
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFine-tuned organic photoredox catalysts for fragmentation-alkynylation cascades of cyclic oxime ethers†
Franck
Le Vaillant
 a,
Marion
Garreau
a,
Stefano
Nicolai
a,
Ganna
Gryn'ova
b,
Clemence
Corminboeuf
b and
Jerome
Waser
*a
a,
Marion
Garreau
a,
Stefano
Nicolai
a,
Ganna
Gryn'ova
b,
Clemence
Corminboeuf
b and
Jerome
Waser
*a
aLaboratory of Catalysis and Organic Synthesis, Institut des Sciences et Ingénierie Chimique, Ecole Polytechnique Fédérale de Lausanne, Ch-1015, Lausanne, Switzerland. E-mail: jerome.waser@epfl.ch
bLaboratory for Computational Molecular Design, Institut des Sciences et Ingénierie Chimique, Ecole Polytechnique Fédérale de Lausanne, Ch-1015, Lausanne, Switzerland
First published on 30th May 2018
Abstract
Fine-tuned organic photoredox catalysts are introduced for the metal-free alkynylation of alkylnitrile radicals generated via oxidative ring opening of cyclic alkylketone oxime ethers. The redox properties of the dyes were determined by both cyclic voltammetry and computation and covered an existing gap in the oxidation potential of photoredox organocatalysts.
1. Introduction
Cascade reactions leading to several bond forming/bond breaking events are important tools to access efficiently the complex carbon skeleton of organic compounds.1 Single electron chemistry is well suited for cascade transformations due to the high reactivity of radical intermediates combined with neutral reaction conditions.2 However, many radical-based cascade reactions still require the stoichiometric use of toxic reagents, such as tin hydrides and/or harsh reaction conditions. In the last decade, visible light driven photoredox catalysis has emerged as a general method for radical generation under mild oxidative or reductive conditions without the need for toxic reagents.3 First successes were met with metal complexes as catalysts.4 More recently organic dyes have been introduced, as they are often cheap, non-toxic and easy to modify.5In the case of reductive quenching cycles for the generation of radical by oxidation, the reduction potential of the photo-activated catalyst is an essential parameter (Fig. 1). The reduction potential of the most often used iridium catalyst (Ir[dF(CF3)ppy]2(dtbbpy))PF6 (1) (+1.21 V vs. SCE in MeCN)6 is lower than frequently used organic dyes, such as Fukuzumi dye (2) and dicyanoanthracene (3) (+2.06 V vs. SCE in MeCN).5,7 To enrich the range of transformations available to organic catalysts, using dyes with lower oxidation values would be highly desirable.
 | ||
| Fig. 1 Bridging the gap in the reduction potential of activated organic photocatalysts. | ||
In this context, 2,4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile (4CzIPN, 4a) has emerged as an alternative organic dye with a lower reduction potential (+1.35 V vs. SCE in MeCN).8 Nevertheless, there is a need for further catalysts with reduction potentials between +1.4 V and + 2.0 V to allow challenging transformations asking for efficient oxidation.
In this context, heteroatom centered radicals initiated transpositions have found widespread applications in synthetic chemistry.9 Zard and coworkers developed iminyl radical initiated cascades using tin reagents and electrophilic olefins to trap the generated intermediate (Scheme 1A).10 Transition metal catalysis (Pd, Ir, Fe) and/or heating at temperature higher than 90 °C were later reported to avoid the use of tin reagents.11 In 2017, Shi and co-workers described one single example of a Heck-like coupling at room temperature under photoredox conditions (Scheme 1B).12 After Shi's pioneering example, Xiao and coworkers developed in 2018 a general method for C–C bond formation after reductive cleavage of cycloalkyloxime esters for the introduction of both alkanes and alkenes.13 Zhou and coworkers reported a multicomponent alkylation etherification of alkenes using a similar strategy.14 These breakthroughs greatly enhanced the synthetic potential of fragmentation cascades, but the exclusive use of reductive conditions limited the types of bond formations possible.
 | ||
| Scheme 1 Existing methods for C–C bonds formation after iminyl radical initiated ring opening (A and B) and our work based on oxidative photoredox catalysis (C) (PC = photoredox catalysis, EBX = ethynyl benziodoxolone). | ||
Recently, Studer and coworkers15 and Leonori and coworkers16 independently reported a new approach for the generation of iminyl radicals based on an oxidative quenching cycle starting from carboxylic acid derived oxime ethers. In 2018, this activation strategy was used by Leonori to develop the fluorination, chlorination and azidation of alkylnitrile radicals generated by fragmentation of iminyl radicals (Scheme 1B).17 However, no example of C–C bond formation was reported. In this respect, our group and others have shown that ethynylbenziodoxolones (EBX) reagents 5 allowed the alkynylation of radicals generated through oxidative photoredox cycles.18 We therefore envisioned that EBX reagents would be ideally suited to overcome these limitations and deliver remotely-functionalized alkynyl nitrile through an oxidative fragmentation C–C bond formation sequence. Such a transformation has never been reported under photoredox conditions.19 However, preliminary results showed that only low yields could be obtained in this reaction using established photoredox catalysts 1–3 (see Results and Discussions).
Herein, we report for the first time the use of the modified 4XCzIPN dyes 4b–d in photoredox catalysis. In particular, 4ClCzIPN dye 4c led to a highly efficient fragmentation alkynylation process. Both theory and experiment showed that 4c has an increased reduction potential compared to 4a, leading to the efficient fragmentation of both four- and five-membered cyclic oxime ethers to give compounds containing versatile alkyne and nitrile functionalities in good yield under mild conditions.
2. Results and Discussions
Dye synthesis and properties
The catalysts 4a–d were easily synthesized from commercially available 2,4,5,6-tetrafluoroisophthalonitrile (6) and the corresponding carbazoles 7a–dvia nucleophilic aromatic substitution (Scheme 2).20 Good yields were obtained, except for fluoro-substituted dye 4b.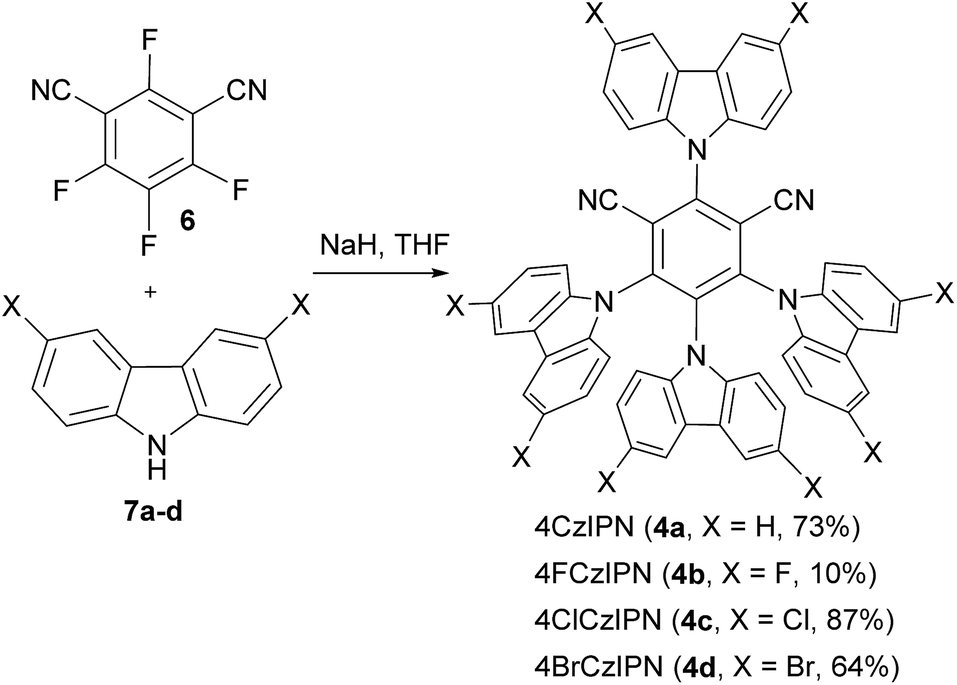 | ||
| Scheme 2 Synthesis of 4XCzIPN dyes 4a–d. | ||
The redox properties of catalysts 4a–d were then determined (Table 1). Ground state reduction and oxidation potentials of −1.21 V and +1.52 V for catalyst 4a in acetonitrile have been reported based on cyclic voltammetry.8a These values combined with the difference in absorbance and emission signals of the dye allowed to estimate the potentials in the excited state: +1.35 V and −1.04 V for reduction and oxidation, respectively (entry 1). In our hand, slightly different values were obtained (entry 2). In particular, a higher reduction potential of +1.59 V was observed in the excited state. We then turned to halogenated dyes. With catalyst 4c, both the cationic and anodic shifts were measured, with reduction and oxidation potentials of −0.97 V and +2.05 V (entry 3). This resulted in an increased reduction potential of +1.71 V of the photoexcited dye. Unfortunately, the cyclic voltammograms of dyes 4b and 4d could not be determined in acetonitrile due to their limited solubility. Only the absorption/emission spectra of dye 4d could be measured. We therefore turned to theory to have a more reproducible and expanded access to redox potential values. At the PCM-UAKS/PBE0-D3BJ/def2-SVP level, the ground state reduction potentials decrease and the oxidation potentials increase in the order of 4a, 4b, 4c, 4d (entries 4–7).21 This leads to a higher reduction potential in the excited state for catalyst 4c and 4d compared to 4a (+1.58 V and + 1.82 V respectively compared to + 1.35 V). Therefore, both measurement and computation confirmed the increased potential for 4c. The same trends but with lower reduction potentials were obtained in dichloromethane as solvent (entries 8–11). In this case, absorption and emission spectra could be measured for all dyes, but no good quality cyclic voltammograms could be acquired. Values in the ground state were therefore again obtained by computation.
| Entry | Catalyst | Solvent | E 1/2(P/P−) | E 1/2(P+/P) | E 0–0 | E 1/2(P*/P−) | E 1/2(P+/P*) |
|---|---|---|---|---|---|---|---|
| a Potentials in V vs. SCE. The excitation energy E0−0 was estimated by the point of intersection of the normalized absorbance and emission signals. E1/2(P+/P*) = E1/2(P+/P) − E0–0 and E1/2(P*/P−) = E0–0 + E1/2(P/P−). See ESI for details. b Experimental values of E0−0 were used. | |||||||
| 1 | 4a(lit) | CH3CN | −1.21 | +1.52 | 2.56 | +1.35 | −1.04 |
| 2 | 4a(mes) | CH3CN | −1.05 | +1.68 | 2.64 | +1.59 | −0.96 |
| 3 | 4c(mes) | CH3CN | −0.97 | +2.05 | 2.68 | +1.71 | −0.63 |
| 4 | 4a(comp) | CH3CN | −1.29 | +1.56 | 2.64b | +1.35 | −1.08 |
| 5 | 4b(comp) | CH3CN | −1.18 | +1.67 | — | — | — |
| 6 | 4c(comp) | CH3CN | −1.10 | +1.76 | 2.68b | +1.58 | −0.92 |
| 7 | 4d(comp) | CH3CN | −0.83 | +1.89 | 2.65b | +1.82 | −0.76 |
| 8 | 4a(comp) | CH2Cl2 | −1.29 | +1.67 | 2.59b | +1.30 | −0.92 |
| 9 | 4b(comp) | CH2Cl2 | −1.18 | +1.79 | 2.60b | +1.42 | −0.81 |
| 10 | 4c(comp) | CH2Cl2 | −1.10 | +1.87 | 2.59b | +1.49 | −0.72 |
| 11 | 4d(comp) | CH2Cl2 | −0.85 | +1.98 | 2.58b | +1.73 | −0.60 |
The trend in reduction potentials increasing in the order of 4a < 4b < 4c < 4d (substituents on catalyst 4: H < F < Cl < Br) can be rationalized as follows. Upon reduction, an electron is added to the lowest unoccupied molecular orbital (LUMO), located mostly on the central isophthalonitrile ring (Fig. 2). However, the LUMO also involves the carbazole moieties in the 4 and 6 positions of the isophthalonitrile ring and is potentially stabilized by them to a greater extent in the case of Cl and Br substituents (resonance donors) compared to H and F. We also note that while molecules 4a and 4b are fairly symmetric, systems 4c and 4d feature noticeable distortion of the 4-carbazole (Fig. 2). This peculiar structure, observed both in the gas-phase optimized geometries, obtained at several dispersion-corrected DFT levels, and experimental crystal structures,22 is due to strong Cl⋯Cl and Br⋯Br contacts in 4c and 4d, respectively.23
 | ||
| Fig. 2 Optimized structures and LUMO plots (isovalue 0.02) of the dyes 4a and 4c at the PBE0/def2-SVP level, as well as the crystal structure of 4c. See the SI for the plots of 4b and 4d. | ||
Optimization of the fragmentation cascade
Our investigations began with the gram scale synthesis of hydroxylamine 10 (Scheme 3, eqn (1)).24 Nucleophilic substitution of commercially available N-hydroxyphthalimide (8) and ethyl 2-bromo-2-methylpropanoate (9), followed by acidic hydrolysis of the phthalimide, gave 5.8 g of 10 in 86% yield over 2 steps. Condensation of 10 with cyclobutanone (11) led to oxime ether 12a in 90% yield as a crystalline solid (Scheme 3, eqn (2)). | ||
| Scheme 3 Gram scale synthesis of 1-carboxy-1-methylethoxyammonium chloride (10), and condensation with cyclobutanone (11) to give model substrate 12a. | ||
A cyclic voltammetry measurement on the corresponding potassium carboxylate 12a′ showed a clear oxidation peak at +1.48 V vs. SCE in DMF. We decided then to first test the best catalysts reported by Studer:15 (Ir[dF(CF3)ppy]2(dtbbpy))PF6 (1) and Leonori:16,17 Fukuzumi's catalyst 2 (Table 2). Based on our previous studies on photoredox catalysis using EBX reagents,18a,b we used DCE as solvent and an excess of Ph-EBX reagent 5a (2.0 equiv.). With 1 mol% of 1a, full conversion and 55% NMR yield of alkyne 13a were reached after 2 h, whereas only 50% conversion and 20% NMR yield was obtained with 2 (Table 2, entries 1–2). In both cases, significant side products formation was observed. DiCyanoAnthracene (DCA, 3) gave only 5% NMR yield of 13a (entry 3). With 4CzIPN (4a) we were pleased to observe 60% conversion of 12a and 50% NMR yield of 13a after 2 h (entry 4).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Base | Time | Conversiona | Yieldb |
| a Reaction conditions: using 0.1 mmol 12a (1 equiv.), 0.2 mmol 5a (2.0 equiv.), 5 mol% 1–4 (0.05 equiv.) in DCE (2.0 mL) for 2 h at RT. The conversion of 12a by NMR is given. NR = no reactivity. b NMR yield using dibromomethane as internal standard. c Using 1 mol% of 1. d Using 1.0 equiv. of base. e Using 1.5 equiv. of 5a. f Using 3.0 mol% of organic dye 4c. g Without irradiation. | |||||
| 1c,d | 1 | Cs2CO3 | 2 h | >95% | 55% |
| 2d | 2 | Cs2CO3 | 2 h | 50% | 20% |
| 3d | 3 | Cs2CO3 | 2 h | 90% | 5% |
| 4 | 4a | Cs2CO3 | 2 h–6 h | 60% | 50% |
| 5 | 4c | Cs2CO3 | 2 h | >95% | 70% |
| 6 | 4c | K2CO3 | 2 h | >95% | 75% |
| 7 | 4c | K2CO3 | 1 h | >95% | 80% |
| 8 | 4d | K2CO3 | 1 h | >95% | 75% |
| 9 | 4b | K2CO3 | 1 h | >95% | 75% |
| 10e | 4c | K2CO3 | 1 h | >95% | 70% |
| 11f | 4c | K2CO3 | 1 h | >95% | 80% |
| 12 | 4c | K2CO3 | 0.5 h | >95% | 80% |
| 13 | — | K2CO3 | 1 h | NR | NR |
| 14 | 4c | — | 1 h | <10% | 5% |
| 15g | 4c | K2CO3 | 1 h | NR | NR |
Increasing reaction time (6 h) and/or temperature (50 °C) was not successful to improve conversion. Speculating that stronger oxidizing dyes could lead to better results, catalyst 4c was then examined. Full conversion of 12a was observed and 70% of 13a could be isolated (entry 5). K2CO3 was found to be slightly better than Cs2CO3 as a base (entry 6), allowing us to reduce the reaction time to 1 h (entry 7). The other new photocatalysts 4b and 4d were then tested, and provided similar photocatalytic activity as 4c (entries 8–9). Due to its better solubility and ease of synthesis, 4c was selected as photocatalyst to continue the study. Decreasing the number of equivalents of 5a to 1.5 resulted in lower yield and more side reactions (entry 10). Interestingly, the catalyst loading could be decreased to 3 mol% (entry 11) or the reaction time reduced to only 30 min (entry 12) without any effect on the yield. Finally, control experiments showed that photocatalyst, base and light were all required for the reaction to proceed (entries 13–15).
Scope of the fragmentation cascade
We then investigated the scope of oximes at 0.30 mmol scale using 3 mol% of 4c (Scheme 4A). Oxime 12a afforded 13a in 79% isolated yield. Phenyl, alkyl and protected amines were tolerated at the β-position (13b–d, 41–80% yield). Benzocyclobutenone oxime 12e afforded propargylic benzocarbonitrile 13e in 40% yield. Bicyclic compound 13f was obtained in 65% yield and excellent diastereoselectivity. Oxetanone and azetidinones oximes ethers were also excellent substrates (13g–i). Cyclopentanones derivatives were also successful. Using a gem-dimethyl substituent for the generation of a δ-tertiary alkylnitrile radical allowed the isolation of 13j in 77% yield. In the work of Leonori and coworkers, a benzylic position was used to promote ring opening.17 In our case, an α-heteroatom was exploited. With an oxygen linker, product 13k was obtained in 65% yield, whereas a nitrogen group led to a modest yield (30%) of 13l. In addition, compounds 13a and 13h were obtained in similar yields using sunlight instead of LEDs.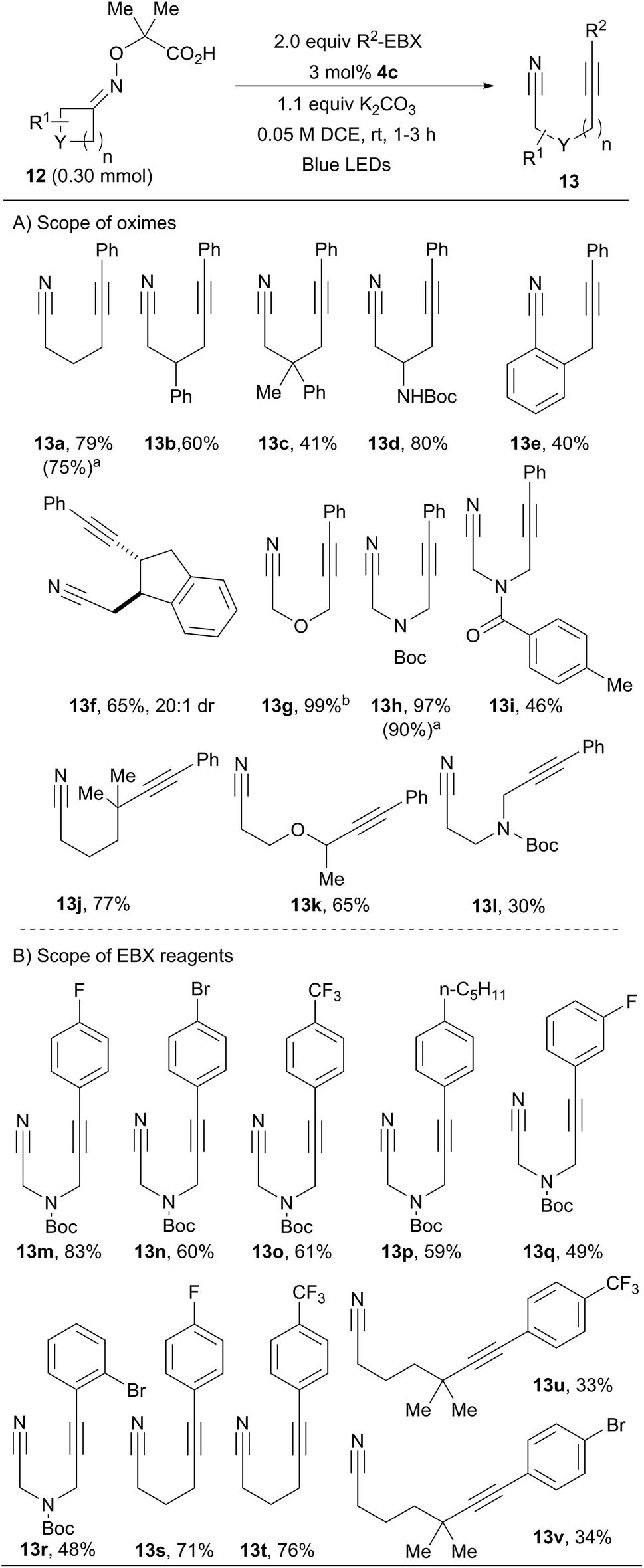 | ||
| Scheme 4 Scope of the reaction. Using oximes 12 (1.0 equiv.), photocatalyst 4c (3 mol%), EBX 5 (2 equiv.) and K2CO3 (1.1 equiv.) in DCE (0.05 M) at rt, 1–3 h. aUsing sunlight instead of blue LEDs. NMR yield is given. bOn 0.10 mmol scale. | ||
We then turned our attention to the scope of the reagents (Scheme 4B). The conversion of azetidinone oxime ether 12h into α-α′-cyanoalkynylamines was achieved in good yields, tolerating electron-withdrawing groups (13m–o) and electron-donating groups (13p) in para position of the benzene ring.25 Good yields were also obtained with a fluorine in meta or a bromine in ortho positions (13q and 13r). Cyclobutanone oxime 12a could also be used with fluorinated arene substituents on the EBX (13s–t). Products 13u and 13v bearing a trifluoromethyl and bromine at the para-position were obtained in moderate yields from cyclopentanone oxime 12j. Alkynyl nitriles are important building blocks combining two highly useful functional groups.26 For example, compounds such as 13a have been used in cyclization reactions with alkynes,26a–c vinyl iodonium salts,26d or benzyne equivalents.26e
Preliminary investigations were then conducted for other types of C–C bond formation (Scheme 5). A silyl EBX reagent gave a lower yield of product 13w and alkyl EBX could not be used. Preliminary results for an alkenylation cascade were also obtained using Phenyl Vinyl BenziodoXolone (PhVBX, 14), recently introduced by Olofsson and co-workers.27 Under the optimized reaction conditions, alkene 15 was isolated in 50% yield with a 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 E/Z selectivity.28 Changing the base from K2CO3 to CsOBz afforded lower yield (40%), but with enhanced selectivity (17:1 E/Z). Using two equivalents of cyanobenziodoxolone (CBX, 16),18b bisnitrile 17 was obtained in 51% yield. With Togni reagent 18,29 no desired product was formed.
:1 E/Z selectivity.28 Changing the base from K2CO3 to CsOBz afforded lower yield (40%), but with enhanced selectivity (17:1 E/Z). Using two equivalents of cyanobenziodoxolone (CBX, 16),18b bisnitrile 17 was obtained in 51% yield. With Togni reagent 18,29 no desired product was formed.
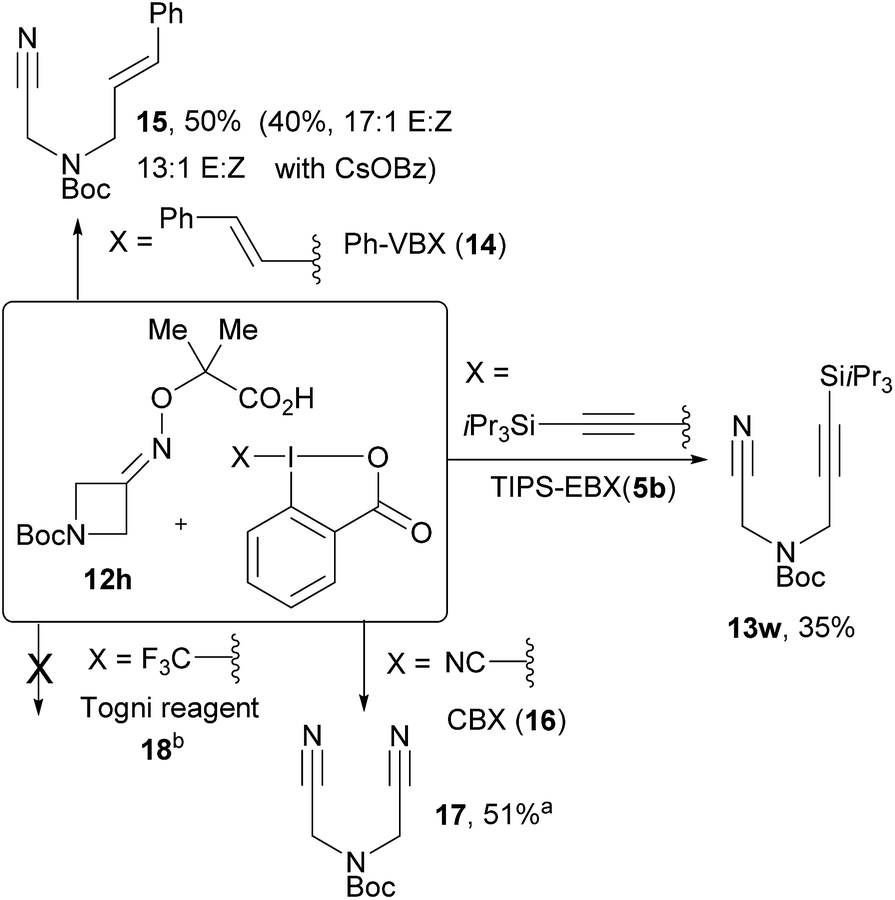 | ||
| Scheme 5 Extension to other C–C bond forming reactions. Reaction conditions: 5 mol% 4c, K2CO3 (1.1 equiv.), 0.05 M DCE, blue LEDs, rt, 1 h. aReaction run of reagent 16 (2 equiv.) for 14 h. NMR yield is given (isolated yield: 31%) breaction run of reagent 18 (2 equiv.) for 24 h. | ||
A one-pot protocol starting directly from the ketones was then developed (Scheme 6A). After condensation of cyclobutanone (11) with hydroxylamine 10, addition of two equivalents of Ph-EBX (5a) and 5 mol% of the organic dye followed by one hour of irradiation delivered alkyne 13a in 71% yield. A frequent drawback of organic dyes is their limited stability, leading often to low turnover numbers. Nevertheless, 66% of 13h could still be isolated when using only 1 mol% of 4c at a 1.0 mmol scale (Scheme 6B).
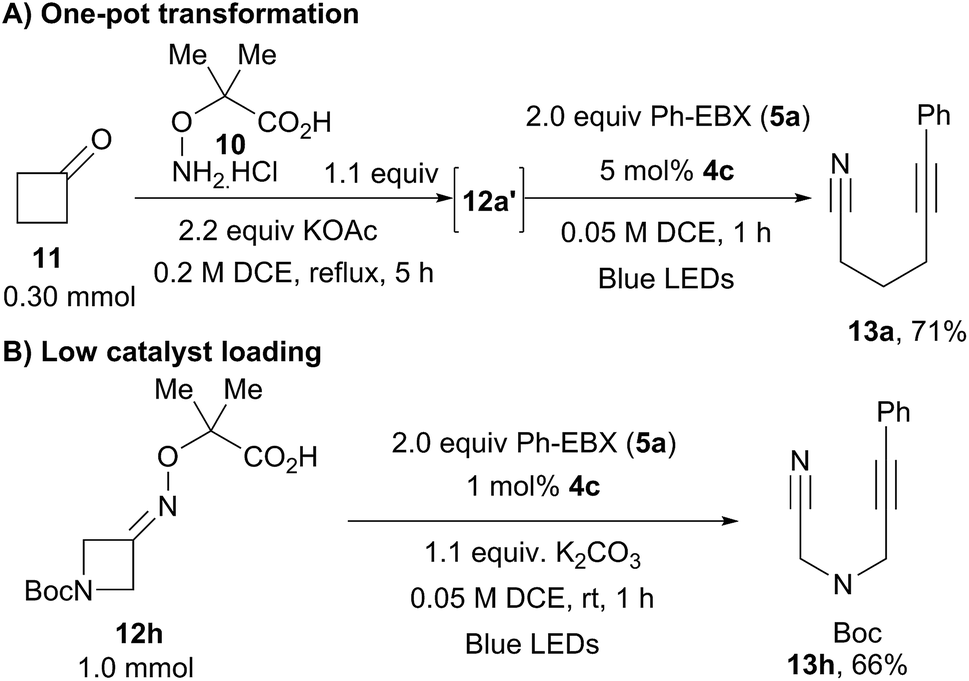 | ||
| Scheme 6 One-pot protocol and low catalyst loading. | ||
According to previous studies on EBX reagents,18a,b and the redox potential of dye 4c, we assume that the reaction starts with the oxidation of the potassium carboxylate 12a′ (E1/2 = +1.48 V vs. SCE in DMF) by the excited state PS* of the organic dye to generate carboxyl radical I and the reduced state PS− (Scheme 7). Fast decarboxylation releases the α-oxy radical II, which can either lead to iminyl radical III after acetone extrusion, or can be directly trapped by EBX reagents forming 19. Although 19 was not detected, its hydrated form 20 was observed by 1H NMR. The iminyl radical III then fragments into a nucleophilic alkyl nitrile radical IV, which reacts with EBX 5. For this step, a concerted mechanism via transition state TS1 can be envisioned according to our previous studies18b to afford the desired alkynylnitrile and radical V. A β-addition followed by a 1,2 shift of the EBX substituent could also be considered depending on the migratory aptitude.18b Final reduction of V allows the regeneration of the ground state photocatalyst PS and generates carboxylate 21.
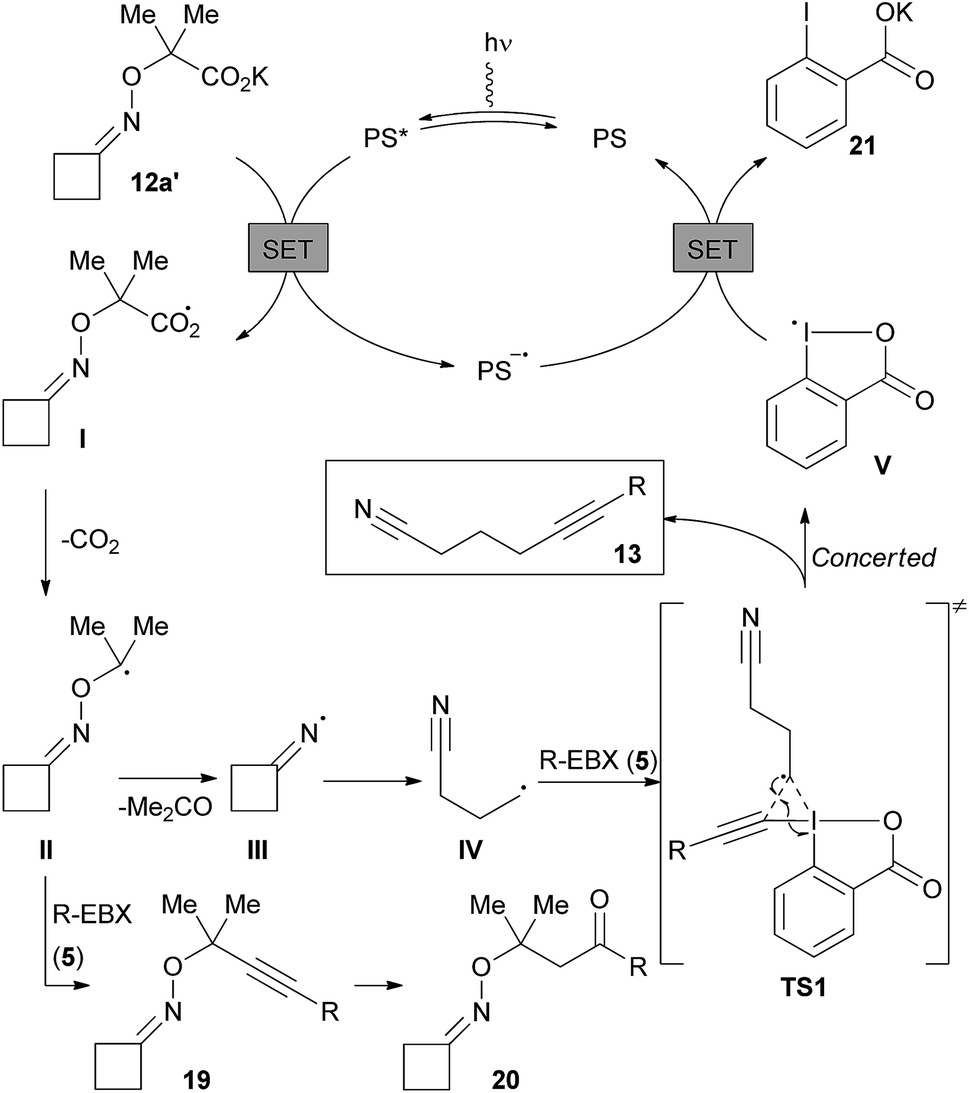 | ||
| Scheme 7 Speculative reaction mechanism. | ||
3. Conclusions
In summary, we have described the fine-tuning of organic dyes for the synthesis of alkynyl nitriles based on the alkynylation of alkylnitrile radicals generated after a visible light-driven oxidative ring fragmentation reaction involving an iminyl radical as intermediate. Preliminary results for the corresponding alkenylation and cyanation were also achieved. High yields were obtained in a short reaction time with low catalyst loading. We envision a bright future for these new dyes and further applications of cascade approaches in challenging C–C bond forming reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank ERC (European Research Council, Starting Grant iTools4MC, number 334840), the NCCR Chemical Biology (funded by the Swiss Science National Foundation) and EPFL for financial support. Ophélie Planes from the Laboratory of Supramolecular Chemistry at EPFL and Murat Alkan-Zambada and Laurent Liardet from the Laboratory of Inorganic Synthesis and Catalysis at EPFL are acknowledged for their help in the measurement of cyclic voltammograms. We thank Dr R. Scopelliti and Dr F. F. Tirani from ISIC at EPFL for X-ray analysis. G. G. acknowledges the funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No. 701885.Notes and references
- (a) L. F. Tietze, Chem. Rev., 1996, 96, 115 CrossRef PubMed; (b) L. F. Tietze, G. Brasche and K. M. Gericke, Domino Reactions in Organic Synthesis, Wiley-VCH, 2006 Search PubMed; (c) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134 CrossRef PubMed.
- M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692 CrossRef PubMed.
- (a) M. H. Shaw, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef PubMed; (b) D. W. C. Macmillan, C. R. J. Stephenson and T. P. Yoon, Visible Light Photocatalysis in Organic Chemistry, Wiley, 2018 Search PubMed; (c) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850 CrossRef PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075 CrossRef PubMed.
- M. S. Lowry, J. I. Goldsmith, J. D. Slinker, R. Rohl, R. A. Pascal, G. G. Malliaras and S. Bernhard, Chem. Mater., 2005, 17, 5712 CrossRef.
- (a) S. Fukuzumi and K. Ohkubo, Org. Biomol. Chem., 2014, 12, 6059 RSC; (b) D. A. Nicewicz and T. M. Nguyen, ACS Catal., 2014, 4, 355 CrossRef; (c) C. Yang, J. D. Yang, Y. H. Li, X. Li and J. P. Cheng, J. Org. Chem., 2016, 81, 12357 CrossRef PubMed.
- (a) J. Luo and J. Zhang, ACS Catal., 2016, 6, 873 CrossRef; (b) J. K. Matsui and G. A. Molander, Org. Lett., 2017, 19, 436 CrossRef PubMed; (c) H. Huang, C. Yu, Y. Zhang, Y. Zhang, P. S. Mariano and W. Wang, J. Am. Chem. Soc., 2017, 139, 9799 CrossRef PubMed.
- (a) S. Z. Zard, Chem. Soc. Rev., 2008, 37, 1603 RSC; (b) J. C. Walton, Molecules, 2016, 21, 63 CrossRef PubMed.
- (a) J. Boivin, E. Fouquet and S. Z. Zard, J. Am. Chem. Soc., 1991, 113, 1057 CrossRef; (b) J. Boivin, E. Fouquet and S. Z. Zard, Tetrahedron, 1994, 50, 1757 CrossRef.
- (a) T. Nishimura and S. Uemura, J. Am. Chem. Soc., 2000, 122, 12049 CrossRef; (b) T. Nishimura, Y. Nishiguchi, Y. Maeda and S. Uemura, J. Org. Chem., 2004, 69, 5342 CrossRef PubMed; (c) T. Nishimura, T. Yoshinaka, Y. Nishiguchi, Y. Maeda and S. Uemura, Org. Lett., 2005, 7, 2425 CrossRef PubMed; (d) H.-B. Yang and N. Selander, Chem.–Eur. J., 2017, 23, 1779 CrossRef PubMed; (e) Y.-R. Gu, X.-H. Duan, L. Yang and L.-N. Guo, Org. Lett., 2017, 19, 5908 CrossRef PubMed; (f) J. Wu, J.-Y. Zhang, P. Gao, S.-L. Xu and L.-N. Guo, J. Org. Chem., 2018, 83, 1046 CrossRef PubMed; (g) M. M. Jackman, S. Im, S. R. Bohman, C. C. L. Lo, A. L. Garrity and S. L. Castle, Chem.–Eur. J., 2018, 24, 594 CrossRef PubMed.
- B. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 12727 CrossRef PubMed.
- X. Y. Yu, J. R. Chen, P. Z. Wang, M. N. Yang, D. Liang and W. J. Xiao, Angew. Chem., Int. Ed., 2018, 57, 738 CrossRef PubMed.
- L. Li, H. Chen, M. Mei and L. Zhou, Chem. Commun., 2017, 53, 11544 RSC.
- (a) H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2017, 56, 12273 CrossRef PubMed; (b) H. Jiang and A. Studer, Angew. Chem., Int. Ed., 2017, 57, 1692 CrossRef PubMed.
- J. Davies, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2017, 56, 13361 CrossRef PubMed.
- E. M. Dauncey, S. P. Morcillo, J. J. Douglas, N. S. Sheikh and D. Leonori, Angew. Chem., Int. Ed., 2018, 57, 744 CrossRef PubMed.
- (a) F. Le Vaillant, T. Courant and J. Waser, Angew. Chem., Int. Ed., 2015, 54, 11200 CrossRef PubMed; (b) F. Le Vaillant, M. D. Wodrich and J. Waser, Chem. Sci., 2017, 8, 1790 RSC; (c) Q.-Q. Zhou, W. Guo, W. Ding, X. Wu, X. Chen, L.-Q. Lu and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 11196 CrossRef PubMed.
- Castle and coworkers reported one single example of alkynylation in 54% yield using microwave irradiation at 90 °C and >3.5 equiv EBX reagent. See ref. 11g.
- A. Kretzschmar, C. Patze, S. T. Schwaebel and U. H. F. Bunz, J. Org. Chem., 2015, 80, 9126 CrossRef PubMed . 4c and 4d were used for OLEDs applications in this work..
- See the ESI† for computational methods..
- (a) Crystal structure of 4a is available at CCDC under number 1052646 (YUGDOV), see S. Wang, Y. Zhang, W. Chen, J. Wei, Y. Liu and Y. Wang, Chem. Commun., 2015, 51, 11972 RSC; (b) Crystal structure of 4c is available at CCDC under number 1838186.
- See the ESI† for a more detailed discussion..
- 10 is commercially available at a high price from a limited number of suppliers. Its synthesis was described in the following patent, which is available only in Chinese: L. Jiang, J. Yang, Z. Shumin, Synthesis of Oxoamino-Aliphatic Carboxylic Acids, 1991, CN1051170 (A).
- Aldehyde and nitrile substituents in para position of the ethynylarene were also tolerated, albeit led to low yield. (see ESI†).
- (a) D. J. Brien, A. Naiman and K. P. C. Vollhardt, J. Chem. Soc., Chem. Commun., 1982, 133 RSC; (b) J. A. Varela, L. Castedo and C. Saa, J. Am. Chem. Soc., 1998, 120, 12147 CrossRef; (c) L.-G. Xie, S. Shaaban, X. Chen and N. Maulide, Angew. Chem., Int. Ed., 2016, 55, 12864 CrossRef PubMed; (d) J. Sheng, Y. Wang, X. Su, R. He and C. Chen, Angew. Chem., Int. Ed., 2017, 56, 4824 CrossRef PubMed; (e) Y. Wang, C. Chen, S. Zhang, Z. Lou, X. Su, L. Wen and M. Li, Org. Lett., 2013, 15, 4794 CrossRef PubMed.
- (a) E. Stridfeldt, A. Seemann, M. J. Bouma, C. Dey, A. Ertan and B. Olofsson, Chem.–Eur. J., 2016, 22, 16066 CrossRef PubMed; (b) A. Boelke, L. D. Caspers and B. J. Nachtsheim, Org. Lett., 2017, 19, 5344 CrossRef PubMed.
- Ph-VBX was first used in photoredox catalysis by Leonori and coworkers (see ref. 16), affording only the E isomer..
- (a) P. Eisenberger, S. Gischig and A. Togni, Chem.–Eur. J., 2006, 12, 2579 CrossRef PubMed; (b) J. Charpentier, N. Frueh and A. Togni, Chem. Rev., 2015, 115, 650 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational data. CCDC 1052646 and 1838186. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01818a |
| This journal is © The Royal Society of Chemistry 2018 |