
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA feasible strategy to prepare quantum dot-incorporated carbon nanofibers as free-standing platforms†
Taeyoung
Song‡
a,
Jun Young
Cheong‡
a,
Ji Yong
Choi‡
 b,
Cheolmin
Park
c,
Chulhee
Lee
a,
Changsoo
Lee
a,
Hyuck Mo
Lee
a,
Sung-Yool
Choi
c,
Hyunjoon
Song
*b,
Il-Doo
Kim
*a and
Duk Young
Jeon
*a
b,
Cheolmin
Park
c,
Chulhee
Lee
a,
Changsoo
Lee
a,
Hyuck Mo
Lee
a,
Sung-Yool
Choi
c,
Hyunjoon
Song
*b,
Il-Doo
Kim
*a and
Duk Young
Jeon
*a
aDepartment of Materials Science and Engineering, Korea Advanced Institute of Science and Technology, 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea. E-mail: dyjeon@kaist.ac.kr; idkim@kaist.ac.kr
bDepartment of Chemistry, Korea Advanced Institute of Science and Technology, 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Republic of Korea. E-mail: hsong@kaist.ac.kr
cSchool of Electrical Engineering, Center for Advanced Materials Discovery towards 3D Display, Graphene/2D Materials Research Center, Korea Advanced Institute of Science and Technology, 291 Daehak-ro, Yuseong-gu, Daejeon 34141, Korea
First published on 26th August 2019
Abstract
Recently, quantum dots (QDs) have often garnered significant attention and have been employed for various applications. Nevertheless, most conventional devices utilize a glass substrate and/or brittle substrate, which is not compatible with next-generation wearable electronics. A suitable method for devising conductive and flexible free-standing platforms that can be combined with various kinds of QDs is thus in great need for next-generation wearable electronics. In this work, we introduce a universal and simple method to coat QDs on carbon nanofibers (CNFs) by a dip-coating process, where many kinds of QDs can be well decorated on the surface of CNFs. As one potential application among many, QD-coated CNFs were examined for their photocatalytic applications and characterization. As a result, it was found that the best performance of CdSe QD-coated CNFs for hydrogen production was 3.8 times higher than that of only QDs with the same 1 mg of QDs. This is an early report on fabricating various kinds of QD-coated CNFs, which can be extended to a myriad set of applications.
Introduction
Quantum dots (QDs) have received considerable attention, not only in the fields of photovoltaic solar cells1 but also in the fields of light emitting diodes,2 neuroscience,3 batteries,4 and photodetectors.5,6 The popularity of QDs can be attributed to their quantum confinement effect based on confinement in three spatial dimensions by the potential barrier,7 as well as the fact that QDs offer the advantages of simple processing, large-scale production, and facile control of energy levels of the junction with the substrate using changeable highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels of QDs.1,5 Much research has already been conducted for using QDs as crucial components of optoelectronics, some of which include, but are not limited to, PbS–MoS2,5 PbS,6 ZnO,8 HgTe,9 and Ge.10With the emergence of the IoT technology, wearable/flexible electronics have received tremendous attention.11,12 For such technology, it is critical to use a conductive free-standing platform. In lieu of these trends, a majority of devised optoelectronics have been based on brittle substrates,4–6,9,10 although some literature has been reported on wearable/flexible substrates.8 A facile strategy to make a QD-conductive free-standing substrate needs to be devised to establish a milestone for next-generation electronic devices.
Among the materials for free-standing substrates, carbon nanofibers (CNFs), simply prepared using electrospinning, have the advantage of being low cost and have been highlighted. CNFs have been studied as a structure coated with various materials such as Co4N, WS2, CdS, ZnO, AgS2, TiO2, and ZnIn2S4 and so on. Most of these coated structures have been formed by allowing materials to grow and crystallize on the surface of CNFs using various methods.13–20 The methods include the solvothermal method, the hydrothermal method, successive ionic layer adsorption and reaction, and heat treatment under specific gas-flowing conditions.13–18,21,22 However, these methods have disadvantages in that they involve a difficult synthesis process, result in problematic surface uniformity, and are not amenable to mass production. The mechanical and physical properties of CNFs also change due to the additional chemical reaction.
In this work, we firstly suggest a highly facile CNF platform coated with various QDs using a simple dip-coating process. The dip-coating is universal and scalable, and it can be used for large-scale production, so that large-scale preparation of QD-coated CNFs is feasible and various kinds of QDs can be readily coated onto CNFs. QDs have changeable band gap properties, and fabricated QD-coated CNFs have the merit of adjustable energy levels of heterojunctions between the QDs and the CNFs, something which is also useful in optoelectronics.5 As one suitable application, QD-coated CNF structures were applied to photocatalytic hydrogen evolution, and the best catalytic efficiency of CdSe QD-coated CNFs was about 3.8 times higher than that of pure CdSe QDs. Additionally, the photosensitivity of the conductive and flexible free-standing structure was simply verified, and this proves useful for IoT devices due to the straightforward applicability to various shapes of reactors or devices.23 Our work remains an early report on a feasible strategy to deposit various kinds of QDs onto a free-standing conducting carbon material, which offers the potential for numerous applications in the near future.
Experimental
Materials
Polyacrylonitrile (PAN, Mw = 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was purchased from Sigma Aldrich (USA). N,N-Dimethylformamide (DMF) was purchased from Sigma Aldrich (USA). CdO (99.9%), oleic acid (OA, 90%), 1-octadecene (1-ODE, 90%), trioctylphosphine (TOP, 97%), and Se powder (99.99%) were purchased from Sigma Aldrich. PbO (99.99%), bis(trimethylsilyl)sulfide ((TMS)2S, synthesis grade), InCl3 (98%), ZnCl2 (97%), oleylamine (70%), tris(dimethylamino)phosphine (P(DMA)3, 97%), 1-dodecanethiol (DDT, 98%), CuI (99.999%), indium acetate (99.99%), zinc acetate (99.99%), sulfur (99.998%), zinc diethyldithiocarbamate (Zn DDTC, 97%), cadmium acetate (99.995%), ZnO (99.999%), sodium sulfide nonahydrate (98+%), and sodium sulfite (≥98%) were purchased from Sigma Aldrich.
000) was purchased from Sigma Aldrich (USA). N,N-Dimethylformamide (DMF) was purchased from Sigma Aldrich (USA). CdO (99.9%), oleic acid (OA, 90%), 1-octadecene (1-ODE, 90%), trioctylphosphine (TOP, 97%), and Se powder (99.99%) were purchased from Sigma Aldrich. PbO (99.99%), bis(trimethylsilyl)sulfide ((TMS)2S, synthesis grade), InCl3 (98%), ZnCl2 (97%), oleylamine (70%), tris(dimethylamino)phosphine (P(DMA)3, 97%), 1-dodecanethiol (DDT, 98%), CuI (99.999%), indium acetate (99.99%), zinc acetate (99.99%), sulfur (99.998%), zinc diethyldithiocarbamate (Zn DDTC, 97%), cadmium acetate (99.995%), ZnO (99.999%), sodium sulfide nonahydrate (98+%), and sodium sulfite (≥98%) were purchased from Sigma Aldrich.
Synthesis of CNFs
The synthesis of CNFs was similar to what was previously reported.24 1200 mg of PAN was dissolved in 10000 mg of DMF, and the mixture was stirred for 24 h at 80 °C to allow complete dissolution. The electrospinning was conducted with the following conditions: 15 kV applied voltage, 20 μL mL−1 flow rate, 25 Gauge needle, and 15 cm distance between the collector and the tip of the syringe. It was stabilized under air at 280 °C for 2 h with a ramp rate of 2 °C min−1. Then, the CNFs were later carbonized under an Ar atmosphere at 900 °C for 2 h with a ramp rate of 5 °C min−1.
Synthesis of CdSe QDs
150 mg of CdO, 1.5 mL of OA and 20 mL of 1-ODE were loaded into a 100 mL three-neck flask. The mixture was degassed under high vacuum for 2 h at 130 °C and then heated to 300 °C under an Ar atmosphere. At this temperature, 20 mg of Se powder dissolved in 1 mL of TOP was quickly injected into the solution. After 3 min, the solution was cooled down to room temperature.Synthesis of PbS QDs
900 mg of PbO, 4 mL of OA, and 20 mL of 1-ODE were loaded into a 100 mL three-neck flask and heated to 120 °C under vacuum for 2 h. After the flask was filled with Ar, 0.4 mL of (TMS)2S diluted in 10 mL of 1-ODE was rapidly injected. Then the flask was separated from the heating mantle and naturally cooled down to room temperature.Synthesis of InP QDs and InP/ZnS QDs
40 mg of InCl3, 300 mg of ZnCl2, and 14 mL of oleylamine were loaded into a 100 mL three-neck flask and heated to 120 °C under vacuum for 2 h. Then the solution was heated to 250 °C under an Ar atmosphere. At 250 °C, 0.3 mL of P(DMA)3 was swiftly injected into the solution. After 5 min, InP QDs were synthesized. For the growth of the ZnS shell, 3 mL of DDT was injected slowly into the solution and the temperature was maintained at 250 °C for 6 h. Then the solution was cooled down to room temperature.Synthesis of CuInS2 QDs and CuInS2/ZnS QDs
38 mg of CuI, 58 mg of indium acetate, 2 mL of DDT, and 16 mL of 1-ODE were loaded into a 100 mL three-neck flask. After being degassed under high vacuum for 2 h at 120 °C, the solution was heated to 210 °C and maintained for 90 min under an Ar atmosphere. Then the solution was cooled down to room temperature. This solution contains CuInS2 QDs. For the growth of the ZnS shell, the crude solution needed a purification process, which is a simple precipitation/dispersion method. After the purification process, 20 mg of CuInS2 QDs, 55 mg of zinc acetate, and 5 mL of DDT were loaded into a 100 mL flask and heated to 60 °C under vacuum for 40 min. Then the flask was heated to 280 °C and maintained for 20 min under Ar gas.Synthesis of CdS QDs
128 mg of CdO, 2 mL of OA and 12 mL of 1-ODE were loaded into a 100 mL three-neck flask and heated to 130 °C for 2 h under high vacuum. Then the temperature was set at 250 °C under an Ar atmosphere. At 250 °C, 16 mg of S powder dissolved in 2 mL of 1-ODE was injected into the solution. After 10 min, the temperature was reduced at room temperature.Synthesis of red-emitting CdSe/ZnS QDs
820 mg of CdO, 6 mL of OA, and 10 mL of 1-ODE were loaded into a three neck flask and heated to 130 °C under high vacuum for 2 h. Then the temperature was adjusted to 300 °C under an Ar atmosphere. At 300 °C, 30 mg of Se powder dissolved in 0.6 mL of TOP was quickly injected into the flask and the temperature was maintained for 10 min. Subsequently, the flask was cooled to 225 °C and 140 mg of Zn DDTC dissolved in 1.4 mL of TOP was injected into the flask. Then the flask was heated to 280 °C and maintained for 20 min.Synthesis of green-emitting CdZnSeS/ZnS QDs
32 mg of cadmium acetate, 277 mg of ZnO, 7 mL of OA and 15 mL of 1-ODE were loaded into a three-neck flask and heated to 130 °C for 2 h under high vacuum. Subsequently, after increasing the temperature to 300 °C under an Ar atmosphere and a mixture of 64 mg of sulfur and 158 mg of Se powder dissolved in 2 mL of TOP was rapidly injected into the flask. After 10 min, the flask was cooled to 225 °C and 200 mg of Zn DDTC dissolved in 2 mL of TOP was injected into the flask. Next, the flask was heated to 280 °C and maintained for 10 min.Synthesis of blue-emitting CdZnS/ZnS QDs
1834 mg of zinc acetate, 128 mg of CdO, 7 mL of OA, and 15 mL of 1-ODE were loaded into a three-neck flask and heated to 120 °C for 2 h under vacuum. Then the solution was heated to 310 °C under an Ar atmosphere and 51 mg of sulfur dissolved in 2.4 mL of 1-ODE was rapidly injected into the solution. After 12 min, 128 mg of sulfur dissolved in 5 mL of OA was injected into the solution and maintained for 3 h at 310 °C.Purification of QDs
The synthesized crude solution of QDs needs a purification process for devices. The purification process is a precipitation/redispersion method. An excess amount of ethanol is added to precipitate the synthesized QDs in the solution. Then the precipitated QDs are dispersed in hexane. This process is repeated 3 times and the final precipitated QDs are redispersed in hexane at specific concentrations.Dip-coating of QDs on CNFs
QDs were coated onto CNFs by simple dip-coating. The QD solution was produced in hexane with various concentrations such as 20, 50 and 100 mg mL−1. The CNFs can be cut into different sizes and dimensions, and they were immersed in each QD solution for 1 min and taken out of the solution.Characterization
Morphological features were obtained by scanning electron microscopy (SEM, SU8230, Hitachi) and field emission transmission electron microscopy (TEM, Tecnai G2 F30 S-Twin). Energy dispersive X-ray spectroscopy (EDS) mapping images were obtained using scanning transmission electron microscopy (STEM, Tecnai F20, FEI company). Component analysis of elements was performed using a SU8230 by EDS. X-ray diffraction (XRD) patterns were recorded by using an X-ray diffractometer (SmartLab, Rigaku). UV-vis absorbance spectra were recorded by a UV-1800 and SolidSpec-3700, Shimadzu. The photoluminescence spectra were recorded by using an F-7000, Hitachi, and FL920, Edinburgh instruments. Time-correlated single photon counting (TCSPC) was performed using an FL920 with a picosecond pulsed diode laser (470 nm), EPL-470, and the luminescence decay time values were calculated by using an F900 program. The decay times of CdSe–CNF, PbS–CNF, InP–CNF and CuInS2–CNF were measured at 582.8 nm, 1068 nm, 671 nm and 730 nm emission wavelengths, respectively, at room temperature. The work function of CNFs was obtained using ultraviolet photoelectron spectroscopy (UPS, Axis-Supra, Kratos) with a He I (21.2 eV) light source. For photocatalytic hydrogen evolution, the as-prepared catalysts were placed in a 0.25 M Na2S/0.35 M Na2SO3 aqueous solution (15 mL), and purged with N2 gas for 15 min before irradiating with visible light by using a Xe lamp (300 W, Oriel) equipped with a 10 cm IR water filter and a UV cut-off filter (λ ≥ 420 nm). The reaction was performed with a homemade quartz flask with a total volume of 36 mL, sealed with a rubber septum, and the product was periodically collected by using a syringe from the head space. Before the photocatalytic reaction, the samples were activated through the irradiation with visible light for 30 min to eliminate the surface organic compounds and purged with N2 gas again, and the product was measured by using a gas chromatograph (GC, YL6100) with a thermal conductivity detector (TCD) using argon as a carrier gas. Quantum efficiency (QE) was measured by using a 10 mW Thorlabs Violet (420 nm) mounted high-power light emitting diode (LED) light (M420L3), and calculated by using the following equation:where ne is the number of electrons utilized in the hydrogen evolution, np is the number of photons irradiated on catalysts, and nH2 is the number of hydrogen molecules generated during the reaction. The ne is twice the nH2 because the hydrogen evolution is a two-electron process.
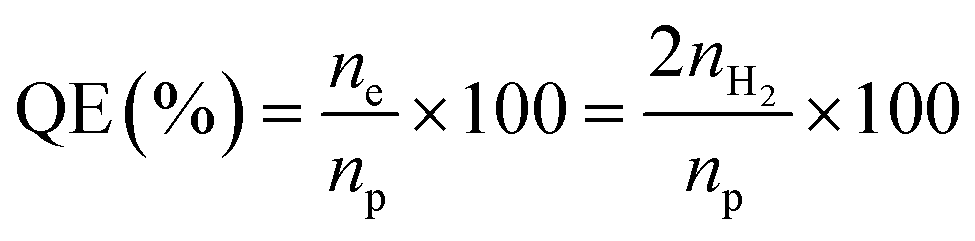
Results and discussion
The QDs were easily coated onto the CNF platform through dip-coating, as schematically illustrated in Fig. 1a. After the electrospinning of the as-spun nanofibers composed of polyacrylonitrile (PAN) and subsequent heat treatments, CNFs can be fabricated easily. The QDs dispersed in either hexane, toluene, or chloroform can be easily coated onto CNFs by simple dip-coating, where QDs with ligands having an alkyl group are physically attached on the surface of CNFs by van der Waals force.25,26 QDs with various average sizes (as shown in Table S1†) can be coated directly onto the CNF scaffold, demonstrating the feasibility of the dip-coating process as a universal method to coat many different kinds of QDs onto CNFs. As suggested in Fig. 1b, various kinds of QDs with different atomic compositions can also be coated onto CNFs, where diverse functionality is expected to arise.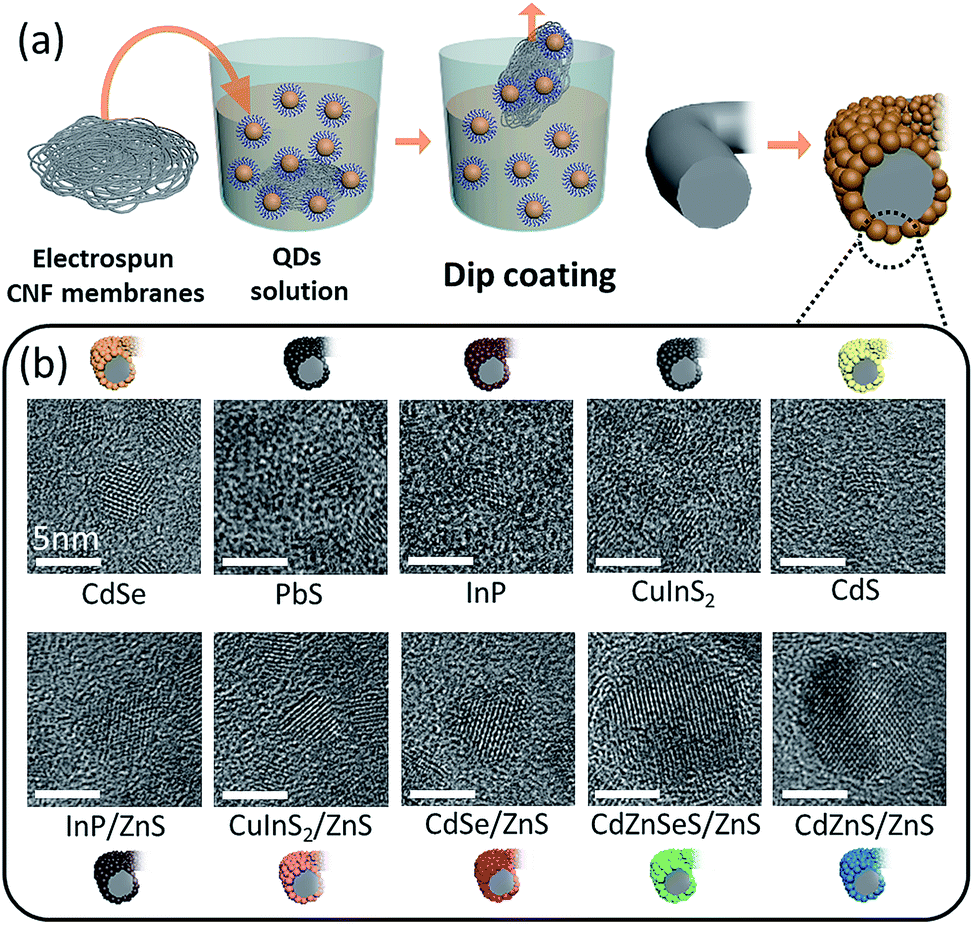 | ||
| Fig. 1 (a) Schematic illustration showing the easy dip-coating process of the QD-coated CNFs and (b) TEM images showing various kinds of QDs with different atomic compositions, which were used to deposit on the surface of CNFs. | ||
Scanning electron microscopy (SEM) images of pristine CNFs and various QD-coated CNFs are presented in Fig. 2. Prior to coating QDs onto CNFs, the intrinsic properties, such as the absorbance and photoluminescence (PL) spectra, of ten different kinds of QDs (CdSe, PbS, InP, CuInS2, CdS, InP/ZnS, CuInS2/ZnS, CdSe/ZnS, CdZnSeS/ZnS, and CdZnS/ZnS) were recorded and confirmed (Fig. S1†). The low and high magnification SEM images of pristine CNFs with a fiber thickness of about 300–500 nm are shown in Fig. 2a and b, respectively. The ten different kinds of QDs dispersed in hexane with identical concentration (20 mg mL−1) were easily coated onto CNFs by simple dip-coating for 1 min (Fig. 2c–l), although some were difficult to detect. Such a discrepancy is dependent on the particle size of individual QDs – when the size of QDs is around 10 nm, they are visible in the SEM image, but the sizes less than 5 nm become difficult to detect. Based on Fig. 2c–l, it is apparent that various kinds of QDs are homogeneously coated onto the CNF surface by dip-coating. In addition, low magnification SEM images (Fig. S2†) confirm that the nanofibrous structure was well maintained after the dip-coating process, in which coating of the QDs did not alter the overall morphology. For the detailed study of electrical junction properties between the QDs and the CNFs, we chose a CdSe QD-coated CNF (denoted as CdSe–CNF) among the aforementioned QDs. To further delve into the concentration effect of QDs on the overall degree of coating, CdSe QDs on CNFs with various coating concentrations (20, 50, and 100 mg mL−1, denoted as CdSe–CNF 20, CdSe–CNF 50, and CdSe–CNF 100, respectively) were visualized through SEM images (Fig. S3†). The morphology of CNFs is not significantly altered by the coating concentration – all of the samples exhibit a similar morphology, where QDs were coated on CNFs. To confirm the presence of QDs coated on CNFs, energy dispersive X-ray spectroscopy (EDS) mapping was conducted for CdSe–CNF 20 (Fig. S4†).
 | ||
| Fig. 2 SEM images of pristine CNFs in (a) low and (b) high magnification. SEM images of CNFs and CNFs coated with various kinds of QDs (20 mg mL−1): (c) CdSe, (d) PbS, (e) InP, (f) CIS, (g) CdS, (h) InP/ZnS, (i) CIS/ZnS, (j) CdSe/ZnS, (k) CdZnSeS/ZnS, and (l) CdZnS/ZnS. Overall surface of the hybrid composite is uniform without aggregated QD particles. | ||
As the size of the QDs is generally less than 10 nm, transmission electron microscopy (TEM) analysis was used to investigate the exact microstructure of CdSe–CNF 20 (Fig. 3). The high-resolution TEM (HRTEM) images and the corresponding selected area diffraction (SAED) pattern of CdSe QDs are shown in Fig. 3a, b, and e, revealing the successful synthesis of QDs. The low and high magnification TEM images of CdSe–CNF further confirm the uniform deposition of QDs, in which a number of QDs are deposited on the surface of the CNFs (Fig. 3c and d). The CdSe–CNF SAED patterns have also shown that the crystal structures of QDs coated on the CNFs are similar to those of QDs only (Fig. 3e). To investigate the overall morphologies of the QD-coated surface, TEM images of CdSe–CNFs with different coating concentrations were obtained, and they also exhibit similar fibrous morphologies (Fig. S5†). In Fig. 3f, TEM-EDS mapping was further conducted to confirm the deposition of CdSe–CNF. The Cd and Se are more concentrated on the edge site of the surface in the images, which can be attributed to the characteristics of EDS being more strongly influenced by the edge site. From the EDS study, we clearly confirmed the effective surface coating of QDs on the CNF surface, which was enabled by the dip-coating process. A similar trend is also observed in other QD-coated CNFs (Fig. S6†).
 | ||
| Fig. 3 (a) TEM image and (b) HRTEM image of CdSe QDs. (c) TEM image and (d) HRTEM image of CdSe–CNF 20. (e) SAED patterns of (left) CdSe QDs and (right) CdSe–CNF 20. (f) TEM-EDS mapping of CdSe–CNF 20. | ||
We also conducted quantitative and structural analyses regarding the coating concentrations of CdSe QDs. Based on the inductively coupled plasma mass spectrometry (ICP) analysis of CdSe–CNF (Table S2†) and the trend in the average amount of CdSe QDs with respect to the weight of CdSe–CNF (Fig. 4a), it is clear that both the atomic and weight ratios of Cd and Se increase with respect to C with the increase of the coating concentration. Based on the X-ray diffraction (XRD) patterns, when the coating concentration of CdSe QDs increases, the intensities of crystalline peaks for CdSe (ICDD no. 01-073-6987) also proportionally increase (Fig. 4b). CdSe–CNF 100 exhibits the most pronounced crystalline peaks, followed by CdSe–CNF 50 and CdSe–CNF 20. The presence of other QDs (such as InP and CuInS2) on the CNFs is also clearly confirmed by the XRD patterns at a coating concentration of 20 mg mL−1 (Fig. S7†). Furthermore, Fig. 4c shows that the coated QD layer becomes thicker with the increase of the coating loaded amount of CdSe QDs. This can be inferred from the elemental intensity of TEM-EDS mapping at the edge site of the CdSe–CNFs. As mentioned previously, from the point of view of the 2D EDS mapping images, the CdSe QDs are densely located at the edge site due to the cylinder shape of the CNFs. The intensity of the Cd element is higher at the edge site than at the inner site, and Fig. 4c shows that the thickness of the dense Cd element region increases with the coating amount at the edge site. Other elemental mapping images are shown in Fig. S8.†Fig. 4d shows the intensity of the Cd element from the edge site to the inner site. As a criterion of full width at half maximum, the thicknesses of CdSe QD layers of CdSe–CNF 20, CdSe–CNF 50, and CdSe–CNF 100 are 5.4 nm, 11.7 nm, and 18.8 nm, respectively.
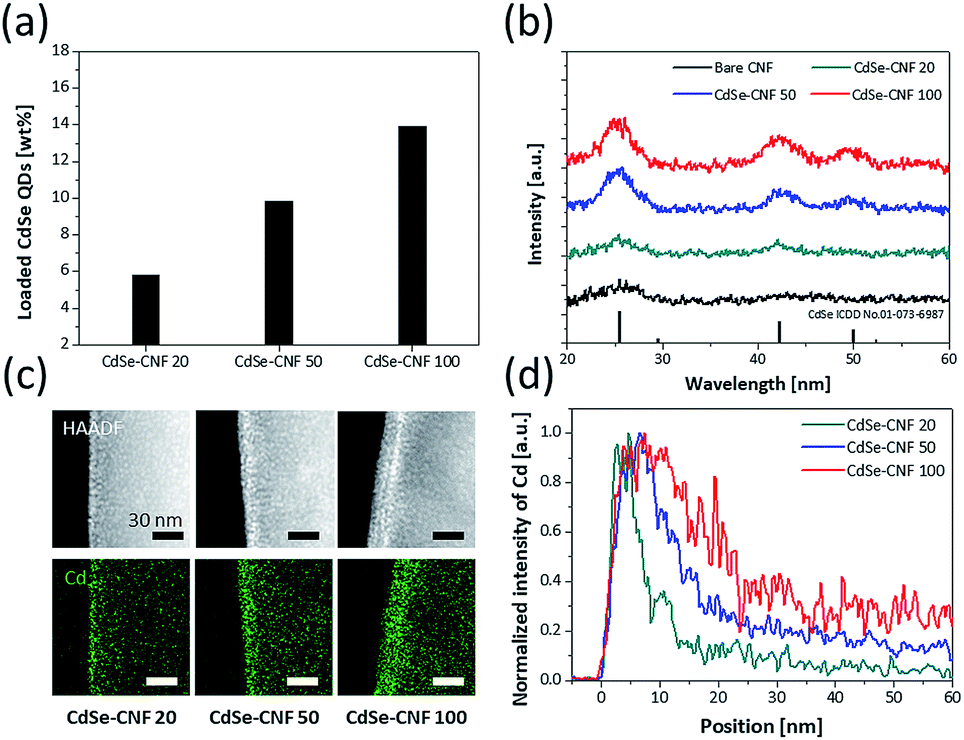 | ||
| Fig. 4 (a) Ratio of CdSe QDs with respect to CdSe–CNF for different CdSe QD coating conditions. (b) XRD patterns of bare CNFs and CdSe–CNFs. (c) TEM-EDS cadmium element mapping images of CdSe–CNFs at the edge site. (d) Normalized EDS mapping intensity of cadmium from the edge site. | ||
The absorbance and PL spectra of CdSe QDs dispersed in hexane are shown in Fig. 5a. The PL peak of the synthesized CdSe QDs is at 582.8 nm. In addition, Fig. 5a shows the PL spectra of the CdSe–CNFs and a CdSe QD layer on glass. The PL intensity and PL peak of the CdSe QD layer on glass were lower and shifted to a longer wavelength, respectively, when compared with those of CdSe QDs dispersed in hexane. In the latter case, the distance between QDs is relatively shorter than the distance between QDs dispersed in hexane solution. As the distance becomes shorter, non-radiative Förster resonance energy transfer (FRET) becomes more activated between QDs, and the PL peak shifts from 583.2 nm to 591.1 nm. The FRET enables excited electron–hole pairs in QDs not to emit light but to excite the other QDs in close proximity, and generates a red-shifted PL peak and lower PL intensity.27–29 In CdSe–CNFs, as the coating concentration increases, the PL intensity becomes relatively larger due to the increased amount of coated CdSe QDs on CNFs. This trend fits the ICP analysis and the XRD patterns (Fig. 4a and b). However, the PL intensities of all CdSe–CNFs are lower than the intensity of CdSe QDs in hexane, and even lower than the intensity of the CdSe QD layer on the glass substrate. In this regard, the reduced intensities of CdSe–CNFs mean that not only interaction between QDs but also interaction between QDs and CNF occurred.
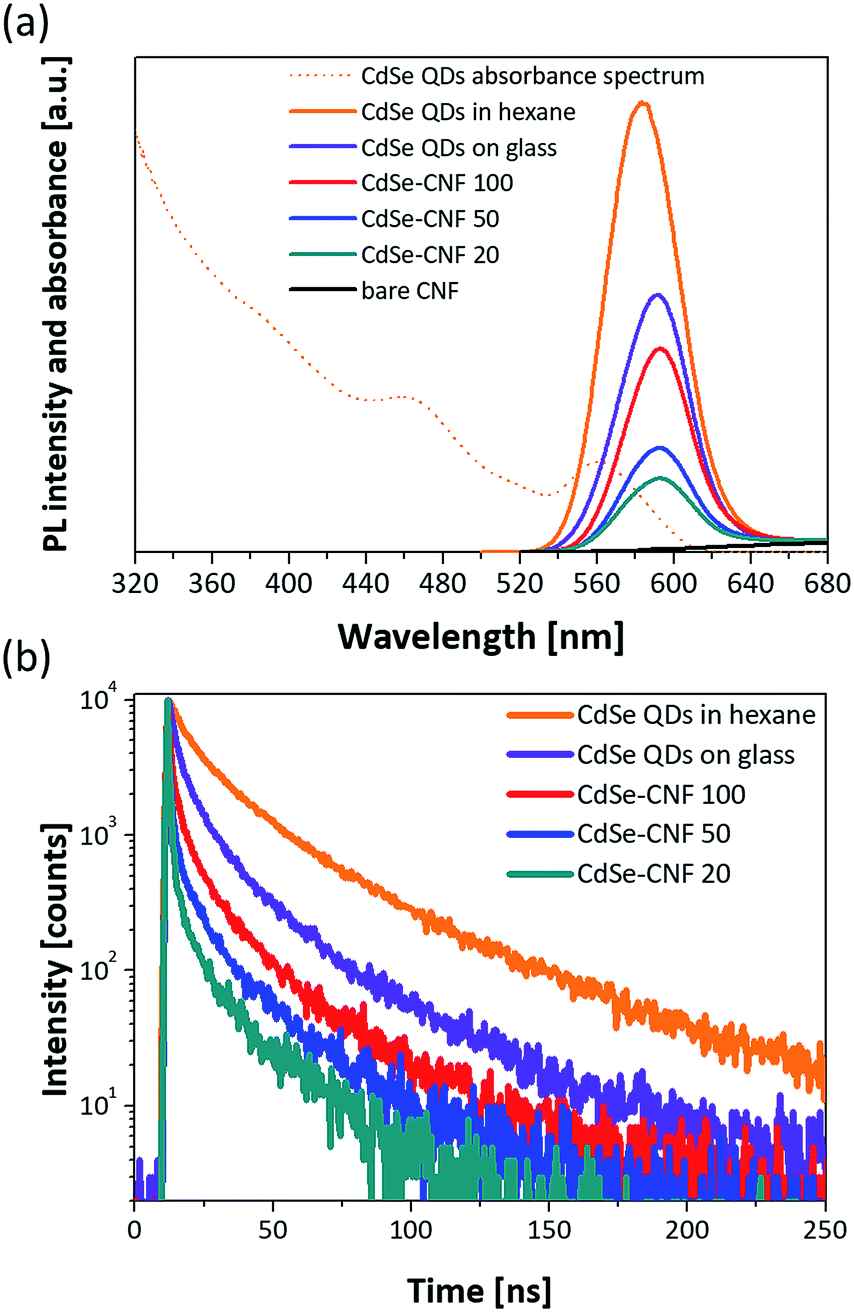 | ||
| Fig. 5 (a) Absorbance and PL spectra of the synthesized CdSe QDs dispersed in hexane and PL spectra of CdSe–CNFs and a CdSe QD layer on glass. (b) Transient PL decay curves of CdSe QDs in hexane, a CdSe QD layer on glass, and CdSe–CNFs. Every characterization is performed using a source of light (excitation wavelength = 470 nm). For the CdSe QD film on glass, the CdSe QDs (20 mg mL−1 in hexane) were spin coated on glass at 2000 rpm for 30 s. | ||
To verify this, the transient PL decay times of each sample were measured. Fig. 5b shows the transient PL decay curves for each condition. In Table 1, although the decay time of the CdSe QDs dispersed in hexane is 29.93 ns, the decay time of the CdSe QD layer on glass is 16.64 ns due to the faster non-radiative transitions of the FRET than the radiative recombination rate of the electron–hole pairs.28,30,31 Then, the decay times of all the CdSe–CNFs are shorter than that of the CdSe QD layer on glass with insulating properties. This means that non-radiative energy transfer and/or charge transfer occur at the junction of the CdSe QDs and the CNFs.32 The charge transfer is generated by different energy level between the CdSe QD and the CNF.33–39Fig. 6a shows the their energy levels. The LUMO and HOMO levels of CdSe QDs are −3.44 eV and −5.52 eV, respectively, as obtained by cyclic voltammetry measurements (Fig. S9†).40,41 The work function of CNFs is 4.33 eV obtained using ultraviolet photoelectron spectroscopy (Fig. S10†). Such a phenomenon can also be shown for CuInS2–CNF and InP–CNF, in which charge transfer also takes place (Fig. S11† and Table S3†).
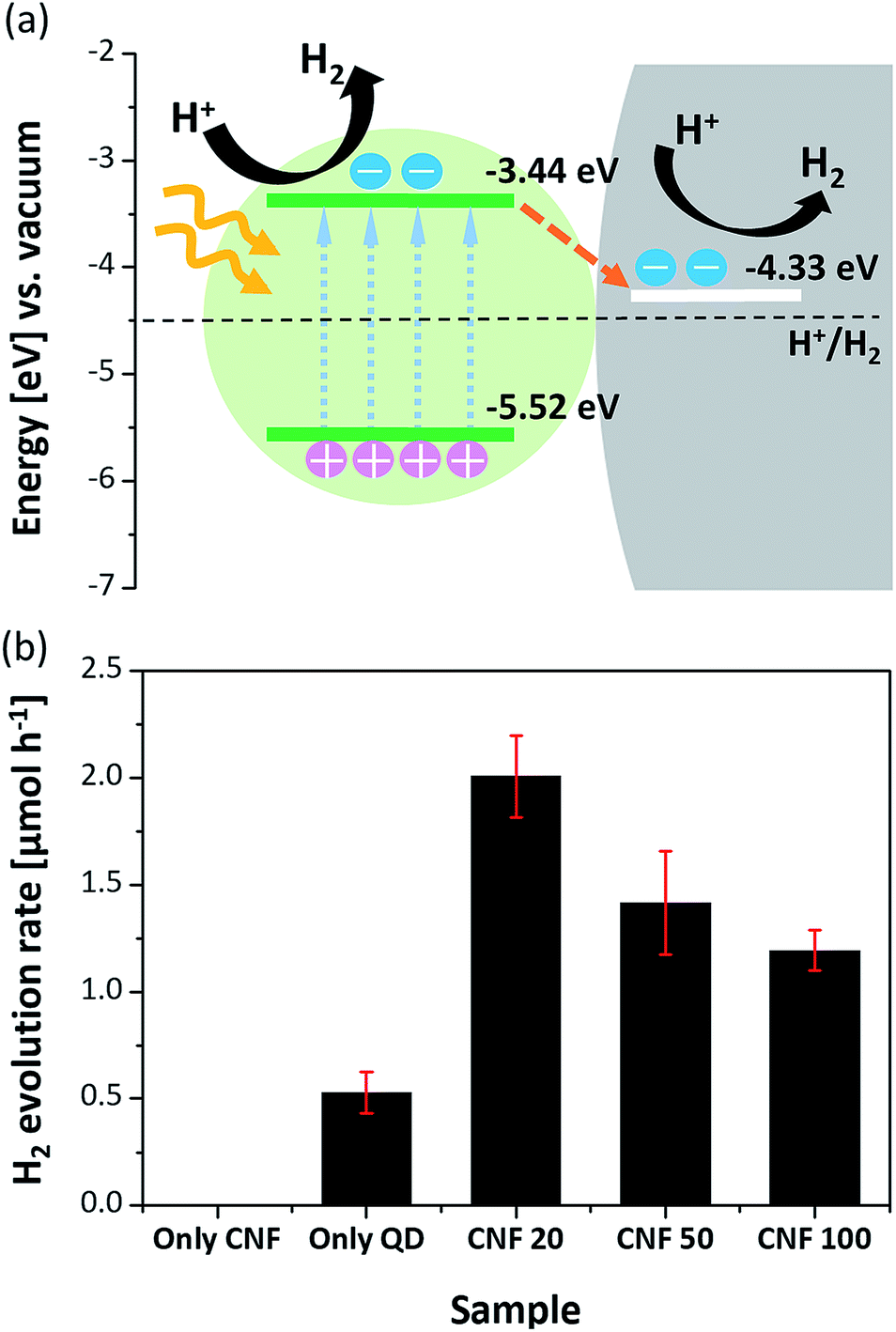 | ||
| Fig. 6 (a) Energy band alignment diagram and proposed photocatalytic mechanism for CdSe–CNF. (b) Photocatalytic hydrogen evolution rates of the CNFs, CdSe QDs, and CdSe-CNFs in 0.25 M Na2S/0.35 M Na2SO3 aqueous solution under visible light (λ ≥ 420 nm). | ||
The values in Table 1 were fitted with a biexponential function composed of fast (τ1) and slow (τ2) decay components assigned to a radiative and a non-radiative recombination process, respectively. The fast decay component of CdSe QD solution shifts from 10.00 ns to 0.97 ns for CdSe–CNF 20, and its proportion increases from 41.64% to 58.53%. The fast decay component, which is assigned to non-radiative recombination, means that the PL of CdSe QDs is quenched by the junctions.32,42 In addition, with decreasing proportion of the slow decay time component assigned to the radiative process, the PL intensity was reduced. While CdSe–CNF 20 has the shortest decay time (7.34 ns), CdSe–CNF 100 has a longer decay time of 12.15 ns. When the coating concentration is increased, the amount of coated CdSe QDs increases, and the CdSe QD layer on the CNF obviously becomes thicker. Then, the ratio of additional charge transfer at the interface between the QDs and the CNF is relatively reduced and the decay time is recovered. It is expected that the charge transfer is saturated at around 10 mg mL−1 CdSe QD concentration for dip-coating because the change of decay time from CdSe–CNF 10 to CdSe–CNF 20 is significantly larger compared with the change of decay time from CdSe–CNF 5 to CdSe–CNF 10. This means that most of the CNF surface is covered with CdSe QDs at a concentration of around 10 mg mL−1 (Fig. S12 and Table S4†). The junction between the QDs and the CNF, formed by the dip-coating process, is electrically well conductive and charges that are transferred to the CNF contribute to a superior performance of optoelectronics such as photocatalysts, photodetectors, sensors, and solar cells.22,32,38,42–49
To investigate the catalytic roles of the junctions that are present in CdSe–CNFs, photocatalytic hydrogen evolution was performed in an aqueous solution containing 0.25 M Na2S/0.35 M Na2SO3 as hole scavengers. CdSe QDs were chosen as the model catalyst, and the photocatalytic evolution rates were measured under the illumination of visible light (λ ≥ 420 nm) using a 300 W Xe lamp with a UV cut-off filter. To reveal the possible effect of the hybrid system, experiments with three distinct CdSe–CNFs with different QD contents (CdSe–CNF 20, CdSe–CNF 50, and CdSe–CNF 100) and control experiments with CNFs alone and QDs alone were performed. The amount of catalysts was standardized to be equal based on the 1 mg CdSe amount by ICP. As shown in Fig. 6b, the CdSe–CNFs exhibited activity superior to that of pure CdSe QDs, and the CNF alone showed no activity under visible light irradiation.13,50 In particular, CdSe–CNF 20 showed the highest hydrogen evolution rate of 2.01 μmol h−1, which was more than 3.8 times higher than that of the pure CdSe QDs (0.53 μmol h−1). The quantum yield of CdSe–CNF 20 was estimated to be 0.39% using a 420 nm LED light with an intensity value of 10 mW. In addition to measuring photocatalytic activity, a stability test was also undertaken, and it maintained the activity for three cycles of a 2 h reaction (Fig. S13†). Therefore, the result supported the idea that the CNF alone cannot play the role of a photocatalyst, but it can remarkably improve the photocatalytic activity. This improvement with the CNF can be explained by that the fact that the CNF induced rapid charge separation, which retarded the radiative recombination of excitons in CdSe QDs, and increased the catalytic efficiency. The expected mechanism for photocatalytic hydrogen evolution on CdSe QD-coated CNFs is as follows. Fig. 6a shows the difference in energy levels at the junction between the QDs and the CNF, and that the structure splits the photo-induced electron–hole pairs and causes electron transfer from CdSe QDs to the CNF under the illumination state.17,38 This rapid charge transfer decreases the probability of electron–hole recombination, and the remaining electrons are used for hydrogen reduction.
The ratio between QDs and CNFs significantly affects the catalytic efficiency. When comparing the three distinct samples, the amount of hydrogen production during 1 h irradiation was in the order of CdSe–CNF 20 (2.01 μmol h−1) > CdSe–CNF 50 (1.42 μmol h−1) > CdSe–CNF 100 (1.19 μmol h−1). The activity was relatively lower when the CdSe QD content was higher in the hybrid system. This was mainly caused by the thickness of the coated QD layer. As shown in Fig. 4, the thickness of the QD layer increases from about 5.4 nm to 18.8 nm with to the increase of the QD ratio, which means that the junction surface between the QDs and the CNF becomes saturated with the thicker layer. Therefore, it can be expected that in the case of CdSe–CNF 100, following visible light irradiation, a large amount of charge will remain in the QD layer instead of transferring to the CNF. Then, the electron–hole recombination amount will be larger when it is compared with that of CdSe–CNF 20. This expectation can be verified through the PL decay results. As indicated in Fig. 5b, CdSe–CNF 20 had the shortest decay time and that means the largest prevalence of charge separation. Therefore, CdSe–CNF 20 had the highest photocatalytic efficiency among the CdSe–CNFs due to the optimum ratio of QDs and CNFs that reduced the electron–hole recombination and increased the active electrons for catalytic reactions.
Additionally, photosensitivity was measured using the free-standing structure of CdSe–CNF 20, and it was significantly enhanced, with the augmentation of photo-current (Iph) with respect to dark-current (Idark) being almost 1.7 times greater with the introduction of CdSe QDs on CNFs (10.4%) compared with bare CNFs (6.3%) (Fig. S14†). Although the Idark is in a mA range which is not suitable for application in photodetectors at this stage, this structure demonstrates applicability to various optoelectronics. In accordance with the emerging trends of IoT technology, the uncomplicated coating of QDs on CNFs that can be used as a flexible/conductive free-standing substrate is tremendously expected to broaden the technical spectrum of QDs. Furthermore, the platform would be applicable to photodetectors, solar cells, and gas sensors along with photocatalysts. For these applications, it is critical to use a conductive free-standing platform.
Conclusions
In this work, for the first time, we have suggested a very comprehensive and feasible strategy to prepare a conductive and free-standing QD-coated platform by a dip-coating process. The method was tried for various kinds of QDs, and morphological analysis confirms that they were well decorated on the surface of CNFs. Compositional analyses further confirm that the deposited amount of CdSe QDs on CNFs can be tuned by the concentration of QD solution during the dip-coating. The junction properties between CdSe QDs and CNFs were investigated by PL analyses and induced charge transfer from CdSe QDs to CNFs. As one of the possible applications based on the junction, various QD-coated CNFs were tested for photocatalytic hydrogen production from water splitting. As a result, this structure shows that at the same amount of CdSe QDs (1 mg), the CdSe–CNFs exhibited higher efficiency for hydrogen production than using CdSe QDs only. Especially, CdSe–CNF 20 showed the highest hydrogen production rate of 2.01 μmol h−1, more than 3.8 times compared with using the CdSe QDs only. Additionally, we verified that the free-standing CdSe–CNF platform has potential for photosensitive applications. The method suggested in this paper can be applied to various QDs, which can be extended for a number of other applications utilizing QDs as a unique physicochemical medium, such as solar cells, photodetectors, and gas sensors as well as photocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Creative Materials Discovery Program through the National Research Foundation (NRF) of Korea funded by the Ministry of Science and ICT (NRF-2016M3D1A1900035). And this work was supported by the National Research Foundation of Korea (NRF), grant no. 2014R1A4A1003712 (BRL Program), the funding from the Korea CCS R&D Center (KCRC) grant by the Korean Government (Ministry of Science, ICT & Future Planning) (No. NRF-2014M1A8A1049303), the Wearable Platform Materials Technology Center (WMC) (NR-2016R1A5A1009926), and the NRF (National Research Foundation of Korea) Grant funded by the Korean Government (NRF-2017H1A2A1042006-Global Ph.D. Fellowship Program). This work was also supported by the National Research Foundation of Korea (NRF) funded by the Korean Government (MSIP) (NRF-2018R1A2B3004096 and NRF-2018R1A5A1025208).References
- M. Nam, J. Park, S.-W. Kim and K. Lee, J. Mater. Chem. A, 2014, 2, 3978–3985 RSC.
- Y. Shirasaki, G. J. Supran, M. G. Bawendi and V. Bulović, Nat. Photonics, 2013, 7, 13 CrossRef CAS.
- S. Pathak, E. Cao, M. C. Davidson, S. Jin and G. A. Silva, J. Neurosci., 2006, 26, 1893–1895 CrossRef CAS PubMed.
- R. Mo, Z. Lei, K. Sun and D. Rooney, Adv. Mater., 2014, 26, 2084–2088 CrossRef CAS PubMed.
- D. Kufer, I. Nikitskiy, T. Lasanta, G. Navickaite, F. H. Koppens and G. Konstantatos, Adv. Mater., 2015, 27, 176–180 CrossRef CAS PubMed.
- S. A. McDonald, G. Konstantatos, S. Zhang, P. W. Cyr, E. J. Klem, L. Levina and E. H. Sargent, Nat. Mater., 2005, 4, 138 CrossRef CAS PubMed.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CrossRef CAS.
- L. Li, L. Gu, Z. Lou, Z. Fan and G. Shen, ACS Nano, 2017, 11, 4067–4076 CrossRef CAS PubMed.
- M. Chen, H. Lu, N. M. Abdelazim, Y. Zhu, Z. Wang, W. Ren, S. V. Kershaw, A. L. Rogach and N. Zhao, ACS Nano, 2017, 11, 5614–5622 CrossRef CAS PubMed.
- X. Liu, X. Ji, M. Liu, N. Liu, Z. Tao, Q. Dai, L. Wei, C. Li, X. Zhang and B. Wang, ACS Appl. Mater. Interfaces, 2015, 7, 2452–2458 CrossRef CAS PubMed.
- W. Zeng, L. Shu, Q. Li, S. Chen, F. Wang and X. M. Tao, Adv. Mater., 2014, 26, 5310–5336 CrossRef CAS PubMed.
- J. S. Heo, J. Eom, Y. H. Kim and S. K. Park, Small, 2018, 14, 1703034 CrossRef PubMed.
- Y. Chen, G. Tian, Z. Ren, K. Pan, Y. Shi, J. Wang and H. Fu, ACS Appl. Mater. Interfaces, 2014, 6, 13841–13849 CrossRef CAS PubMed.
- X. Zhang, Y. Chen, Y. Xiao, W. Zhou, G. Tian and H. Fu, Nanoscale, 2018, 10, 4041–4050 RSC.
- Y. K. Kim, M. Kim, S.-H. Hwang, S. K. Lim, H. Park and S. Kim, Int. J. Hydrogen Energy, 2015, 40, 136–145 CrossRef CAS.
- S. Yue, B. Wei, X. Guo, S. Yang, L. Wang and J. He, Catal. Commun., 2016, 76, 37–41 CrossRef CAS.
- J. Mu, C. Shao, Z. Guo, Z. Zhang, M. Zhang, P. Zhang, B. Chen and Y. Liu, ACS Appl. Mater. Interfaces, 2011, 3, 590–596 CrossRef CAS PubMed.
- K. R. Yoon, K. Shin, J. Park, S.-H. Cho, C. Kim, J.-W. Jung, J. Y. Cheong, H. R. Byon, H. M. Lee and I.-D. Kim, ACS Nano, 2017, 12, 128–139 CrossRef PubMed.
- C.-L. Lee, C. Kim and I.-D. Kim, RSC Adv., 2017, 7, 44804–44808 RSC.
- J.-H. Cha, S.-J. Choi, S. Yu and I.-D. Kim, J. Mater. Chem. A, 2017, 5, 8725–8732 RSC.
- J. Y. Cheong, J.-W. Jung, D.-Y. Youn, C. Kim, S. Yu, S.-H. Cho, K. R. Yoon and I.-D. Kim, J. Power Sources, 2017, 360, 434–442 CrossRef CAS.
- H. Li, H. Yu, S. Chen, H. Zhao, Y. Zhang and X. Quan, Dalton Trans., 2014, 43, 2888–2894 RSC.
- J. Oliva, C. Gomez-Solis, L. Diaz-Torres, A. Martinez-Luevanos, A. Martinez and E. Coutino-Gonzalez, J. Phys. Chem. C, 2018, 122, 1477–1485 CrossRef CAS.
- K. R. Yoon, J. Choi, S.-H. Cho, J.-W. Jung, C. Kim, J. Y. Cheong and I.-D. Kim, J. Power Sources, 2018, 380, 174–184 CrossRef CAS.
- C. Murray, C. Kagan and M. Bawendi, Science, 1995, 270, 1335–1338 CrossRef CAS.
- D. Jariwala, T. J. Marks and M. C. Hersam, Nat. Mater., 2017, 16, 170 CrossRef CAS PubMed.
- J. J. Choi, J. Luria, B.-R. Hyun, A. C. Bartnik, L. Sun, Y.-F. Lim, J. A. Marohn, F. W. Wise and T. Hanrath, Nano Lett., 2010, 10, 1805–1811 CrossRef CAS PubMed.
- R. Koole, P. Liljeroth, C. de Mello Donegá, D. Vanmaekelbergh and A. Meijerink, J. Am. Chem. Soc., 2006, 128, 10436–10441 CrossRef CAS PubMed.
- L. Sun, J. J. Choi, D. Stachnik, A. C. Bartnik, B.-R. Hyun, G. G. Malliaras, T. Hanrath and F. W. Wise, Nat. Nanotechnol., 2012, 7, 369 CrossRef CAS PubMed.
- M. Achermann, M. A. Petruska, S. A. Crooker and V. I. Klimov, J. Phys. Chem. B, 2003, 107, 13782–13787 CrossRef CAS.
- S. Crooker, J. Hollingsworth, S. Tretiak and V. I. Klimov, Phys. Rev. Lett., 2002, 89, 186802 CrossRef CAS PubMed.
- I. V. Lightcap and P. V. Kamat, J. Am. Chem. Soc., 2012, 134, 7109–7116 CrossRef CAS PubMed.
- J. Lim, B. G. Jeong, M. Park, J. K. Kim, J. M. Pietryga, Y. S. Park, V. I. Klimov, C. Lee, D. C. Lee and W. K. Bae, Adv. Mater., 2014, 26, 8034–8040 CrossRef CAS PubMed.
- P. P. Jha and P. Guyot-Sionnest, J. Phys. Chem. C, 2010, 114, 21138–21141 CrossRef CAS.
- X. Dai, Z. Zhang, Y. Jin, Y. Niu, H. Cao, X. Liang, L. Chen, J. Wang and X. Peng, Nature, 2014, 515, 96 CrossRef CAS PubMed.
- K.-T. Kuo, D.-M. Liu, S.-Y. Chen and C.-C. Lin, J. Mater. Chem., 2009, 19, 6780–6788 RSC.
- B. S. Mashford, M. Stevenson, Z. Popovic, C. Hamilton, Z. Zhou, C. Breen, J. Steckel, V. Bulovic, M. Bawendi and S. Coe-Sullivan, Nat. Photonics, 2013, 7, 407 CrossRef CAS.
- Z. Zheng, L. Gan, J. Zhang, F. Zhuge and T. Zhai, Adv. Sci., 2017, 4, 1600316 CrossRef PubMed.
- J. Yu, T. Ma and S. Liu, Phys. Chem. Chem. Phys., 2011, 13, 3491–3501 RSC.
- S. N. Inamdar, P. P. Ingole and S. K. Haram, ChemPhysChem, 2008, 9, 2574–2579 CrossRef CAS PubMed.
- S. K. Haram, A. Kshirsagar, Y. D. Gujarathi, P. P. Ingole, O. A. Nene, G. B. Markad and S. P. Nanavati, J. Phys. Chem. C, 2011, 115, 6243–6249 CrossRef CAS.
- K.-A. Tsai and Y.-J. Hsu, Appl. Catal., B, 2015, 164, 271–278 CrossRef CAS.
- S. Min, J. Hou, Y. Lei, X. Ma and G. Lu, Appl. Surf. Sci., 2017, 396, 1375–1382 CrossRef CAS.
- K. Woan, G. Pyrgiotakis and W. Sigmund, Adv. Mater., 2009, 21, 2233–2239 CrossRef CAS.
- Y.-X. Pan, J.-B. Peng, S. Xin, Y. You, Y.-L. Men, F. Zhang, M.-Y. Duan, Y. Cui, Z.-Q. Sun and J. Song, ACS Sustainable Chem. Eng., 2017, 5, 5449–5456 CrossRef CAS.
- G. Konstantatos, M. Badioli, L. Gaudreau, J. Osmond, M. Bernechea, F. P. G. De Arquer, F. Gatti and F. H. Koppens, Nat. Nanotechnol., 2012, 7, 363 CrossRef CAS PubMed.
- Z. Yang, M. Wang, X. Song, G. Yan, Y. Ding and J. Bai, J. Mater. Chem. C, 2014, 2, 4312–4319 RSC.
- D. Zhang, L. Gan, Y. Cao, Q. Wang, L. Qi and X. Guo, Adv. Mater., 2012, 24, 2715–2720 CrossRef CAS PubMed.
- Z. Sun, Z. Liu, J. Li, G. a. Tai, S. P. Lau and F. Yan, Adv. Mater., 2012, 24, 5878–5883 CrossRef CAS PubMed.
- Y. Lei, C. Yang, J. Hou, F. Wang, S. Min, X. Ma, Z. Jin, J. Xu, G. Lu and K.-W. Huang, Appl. Catal., B, 2017, 216, 59–69 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9na00423h |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |