
Lipid-based mesophases as matrices for nanoscale reactions
Livia
Salvati Manni
 *ab,
Wye-Khay
Fong
*c and
Raffaele
Mezzenga
*a
*ab,
Wye-Khay
Fong
*c and
Raffaele
Mezzenga
*a
aDepartment of Health Sciences and Technology, Swiss Federal Institute of Technology in Zurich, 8092 Zurich, Switzerland. E-mail: raffaele.mezzenga@hest.ethz.ch
bSchool of Chemistry and University of Sydney Nano Institute, The University of Sydney, NSW 2006, Australia. E-mail: livia.salvatimani@sydney.edu.au
cDiscipline of Chemistry, School of Environmental and Life Sciences, University of Newcastle, Callaghan 2308, NSW, Australia. E-mail: khay.fong@newcastle.edu.au
First published on 16th April 2020
Abstract
Lipidic mesophases are versatile bioorganic materials that have been effectively employed as nanoscale matrices for membrane protein crystallization, drug delivery and as food emulsifiers over the last 30 years. In this review, the focus is upon studies that have employed non-lamellar lipid mesophases as matrices for organic, inorganic and enzymatic reactions. The ability of lipidic mesophases to incorporate hydrophilic, amphiphilic and hydrophobic molecules, together with the high interfacial area of the lipidic cubic and inverse hexagonal phases has been exploited in heterogeneous catalysis as well as for enzyme immobilization. The unique nanostructure of these mesophases is the driving force behind their ability to act as templates for synthesis, resulting in the creation of highly ordered polymeric and inorganic materials with complex geometries.
 Livia Salvati Manni | Livia Salvati Manni's research interests focus on the self-assembly of amphiphilic molecules in water and ionic liquids. She obtained her PhD from University of Zurich in 2017, where she was funded by the Forschungskredit grant. Between 2017 and 2019 she was a postdoctoral researcher at ETH Zurich. Currently, she is a postdoctoral researcher in Chemistry at the University of Sydney. She has recently been awarded a SNF early postdoc mobility grant to develop her research program into the use of lipidic self-assembled nanostructures in biochemistry. |
 Wye-Khay Fong | Dr Wye-Khay Fong is a Lecturer in Chemistry in the Faculty of Science at the University of Newcastle. She obtained a PhD in Pharmacy at Monash University in 2013 and subsequently held two prestigious postdoctoral fellowships at ETH Zurich and Monash, and later at the Adolphe Merkle Institute. Her research focuses on the self-assembly of lipids and their use as novel matrices for biomedical applications, where she exploits the brightness of synchrotron science, light scattering and synthesis of novel amphiphiles. |
 Raffaele Mezzenga | Raffaele Mezzenga research focuses on the fundamental understanding of self-assembly principles in proteins, polymers, liquid crystals, food and biological colloidal systems. He has co-authored more than 300 publications. He has been a visiting Professor at the Helsinki University of Technology (now Aalto University), RMIT Melbourne, Monash University and NTU Singapore. His work has been recognized by several prestigious international distinctions such as the 2017 Fellowship and the 2013 John H. Dillon Medal of the of the American Physical Society, the 2013 Biomacromolecules/Macromolecules Young Investigator Award of the American Chemical Society, the 2011 American Oil Chemists’ Society Young Scientist Research Award, and the 2004 Swiss Science National Foundation Professorship Award. |
Introduction
Lipid molecules are a key component of life and are involved in a large number of natural phenomena. Lipid molecules are characterised by an amphiphilic nature and consequently are subject to a hydrophobic force in presence of water, resulting in their self-assembly into different mesophases.1Most importantly, lipid bilayers are the main constituent of cell membranes and are responsible for creating compartments that differentiate organelles and vesicles in cells. For this reason, artificially formed lipidic bilayers are good models for biological membranes. As biomimetic materials, lipid-based matrices constitute powerful tools to simulate cell environments in hosting, stabilising, and compartmentalising biomacromolecules like proteins or DNA. Consequently, these nanomaterials have been utilised as matrices for applications such as protein crystallisation,2,3 drug delivery4,5 and as reactions on the nanoscale.6,7
The use of microemulsions in organic and inorganic synthesis was established in the 1980s, where microemulsions were shown to improve reaction rates and yield by firstly, enhancing the solubility of reagents and secondly, compartmentalising reagents into hydrophilic and hydrophobic phases.8 Compartmentalisation significantly increases the surface area available for reaction and can favourably orient reagents; additionally, surfactants can act as phase transfer agents, allowing for selective partitioning between these compartments.9
Over the past few decades, scientists have discovered new methods to synthesise new mesoscopic materials with regular nanostructures in processes inspired by nature such as self-organisation, replication and metamorphosis.10,11 These bioinspired methods are largely based on using the elaborate structures formed by the self-assembly of surfactants as a templates to create complex, two and three-dimensional materials.
The focus of this review is on the use of lipidic mesophases as matrices for reactions on the nanoscale. The large majority of research reported in the literature has utilised non-lipidic surfactants. The physicochemical behaviour and the applications of these surfactants are outside the scope of this review, however some of these studies will be mentioned in order to contextualise and generalise the studies pertinent to lipidic matrices. Studies that utilise mesophases formed by other types of surfactants are excellently reviewed in ref. 8 and 12–16.
Lipid self-assembly in water is mediated by weak intermolecular forces: hydrogen bonds, electrostatic interactions, hydrophobic interactions, van der Waals interactions, and water-mediated hydrogen bonds.17–19 In isolation, these forces are relatively insignificant, however, when combined, they influence the interaction between lipidic molecules and water and so govern their mesoscopic structural conformation.18 Lipidic mesophases are lyotropic liquid crystals that have ordered long-range nanostructure of different morphologies. The formation of different morphologies with high surface area at the hydrophilic–hydrophobic interface, together with their biomimetic nature makes lipidic nanostructures particularly appealing for heterogeneous catalysis and as templating matrices. In recent years, a better understanding of the behaviour of lipids in water has enhanced their use as nanoreactors for a plethora of reactions: organic and inorganic reactions, as well as model systems to study natural biomineralisation and enzymatic reactions.
Depending of the chemical structure of the lipid20–22 and external conditions such as temperature, hydration level, pH, pressure etc., lipids can assemble into different geometries: from flat lipidic bilayers to curved and more complex topologies (Fig. 1).23 The most well characterised and utilised lipid mesophase is the lamellar phase (Lα). For example, single lipid bilayers arranged in closed vesicles (liposomes) were the first matrix utilised as a nanomedicine in biomedical applications.24,25 The lamellar phase can also exist as multilamellar systems, i.e. a sequence of planar layers intercalated with water slabs.26
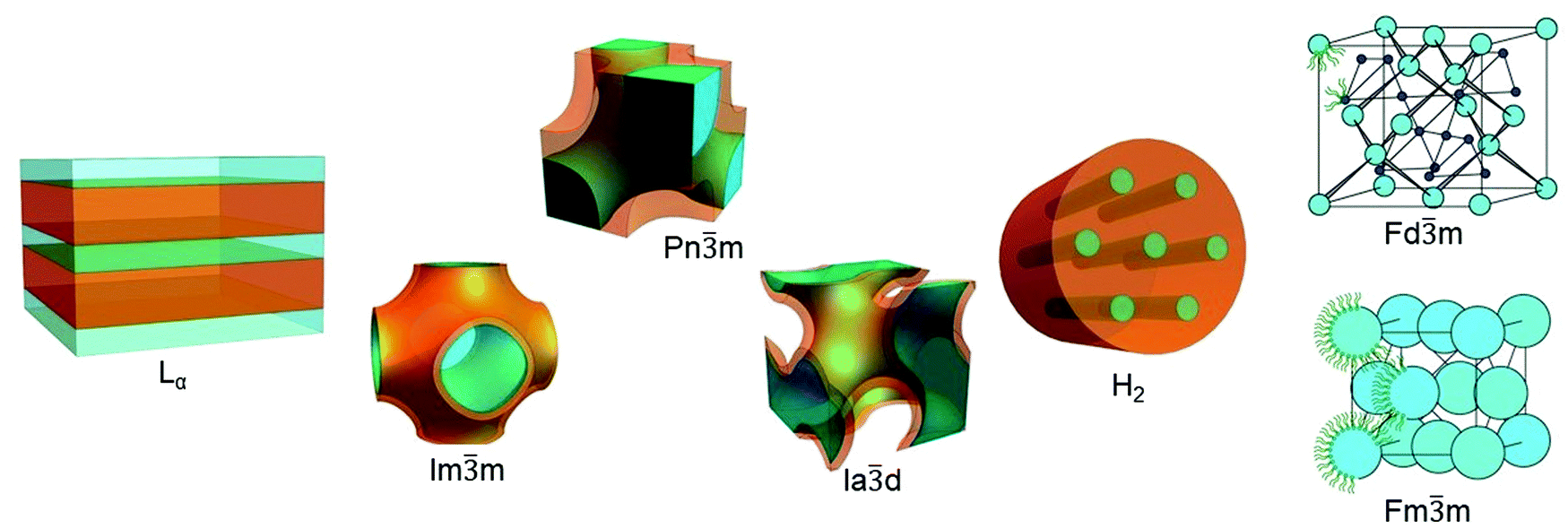 | ||
Fig. 1 Lipidic mesophases. From left to right lamellar phase Lα (adapted with permission from ref. 35), cubic phases Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, Pnm, Iad (adapted with permission from ref. 36), inverse hexagonal phase H2 (adapted with permission from ref. 35), and micellar cubic phases Fdm and Fmm (adapted with permission from ref. 37). m, Pnm, Iad (adapted with permission from ref. 36), inverse hexagonal phase H2 (adapted with permission from ref. 35), and micellar cubic phases Fdm and Fmm (adapted with permission from ref. 37). | ||
More recently, non-lamellar phases have attracted significant attention for their higher surface area to volume ratio and for their complex topologies; this review will focus on the use of the bicontinuous cubic phases and hexagonal phases as templates for nanoscale reactions. In the bicontinuous lipidic cubic phases (LCP), the lipid bilayer is organised following minimal surface topologies that delineate two identical and not interconnected water channels.27 The most common bicontinuous surface topologies used as matrices for nanoreactors are described by the crystallographic space groups Pnm, Imm and Iad.28 Sponge phases, which can be considered as a ‘disordered cubic phase’ as they are formed of water channels which have no long-range order,18,29 have also been used but are less common. In hexagonal phases, lipidic monolayers are packed in micellar cylinders organised in a hexagonal pattern. They can be distinguished into direct and inverse phases according with the relative orientation of the polar head and the lipidic tail: if the tail is inside of the micelle and the polar head group is outside, the hexagonal phase is direct (H1 phase);30 if the lipidic tails are on the outside of the micelle, resulting in the formation of water cylinders packed in the hexagonal array, the phase is defined inverse (H2 phase).31 Additionally, the LCP and H2 mesophases are stable in an excess of water, which allows for the formation of structured particles with similar nanoscopic properties to the bulk phase. Particles which have a bicontinuous cubic structure or inverse hexagonal structure are known as cubosomes and hexosomes respectively and have been proposed as matrices for biomedical applications.32–34
Lipidic mesophases have complex nanostructures which display long range order. Techniques for their identification is outside the scope of this review, however, readers are referred to the following references. By far, the most common technique for the identification of the structure and dimension of mesophases has been small angle X-ray scattering (SAXS) and cryogenic electron microscopy.38–41 Synchrotron light sources have provided the advantage of identifying kinetics of dynamic phase changes.42,43 Small angle neutron scattering (SANS)43,44 and nuclear magnetic resonance (NMR) spectroscopy45–48 techniques49 provide the ability to identify the location of incorporated molecules around the interface of the mesophases. Rheology has also been shown to identify different symmetries depending on their viscoelastic signatures.50
Controlling the dimensions of both the hydrophilic and hydrophobic domains affords exquisite control over the movements of the reactants, thus providing a unique matrix for hosting both organic and inorganic reactions.
The nanostructures of the different mesophases determine the properties of the material, for example, rate of diffusion, rheological properties and geometries, which gives them distinctive advantages for application in structural biology, materials science and functional foods.5,35,51 Structure determines direction, partitioning and rate of flow of reagents and products in the mesophase.36,52,53 This property has been manipulated for the controlled release of drugs from both bulk54–56 and dispersed33,57,58 lipidic mesophases. Numerous studies have shown how lipid shape, composition and external environment influence the self-assembly, which has been used to great effect in on-demand drug delivery.59
Control over the phase behaviour of these self-assembled matrices is difficult to achieve, but critical when employing these materials as reaction matrices. Lipid formulations have been designed to stabilise certain phases over a large range of temperature or composition, and different additives have been optimized to respond to defined stimuli to switch between structures and to control the lattice parameters.4,60,61 Synthetic lipids have been designed to enlarge the library of available lipids that assemble in not lamellar phases. Unfortunately, a priori phase behaviour is still not achievable.62
The control over the hydration level is of pivotal importance in controlling the size of the water channel. In the last few years, LCP with both swollen, highly hydrated water channels, as well as LCP with water channels with diameters smaller than 1 nm have been reported. This exquisite control over the size of the water channels has been achieved using charged lipidic additives,63,64 controlled evaporation rate,65 and the employment of synthetic lipids.49 These advances have the potential to result in the creation of increasingly sophisticated systems where the confinement effect of the matrix on the reagents can be finely tuned spanning several orders of magnitude.
The review is divided into section which reflect the main types of reactions which have been achieved in lipidic mesophases: organic reactions, polymerisation, enzymatic reactions, ordered mesoporous materials (silica) and metallic nanoparticles and films. The final section will highlight advances in the field of self-assembled lipid systems and how they can be utilised to enhance reactions confined in lipidic mesophases.
Organic reactions
Surfactant self-assembly has been exploited to achieve new heterogeneous catalytic systems able to compartmentalise reagents in order to create nano- and microreactors for organic synthesis.8 While vesicles (liposomes) and lipid microemulsions have been largely used as reaction matrices,8,66–69 non-lamellar phases have been largely unexplored.In 2000, Vauthey et al.70 studied Maillard reactions in structured fluids composed of monoacylglycerols, using these self-assembled structures as microreactors. It was shown that the type of reactions that could progress in LCP were more wide-ranging and achieved a higher yield compare to both inverse micellar (L2) microemulsions and pure water. This “cubic catalyst” effect was attributed to: firstly, the partitioning of reagents to the water–lipid interface in both the LCP and L2 resulting in the compartmentation of the reagents into a surfactant “cage”; secondly, the higher interfacial area of LCP resulted in increased yield and rate over the microemulsions, but the authors assert that is difficult to establish the role of the lipidic layer curvature in these two different models.
More recently, Duss et al.71 reported an asymmetric aldol reaction in LCP where the lipidic bilayer was used as a scaffold doped with a molecular catalyst. The guest catalyst was an amphiphilic molecule with a soluble active site attached to a lipidic tail, designed and synthesised in order to be semi-immobilised at the interface (Fig. 2). Bulk cubic phases (Pnm) with different water channel sizes, and cubosomes with different geometry i.e. Pnm and Imm were tested as reactive recyclable scaffolds. An aldol reaction between water-soluble aldehydes and cyclohexanone was employed to achieve good organo-catalytic efficiency as well as modulation of the diastereo- and enantioselectivity. The reaction in LCP proceeded with higher yields than in water with a water-soluble catalyst. However, the best results were achieved with a lipidic catalyst that self-assembled into polydisperse micelles.
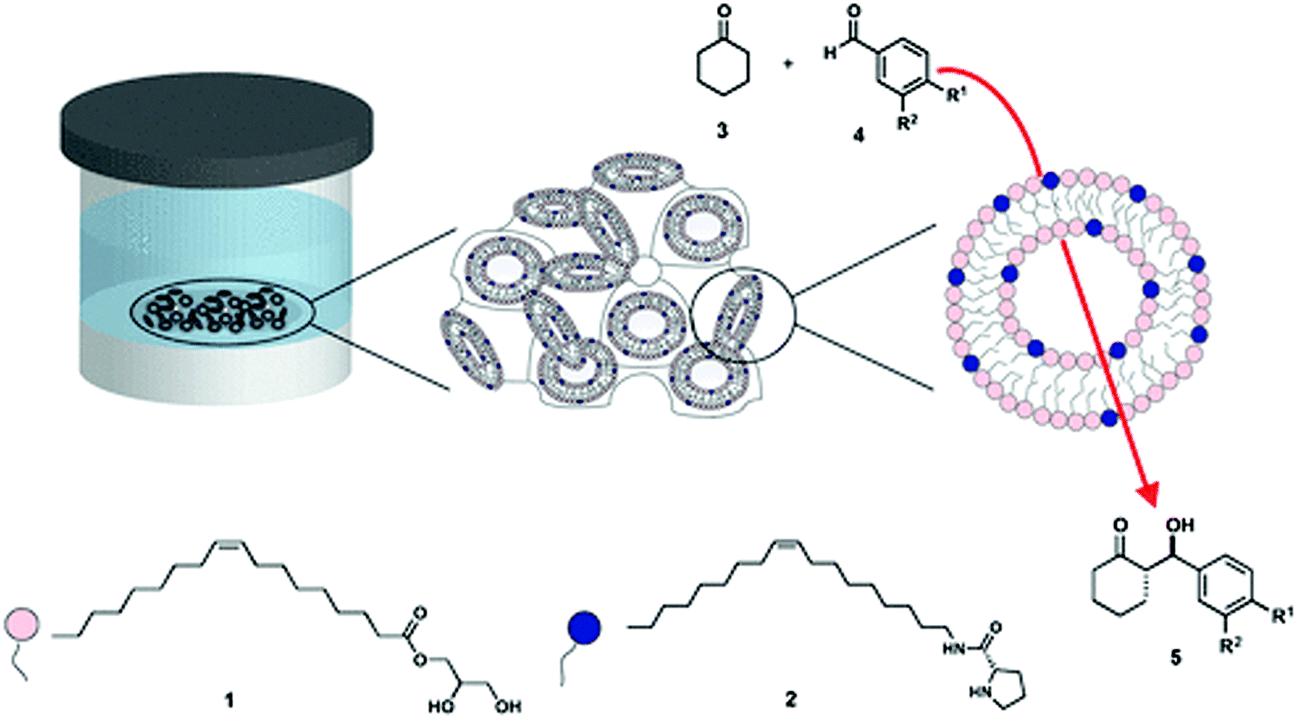 | ||
| Fig. 2 Schematic representation of the LCP scaffold constituted of monoolein (1, pink) in excess of water, doped with a catalytic lipid (2, dark blue) developed by Duss et al.71 to achieve diastereo- and enantioselectivity in model aldol reactions (3 and 4 to yield 5). Figure reproduced with permission from ref. 71. | ||
The same authors in a different study,6 inspired by the pioneering work of Puvvada et al.,72 employed palladium nanoparticles grown and embedded in different bulk and dispersed mesophases as catalysts in Suzuki–Miyaura cross-coupling reactions. In this case, the lipidic scaffold had three roles: as a reducing agent for Pd2+, as a template to grow monodispersed particles, and as nanostructured support for the organic reaction. The addition of cholesterol swelled the water channels of the mesophase, thereby enhancing the organic reaction. These innovative studies confirm that non-lamellar mesophase have a consistent potential as “green” reaction scaffolds and for catalysis in soft nanoconfined space as they are formed by cheap, biocompatible materials.
Polymerisation
Chemically engineered lipids have been synthesised to control their assembly into different geometries. The dynamic ability to switch between mesophases in response to an external stimulus is of fundamental importance for several applications but, at the same time, could impede the use of such systems in situations in which one phase is desired and it is difficult or impossible to control the environmental conditions, such as pH, temperature or hydration level. Thus, the polymerisation of self-assembled lipids would preserve qualities such as topology of the lipidic bilayer, nanostructure and diffusion of solutes within the mesophase. A few reviews address the molecular design of lipids with different polymerisable motifs and their position within the lipidic tail.73,74Similar materials have been successfully prepared via polymerisation of detergents that form cubic or hexagonal structures, and that were cross linked retaining the geometry and resulting in stable polymeric materials with an organized template.13,14,75,76 In recent years, the development of new applications of nanostructured organic materials has increased the popularity of polymerisable surfactants (mostly detergents) to create films77 and membranes78,79 with high surface area and uniform pore sizes. Examples of possible applications are in water filtration,80,81 heterogenous catalysis,82 enhancement of photoluminescence83 and the creation of breathable, vapor-resistant membranes.84
In lipid-based nanomaterials, two main types of reactions have been demonstrated: the polymerisation of the lipidic tails of lipids in the non-lamellar mesophases, and the polymerisation of soluble compounds within the water channel.
The stabilisation of different lipidic mesophases through the polymerisation of lipidic chains has been shown to preserve the topology of the self-assembled systems. O’Brien et al. succeeded in cross-linking the lipids around the water channels of both LCP and H2, thereby maintaining the desired morphology in a broad range of temperatures. For this purpose, lipids like phosphoethanolamines,85,86 mono- and diacylglycerols87,88 were designed and synthesised with a polymerisable dienoyl group on the lipidic chain in proximity to the polar headgroup. In cases in which the polymerisation was overly efficient, the cross liked material was insoluble in solvents.85,86 In other cases where the lipids were not completely cross linked, the thermal stability of the original Iad phase was improved.87 It is expected that the ability of the crossed-linked mesophases to be solvent resistant and stable in refluxing organic solvents, opens up the possibility to use these structures as reactors in presence of different organic solvents, exploiting their innate high surface area and ability to confine substrates.82,85,86
A polymerised hexagonal columnar nanostructure (Fig. 3A) was achieved by Feng et al.,89 utilising a core molecular template of plant-derived polymerisable fatty acids. After the vertical alignment of the cylinders by a surface-confinement method, the lipids were polymerised into an oriented hexagonal phase, in order to create membranes with a controlled porosity, long-range order and a nanometric channel size.
 | ||
| Fig. 3 (A) Core templated, oriented H2 phase, successfully polymerised in a stable matrix. The orientation and the geometry are retained after the removal of the core templating agent as visible from the SAXS patterns. Figure adapted with permission from ref. 89. (B) TEM picture with scale bar of 100 nm of a portion of a polymerised cubosome. Figure adapted with permission from ref. 90. | ||
Polymerised cubosomes were achieved by Yang et al. utilising a synthesised polymerisable monoacylglycerol, 3-(2,4,13-(E,E)-tetradecatrienoyl)-sn-glycerol (Fig. 3B). The bulk phase was dispersed in the presence of Poloxamer 407 in excess of water to form cubosomes and then polymerised via photo or redox initiators. The resulting crossed linked cubosomes were resistant to excess of surfactant solutions such as Triton X-100.90 This stability is the main innovation of these crossed linked particles as they are expected to be useful nanostructured materials for applications that require stability in solvents or in broad temperature ranges.91
In order to capture the complex topology of the liquid crystalline mesophases, a few studies have focused on the polymerisation of soluble monomer within the water channels. The attempts to preserve the structure and obtain a polymer with the geometry and the characteristics of the water channels of the original liquid crystalline moulds have led to new materials with complex nanostructures. The use of lyotropic liquid crystals as templating matrices for the polymerisation of monomers within the water channels has been largely explored for non-lipidic surfactant based mesophases.14,92,93 We hypothesise that this can be easily extrapolated for lipidic matrices as many of the general finding valid for surfactants can be applied to lipid-based amphiphiles. A few studies report the photochemical94,95 and thermal96 polymerisation of acrylamide in detergent based mesophases with different nanostructure i.e. H2, LCP and Lα.
Ström et al.97,98 added hydrophobic and hydrophilic polymerisable additives, divinylbenzene or acrylamide, to the surfactant–water mixture. The polymerisation was induced by photoinitiation and produced cubic polymers, with retention of the structure and the water mobility, as confirmed by SAXS and NMR self-diffusion studies. Quantification of the amount of reactant polymerised by dissolution of the lipidic phase and the isolation of the polymer were successfully achieved, however structure identification of the surfactant free polymer by SAXS was unsuccessful.
Additionally, studies on surfactant phase behaviour suggest that the aggregation of the polymer is localised in the grain boundaries.94 For this reason, it is essential to find an area of the phase diagram in which the chosen liquid crystalline phase is stable in presence and absence of the monomer in order to macroscopically avoid phase separation. Systematic studies show that the structure of the obtained polymer depends on the type of monomer, crosslinking density, kinetics, monomer and surfactant concentration, and can be varied systematically to result in different pore morphologies and a broad size range varying from hundreds of nanometers to micrometers.95,96,99 As can be seen, many molecular and macroscopic aspects must be taken into account in order to obtain a nanostructured material with long-range order.
Enzymatic reactions
The immobilisation of biological enzymes onto substrates has been used to control and improve the preservation, locality, recovery, observed activity, specificity of selectivity of an enzyme for application in both synthesis and biosensing.100 Many enzymes that are central to physiological functions are centred at the interface or within the cell membrane.101,102 As such, it has been established that the nanostructure, fluidity and symmetry of the lipid membrane influence enzyme functionality,103–105 where more open structures (i.e. LCPs) allow for a higher specific activity of the enzyme.106 The viability of enzymes has been established in LCPs. A wide range of hydrophilic107–110 and membrane111–118 proteins in their native form have been incorporated into the cubic phase, whilst maintaining the bicontinuous cubic nanostructure and protein activity for prolonged times.119,120 Native proteins have also been successfully reconstituted in inverse hexagonal phases.121 In most cases, enzymes immobilised in LCPs maintain classical Michaelis–Menten kinetics, but can differ due to the confinement of enzyme in the mesophase (Fig. 4A).7 The ability to maintain the function of these catalytic centres led to their development as matrices for the detection of biological components.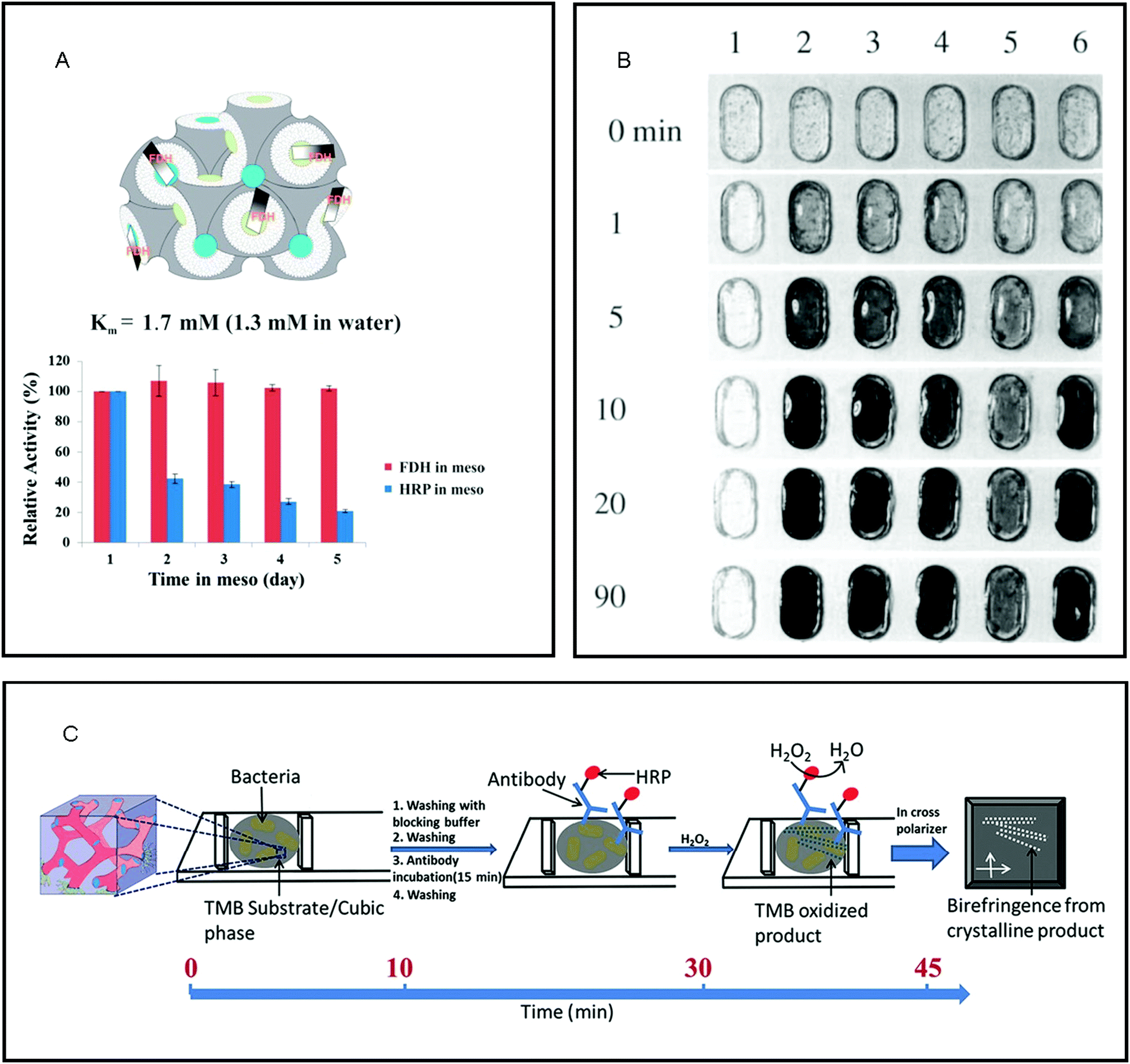 | ||
| Fig. 4 The properties of lipid mesophases makes them ideal matrices for the immobilisation of enzymes for reactions on the nanoscale. (A) the amphiphilic nature of the lipid bilayer of the cubic phase maintains the native structure and therefore activity of membrane proteins such as fructose dehydrogenase (FDH) as compared to a water soluble enzyme, horseradish peroxidase (HRP). Reproduced with permission from ref. 112. As the lipidic cubic phase is isotropic, reactions catalysed by immobilised enzymes that cause a change in optical properties make them ideal for biosensing for: (B) the detection of glucose where 1 – buffer solution, 2 – 10 mM, 3 – 5 mM, 4 – 2.5 mM, 5 – 1 mM and & 6 – blood plasma (5.1 mM glucose); reproduced with permission from ref. 122, and (C) schematic of bacteria detection via development of birefringent crystals in LCP. Reproduced with permission from ref. 131. | ||
The immobilisation of enzymes into LCP and the detection of the subsequent formation of substrate have been engineered for use as matrices for biosensors. The first use of these materials for biosensing was reported by Wallin et al. who immobilised glucose oxidase into ethoxylated fatty alcohol (C16–18(OCH2CH2)80OH) and water as a simple glucose monitor of both solutions and blood plasma, where the enzyme immobilised within the mesophase maintained a higher amount of its original activity than in solution (Fig. 4B).122 Razumas et al. immobilised various enzymes into a monoolein cubic phase for the amperometric detection of β-D-glucose and L-lactate, and pH-sensitive urea and creatinine.123 In these types of amperometric biosensors, the aqueous domains of the lipidic cubic phase matrix provide a double benefit: firstly, these water-soluble proteins can be confined in a stable manner, and secondly, the substrates and products can easily flow into and out of the bulk water depending on the size and topology of the water channels.112,124 Since their introduction, enzymes confined into LCP have been utilised in a vast range of electrochemical biosensors125 including glucose,126 fructose,127 cholesterol,128 dioxygen monitoring129 and hydrogen peroxide.130
As a rapid, point-of-care analysis tool, the enzyme-catalysed crystallisation of substrate within an optically isotropic cubic phase has provided a simple visual indicator for a positive or negative biochemical reaction. LCP are isotropic in nature and so do not exhibit birefringence when observed in between cross-polarisers, whereas crystallised substrates exhibit birefringence. Vallooran et al. exploited this effect in order to detect biomarkers (glucose and cholesterol) and antigens (E. Coli., Malaria, HIV and Ebola).131 The presence of these biomarkers resulted in the formation of hydrogen peroxide (H2O2) via the presence of oxidase enzymes; H2O2 then activated a cascade reaction via the activation of an additional immobilised enzyme, horseradish peroxidase, which subsequently activated the crystallisation of different organic substrates. The crystallisation of the organic substrates resulted in quantifiable amounts of birefringence and so a positive result. In the case of the detection of antigens, horseradish peroxidase was directly attached to the corresponding antibody, where detection of the antigens in the presence of H2O2 resulted in crystallisation and thus, a quantifiable amount of birefringence (Fig. 4C). As matrices for biosensing, the unique nanostructure of the LCP has three key functions: maintenance of the native structure of enzyme, flow of substrate and product to/from the enzyme and ideal optical properties.112,124,132
Recently, enzymes immobilised in lipidic mesophases have also been used as matrices for the immobilisation of enzymes for synthesis. The synthesis of carbohydrates by aldolase confined in LCP was found to be three-times more efficient than in solution, attributed to both the ability of the protein to maintain its native conformation in the LCP nanostructure, and the compartmentalisation of the substrate and products.133 In this case, the structure of the LCP plays a key role in the maintenance and enhancement of activity of enzymes immobilised within the mesophase.
Reactions catalysed by enzymes encapsulated in dispersed liquid crystalline phases have also been demonstrated. The immobilisation of a large enzyme, β-galactosidase, was achieved within a lipid sponge phase in order to enhance the stability and the hydrolysis of lactose to its component monosaccharides.134 To our knowledge, enzyme immobilisation in lipidic cubosomes has not been achieved, however, La et al. utilised polymeric cubosomes formed by the self-assembly of amphiphilic block copolymers to covalently immobilise maleimide-activated horseradish peroxidase (HRP, 44.1 kDa); the kinetic action of HRP was found to be comparable to values of HRP in free solution.135 In the same way as the bulk, the structure of dispersed mesophases maintains the activity of the enzyme and influences the flow of substrate and product.
Ordered mesoporous materials (silica)
Mesoporous materials possess attractive properties, such as high surface areas, tuneable pore sizes and shapes, various structures, and a multitude of compositions. Of these materials, silica is one of the most frequently studied inorganic frameworks due to their tetra-connected covalent bonds. Mesoporous silica has been extensively studied for application in a wide range of fields, ranging from biomedical (drug delivery and biosensors) to industrial (catalysis, sorption, gas sensing, ion exchange, optics, photovoltaics, coatings and separation materials) as they are chemically inert, thermally stable, inexpensive and have high surface area which makes them promising materials.136–138 Of particular interest are mesoporous silica nanomaterials; these materials contain pores with diameters between 2 and 50 nm, resulting in the augmentation of the surface area thus enabling new functions.139,140In general, mesoporous silica matrices are produced by the following reaction: a solubilised silica source, e.g. tetraethylorthosilicate (TEOS) or silicate solutions, forms the solid silica matrix via the formation of sequential Si–O–Si bonds in a polycondensation process (eqn (1)).
| –Si(OH) + –Si(OH) ⇌ –Si–O–Si–(s) + H2O(l) | (1) |
The ability to form mesoporous silica matrices with long range order was established by using liquid crystal structures formed by concentrated surfactant systems as templates in a patent in 1969 leading to the birth of the field in 1992.141,142 The seminal paper by Kresge et al. in 1992 was highlighted in the collection, “150 years of Nature” as one of the “10 extraordinary Nature papers” as it is considered a significant breakthrough in materials science which triggered an explosion of research into mesoporous materials.142,143 Since then, it has been shown that the structure, thickness and porosity of the mesoporous silica is intimately linked to the physicochemical properties and concentration of the surfactant.142,144–148 The assembly process is controlled by silica precursors condensing around the polar headgroups of the surfactants, resulting in the formation of layers of silica at the interface of the micelles (Fig. 5A). This process is controlled by electrostatic interactions between the inorganic ions in solution, the charged surfactant head groups and inorganic counterions.149–151 As a consequence of the silica forming, phase transitions have also been observed in the templating matrix.152 It has been proposed that the final morphology of the mesoporous materials is influenced by the competition mainly between the free energy of mesostructure self-assembly (ΔG) and the colloidal surface free energy (F).153
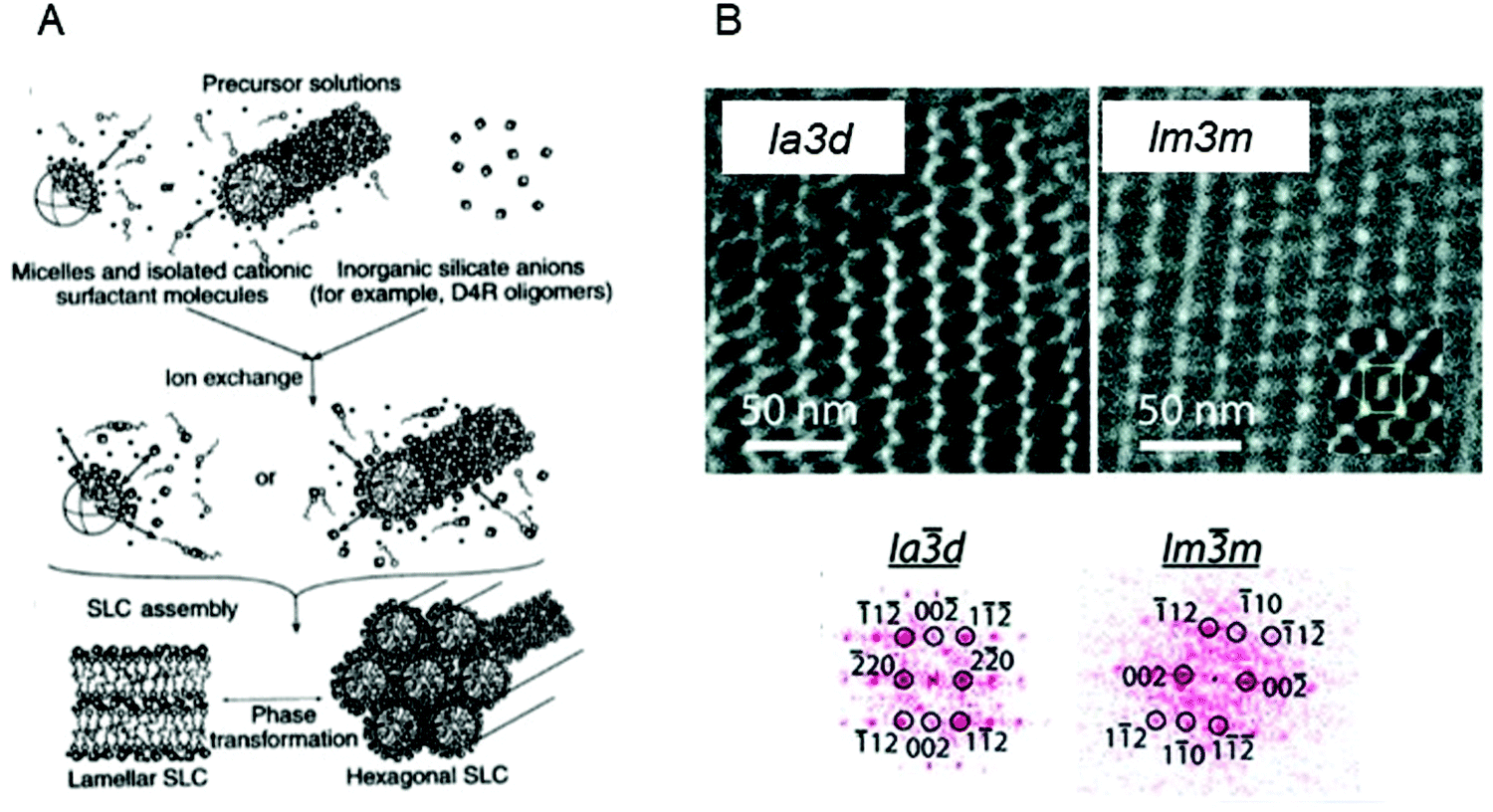 | ||
| Fig. 5 (A) General mechanism of mesoporous silica templating. Adapted with permission from ref. 151. B (top) TEM image of mesoporous silica films formed using MO based templates; (bottom) 2D Fourier transforms for data in top images. Adapted with permission from ref. 172. | ||
Many of the reported syntheses have utilised lyotropic liquid crystals formed by aqueous mixtures of cationic surfactants, e.g. cetyltrimethylammonium,151,154–156 amphiphilic block copolymers,157,158 non-ionic surfactants,159–163 and anionic surfactants.164 The resulting structure of the mesoporous silica can be tuned through the use of cosolvents, surfactant blends and by changing the aqueous environment such as pH and salt concentration.165,166 The nanostructured silica matrices can subsequently be used as a support matrix for metallic nanocomposites.167 A number of very comprehensive reviews on the formation of mesoporous silica via surfactant templating provide a good insight into their formation.168–170
Few studies have utilised lipid-based liquid crystals as a template for creating mesoporous materials. To our knowledge, the first mesoporous material formed by lipidic cubic phase templating was reported in 2001: the formation of mesoporous alumina with a pore size of 38 Å, which corresponds well to the pore size of the aqueous channels formed by monoolein and water (35 Å).171 Spicer et al. report large 100 μm silica cubosomes formed by coating a large cubosome with silica sol, evaporating the water to consolidate the silica, and dissolving the cubosome, though, the composition of the cubosome template remains unclear.32 Moreover, in both these brief communications, it is not clear whether the resulting material maintained the original symmetry of the lipidic mesophase. In 2011, mesoporous silica films were achieved by Dunphry et al. through the use of LCP (MO) templates and evaporation-induced self-assembly (Fig. 5B), where the pore size of both the LCP and subsequent silica film was enlarged through the use of cetyltrimethylammonium bromide.172 The ability to modulate the water channel size by the incorporation of charged additives resulting in increased mesoporous silica pore size demonstrates the advantage of using LCP as matrices for templating.
Phospholipid-based mesophases have also been utilised. A twisted multilamellar hybrid silica mesophase was obtained by Seddon et al., in which the lipid bilayers were intercalated with thin sheets of amorphous silica.173 This hybrid mesostructure involved the co-assembly of phospholipids containing polymerizable diacetylinic groups with the in situ synthesis of silica by acid hydrolysis and condensation of TEOS. In a systematic study, a wide range of phospholipids of varying lipid tail length, number of acyl groups and headgroup type, was utilised to produce silica and hybrid thin films with 1D, 2D and 3D long range order.174 The acyl tails were found to be the main determinant of whether total phase separation between silica and lipid during film assembly occurred; and the headgroups determined the suppression or enhancement of siloxane framework reactivity. The phase separation issue was overcome through the use of a co-surfactant175 or lipids without a phosphatidyl headgroup.172 Mixed phospholipid/surfactant systems have also been used to form mesoporous silica with sponge phase structure for hosting enzymes.176
As can be seen by these studies, controlling the composition of the lipidic mesophase ultimately determines the structure and porosity of the resulting mesoporous silica matrices.
Metallic nanoparticles and films
As seen in the preceding sections, the biomimetic method of utilising surfactant-based soft templating for the crystallisation of inorganic composites in order to generate new organic–inorganic materials was established in the late 80's and early 90's.10,177 Most of the work in the field has employed non-lipidic amphiphiles to generate the lyotropic liquid crystalline templates.178–180 Possibly the most explored method is the formation of films via electrodeposition of metal salts in the aqueous compartment of non-lipidic surfactant based liquid crystalline phases, resulting in metal films with uniform nanostructures and high specific surface areas; these materials have been employed for catalysis,181 enhanced electrical connectivity,179 and for the formation of semiconducting superlattices.180 In some cases, a double templating strategy was employed to produce highly ordered materials, where a hard silica template was generated from a surfactant template and then used to create mesoporous metals through deposition.182–184 For a more focused review on mesoporous metals by soft templating the reader is directed to Yamauchi et al.16Lipidic mesophases have been less explored. In 1994, Puvvada et al.72 employed LCP as a reactive matrix to produce palladium (Pd) nanoparticles. The particles were synthesised in the lipidic matrix via the reduction of a Pd salt to Pd0 by the glycerol head group of monoolein. In this way, the resulting Pd nanoparticles that were formed inside the water channels possessed a diameter similar to the water channel size, with excellent monodispersity. In two recent studies, Akbar et al.185,186 developed two methods for producing platinum (Pt) nanowires (Fig. 5). In the first study, an electrodeposition method in a Fdm cubic phase was used to create a highly homogenous nanowire network with a high electrochemical surface area as determined by cyclovoltammetry (Fig. 6A–C).185 In a subsequent study, ultrathin uniform platinum nanowires were obtained by using chemical reduction via galvanic displacement in a H2 phase lipidic matrix composed mainly of phytantriol (Fig. 6D).186
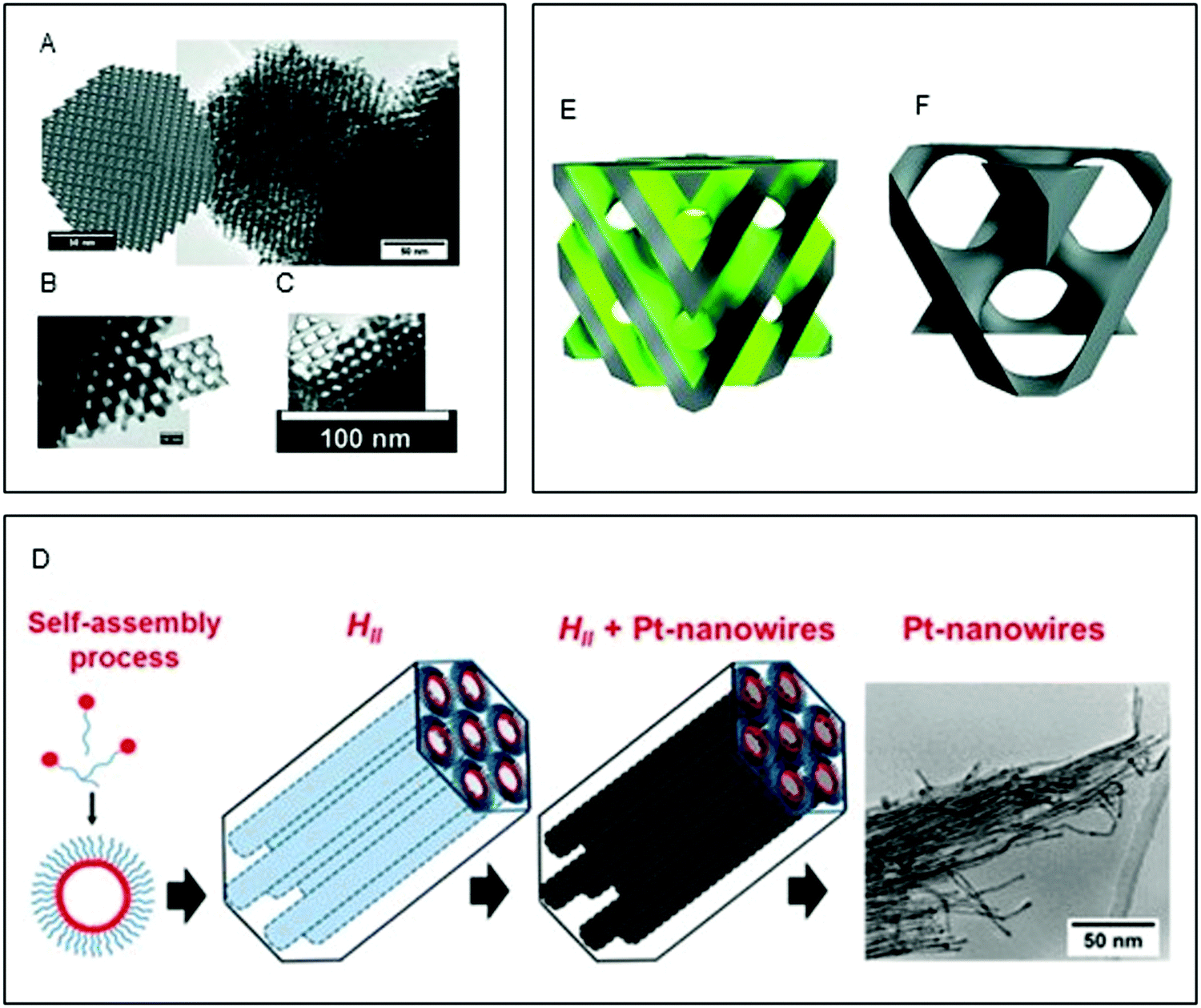 | ||
| Fig. 6 (A–C) Simulated Fdm phases superimposed onto TEM images of nanostructured Pt Fdm single diamond electrodes with scale bars of 50 nm (a), 10 nm (b), and 100 nm (c). Adapted with permission from Akbar et al.185 (D) schematic of the templating mechanism. Formation of the self-assembled lipidic mesophase, followed by metal electrodeposition and formation of the Pt nanowires. Reproduced with permission from ref. 186; (E and F) structure of the Pnm templating mesophase, where the grey region is occupied by the lipid bilayer and the green one by water, and corresponding Imm platinum structure obtained by electrodeposition. Reproduced with permission from ref. 187. | ||
Lipidic mesophases on solid supports have been used to produce nanostructured metallic films. By using a Pnm lipidic cubic phase template on a gold foil support, Richardson et al.187 obtained a Fdm nanostructured aligned metallic film via the deposition of Pt. The authors attribute this change in geometry to the capping of one of the channels of the cubic phase near the gold surface, and the subsequent asymmetric deposition of Pt in one of the two water channels as confirmed by their in situ SAXS study.188 They also explain the alignment of the resulting platinum film as an interfacial effect on the lipidic cubic phase deposited on the solid support: while the lipids domains are disordered, an alignment is achieved at the interface, resulting in aligned platinum films.187 The same change in geometry from a Pnm lipidic cubic phase to a Fdm templated metallic (Bi2Te3) nanostructure was observed from Burton et al. in the formation of bismuth sulphide 3D structures.189
Perspectives
Lipidic mesophases are readily available and environmentally friendly matrices that can be implemented as natural and reusable matrices for reactions on the nanoscale. Recent advances in the field have demonstrated that the structure of mesophase is easily tunable by controlling environmental factors (temperature, hydration, pH) as well as molecular dimensions of the surfactant; a trait that can be used advantageously to enhance nanoscale reactions. It is envisaged that the exquisite control over structure and size of the water channels will lead to the creation of new nanostructured materials and consequently, better control over organic and enzymatic reactions. The highly regular structure of the matrix will allow also for mass production with fewer defects and a low impact on environment.One limitation to the development of nanostructured particles is the control over particle size. It is envisaged that the use of templating will overcome this shortcoming.
Proof of concept studies have shown the effect of different diffusion rates and mesophase geometry on reactions in lipidic mesophases, however, the effect of the water channel size and lipidic bilayer curvature on the rate and yield of in-meso reactions is still to be optimised and understood, especially for systems where confinement plays an important role. The employment of these matrices for enantioselective reactions opens the question: what is the effect of confinement on enantio- and diasteroselectivity? The generalisability of these rules is of pivotal importance for the implementation of mesophases in scalable reactions.
The implementation of lipids for the creation of nanostructured polymers is still under development, and the optimisation of the process to avoid phase separation during polymerisation seems to be the most important factor to achieve regular structures that can conserve the template geometry. On the other hand, the possibility to cross-link lipids in a certain geometry opens up the opportunity to implement these materials in applications that require fixed geometry and lattice parameters, including their use as reactors for reactions in organic solvents.
Cubic and hexagonal structures have unique optical and electrical properties. The development of structurally aligned mesophases that have defined geometry, high porosity and surface area pave the way to the possibility of direct fabrication of novel 2D and 3D structures, and thus, may bring new opportunities and advances in the fields of electrocatalysis and optoelectronics.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Livia Salvati Manni acknowledges financial support from the Professor J. A. Schofield fund.References
- M. B. Goldlust, A. Luzzati and L. Levine, J. Bacteriol., 1968, 96, 1961–1968 CrossRef CAS PubMed.
- E. M. Landau and J. P. Rosenbusch, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14532–14535 CrossRef CAS PubMed.
- M. Caffrey and V. Cherezov, Nat. Protoc., 2009, 4, 706–731 CrossRef CAS PubMed.
- W.-K. Fong, T. Hanley and B. J. Boyd, J. Controlled Release, 2009, 135, 218–226 CrossRef CAS PubMed.
- R. Mezzenga, J. M. Seddon, C. J. Drummond, B. J. Boyd, G. E. Schröder-Turk and L. Sagalowicz, Adv. Mater., 2019, 31, 1900818 CrossRef PubMed.
- M. Duss, J. J. Vallooran, L. Salvati Manni, N. Kieliger, S. Handschin, R. Mezzenga, H. J. Jessen and E. M. Landau, Langmuir, 2019, 35, 120–127 CrossRef CAS PubMed.
- W. Sun, J. J. Vallooran, A. Zabara and R. Mezzenga, Nanoscale, 2014, 6, 6853–6859 RSC.
- K. Holmberg, Adv. Colloid Interface Sci., 1994, 51, 137–174 CrossRef CAS.
- A. S. Chhatre, R. A. Joshi and B. D. Kulkarni, J. Colloid Interface Sci., 1993, 158, 183–187 CrossRef CAS.
- S. Mann and G. A. Ozin, Nature, 1996, 382, 313–318 CrossRef CAS.
- S. I. Stupp and P. V. Braun, Science, 1997, 277, 1242–1248 CrossRef CAS PubMed.
- B. H. H. Jones and T. P. P. Lodge, Polym. J., 2012, 44, 131–146 CrossRef CAS.
- S. A. Miller, J. H. Ding and D. L. Gin, Curr. Opin. Colloid Interface Sci., 1999, 4, 338–347 CrossRef CAS.
- F. Yan and J. Texter, Adv. Colloid Interface Sci., 2006, 128–130, 27–35 CrossRef CAS PubMed.
- C. Wang, D. Chen and X. Jiao, Sci. Technol. Adv. Mater., 2009, 10, 023001 CrossRef PubMed.
- Y. Yamauchi and K. Kuroda, Chem. – Asian J., 2008, 3, 664–676 CrossRef CAS PubMed.
- C. Tanford, Science, 1978, 200, 1012–1018 CrossRef CAS PubMed.
- J. M. Seddon and R. H. Templer, in Handbook of Biological Physics, ed. R. Lipowski and E. Sackman, Elsevier, Amsterdam, 1995, pp. 97–160 Search PubMed.
- W. B. Lee, R. Mezzenga and G. H. Fredrickson, Phys. Rev. Lett., 2007, 99, 187801 CrossRef PubMed.
- M. E. Kimchi-Schwartz, L. Martin, E. Flurin, C. Aron, M. Kulkarni, H. E. Tureci and I. Siddiqi, Phys. Rev. Lett., 2016, 116, 240503 CrossRef CAS PubMed.
- C. V. Kulkarni, T. Y. Tang, A. M. Seddon, J. M. Seddon, O. Ces and R. H. Templer, Soft Matter, 2010, 6, 3191–3194 RSC.
- C. Fong, T. Le and C. J. Drummond, Chem. Soc. Rev., 2012, 41, 1297–1322 RSC.
- C. V. Kulkarni, W. Wachter, G. Iglesias-Salto, S. Engelskirchen and S. Ahualli, Phys. Chem. Chem. Phys., 2011, 13, 3004–3021 RSC.
- U. Bulbake, S. Doppalapudi, N. Kommineni and W. Khan, Pharmaceutics, 2017, 9, 12 CrossRef PubMed.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- M. Rappolt, Advances in Biomembranes and Lipid Self-Assembly, Academic Press, 2019, vol. 29, pp. 1–21 Search PubMed.
- S. T. Hyde, Curr. Opin. Solid State Mater. Sci., 1996, 1, 653–662 CrossRef CAS.
- S. T. Hyde, Handbook of Applied Surface and Colloid Chemistry, Wiley, 2001, ch.16, pp. 299–321 Search PubMed.
- V. Luzzati, H. Delacroix, A. Gulik, T. Gulik-Krzywicki, P. Mariani and R. Vargas, Stud. Surf. Sci. Catal., 2004, 148, 17–40 CrossRef CAS.
- P. A. Winsor, Chem. Rev., 2002, 68, 1–40 CrossRef.
- J. M. Seddon, Biochim. Biophys. Acta, Rev. Bioenerg., 1990, 1031, 1–69 CrossRef CAS.
- P. T. Spicer, Curr. Opin. Colloid Interface Sci., 2005, 10, 274–279 CrossRef CAS.
- J. Zhai, C. Fong, N. Tran and C. J. Drummond, ACS Nano, 2019, 13, 6178–6206 CrossRef CAS PubMed.
- H. M. G. Barriga, M. N. Holme and M. M. Stevens, Angew. Chem., Int. Ed., 2019, 58, 2958–2978 CrossRef CAS PubMed.
- S. Assenza and R. Mezzenga, Nat. Rev. Phys., 2019, 1, 551–566 CrossRef.
- S. Assenza and R. Mezzenga, J. Chem. Phys., 2018, 148, 054902 CrossRef PubMed.
- I. Martiel, L. Sagalowicz and R. Mezzenga, Langmuir, 2013, 29, 15805–15812 CrossRef CAS PubMed.
- Y. Da Dong and B. J. Boyd, Int. J. Pharm., 2011, 417, 101–111 CrossRef PubMed.
- B. J. Boyd, Y. Da Dong and T. Rades, J. Liposome Res., 2009, 19, 12–28 CrossRef CAS.
- A. Yaghmur and O. Glatter, Adv. Colloid Interface Sci., 2009, 147–148, 333–342 CrossRef CAS PubMed.
- D. Demurtas, P. Guichard, I. Martiel, R. Mezzenga, C. Hébert and L. Sagalowicz, Nat. Commun., 2015, 6, 8915 CrossRef CAS PubMed.
- D. B. Warren, M. U. Anby, A. Hawley and B. J. Boyd, Langmuir, 2011, 27, 9528–9534 CrossRef CAS PubMed.
- B. Angelov, A. Angelova, V. M. Garamus, G. Lebas, S. Lesieur, M. Ollivon, S. S. Funari, R. Willumeit and P. Couvreur, J. Am. Chem. Soc., 2007, 129, 13474–13479 CrossRef CAS PubMed.
- L. Van’t Hag, L. De Campo, N. Tran, A. Sokolova, R. Trenker, M. E. Call, M. J. Call, C. J. Garvey, A. E. Leung, T. A. Darwish, A. Krause-Heuer, R. Knott, T. G. Meikle, C. J. Drummond, R. Mezzenga and C. E. Conn, Langmuir, 2019, 35, 8344–8356 Search PubMed.
- P. O. Eriksson and G. Lindblom, Biophys. J., 1993, 64, 129–136 CrossRef CAS PubMed.
- T. G. Meikle, A. Sethi, D. W. Keizer, J. J. Babon, F. Separovic, P. R. Gooley, C. E. Conn and S. Yao, J. Magn. Reson., 2019, 305, 146–151 CrossRef CAS PubMed.
- Y. Yang, H. Yao and M. Hong, J. Phys. Chem. B, 2015, 119, 4993–5001 CrossRef CAS PubMed.
- M. F. Etter, D. Dellenbach, A. Petri-fink, B. Rothen-rutishauser, E. M. Landau and W. Fong, J. Colloid Interface Sci., 2019, 562, 502–510 CrossRef PubMed.
- L. Salvati Manni, S. Assenza, M. Duss, J. J. Vallooran, F. Juranyi, S. Jurt, O. Zerbe, E. M. Landau and R. Mezzenga, Nat. Nanotechnol., 2019, 14, 609–615 CrossRef CAS PubMed.
- R. Mezzenga, C. Meyer, C. Servais, A. I. Romoscanu, L. Sagalowicz and R. C. Hayward, Langmuir, 2005, 21, 3322–3333 CrossRef CAS PubMed.
- R. Mezzenga, P. Schurtenberger, A. Burbidge and M. Michel, Nat. Mater., 2005, 4, 729–740 CrossRef CAS PubMed.
- R. Ghanbari, S. Assenza and R. Mezzenga, J. Chem. Phys., 2019, 150, 094901 CrossRef PubMed.
- L. M. Antognini, S. Assenza, C. Speziale and R. Mezzenga, J. Chem. Phys., 2016, 145, 084903 CrossRef PubMed.
- J. C. Shah, Y. Sadhale and D. M. Chilukuri, Adv. Drug Delivery Rev., 2001, 47, 229–250 CrossRef CAS PubMed.
- S. B. Rizwan, B. J. Boyd, T. Rades and S. Hook, Expert Opin. Drug Delivery, 2010, 7, 1133–1144 CrossRef CAS PubMed.
- Y. Chen, P. Ma and S. Gui, Biomed Res. Int., 2014, 2014, 815981 Search PubMed.
- X. Mulet, B. J. Boyd and C. J. Drummond, J. Colloid Interface Sci., 2013, 393, 1–20 CrossRef CAS PubMed.
- K. Khaliqi, A. Ghazal, I. D. M. Azmi, H. Amenitsch, K. Mortensen, S. Salentinig and A. Yaghmur, Analyst, 2017, 142, 3118–3126 RSC.
- W. K. Fong, R. Negrini, J. J. Vallooran, R. Mezzenga and B. J. Boyd, J. Colloid Interface Sci., 2016, 484, 320–339 CrossRef CAS PubMed.
- R. Negrini and R. Mezzenga, Langmuir, 2011, 27, 5296–5303 CrossRef CAS PubMed.
- A. Yaghmur, L. de Campo, L. Sagalowicz, M. E. Leser and O. Glatter, Langmuir, 2006, 22, 9919–9927 CrossRef CAS PubMed.
- L. Van’t Hag, S. L. Gras, C. E. Conn and C. J. Drummond, Chem. Soc. Rev., 2017, 46, 2705–2731 RSC.
- A. I. Tyler, H. M. Barriga, E. S. Parsons, N. L. McCarthy, O. Ces, R. V. Law, J. M. Seddon and N. J. Brooks, Soft Matter, 2015, 11, 3279–3286 RSC.
- A. Zabara, J. T. Y. Chong, I. Martiel, L. Stark, B. A. Cromer, C. Speziale, C. J. Drummond and R. Mezzenga, Nat. Commun., 2018, 9, 554 CrossRef PubMed.
- H. Kim, Z. Song and C. Leal, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 10834–10839 CrossRef CAS PubMed.
- A. Madej, D. Paprocki, D. Koszelewski, A. Zadło-Dobrowolska, A. Brzozowska, P. Walde and R. Ostaszewski, RSC Adv., 2017, 7, 33344–33354 RSC.
- D. Paprocki, D. Koszelewski, P. Walde and R. Ostaszewski, RSC Adv., 2015, 5, 102828 RSC.
- S. Otto, J. B. F. N. Engberts and J. C. T. Kwak, J. Am. Chem. Soc., 1998, 120, 9517–9525 CrossRef CAS.
- E. J. Fendler and J. H. Fendler, Adv. Phys. Org. Chem., 1970, 8, 271–406 CrossRef CAS.
- S. Vauthey, C. Milo, P. Frossard, N. Garti, M. E. Leser and H. J. Watzke, J. Agric. Food Chem., 2000, 48, 4808–4816 CrossRef CAS PubMed.
- M. Duss, L. Salvati Manni, L. Moser, S. Handschin, R. Mezzenga, H. J. Jessen and E. M. Landau, ACS Appl. Mater. Interfaces, 2018, 10, 5114–5124 CrossRef CAS PubMed.
- S. Puvvada, S. Baral, G. M. Chow, S. B. Qadri and B. R. Ratna, J. Am. Chem. Soc., 1994, 116, 2135–2136 CrossRef CAS.
- M. P. Cashion and T. E. Long, Acc. Chem. Res., 2009, 42, 1016–1025 CrossRef CAS PubMed.
- D. F. O’Brien, B. Armitage, A. Benedicto, D. E. Bennett, H. G. Lamparski, Y.-S. Lee, W. Srisiri and T. M. Sisson, Acc. Chem. Res., 1998, 31, 861–868 CrossRef.
- B. A. Pindzola, J. Jin, D. L. Gin, B. A. Pindzola, J. Jin and D. L. Gin, J. Am. Chem. Soc., 2003, 125, 2940–2949 CrossRef CAS PubMed.
- B. A. Pindzola, B. P. Hoag and D. L. Gin, J. Am. Chem. Soc., 2001, 123, 4617–4618 CrossRef CAS PubMed.
- D. L. Gin, W. Gu, B. A. Pindzola and W.-J. Zhou, Acc. Chem. Res., 2001, 34, 973–980 CrossRef CAS PubMed.
- J. Zhang, Z. Xie, M. Hoang, A. J. Hill, W. Cong, F. H. She, W. Gao and L. X. Kong, Soft Matter, 2014, 10, 5192–5200 RSC.
- X. Feng, M. E. Tousley, M. G. Cowan, B. R. Wiesenauer, S. Nejati, Y. Choo, R. D. Noble, M. Elimelech, D. L. Gin and C. O. Osuji, ACS Nano, 2014, 8, 11977–11986 CrossRef CAS PubMed.
- M. Zhou, P. R. Nemade, X. Lu, X. Zeng, E. S. Hatakeyama, R. D. Noble, D. L. Gin, P. R. Nemade, X. Lu, X. Zeng, E. S. Hatakeyama, R. D. Noble and D. L. Gin, J. Am. Chem. Soc., 2007, 129, 9574–9575 CrossRef CAS PubMed.
- M. Zhou, T. J. Kidd, R. D. Noble and D. L. Gin, Adv. Mater., 2005, 17, 1850–1853 CrossRef CAS.
- S. A. Miller, E. Kim, D. H. Gray and D. L. Gin, Angew. Chem., Int. Ed., 1999, 38, 3021–3026 CrossRef PubMed.
- R. C. Smith, W. M. Fischer and D. L. Gin, J. Am. Chem. Soc., 1997, 119, 4092–4093 CrossRef CAS.
- X. Lu, V. Nguyen, M. Zhou, X. Zeng, J. Jin, B. J. Elliott and D. L. Gin, Adv. Mater., 2006, 18, 3294–3298 CrossRef CAS.
- W. Srisiri, T. M. Sisson, D. F. O’Brien, K. M. McGrath, Y. Han and S. M. Gruner, J. Am. Chem. Soc., 1997, 119, 4866–4873 CrossRef CAS.
- Y.-S. Lee, J.-Z. Yang, T. M. Sisson, D. A. Frankel, J. T. Gleeson, E. Aksay, S. L. Keller, S. M. Gruner and D. F. O’Brien, J. Am. Chem. Soc., 1995, 117, 5573–5578 CrossRef CAS.
- W. Srisiri, A. Benedicto, D. F. O’Brien, T. P. Trouard, G. Orädd, S. Persson and G. Lindblom, Langmuir, 1998, 14, 1921–1926 CrossRef CAS.
- W. Srisiri, H. G. Lamparski, D. F. O’Brien, H. G. Lamparski and D. F. O’Brien, J. Org. Chem., 1996, 61, 5911–5915 CrossRef CAS.
- X. Feng, K. Kawabata, G. Kaufman, M. Elimelech and C. O. O. Osuji, ACS Nano, 2017, 11, 3911–3921 CrossRef CAS PubMed.
- D. Yang, D. F. O’Brien and S. R. Marder, J. Am. Chem. Soc., 2002, 124, 13388–13389 CrossRef CAS PubMed.
- D. Yang, B. Armitage and S. R. Marder, Angew. Chem., Int. Ed., 2004, 43, 4402–4409 CrossRef CAS PubMed.
- A. Mueller and D. F. O’Brien, Chem. Rev., 2002, 102, 727–757 CrossRef CAS PubMed.
- H.-P. Hentze and E. W. Kaler, Curr. Opin. Colloid Interface Sci., 2003, 8, 164–178 CrossRef CAS.
- R. Laversanne, Macromolecules, 1992, 25, 489–491 CrossRef CAS.
- M. Antonietti, R. A. Caruso, C. G. Göltner and M. C. Weissenberger, Macromolecules, 1999, 32, 1383–1389 CrossRef CAS.
- M. A. DePierro, K. G. Carpenter and C. A. Guymon, Chem. Mater., 2006, 18, 5609–5617 CrossRef CAS.
- P. Ström and D. M. M. Anderson, Langmuir, 1992, 8, 691–709 CrossRef.
- P. Ström, J. Colloid Interface Sci., 1992, 154, 184–193 CrossRef.
- B. S. Forney, C. A. Guymon, B. S. Forney and C. Allan Guymon, Macromolecules, 2010, 43, 8502–8510 CrossRef CAS.
- R. C. Rodrigues, C. Ortiz, Á. Berenguer-Murcia, R. Torres and R. Fernández-Lafuente, Chem. Soc. Rev., 2013, 42, 6290–6307 RSC.
- H. Wombacher, Mol. Cell. Biochem., 1983, 56, 155–164 CrossRef CAS PubMed.
- G. R. Welch, Prog. Biophys. Mol. Biol., 1978, 32, 103–191 CrossRef.
- W. Qiu, Z. Fu, G. G. Xu, R. A. Grassucci, Y. Zhang, J. Frank, W. A. Hendrickson and Y. Guo, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 12985–12990 CrossRef CAS PubMed.
- F. Snyder, T. Lee, M. L. Blank and C. Moore, Membrane Fluidity, Humana Press, 1980, pp. 307–323 Search PubMed.
- A. Vieler, H. A. Scheidt, P. Schmidt, C. Montag, J. F. Nowoisky, M. Lohr, C. Wilhelm, D. Huster and R. Goss, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 1027–1034 CrossRef CAS PubMed.
- N. L. Klyachko, A. V. Levashov, A. V. Pshezhetsky, N. G. Bogdanova, I. V. Berezin and K. Martinek, Eur. J. Biochem., 1986, 161, 149–154 CrossRef CAS PubMed.
- B. Ericsson, P. O. Eriksson, J. E. Löfroth and S. Engström, Cubic Phases as Delivery Systems for Peptide Drugs, 2009 Search PubMed.
- E. Nazaruk and R. Bilewicz, Bioelectrochemistry, 2007, 71, 8–14 CrossRef CAS PubMed.
- M. H. Shah and A. Paradkar, Int. J. Pharm., 2005, 294, 161–171 CrossRef CAS PubMed.
- Y. Sadhale and J. C. Shah, Int. J. Pharm., 1999, 191, 51–64 CrossRef CAS PubMed.
- Y. Corvis, G. Brezesinski, R. Rink, A. Walcarius, A. Van Der Heyden, F. Mutelet and E. Rogalska, Anal. Chem., 2006, 78, 4850–4864 CrossRef CAS PubMed.
- W. Sun, J. J. Vallooran, W. K. Fong and R. Mezzenga, J. Phys. Chem. Lett., 2016, 7, 1507–1512 CrossRef CAS PubMed.
- C. Speziale, L. Salvati Manni, C. Manatschal, E. M. Landau and R. Mezzenga, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7491–7496 CrossRef CAS PubMed.
- C. Speziale, A. F. Zabara, C. J. Drummond and R. Mezzenga, ACS Nano, 2017, 11, 11687–11693 CrossRef CAS PubMed.
- S. B. Leslie, S. Puvvada, B. R. Ratna and A. S. Rudolph, Biochim. Biophys. Acta, Biomembr., 1996, 1285, 246–254 CrossRef CAS.
- P. Nogly, T. Weinert, D. James, S. Carbajo, D. Ozerov, A. Furrer, D. Gashi, V. Borin, P. Skopintsev, K. Jaeger, K. Nass, P. Båth, R. Bosman, J. Koglin, M. Seaberg, T. Lane, D. Kekilli, S. Brünle, T. Tanaka, W. Wu, C. Milne, T. White, A. Barty, U. Weierstall, V. Panneels, E. Nango, S. Iwata, M. Hunter, I. Schapiro, G. Schertler, R. Neutze and J. Standfuss, Science, 2018, 361, eaat0094 CrossRef PubMed.
- K. Edman, P. Nollert, A. Royant, H. Beirhali, E. Pebay-Peyroula, J. Hajdu, R. Neutze and E. M. Landau, Nature, 1999, 401, 822–826 CrossRef CAS PubMed.
- M. Rakotoarisoa, B. Angelov, S. Espinoza, K. Khakurel, T. Bizien and A. Angelova, Molecules, 2019, 24, 3058 CrossRef CAS PubMed.
- D. Li and M. Caffrey, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8639–8644 CrossRef CAS PubMed.
- C. E. Conn and C. J. Drummond, Soft Matter, 2013, 9, 3449–3464 RSC.
- A. Zabara, R. Negrini, P. Baumann, O. Onaca-Fischer and R. Mezzenga, Chem. Commun., 2014, 50, 2642–2645 RSC.
- R. Wallin, S. Engström and C. F. Mandenius, Biocatal. Biotransform., 1993, 8, 73–80 CrossRef CAS.
- V. Razumas, J. Kanapieniené, T. Nylander, S. Engström and K. Larsson, Anal. Chim. Acta, 1994, 289, 155–162 CrossRef CAS.
- W. Sun, J. J. Vallooran and R. Mezzenga, Langmuir, 2015, 31, 4558–4565 CrossRef CAS PubMed.
- E. Nazaruk, R. Bilewicz, G. Lindblom and B. Lindholm-Sethson, Anal. Bioanal. Chem., 2008, 391, 1569–1578 CrossRef CAS PubMed.
- P. Rowinski, M. Rowinska and A. Heller, Anal. Chem., 2008, 80, 1746–1755 CrossRef CAS PubMed.
- E. Nazaruk, E. M. Landau and R. Bilewicz, Electrochim. Acta, 2014, 140, 108–115 CrossRef.
- M. H. Ropers, R. Bilewicz, M. J. Stébé, A. Hamidi, A. Miclo and E. Rogalska, Phys. Chem. Chem. Phys., 2001, 3, 240–245 RSC.
- P. Rowińki, R. Bilewicz, M. J. Stébé and E. Rogalska, Anal. Chem., 2004, 76, 283–291 CrossRef PubMed.
- F. Gao, Z. Yao, Q. Huang, X. Chen, X. Guo, Q. Ye and L. Wang, Colloids Surf., B, 2011, 82, 359–364 CrossRef CAS PubMed.
- J. J. Vallooran, S. Handschin, S. M. Pillai, B. N. Vetter, S. Rusch, H. P. Beck and R. Mezzenga, Adv. Funct. Mater., 2016, 26, 181–190 CrossRef CAS.
- J. J. Vallooran, S. Assenza and R. Mezzenga, Angew. Chem., Int. Ed., 2019, 58, 7289–7293 CrossRef CAS PubMed.
- T. Zhou, J. J. Vallooran, S. Assenza, A. Szekrenyi, P. Clapés and R. Mezzenga, ACS Catal., 2018, 8, 5810–5815 CrossRef CAS.
- J. Gilbert, M. Valldeperas, S. K. Dhayal, J. Barauskas, C. Dicko and T. Nylander, Nanoscale, 2019, 11, 21291–21301 RSC.
- Y. La, C. Park, T. J. Shin, S. H. Joo, S. Kang and K. T. Kim, Nat. Chem., 2014, 6, 534–541 CrossRef CAS PubMed.
- E. Da’na, Microporous Mesoporous Mater., 2017, 247, 145–157 CrossRef.
- R. Narayan, U. Y. Nayak, A. M. Raichur and S. Garg, Pharmaceutics, 2018, 10, 118 CrossRef CAS PubMed.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- L. B. McCusker, Micro- and Mesoporous Mineral Phases, Walter de Gruyter GmbH, 2018, vol. 57, pp. 1–16 Search PubMed.
- F. Liebau, Microporous Mesoporous Mater., 2003, 58, 15–72 CrossRef CAS.
- F. Di Renzo, H. H. Cambon and R. Dutartre, Microporous Mater., 1997, 10, 283–286 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- R. Ryoo, Nature, 2019, 575, 40–41 CrossRef CAS PubMed.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C.-W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- J. C. Vartuli, K. D. Schmitt, C. T. Kresge, W. J. Roth, M. E. Leonowicz, S. B. McCullen, S. D. Hellring, J. S. Beck, J. L. Schlenker, D. H. Olson and E. W. Sheppard, Chem. Mater., 2005, 6, 2317–2326 CrossRef.
- K. J. Edler, T. Brennan, S. J. Roser, S. Mann and R. M. Richardson, Microporous Mesoporous Mater., 2003, 62, 165–175 CrossRef CAS.
- T. Yanagisawa, T. Shimizu, K. Kuroda and C. Kato, Bull. Chem. Soc. Jpn., 1990, 63, 988–992 CrossRef CAS.
- S. Inagaki, Y. Fukushima and K. Kuroda, J. Chem. Soc., Chem. Commun., 1993, 680–682 RSC.
- Q. Huo, D. I. Margolese, U. Cleslat, P. Feng, T. E. Gler, P. Sieger, R. Leont, P. M. Petrofft, F. Schiith, G. D. Stucky, U. Ciesla, P. Feng, T. E. Gier, P. Sieger, R. Leon, P. M. Petroff, F. Schüth and G. D. Stucky, Nature, 1994, 368, 317–321 CrossRef CAS.
- A. Monnier, F. Schüth, Q. Huo, D. Kumar, D. Margolese, R. S. S. Maxwell, G. D. D. Stucky, M. Krishnamurty, P. Petroff, A. Firouzi, M. Janicke and B. F. F. Chmelka, Science, 1993, 261, 1299–1303 CrossRef CAS PubMed.
- D. Kumar, L. Bull, T. Besier, P. Sieger, Q. Huo, S. Walker, J. Zasadzinski, C. Glinka, J. Nicol, D. Margolese, G. D. Stucky and B. F. Chmelka, Science, 1995, 267, 1138–1143 CrossRef PubMed.
- L. Omer, S. Ruthstein, D. Goldfarb and Y. Talmon, J. Am. Chem. Soc., 2009, 131, 12466–12473 CrossRef CAS.
- C. Yu, J. Fan, B. Tian and D. Zhao, Chem. Mater., 2004, 16, 889–898 CrossRef CAS.
- W. Tanglumlert, T. Imae, T. J. White and S. Wongkasemjit, J. Am. Ceram. Soc., 2007, 90, 3992–3997 CAS.
- V. Alfredsson, M. W. Anderson, T. Ohsuna, O. Terasaki, M. Jacob and M. Bojrup, Chem. Mater., 1997, 9, 2066–2070 CrossRef CAS.
- L. Travaglini and L. De Cola, Chem. Mater., 2018, 30, 4168–4175 CrossRef CAS.
- L. Han, D. Xu, Y. Liu, T. Ohsuna, Y. Yao, C. Jiang, Y. Mai, Y. Cao, Y. Duan and S. Che, Chem. Mater., 2014, 26, 7020–7028 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS PubMed.
- S. A. El-Safty and T. Hanaokat, Chem. Mater., 2004, 16, 384–400 CrossRef CAS.
- G. S. Attard, J. C. Glyde and C. G. Göltner, Nature, 1995, 378, 366–368 CrossRef CAS.
- J. L. Blin, A. Léonard and B. L. Su, Chem. Mater., 2001, 13, 3542–3553 CrossRef CAS.
- S. A. Bagshaw, E. Prouzet and T. J. Pinnavaia, Science, 1995, 269, 1242–1244 CrossRef PubMed.
- P. C. A. Alberius, K. L. Frindell, R. C. Hayward, E. J. Kramer, G. D. Stucky and B. F. Chmelka, Chem. Mater., 2002, 14, 3284–3294 CrossRef CAS.
- S. Che, A. E. Garcia-Bennett, T. Yokoi, K. Sakamoto, H. Kunieda, O. Terasaki and T. Tatsumi, Nat. Mater., 2003, 2, 801–805 CrossRef CAS PubMed.
- L. Sierra, B. Lopez and J. L. Guth, Microporous Mesoporous Mater., 2000, 39, 519–527 CrossRef CAS.
- Q. Huo, D. I. Margolese and G. D. Stucky, Chem. Mater., 1996, 8, 1147–1160 CrossRef CAS.
- D. Carta, S. Bullita, M. F. Casula, A. Casu, A. Falqui and A. Corrias, ChemPlusChem, 2013, 78, 364–374 CrossRef CAS.
- A. Berggren, A. E. C. Palmqvist and K. Holmberg, Soft Matter, 2005, 1, 219–226 RSC.
- L. Han and S. Che, Adv. Mater., 2018, 30, 1705708 CrossRef PubMed.
- E. Ruiz-Hitzky, P. Aranda, M. Darder and M. Ogawa, Chem. Soc. Rev., 2011, 40, 801–828 RSC.
- N. Cruise, K. Jansson and K. Holmberg, J. Colloid Interface Sci., 2001, 241, 527–529 CrossRef CAS.
- D. R. Dunphy, F. L. Garcia, B. Kaehr, C. Y. Khripin, A. D. Collord, H. K. Baca, M. P. Tate, H. W. Hillhouse, J. W. Strzalka, Z. Jiang, J. Wang and C. J. Brinker, Chem. Mater., 2011, 23, 2107–2112 CrossRef CAS PubMed.
- A. M. Seddon, H. M. Patel, S. L. Burkett and S. Mann, Angew. Chem., Int. Ed., 2002, 41, 2988–2991 CrossRef CAS.
- D. R. Dunphy, T. M. Alam, M. P. Tate, H. W. Hillhouse, B. Smarsly, A. D. Collord, E. Carnes, H. K. Baca, R. Köhn, M. Sprung, J. Wang and C. J. Brinker, Langmuir, 2009, 25, 9500–9509 CrossRef CAS PubMed.
- D. R. Dunphy, F. L. Garcia, Z. Jiang, J. Strzalka, J. Wang and C. J. Brinker, Chem. Commun., 2011, 47, 1806–1808 RSC.
- A. Galarneau, G. Renard, M. Mureseanu, A. Tourrette, C. Biolley, M. Choi, R. Ryoo, F. Di Renzo and F. Fajula, Microporous Mesoporous Mater., 2007, 104, 103–114 CrossRef CAS.
- D. D. Archibald and S. Mann, Nature, 1993, 364, 430–433 CrossRef CAS.
- G. S. Attard, C. G. Göltner, J. M. Corker, S. Henke and R. H. Templer, Angew. Chem., Int. Ed. Engl., 1997, 36, 1315–1317 CrossRef CAS.
- G. S. Attard, P. N. Bartlett, N. R. B. Coleman, J. M. Elliott, J. R. Owen and J. H. Wang, Science, 1997, 278, 838–840 CrossRef CAS.
- P. V. Braun, P. Osenar and S. I. Stupp, Nature, 1996, 380, 325–328 CrossRef CAS.
- J. Kibsgaard, Y. Gorlin, Z. Chen and T. F. Jaramillo, J. Am. Chem. Soc., 2012, 134, 7758–7765 CrossRef CAS PubMed.
- D. Wang, H. Luo, R. Kou, M. P. P. Gil, S. Xiao, V. O. O. Golub, Z. Yang, C. J. J. Brinker and Y. Lu, Angew. Chem., Int. Ed., 2004, 43, 6169–6173 CrossRef CAS PubMed.
- D. Wang, W. L. Zhou, B. F. McCaughy, J. E. Hampsey, X. Ji, Y. B. Jiang, H. Xu, J. Tang, R. H. Schmehl, C. O’Connor, C. J. Brinker and Y. Lu, Adv. Mater., 2003, 15, 130–133 CrossRef CAS.
- H. J. Shin, R. Ryoo, Z. Liu and O. Terasaki, J. Am. Chem. Soc., 2001, 123, 1246–1247 CrossRef CAS PubMed.
- S. Akbar, J. M. Elliott, M. Rittman and A. M. Squires, Adv. Mater., 2013, 25, 1160–1164 CrossRef CAS PubMed.
- S. Akbar, J. Boswell, C. Worsley, J. M. Elliott and A. M. Squires, Langmuir, 2018, 34, 6991–6996 CrossRef CAS.
- S. J. Richardson, M. R. Burton, P. A. Staniec, I. S. Nandhakumar, N. J. Terrill, J. M. Elliott and A. M. Squires, Nanoscale, 2016, 8, 2850–2856 RSC.
- S. J. Richardson, M. R. Burton, X. Luo, P. A. Staniec, I. S. Nandhakumar, N. J. Terrill, J. M. Elliott and A. M. Squires, Nanoscale, 2017, 9, 10227–10232 RSC.
- M. R. Burton, C. Lei, P. A. Staniec, N. J. Terrill, A. M. Squires, N. M. White and I. S. Nandhakumar, Sci. Rep., 2017, 7, 6405 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |