 2017, 60, 7199–7205 CrossRef CAS.
- K. Mizushige, T. Ueda, K. Yukiiri and H. Suzuki, Cardiovasc. Drug Rev., 2002, 20, 163–174 CrossRef CAS.
- C. W. McNamara, M. C. Lee, C. S. Lim, S. H. Lim, J. Roland, O. Simon, B. K. Yeung, A. K. Chatterjee, S. L. McCormack, M. J. Manary, A.-m. Zeeman, K. J. Dechering, T. S. Kumar, P. P. Henrich, K. Gagaring, M. Ibanez, N. Kato, K. L. Kuhen, C. Fischli, A. Nagle, M. Rottmann, D. M. Plouffe, B. Bursulaya, S. Meister, L. Rameh, J. Trappe, D. Haasen, M. Timmerman, R. W. Sauerwein, R. Suwanarusk, B. Russell, L. Renia, F. Nosten, D. C. Tully, C. H. Kocken, R. J. Glynne, C. Bodenreider, D. A. Fidock, T. T. Diagana and E. A. Winzeler, Nature, 2013, 504, 248–253 CrossRef CAS.
- N. Desroy, B. Heckmann, R. C. X. Brys, A. M. Joncour, C. Peixoto, X. M. Bock and C. G. Housseman, WO2014/139882A1, 2014.
- C. Wallrapp, E. Thoenes and P. Geigle, WO2008/116648A2, 2008.
- S. Régnier, W. S. Bechara and A. B. Charette, J. Org. Chem., 2016, 81, 10348–10356 CrossRef.
- Z. Fei, Y.-p. Zhu, M.-c. Liu, F.-c. Jia and A.-x. Wu, Tetrahedron Lett., 2013, 54, 1222–1226 CrossRef CAS.
- P. R. Adiyala, G. S. Mani, J. B. Nanubolu, K. C. Shekar and R. A. Maurya, Org. Lett., 2015, 17, 4308–4311 CrossRef CAS.
- A. R. Katritzky, Y.-j. Xu and H. Tu, J. Org. Chem., 2003, 68, 4935–4937 CrossRef CAS.
- K. Groebke, L. Weber and F. Mehlin, Synlett, 1998, 661–663 CrossRef CAS.
- C. Blackburn, B. Guan, P. Fleming, K. Shiosaki and S. Tsai, Tetrahedron Lett., 1998, 39, 3635–3638 CrossRef CAS.
- H. Bienaymé and K. A. Bouzid, Angew. Chem., Int. Ed., 1998, 37, 2234–2237 CrossRef.
- For a review of the GBBR, see: A. Boltjes and A. Dömling, Eur. J. Org. Chem., 2019, 42, 7007–7049 CrossRef.
- For a review of the GBBR, see: N. Devi, R. K. Rawal and V. Singh, Tetrahedron, 2015, 71, 183–232 CrossRef CAS.
- NH4Cl: A. Shaabani, F. Rezazadeh and E. Soleimani, Monatsch. Chem., 2008, 139, 931–933 CrossRef CAS.
- PTSA: J. J. Chen, A. Golebiowski, J. McClenaghan, S. R. Klopfenstein and L. West, Tetrahedron Lett., 2001, 42, 2269–2271 CrossRef CAS.
- Sc(OTf)3: S. M. Ireland, H. Tye and M. Whittaker, Tetrahedron Lett., 2003, 44, 4369–4371 CrossRef CAS.
- Sc(OTf)3: M. Konstantinidou, Z. Boiarska, R. Butera, C. G. Neochoritis, K. Kurpiewska, J. Kalinowska-Tłuscik and A. Dömling, Eur. J. Org. Chem., 2020, 34, 5601–5605 Search PubMed.
- ZnCl2: A. L. Rousseau, P. Matlaba and C. J. Parkinson, Tetrahedron Lett., 2007, 48, 4079–4082 CrossRef CAS.
- ZrCl4: A. J. E. Butler, M. J. Thompson, P. J. Maydom, J. A. Newby, K. Guo, H. Adams and B. Chen, J. Org. Chem., 2014, 79, 10196–10202 CrossRef CAS.
- AgOAc: M. Hussein, J. Liu, L. Fu and M. Hasan, J. Heterocycl. Chem., 2020, 57, 955–964 CrossRef.
- B. Maiti, K. Chanda, M. Selvaraju, C.-c. Tseng and C.-m. Sun, ACS Comb. Sci., 2013, 15, 291–297 CrossRef CAS.
- S. Vidyacharan, A. H. Shinde, B. Satpathi and D. S. Sharada, Green Chem., 2014, 16, 1168–1175 RSC.
- M. Budhiraja, R. Kondabala, A. Ali and V. Tyagi, Tetrahedron, 2020, 76, 131643 CrossRef.
- S. Mathavan and R. B. R. D. Yamajala, ChemistrySelect, 2020, 5, 10637–10642 CrossRef CAS.
- A. Shaabani, E. Soleimani and A. Maleki, Tetrahedron Lett., 2006, 47, 3031–3034 CrossRef CAS.
- R. S. Varma and D. Kumar, Tetrahedron Lett., 1999, 40, 7665–7669 CrossRef CAS.
- M. Reutlinger, T. Rodrigues, P. Schneider and G. Schneider, Angew. Chem., Int. Ed., 2014, 53, 582–585 CrossRef CAS.
- H. E. Gottlieb, G. Graczyk-Millbrandt, G. G. A. Inglis, A. Nudelman, D. Perez, Y. Qian, L. E. Shuster, H. F. Sneddon and R. J. Upton, Green Chem., 2016, 18, 3867–3878 RSC.
- G. S. Mandair, M. Light, A. Russell, M. Hursthouse and M. Bradley, Tetrahedron Lett., 2002, 43, 4267–4269 CrossRef CAS.
- V. Z. Parchinsky, O. Shuvalova, O. Ushakova, D. V. Kravchenko and M. Krasavin, Tetrahedron Lett., 2006, 47, 947–951 CrossRef CAS.
- R. P. Law, S. Ukuser, D. T. Tape and E. P. A. Talbot, Synthesis, 2017, 49, 3775–3793 CrossRef CAS.
- C. Blackburn and B. Guan, Tetrahedron Lett., 2000, 41, 1495–1500 CrossRef CAS.
- Y. Sandulenko, A. Komarov, K. Rufanov and M. Krasavin, Tetrahedron Lett., 2008, 49, 5990–5993 CrossRef CAS.
- S. Allen, B. J. Brandhuber, K. R. Condroski, L. Huang, T. Kercher and S. L. Winski, WO2014/078408A1/, 2014.
- R. Gladysz, Y. Adriaenssens, H. D. Winter, J. Joossens, A.-m. Lambeir, K. Augustyns and P. V. d. Veken, J. Med. Chem., 2015, 58, 9238–9257 CrossRef CAS.
- A. Demjén, M. Gyuris, J. Wölfling, L. G. Puskás and I. Kanizsai, Beilstein J. Org. Chem., 2014, 10, 2338–2344 CrossRef.
- M. L. Bode, D. Gravestock, S. S. Moleele, C. W. v. d. Westhuyzen, S. C. Pelly, P. A. Steenkamp, H. C. Hoppe, T. Khan and L. A. Nkabinde, Bioorg. Med. Chem., 2011, 19, 4227–4237 CrossRef CAS.
- M. A. Lyon and T. S. Kercher, Org. Lett., 2004, 6, 4989–4992 CrossRef CAS.
- A. Sharma and H.-y. Li, Synlett, 2011, 1407–1412 CAS.
- P. Surkar, T. Kaur and A. Sharma, Synlett, 2015, 26, 1403–1407 CrossRef CAS.
- K. K. Georgieff, J. Appl. Polym. Sci., 1966, 10, 1305–1313 CrossRef CAS.
- M. Baenziger, E. Durantie and C. Mathes, Synthesis, 2017, 49, 2266–2274 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc03675g |
| This journal is © The Royal Society of Chemistry 2021 |