
Visible-light-active g-C3N4/N-doped Sr2Nb2O7 heterojunctions as photocatalysts for the hydrogen evolution reaction†
Shiba P.
Adhikari‡
 ab,
Zachary D.
Hood§
cd,
Vincent W.
Chen
c,
Karren L.
More
d,
Keerthi
Senevirathne
e and
Abdou
Lachgar
*ab
ab,
Zachary D.
Hood§
cd,
Vincent W.
Chen
c,
Karren L.
More
d,
Keerthi
Senevirathne
e and
Abdou
Lachgar
*ab
aDepartment of Chemistry, Wake Forest University, Winston-Salem, NC 27109, USA. E-mail: lachgar@wfu.edu; Fax: +1-336-758-4656; Tel: +1-336-758-4676
bCenter for Energy, Environment and Sustainability (CEES), Wake Forest University, Winston-Salem, NC 27109, USA
cSchool of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA
dCenter for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, TN 37831, USA
eDepartment of Chemistry, Florida Agricultural and Mechanical University, Tallahassee, FL 32307, USA
First published on 29th August 2018
Abstract
Different visible-light-active g-C3N4/nitrogen-doped Sr2Nb2O7 heterojunction photocatalysts were fabricated by deposition of graphitic carbon nitride over N-doped strontium pyro-niobate prepared by ammonolysis at different temperatures of solvothermally synthesized Sr2Nb2O7 nanorods. Their photocatalytic performance was determined by the amount of hydrogen generated from water reduction under visible light irradiation. The best performing heterojunction was found to be the one formed by g-C3N4 and N-doped Sr2Nb2O7 obtained at 700 °C. The enhanced activity of the heterojunction can be explained by better charge separation due to proper bands alignment and intimate contact between the heterojunction components as revealed by electron microscopy. A mechanism for the observed enhanced photocatalytic activity is proposed and supported by band position calculations and photoluminescence data.
1. Introduction
Semiconductor-based heterogeneous photocatalysis utilizing solar energy is a green technology that has the potential to address the environmental impacts of fossil fuels. The discovery of photo-electrochemical splitting of water using n-TiO2 anode in the early 1970s led to the study of numerous semiconductors such as oxides, sulfides, and oxynitrides as potential photocatalysts for solar fuels generation and environmental remediation.1–5 However, most photocatalysts studied have wide band gaps and therefore are active only under UV irradiation. To maximize the use of solar radiation, the photo-response of the catalysts should be in the visible region of the electromagnetic spectrum since visible light accounts for about half of the solar energy spectrum while UV radiation makes up approximately 4%. Therefore, developing photocatalysts with a high activity and good photochemical stability under visible light is highly desirable.6–9To overcome the limitation that most oxides are only UV responsive, different methods have been developed to extend their light absorption range into the visible. These include doping with metal and/or non-metal ions and surface modifications such as the formation of heterojunctions by combining the oxide with metals or other semiconductors. Cation doping creates acceptor states below the conduction band, and anion doping creates donor states above the valence band. The net result is smaller band gaps. The study of N-doped TiO2 by Asahi and co-workers in 2001,7 led to extensive studies on visible-light-active semiconductors doped with non-metallic elements such as nitrogen, sulfur, and carbon. Irie et al. described the effect of the amount of nitrogen concentration on the photocatalytic activity of TiO2−xNx microcrystalline powders prepared by annealing TiO2 anatase in ammonia.10 Anion (nitrogen and sulfur) doping was extended to oxides with layered perovskite or pyrochlore structure-types. Thus, several nitrogen-doped (N-doped) perovskites and pyrochlores including NaTaO3, SrTiO3, Sr2Ta2O7, Ba5Ta4O15, CsTaWO6, Ca2Nb3O10, CsCa2Ta3O10 have been reported to possess wide visible light absorption.11–18
Semiconductor heterojunctions have emerged as an effective approach to inhibit charge carrier recombination and maximize visible light absorption.19–22 Properly designed heterojunctions have been shown to lead to separation of photogenerated electron–hole pairs, thus increasing their lifetime so they can participate in surface redox processes. A number of heterojunctions were fabricated and their photocatalytic performances were examined in a variety of applications including water splitting, CO2 reduction, and toxic organics degradation.23–27
In the search for robust and stable visible light active photocatalysts, graphitic carbon nitride (g-C3N4) with a band-gap of about 2.7 eV was studied extensively.28,29 In comparison to metal-based photocatalysts, g-C3N4 can be easily prepared by thermal polycondensation of nitrogen-rich organic precursors such as cyanamide, dicyandiamide, melamine, and urea.28,30–33 However, the photocatalytic efficiency of g-C3N4 is unsatisfactory due to high recombination rate of photogenerated electron–hole pairs, thus several g-C3N4 containing heterojunctions have been successfully fabricated and found to display significantly higher photocatalytic activity under visible light irradiation.34–48
Taking into account the benefits of designing and constructing heterojunction photocatalysts, we recently reported the photocatalytic activity of g-C3N4/Sr2Nb2O7.49 The incorporation of g-C3N4 in Sr2Nb2O7 not only extends light absorption range of Sr2Nb2O7 but more importantly, significantly enhances its photocatalytic activity. The rate of photocatalytic hydrogen production from water reduction under visible light was found to be 11 times larger for the g-C3N4/Sr2Nb2O7 heterojunction compared to g-C3N4. The choice of Sr2Nb2O7 for the construction of g-C3N4/Sr2Nb2O7 heterojunction was purely based on the presence of layered structure, which has attracted wide interest owing to its high quantum yield for photocatalytic water splitting.50,51 We have extended this work by studying the photocatalytic activity of heterojunctions based on g-C3N4 and N-doped Sr2Nb2O7 synthesized by ammonolysis at different temperatures of solvothermally prepared Sr2Nb2O7 in order to further enhance the photocatalytic performance of the semiconductor heterojunction. This study gives the comparative outlook of the effect of N-doping on photocatalytic activities of heterojunctions by taking into the account of N doped and undoped Sr2Nb2O7 while forming the heterojunction with g-C3N4. Although it has been reported that the N doping of niobate based semiconductors suffers from damage due to nitridization during ammonolysis,52,53 there is not any report out there comparing the effect of N doping on Sr2Nb2O7 and studying the photocatalytic effects by creating the heterojunction photocatalysts. The heterojunctions and their individual components were characterized by a number of analytic techniques, including PXRD, FTIR, TGA/DSC, STEM, SEM-EDS, TEM, DRS, PL, EIS, and BET surface area. Their photocatalytic performance was evaluated using photocatalytic reduction of water under visible light irradiation in the presence of methanol as a hole scavenger. The heterojunctions displayed better performance than their individual components. The heterojunction CN/SNON-700 was found to exhibit a remarkably enhanced photocatalytic activity of 128.8 mmol H2 h−1 mol−1 of CN, which is ∼14× times larger than the amount of hydrogen generated from one mole of pristine CN. The enhancement factor associated for this N-doped heterojunction is significantly larger than that of the g-C3N4/Sr2Nb2O7 heterojunction.49 A mechanism for the observed enhanced photocatalytic activity is proposed based on the observed activity and band position calculations.
2. Experimental section
2.1 Synthesis of strontium niobate (SNO) and N-doped strontium niobate (SNON-X; ‘X’ indicates the temperature used for ammonolysis)
Strontium pyroniobate (SNO) was prepared hydrothermally using niobium chloride (NbCl5, Alfa Aesar, 99.9%) and strontium nitrate (Sr(NO3)2, Sigma-Aldrich, 99.0%) as reported in our previous work.49 In a typical reaction, a solution of NbCl5 in ethanol (5 mmol (1.350 g) dissolved in 10 mL of ethanol) was added dropwise to an aqueous solution of Sr(NO3)2 (5 mmol (1.058 g) of Sr(NO3)2 dissolved in 10 mL of deionized water). An aqueous solution of NaOH (2 M) was added to maintain the pH of the mixture at 10. A white precipitate formed after the mixture was stirred for 30 min at RT. The mixture was transferred to a 100 mL autoclave, sealed, and heated at 220 °C for 24 h (heating and cooling rate of 5 °C min−1). The strontium pyroniobate obtained (white powder, 600 mg in each case) was subjected to ammonolysis using dry NH3 (flow rate: 200 mL min−1) for 2 hours in a tube furnace at 600 °C, 700 °C, 800 °C, and 950 °C (heating and cooling rates of 10 °C min−1). The color of N-doped products ranged from yellow-white to brown as the ammonolysis temperature increased. The N-doped samples were labeled as SNON-X where X represents the temperature at which they were prepared (600, 700, 800, or 950 °C).2.2 Synthesis of graphitic carbon nitride (g-C3N4) and heterojunctions
Graphitic carbon nitride (CN) was prepared by thermal decomposition of melamine at 525 °C in a muffle furnace for 4 hours at a heating rate of 5 °C min−1. The g-C3N4/N-doped Sr2Nb2O7 heterojunctions were prepared by mixing a suspension of g-C3N4 (200 mg of g-C3N4 sonicated in 25 mL of methanol for 1 hour) and a suspension of SNON-X 200 mg of SNON-X sonicated in 25 mL of methanol for 1 hour. The two suspensions were mixed and magnetically stirred at 50 °C. After 5 hours, the product was filtered and dried, thoroughly ground, then calcined at 400 °C for 4 hours under argon flow (100 mL min−1).2.3 Characterization
Powder X-ray diffraction (PXRD) data were collected on a Bruker-D2 Phaser diffractometer, with Cu-Kα radiation (λ = 0.15418 nm, 30 kV, and 10 mA). Scanning electron microscopy (SEM) images were collected on a Zeiss Ultra60 FE-SEM with an operating voltage at 5.0 kV. Energy-dispersive X-ray spectroscopy (EDS) with elemental mappings were completed on an Oxford Instruments X-MaxN at 10.0 kV. The microstructure and elemental mapping were performed on a scanning transmission electron microscope (STEM, Hitachi HF3300) at 300 kV. Differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) was completed using an SDT-Q600 (TA instrument, USA) from 32–800 °C under constant airflow (100 mL min−1) and a heating rate of 10 °C min−1. X-ray photoelectron spectroscopy (XPS) was performed using Kratos Axis Ultra DLD X-ray Photoelectron Spectrometer. The samples were analyzed as powders mounted on small pieces of indium metal. Brunauer–Emmett–Teller (BET) surface areas were determined from nitrogen adsorption isotherms at 77 K using Autosorb-iQ from Quantachrome. Diffuse reflectance UV-Vis spectra (DRS) were collected on an Ocean Optics FLAME-S-XR1-ES spectrophotometer equipped with an integrating sphere. Fourier transform infrared spectra (FTIR) were recorded on Spectrum 100 spectrometer (PerkinElmer). Photoluminescence (PL) and time-resolved fluorescence emission decay measurements were collected on a Horiba JobinYvon Fluorolog Fluorimeter (Model no. FL-1057) using a 450 W Xe lamp/monochromator combination and a pulsed LED (Model NanoLED N-330), respectively, as the excitation sources. The excitation monochromator was set at 336 nm to match the output of the pulsed LED for all samples for both emission and lifetime measurements. The powder samples were compacted into the sample holder (Model no. 1933) which was oriented at 60° with respect to the excitation source to minimize scattering. All measurements were performed at room temperature. The emission signals were fitted using tri-exponential decay. The average lifetime (τavg) was determined by the following equation: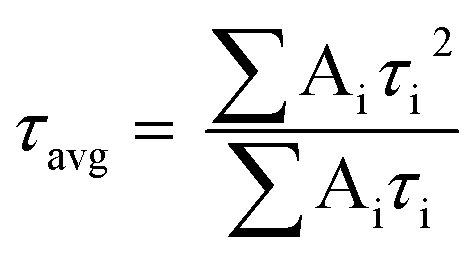 | (1) |
Electrochemical impedance spectra (EIS) were collected with a Gamry 3000 electrochemical workstation using a conventional three-electrode system consisting of a fluorine-doped tin oxide (FTO) glass as the working electrode, Ag/AgCl as the reference electrode, and a platinum wire as the counter electrode. The measurements were performed using a 5.0 mV AC voltage signal at 1 V vs. Ag/AgCl between 100 kHz to 0.1 Hz in 0.5 M Na2SO4 solution. The FTO electrode was prepared by spin coating a slurry of photocatalyst on FTO glass (dimensions: 1 cm × 1 cm) and drying in an oven at 60 °C.
2.4 Photocatalytic tests
The photocatalytic performance was determined using a 300 W Xenon lamp (Newport Corporation) was used as the light source with a 420 nm cut-off filter to provide visible light irradiation only. All experiments were performed at 25 °C using a water-jacketed photocatalytic cell. In a typical photocatalytic reaction, 100 mg of the photocatalyst was dispersed in an aqueous solution of methanol (5 mL MeOH and 45 mL of deionized H2O). Before illumination, the suspension was degassed with nitrogen gas for 30 min. The solution was exposed to visible light irradiation (λ ≥ 420 nm) under magnetic stirring. The amount of hydrogen produced was quantified by gas chromatography (HP 6890 with TCD, argon carrier). For recyclability and stability tests, the suspension was degassed with nitrogen gas for 30 min before each cycle. In all cases, Pt co-catalyst (2.5% by weight) was photo-deposited on the heterojunction particles using chloroplatinic acid hexahydrate (H2PtCl6·6H2O, Sigma-Aldrich) as the Pt source. In a typical reaction, 100 mg of catalyst and 6.63 mg of chloroplatinic acid hexahydrate were introduced into a pyrex photocatalytic cell with 50 mL of aqueous solution containing 10 mL of methanol. The suspension was irradiated with UV light for 4 hours with constant magnetic stirring. The gray colored Pt-coated samples were recovered by centrifugation and dried in vacuum at 60 °C for 4 hours before subsequent use. The Pt co-catalyst was loaded in all photocatalyst samples including the heterogeneous mixture (blend sample), which was used to check the photocatalytic enhancement as a control experiment. The apparent quantum yield (AQY) was calculated using the following equation: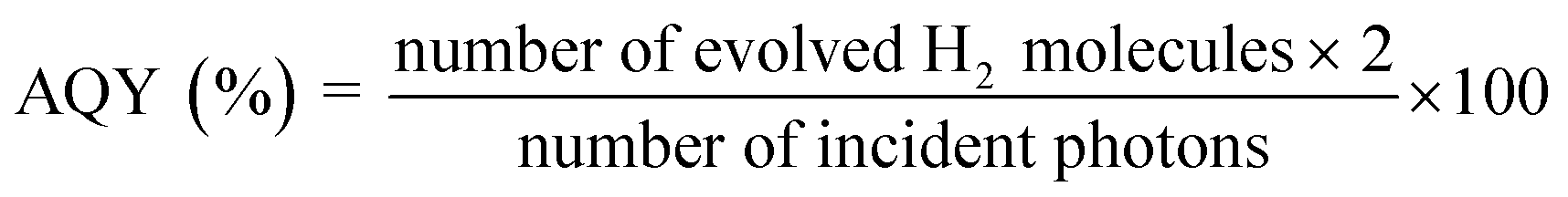 | (2) |
The average irradiation intensity (with bandpass filter at λ = 420 nm) was determined to be 0.45 W cm−2 with the irradiation area of 4.12 cm2. The numbers of incident photons (N) were calculated to be 2.80 × 1021 for one hour of irradiation. The turnover number (TON) and turnover frequencies (TOF) were calculated using the following equations.
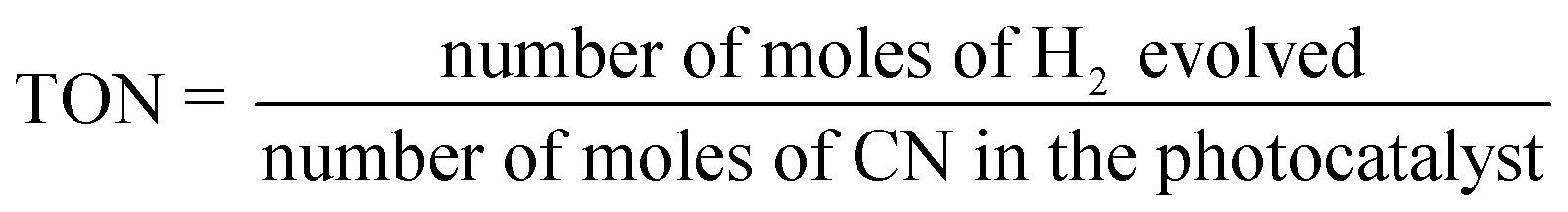 | (3) |
 | (4) |
To investigate the photocatalytic recyclability, three runs of the photocatalytic hydrogen generation were performed for 15 h. The water solution containing the heterojunction photocatalyst (CN/SNON-700) used for the hydrogen generation was degassed after each run by bubbling argon gas through the solution.
3. Results and discussions
3.1 Characterizations
Powder X-ray diffraction (PXRD) patterns of Sr2Nb2O7 and N-doped Sr2Nb2O7 (SNON) at different temperature are shown in Fig. 1a. The PXRD pattern for Sr2Nb2O7 obtained by the solvothermal route is consistent with that of layered perovskite prepared by high-temperature solid–state reactions.50,54 The PXRD pattern of SNON at temperatures below 800 °C are similar to the parent oxide. The PXRD of SNON-950 indicates the formation of the cubic oxynitride SrNbO2N,55 as previously reported by Guo et al.56 Elemental analysis of different SNON-X showed that nitrogen content increases with increasing ammonolysis temperature (Table 1).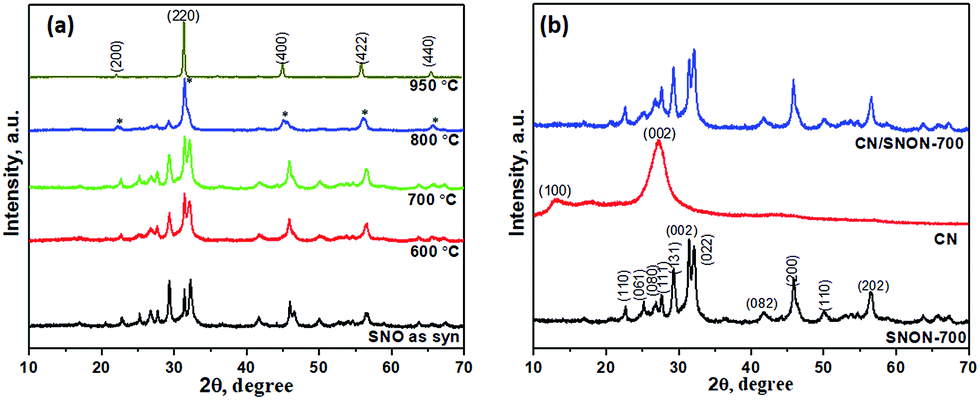 | ||
| Fig. 1 (a) PXRD patterns of SNO and different SNON-X. (b) The PXRD pattern of C3N4 and heterojunctions based on C3N4 and SNON-700 (* represents the peaks due to oxynitride phase which appears as low as 800 °C). | ||
| Sample SNON-X | Band gap, eV | Band positions based on VB XPS, eV vs. NHE | Nitrogen content, wt% | Color | Surface area, m2 g−1 | |
|---|---|---|---|---|---|---|
| VB | CB | |||||
| SNO | 3.58 | +2.65 | −0.93 | 0 | White | 64.7 |
| SNON-600 | 2.85 | +1.93 | −0.92 | 0.23 | Light yellow | 25.1 |
| SNON-700 | 2.60 | +1.70 | −0.90 | 0.73 | Yellow | 20.3 |
| SNON-800 | 2.20 | +1.30 | −0.90 | 1.72 | Green yellow | 11.8 |
| SNON-950 | 2.00 | +1.10 | −0.90 | 5.19 | Brown | 8.2 |
PXRD patterns of the SNON-700, CN and CN/SNON-700 are shown in Fig. 1b. The diffraction peaks at 13.0° and 27.3° for pure CN were indexed in the hexagonal unit cell (cell parameters: a = 6.498(5) Å, c = 6.702(4) Å, and space group: P![[6 with combining macron]](https://www.rsc.org/images/entities/char_0036_0304.gif) ) as (100) and (002) (JCPDS 87-1526).29,57,58 The PXRD of SNON-700 was indexed in the orthorhombic unit cell of Sr2Nb2O7 (JCPDS 70-9937) (a = 3.94269(5) Å, b = 26.2057(1) Å, and c = 5.6821(9) Å, and space group: Cmc21).49 Since diffraction peaks of CN could not be clearly observed in the CN/SNON-700 heterojunction due to peak overlap, FTIR of CN/SNON-700 was collected and showed strong absorption bands in the 1200–1650 cm−1 region ascribed to stretching modes of CN heterocycles, and a strong band at ∼804 cm−1 that corresponds to the characteristic stretching band of tris-triazine units, confirming the presence of CN (Fig. S1, ESI†).41,59
) as (100) and (002) (JCPDS 87-1526).29,57,58 The PXRD of SNON-700 was indexed in the orthorhombic unit cell of Sr2Nb2O7 (JCPDS 70-9937) (a = 3.94269(5) Å, b = 26.2057(1) Å, and c = 5.6821(9) Å, and space group: Cmc21).49 Since diffraction peaks of CN could not be clearly observed in the CN/SNON-700 heterojunction due to peak overlap, FTIR of CN/SNON-700 was collected and showed strong absorption bands in the 1200–1650 cm−1 region ascribed to stretching modes of CN heterocycles, and a strong band at ∼804 cm−1 that corresponds to the characteristic stretching band of tris-triazine units, confirming the presence of CN (Fig. S1, ESI†).41,59
Diffuse reflectance UV-Vis spectra of all compounds studied are shown in Fig. 2a. The parent oxide, Sr2Nb2O7, possess strong absorption in UV (λ ∼ 350 nm), while all SNON-X possess strong absorption in the visible (λ > 420 nm). Optical band gap energy of the semiconductors studied was determined by fitting the absorption spectra with the following equation:60
| αhν = A(hν − Eg)n/2 | (5) |
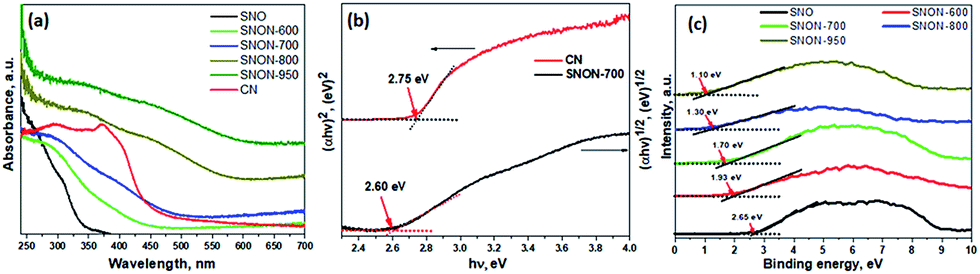 | ||
| Fig. 2 (a) UV-vis diffuse reflectance spectroscopy for different samples; (b) corresponding Tauc plots for CN and SNON-700; (c) VB-XPS analysis for Sr2Nb2O7 and all N-doped samples. | ||
The band gap of SNON-X (Table 1) decreases with increasing nitrogen content and temperatures and are consistent with the colors of the samples. The band positions were calculated based on band gaps, and VB positions obtained from VB-XPS data (Fig. 2c). The CB positions were calculated using the following equation:
| ECB = EVB − Eg | (6) |
As expected, the CB positions are virtually the same for all SNON-X, while the top of the VB is significantly affected by different N-doping resulting from ammonolysis at different temperatures (Table 1). Surface areas of SNON-X decreased with increasing sintering temperatures as shown in Table 1. Hydrothermally synthesized SNO has larger surface area than that of Sr2Nb2O7 synthesized by solid–state reactions at high temperatures.54,67–69 Additionally, surface areas of SNON-X were found to be larger than nitridized SNO reported by Guo et al.56 Similar trend in surface area was observed for the heterojunctions (Table S1, ESI†).
SEM images of SNON-X (Fig. S2†) clearly show that the nanoplate morphology of the parent oxide (SNO) is maintained in SNON-600 and SNON-700, while larger particles were observed in SNON-800 and SNON-950 due to higher sintering temperatures. SEM images EDS mapping of different heterojunctions (Fig. S3†) clearly show that SNON-X were uniformly coated with g-C3N4.49 High-resolution STEM images of the heterojunction made by g-C3N4 and SNON-700 (Fig. 3) show that CN crystallizes as fibers (Fig. 3a), and SNON-700 possesses nanoplate like morphology similar to the parent oxide, Sr2Nb2O7. On the other hand, the CN/SNON-700 heterojunction sample showed that the SNON-700 nanoplates are uniformly covered by flake-like CN particles (Fig. 3c). High-resolution STEM images clearly reveal the nanostructure of this particular heterojunction (Fig. 3d–f), showing that intimate contact is present between its two components. SEM-EDS elemental mapping of CN/SNON-700 heterojunction (Fig. 4), confirms that CN is uniformly distributed over SNON. Similarly, EDS mapping of all heterojunctions (Fig. S3†) clearly showed that the SNON-X samples are uniformly covered with CN particles.
 | ||
| Fig. 3 STEM images for (a) CN, (b) SNON-700, (c) CN/SNON-700, and (d, e and f) high-resolution STEM images for CN/SNON-700. | ||
 | ||
| Fig. 4 CN/SNON-700 heterojunction; (a) SEM and (b, c, d, and e) EDS mapping for N, Sr, O, and Nb present in the heterojunction sample, demonstrating a tight heterojunction present between the CN and SNON-700 particles. | ||
The chemical states and compositions were further investigated using X-ray photoelectron spectroscopy (XPS). The survey XPS spectra for CN, SNON-700, and the CN/SNON-700 heterojunction (Fig. 5a) show the presence of C, N, Sr, Nb, and O in CN/SNON-700. The binding energy values for the elements in CN, SNON-700, and CN/SNON-700 heterojunction are reported in Table 2. The high-resolution XPS spectrum of C1s (Fig. 5b) in CN displays two major peaks around 284.3 and 288.1 eV, which can be attributed to sp2 hybridized carbon from aromatic rings and sp2 hybridized carbon in the N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group, respectively.26,41,62,70,71 Similarly, three N1s peaks (at 398.4 eV, 400.8 eV, and 404.2 eV) are observed for CN (Fig. 5c). The small peak observed around 395.6 eV in the XPS spectrum of SNON-700 is assigned to N1s, confirming the N-doping. The C1s and N1s peaks in CN/SNON-700 heterojunction are found at lower binding energies than that of CN, confirming CN hybridization with SNON-700 upon heterojunction fabrication.26,41,72,73 A similar conclusion can be made by comparing the corresponding binding energy values of Sr3d, Nb3d and O1s in SNON-700 and CN/SNON-700 heterojunction samples (Table 2 and Fig. 5d–f). Furthermore, as shown in Table 2 and Fig. S4 (ESI†), the binding energy values for C1s, N1s, Sr3d, Nb3d and O1s in blend sample (heterogeneous mixture) are the same as those for individual components (CN and SNON-700). This result further supports the fact that CN and SNON-700 in the heterojunction are hybridized as observed in electron microscopy studies and other characterizations tools used in this study.
N group, respectively.26,41,62,70,71 Similarly, three N1s peaks (at 398.4 eV, 400.8 eV, and 404.2 eV) are observed for CN (Fig. 5c). The small peak observed around 395.6 eV in the XPS spectrum of SNON-700 is assigned to N1s, confirming the N-doping. The C1s and N1s peaks in CN/SNON-700 heterojunction are found at lower binding energies than that of CN, confirming CN hybridization with SNON-700 upon heterojunction fabrication.26,41,72,73 A similar conclusion can be made by comparing the corresponding binding energy values of Sr3d, Nb3d and O1s in SNON-700 and CN/SNON-700 heterojunction samples (Table 2 and Fig. 5d–f). Furthermore, as shown in Table 2 and Fig. S4 (ESI†), the binding energy values for C1s, N1s, Sr3d, Nb3d and O1s in blend sample (heterogeneous mixture) are the same as those for individual components (CN and SNON-700). This result further supports the fact that CN and SNON-700 in the heterojunction are hybridized as observed in electron microscopy studies and other characterizations tools used in this study.
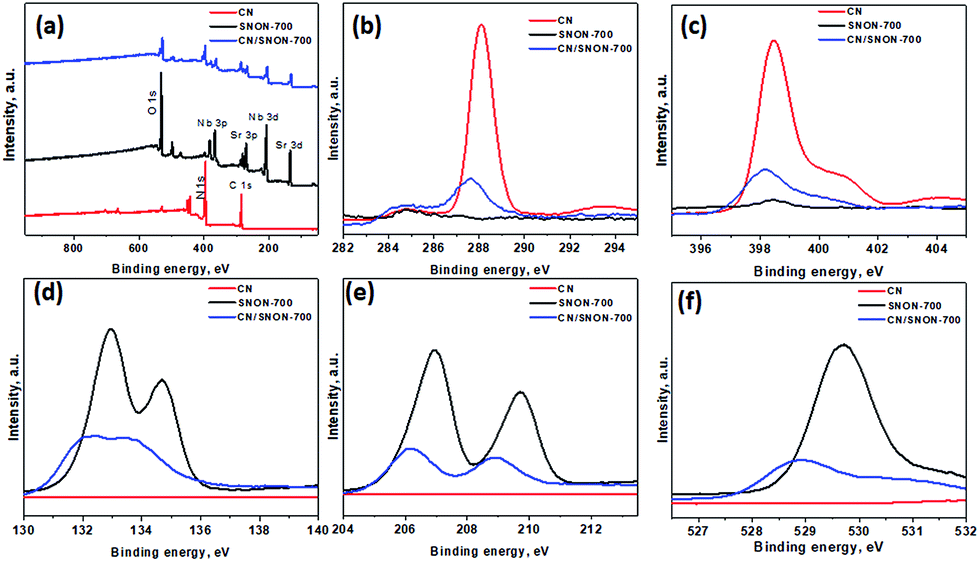 | ||
| Fig. 5 (a) XPS for CN, SNON-700, and CN/SNON-700. High resolution XPS spectra of (b) C 1s, (c) N1s, (d)Sr3d, (e) Nb3d, and (f) O1s for CN, SNON-700 and CN/SNON-700. | ||
| Sample | Binding energy values, eV | ||||
|---|---|---|---|---|---|
| C1s | N1s | Sr3d | Nb3d | O1s | |
| CN | 284.6 | 398.4 | — | — | — |
| 288.1 | 400.8 | ||||
| 404.0 | |||||
| SNON-700 | — | 398.5 | 132.9 | 206.9 | 529.7 |
| 134.6 | 209.7 | ||||
| CN/SNON-700 | 284.3 | 398.2 | 132.3 | 206.1 | 528.9 |
| 287.7 | 400.6 | 133.8 | 208.9 | ||
| — | |||||
| Blend sample of CN and SNON-700 | 284.7 | 398.4 | 132.9 | 206.9 | 529.7 |
| 288.1 | 400.7 | 134.7 | 209.7 | ||
| 404.1 | |||||
3.2 Photocatalytic performance
The photocatalytic hydrogen production was examined under visible light irradiation (λ ≥ 420 nm) in the presence of CH3OH (10 vol%) as a hole scavenger and 2.5 wt% of Pt co-catalyst (Fig. 6). SNON-X exhibited lower activities in comparison with their corresponding heterojunctions. Pure SNO does not show any activity under visible light due to its large band gap. Hydrogen evolution rates are rather low for SNON-X. This result could be attributed to high recombination rate of photo-generated charge carriers. The decreased activities observed for SNON-800 and SNON-950 could be due to the introduction of trapped states generated during harsh nitridation conditions74,75 H2 evolution rate for pristine CN is also low, consistent with literature values.27,28,33,49 In contrast, all heterojunctions show higher photocatalytic activity than their individual components. The CN/SNON-700 heterojunction exhibited the best photocatalytic activity, with hydrogen evolution rate of 70 μmol h−1, significantly higher than that of its individual components SNON-700 1(5.6 μmol h−1) and CN (9.8 μmol h−1) (Fig. 6). H2 evolution rate observed for a blend sample, prepared by mechanically grinding CN and SNON-700, was found to be 3.6 μmol h−1, which is similar to that of the pristine CN. Thus, the visible-light-induced hydrogen evolution rate for CN/SNON-700 heterojunction (128.8 mmol h−1 mol−1 of CN) is ∼14× times larger than that of one mole of pristine CN slightly larger than that of our previous report on g-C3N4/Sr2Nb2O7.49 This result may be due to the involvement of both components in the absorption and charge separation.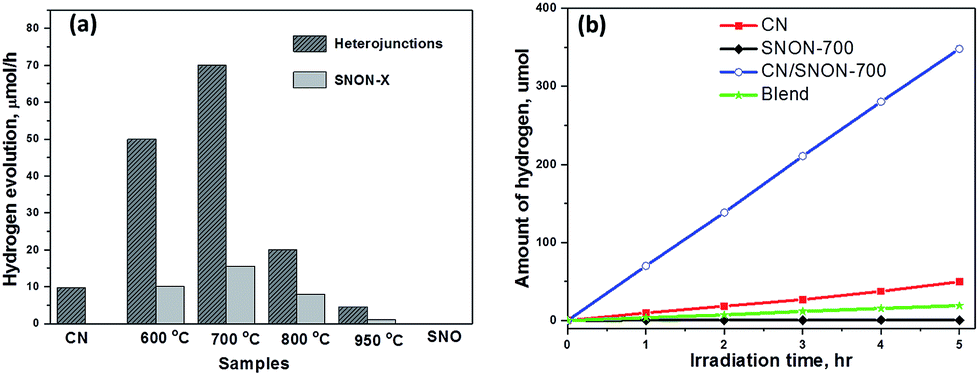 | ||
| Fig. 6 (a) Visible light-induced hydrogen evolution rates for different photocatalysts. (b) Amounts of hydrogen generated in the presence of CN, SNON-700, CN/SNON-700 heterojunction, and blend samples. Conditions: 100 mg catalyst, 50 mL 10 vol% methanol aqueous solution, 300 W Xe lamp with filter for visible light irradiation (λ ≥ 420 nm). | ||
The apparent quantum yields (AQY) were calculated to be 0.38% for CN and 3% for the CN/SNON-700 heterojunction based on the observed activity of 100 mg of the sample for 1 hour visible light irradiation (λ = 420 nm). The turnover number (TON) for CN/SNON-700 reached ∼1.9 after 15 hours with a turnover frequency (TOF) of 0.12. TON and TOF were determined using the amount of hydrogen generated after 15 hours of visible light irradiation which was found to be 1033.8 μmol by 100 mg of CN/SNON-700.
Three consecutive photocatalytic water reduction experiments using the same CN/SNON-700 heterojunction sample were performed in the course of 15 hours to study its photocatalytic recyclability and stability. Aqueous suspensions containing the photocatalyst were degassed after each run by bubbling argon gas through the suspension. The total amount of hydrogen produced in the third 5 hour cycle was found to be 340 μmol, which is similar to the amount produced in the first 5 hours, 348.2 μmol. Hence, the rate of photocatalytic hydrogen production of the CN/SNON-700 sample was virtually unchanged after three cycles over a period of 15 hours (Fig. S5a, ESI†), indicating that the heterojunction is stable under visible light irradiation. PXRD patterns of CN/SNON-700 before and after photocatalytic reactions are also similar (Fig. S5b, ESI†), indicating the photochemical stability of the heterojunction under visible light irradiation.
3.3 Proposed mechanism
The improved activity of the CN/SNON heterojunction over C3N4 could be due to a more efficient charge separation of photogenerated electron–hole pairs. To elucidate the mechanism of the enhanced photocatalytic activity of the CN/SNON heterojunction, the relative band positions of the two semiconductors components were considered since the band-edge potential levels play a crucial role in determining the migration pathway for the photogenerated charge carriers. The band positions were determined using VB-XPS and diffuse reflectance spectroscopy and are summarized in Table 1. The CB and VB positions for SNON-700 are found to be −0.90 eV and +1.70 eV respectively, and the CB and VB potentials for CN were determined to be −1.10 and +1.65 eV, respectively (Table 1).49 Thus, the CB and VB of CN are higher than that of SNON-700. Under visible light irradiation, both CN and SNON-700 generate electron–hole pairs. The photogenerated electrons in the CB of CN can be transferred to the CB of SNON-700, and then injected in the Pt co-catalyst to generate hydrogen through water reduction (Fig. 7). Simultaneously, the photogenerated holes in the VB of SNON-700 can be transferred to the VB of CN and then used to oxidize hole scavengers such as methanol. Thus, the better charge separation leads to a reduction of the recombination rate and results in a remarkable enhancement in the photocatalytic activity observed for the CN/SNON-700 heterojunction. To understand the effect of N-doping on visible-light-driven water splitting, we have compared the above-mentioned mechanism with the mechanism that we proposed for CN/SNO (Fig. S6, ESI†). Because of the different band gaps for SNO and SNON, only one component is active in visible light for CN/SNO heterojunction while both components are active in CN/SNON-700 heterojunction sample.49,76 Hence, the mechanism for enhanced activity is totally different. In the case of CN/SNO, only CN is responsible for the generation of photogenerated electron and holes while both CN and SNON-700 are responsible in CN/SNON heterojunction, further explaining the observed enhancement in photocatalytic activity.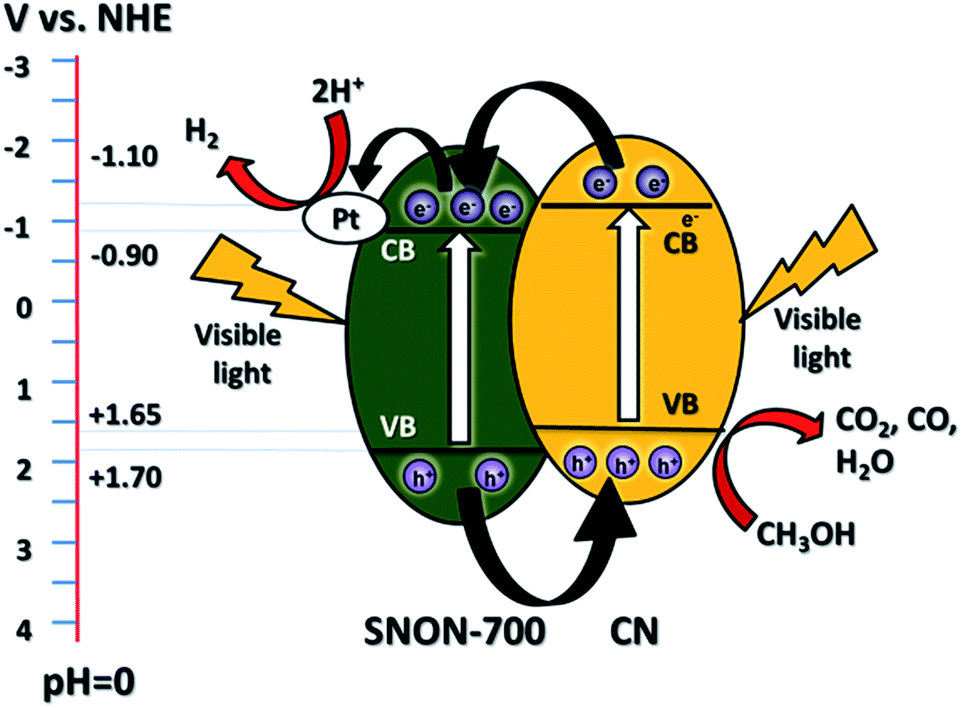 | ||
| Fig. 7 Schematic diagram of the mechanism of separation and transfer of photogenerated carriers in the CN/SNON-700 heterojunction under visible light irradiation. | ||
To confirm the above-mentioned mechanism, the lifetime of photogenerated charge carriers was studied by photoluminescence (PL) experiments. The intensity of the PL emission peak centered at ∼ 460 nm (Fig. 8a) is in the order CN > CN/SNON-950 > CN/SNON-800 > CN/SNON-600 > CN/SNON-700.76 In comparison to the heterojunctions, the SNON-700 sample gave no significant PL emission under the same experimental conditions (See Fig. S7, ESI†). This result suggests that the heterojunction made with CN and SNON-700 has lower recombination rate, consistent with the observed higher photocatalytic activity. The observed peak shape and a small blue shift in the PL emission spectrum can be explained based on nanostructured deposition of CN on SNON surfaces during heterojunction synthesis. It has been already reported that emission peak for CN in the bulk form, occurs at wavelengths between 460 to 475 nm; however, the PL peaks nay shift up to 450 nm in CN with nanosheet.48,49,75 So, in the case of CN/SNON-700 and other heterojunctions, CN may have been deposited in the form of nanosheet. This is supported by the STEM images as shown in Fig. 3.
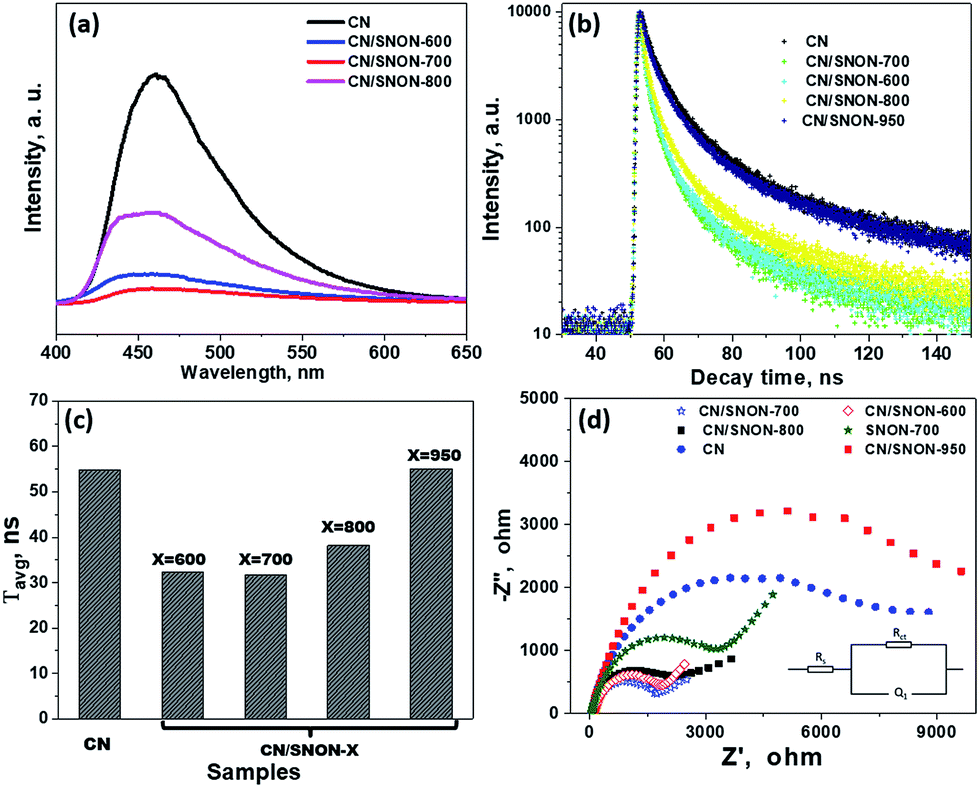 | ||
| Fig. 8 (a) Photoluminescence (PL) spectra of CN and CN/SNON-X at an excitation wavelength of 336 nm; (b) time-resolved fluorescence emission decay curves for g-C3N4 and CN/SNON-X. The emission wavelength was set at 460 nm, with the excitation wavelength at 336 nm; (c) weighted average lifetimes (τavg) for g-C3N4 and heterojunctions CN/SNON-X; (d) electrochemical impedance spectra (EIS) of photocatalysts studied. | ||
To determine the lifetime of photogenerated electron–hole pairs, time-resolved fluorescence emission spectra were collected for CN and all heterojunctions samples (Fig. 8b). The fluorescence intensities were best fitted using a tri-exponential curve.32,77,78 The kinetic parameters for the materials studied are summarized in Table S2, ESI,† where three radiative lifetimes and their relative amplitudes are reported.
The weighted average lifetimes (τavg) show that the radiative lifetime of the charge carriers is higher for CN (54.8 ns). The average lifetimes for CN/SNON-700 (31.6 ns) and CN/SNON-600 (32.4 ns) are lower than that of other heterojunctions. The lower lifetime of electrons suggests that photo-generated electrons in the conduction band of CN undergo rapid transfer to CB of SNON-700 or SNON-600, which result in a better separation of charge carriers. The fast electron injection confirms that the formation of the heterojunction promotes charge transfer efficiency, which enhances the photocatalytic generation of hydrogen.79,80 The PL data (Fig. 8a–c) show that CN/SNON-800 and SN/SNON-950 heterojunctions exhibit more charge recombination than CN/SNON-600 and CN/SNON-700. This property may be explained by the introduction of new trapped states during the N-doping for these high-temperature nitridized samples (800 and 950 °C).
Electrochemical impedance spectra (EIS) showed better charge transfer between the photocatalysts and the electrolyte. The high-frequency region denotes charge-transfer resistance at the CN/SNON-700/FTO interfaces, while the low-frequency region is associated with the electron transport kinetics at the CN/SNON-700 interface. The Nyquist plots can be interpreted based on the equivalent circuits, as shown as an inset in Fig. 8d. A smaller arc radius in EIS Nyquist plots indicates a higher reaction rate.72,81–83 The arc radius for the CN/SNON-700 heterojunction was found to be significantly smaller than that of its components CN and SNON-700, suggesting lower resistance to charge transfer at the interface between the electrode and electrolyte. The observed decrease of impedance in EIS suggests that photogenerated electrons have a higher probability to participate in redox reactions at the surface of photocatalysts obtained at temperatures between 600–800 °C (especially for CN/SNON-700). The reduction in the charge-transfer resistance further supports experimental evidence that CN/SNON-700 has a higher photocatalytic hydrogen evolution compared to pristine CN and SNON.
4. Conclusions
In summary, CN/SNON-X heterojunctions were fabricated and found to be photocatalytically active under visible light irradiation. The heterojunctions were found to exhibit higher photocatalytic efficiency than their individual components. The optimum activity was found for the CN/SNON-700, where the rate of photocatalytic hydrogen production under visible light was found to be ∼14× larger than that of pristine CN. The mechanism of the charge carriers pathways at the interface between CN and SNON-700 was proposed and experimentally confirmed. The observed higher photocatalytic activity of the CN/SNON-700 heterojunction is due to suitable band positions, which allows for better charge separation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Wake Forest University Center for Energy, Environment, and Sustainability and by NSF MRI 1040264. Support from the WFU Science Research Fund is also acknowledged. The authors would like to thank Dr Scott Geyer and Hui Li from the Department of Chemistry at WFU for their assistance with the collection of DRS and EIS data. A portion of this research was completed at the Center for Nanophase Materials Sciences, which is a DOE Office of Science User Facility. ZDH gratefully acknowledges a Graduate Research Fellowship from the National Science Foundation (No. DGE-1650044) and the Georgia Tech-ORNL Fellowship.Notes and references
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef PubMed.
- M. Ahmed and G. Xinxin, Inorg. Chem. Front., 2016, 3, 578–590 RSC.
- W. Jiao, L. Wang, G. Liu, G. Q. (Max) Lu and H.-M. Cheng, ACS Catal., 2012, 2, 1854–1859 CrossRef.
- A. Kumar, A. S. Patel and T. Mohanty, J. Phys. Chem. C, 2012, 116, 20404–20408 CrossRef.
- A. Mills and S. Le Hunte, J. Photochem. Photobiol., A, 1997, 108, 1–35 CrossRef.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401–425 CrossRef.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef PubMed.
- P. Kanhere and Z. Chen, Molecules, 2014, 19, 19995–20022 CrossRef PubMed.
- C.-H. Liao, C.-W. Huang and J. C. S. Wu, Catalysts, 2012, 2, 490–516 CrossRef.
- H. Irie, Y. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 5483–5486 CrossRef.
- Y. Matsumoto, M. Koinuma, Y. Iwanaga, T. Sato and S. Ida, J. Am. Chem. Soc., 2009, 131, 6644–6645 CrossRef PubMed.
- A. Mukherji, R. Marschall, A. Tanksale, C. Sun, S. C. Smith, G. Q. Lu and L. Wang, Adv. Funct. Mater., 2011, 21, 126–132 CrossRef.
- A. Mukherji, B. Seger, G. Q. (Max) Lu and L. Wang, ACS Nano, 2011, 5, 3483–3492 CrossRef PubMed.
- A. Mukherji, C. Sun, S. C. Smith, G. Q. Lu and L. Wang, J. Phys. Chem. C, 2011, 115, 15674–15678 CrossRef.
- X. Wang, G. Liu, Z.-G. Chen, F. Li, G. Q. Lu and H.-M. Cheng, Chem. Lett., 2009, 38, 214–215 CrossRef.
- J. Xu, Y. Wei, Y. Huang, J. Wang, X. Zheng, Z. Sun, L. Fan and J. Wu, Ceram. Int., 2014, 40, 10583–10591 CrossRef.
- X. Zong, C. Sun, Z. Chen, A. Mukherji, H. Wu, J. Zou, S. C. Smith, G. Q. Lu and L. Wang, Chem. Commun., 2011, 47, 6293–6295 RSC.
- F. Zou, Z. Jiang, X. Qin, Y. Zhao, L. Jiang, J. Zhi, T. Xiao and P. P. Edwards, Chem. Commun., 2012, 48, 8514–8516 RSC.
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421–2440 CrossRef.
- H. Wang, L. Zhang, Z. Chen, J. Hu, S. Li, Z. Wang, J. Liu and X. Wang, Chem. Soc. Rev., 2014, 43, 5234–5244 RSC.
- S. J. A. Moniz, S. A. Shevlin, D. J. Martin, Z.-X. Guo and J. Tang, Energy Environ. Sci., 2015, 8, 731–759 RSC.
- P. Chowdhury, G. Hassan and Ajay K. Ray, in Sustainable Nanotechnology and the Environment: Advances and Achievements, American Chemical Society, 2013, vol. 1124, pp. 231–266 Search PubMed.
- S. P. Adhikari, H. Dean, Z. D. Hood, R. Peng, K. L. More, I. Ivanov, Z. Wu and A. Lachgar, RSC Adv., 2015, 5, 91094–91102 RSC.
- S. P. Adhikari, Z. D. Hood, K. L. More, I. Ivanov, L. Zhang, M. Gross and A. Lachgar, RSC Adv., 2015, 5, 54998–55005 RSC.
- S. P. Adhikari, L. Zhang, M. Gross and A. Lachgar, MRS Online Proc. Libr., 2015, 1738, mrsf14-1738-v05-52 CrossRef.
- H. Shi, G. Chen, C. Zhang and Z. Zou, ACS Catal., 2014, 4, 3637–3643 CrossRef.
- S. P. Adhikari, Z. D. Hood, H. Wang, R. Peng, A. Krall, H. Li, V. W. Chen, K. L. More, Z. Wu, S. Geyer and A. Lachgar, Appl. Catal., B, 2017, 217, 448–458 CrossRef.
- S. Cao and J. Yu, J. Phys. Chem. Lett., 2014, 5, 2101–2107 CrossRef PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef PubMed.
- D. J. Martin, K. Qiu, S. A. Shevlin, A. D. Handoko, X. Chen, Z. Guo and J. Tang, Angew. Chem., 2014, 126, 9394–9399 CrossRef.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef PubMed.
- Y. Zhang, Q. Pan, G. Chai, M. Liang, G. Dong, Q. Zhang and J. Qiu, Sci. Rep., 2013, 3, 1943 CrossRef PubMed.
- S. Martha, A. Nashim and K. M. Parida, J. Mater. Chem. A, 2013, 1, 7816–7824 RSC.
- S.-W. Cao, X.-F. Liu, Y.-P. Yuan, Z.-Y. Zhang, Y.-S. Liao, J. Fang, S. C. J. Loo, T. C. Sum and C. Xue, Appl. Catal., B, 2014, 147, 940–946 CrossRef.
- L.-Y. Chen and W.-D. Zhang, Appl. Surf. Sci., 2014, 301, 428–435 CrossRef.
- S. Chengjie, F. Mingshan, H. Bo, C. Tianjun, W. Liping and S. Weidong, CrystEngComm, 2015, 17, 4575–4583 RSC.
- Y. Cui, Chin. J. Catal., 2015, 36, 372–379 CrossRef.
- F. Dong, Z. Zhao, T. Xiong, Z. Ni, W. Zhang, Y. Sun and W.-K. Ho, ACS Appl. Mater. Interfaces, 2013, 5, 11392–11401 CrossRef PubMed.
- J. Fu, B. Chang, Y. Tian, F. Xi and X. Dong, J. Mater. Chem. A, 2013, 1, 3083–3090 RSC.
- L. Ge, C. Han and J. Liu, Appl. Catal., B, 2011, 108–109, 100–107 CrossRef.
- C. Han, Y. Wang, Y. Lei, B. Wang, N. Wu, Q. Shi and Q. Li, Nano Res., 2015, 8, 1199–1209 CrossRef.
- H. W. Kang, S. N. Lim, D. Song and S. B. Park, Int. J. Hydrogen Energy, 2012, 37, 11602–11610 CrossRef.
- W. Liu, M. Wang, C. Xu and S. Chen, Chem. Eng. J., 2012, 209, 386–393 CrossRef.
- C. Miranda, H. Mansilla, J. Yáñez, S. Obregón and G. Colón, J. Photochem. Photobiol., A, 2013, 253, 16–21 CrossRef.
- C. Pan, J. Xu, Y. Wang, D. Li and Y. Zhu, Adv. Funct. Mater., 2012, 22, 1518–1524 CrossRef.
- J.-X. Sun, Y.-P. Yuan, L.-G. Qiu, X. Jiang, A.-J. Xie, Y.-H. Shen and J.-F. Zhu, Dalton Trans., 2012, 41, 6756–6763 RSC.
- X. Wang, L. Zhang, H. Lin, Q. Nong, Y. Wu, T. Wu and Y. He, RSC Adv., 2014, 4, 40029–40035 RSC.
- Z. Zhang, K. Liu, Z. Feng, Y. Bao and B. Dong, Sci. Rep., 2016, 6, 19221 CrossRef PubMed.
- S. P. Adhikari, Z. D. Hood, K. L. More, V. W. Chen and A. Lachgar, ChemSusChem, 2016, 9, 1869–1879 CrossRef PubMed.
- A. Kudo, H. Kato and S. Nakagawa, J. Phys. Chem. B, 2000, 104, 571–575 CrossRef.
- L. Botao, M. Junfeng, Y. Yan, L. Jun, R. Yang, J. Xiaohui, S. Yong and L. Zhensen, J. Am. Ceram. Soc., 2008, 91, 1329–1331 CrossRef.
- M. Kodera, M. Katayama, T. Hisatomi, T. Minegishi and K. Domen, CrystEngComm, 2016, 18, 3186–3190 RSC.
- H. Suzuki, O. Tomita, M. Higashi, A. Nakada and R. Abe, Appl. Catal., B, 2018, 232, 49–54 CrossRef.
- S. M. Ji, P. H. Borse, H. G. Kim, D. W. Hwang, J. S. Jang, S. W. Bae and J. S. Lee, Phys. Chem. Chem. Phys., 2005, 7, 1315–1321 RSC.
- K. Maeda, M. Higashi, B. Siritanaratkul, R. Abe and K. Domen, J. Am. Chem. Soc., 2011, 133, 12334–12337 CrossRef PubMed.
- J. Guo, H. Zhou, S. Ouyang, T. Kako and J. Ye, Nanoscale, 2014, 6, 7303–7311 RSC.
- Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68–89 CrossRef PubMed.
- H. Yan, Chem. Commun., 2012, 48, 3430–3432 RSC.
- J. Yu, S. Wang, J. Low and W. Xiao, Phys. Chem. Chem. Phys., 2013, 15, 16883–16890 RSC.
- A. B. Murphy, Sol. Energy Mater. Sol. Cells, 2007, 91, 1326–1337 CrossRef.
- V. V. Atuchin, J.-C. Grivel, A. S. Korotkov and Z. Zhang, J. Solid State Chem., 2008, 181, 1285–1291 CrossRef.
- M. Tahir, C. Cao, N. Mahmood, F. K. Butt, A. Mahmood, F. Idrees, S. Hussain, M. Tanveer, Z. Ali and I. Aslam, ACS Appl. Mater. Interfaces, 2014, 6, 1258–1265 CrossRef PubMed.
- K. Shimizu, Y. Tsuji, T. Hatamachi, K. Toda, T. Kodama, M. Sato and Y. Kitayama, Phys. Chem. Chem. Phys., 2004, 6, 1064–1069 RSC.
- K. Shimizu, Y. Tsuji, M. Kawakami, K. Toda, T. Kodama, M. Sato and Y. Kitayama, Chem. Lett., 2002, 31, 1158–1159 CrossRef.
- S. Liang, L. Shen, J. Zhu, Y. Zhang, X. Wang, Z. Li, L. Wu and X. Fu, RSC Adv., 2011, 1, 458–467 RSC.
- X. Xin, J. Lang, T. Wang, Y. Su, Y. Zhao and X. Wang, Appl. Catal., B, 2016, 181, 197–209 CrossRef.
- B. Brahmaroutu, G. L. Messing and S. Trolier-McKinstry, J. Am. Ceram. Soc., 1999, 82, 1565–1568 CrossRef.
- N. Ishizawa, F. Marumo, T. Kawamura and M. Kimura, Acta Crystallogr., Sect. B, 1975, 31, 1912–1915 CrossRef.
- S. Khademinia and M. Behzad, Adv. Powder Technol., 2015, 26, 644–649 CrossRef.
- Y. Cui, Z. Ding, X. Fu and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 11814–11818 CrossRef PubMed.
- S. Wang, C. Li, T. Wang, P. Zhang, A. Li and J. Gong, J. Mater. Chem. A, 2014, 2, 2885–2890 RSC.
- L. Huang, H. Xu, Y. Li, H. Li, X. Cheng, J. Xia, Y. Xu and G. Cai, Dalton Trans., 2013, 42, 8606–8616 RSC.
- T. Yan, Q. Yan, X. Wang, H. Liu, M. Li, S. Lu, W. Xu and M. Sun, Dalton Trans., 2015, 44, 1601–1611 RSC.
- T. Takata, C. Pan and K. Domen, ChemElectroChem, 2016, 3, 31–37 CrossRef.
- T. Takata, C. Pan and K. Domen, Sci. Technol. Adv. Mater., 2015, 16, 033506 CrossRef PubMed.
- S. P. Adhikari, Z. D. Hood and A. Lachgar, MRS Adv., 2018, 1–8 Search PubMed.
- P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef.
- S. Nayak, L. Mohapatra and K. Parida, J. Mater. Chem. A, 2015, 3, 18622–18635 RSC.
- Z. Sun, H. Zheng, J. Li and P. Du, Energy Environ. Sci., 2015, 8, 2668–2676 RSC.
- S.-W. Cao, X.-F. Liu, Y.-P. Yuan, Z.-Y. Zhang, Y.-S. Liao, J. Fang, S. C. J. Loo, T. C. Sum and C. Xue, Appl. Catal., B, 2014, 147, 940–946 CrossRef.
- H. Huang, L. Liu, Y. Zhang and N. Tian, RSC Adv., 2015, 5, 1161–1167 RSC.
- Z. Hosseini, N. Taghavinia, N. Sharifi, M. Chavoshi and M. Rahman, J. Phys. Chem. C, 2008, 112, 18686–18689 CrossRef.
- X. Bai, L. Wang and Y. Zhu, ACS Catal., 2012, 2, 2769–2778 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Supporting information includes FTIR, surface area data, SEM images, XPS, recyclability tests, proposed mechanisms, PL and Kinetic parameters of the emission decay for different catalysts. See DOI: 10.1039/c8se00319j |
| ‡ Current address: Materials Science and Technology Division (MSTD), Oak Ridge National Laboratory, Oak Ridge, Tennessee, 37831, USA. |
| § Current address: Electrochemical Materials Laboratory, Department of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. |
| This journal is © The Royal Society of Chemistry 2018 |