
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-dimensional LC-MS: the next generation characterization of antibody-based therapeutics by unified online bottom-up, middle-up and intact approaches
Julien
Camperi†
 *a,
Alexandre
Goyon†
b,
Davy
Guillarme
*cd,
Kelly
Zhang
b and
Cinzia
Stella
a
*a,
Alexandre
Goyon†
b,
Davy
Guillarme
*cd,
Kelly
Zhang
b and
Cinzia
Stella
a
aDepartment of Protein Analytical Chemistry, Genentech Inc., 1 DNA Way, South San Francisco, CA 94080, USA
bDepartment of Small Molecule Analytical Chemistry, Genentech Inc., 1 DNA Way, South San Francisco, CA 94080, USA
cSchool of Pharmaceutical Sciences, University of Geneva, CMU - Rue Michel-Servet, 1, 1206 Geneva, Switzerland. E-mail: davy.guillarme@unige.ch
dInstitute of Pharmaceutical Sciences of Western Switzerland (ISPSO), University of Geneva, CMU-Rue Michel Servet 1, 1211 Geneva 4, Switzerland
First published on 15th December 2020
Abstract
Accelerated development of new therapeutics in an increasingly competitive landscape requires the use of high throughput analytical platforms. In addition, the complexity of novel biotherapeutic formats (e.g. fusion proteins, protein-polymer conjugates, co-formulations, etc.) reinforces the need to improve the selectivity and resolution of conventional one-dimensional (1D) liquid chromatography (LC). Liquid chromatography-mass spectrometry (LC-MS)-based technologies such as native LC-MS for intact mass analysis or peptide mapping (also called bottom-up approach)-based multi-attribute methods (MAM) have already demonstrated their potential to complement the conventional analytical toolbox for monoclonal antibody (mAb) characterization. Two-dimensional liquid-chromatography (2D-LC-MS) methods have emerged in the last ten years as promising approaches to address the increasing analytical challenges faced with novel antibody formats. However, off-line sample preparation procedures are still required for conventional 1D and 2D-LC-MS methods for the in-depth variant characterization at the peptide level. Multi-dimensional LC-MS (mD-LC-MS) combine sample preparation and multi-level (i.e. intact, reduced, middle-up and peptide) analysis within the same chromatographic set-up. This review presents an overview of the benefits and limitations of mD-LC-MS approaches in comparison to conventional chromatographic methods (i.e. 1D-LC-UV methods at intact protein level and 1D-LC-MS methods at peptide level). The current analytical trends in antibody characterization by mD-LC-MS approaches, beyond the 2D-LC-MS workhorse, are also reviewed, and our vision on a more integrated multi-level mD-LC-MS characterization platform is shared.
 Julien Camperi | Dr Julien Camperi received his Ph.D. in Analytical Chemistry from the Paris Sciences et Lettres University, France. He is currently a Postdoctoral scientist in the Department of Analytical Protein Chemistry (PAC) at Genentech. His current research involves the development and implementation of novel analytical methodologies for the characterization of biomacromolecules. |
 Alexandre Goyon | Dr Alexandre Goyon is a Scientist in the Small Molecule Pharmaceutical Science organization of Genentech. He received a Ph.D. degree in Pharmaceutical Analytical Chemistry in 2019 from the University of Geneva, Switzerland. His team supports early & late stage research. |
 Davy Guillarme | Dr Davy Guillarme holds a Ph.D. degree in analytical chemistry from the University of Lyon, France. He is now senior lecturer and research associate at the University of Geneva in Switzerland. He authored more than 270 journal articles related to pharmaceutical analysis. He is an associate editor of Journal of chromatography B and editorial advisory board member of several journals (Analytical chemistry, Journal of Chromatography A, Journal of Separation Science, LC-GC North America…). He was elected as one of the world's most influential analytical scientists by “Analytical Science” magazine in 2013, 2014, 2015, 2017, 2019 and 2020. |
 Kelly Zhang | Dr Kelly Zhang is a Principal Scientist & Director at Genentech (A member of Roche group) in South San Francisco, California. She leads analytical chemistry R&D teams for drug substance and drug formulation, moving novel therapeutic molecules from research to clinical trials through collaboration with cross-disciplinary teams. She is a Permanent Scientific Committee member of the international HPLC Conferences, and the international Pharmaceutical Biomedical Analysis Conferences. She was named one of the top 50 most influential women in analytical science in 2016 and one of the top 100 most influential analytical scientists in 2019 by The Analytical Scientist. |
 Cinzia Stella | Dr Cinzia Stella received her Ph.D. in Pharmaceutical Analytical Chemistry from the University of Geneva (Switzerland), after her Master's Degree in Pharmaceutical Sciences from the University of Pavia (Italy). Following a post-doctoral fellowship at Imperial College London (UK) on Metabolomics funded by Unilever, she held a position at the University of Geneva in the School of Pharmacy, where she was responsible for the development and optimization of protein based pharmaceutical formulations in collaborations with industrial partners. She is currently a Sr Scientist and Functional Leader at Genentech and she is responsible for the development and implementation of analytical platforms to support the pipeline. |
1. Introduction
Therapeutic antibodies have been effectively used for treatment of a wide range of diseases, including oncology and auto-immune conditions.1 The acceleration of biologics license applications (BLAs) approved in the last ten years is fueled by a robust late stage pipeline with a number of antibody candidates that has more than tripled between 2010 and 2019, with 79 antibody candidates evaluated just in 2019.2 Only 40% of antibody-based therapeutics approved in 2019 were monoclonal antibody (mAb) products, indicating the surge of alternative antibody formats. For example, a bispecific antibody was approved in 2017 for the treatment of hemophilia (Helimbra®/emicizumab) and one new nanobody modality was approved for the first time in 2019 (Cablivi®/caplacizumab)1 for the treatment of thrombotic thrombocytopenic purpura (aTTP). The biopharmaceutical landscape is a highly competitive environment and the immunotherapy field in particular, where for example 2975 clinical trials involving only PD-1/PD-L1 mAbs were active in September 2019.3The quest to drug the undruggable biological targets and bring innovative first-in class therapeutics faster to the patients is constantly challenging conventional analytical methods. These analytical methods are routinely used in quality control environments (QC) to support batch release4 and to assess potential critical quality attributes (pCQAs) at various stages of the drug lifecycle, during production and shelf-life. Antibody size and charge variants are usually quantified by size exclusion chromatography (SEC) and ion exchange chromatography (IEC), respectively. Ideally, size variants are separated according to their difference in hydrodynamic radius in SEC,5 while the surface charge distribution of charge variants allows their separation by IEC.6,7 Capillary electrophoresis sodium dodecyl sulfate (CE-SDS)8,9 and imaged capillary isoelectric focusing (icIEF)10,11 techniques provide an orthogonal separation to SEC and IEC, respectively. Oxidations and glycoforms are commonly characterized using hydrophobic interaction chromatography (HIC)12 and hydrophilic interaction liquid chromatography (HILIC),13 respectively.
The rise of MS-based techniques for the characterization of antibody-based therapeutics has driven the development of MS-compatible mobile phases in order to couple native MS with non-denaturing SEC, IEC, and HIC modes.14 2D-LC-MS approaches have been developed to mimic the chromatographic separations achieved by QC methods with non-volatile salts in the first dimension and the use of a second dimension to desalt and remove non-volatile salts before MS analysis. The characterization of antibody variants at the bottom-up level is performed by peptide mapping, which involves multiple manual sample preparation steps, such as (i) chemical reduction of the antibody; (ii) alkylation of the reduced species, (iii) buffer exchange and (iv) an enzymatic digestion. The coupling with high resolution mass spectrometry (HRMS) allows the simultaneous sequence confirmation and determination of multiple post-translational modifications (PTMs) in a single analysis.15
However, conventional peptide mapping-based MAM can be time consuming and the multiple sample preparation steps can affect the reproducibility of the method.16 In order to characterize the peaks separated at the intact level (by IEC or SEC for example) and identify corresponding PTMs, an additional off-line fractionation step is needed. As an example, the characterization of five charge variants separated by IEC using off-line procedures can require up to 52 hours.17 In order to streamline this process, multi-dimensional LC-MS approaches have been developed and implemented to perform online fractionation followed by an in-line peptide mapping to characterize antibody variants separated by IEC or SEC.17–20 The automated characterization of antibody variants by mD-LC-MS can be performed with a much faster turnaround (typically 9 vs. 52 hours17) compared to conventional procedures (manual and/or off-line sample preparation and fraction collection), and has the potential to be used as a more integrated analytical platform for both upstream and downstream applications.
In the first part of this review, the identification of antibody variants at the intact level by native MS and 2D-LC-MS is described. Promising 2D-LC applications for the characterization of antibody higher order structures, antibodies present in cell culture supernatant or process analytical technology (PAT) are provided. In the second part, the characterization of PTMs at the peptide level using MAM peptide mapping, is discussed. In the last part, automated mD-LC-MS approaches allowing the online characterization of mAb variants at multiple levels, i.e. combination of intact and peptide levels, are reviewed.
2. Intact mass analysis
2.1 Native mass spectrometry (nMS)
Electrospray ionization (ESI)-MS analysis performed in native conditions (native MS) is of particular interest for the characterization of recombinant mAbs.21,22 Native MS (nMS) technology minimizes sample preparation and preserves the protein higher-order structure and noncovalent interactions.23,24 For instance, nMS is commonly used for the characterization of cysteine-linked ADCs, which consist of a broad population of noncovalent assemblies.25–29 Some of the typical critical quality attributes (CQAs) such as (i) the average drug-to-antibody ratio (DAR); (ii) the distribution of drug-loaded species; and (iii) the amount of unconjugated mAbs can be determined by nMS.30The use of volatile mobile phases allows the hyphenation of IEC,31–34 SEC,35–37 HIC,38,39 RPLC,40 and affinity chromatography (AC)41–44 techniques to native MS. LC-nMS separates mAb variants prior to the entrance into the MS system, significantly improving sensitivity, dynamic range, and thus the detection of minor variants by limiting competition effects during the MS ionization process. For instance, the direct hyphenation of IEC to MS utilizing low ionic strength eluents on a strong cation exchange (SCX) stationary phase enabled MS data generation from minor mAb variants, and intact mass analysis of various PTMs, such as lysine truncation, glycosylation, and deamidation (mass shift of +1 Da relative to the main peak).33 To perform analysis under native conditions, the authors highlighted the importance of optimizing MS parameters since the ESI process is performed with an aqueous mobile phase, and recommended a low MS resolution to capture the protein signal with the highest sensitivity. However, adduct species related to the presence of salt required by the IEC chromatography are not resolved with a low resolution, which results in broader and asymmetrical spectral peaks and decreased mass accuracy following deconvolution, further limiting the applications to complex mixture samples.
Direct hyphenation of SEC to native MS has been facilitated by the development of SEC columns with improved chemical inertness14 and adopted for the characterization of mAb size variants by simply replacing non-volatile, high ionic strength buffers (e.g., phosphate salts) with volatile (e.g., ammonium acetate) mobile phase conditions.35–37 For instance, Yan et al. reported a mixed-mode SEC-nMS method using an ammonium acetate and ammonium bicarbonate mixture for highly sensitive detection and quantitation of homodimer impurities for four BsAbs with limits of detection (LOD) from 0.1 to 0.01% relative abundance.35 However, the use of volatile salt mobile phases at relatively low concentrations resulting in poor peak resolution of the different variants. In a comparison study performed on 30 therapeutic proteins including 24 mAbs, 2 ADCs, and 4 Fc-fusion proteins, it was demonstrated that only acidic therapeutic proteins (pI < 7) could be successfully analyzed with a MS-compatible mobile phase containing 100 mM ammonium acetate for SEC.36 More recently, Ventouri et al., reported the potential risk that the separation conditions used for SEC-nMS can impact the protein structure.45 The authors have demonstrated that ammonium acetate (kosmotropic) at ionic strengths above 0.1 M effectively eliminated undesired secondary interactions with the stationary phase and did not induce denaturation, whereas ammonium formate and bicarbonate (both chaotropic) caused structural changes. These findings highlight the limited choice of volatile buffers that can be used for SEC-nMS analysis, as well as the challenge of having to operate at high salt concentrations (i.e. above 0.1 M), whereas MS manufacturers typically recommend the use of a 10–50 mM concentration range in order to reduce ion suppression and prevent the MS source contamination.
The introduction of new hydrophobic resins enabled HIC to be coupled to MS using ammonium acetate in combination with one or more organic modifiers in the mobile phase, for the characterization of both mAbs46 and ADCs.40 As an example, Wei et al. reported the development of a HIC-nMS method for the determination of minor mAb variants including oxidation, glycation, and free thiol variants by utilizing 150 mM ammonium acetate on a polypropyl UHPLC wide-pore HIC column, which both ensure retention and adequate MS sensitivity for the characterization of low-abundance peaks.38 In another recent study, Chen et al. described the development of a native RPLC-nMS method using a 50 mM ammonium acetate and water/isopropanol mobile phase for the separation of intact ADCs.40 The stationary phase used in this study provides weaker drug–surface interactions compared to the non-covalent interactions holding the ADC conjugates together. The hydrophobicity of the bonded phase is varied, and the least hydrophobic bonded phase in the series, poly(methyl methacrylate), resolved the intact species of two ADCs. However, the dissociation of noncovalent ADC complexes (some with DAR-6 and all with DAR-8) is substantial and extensive under these conditions.14
Affinity chromatography hyphenated to native MS (AC-nMS) has been also implemented for the characterization of therapeutic mAbs.41,43,44 Gahoual et al., reported the hyphenation of a FcRn stationary phase with nMS to investigate methionine oxidation41 (Fig. 1). This FcRn affinity LC-nMS method proved to be suitable to identify and quantify PTMs and their impact on the binding to the FcRn receptor. In another study, the hyphenation of FcγRIIIa AC columns to nMS enabled the assessment of the major Fc glycoforms at the intact level, which is highly relevant to characterize antibody-dependent cellular cytotoxicity (ADCC).43,44
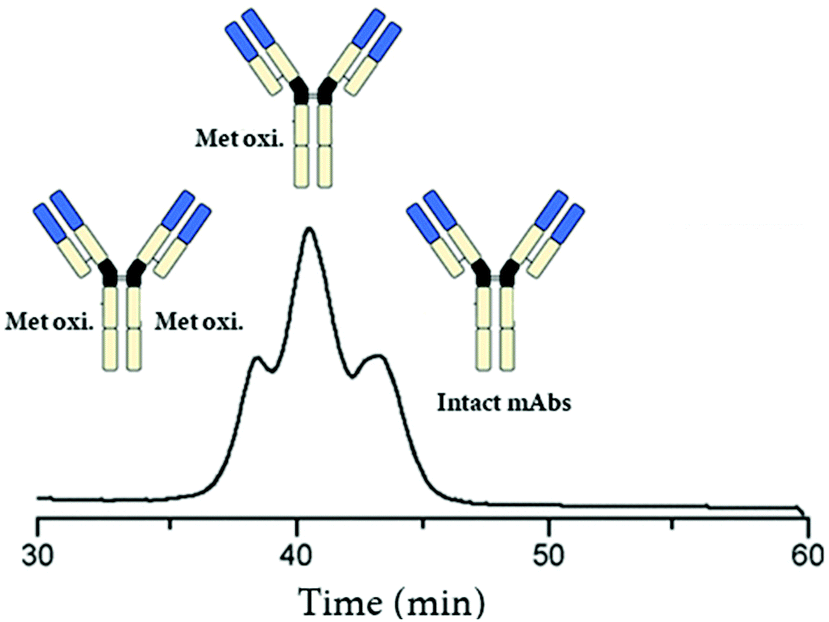 | ||
| Fig. 1 Total ion chromatogram (TIC) recorded during analysis of oxidized mAb1 by native FcRn AC-nMS. Reproduced from R. Gahoual, A.-K. Heidenreich, G. W. Somsen, P. Bulau, D. Reusch, M. Wuhrer, et al., Detailed Characterization of Monoclonal Antibody Receptor Interaction Using Affinity Liquid Chromatography Hyphenated to Native Mass Spectrometry, Anal. Chem., 2017, 89, 105404–5412. With permission from American Chemical Society, copyright 2017. | ||
Overall, the hyphenation of conventional LC methods with native MS for intact mass analysis has been implemented over the last few years to complement the existing analytical toolbox for mAb characterization. However, some challenges still need to be overcome, such as avoiding the formation of adducts, enhancing sensitivity, and the optimization of the chromatographic separation for a broader implementation of this technology in the biopharmaceutical field.
2.2. Two-dimensional liquid chromatography/mass spectrometry (2D-LC-MS)
The value of 2D-LC in the biopharmaceutical industry became obvious in the last ten years and its implementation has been facilitated by the efforts of various research groups.47–51 Schoenmakers, Stoll, and Pirok have listed 160 2D-LC methods between 2016 and 2018 time frame for example.47 According to the FDA, there are 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 potential variants for one single mAb product.52 The greater complexity of large molecules in comparison to small ones has accelerated the development of 2D-LC methods to overcome the limits of (i) peak capacity with 1D-LC method; and (ii) nMS with regards to solvent compatibility. There are several beneficial aspects when analyzing large molecules by 2D-LC in comparison to small ones. It has been reported that resolution is independent from column length for RPLC gradient elution of proteins.53,54 Therefore, the gradient slope has a greater contribution than column length on the chromatographic separation, allowing the use of short columns and analyte peak focusing on the 2D column inlet.54 Second, the under sampling issue, which is well known with small molecules,48 is less critical with large ones due to broader peaks caused by slower mass transport due to their larger size.55 Overall, 2D-LC is an elegant approach to improve the chromatographic peak capacity within a reasonable analysis time. Pirok et al. described a variety of orthogonal chromatographic modes that can be coupled, and weighted associated challenges in ensuring a solvent compatibility between the two dimensions.56
000000 potential variants for one single mAb product.52 The greater complexity of large molecules in comparison to small ones has accelerated the development of 2D-LC methods to overcome the limits of (i) peak capacity with 1D-LC method; and (ii) nMS with regards to solvent compatibility. There are several beneficial aspects when analyzing large molecules by 2D-LC in comparison to small ones. It has been reported that resolution is independent from column length for RPLC gradient elution of proteins.53,54 Therefore, the gradient slope has a greater contribution than column length on the chromatographic separation, allowing the use of short columns and analyte peak focusing on the 2D column inlet.54 Second, the under sampling issue, which is well known with small molecules,48 is less critical with large ones due to broader peaks caused by slower mass transport due to their larger size.55 Overall, 2D-LC is an elegant approach to improve the chromatographic peak capacity within a reasonable analysis time. Pirok et al. described a variety of orthogonal chromatographic modes that can be coupled, and weighted associated challenges in ensuring a solvent compatibility between the two dimensions.56
Multiple modulation strategies have been described in order to ensure solvent compatibility between chromatographic dimensions (typically RPLC-HILIC), including the popular active solvent modulation (ASM) introduced by Stoll,57 or the stationary-phase-assisted modulation (SPAM).58 While the compatibility of aqueous SEC to RPLC is ideal, the use of HILIC in the second dimension generally requires the use of active modulation strategies.56 Several 2D-LC modes can be selected depending on the complexity of the sample, such as the heart-cutting (LC-LC), multiple-heart cutting (LC-LC), selective comprehensive (sLC × LC) and comprehensive (LC × LC) modes.48 The heart cutting and multiple heart cutting modes are simple 2D-LC strategies allowing the fractionation of a limited number of peaks in the first dimension. The multiple-heart cutting core is largely used in the biopharmaceutical industry at the protein intact level as it allows the simultaneous characterization of multiple impurities. The sLC × LC and LC × LC separation modes are generally preferred for complex mixtures because they provide a more complete picture of the sample. The sLC × LC allows the collection of multiple fractions across one chromatographic peak, while the full 1D separation will be fractionated in the LC × LC mode, which allows the fractionation and analysis of untargeted impurities. However, with the comprehensive 2D-LC mode, the 2D separation must be fast (less than 1 min) to allow collecting enough fractions in the first dimension, which may affect the 2D separation performance. In addition, the data treatment can become tedious and time consuming.
Non-denaturing LC techniques are often incompatible with MS due to the non-volatile salts (e.g. phosphate for SEC) used with these LC separations. Numerous 2D-LC applications for biopharmaceutical proteins have been described, where RPLC was used for desalting of samples coming from the first dimension separations that involve the use of non-volatile salts or high volatile salt concentrations.59–62 In particular, SEC-RPLC and IEX-RPLC methods have been published for the characterization of size and charge variants of antibody at intact level using denaturing conditions in the second dimension.63,64 In addition, online on-column reduction strategies have been developed to generate antibody fragments in order to improve the mass accuracy when compared to intact mass analysis.64,65 Beside the use of 2D RPLC denaturing conditions, the use of 2D SEC columns allowed an online desalting using ammonium acetate under non-denaturing conditions, which are required for the analysis of samples with weak inter-molecular interactions (non-covalent aggregates)60 or inter-molecular interactions (cysteine-linked ADCs).59,65
Beyond the improvement in the peak capacity, 2D-LC has also gained a major interest for PAT, enabling the integration of this analytical technology in production and purification environments. Since recombinant mAbs are mainly produced in mammalian host cell lines,66 the presence of process-related impurities such as host cell proteins (HCPs) in the harvested cell culture fluid (HCCF) may interfere with the analysis of mAb PTMs.67,68 Therefore, the targeted mAb must be extracted from cell culture media prior to analysis by LC. Affinity chromatography is the most selective technique used in the biopharmaceutical field as it enables the separation of a protein based on a reversible interaction between the protein and a specific ligand.67,69 Due to the high-affinity of Protein-A (ProA) for the Fc region of IgG-type antibodies, the affinity chromatography using Protein-A (ProA-AC) is conventionally used as an effective capture step in purification processes.69,70 However, the isolation of antibody in cell culture by off-line procedures, prior to analytical testing, presents a considerable workload. The use of 2D-LC to fractionate and isolate the target antibody from process impurities such as HCPs before their in-line analysis represent a promising technology to streamline the purification and characterization process in a time efficient way.71–73 For instance, Dunn et al., have recently reported the development of a 2D-LC ProA–SEC method to measure titer and aggregation of a target mAb from HCCF in under 5 min.71 Similarly, a fully automated 2D-HPLC method was developed for characterizing protein aggregation of crude in-process bioreactor samples, by combining ProA purification and SEC separation, which has the potential to be employed at-line within a bioprocessing system. The authors utilizes an in-line fraction collection device that can collect up to twelve fractions from a single sample.73 Alternatively, Sandra et al. described several 2D-LC approaches, combining ProA in the first dimension with either SEC-UV, IEC-UV or RPLC-MS in a heart-cutting mode in the second dimension.72 These strategies enable performing clone selection based on titer and structural aspects such as aggregation and fragmentation (SEC), charge variants (IEX). In addition, molecular weight, amino acid sequence, and glycosylation can be determined by RPLC-MS. Fig. 2 shows an example of the use of heart-cutting 2D-LC (Protein A-SEC) method for the analysis of size product variants for (a and b) Herceptin (trastuzumab) and (c and d) trastuzumab in the supernatant derived from different CHO clones. The 2D-LC method allowed the quick identification of high producing clones with a low tendency to produce aggregate.
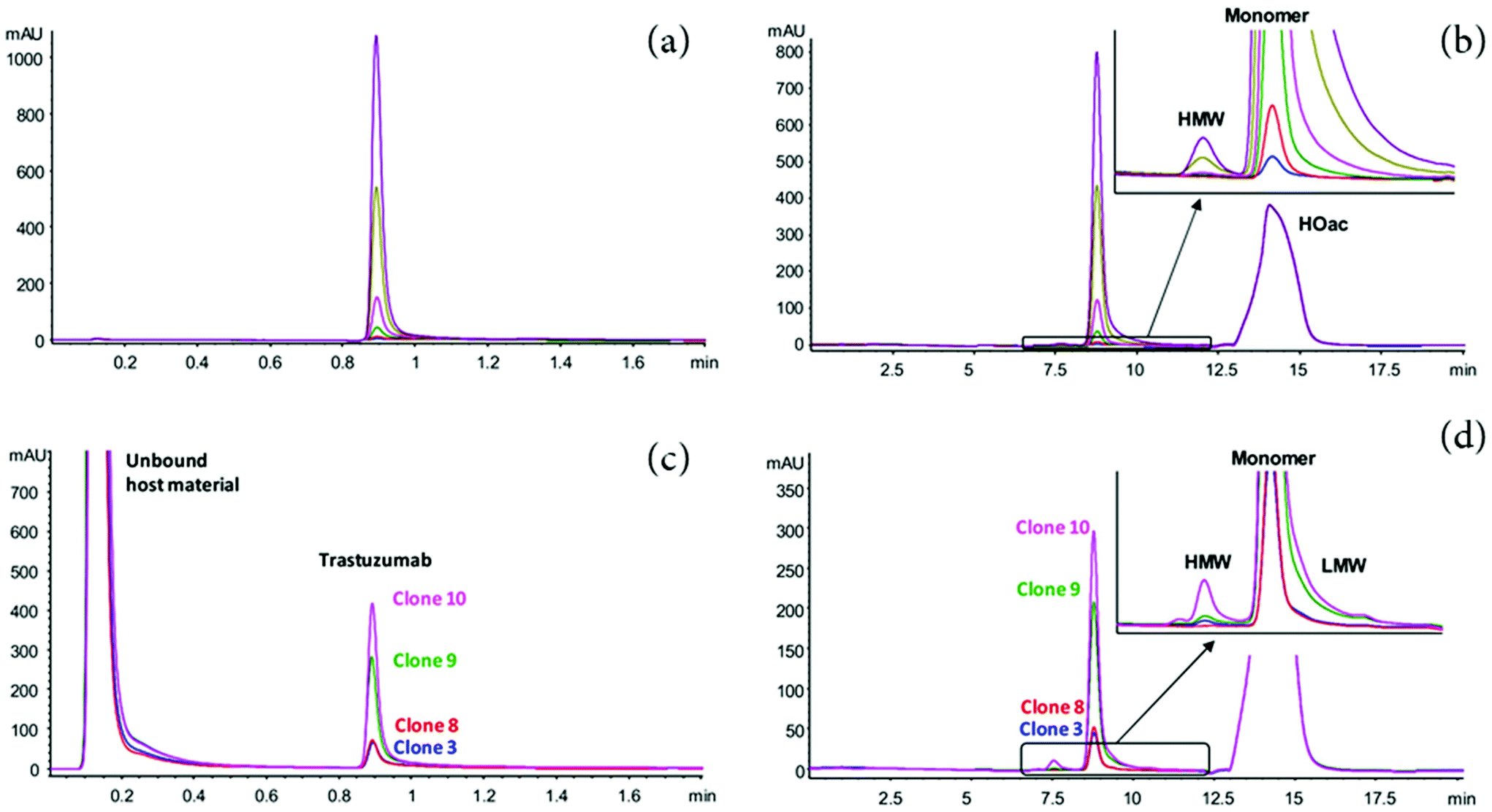 | ||
| Fig. 2 2D-LC (Protein A-SEC) analysis of a dilution series of trastuzumab (a and b) and supernatant collected from CHO clones producing trastuzumab (c and d). (a) 1D Protein A chromatograms at UV 280 nm of dilution series (0.02*, 0.05, 0.1, 0.2, 0.5 and 1 mg mL−1). (b) 2D SEC chromatograms at UV 214 nm of dilution series of trastuzumab and zoomed view showing the detection of HMW variants. (c) 1D Protein A chromatograms of the supernatant collected from four CHO clones producing trastuzumab. (d) 2D SEC chromatograms of fractionated trastuzumab and zoomed view showing the detection of HMW and LMW variants. Reproduced from K. Sandra, M. Steenbeke, I. Vandenheede, G. Vanhoenacker, P. Sandra, The versatility of heart-cutting and comprehensive two-dimensional liquid chromatography in monoclonal antibody clone selection, J. Chromatogr. A., 2017, 1523, 283–292. With permission from Elsevier, copyright 2017. | ||
The success of 2D-LC in the biopharmaceutical industry was first fueled by its inherent improvement of separation performance and more recently boosted by its possibility to be integrated into production and purification environment, allowing the automation of analytical characterization. However, 2D-LC approaches can only provide complementary information at the intact and sometimes reduced levels of mAbs, but more detailed information at the peptide level is often needed at the very early stages of development.
Therefore, the automation of the off-line sample preparation steps within the same chromatographic system is highly desirable. Despite the success of 2D-LC and bio-compatible 1D-LC systems in the biopharmaceutical industry, there is currently no commercial fully biocompatible 2D-LC system. When running SEC methods in the first dimension of 2D-LC setups with mobile phases containing a high amount of chloride anions, regular stainless steel systems will be corroded over time and release metals in the sample flow path. In the meantime, the release of iron has been shown to induce mAb adsorption on CEX columns.74 In addition, non-specific interactions between the stainless steel parts of LC systems and phosphate-containing buffers are well-known, but may occur with proteins too.75 The lack of fully-biocompatible 2D-LC system might be explained by the limited mechanical stability of the truly biocompatible PEEK material, and only the partial removal of non-specific interactions with titanium or MP35N materials (iron free but not metal free). However, PEEK-lined titanium capillaries have been recently commercialized. These materials present the advantage of tolerating high pressure and being truly biocompatible. The improvement of chromatography data systems (CDS) software will further push 2D-LC to the next level with regards to its adoption beyond 2D-LC experts as discussed elsewhere.51,76 For example, some CDS software now allows the control of both mD-LC setups and HRMS instrument using a single software, preventing possible mismatch between the LC and MS sequences.
3. Bottom-up analysis
In 2009, the FDA recommended a quality-by-design (QbD) approach for manufacturing therapeutics.77 The QbD guidelines require direct monitoring of pCQAs at the peptide level, thus ensuring the desired product quality is achieved at the end of the production process.77,78 This involves a better understanding early-on of both the product and process, as well as the deployment of analytical strategies to monitor and maintain high quality throughout the lifecycle of biotherapeutic products. As a result, despite the challenges posed by manual off-line steps and data analysis, LC-MS/MS peptide mapping techniques for quality testing into regulated (GxP) laboratories have generated significant interest in recent years.69,793.1 Conventional multi-attribute methods (MAM)
The LC-MS/MS peptide mapping-based approach is a multi-attribute method (MAM) as it can measure simultaneously multiple protein modifications in a single analysis. This approach was recently introduced for quality testing during mAb development.15,80,81Fig. 3 presents the standard peptide mapping-based MAM workflow for therapeutic mAb analysis. Briefly, mAb is digested using a protease, generally using trypsin, followed by an in-depth characterization of obtained peptides using MS/MS experiments. Afterward, wild-type peptides as well as site-specific modifications including PTMs, sequence variants, clippings, etc. are identified from protein database search. Product quality attributes (PQAs) of interest are then selected, acceptance criteria are set for each attribute, and an MS1-based extracted ion chromatogram (EIC), together with the automated processing method is created. Relative abundance values for each PQAs are calculated based on the ratio of modified to total peptide abundance.69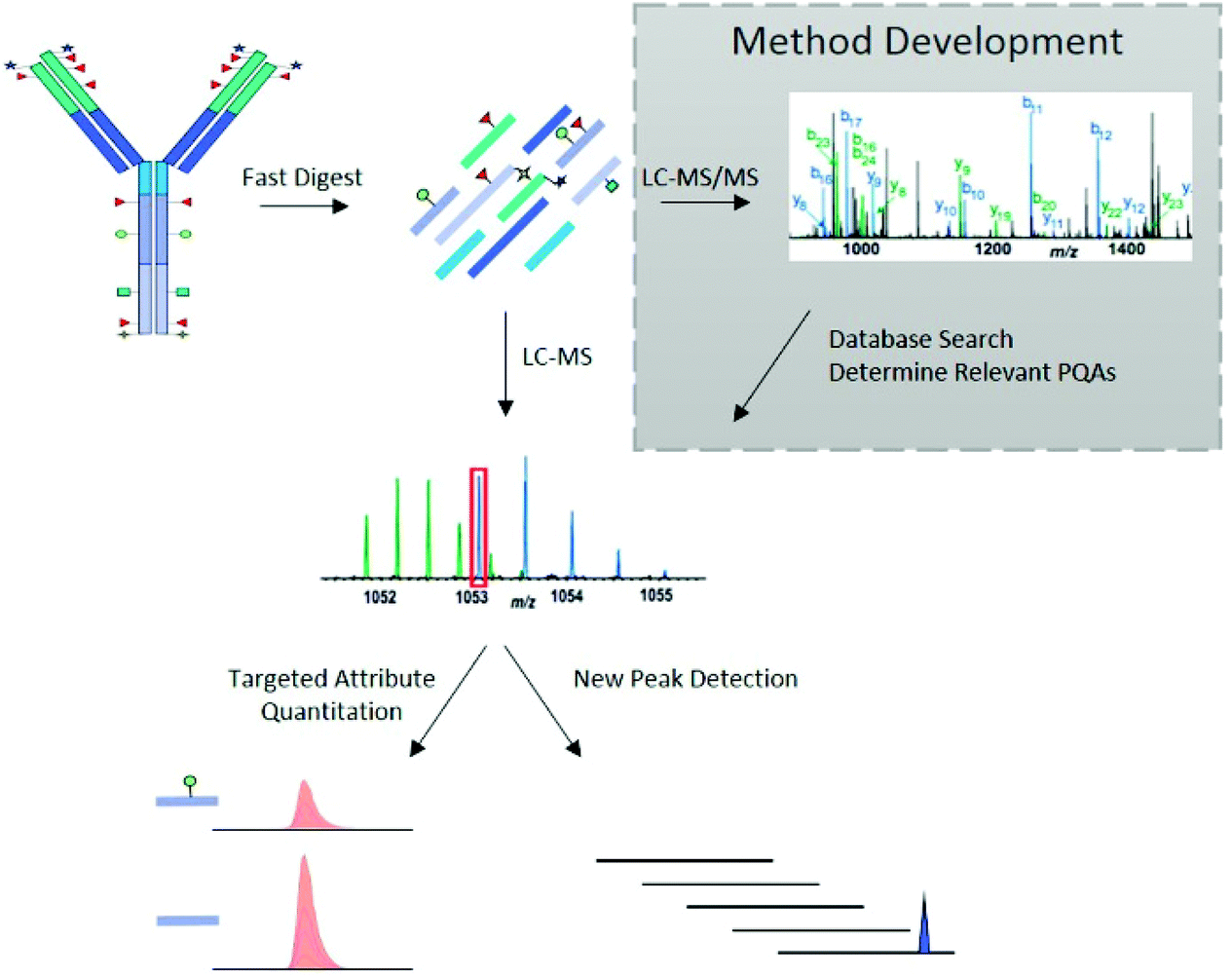 | ||
| Fig. 3 Standard peptide mapping-based MAM workflow for therapeutic mAb analysis. After digestion of mAb, the obtained peptides are analyzed by LC-MS/MS, followed by a database searching, to characterize the mAb molecules and determine relevant PQAs for further testing. Reproduced from S. Rogstad, H. Yan, X. Wang, D. Powers, K. Brorson, B. Damdinsuren, et al., Multi-Attribute Method for Quality Control of Therapeutic Proteins, Anal. Chem., 2019, 91, 2214170–14177 with permission from American Chemical Society, copyright 2019. | ||
In addition to the targeted attribute quantification, new peak detection (NPD), also referred to as Non-Targeted MS Processing (NTMS), is a data processing commonly used in MAM approach, allowing to compare two LC-HRMS data files.78 From this, it is possible to determine any differences between a test sample and that of a reference standard, which may indicate unexpected changes in product quality. NPD can also detect both the presence of new peaks and the loss of existing peaks.
Compared to historical methods (Table 1), which typically assess the intact molecule or its subunits, this LC-MS/MS peptide mapping-based approach provides information on site-specific CQAs (e.g., charged modifications) and a more detailed understanding of the product.
| Attribute | Historical technique | Peptide mapping-based MAM |
|---|---|---|
| • Charge variant | • cIEF and IEX | • Specific post-translational modifications (asparagine deamidation, Lys-glycation, proline amidation, isomerization) and N- and C-terminal variants |
| • Size variant | • SEC (aggregates) | • Specific clipped species |
| • CE-SDS (fragments) | ||
| • Glycan | • HILIC (released N-glycans profiling) | • Both N- and O-glycopeptides |
| • Oxidation | • HIC | • Specific methionine, tryptophan and cysteine oxidation |
Because of these benefits, peptide mapping-based MAM approach has recently been suggested as a replacement of conventional batch release methods, which typically are designed to test one attribute at a time (e.g. IEX for charge variant analysis, reducing gel electrophoresis (rCE-SDS) for clipped variant analysis, or HILIC for glycan profiling). In this context, multiple comparisons of peptide mapping-based MAM workflows with conventional methods were performed.15,80,82,83 For instance, Rogers et al. demonstrated the performance of a MAM based method using a Orbitrap MS for automated detection and relative quantification of PTMs including oxidation, deamidation, glycosylation, and clipped variant, by showing consistent results with the conventional QC methods.15 In another study, Wang et al. reported the application of a MAM method for the assessment of multiple glycosylation sites of an IgG1 and compared with the traditional HILIC analysis of released and labeled glycans.83 The quantification of the glycopeptides using the MAM approach has shown a good correlation with the conventional method with similar accuracy and precision. Overall, these studies demonstrated the broad interest in MAM across the MS and biopharmaceutical fields, but still, some challenges need to be further addressed for implementing MAM for QC testing of therapeutic proteins.
One main challenge of MAM is to correlate peptide level with protein level information. For example, the quantification of low abundant PTMs such as glycated and oxidized amino acids at the protein intact level might not be confirmed at the peptide level due to the lower sensitivity of the MAM approach.84 In addition, the analysis at the peptide level usually requires off-line and time-consuming sample preparation procedures, which may generate artifacts such as oxidation, isomerization, and deamidation.85–87 To limit possible artificial degradation, Wang et al. reported the development of an ultrafast peptide mapping-based approach (uLC-MS/MS) with a 5 min tryptic digestion.88 Although these results are promising, some improvements still need to be done such as the reduction of missed cleavages.
3.2 Correlating peptide with protein level information using mD-LC-MS methods
Automated peptide mapping approaches have been investigated via two main strategies with (i) the use of robotic liquid handling systems89–91 and; (ii) the use of HPLC systems.92–95 The use of robotic liquid handling systems allows the simultaneous preparation of samples in the 96-well plate format using in-solution reduction and digestion,90,91 while the use of HPLC systems allow generally the preparation of one sample at a time when the digestion is performed using an immobilized trypsin cartridge.93–96 Robotic liquid handling systems are typically used for the sample preparation of a large number of samples, whereas HPLC-based methods present a valuable approach with a smaller number of samples as the digestion time is significantly reduced when it is performed on-cartridge (minutes) compared to in solution (hours). HPLC-based methods allow the automated online fractionation of antibody variants by conventional LC methods, followed by sample preparation and analysis.92 Various automated approaches, using either on-cartridge95 or liquid handling platforms,91 have been developed. These automated approaches aim at improving the throughput of conventional off-line peptide mapping procedure. However, the characterization of antibody variants separated at the intact level by SEC, CEX, or HIC or the isolation of a targeted antibody from complex matrices such as cell cultures still require a time-consuming manual off-line fractionation.In 2018, Gstöttner et al. developed a 4D-LC-MS/MS workflow for the automated characterization of antibody charge variants by an online peptide mapping procedure involving in-line fraction collection17 (see Table 2). The multiple-heart cutting 4D-LC-MS approach allowed to characterize up to 9 peaks with the use of a home-made 4D-LC-MS system with commercial LC modules and controlled via two software (one for the 4D-LC system and another one for the Q-TOF instrument) and an additional in-house macro.17 The 4D-LC-MS setup allowed a significant time saving when characterizing 5 charge variants (9 hours in total) in comparison to 52 hours using the conventional off-line procedure. In addition, the authors reported almost similar sequence coverage achieved by the online and off-line approaches (94% LC, 86% HC vs. 94% LC, 94% HC). A schematic representation of 4D-LC-MS setup is reported in Fig. 4.
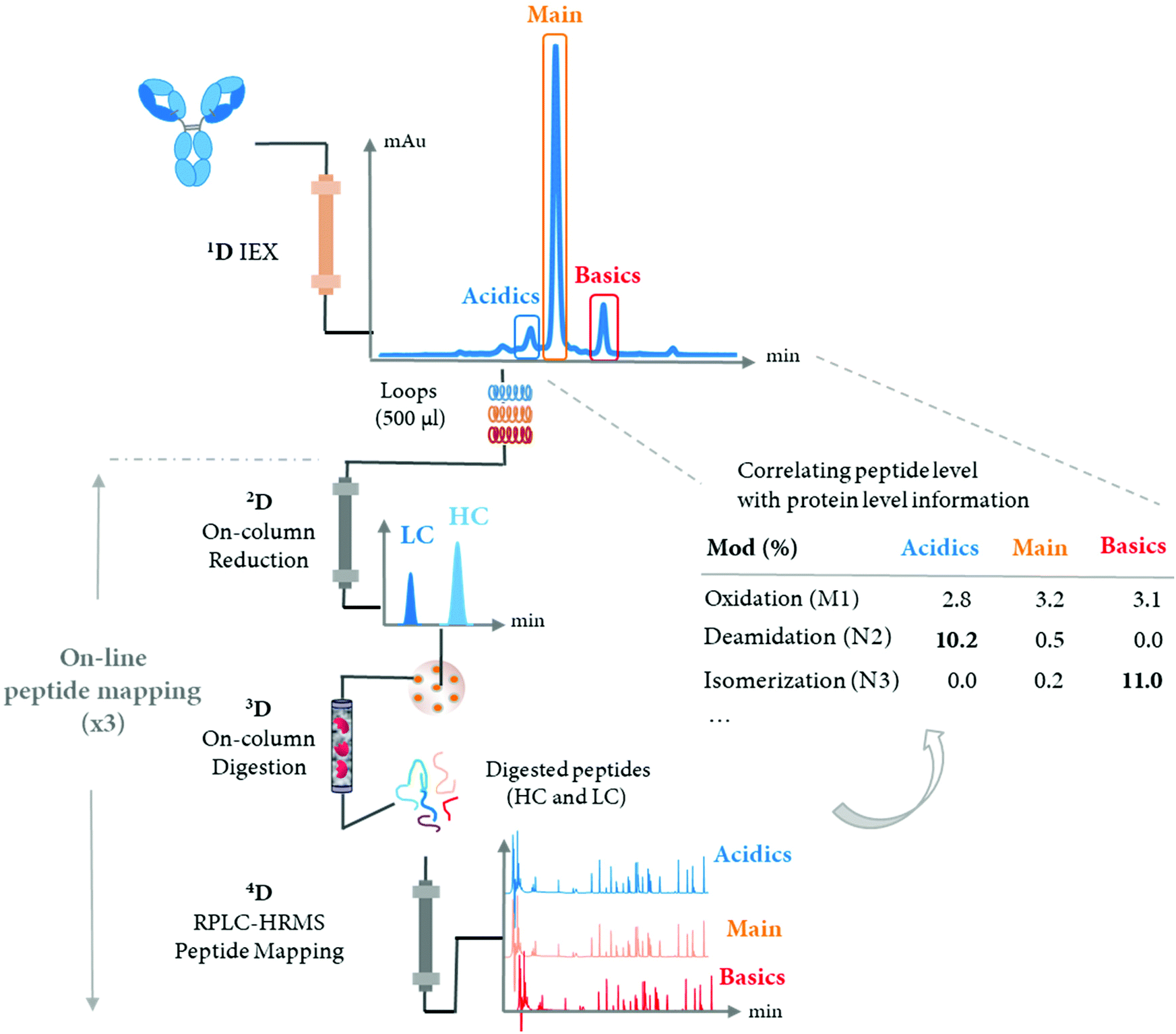 | ||
| Fig. 4 Schematic representation of 4D-LC-MS setup for the automated characterization of antibody charge variants by an online peptide mapping procedure, enabling the separation and fractionation of charge variants by IEX (1D) followed by on-column reduction by RPLC (2D), trypsin digestion in flow-through mode (3D), and peptide mapping analysis by RPLC (4D). IEX peaks assigned in one single analysis without off-line fraction collection and/or sample prep, while multiple online peptide mapping analyses are performed allowing the identification and quantification of PTMs under each IEX peaks (i.e. acidics, main, and basics). | ||
| mAb format | Injection (μg) | mD-LC-MS method | Analysis level | Analytical columns (dimensions) | Analysis time (min) | Targeted by mD-LC-MS | General comments | Ref. |
|---|---|---|---|---|---|---|---|---|
| Abbreviations: ADC, anti-drug conjugate; BsAb, bispecific monoclonal antibody; DAR, drug antibody ratio; Dig., digestion; nD; dimension; Frac., fraction; HILIC, hydrophilic interaction liquid chromatography; IEX, ion-exchange chromatography; IgG, immunoglobulin G; Red., reduction; RPLC, reversed-phase liquid chromatography; LC, liquid chromatography; mAb, monoclonal antibody; MS, mass spectrometry; Pep. Map, peptide mapping; ProA, protein A; SEC, size exclusion chromatography. All mD-LC-MS instruments are home-made and configured with multiple standard modules.a Intact – peptide, correlation peptide level with protein level information. | ||||||||
| mAb (IgG1) | 175 | 4D – (IEX × Red. × Dig. × RPLC)/impact II ESI Q-TOF MS | Intact – peptidea | 1D ProPac WCX-10 (4.0 × 250 mm, 10.0 μm) | 1D IEX: 56 min | Oxidation, deamidation, glycation | Online vs. offline | 17 |
| 2D AdvancedBio Desalting-RP (2.1 × 12.5 mm) | 2D, 3D, 4D Pep. Map | Sequence coverages: 94% LC, 86% HC vs. 94% LC, 94% HC | ||||||
| 3D StyrosZyme TPCK-Trypsin (2.1 × 100 mm) | 96 min frac−1. For 5 charge variants | |||||||
| 4D UPLC ACQUITY BEH C18 (2.1 × 50 mm, 1.7 μm) | ∼9 hours | 9 hours vs. 52 hours | ||||||
| 3 mAbs (IgG1, IgG2, IgG4), BsAb | 60 | 4D - (IEX × Red. × Dig. × RPLC)/QExactive™ Orbitrap MS | Intact – peptidea | 1D MAbPac SCX-10 (4.0 × 50 mm, 5.0 μm) | 1D IEX: 30 min | Oxidation, deamidation, isomerization | Reduction of artifact formation with the online approach (deamidation and isomerization) | 18 |
| 2D BioResolve™ RP mAb Polyphenyl (2.1 × 50 mm, 2.7 μm, 450 Å) | 2D, 3D, 4D | |||||||
| 3D Poroszyme® Immobilized Trypsin (2.1. × 30 mm) | Pep. Map.: 80 min per fraction | |||||||
| 4D RPLC XSelect™ CSH C18 (2.1. × 100 mm, 3.5 μm, 130 Å) | For 5 charge variants ∼7 hours | |||||||
| ADC | 60 | 4D – (SEC × Red. × Dig. × RPLC)/QExactive™ Orbitrap MS | Intact – peptidea | 1D TSKgel UP-SW3000 UHP-SEC (4.6. × 300 mm, 2.0 μm, 250 Å) | 1D SEC: 20 min | Size variants, aggregation, average DAR, oxidation, deamidation | Characterization of the ADC molecule. Complementarity information between the 2 approaches | 19 |
| 2D BioResolve™ RP mAb Polyphenyl (2.1 × 50 mm, 2.7 μm, 450 Å) | 2D, 3D, 4D Pep. Map.: 75 min per fraction | |||||||
| 3D Poroszyme® Immobilized Trypsin (2.1. × 30 mm) | ||||||||
| 4D RPLC XSelect™ CSH C18 (2.1. × 100 mm, 3.5 μm, 130 Å) | ||||||||
| mAb (IgG1) | 50 | 2D – (HIC × SEC)/UV | Intact | 1D TSKgel Butyl-NPR (4.6. × 35 mm, 2.5 μm) | 1D SEC: 20 min 1D HIC: 40 min | 103 | ||
| 2D TSKgel UP-SW3000 UHP-SEC (4.6. × 300 mm, 2.0 μm, 250 Å) | ||||||||
| 10 | 4D – (IdeS × Red. × Dig. × RPLC)/QExactive™ Orbitrap MS | Intact – peptidea | 1D IdeS-HPLC (2.1 × 50 mm) | 1D IdeS: 10 min | Oxidation, deamidation, isomerization, glycosylation | Targeted bottom-up: improvement of both online digestion and peptide separation Sequence coverages: 98.5% LC, 97% HC Characterization of N-glycopeptides | ||
| 2D BioResolve™ RP mAb Polyphenyl (2.1 × 50 mm, 2.7 μm, 450 Å) | 2D, 3D, 4D Pep. Map.: 105 min per fraction | |||||||
| 3D Poroszyme® Immobilized Trypsin (2.1. × 30 mm) | ||||||||
| 4D RPLC XSelect™ CSH C18 (2.1. × 150 mm, 3.5 μm, 130 Å) | ||||||||
| mAb (IgG1) | 10 | 4D – (ProA × Red. × Dig. × RPLC) /QExactive™ Orbitrap MS | Peptide | 1D Bio-Monolith Protein-A (4.95 × 5.2 mm) | Total analysis time: 100 min | Oxydation, succinimide formation, deamidation | Characterization of mAbs directly from HCCF materials 2 workflows (reduced and peptide mapping) within the same system | 102 |
| 2D BioResolve™ RP mAb Polyphenyl (2.1 × 50 mm, 2.7 μm, 450 Å) | ||||||||
| 3D Poroszyme® Immobilized Trypsin (2.1. × 30 mm) | ||||||||
| 4D RPLC XSelect™ CSH C18 (2.1. × 150 mm, 3.5 μm, 130 Å) | ||||||||
| 3D – (ProA × Red. × HILIC)/QExactive™ Orbitrap MS | Reduced | 1D Bio-Monolith Protein-A (4.95 × 5.2 mm) | Total analysis time: 80 min | Glycosylation | ||||
| 2D BioResolve™ RP mAb Polyphenyl (2.1 × 50 mm, 2.7 μm, 450 Å) | ||||||||
| 3D ACQUITY UPLC glycoprotein BEH Amide (2.1 × 150 mm, 1.7 μm, 300 Å) | ||||||||
| 2 mAbs (IgG1) | 10 | 3D – (IdeS × Red. × HILIC)/QExactive™ Orbitrap MS | Middle-up | 1D IdeS-HPLC (2.1 × 50 mm) | Total analysis time: 85 min | Glycosylation, glycation | Characterization of the N-glycosylation patterns | 20 |
| 2D BioResolve™ RP mAb Polyphenyl (2.1 × 50 mm, 2.7 μm, 450 Å) | ||||||||
| 3D ACQUITY UPLC Glycoprotein BEH Amide (2.1 × 150 mm, 1.7 μm, 300 Å) | ||||||||
More recently, an optimized 4D-LC setup based on the work by Gstöttner et al.17 was performed using bio-compatible LC modules from another instrument supplier.18 With the bio-compatible 4D-LC setup, a maximum of five fractions could be collected, which is sufficient for most CEX separations, and two software were also needed to control the various modules, including a QExactive Orbitrap instrument. A washing procedure was developed with the optimized 4D-LC system to limit sample carry-over issues,18 while the published 4D-LC setup in 2018 is based on a commercial 2D-LC system that automatically washes the loops. Thorough optimization of the different steps of the online peptide mapping was performed with (i) a BioResolve RP mAb Polyphenyl column known to limit antibody adsorption issues in comparison to C4 and C18 RPLC columns97 and increased length (50 mm vs. 12.5 mm) to limit 2D column overloading issues, (ii) a Poroszyme cartridge that increased sequence coverage in comparison to the StyrosZyme TPCK-Trypsin cartridge, and (iii) alternative RPLC column chemistry (CSH C18 vs. BEH C18) and dimensions to improve the chromatographic separation of peptides. In order to use the system for multiple days, a thorough optimization of the 2D on-column reduction conditions was performed with the use of TCEP, which is known to be more efficient and stable than the DTT reagent previously employed17,98 (50% of 2 mM DTT prepared in 50 mM Tris at pH 8.0 is oxidized in 2.5 hours98). Deamidation, oxidation, and glycation could be quantified using lower amounts of protein in comparison to the conventional off-line approach (60 μg online vs. 250 μg off-line). Overall, the online approach showed similar sequence coverage of 90–95% in comparison to the previous study17 and measured similar amount of oxidations in comparison to off-line approaches. The precision of the 4D-LC method for the measurement of PTMs was similar to the one achieved by the conventional off-line approach.18 Interestingly, a difference in the deamidation level of ∼16% was observed for an asparagine residue between the off-line and online procedure.18 The authors concluded on an artificial deamidation induced by the off-line procedure due to longer digestion times (4 hours vs. 0.4 min).18,99
In another study, a 4D-LC-MS/MS workflow for the characterization of antibody–drug conjugate (ADC) size variants was developed using the same 4D-LC setup previously published, demonstrating its versatility18,19 (Fig. 5). An optimization of the 2D RPLC gradient allowed the separation of various LC and HC drug-loaded species, which enabled the determination of the drug to antibody ratio (DAR) of the size variants (see Table 2). In addition to proving the presence of higher DAR species in the fraction containing high-molecular weight species (HMWS), the online peptide mapping also showed different amounts of an oxidized methionine for the HMWS in comparison to the monomeric species when the ADC was submitted to oxidative and light stress conditions. Deamidated asparagine residues have been shown to possibly compromise antibody colloidal stability, and enhance antibody aggregation.100 The authors further studied the impact of methionine and tryptophan oxidation on antibody stability and aggregation under different conditions.101 To our knowledge, the SEC-Reduction-Digestion-RPLC 4D-LC-MS method was the first to characterize ADC by online peptide mapping and provides a specific example on how this analytical tool could be used to perform a high-throughput screening of clinical candidates and eventually use these data to assess whether a specific amino acidic residue is responsible for protein instability (e.g. aggregation) and reengineer the antibody based on this information.
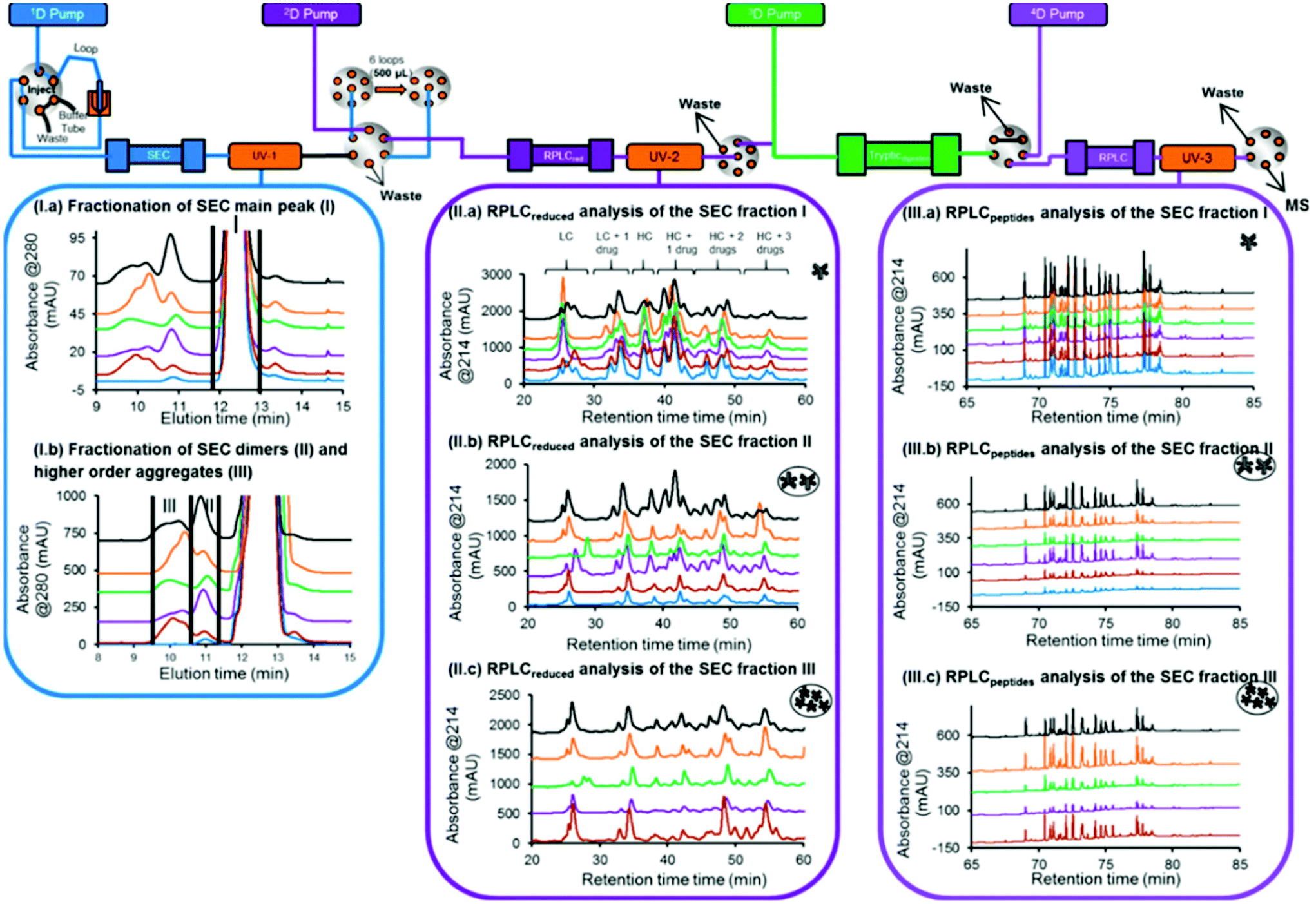 | ||
| Fig. 5 Schematic representation of an automated 4D-LC-MS workflow for SEC coupled to online peptide mapping, enabling the separation and fractionation of size variants by SEC (1D) followed by on-column reduction by RPLC (2D), trypsin digestion in flow-through mode (3D), and peptide mapping analysis by RPLC (4D). (I, a, b) SEC of ADC product reference, and samples stressed thermally, by light, at basic pH, at acidic pH, and under oxidative conditions (blue, brown, purple, green, orange, gray, and black traces, respectively). (II, a–c) 2D RPLC of the SEC heart-cut fractions that have been reduced online and then eluted. (III, a–c) 4D RPLC of the SEC heart-cut fractions that have been reduced and trypsin digested online. Reproduced from A. Goyon, M. Kim, L. Dai, C. Cornell, F. Jacobson, D. Guillarme, et al., Streamlined Characterization of an Antibody–Drug Conjugate by Two-Dimensional and Four-Dimensional Liquid Chromatography/Mass Spectrometry, Anal. Chem., 2019, 91, 2314896–14903. With permission from American Chemical Society, copyright 2019. | ||
Overall, the digestion performed using an immobilized trypsin cartridge in flow through mode significantly decreases the digestion time in comparison to off-line, in-solution protocols (minutes vs. hours). However, these shortened digestion-time for an intact mAb can cause incomplete peptide cleavages, thereby resulting in lower sequence coverage and poor repeatability. A study evidenced a higher number of miss-cleaved peptides with the online procedure digestion, approximately 30% (compared to 10% generally obtained with conventional off-line digestion), and this can complicate the data treatment for few PTMs.102
Recently, a novel strategy aimed at minimizing the missed cleavages was developed (see Table 2).103 This innovative approach decreases the complexity of intact mAbs, by digesting and reducing the product into small fragments of around 25 kDa, followed by an online peptide mapping analysis of each fragment. This approach considerably improved digestion efficiency with a higher sequence coverage in comparison to the previously published online workflow (>97% vs. 94%) (Fig. 6a). This improvement can also be explained by a better chromatographic separation of the peptides, since only a fragment of the total mAb is digested and analyzed at one time, and a better ionization (undesirable ionization competition may occur for co-eluting peptides). Notably, the sequence coverage obtained by the novel approach was also slightly higher in comparison to the conventional off-line peptide mapping procedure (>97% vs. 96%) (Fig. 6a). Based on these features, the strategy was successfully applied for the quantification of common PTMs such as oxidation, deamidation, and isomerization located in CDR regions, with similar amounts measured in comparison to the standard off-line approach (see Fig. 6b). The authors also evidenced that by digesting separately the various mAb fragments, the missed cleavages due the steric hindrance of the N-glycans are minimized and their relative abundance levels are similar to the ones obtained with the standard off-line peptide mapping approach (Fig. 6c).
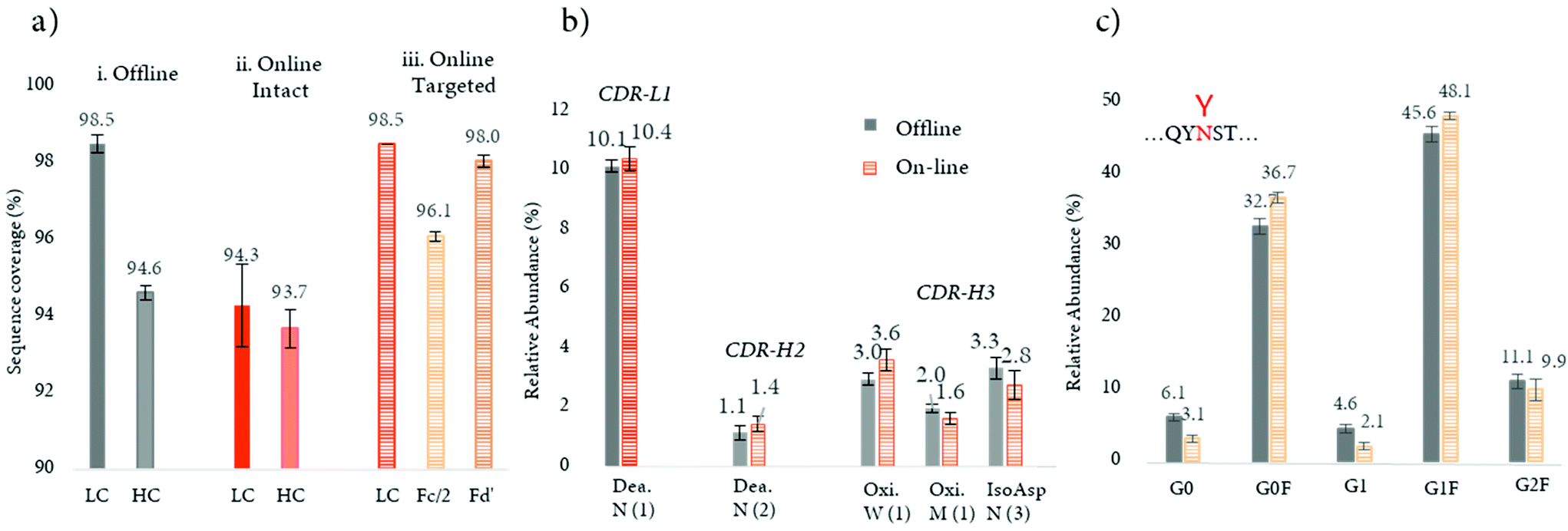 | ||
| Fig. 6 Targeted bottom-up approach for the improvement of online digestion. (a) Sequence coverages for LC and HC subunits obtained for a mAb with (a) a standard off-line and (b) online approaches for intact mAb, and (c) LC, Fc/2, and Fd’ obtained with the targeted approach. (b) Average levels (n = 3) of deamidation (Dea.), oxidation (Oxi.), and isomerization (IsoAsp) located in the CDRs; CDR-L1, CDR-H2, and CDR-H3 (from right to left), were depicted in full and hachured bars for the off-line and online approaches, respectively. Asparagine (N), tryptophan (W), and methionine (M) single codes are followed by numbers to distinguish the different residues. (c) Average levels (n = 3) of N-glycopeptides located in the Fc/2 fragment. Reproduced from J. Camperi, D. Guillarme, C. Stella, Targeted bottom-up characterization of recombinant monoclonal antibodies by multi-dimensional LC/MS, Anal. Chem., 2020, acs.analchem.0c02780. With permission from American Chemical Society, copyright 2020. | ||
In another study, a 4D-LC-MS (Protein-A × reduction × digestion × RPLC-MS) was developed for the analysis of mAbs in cell culture media, further demonstrating the versatility of this analytical approach.102 In this proof of concept study, a Protein-A column was used in the first dimension for the isolation of the targeted mAb from HCCF materials, followed by an online peptide mapping analysis using an MS/MS experiment for the determination of multiple common PTMs including oxidation, succinimide formation, and deamidation (see Table 2). The data generated through this 4D-LC-MS setup showed the increase of most of PTM levels during production process. Fig. 7a shows the difference of PTM levels at two different time points (7 and 12 days).
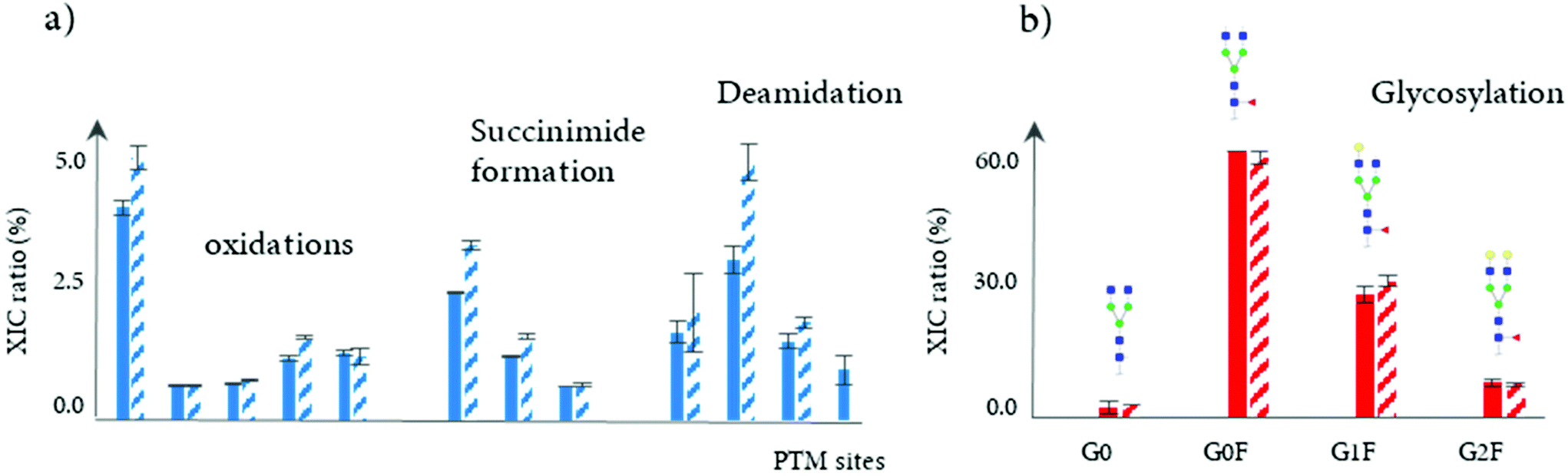 | ||
| Fig. 7 Analysis of mAbs directly from HCCF materials using of a Protein-A affinity chromatography column (1D), followed by online bottom-up analysis or HILIC/MS for subunit separation. (a) Determination and quantitation of oxidation, succinimide formation, and deamidation levels of a mAb for different modification sites, obtained by the online peptide analysis (n = 3). (b) Relative abundance of main N-glycans obtained by the reduced level analysis (n = 3), with the assignment of different N-glycans structures. The full and hatched bars correspond to two different days of the production process, days 7 and 12. Reproduced from J. Camperi, L. Dai, D. Guillarme, C. Stella, Fast and automated characterization of monoclonal antibody minor variants from cell cultures by combined Protein-A and multi-dimensional LC/MS methodologies, Anal. Chem., 2020, acs.analchem.0c01250. With permission from American Chemical Society, copyright 2020. | ||
In addition, a 3D-LC-MS (protein A – reduction – HILIC/MS) workflow was incorporated into the same chromatographic system to measure glycation and glycosylation at the reduced level. An accurate and comprehensive profiling of glycosylation patterns was obtained, showing the relative intensities of mAb glycoforms throughout the production process (see Fig. 7b). Overall, the total acquisition time using both mD-LC setups (3D and 4D-LC-MS) for a multi-characterization level can be completed in just 3 hours. Based on these results, mD-LC-MS can be successfully implemented to perform fast and reliable monitoring of PTMs during the manufacturing.
4. Middle-up analysis
4.1 Conventional middle-down/up analysis
The middle-up level analysis is gaining interest due to its complementarity to the intact mass and bottom-up analysis.85,104–106 The middle-up approach refers to the analysis of mAbs after their cleavage into large fragments or subunits following limited proteolysis. IgG-degrading enzyme of Streptococcus pyogenes (IdeS) protease, which specifically cleaves between the two consecutive glycine residues under the hinge region yielding (Fab′)2 and Fc/2 fragments, is typically used.104 The subsequent chemical reduction of IdeS-generated fragments results in smaller species of around 25 kDa: the LC and Fd. The off-line sample preparation comprising a digestion and reduction steps can be performed within one hour, and requires a low amount of material.104 The 25 kDa fragments can then be easily separated by LC107–109 or CE110–113 and simultaneously identified by HRMS instruments such as QTOFs and Orbitrap analyzers that can resolve the isotopic pattern of 25 kDa proteins with a mass accuracy around 5 ppm. The middle-down approach, which refers to MS/MS-based experiments, can also be performed for sequencing these fragments.114 Middle-up/down approaches have been successfully applied for: (i) the characterization of mAb PTMs;84,113 (ii) the study of specific molecular properties such as mispairing of subunits for BsAb molecules;115 (iii) the mapping of multiple modifications disulfide linkages;116 and (iv) the determination of drug load profile and distribution of ADCs.117 The middle-up approach was particularly interesting for the qualitative profiling of mAb glycosylation patterns.108,118,119Fornelli et al. reported an effective middle-down analysis approach for the identification of oxidized mAb methionine residues using an IdeS digestion step, followed by a RPLC-ETD analysis using an high-field Orbitrap Fourier transform mass spectrometry (FTMS).109 The developed method allowed the quick detection and localization of IgG oxidation sites, making it ideal for mAbs’ oxidation assessment. A similar strategy was applied to investigate both cysteine and lysine conjugated ADCs (i.e., BV; Adcetris™ and T-DM1; Kadcyla™) at the subunit level, and evaluate multiple analytical attributes including average DAR, positional isomers, conjugation sites, site occupancy and microvariants.117 D'Atri et al. described a middle-up approach to compare the glycosylation patterns of originators and biosimilars mAbs by using HILIC coupled to HRMS.108 The HILIC mode was the most suitable for the elucidation of glycosylation patterns, due to the high resolving power and compatibility with MS.87,120,121 Similar strategies were successfully applied for the characterization of an ADC,122 Brentuximab Vedotin, and a recombinant Fc fusion protein, Etanercept.107
In this context, the use of 2D-LC can be particularly beneficial for the analysis of complex mixtures such as antibodies digested at the middle-up or bottom-up levels. Several 2D-LC methods have been published for the comparison of originator and biosimilar antibodies,123 high-resolution characterization of Fc-fusion proteins107 or more fundamental applications.124 For example, Sorensen et al. reported the use of a CEX × RPLC 2D-LC method coupled to a TOF instrument in order to compare the originator and biosimilar products of 3 mAbs at the middle-up level.123 The authors indicated an excellent retention time precision achieved with the 2D-LC-MS method, which could allow to track characteristic mAb fragment and quickly identify minor differences between a biosimilar and originator products.
Other IgG hinge-specific proteases such as IgdE (FabALACTICA©) and Kgp (GingisKHAN©) are also available for in-solution digestions, in order to generate Fab fragments.116,125–127 In contrast with IdeS protease, which cleaves below the hinge region, IgdE and Kgp cleaves above the hinge region, generating intact Fab fragments from human IgG1.104 The complementarity of both IdeS- and IgdE/Kgp-digested mAbs was demonstrated in many applications.34,116,125 For instance, Faid et al. reported a middle-up analysis of mAbs after combined IgdE and IdeS hinge proteolysis for the analysis of mAbs disulfide bridges by LC-MS, where the three fragments as well as their corresponding free sulfhydryl forms were well separated.116 Although the digestion sites of the three proteases IgdE, Kgp, and IdeS are in close proximity in the hinge region of mAbs, different retention profiles can be expected for the differently cleaved Fc and Fd’ fragments. As an example, a middle-up approach using a 2D-LC-MS (RPLC-RPLC) method, where the first dimension was merely used for online sample reduction, followed by RPLC-MS analysis of protein fragment separation, was performed for the analysis of a mAb and an ADC (unpublished results). While only minor retention time shifts for Fc/2 and Fd’ fragments were observed for the mAb (Fig. 8a), the retention profiles for ADC fragments were completely different due to the distribution of drugs on the Fc/2 and Fd’ fragments (Fig. 8b).
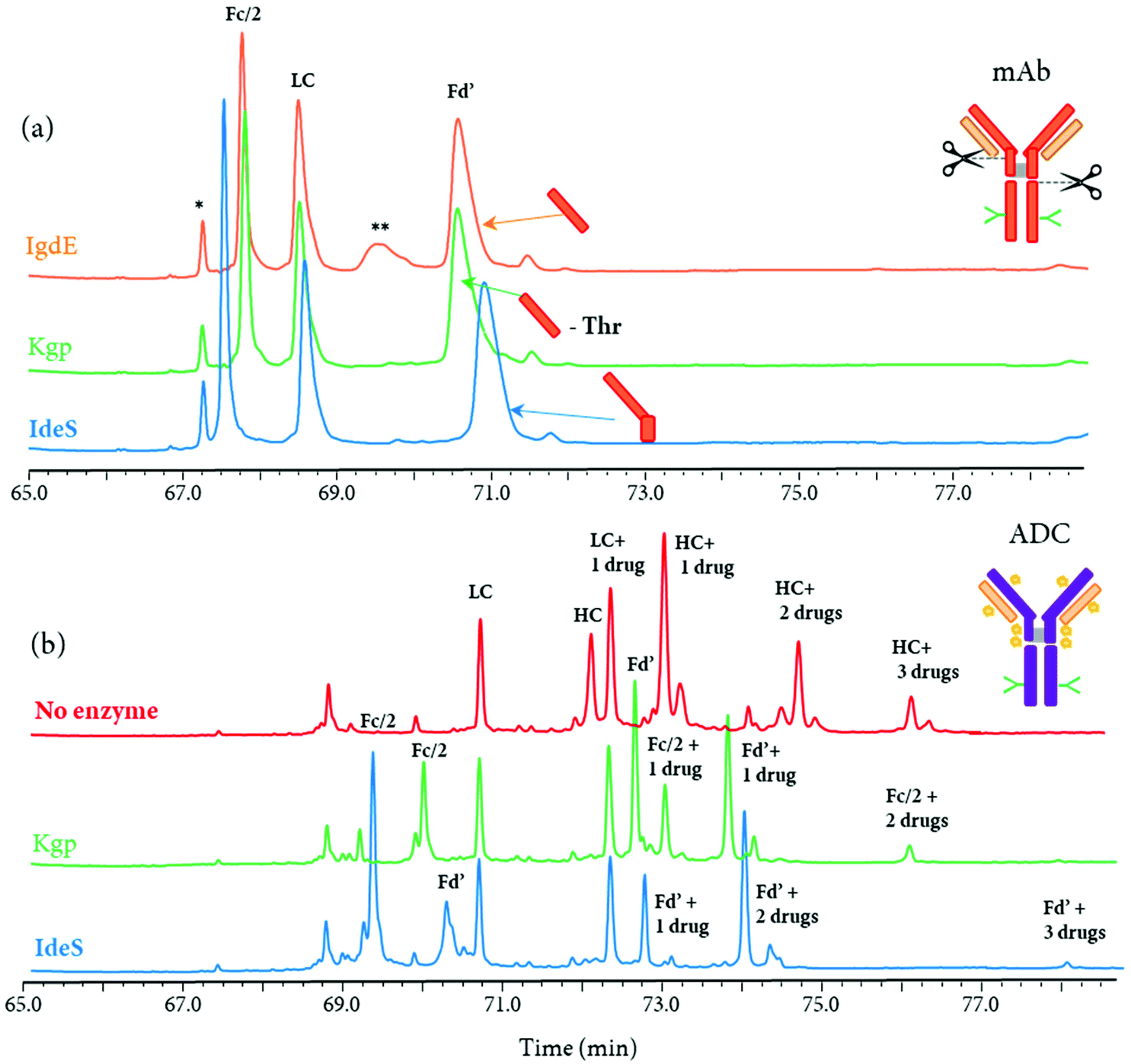 | ||
| Fig. 8 Comparison of 2D-LC (RP × RP) separation profiles (MS detection) at the middle-up level obtained after a (a) mAb and (b) ADC digestion by different IgG hinge-specific proteases. For the in-solution digestion of mAb, IgdE, KgP, and IdeS-treatment were performed (from top to bottom). The indicated ‘Thr’ corresponds to the one amino acid difference on the Fd’ fragment between IgdE- and KgP-generated fragments. For the ADC, an reduced analysis, as well as middle-up analysis using KgP- and IdeS-protease treatment were represented. mAb, ADC and relative fragments are represented. Incomplete digestion is represented by (**) and peak related to the gradient conditions by (*). Unpublished results. | ||
4.2 Automated middle-up analysis and multi-level characterization
An immobilized IdeS-HPLC column was recently commercialized, enabling the full automation of mAb digestion into (Fab′)2 and Fc/2 fragments within only 10 min. An innovative 2D-LC-MS workflow was developed to characterize a mAb and a BsAb using the immobilized IdeS-HPLC cartridge in the first dimension to generate fragments subsequently separated on a 2D RP column and identified by HRMS.115 The reduction of spectral complexity of IdeS-generated ∼25 kDa fragments allowed an accurate profiling of Fc/2 N-glycans, as well as the identification of mAb variants including C-terminal lysine cleavage, N-terminal pyroglutamination, and glycation. For the BsAb, the same 2D-LC-MS setup also enabled the detection of product impurities due to chain mispairing in one single analysis, with a total run time of only 85 min. This is an example of how a 2D-LC-MS based middle-up analysis can be used to successfully characterize both mAbs and BsAbs.The immobilized IdeS-HPLC cartridge was also implemented in a 3D-LC-MS setup to automatize the off-line sample preparation steps, allowing a fast (85 min per run) and effective characterization of N-glycosylation patterns of mAbs at the middle-up level20 (Table 2). The schematic representation of the 3D-LC-MS workflow is represented in Fig. 9.
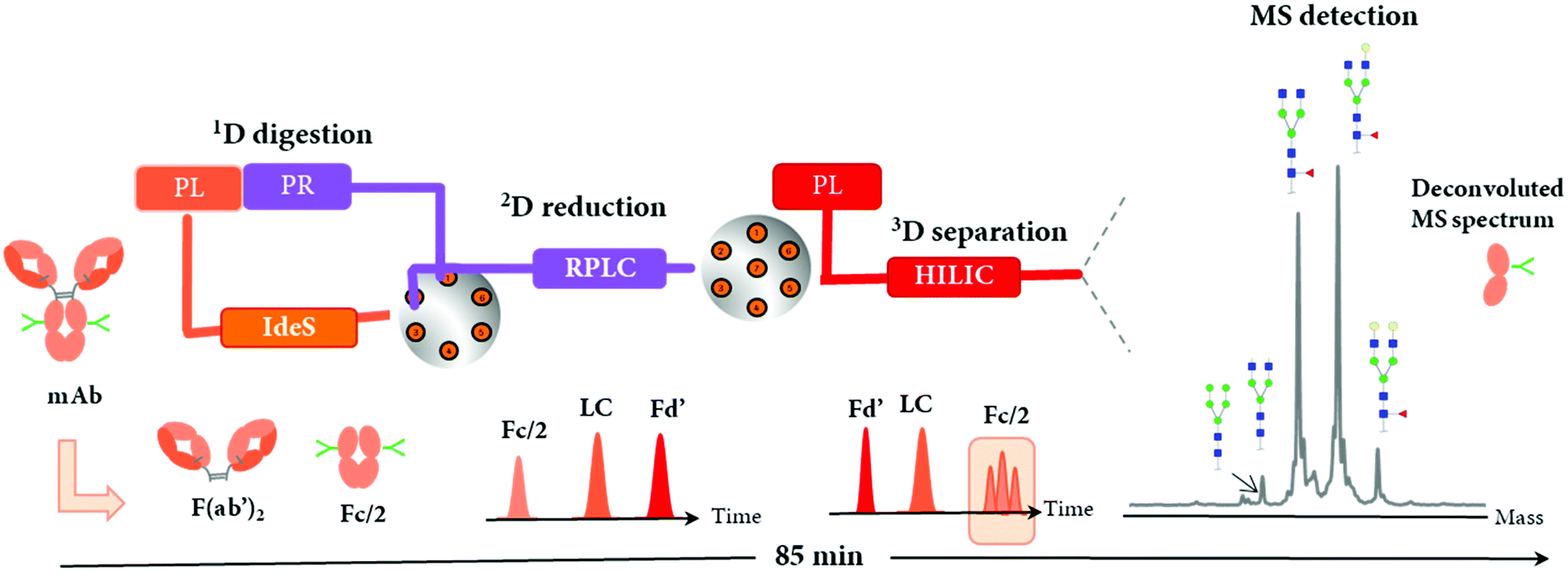 | ||
| Fig. 9 Schematic representation of the online 3D-HPLC workflow for automated middle-up analysis of mAbs and assignment of major glycoforms of Fc/2 fragment. An IdeS-HPLC column was used as first dimension (1D) followed by on-column reduction by RP-HPLC (2D) and HILIC (3D)/HRMS analysis of the proteolytic fragments Fd’, LC, and Fc/2. Reproduced from J. Camperi, L. Dai, D. Guillarme, C. Stella, Development of a 3D–LC/MS workflow for fast, automated and effective characterization of glycosylation patterns of biotherapeutic products, Anal. Chem., 2020, acs.analchem.9b05193 with permission from American Chemical Society, copyright 2019. | ||
Following online 1D IdeS-digestion combined with a 2D reduction step, the three ∼25 kDa fragments including Fc/2, LC, and Fd’ were successfully analyzed by 3D HILIC coupled to a QExactive MS instrument. As expected, the elution profile of mAb fragments by HILIC was reversed compared to 2D RPLC separation, and enabled the separation of N-glycoforms present on the Fc/2, thus facilitating the glycosylation patterns assessment by MS. Perchepied et al. demonstrated that increased level of confidence can be achieved for the identification of N-glycoforms when they are chromatographically separated since artificial fragmentation may occur during the ESI process.128 In addition, other PTMs including C-terminal lysine truncation or glycation could also be identified. A mixing tee was required before the HILIC separation in order to improve the solvent compatibility by diluting the flow coming from the 2D RP column containing ∼60% aqueous mobile phase with a high amount of acetonitrile (from the 3D pump) to inject mAbs under HILIC conditions (acetonitrile ≈ 80%). This reported 3D-LC-MS setup could potentially characterize other mAb or complex antibody formats, especially those with a high number of glycosylation forms such as Cetuximab, where different N-glycosylation sites are present on both Fc and Fd’ fragments,129 or Fc-fusion proteins.130
Several studies highlighted the advantages of combining the analysis of antibodies at multiple levels, i.e. intact, reduced, middle-up, and peptide level, to allow a fast and extensive characterization.111,131–133 For instance, Giorgetti et al. reported the combination of intact, middle-up, and bottom-up analysis using a CE-MS instrument for the analysis of 7 FDA-approved mAbs111 with: (i) the identification of major N-glycoforms at the intact level; (ii) additional PTMs as K-clip, oxidations or deamidations observed at the middle-up level; and (iii) a precise location and relative quantitation of each PTM at the bottom-up level. This study demonstrated the utility of profiling a mAb at multiple levels to achieve a comprehensive identification of the CQAs. Developed CE methods for the characterization of therapeutic antibodies at multiple levels typically involve similar experimental conditions (use of same separation buffer, of capillary coatings, injection buffer etc.) and can be used with a single instrument.
Multi-dimensional LC-MS technologies have the potential to combine the analysis of therapeutic proteins at multiple levels too. As an example, the 3D-HPLC-MS workflow previously described (Fig. 9) can be used for mAb characterization at both middle-up and reduced level within the same system, by bypassing the IdeS-HPLC column. Fig. 10 presents the LC separation profiles obtained for the analysis of an aglycosylated BsAb at reduced and middle-up levels, represented in blue and orange colors, respectively. Using the same mobile phases i.e. 0.1% formic acid and 0.05% trifluoroacetic acid in water (A) and acetonitrile (B), two RPLC and HILIC modes can be simultaneously used in the third dimension to separate the fragments. At the reduced antibody level, RPLC selectivity was not sufficient to separate the two pairs of light (L1 and L2) and heavy (H1 and H2) chains (Fig. 10a), while that HILIC mode allowed to resolve the four BsAb species (Fig. 10b), enabling their assignment by HRMS. Additional middle-up characterization was performed for a deeper BsAb characterization. Fig. 10c shows the 3D RPLC separation of most of the 25 kDa fragments, including the two Fc/2 and Fd’, but the two LCs were co-eluting. Conversely, the Fd and Fc/2 fragments could not be separated on the 3D HILIC column, but the two LCs were partially resolved (Fig. 10d). The results demonstrate the high complementary of the HILIC and RPLC separation modes and how the reduced and middle-up level analysis of this BsAb can be performed on such analytical workflow.
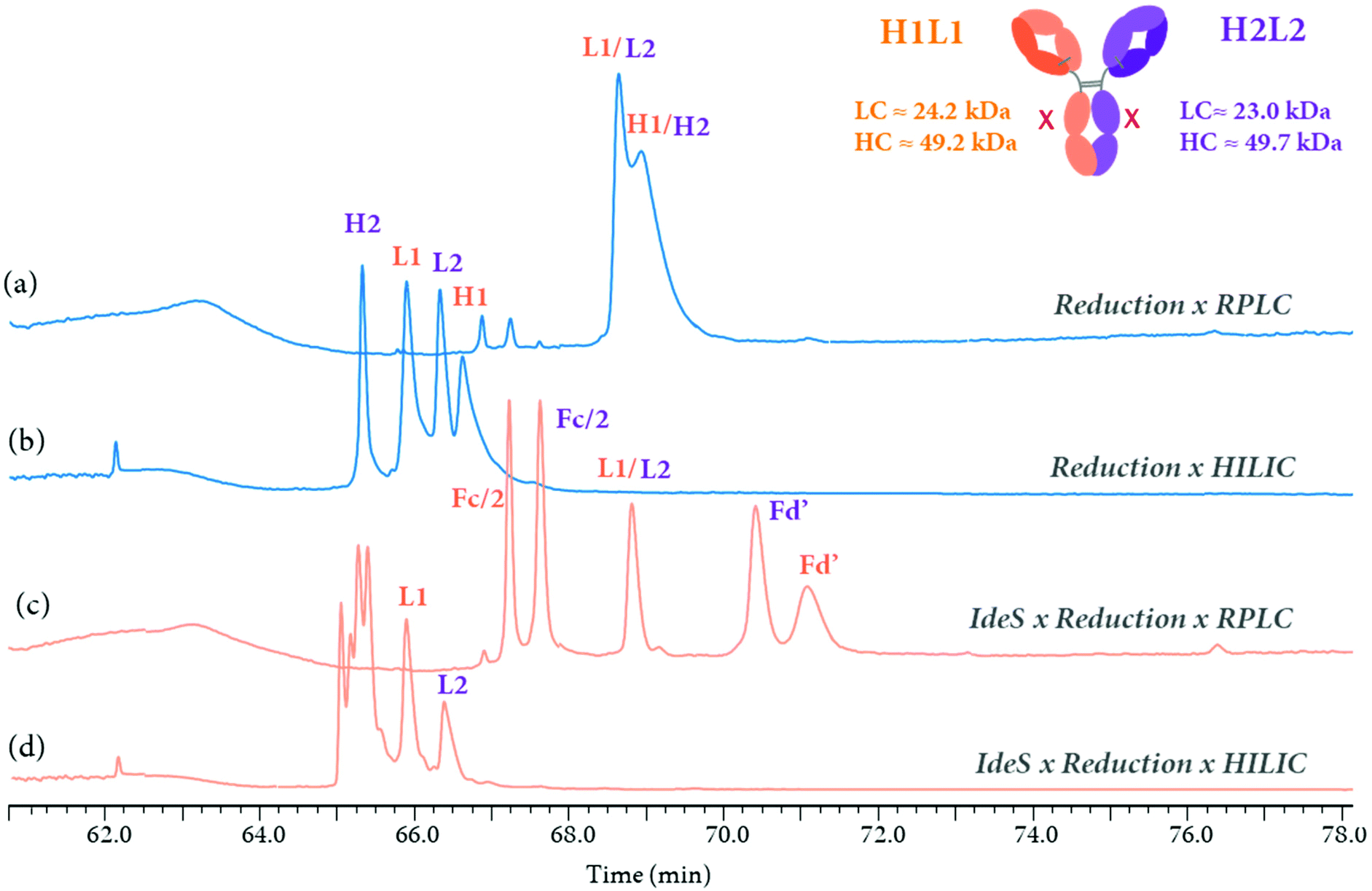 | ||
| Fig. 10 Reduced and middle-up analysis of an aglycosylated BsAb performed by an online 3D-HPLC (IdeS × reduction × HILIC or RPLC-MS) workflow. UV chromatograms obtained at the reduced level (in blue) by (a) RPLC or (b) HILIC by bypassing the IdeS-HPLC column. Middle-up analysis (in orange) using a (3D) (c) RPLC or (d) HILIC separation (from top to bottom). H1L1 and H2L2 were represented in orange and purple, respectively. The molecular weight of each subunits (LC and HC) were added. The corresponding fragments Fc/2, LC, and Fd’ were represented on each chromatogram. Unpublished results. | ||
An alternative 3D-LC-MS setup was developed with the addition of an immobilized trypsin cartridge to perform PTM analysis at the peptide level. This novel 4D-LC-MS method can be used to localize and quantify PTMs such as oxidation, deamidation, or isomerization present in the CDRs.103
Overall, the versatility of the mD-LC-MS setup offers the possibility to perform multi-level of analysis, including reduced, middle-up, and peptide levels for a full comprehensive characterization of therapeutic protein with rapid data acquisition times, within 90 min and without any tedious off-line steps. In the future, the immobilization of additional specific enzymes such as the IgdE, Kgp ones presented in the section 4.1 would be beneficial for the automation of the middle-up/down approach using mD-LC-MS systems. A wider choice of immobilized enzymes would bring the middle-up approach to the next level, with a wider implementation and the possibility to solve analytical challenges inherent to the introduction of novel therapeutic antibody formats.
5. Conclusion
The present review highlighted applications where multi-dimensional LC-MS complement and automatize conventional methods for the characterization of therapeutic antibodies. State of the art LC-MS technologies used to characterize therapeutic proteins at the intact, middle-up and bottom-up levels have been discussed. Native MS and 2D-LC methods are now well established approaches and are implemented throughout the development of conventional therapeutic proteins. In the meantime, automation and high throughput analytical platforms have become a necessity in the fast-paced biopharmaceutical environment. The need to automatize conventional, time consuming off-line fraction collection and multi-steps peptide mapping has never been that urgent. To overcome the limits of conventional sample preparation and analytical methods, the recent development of mD-LC-MS methods, beyond the use of 2D-LC instruments, has brought the technique to the next level. With the data generated by the mD-LC-MS platforms, a direct correlation between peptide level and protein level information can be obtained. The capability of mD-LC-MS methods has been demonstrated with the online characterization of charge and size variants by in-line fraction collection and peptide mapping. Due to the minimal amount of required sample and significant time-saving, the mD-LC-MS technology can be also potentially integrated into production and purification environments for earlier characterization of antibody variants. As an example, the use of protein A chromatography in the first dimension of a multidimensional LC-MS setup allows the online characterization of PTMs, and could therefore enable a continuous monitoring of cell cultures. Therefore, mD-LC-MS has the potential to be an in-process control (IPC) method to better understand and improve biological processes. The versatility of mD-LC-MS platforms also offers the possibility to perform antibody characterization at multiple levels, i.e. reduced, middle-up, and bottom-up levels, within the same chromatographic system. The recent commercialization of a protease-immobilized HPLC cartridge has unleashed the potential of online middle-up analysis to provide complementary information to the conventional intact and bottom-up approaches. The development of additional immobilized enzyme cartridges would provide analytical scientists a complete toolbox to overcome future analytical challenges with more complex therapeutics.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors acknowledge David A. Michels, Feng Yang, and Vikas Sharma from Genentech (South San Francisco, CA, USA) for providing a critical review for this manuscript.References
- A. Mullard, 2019 FDA drug approvals, Nat. Rev. Drug Discovery, 2020, 19(2), 79–84 CrossRef CAS.
- H. Kaplon, M. Muralidharan, Z. Schneider and J. M. Reichert, Antibodies to watch in 2020, mAbs, 2020, 12(1), 1703531 CrossRef.
- J. X. Yu, J. P. Hodge, C. Oliva, S. T. Neftelinov, V. M. Hubbard-Lucey and J. Tang, Trends in clinical development for PD-1/PD-L1 inhibitors, Nat. Rev. Drug Discovery, 2019, 19(3), 163–164 Search PubMed.
- T. Graf, K. Heinrich, I. Grunert, H. Wegele, M. Haberger and P. Bulau, et al., Recent advances in LC–MS based characterization of protein-based bio-therapeutics – mastering analytical challenges posed by the increasing format complexity, J. Pharm. Biomed. Anal., 2020, 186, 113251 CrossRef CAS.
- A. Goyon, S. Fekete, A. Beck, J.-L. Veuthey and D. Guillarme, Unraveling the mysteries of modern size exclusion chromatography - the way to achieve confident characterization of therapeutic proteins, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1092, 368–378 CrossRef CAS.
- S. Fekete, A. Beck, J. Fekete and D. Guillarme, Method development for the separation of monoclonal antibody charge variants in cation exchange chromatography, Part II: pH gradient approach, J. Pharm. Biomed. Anal., 2015, 102, 282–289 CrossRef CAS.
- S. Fekete, A. Beck, J. Fekete and D. Guillarme, Method development for the separation of monoclonal antibody charge variants in cation exchange chromatography, Part I: salt gradient approach, J. Pharm. Biomed. Anal., 2015, 102, 33–44 CrossRef CAS.
- O. Salas-Solano, K. Babu, S. Park, X. Zhang, L. Zhang and Z. Sosic, et al., Intercompany Study to Evaluate the Robustness of Capillary Isoelectric Focusing Technology for the Analysis of Monoclonal Antibodies, Chromatographia, 2011, 73, 1137–1144 CrossRef CAS.
- E. Wagner, O. Colas, S. Chenu, A. Goyon, A. Murisier and S. Cianferani, et al., Determination of size variants by CE-SDS for approved therapeutic antibodies: Key implications of subclasses and light chain specificities, J. Pharm. Biomed. Anal., 2020, 184, 113166 CrossRef CAS.
- O. Salas-Solano, B. Kennel, S. S. Park, K. Roby, Z. Sosic and B. Boumajny, et al., Robustness of iCIEF methodology for the analysis of monoclonal antibodies: an interlaboratory study, J. Sep. Sci., 2012, 35(22), 3124–3129 CrossRef CAS.
- A. Goyon, M. Excoffier, M.-C. Janin-Bussat, B. Bobaly, S. Fekete and D. Guillarme, et al., Determination of isoelectric points and relative charge variants of 23 therapeutic monoclonal antibodies, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1065–1066, 119–128 CrossRef CAS.
- S. Fekete, J.-L. Veuthey, A. Beck and D. Guillarme, Hydrophobic interaction chromatography for the characterization of monoclonal antibodies and related products, J. Pharm. Biomed. Anal., 2016, 130, 3–18 CrossRef CAS.
- T. Ikegami, Hydrophilic interaction chromatography for the analysis of biopharmaceutical drugs and therapeutic peptides: A review based on the separation characteristics of the hydrophilic interaction chromatography phases, J. Sep. Sci., 2019, 42(1), 130–213 CrossRef CAS.
- E. Farsang, D. Guillarme, J.-L. Veuthey, A. Beck, M. Lauber and A. Schmudlach, et al., Coupling non-denaturing chromatography to mass spectrometry for the characterization of monoclonal antibodies and related products, J. Pharm. Biomed. Anal., 2020, 185, 113207 CrossRef CAS.
- R. S. Rogers, N. S. Nightlinger, B. Livingston, P. Campbell, R. Bailey and A. Balland, Development of a quantitative mass spectrometry multi-attribute method for characterization, quality control testing and disposition of biologics, mAbs, 2015, 7(5), 881–890 CrossRef CAS.
- S. Magdeldin, J. J. Moresco, T. Yamamoto and J. R. Yates, Off-Line Multidimensional Liquid Chromatography and Auto Sampling Result in Sample Loss in LC/LC–MS/MS, J. Proteome Res., 2014, 13(8), 3826–3836 CrossRef CAS.
- C. Gstöttner, D. Klemm, M. Haberger, A. Bathke, H. Wegele and C. Bell, et al., Fast and Automated Characterization of Antibody Variants with 4D HPLC/MS, Anal. Chem., 2018, 90(3), 2119–2125 CrossRef.
- A. Goyon, L. Dai, T. Chen, B. Wei, F. Yang and N. Andersen, et al., From proof of concept to the routine use of an automated and robust multi-dimensional liquid chromatography mass spectrometry workflow applied for the charge variant characterization of therapeutic antibodies, J. Chromatogr., A, 2020, 1615, 460740 CrossRef CAS.
- A. Goyon, M. Kim, L. Dai, C. Cornell, F. Jacobson and D. Guillarme, et al., Streamlined Characterization of an Antibody–Drug Conjugate by Two-Dimensional and Four-Dimensional Liquid Chromatography/Mass Spectrometry, Anal. Chem., 2019, 91(23), 14896–14903 CrossRef CAS.
- J. Camperi, L. Dai, D. Guillarme and C. Stella, Development of a 3D–LC/MS workflow for fast, automated and effective characterization of glycosylation patterns of biotherapeutic products, Anal. Chem., 2020, 92, 4357–4363 CrossRef CAS.
- G. Terral, A. Beck and S. Cianférani, Insights from native mass spectrometry and ion mobility-mass spectrometry for antibody and antibody-based product characterization, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1032, 79–90 CrossRef CAS.
- H. Zhang, W. Cui and M. L. Gross, Mass spectrometry for the biophysical characterization of therapeutic monoclonal antibodies, FEBS Lett., 2014, 588(2), 308–317 CrossRef CAS.
- A. C. Leney and A. J. R. Heck, Native Mass Spectrometry: What is in the Name?, J. Am. Soc. Mass Spectrom., 2017, 28(1), 5–13 CrossRef CAS.
- J. Snijder, M. van de Waterbeemd, E. Damoc, E. Denisov, D. Grinfeld and A. Bennett, et al., Defining the Stoichiometry and Cargo Load of Viral and Bacterial Nanoparticles by Orbitrap Mass Spectrometry, J. Am. Chem. Soc., 2014, 136(20), 7295–7299 CrossRef CAS.
- M. Källsten, R. Hartmann, K. Artemenko, S. B. Lind, F. Lehmann and J. Bergquist, Qualitative analysis of antibody–drug conjugates (ADCs): an experimental comparison of analytical techniques of cysteine-linked ADCs, Analyst, 2018, 143(22), 5487–5496 RSC.
- F. Debaene, A. Bœuf, E. Wagner-Rousset, O. Colas, D. Ayoub and N. Corvaïa, et al., Innovative Native MS Methodologies for Antibody Drug Conjugate Characterization: High Resolution Native MS and IM-MS for Average DAR and DAR Distribution Assessment, Anal. Chem., 2014, 86(21), 10674–10683 CrossRef CAS.
- J. R. McCombs and S. C. Owen, Antibody Drug Conjugates: Design and Selection of Linker, Payload and Conjugation Chemistry, AAPS J., 2015, 17(2), 339–351 CrossRef CAS.
- J. Chen, S. Yin, Y. Wu and J. Ouyang, Development of a Native Nanoelectrospray Mass Spectrometry Method for Determination of the Drug-to-Antibody Ratio of Antibody–Drug Conjugates, Anal. Chem., 2013, 85(3), 1699–1704 CrossRef CAS.
- J. Jones, L. Pack, J. H. Hunter and J. F. Valliere-Douglass, Native size-exclusion chromatography-mass spectrometry: suitability for antibody–drug conjugate drug-to-antibody ratio quantitation across a range of chemotypes and drug-loading levels, mAbs, 2020, 12(1), 1682895 CrossRef.
- R. Fleming, ADC Analysis by Hydrophobic Interaction Chromatography, in Antibody-Drug Conjugates, ed. L. N. Tumey, Springer USNew York, NY, 2020, pp. 147–161. (Methods in Molecular Biology; vol. 2078), DOI: DOI:10.1007/978-1-4939-9929-3_10.
- A. O. Bailey, G. Han, W. Phung, P. Gazis, J. Sutton and J. L. Josephs, et al., Charge variant native mass spectrometry benefits mass precision and dynamic range of monoclonal antibody intact mass analysis, mAbs, 2018, 10(8), 1214–1225 CrossRef CAS.
- Y. Yan, A. P. Liu, S. Wang, T. J. Daly and N. Li, Ultrasensitive Characterization of Charge Heterogeneity of Therapeutic Monoclonal Antibodies Using Strong Cation Exchange Chromatography Coupled to Native Mass Spectrometry, Anal. Chem., 2018, 90(21), 13013–13020 CrossRef CAS.
- F. Füssl, K. Cook, K. Scheffler, A. Farrell, S. Mittermayr and J. Bones, Charge Variant Analysis of Monoclonal Antibodies Using Direct Coupled pH Gradient Cation Exchange Chromatography to High-Resolution Native Mass Spectrometry, Anal. Chem., 2018, 90(7), 4669–4676 CrossRef.
- Y. Leblanc, C. Ramon, N. Bihoreau and G. Chevreux, Charge variants characterization of a monoclonal antibody by ion exchange chromatography coupled on-line to native mass spectrometry: Case study after a long-term storage at + 5 °C, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1048, 130–139 CrossRef CAS.
- Y. Yan, T. Xing, S. Wang, T. J. Daly and N. Li, Coupling Mixed-Mode Size Exclusion Chromatography with Native Mass Spectrometry for Sensitive Detection and Quantitation of Homodimer Impurities in Bispecific IgG, Anal. Chem., 2019, 91(17), 11417–11424 CrossRef CAS.
- A. Goyon, V. D'Atri, O. Colas, S. Fekete, A. Beck and D. Guillarme, Characterization of 30 therapeutic antibodies and related products by size exclusion chromatography: Feasibility assessment for future mass spectrometry hyphenation, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1065–1066, 35–43 CrossRef CAS.
- M. Haberger, M. Leiss, A.-K. Heidenreich, O. Pester, G. Hafenmair and M. Hook, et al., Rapid characterization of biotherapeutic proteins by size-exclusion chromatography coupled to native mass spectrometry, mAbs, 2016, 8(2), 331–339 CrossRef CAS.
- B. Wei, G. Han, J. Tang, W. Sandoval and Y. T. Zhang, Native Hydrophobic Interaction Chromatography Hyphenated to Mass Spectrometry for Characterization of Monoclonal Antibody Minor Variants, Anal. Chem., 2019, 91(24), 15360–15364 CrossRef CAS.
- B. Chen, Y. Peng, S. G. Valeja, L. Xiu, A. J. Alpert and Y. Ge, Online Hydrophobic Interaction Chromatography–Mass Spectrometry for Top-Down Proteomics, Anal. Chem., 2016, 88(3), 1885–1891 CrossRef CAS.
- T.-H. Chen, Y. Yang, Z. Zhang, C. Fu, Q. Zhang and J. D. Williams, et al., Native Reversed-Phase Liquid Chromatography: A Technique for LCMS of Intact Antibody–Drug Conjugates, Anal. Chem., 2019, 91(4), 2805–2812 CrossRef CAS.
- R. Gahoual, A.-K. Heidenreich, G. W. Somsen, P. Bulau, D. Reusch and M. Wuhrer, et al., Detailed Characterization of Monoclonal Antibody Receptor Interaction Using Affinity Liquid Chromatography Hyphenated to Native Mass Spectrometry, Anal. Chem., 2017, 89(10), 5404–5412 CrossRef CAS.
- T. Schlothauer, P. Rueger, J. O. Stracke, H. Hertenberger, F. Fingas and L. Kling, et al., Analytical FcRn affinity chromatography for functional characterization of monoclonal antibodies, MAbs, 2013, 5(4), 576–586 CrossRef.
- S. Lippold, S. Nicolardi, M. Wuhrer and D. Falck, Proteoform-Resolved FcγRIIIa Binding Assay for Fab Glycosylated Monoclonal Antibodies Achieved by Affinity Chromatography Mass Spectrometry of Fc Moieties, Front. Chem., 2019, 7, 698 CrossRef CAS.
- S. Lippold, S. Nicolardi, E. Domínguez-Vega, A.-K. Heidenreich, G. Vidarsson and D. Reusch, et al., Glycoform-resolved FcγRIIIa affinity chromatography–mass spectrometry, mAbs, 2019, 11(7), 1191–1196 CrossRef CAS.
- I. K. Ventouri, D. B. A. Malheiro, R. L. C. Voeten, S. Kok, M. Honing and G. W. Somsen, et al., Probing Protein Denaturation during Size-Exclusion Chromatography Using Native Mass Spectrometry, Anal. Chem., 2020, 92(6), 4292–4300 CrossRef CAS.
- B. Chen, Z. Lin, A. J. Alpert, C. Fu, Q. Zhang and W. A. Pritts, et al., Online Hydrophobic Interaction Chromatography–Mass Spectrometry for the Analysis of Intact Monoclonal Antibodies, Anal. Chem., 2018, 90(12), 7135–7138 CrossRef CAS.
- B. W. J. Pirok, D. R. Stoll and P. J. Schoenmakers, Recent Developments in Two-Dimensional Liquid Chromatography: Fundamental Improvements for Practical Applications, Anal. Chem., 2019, 91(1), 240–263 CrossRef CAS.
- D. R. Stoll and P. W. Carr, Two-Dimensional Liquid Chromatography: A State of the Art Tutorial, Anal. Chem., 2017, 89(1), 519–531 CrossRef CAS.
- D. Stoll, J. Danforth, K. Zhang and A. Beck, Characterization of therapeutic antibodies and related products by two-dimensional liquid chromatography coupled with UV absorbance and mass spectrometric detection, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1032, 51–60 CrossRef CAS.
- D. R. Stoll, K. Zhang, G. O. Staples, A. Beck, K. Zhang and G. O. Staples, et al., Recent Advances in Two-Dimensional Liquid Chromatography for the Characterization of Monoclonal Antibodies and Other Therapeutic Proteins, Adv. Chromatogr., 2019, 29–70 CrossRef CAS , Available from: https://www.taylorfrancis.com/.
- A. C. A. Ehkirch, The Current Status and Future of Two- and Multidimensional Liquid Chromatography in Pharmaceutical R&D and QC. [cited 2020 Apr 28]. Available from: http://www.chromatographyonline.com/current-status-and-future-two-and-multidimensional-liquid-chromatography-pharmaceutical-rd-and-qc.
- S. Kozlowski and P. Swann, Current and future issues in the manufacturing and development of monoclonal antibodies, Adv. Drug Delivery Rev., 2006, 58(5–6), 707–722 CrossRef CAS.
- M. Gilar and U. D. Neue, Peak capacity in gradient reversed-phase liquid chromatography of biopolymers: Theoretical and practical implications for the separation of oligonucleotides, J. Chromatogr., A, 2007, 1169(1), 139–150 CrossRef CAS.
- L. R. Snyder, M. A. Stadalius and M. A. Quarry, Gradient elution in reversed-phase HPLC-separation of macromolecules, Anal. Chem., 1983, 55(14), 1412A–1430A CAS.
- C. R. Bupp and M. J. Wirth, Making Sharper Peaks for Reverse-Phase Liquid Chromatography of Proteins, Annu. Rev. Anal. Chem., 2020, 13(1), 363–380 CrossRef CAS.
- B. W. J. Pirok, A. F. G. Gargano and P. J. Schoenmakers, Optimizing separations in online comprehensive two-dimensional liquid chromatography, J. Sep. Sci., 2018, 41(1), 68–98 CrossRef CAS.
- D. R. Stoll, K. Shoykhet, P. Petersson and S. Buckenmaier, Active Solvent Modulation: A Valve-Based Approach To Improve Separation Compatibility in Two-Dimensional Liquid Chromatography, Anal. Chem., 2017, 89(17), 9260–9267 CrossRef CAS.
- R. J. Vonk, A. F. G. Gargano, E. Davydova, H. L. Dekker, S. Eeltink and L. J. de Koning, et al., Comprehensive Two-Dimensional Liquid Chromatography with Stationary-Phase-Assisted Modulation Coupled to High-Resolution Mass Spectrometry Applied to Proteome Analysis of Saccharomyces cerevisiae, Anal. Chem., 2015, 87(10), 5387–5394 CrossRef CAS.
- A. Ehkirch, V. D'Atri, F. Rouviere, O. Hernandez-Alba, A. Goyon and O. Colas, et al., An Online Four-Dimensional HIC×SEC-IM×MS Methodology for Proof-of-Concept Characterization of Antibody Drug Conjugates, Anal. Chem., 2018, 90(3), 1578–1586 CrossRef CAS.
- A. Ehkirch, A. Goyon, O. Hernandez-Alba, F. Rouviere, V. D'Atri and C. Dreyfus, et al., A Novel Online Four-Dimensional SEC×SEC-IM×MS Methodology for Characterization of Monoclonal Antibody Size Variants, Anal. Chem., 2018, 90(23), 13929–13937 CrossRef CAS.
- M. Sarrut, A. Corgier, S. Fekete, D. Guillarme, D. Lascoux and M.-C. Janin-Bussat, et al., Analysis of antibody-drug conjugates by comprehensive on-line two-dimensional hydrophobic interaction chromatography x reversed phase liquid chromatography hyphenated to high resolution mass spectrometry. I - Optimization of separation conditions, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1032, 103–111 CrossRef CAS.
- K. Sandra and P. Sandra, The opportunities of 2D-LC in the analysis of monoclonal antibodies, Bioanalysis, 2015, 7(22), 2843–2847 CrossRef CAS.
- A. Delobel, 2D-LC–MS for the Analysis of Monoclonal Antibodies and Antibody–Drug Conjugates in a Regulated Environment. [cited 2020 Apr 30]. Available from: http://www.spectroscopyonline.com/2d-lc-ms-analysis-monoclonal-antibodies-and-antibody-drug-conjugates-regulated-environment-0.
- M. Alvarez, G. Tremintin, J. Wang, M. Eng, Y.-H. Kao and J. Jeong, et al., On-line characterization of monoclonal antibody variants by liquid chromatography–mass spectrometry operating in a two-dimensional format, Anal. Biochem., 2011, 419(1), 17–25 CrossRef CAS.
- J. J. Gilroy and C. M. Eakin, Characterization of drug load variants in a thiol linked antibody-drug conjugate using multidimensional chromatography, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1060, 182–189 CrossRef CAS.
- J. H. Thompson, W. K. Chung, M. Zhu, L. Tie, Y. Lu and N. Aboulaich, et al., Improved detection of host cell proteins (HCPs) in a mammalian cell-derived antibody drug using liquid chromatography/mass spectrometry in conjunction with an HCP-enrichment strategy: LC/MS for host cell protein analysis, Rapid Commun. Mass Spectrom., 2014, 28(8), 855–860 CrossRef CAS.
- F. Li, A. Shen and A. Amanullah, Cell Culture Processes in Monoclonal Antibody Production, in Pharmaceutical Sciences Encyclopedia, ed. S. C. Gad, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2013, p. 1–38. DOI:10.1002/9780470571224.pse506.
- N. Alt, T. Y. Zhang, P. Motchnik, R. Taticek, V. Quarmby and T. Schlothauer, et al., Determination of critical quality attributes for monoclonal antibodies using quality by design principles, Biologicals, 2016, 44(5), 291–305 CrossRef CAS.
- V. Háda, A. Bagdi, Z. Bihari, S. B. Timári, Á. Fizil and C. Szántay, Recent advancements, challenges, and practical considerations in the mass spectrometry-based analytics of protein biotherapeutics: A viewpoint from the biosimilar industry, J. Pharm. Biomed. Anal., 2018, 161, 214–238 CrossRef.
- S. Hober, K. Nord and M. Linhult, Protein A chromatography for antibody purification, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 848(1), 40–47 CrossRef CAS.
- Z. D. Dunn, J. Desai, G. M. Leme, D. R. Stoll and D. D. Richardson, Rapid two-dimensional Protein-A size exclusion chromatography of monoclonal antibodies for titer and aggregation measurements from harvested cell culture fluid samples, mAbs, 2020, 12(1), 1702263 CrossRef.
- K. Sandra, M. Steenbeke, I. Vandenheede, G. Vanhoenacker and P. Sandra, The versatility of heart-cutting and comprehensive two-dimensional liquid chromatography in monoclonal antibody clone selection, J. Chromatogr., A, 2017, 1523, 283–292 CrossRef CAS.
- A. Williams, E. K. Read, C. D. Agarabi, S. Lute and K. A. Brorson, Automated 2D-HPLC method for characterization of protein aggregation with in-line fraction collection device, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2017, 1046, 122–130 CrossRef CAS.
- S. Rao and C. Pohl, Reversible interference of Fe3 + with monoclonal antibody analysis in cation exchange columns, Anal. Biochem., 2011, 409(2), 293–295 CrossRef CAS.
- B. Rivera, Bioinert Versus Biocompatible: The Benefits of Different Column Materials in Liquid Chromatography Separations. [cited 2020 Apr 30]. Available from: http://www.chromatographyonline.com/bioinert-versus-biocompatible-benefits-different-column-materials-liquid-chromatography-separations?pageID=2.
- Phenomenex. Seeing the Big Picture: Multidimensional Liquid Chromatography for Biotherapeutic Characterization. [cited 2020 Apr 30]. Available from: http://www.chromatographyonline.com/seeing-big-picture-multidimensional-liquid-chromatography-biotherapeutic-characterization.
- A. S. Rathore, Roadmap for implementation of quality by design (QbD) for biotechnology products, Trends Biotechnol., 2009, 27(9), 546–553 CrossRef CAS.
- S. Rogstad, H. Yan, X. Wang, D. Powers, K. Brorson and B. Damdinsuren, et al., Multi-Attribute Method for Quality Control of Therapeutic Proteins, Anal. Chem., 2019, 91(22), 14170–14177 CAS.
- Characterization of Protein Therapeutics using Mass Spectrometry, ed. G. Chen, Springer US, Boston, MA, 2013. DOI:10.1007/978-1-4419-7862-2.
- R. S. Rogers, M. Abernathy, D. D. Richardson, J. C. Rouse, J. B. Sperry and P. Swann, et al., A View on the Importance of “Multi-Attribute Method” for Measuring Purity of Biopharmaceuticals and Improving Overall Control Strategy, AAPS J., 2018, 20(1), 7 CrossRef.
- Y. Zhang and J. Guo, Characterization and QC of biopharmaceuticals by MS-based “multi-attribute method”: advantages and challenges, Bioanalysis, 2017, 9(6), 499–502 CrossRef CAS.
- Y. Li, Y. Huang, J. Ferrant, Y. Lyubarskaya, Y. Zhang and S. Li, et al., Assessing in vivo dynamics of multiple quality attributes from a therapeutic IgG4 monoclonal antibody circulating in cynomolgus monkey, mAbs, 2016, 8(5), 961–968 CrossRef CAS.
- T. Wang, L. Chu, W. Li, K. Lawson, I. Apostol and T. Eris, Application of a Quantitative LC–MS Multiattribute Method for Monitoring Site-Specific Glycan Heterogeneity on a Monoclonal Antibody Containing Two N-Linked Glycosylation Sites, Anal. Chem., 2017, 89(6), 3562–3567 CrossRef CAS.
- I. Sokolowska, J. Mo, J. Dong, M. J. Lewis and P. Hu, Subunit mass analysis for monitoring antibody oxidation, mAbs, 2017, 9(3), 498–505 CrossRef CAS.
- B. Bobály, S. Fleury-Souverain, A. Beck, J.-L. Veuthey, D. Guillarme and S. Fekete, Current possibilities of liquid chromatography for the characterization of antibody-drug conjugates, J. Pharm. Biomed. Anal., 2018, 147, 493–505 CrossRef.
- J. Camperi, A. J. Schick, D. Guillarme and A. T. Wecksler, Utilizing Multidimensional LC-MS for Hydroxyl Radical Footprinting Analysis, LCGC North Am., 2019, 37(11), 36–39 CAS.
- J. Camperi, V. Pichon and N. Delaunay, Separation Methods hyphenated to Mass Spectrometry for the Characterization of the Protein Glycosylation at the Intact Level, J. Pharm. Biomed. Anal., 2019, 112921 Search PubMed.
- Y. Wang, X. Li, Y.-H. Liu, D. Richardson, H. Li and M. Shameem, et al., Simultaneous monitoring of oxidation, deamidation, isomerization, and glycosylation of monoclonal antibodies by liquid chromatography-mass spectrometry method with ultrafast tryptic digestion, mAbs, 2016, 8(8), 1477–1486 CrossRef CAS.
- S. Millán-Martín, C. Jakes, S. Carillo, T. Buchanan, M. Guender and D. B. Kristensen, et al., Inter-laboratory study of an optimised peptide mapping workflow using automated trypsin digestion for monitoring monoclonal antibody product quality attributes, Anal. Bioanal. Chem., 2020, 412(25), 6833–6848 CrossRef.
- D. Chelius, G. Xiao, A. C. Nichols, A. Vizel, B. He and T. M. Dillon, et al., Automated tryptic digestion procedure for HPLC/MS/MS peptide mapping of immunoglobulin gamma antibodies in pharmaceutics, J. Pharm. Biomed. Anal., 2008, 47(2), 285–294 CrossRef CAS.
- C. Qian, B. Niu, R. B. Jimenez, J. Wang, M. Albarghouthi and X. Chen, Fully Automated Peptide Mapping Protocol for Multi-Attribute Method by Liquid Chromatography–Tandem Mass Spectroscopy with a High-Throughput Robotic Liquid Handling System, bioRxiv, 2020, 2020.01.10.902338.
- J. Richardson, B. Shah, G. Xiao, P. V. Bondarenko and Z. Zhang, Automated in-solution protein digestion using a commonly available high-performance liquid chromatography autosampler, Anal. Biochem., 2011, 411(2), 284–291 CrossRef CAS.
- J. Lippincott, E. Hess and I. Apostol, Mapping of Recombinant Hemoglobin Using Immobilized Trypsin Cartridges, Anal. Biochem., 1997, 252(2), 314–325 CrossRef CAS.
- J. Samskog, D. Bylund, S. P. Jacobsson and K. E. Markides, Miniaturized on-line proteolysis–capillary liquid chromatography–mass spectrometry for peptide mapping of lactate dehydrogenase, J. Chromatogr., A, 2003, 998(1), 83–91 CrossRef CAS.
- T. Nadler, C. Blackburn, J. Mark, N. Gordon, F. E. Regnier and G. Vella, Automated proteolytic mapping of proteins, J. Chromatogr., A, 1996, 743(1), 91–98 CrossRef CAS.
- K. Sandra, Further pushing the limits of LC and LC-MS in biopharmaceutical analysis. ISC 2018•32nd International Symposium on Chromatography, 2018 Sep 24, Cannes Mandelieu, France.
- B. Bobály, M. Lauber, A. Beck, D. Guillarme and S. Fekete, Utility of a high coverage phenyl-bonding and wide-pore superficially porous particle for the analysis of monoclonal antibodies and related products, J. Chromatogr., A, 2018, 1549, 63–76 CrossRef.
- J. C. Han and G. Y. Han, A Procedure for Quantitative Determination of Tris(2-Carboxyethyl)phosphine, an Odorless Reducing Agent More Stable and Effective Than Dithiothreitol, Anal. Biochem., 1994, 220(1), 5–10 CrossRef CAS.
- A. Xu, H. S. Kim, S. Estee, S. ViaJar, W. J. Galush and A. Gill, et al., Susceptibility of Antibody CDR Residues to Chemical Modifications Can Be Revealed Prior to Antibody Humanization and Aid in the Lead Selection Process, Mol. Pharmaceutics, 2018, 15(10), 4529–4537 CrossRef CAS.
- M. E. Alam, G. V. Barnett, T. R. Slaney, C. G. Starr, T. K. Das and P. M. Tessier, Deamidation Can Compromise Antibody Colloidal Stability and Enhance Aggregation in a pH-Dependent Manner, Mol. Pharmaceutics, 2019, 16(5), 1939–1949 CrossRef CAS.
- M. E. Alam, T. R. Slaney, L. Wu, T. K. Das, S. Kar and G. V. Barnett, et al., Unique Impacts of Methionine Oxidation, Tryptophan Oxidation, and Asparagine Deamidation on Antibody Stability and Aggregation, J. Pharm. Sci., 2020, 109(1), 656–669 CrossRef CAS.
- J. Camperi, L. Dai, D. Guillarme and C. Stella, Fast and automated characterization of monoclonal antibody minor variants from cell cultures by combined Protein-A and multi-dimensional LC/MS methodologies, Anal. Chem., 2020, 92, 8506–8513 CrossRef CAS.
- J. Camperi, D. Guillarme and C. Stella, Targeted bottom-up characterization of recombinant monoclonal antibodies by multi-dimensional LC/MS, Anal. Chem., 2020, 92, 13420–13426 CrossRef CAS.
- J. Sjögren, F. Olsson and A. Beck, Rapid and improved characterization of therapeutic antibodies and antibody related products using IdeS digestion and subunit analysis, Analyst, 2016, 141(11), 3114–3125 RSC.
- A. Beck, F. Debaene, H. Diemer, E. Wagner-Rousset, O. Colas and A. V. Dorsselaer, et al., Cutting-edge mass spectrometry characterization of originator, biosimilar and biobetter antibodies: Biosimilar antibodies, J. Mass Spectrom., 2015, 50(2), 285–297 CrossRef CAS.
- A. Beck, E. Wagner-Rousset, D. Ayoub, A. Van Dorsselaer and S. Sanglier-Cianférani, Characterization of Therapeutic Antibodies and Related Products, Anal. Chem., 2013, 85(2), 715–736 CrossRef CAS.
- V. D'Atri, L. Nováková, S. Fekete, D. Stoll, M. Lauber and A. Beck, et al., Orthogonal Middle-up Approaches for Characterization of the Glycan Heterogeneity of Etanercept by Hydrophilic Interaction Chromatography Coupled to High-Resolution Mass Spectrometry, Anal. Chem., 2019, 91(1), 873–880 CrossRef.
- V. D'Atri, S. Fekete, A. Beck, M. Lauber and D. Guillarme, Hydrophilic Interaction Chromatography Hyphenated with Mass Spectrometry: A Powerful Analytical Tool for the Comparison of Originator and Biosimilar Therapeutic Monoclonal Antibodies at the Middle-up Level of Analysis, Anal. Chem., 2017, 89(3), 2086–2092 CrossRef.
- L. Fornelli, D. Ayoub, K. Aizikov, A. Beck and Y. O. Tsybin, Middle-Down Analysis of Monoclonal Antibodies with Electron Transfer Dissociation Orbitrap Fourier Transform Mass Spectrometry, Anal. Chem., 2014, 86(6), 3005–3012 CrossRef CAS.
- J. Dai and Y. Zhang, A Middle-Up Approach with Online Capillary Isoelectric Focusing/Mass Spectrometry for In-Depth Characterization of Cetuximab Charge Heterogeneity, Anal. Chem., 2018, 90(24), 14527–14534 CrossRef CAS.
- J. Giorgetti, A. Beck, E. Leize-Wagner and Y.-N. François, Combination of intact, middle-up and bottom-up levels to characterize 7 therapeutic monoclonal antibodies by capillary electrophoresis – Mass spectrometry, J. Pharm. Biomed. Anal., 2020, 182, 113107 CrossRef CAS.
- A. M. Belov, L. Zang, R. Sebastiano, M. R. Santos, D. R. Bush and B. L. Karger, et al., Complementary middle-down and intact monoclonal antibody proteoform characterization by capillary zone electrophoresis - mass spectrometry, Electrophoresis, 2018, 39(16), 2069–2082 CrossRef CAS.
- M. Biacchi, R. Gahoual, N. Said, A. Beck, E. Leize-Wagner and Y.-N. François, Glycoform Separation and Characterization of Cetuximab Variants by Middle-up Off-Line Capillary Zone Electrophoresis-UV/Electrospray Ionization-MS, Anal. Chem., 2015, 87(12), 6240–6250 CrossRef CAS.
- D. Ayoub, W. Jabs, A. Resemann, W. Evers, C. Evans and L. Main, et al., Correct primary structure assessment and extensive glyco-profiling of cetuximab by a combination of intact, middle-up, middle-down and bottom-up ESI and MALDI mass spectrometry techniques, mAbs, 2013, 5(5), 699–710 CrossRef.
- J. Camperi, D. Guillarme, M. Lei and C. Stella, Automated middle-up approach for the characterization of biotherapeutic products by combining on-line hinge-specific digestion with RPLC-HRMS analysis, J. Pharm. Biomed. Anal., 2020, 113130 CrossRef CAS.
- V. Faid, Y. Leblanc, N. Bihoreau and G. Chevreux, Middle-up analysis of monoclonal antibodies after combined IgdE and IdeS hinge proteolysis: Investigation of free sulfhydryls, J. Pharm. Biomed. Anal., 2018, 149, 541–546 CrossRef CAS.
- B. Chen, Z. Lin, Y. Zhu, Y. Jin, E. Larson and Q. Xu, et al., Middle-Down Multi-Attribute Analysis of Antibody-Drug Conjugates with Electron Transfer Dissociation, Anal. Chem., 2019, 91(18), 11661–11669 CrossRef CAS.
- B. L. Duivelshof, W. Jiskoot, A. Beck, J.-L. Veuthey, D. Guillarme and V. D'Atri, Glycosylation of biosimilars: Recent advances in analytical characterization and clinical implications, Anal. Chim. Acta, 2019, 1089, 1–18 CrossRef CAS.
- B. Q. Tran, C. Barton, J. Feng, A. Sandjong, S. H. Yoon and S. Awasthi, et al., Glycosylation characterization of therapeutic mAbs by top- and middle-down mass spectrometry, Data in Brief, 2016, 6, 68–76 CrossRef.
- J. Camperi, V. Pichon, T. Fournier and N. Delaunay, First profiling in hydrophilic interaction liquid chromatography of intact human chorionic gonadotropin isoforms, J. Pharm. Biomed. Anal., 2019, 174, 495–499 CrossRef CAS.
- J. Camperi, A. Combès, T. Fournier, V. Pichon and N. Delaunay, Analysis of the human chorionic gonadotropin protein at the intact level by HILIC-MS and comparison with RPLC-MS, Anal. Bioanal. Chem., 2020, 412, 4423–4432 CrossRef CAS.
- V. D'Atri, S. Fekete, D. Stoll, M. Lauber, A. Beck and D. Guillarme, Characterization of an antibody-drug conjugate by hydrophilic interaction chromatography coupled to mass spectrometry, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2018, 1080, 37–41 CrossRef.
- M. Sorensen, D. C. Harmes, D. R. Stoll, G. O. Staples, S. Fekete and D. Guillarme, et al., Comparison of originator and biosimilar therapeutic monoclonal antibodies using comprehensive two-dimensional liquid chromatography coupled with time-of-flight mass spectrometry, mAbs, 2016, 8(7), 1224–1234 CrossRef CAS.
- D. R. Stoll, H. R. Lhotka, D. C. Harmes, B. Madigan, J. J. Hsiao and G. O. Staples, High resolution two-dimensional liquid chromatography coupled with mass spectrometry for robust and sensitive characterization of therapeutic antibodies at the peptide level, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2019, 1134–1135, 121832 CrossRef CAS.
- Y. E. M. van der Burgt, D. P. A. Kilgour, Y. O. Tsybin, K. Srzentić, L. Fornelli and A. Beck, et al., Structural Analysis of Monoclonal Antibodies by Ultrahigh Resolution MALDI In-Source Decay FT-ICR Mass Spectrometry, Anal. Chem., 2019, 91(3), 2079–2085 CrossRef CAS.
- J. Moelleken, M. Endesfelder, C. Gassner, S. Lingke, S. Tomaschek and O. Tyshchuk, et al., GingisKHAN™ protease cleavage allows a high-throughput antibody to Fab conversion enabling direct functional assessment during lead identification of human monoclonal and bispecific IgG1 antibodies, mAbs, 2017, 9(7), 1076–1087 CrossRef CAS.
- J. Sjögren, L. Andersson, M. Mejàre and F. Olsson, Generating and Purifying Fab Fragments from Human and Mouse IgG Using the Bacterial Enzymes IdeS, SpeB and Kgp, in Bacterial Pathogenesis, ed. P. Nordenfelt and M. Collin, Springer New York, New York, NY, 2017, pp. 319–329. (Methods in Molecular Biology, vol. 1535). DOI: DOI:10.1007/978-1-4939-6673-8_21.
- S. Perchepied, N. Eskenazi, C. Giangrande, J. Camperi, T. Fournier and J. Vinh, et al., Development of Immobilized Enzyme Reactors for the characterization of the glycosylation heterogeneity of a protein, Talanta, 2020, 206, 120171 CrossRef CAS.
- M.-C. Janin-Bussat, L. Tonini, C. Huillet, O. Colas, C. Klinguer-Hamour and N. Corvaïa, et al., Cetuximab Fab and Fc N-Glycan Fast Characterization Using IdeS Digestion and Liquid Chromatography Coupled to Electrospray Ionization Mass Spectrometry, in Glycosylation Engineering of Biopharmaceuticals, ed. A. Beck, Humana Press, Totowa, NJ, 2013, pp. 93–113. DOI:10.1007/978-1-62703-327-5_7.
- B. L. Duivelshof, A. Murisier, J. Camperi, S. Fekete, A. Beck and D. Guillarme, et al., Therapeutic Fc–fusion proteins: Current analytical strategies, J. Sep. Sci., 2020 DOI:10.1002/jssc.202000765.
- Y. Leblanc, V. Faid, M. A. Lauber, Q. Wang, N. Bihoreau and G. Chevreux, A generic method for intact and subunit level characterization of mAb charge variants by native mass spectrometry, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2019, 121814 CrossRef CAS.
- R. Haselberg, T. De Vijlder, R. Heukers, M. J. Smit, E. P. Romijn and G. W. Somsen, et al., Heterogeneity assessment of antibody-derived therapeutics at the intact and middle-up level by low-flow sheathless capillary electrophoresis-mass spectrometry, Anal. Chim. Acta, 2018, 1044, 181–190 CrossRef CAS.
- N. Said, R. Gahoual, L. Kuhn, A. Beck, Y.-N. François and E. Leize-Wagner, Structural characterization of antibody drug conjugate by a combination of intact, middle-up and bottom-up techniques using sheathless capillary electrophoresis – Tandem mass spectrometry as nanoESI infusion platform and separation method, Anal. Chim. Acta, 2016, 918, 50–59 CrossRef CAS.
Footnote |
| † These authors contributed equally to the manuscript. |
| This journal is © The Royal Society of Chemistry 2021 |