
Efficient and accessible silane-mediated direct amide coupling of carboxylic acids and amines†
Melissa C.
D'Amaral
 ,
Nick
Jamkhou
and
Marc J.
Adler
*
,
Nick
Jamkhou
and
Marc J.
Adler
*
Department of Chemistry & Biology, Ryerson University, Toronto, ON M5B 2K3, Canada. E-mail: marcjadler@ryerson.ca
First published on 23rd November 2020
Abstract
A straightforward method for the direct synthesis of amides from amines and carboxylic acids without exclusion of air or moisture using diphenylsilane with N-methylpyrrolidine has been developed. Various amides are made efficiently, and broad functional group compatibility is shown through a Glorius robustness study. A gram-scale synthesis demonstrates the scalability of this method.
Introduction
The amide is a cornerstone functional group in organic chemistry. Amide-containing molecules are crucial in a variety of fields: 25% of drugs on the market contain an amide bond (including all peptidic and each of the top six selling small-molecule drugs) and polyamides represent one of the most versatile and functional classes of synthetic polymers.1–4 In these applications and others, amide bonds are ubiquitous and vital, and because of this amidation reactions are among the most frequently performed transformations in organic chemistry. Although there has been significant effort dedicated to the development of methods for amidation and great advances have been made, the need for greener approaches remain a high priority in the synthetic organic chemistry community.4–6 Accordingly, the ACS Green Chemistry Institute Pharmaceutical Roundtable (GCIPR) listed sustainable direct amide bond formation as one of their ten key green chemistry research areas in 2018.7The most direct way to make amides is through the condensation of a carboxylic acid with an amine. These substrates are the precursors of choice for amide synthesis due to their large abundance and the generation of only water as waste upon forming the amide bond. However, in the absence of other reagents or catalysts, the elimination of water will not proceed except under harsh, forcing conditions due to spontaneous formation of an ammonium carboxylate salt.2,8,9 Such conditions are impractical for the general synthesis of amides or peptides, specifically those with sensitive functionalities.
A variety of alternative approaches to amide bond formation have been developed to circumvent this issue. One route is through the use of more electrophilic carboxylic acid derivatives, including acid chlorides. However, such derivatives are highly reactive2 and require additional synthetic steps to access them from naturally occurring sources. In situ preactivation of the carboxylic acid using stoichiometric coupling reagents is the most widely used method for amide bond formation.2 The most common coupling reagents include carbodiimides, uronium/phosphonium salts, or benzotriazoles. Despite high efficiency, these reagents result in poor atom economy and toxic by-products, and they can require harsh conditions, expensive reagents, and/or challenging purifications.2,6 Furthermore, recent reports have linked widely used uronium coupling reagents (e.g. HATU) for peptide synthesis to the development of severe allergies as a result of extended exposure.10 Catalytic methods have also been explored for direct amidation in an effort to improve atom economy and reduce waste; catalysts include organoboron species9,11 and metal complexes.12 In spite of their promise, various pitfalls – including inefficiency, water-sensitivity, narrow substrate scopes, and metal toxicity – have prevented these methods from being widely adopted and leave stoichiometric methods as the state-of-the-art.6,13,14 Therefore, the development of green, safe, efficient, and practical stoichiometric coupling reactions are of high interest.
Organosilanes are highly attractive reagents for organic synthesis from a green perspective and have historically shown sporadic promise as stoichiometric amide coupling reagents.15 Silicon is the second most abundant surface element on earth and is environmentally benign.16 Silanes are relatively inert, making them safer to handle and easily stored.17 Their unreactive natures renders them compatible with many common functional groups and a wide range of reaction conditions; for this reason they are often used as protecting groups.18 Because of these favourable attributes and versatile reactivity, organosilanes are widely commercially available and well-established as tools in organic synthesis.
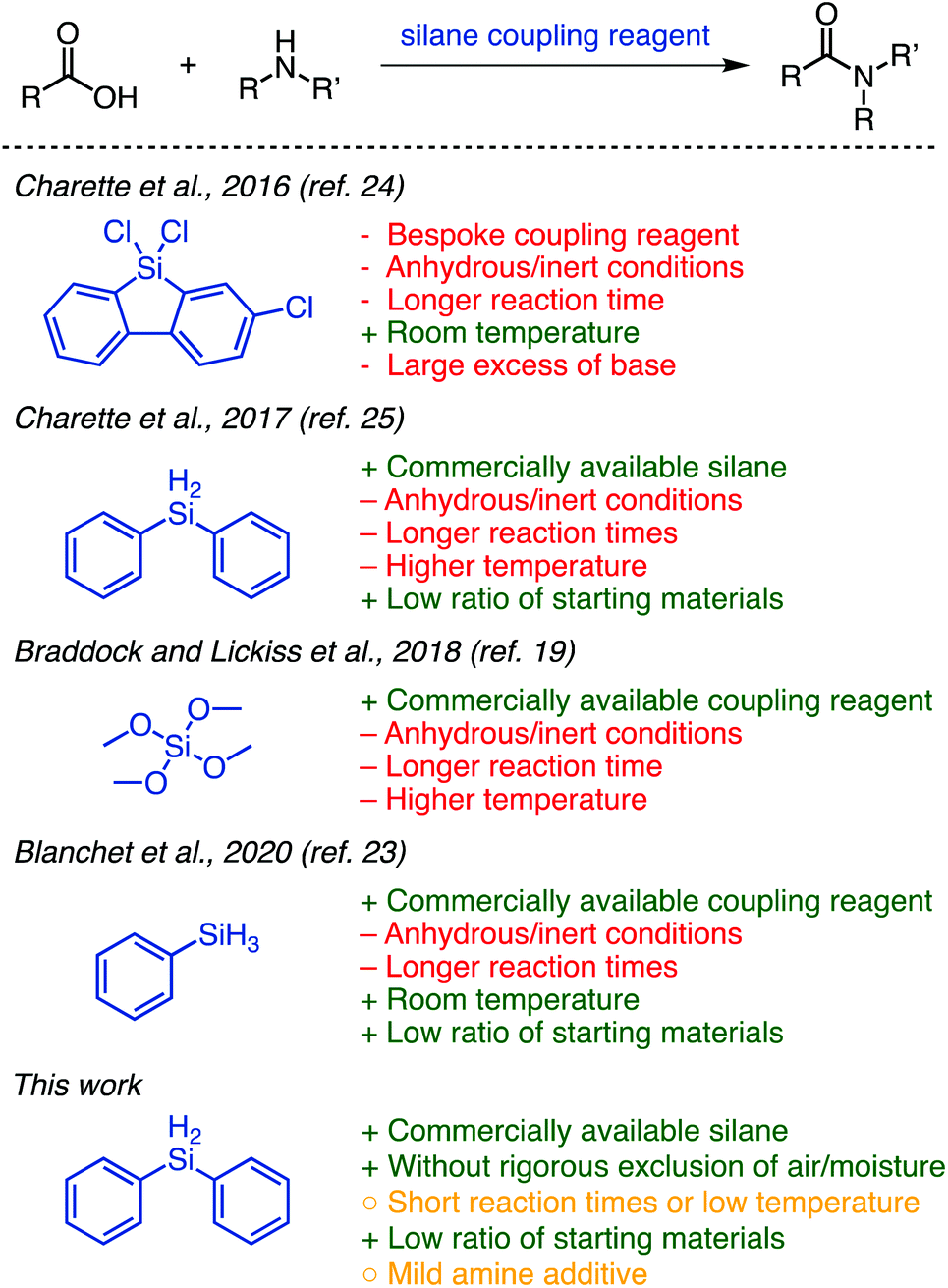
The development of efficient methods that utilize organosilanes in amide coupling reactions could be transformative for the field. For this reason, interest and exploration in the field of silane-mediated coupling has increased over the past few years (Fig. 1).19–23 Tetramethyl orthosilicate (TMOS) was reported by Braddock and Lickiss et al. as a highly efficient coupling reagent for direct amidation.19 Phenylsilane was used by the Denton20–22 and Blanchet23 groups to form amides as products22,23 or intermediates.20,21 Charette and co-workers disclosed two highly efficient methods using 9-silafluorenyl dichlorides24 and diphenylsilane25 (Fig. 1), respectively, as amide coupling reagents. Their advancement from the bespoke silafluorenyl reagent to commercially available diphenylsilane for this protocol improved atom economy and avoided the production of a protic acid, which can be problematic for peptide synthesis. A highlight of these reactions is the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 stoichiometry of amine:carboxylic acid:silane coupling reagent; this waste-minimizing ratio lies in stark contrast to state-of-the-art peptide synthesis that usually requires large excesses of both coupling reagents and one of the two coupling partners to get high conversion.
:1:1 stoichiometry of amine:carboxylic acid:silane coupling reagent; this waste-minimizing ratio lies in stark contrast to state-of-the-art peptide synthesis that usually requires large excesses of both coupling reagents and one of the two coupling partners to get high conversion.
 | ||
| Fig. 1 Recent silane-mediated amide coupling reactions. | ||
The methods disclosed to date show great promise for the approach of using silane coupling reagents; however, they each suffer from drawbacks that preclude their widespread adoption (Fig. 1). The requirement of anhydrous and inert-atmospheric conditions and extended reaction times limits practicality. The use of elevated temperatures (particularly applied for long periods) means it may not be suitable for use with molecules possessing certain sensitive functionalities. Some further suffer from limited reported scope, and it is worth noting in particular that TMOS is a highly dangerous reagent.19 We were, however, inspired by these works and sought to utilize the ideas that were brought forth to develop a greener method for coupling amines and unactivated carboxylic acids using silanes. Herein, we report a straightforward and efficient organosilane-mediated amidation reaction without rigorous exclusion of air or water; this method is metal-free and generates H2 and a siloxane as by-products (Fig. 1). We additionally probe the scope and general compatibility of the proposed method.
Results and discussion
A key, simple issue we noted in our early studies is that when we mixed our trial substrates (benzylamine and 2-phenylacetic acid) an insoluble ammonium carboxylate salt formed. This is problematic for the reaction for two reasons: (1) removing the reactants from solution could hamper their ability to interact with each other and thus react, (2) protonation of the amine substrate decreases the nucleophilicity of the amine. To address these issues and facilitate the reaction, we added an exogenous base, N-methylpyrrolidine (NMPi), which then afforded the amide product in good yield (Table 1). The tertiary amine base serves to deprotonate the carboxylic acid moiety to make the ammonium carboxylate salt more soluble and maintain the nucleophilicity of the amine coupling partner. NMPi is a waste product of the process; however, if desired, it could be recycled after the reaction is complete. We observed that performing the reaction without rigorous exclusion of air or water was optimal under each of the three sets of explored conditions (Table 1), which sets this apart from other methods using organosilicon reagents, where water exclusion was necessary for productive reaction.15g,19,23–25 Trace water seemed to be beneficial,26 but adding one equivalent of water was detrimental under all investigated conditions. Additionally, although microwave irradiation was unsuccessful in reducing reaction times for Charette's diphenylsilane-based method,25 we found that it can be used to reduce reaction times (see ESI, Table S1†). Accordingly, microwave conditions were generally used to optimize this method.|
|
||||
|---|---|---|---|---|
| Conditionsa | Yieldb (%) | |||
| Anhydrous? | +Base? | RT, overnightc | 80 °C, 20 min | 80 °C (MW), 5 min |
| a Reaction conditions: 4 M in MeCN with NMPi as base at indicated temperature and time, unless otherwise stated. b NMR yield using EtOAc as an internal standard, unless otherwise stated. c Reaction run in dichloromethane (CH2Cl2) at 4 M. d Ph-NO2 used as an internal standard. e 1 eq. of water was added to this reaction mixture. | ||||
| No | No | 0 | 41 | 21 |
| Yes | No | 0 | 40 | 14 |
| No | Yes | 63d | 84d | 77d |
| Yes | Yes | 44d | 56 | 52 |
| Noe | Yes | 0 | 31 | 22 |
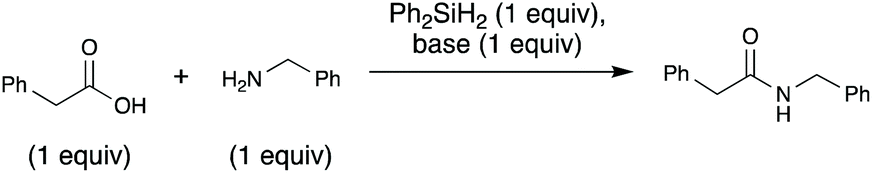
We investigated the effect of various bases on the reaction (see ESI, Table S1†). K2CO3 did not affect the reaction beneficially, but tertiary amine bases did shorten reaction times and improve yields. NMPi provided the highest yield of amide product in the microwave (77%) and can be catalytic: 0.5 equivalents gave the same yield of product as 1.0 equivalents.
Diphenylsilane was the optimal coupling agent of the screened commercially available silanes (Table 2, entry 7).25 No evidence of amide formation was observed using other silanes (entries 1–4 and 6) with triethylamine as the base. A low yield was obtained using 1-hydrosilatrane when pyrrolidine was the amine substrate (Table 1, entry 5). It was determined that a stoichiometric amount of diphenylsilane is needed in this reaction when using NMPi as the base (Table 1, entry 8).
|
|
|||
|---|---|---|---|
| Entry | Silane | Base | Yieldb (%) |
| a Reaction conditions in microwave: 1 M in MeCN and ran for 5 min at 80 °C, unless otherwise noted. b NMR yield using nitrobenzene (Ph-NO2) as an internal standard. c Heated at 100 °C in an oil bath for 20 min. d Pyrrolidine substrate used as the amine and heated at 100 °C in an oil bath for 4 h. e 4 M reaction. f 0.2 eq. of diphenylsilane. g 1.2 eq. of diphenylsilane. | |||
| 1 | PhSiH3 | TEA | <1 |
| 2 | Ph3SiH | TEA | 0 |
| 3 | Hydrosilatrane | TEA | 0 |
| 4 | Hydrosilatrane | NMPi | 0c |
| 5 | Hydrosilatrane | NMPi | 15d |
| 6 | PMHS | TEA | 0 |
| 7 | Ph2SiH2 | TEA | 45 |
| 8 | Ph2SiH2 | NMPi | 77e |
| 9 | Ph2SiH2 | NMPi | 19e,f |
| 10 | Ph2SiH2 | NMPi | 60e,g |
We then probed the reaction solvent, temperature, and time in the microwave. Polar protic and several polar aprotic solvents did not allow the reaction to proceed with any amide product (Table 3, entries 1–4). We tried pyridine (Table 3, entry 5) to explore whether a basic solvent would improve the reaction, but the yield significantly decreased. Other polar aprotic solvents (Table 3, entries 6–8) resulted in moderate yields. Acetonitrile was found to be the optimal solvent (Table 3, entry 9), and increasing concentration (Table 3, entries 9–11) improved yield. Running the reaction neat was not as productive (Table 3, entry 12). Thus, under microwave conditions, the reaction was optimal at 10 M in MeCN and we reached a maximum yield at 120 °C for 5 minutes (entry 13). Running the reaction for just 30 seconds gave a slightly decreased yield of 74% (ESI, Table S8†).
|
|
|||
|---|---|---|---|
| Entry | Solvent | [C] (M) | Yielda (%) |
| a NMR yield using Ph-NO2 as an internal standard, unless otherwise stated. b EtOAc as an internal standard. c 120 °C in the microwave. | |||
| 1 | EtOAc | 1 | 0 |
| 2 | Acetone | 1 | 0 |
| 3 | H2O | 1 | 0 |
| 4 | iPrOH | 1 | 0 |
| 5 | Pyridine | 1 | 21 |
| 6 | THF | 1 | 34 |
| 7 | DMF | 1 | 50 |
| 8 | CH2Cl2 | 1 | 51 |
| 9 | MeCN | 1 | 57 |
| 10 | MeCN | 4 | 77b |
| 11 | MeCN | 10 | 84b |
| 12 | No solvent | — | 42b |
| 13 | MeCN | 10 | 89c |
For the thermal reactions (Table 4), as the temperature increased from 80 °C to 100 °C the yield predictably increased, however the yield at 120 °C (entry 5) was not significantly different from the yield at 100 °C so temperatures higher than that were not explored. The reactions completed in the absence of NMPi had a significantly lower yield compared to the reactions with the amine base even after a longer reaction time, indicating that the inclusion of NMPi is beneficial under thermal conditions. A reaction time of 20 minutes was required for complete reaction (see ESI, Table S12†).
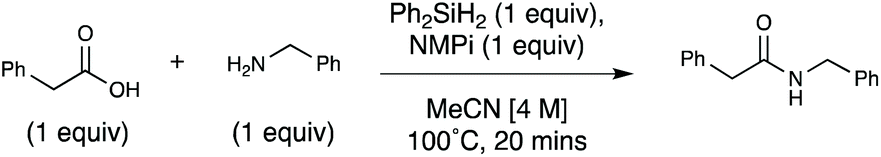
The scope and limitations of this method were then explored (Scheme 1). For accessibility (as not every lab is equipped with a microwave synthesizer) and time-efficiency (as the room temperature reactions must run for an extended period of time), we demonstrated the scope comprehensively using the optimized thermal conditions. Many substrates were further examined using the microwave and/or room temperature methods to establish the generality of these alternative approaches. The reaction was briefly re-optimized for both (1) aromatic carboxylic acid substrates (see ESI, Table S14†) and (2) secondary aliphatic amines (see ESI, Table S15†). Several amides comprising a range of classes were synthesized in good to excellent yield with the optimized reaction conditions.
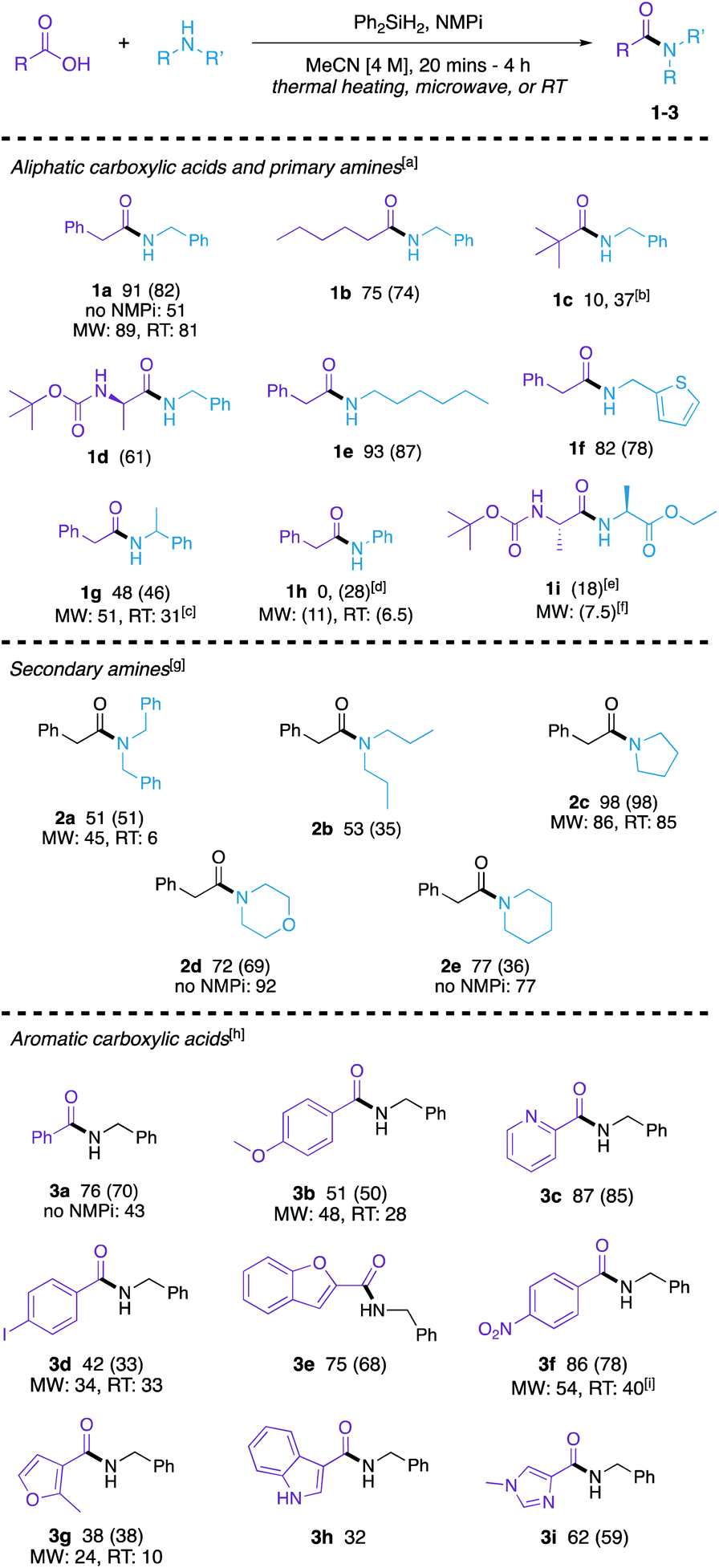 | ||
| Scheme 1 Substrate scope. Yields reported as NMR yield, with isolated yields listed in parentheses, if obtained. aThermal reaction conditions: 1 mmol of acid, amine, silane, and base 100 °C in oil bath for 20 minutes. MW conditions: 120 °C in the microwave, 5 minutes, 10 M, 1.0 mmol of acid, amine, silane, and base. RT conditions: room temperature, 20 h, 4 M, 1.0 mmol of acid, amine, silane, and base. b2.0 mmol of amine and reaction time of 1 h. cReaction run at 1 M. d4 h reaction time. e2 mmol of base and reaction time of 1 h. f2.0 mmol of base at 80 °C. gThermal reaction conditions: 1.0 mmol of acid, amine, silane, and base. 100 °C in an oil bath for 4 h. MW conditions: 120 °C in the microwave, 10 minutes, 4 M, 1.0 mmol of acid, amine, silane, and base. RT conditions: room temperature, 20 h, 4 M, 1.0 mmol of acid, amine, silane, and base. hThermal reaction conditions: 1.5 mmol of acid, and 1.0 mmol of amine, silane, and base. 100 °C in an oil bath for 1 h. MW conditions: 120 °C in the microwave, 5 minutes, 10 M, 1.5 mmol of acid, and 1.0 mmol of amine, silane, and base. RT conditions: room temperature, 20 h, 1 M in CH2Cl2, 1.5 mmol of acid, and 1.0 mmol of amine, silane, and base. i4 M reaction. | ||
Aliphatic carboxylic acids coupled to primary aliphatic amines were produced in moderate to excellent yields (1a–1g). The reaction of 2-phenylacetic acid with benzylamine proceeded smoothly. Bulkier pivalic acid and α-methylbenzylamine were not as efficient substrates under these reaction conditions giving lower yields of their amide product (1c and 1g, respectively) when coupled to benzylamine and phenylacetic acid, respectively. A longer reaction time and 2.0 equivalents of benzylamine was needed to afford amide 1c in a moderate yield of 37%. A long chain carboxylic acid (hexanoic acid) and alkyl amine (hexylamine) were converted to their corresponding amides (1b and 1e, respectively) in good yields. An N-protected amino acid ((tert-butoxycarbonyl)-L-alanine) and 2-aminothiophene afforded their corresponding amides efficiently (1d and 1f, respectively). Aniline was not a productive substrate for this reaction as amide 1h was not able to be made in 20 minutes, however, when reaction time increased to 4 h a modest yield was obtained. The synthesis of a dipeptide (1i) was explored to demonstrate that this method can be used for peptide coupling. This reaction is a proof of concept and further optimization will be needed to establish a genuine protocol for peptide coupling; however, these reactions show that the stereochemical fidelity at the crucial position α to the amide is maintained, as only a single diastereomer is observed in the 1H NMR spectra of the product (see ESI†).
Secondary amines were also coupled to phenylacetic acid (2a–2e) in moderate to good yields, though longer reaction times were needed. Sterically hindered acyclic amines in 2a and 2b resulted in modest yields, while cyclic amines worked well using these reaction conditions (2c–2e).
Finally, the method was expanded to aromatic carboxylic acids (3a–3g). Benzoic acid reacted with benzylamine to form 3a in good yield. Electron-rich p-anisic acid gave 3b in a moderate yield of 51%. Electron-poor 4-nitrobenzoic acid reacted with benzylamine to form amide 3f in 86% yield. Heterocyclic acids of pyridine (3c), furan (3e and 3g), indole (3h), and imidazole (3i) were compatible with this method, and an iodophenyl-derived amide (3d) was also synthesized in moderate yield.
To demonstrate the versatility of this method, we explored complimentary related approaches to several of the synthesized amides both (1) in the microwave and (2) at room temperature. Microwave reactions to synthesize amides 1a, 1g, 1h, 1i, 2a, 2c, 3b, 3d, 3f, and 3g were explored to improve yield (ESI, Table S9†). Good yields were obtained in synthesizing 1a, 1g, 2a, 2c, 3b, and 3d in the microwave and the yields compare favourably to the thermal reactions. Microwave reaction of phenylacetic acid with aniline afforded the corresponding amide 1h in low yield, however, it did enhance the reaction compared to the thermal reaction for 20 minutes. In the synthesis of 1i, 3f, and 3g, though products were formed in relatively reasonable yields in the microwave, it did not enhance the reaction. As room temperature reactions provide mild conditions, we were interested in seeing if the developed method can be used to synthesize amides at room temperature overnight (ESI, Table S16†). These reactions gave low-yielding results for 1h, 2a and 3g; however, 1a and 2c were synthesized in excellent yields and 3d resulted in a 33% yield, which is comparable to the microwave reaction that yielded 34% of product. Moderate yields were also obtained in the synthesis of 1g, 3b, and 3f.
The tertiary amine base is a key aspect to this method and has shown to improve the efficiency of this reaction. To further demonstrate the role of the tertiary amine base in improving the reaction, we chose four amides (1a, 2d, 2e, and 3a) from Scheme 1 and ran the reaction without base under complimentary reaction conditions. Amides 1a and 3a showed an improved yield when NMPi was added. However, when more nucleophilic amines like piperidine and morpholine were used (to make 2d and 2e, respectively), the amine base did not improve yields.
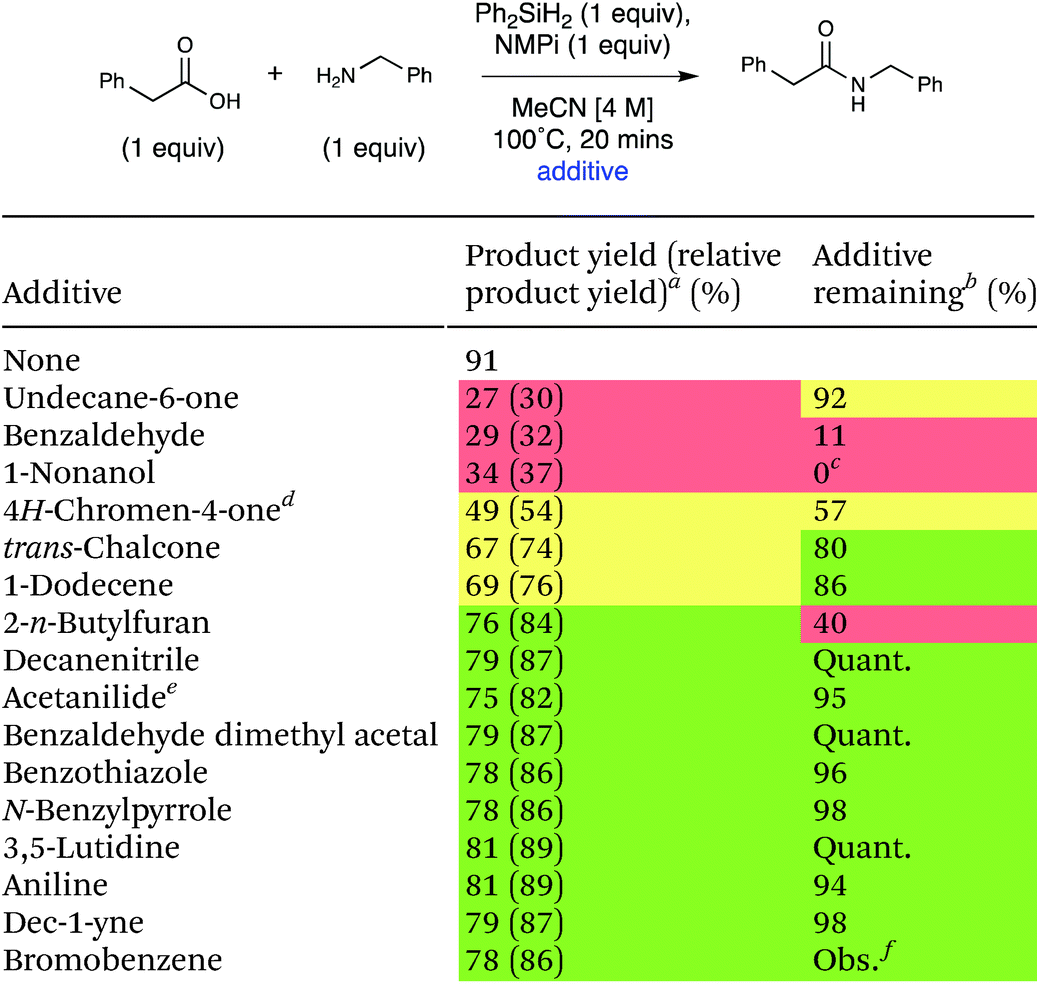
Many important amides exist in molecules that are large and complex with diverse functionality. A general coupling reaction to synthesize amide-containing molecules requires a method that is unreactive toward a wide range of functional groups. To examine the functional group tolerance of this method we conducted a Glorius robustness study.27 16 additives that each contain a different functional group were added into the reaction each at 1 equivalent to probe any limitations with respect to functional group compatibility of the developed method.27 The experimental results are presented in Table 5, and a table of all the experimental results and spectra can be seen in the ESI (Table S19).† The reaction was robust in the presence of most of the additives, demonstrating good general applicability of this method. Lower yields of the amide product were observed upon the addition of 4H-chromen-4-one, undecane-6-one, 1-nonanol, and benzaldehyde, and the amount of additive remaining varied significantly. The crude reaction mixture with 4H-chromen-4-one was subjected to column chromatography and 1H NMR analysis of the separated fractions indicated that conjugate addition of benzylamine to 4H-chromen-4-one occurred.28 The 1H NMR of the reaction mixture of undecane-6-one shows some evidence of imine formation. Finally, the 1H NMR spectra of the reaction with benzaldehyde showed imine formation had occurred.29 In each of these cases, the deleterious reactivity was due to the amine coupling partner, which is a general feature of amidation reactions and not specific to this method. The one additive that seemed to be fully incompatible with the actual method was 1-nonanol; the GC-MS of this crude reaction mixture revealed silyl ether formation due to the high oxophilicity of silicon.
| a Yield of product as 1H NMR yield using EtOAc as an internal standard, unless otherwise stated. Relative 1H NMR yield to 91% of amide product with no additive shown in parentheses (green: 80–100%, yellow: 50–80%, red: <50%), EtOAc internal standard, unless otherwise stated. b 1H NMR yield using EtOAc as an internal standard, unless otherwise stated. c Yield determined by GC-MS. d Average of 2 runs. e Ph-NO2 used as an internal standard. f There is no evidence to suggest that bromobenzene was consumed; however, we were not able to quantify the amount of bromobenzene. It was present in GC-MS in significant amount, and no new products were observed. |
|---|
|
Large-scale production of amides is vital due to their high demand in the pharmaceutical industry.14 To demonstrate the scalability of this diphenylsilane-mediated amide coupling, a gram-scale synthesis of a target amide was performed. Using 1.36 g of phenylacetic acid and 1.09 mL of benzylamine, 1.63 grams of product 1a were made in 20 minutes (Scheme 2). This resulted in a 72% isolated yield, which compares favourably to the small-scale (1 mmol) optimization reaction to synthesize 1a (82% isolated yield).
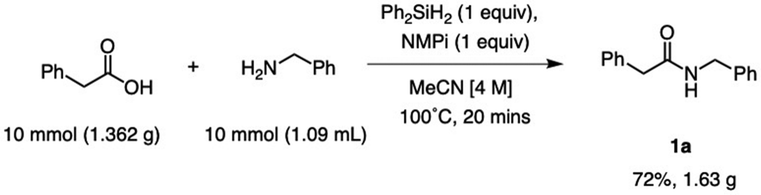 | ||
| Scheme 2 Gram-scale synthesis of 1a. | ||
Conclusions
We have developed a simple and fast organosilane-mediated amide coupling reaction of unactivated carboxylic acids and amines. Using diphenylsilane in the presence of NMPi in a setup that does not require rigorous exclusion of air or water, amidation is enabled and a diverse range of amides can be synthesized in good to high yields. The reaction is highly practical and advantageous over previously reported diphenylsilane-mediated coupling as it (1) is optimal without energy- and time-consuming exclusion of air or water, (2) generates little waste, and (3) can either be fast or run at room temperature. In the present work, the method is readily scalable up to at least 10 mmol and it is robust towards various common organic functional groups. Due to its simplicity in execution, reliance on readily available reagents, and the other aforementioned positive attributes, this method will be of immediate utility to those seeking to make small-molecule amides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Ryerson University and the Natural Sciences and Engineering Research Council of Canada (NSERC) for supporting this work.Notes and references
- (a) N. A. McGrath, M. Brichacek and J. T. Njardarson, A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives, J. Chem. Educ., 2010, 87, 1348 CrossRef CAS; (b) J. Njardarson, et al., Top 200 Pharmaceuticals by Retail Sales in 2019, University of Arizona, 2019 Search PubMed.
- (a) E. Valeur and M. Bradley, Amide bond formation: beyond the myth of coupling reagents, Chem. Soc. Rev., 2009, 38(2), 606–631 RSC; (b) A. El-Faham and F. Albericio, Peptide coupling reagents, more than a letter soup, Chem. Rev., 2011, 111(11), 6557–6602 CrossRef CAS; (c) R. M. De Figueiredo, J. S. Suppo and J. M. Campagne, Nonclassical Routes for Amide Bond Formation, Chem. Rev., 2016, 116(19), 12029–12122 CrossRef CAS.
- K. Marchildon, Polyamides - Still strong after seventy years, Macromol. React. Eng., 2011, 5, 22–54 CrossRef CAS.
- V. R. Pattabiraman and J. W. Bode, Rethinking amide bond synthesis, Nature, 2011, 480(7378), 471–479 CrossRef CAS.
- A. S. Santos, A. M. S. Silva and M. M. B. Marques, Sustainable Amidation Reactions – Recent Advances, Eur. J. Org. Chem., 2020, 2020(17), 2501–2516 CrossRef CAS.
- E. Massolo, M. Pirola and M. Benaglia, Amide bond formation strategies: latest advances on a dateless transformation, Eur. J. Org. Chem., 2020, 2020(30), 4641–4651 CrossRef CAS.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach and G. Moine, et al., Key Green Chemistry research areas from a pharmaceutical manufacturers’ perspective revisited, Green Chem., 2018, 20, 5082–5103 RSC.
- H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Catalytic amide formation from non-activated carboxylic acids and amines, Chem. Soc. Rev., 2014, 43(8), 2714–2742 RSC.
- R. M. Al-Zoubi, O. Marion and D. G. Hall, Direct and waste-free amidations and cycloadditions by organocatalytic activation of carboxylic acids at room temperature, Angew. Chem., Int. Ed., 2008, 47(15), 2876–2879 CrossRef CAS.
- K. J. McKnelly, W. Sokol and J. S. Nowick, Anaphylaxis Induced by Peptide Coupling Agents: Lessons Learned from Repeated Exposure to HATU, HBTU, and HCTU, J. Org. Chem., 2020, 85(3), 1764–1768 CrossRef CAS.
- (a) N. Gernigon, R. M. Al-Zoubi and D. G. Hall, Direct Amidation of Carboxylic Acids Catalyzed by ortho-Iodo Arylboronic Acids: Catalyst Optimization, Scope, and Preliminary Mechanistic Study Supporting a Peculiar Halogen Acceleration Effect, J. Org. Chem., 2012, 77, 8386–8400 CrossRef CAS; (b) K. Ishihara, S. Ohara and H. Yamamoto, 3,4,5-Trifluorobenzeneboronic acid as an extremely active amidation catalyst, J. Org. Chem., 1996, 61(13), 4196–4197 CrossRef CAS; (c) D. N. Sawant, D. B. Bagal, S. Ogawa, K. Selvam and S. Saito, Org. Lett., 2018, 20(15), 4397–4400 CrossRef CAS; (d) T. Maki, K. Ishihara and H. Yamamoto, N-alkyl-4-boronopyridinium salts as thermally stable and reusable amide condensation catalysts, Org. Lett., 2005, 7(22), 5043–5046 CrossRef CAS; (e) T. Maki, K. Ishihara and H. Yamamoto, New boron(III)-catalyzed amide and ester condensation reactions, Tetrahedron, 2007, 63(35), 8645–8657 CrossRef CAS; (f) K. Arnold, A. S. Batsanov, B. Davies and A. Whiting, Synthesis, evaluation and application of novel bifunctional N, N-di-isopropylbenzylamineboronic acid catalysts for direct amide formation between carboxylic acids and amines, Green Chem., 2008, 10(1), 124–113 RSC; (g) T. M. El Dine, J. Rouden and J. Blanchet, Borinic acid catalysed peptide synthesis, Chem. Commun., 2015, 51(89), 16084–16087 RSC; (h) T. Mohy El Dine, W. Erb, Y. Berhault, J. Rouden and J. Blanchet, Catalytic chemical amide synthesis at room temperature: One more step toward peptide synthesis, J. Org. Chem., 2015, 80(9), 4532–4544 CrossRef CAS; (i) Y. Lu, K. Wang and K. Ishihara, Design of Boronic Acid–Base Complexes as Reusable Homogeneous Catalysts in Dehydrative Condensations between Carboxylic Acids and Amines, Asian J. Org. Chem., 2017, 6(9), 1191–1194 CrossRef CAS; (j) K. Ishihara and Y. Lu, Boronic acid-DMAPO cooperative catalysis for dehydrative condensation between carboxylic acids and amines, Chem. Sci., 2016, 7(2), 1276–1280 RSC; (k) P. W. Tang, Boric Acid Catalyzed Amide Formation from Carboxylic Acids and Amines: N-benzyl-4-phenylbutyramide, Org. Synth., 2005, 81, 262 CrossRef CAS; (l) R. Latta, G. Springsteen and B. Wang, Development and synthesis of an arylboronic acid-based solid-phase amidation catalyst, Synthesis, 2001,(11), 1611–1613 CrossRef CAS.
- (a) S. Roy, S. Roy and G. W. Gribble, Metal-catalyzed amidation, Tetrahedron, 2012, 68(48), 9867–9923 CrossRef CAS; (b) S. Y. Lu, S. S. Badsara, Y. C. Wu, D. M. Reddy and C. F. Lee, CuCl/TBHP catalyzed synthesis of amides from aldehydes and amines in water, Tetrahedron Lett., 2016, 57(6), 633–636 CrossRef CAS; (c) W. Zhang, H. Deng and H. Li, Ir(III)-Catalyzed site-selective amidation of azoxybenzenes and late-stage transformation, Org. Chem. Front., 2017, 4(11), 2202–2206 RSC; (d) Y. Park, K. T. Park, J. G. Kim and S. Chang, Mechanistic Studies on the Rh(III)-Mediated Amido Transfer Process Leading to Robust C-H Amination with a New Type of Amidating Reagent, J. Am. Chem. Soc., 2015, 137(13), 4534–4542 CrossRef CAS; (e) S. Y. Chow, M. Y. Stevens, L. Åkerbladh, S. Bergman and L. R. Odell, Mild and Low-Pressure fac-Ir(ppy)3-Mediated Radical Aminocarbonylation of Unactivated Alkyl Iodides through Visible-Light Photoredox Catalysis, Chem. – Eur. J., 2016, 22(27), 9155–9161 CrossRef CAS; (f) L. Wu, X. Fang, Q. Liu, R. Jackstell, M. Beller and X. F. Wu, Palladium-catalyzed carbonylative transformation of C(sp3)-X bonds, ACS Catal., 2014, 4(9), 2977–2989 CrossRef CAS; (g) A. E. Hande, V. B. Ramesh and K. R. Prabhu, Rh(III)-Catalyzed: Ortho -C-(sp2)-H amidation of ketones and aldehydes under synergistic ligand-accelerated catalysis, Chem. Commun., 2018, 54(85), 12113–12116 RSC; (h) A. Álvarez-Pérez, M. A. Esteruelas, S. Izquierdo, J. A. Varela and C. Saá, Ruthenium-Catalyzed Oxidative Amidation of Alkynes to Amides, Org. Lett., 2019, 21(13), 5346–5350 CrossRef; (i) P. W. Tan, A. M. Mak, M. B. Sullivan, D. J. Dixon and J. Seayad, Thioamide-Directed Cobalt(III)-Catalyzed Selective Amidation of C(sp3)−H Bonds, Angew. Chem., Int. Ed., 2017, 56(52), 16550–16554 CrossRef CAS; (j) H. Lundberg and H. Adolfsson, Hafnium-catalyzed direct amide formation at room temperature, ACS Catal., 2015, 5(6), 3271–3277 CrossRef CAS.
- M. T. Sabatini, L. T. Boulton, H. F. Sneddon and T. D. Sheppard, A green chemistry perspective on catalytic amide bond formation, Nat. Catal., 2019, 2(1), 10–17 CrossRef CAS.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Large-Scale Applications of Amide Coupling Reagents for the Synthesis of Pharmaceuticals, Org. Process Res. Dev., 2016, 20(2), 140–177 CrossRef CAS.
- (a) W. C. Chou, M. C. Chou, Y. Y. Lu and S. F. Chen, S. HMDS-promoted in situ amidation reactions of carboxylic acids and amines, Tetrahedron Lett., 1999, 40, 3419–3422 CrossRef CAS; (b) S. H. Van Leeuwen, P. J. L. M. Quaedflieg, Q. B. Broxterman and R. M. J. Liskamp, Synthesis of amides from unprotected amino acids by a simultaneous protection-activation strategy using dichlorodialkyl silanes, Tetrahedron Lett., 2002, 43(50), 9203–9207 CrossRef CAS; (c) T. Tozawa, Y. Yamane and T. Mukaiyama, A convenient method for preparations of 1-acylimidazoles and carboxamides by using novel imidazolylsilane derivatives, Chem. Lett., 2005, 34(5), 734–735 CrossRef CAS; (d) T. Tozawa, Y. Yamane and T. Mukaiyama, An effective method for the synthesis of carboxamides by using tetrakis(pyridine-2-yloxy)silane as a mild coupling reagent, Chem. Lett., 2005, 34(10), 1334–1335 CrossRef CAS; (e) T. Tozawa, Y. Yamane and T. Mukaiyama, An efficient method for the preparation of carboxamides by dehydration condensation using tetrakis(1,1,1,3,3,3-hexafluoro-2-propoxy)silane, Chem. Lett., 2005, 34(12), 1586–1587 CrossRef CAS; (f) T. Tozawa, Y. Yamane and T. Mukaiyama, A convenient method for the synthesis of carboxamides and thioesters by using tetrakis(2-methylimidazol-1-yl)silane, Heterocycles, 2006, 67, 629–641 CrossRef CAS; (g) Z. Ruan, R. M. Lawrence and C. B. Cooper, Phenylsilane as an active amidation reagent for the preparation of carboxamides and peptides, Tetrahedron Lett., 2006, 47(43), 7649–7651 CrossRef CAS; (h) T. H. Chan and L. T. L. Wong, Silicon Tetrachloride as a Coupling Reagent for Amide Formation, J. Org. Chem., 1969, 34(9), 2766–2767 CrossRef CAS; (i) T. H. Chan and L. T. L. Wong, Evaluation of Acyloxysilane as an Acylating Agent for Peptide Synthesis, J. Org. Chem., 1971, 36(6), 850–853 CrossRef CAS.
- E. W. Colvin, Introduction. In Silicon in Organic Synthesis, 1981, pp. 1–3 Search PubMed.
- Y. Nagai, Hydrosilanes as Reducing Agents. A Review, Org. Prep. Proced. Int. New J. Org. Synth., 1989, 21(4), 501–504 CrossRef.
- P. G. M. Wuts and T. W. Greene, in Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, Incorporated, 2014, pp. 533–646 Search PubMed.
- D. C. Braddock, P. D. Lickiss, B. C. Rowley, D. Pugh, T. Purnomo, G. Santhakumar and S. J. Fussell, Tetramethyl Orthosilicate (TMOS) as a Reagent for Direct Amidation of Carboxylic Acids, Org. Lett., 2018, 20(4), 950–953 CrossRef CAS.
- (a) K. G. Andrews, D. M. Summers, L. J. Donnelly and R. M. Denton, Catalytic reductive N-alkylation of amines using carboxylic acids, Chem. Commun., 2016, 52(9), 1855–1858 RSC; (b) E. L. Stoll, T. Tongue, K. G. Andrews, D. Valette, D. J. Hirst and R. M. Denton, A practical catalytic reductive amination of carboxylic acids, Chem. Sci., 2020, 11(35), 9494–9500 RSC.
- K. G. Andrews, R. Faizova and R. M. Denton, A practical and catalyst-free trifluoroethylation reaction of amines using trifluoroacetic acid, Nat. Commun., 2017, 8, 1–6 Search PubMed.
- K. G. Andrews and R. M. Denton, A more critical role for silicon in the catalytic Staudinger amidation: Silanes as non-innocent reductants, Chem. Commun., 2017, 53, 7982–7985 RSC.
- E. Morisset, A. Chardon, J. Rouden and J. Blanchet, Phenysilane and Silicon Tetraacetate: Versatile Promotors for Amide Synthesis, Eur. J. Org. Chem., 2020,(3), 388–392 CrossRef CAS.
- S. J. Aspin, S. Taillemaud, P. Cyr and A. B. Charette, 9-Silafluorenyl Dichlorides as Chemically Ligating Coupling Agents and Their Application in Peptide Synthesis, Angew. Chem., Int. Ed., 2016, 128, 14037–14041 CrossRef.
- M. Sayes and A. B. Charette, Diphenylsilane as a coupling reagent for amide bond formation, Green Chem., 2017, 19, 5060–5064 RSC.
- P. Goswami, J. Park and H. Bae, Water plays a crucial role: Small molecule catalyzed C–C/C–X bond forming reactions using organosilicon reagents under “wet” conditions, Tetrahedron, 2020, 76(25), 131254 CrossRef CAS.
- (a) K. D. Collins and F. Glorius, A robustness screen for the rapid assessment of chemical reactions, Nat. Chem., 2013, 5, 597–601 CrossRef CAS; (b) T. Gensch, M. Teders and F. Glorius, Approach to Comparing the Functional Group Tolerance of Reactions, J. Org. Chem., 2017, 82(17), 9154–9159 CrossRef CAS.
- A. G. Neo, J. Díaz, S. Marcaccini and C. F. Marcos, Conjugate addition of isocyanides to chromone 3-carboxylic acid: An efficient one-pot synthesis of chroman-4-one 2-carboxamides, Org. Biomol. Chem., 2012, 10(17), 3406–3416 RSC.
- L. G. Marinescu, C. M. Pedersen and M. Bols, Safe radical azidonation using polystyrene supported diazidoiodate(I), Tetrahedron, 2005, 61(1), 123–127 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc02833a |
| This journal is © The Royal Society of Chemistry 2021 |