
Thermo-responsive phase-transition polymer grafted magnetic FePt nanoparticles with tunable critical temperature for controlled drug release†
Zhuo
Kang‡
a,
Yushan
Peng‡
a,
Lixin
Zhou‡
a,
Zhimin
Li
b,
Tianyu
Wang
b,
Zheng
Zhang
a,
Qingliang
Liao
a,
Jun
Gao
b,
Yongning
Li
*b and
Yue
Zhang
 *ac
*ac
aState Key Laboratory for Advanced Metals and Materials, School of Materials Science and Engineering, University of Science and Technology Beijing, Beijing 100083, China. E-mail: yuezhang@ustb.edu.cn
bDepartment of Neurosurgery, Peking Union Medical College Hospital, Chinese Academy of Medical Sciences, No. 1 Shuaifuyuan Hutong of Dongcheng District, Beijing, 100730, P. R. China. E-mail: 13901074129@139.com
cKey Laboratory of New Energy Materials and Technologies, University of Science and Technology Beijing, Beijing 100083, China
First published on 15th May 2018
Abstract
A temperature stimuli-responsive drug release system is presented in this work. Polymer (poly(N-isopropylacrylamide), PNIPAM based polymer) grafted FePt nanoclusters were fabricated, tethered with folic acid (FA) on their surfaces for cancer-cell specific targeting. The reversible thermal response of the prepared nanoclusters was successfully verified, and the threshold temperature for drug release is tunable as the lower critical solution temperature (LCST) of the polymer could be modulated from 32 to 45 °C by adjusting comonomers and their relative composition ratios. The resultant nanoclusters realized both amphipathic and hydrophobic cargo release resulting from the shrinkage of the polymer responding to temperature rise. Fluorescence spectroscopic analysis indicates that amphipathic molecules can be released out of the nanoclusters more efficiently than hydrophobic molecules. Additionally, superparamagnetic FePt NPs have the potential to serve as the heat source of the system through the magnetocaloric effect. And the nanoclusters can respond to pH as well, which holds promise for the therapy of various tissues with different pH values. With excellent cytocompatibility and flexible composition design, these thermal responsive nanoclusters have great prospects for controlled drug release to address a series of clinical indications.
Introduction
Encouraged by recent impressive progress in nanotechnology, temporally, spatially and dosage-controlled drug release in response to specific stimuli has become possible in the past decade, and drug delivery systems based on nanomaterials have received significant attention in cell therapy.1–6 However, there are still various problems to be solved, including poor cellular internalization, inefficient intracellular drug release, and drugs being rapidly metabolized or excreted from the body. To overcome the aforementioned limitations, techniques like an active targeting method and stimuli-triggered drug release have been proposed.Nanoparticles modified with cell-specific ligands can bind precisely to the targeted cells that possess the complementary receptors, which can be used for active targeting. For instance, cancer-associated biomarker molecules contain folic acid (FA) receptors (FAR-α, FAR-β),7–10 biotin receptors and so on. Gradually, a broad range of nanocarriers with different surface functions and modifications, core–shell architectures, sizes, and shapes have been developed. They include inorganic nanoparticles (NPs) made of carbon nanotubes, graphene, iron oxide NPs, quantum dots, gold, and metal oxide frameworks11–13 as well as organic-based ones such as dendrimers, liposomes, polymers, and polymer-based micelles, which could be involved in different stimuli-responsive drug delivery systems.
Generally, nanoscale stimuli-responsive drug delivery systems are divided into two classes: endogenous and exogenous. Endogenous stimuli are changes in pH7,14–16 or enzyme concentration,17–19 or redox gradients;20–22 and externally applied stimuli are changes in temperature,23,24 ultrasound intensity,25 magnetic fields,26–29 light,30 or electric pulses.31 Among these stimuli, extensive research on magnetic field and temperature stimuli-responsive systems has been reported. Poly(N-isopropylacrylamide) (PNIPAM), one of the most investigated thermo-responsive polymers, goes through a reversible phase transition from hydrophilic to hydrophobic (swelling to shrinking) in aqueous solution in response to a temperature change, corresponding to the formation and breakage of hydrogen bonds between the polymer and water. PNIPAM has a lower critical solution temperature (LCST) of 32 °C below normal body temperature, which is unsuitable for biomedical application for the human body but could be improved.32–39 Teresa Pellegrino changed the phase transition temperature of polymers from 26 to 47 °C by varying the comonomers and their relative molar ratio.40 In this case, drug release at a specific temperature below or above body temperature may come into existence.41
In this study, we demonstrate a magnetic and thermal responsive targeted drug delivery system based on polymer grafted FePt nanoclusters tethered with FA for cancer-cell specific targeting. By adjusting the monomers incorporated into the polymers, a series of LCSTs near body temperature were achieved, and the biocompatibility and water solubility of the nanoclusters increased as well. At the same time, fluorescent molecules were utilized to conduct the cargo release test of the nanoclusters. Our research also suggested that the system realized both hydrophilic and hydrophobic content release in different release kinetics.
Experimental
Reagents were obtained from commercial suppliers and used without further purification. Iron(III) acetylacetonate (Fe(acac)3), oleic acid and platinum acetylacetonate (Pt(acac)2) were purchased from Sigma. 1,2-Hexadecanediol, oleylamine, ethanol, and 2-aminoethanethiol were purchased from Alfa Aesar. Sodium hydroxide (NaOH, 98%) and acryloyl chloride were purchased from J&K Scientific Ltd. Ammonium persulfate (APS), N-isopropylacrylamide (NIPAM), poly(ethylene glycol)methyl ether methacrylate (PEGMA), allylamine (AA), FA, Nile red and Rhodamine B were purchased from Aladdin-Reagent. Deionized water with a resistivity of 16 MΩ cm−1 was used in all the experiments.General methods
X-ray diffraction (XRD) was performed on a Bruker-Nonius D8 Advance Diffractometer using Cu Kα radiation (λ = 1.5418 Å) with samples on quartz substrates. Particle size and size distribution were observed using a Philips Tecnai12 transmission electron microscope (TEM) operated at 100 kV equipped with a Gatan model 782 CCD camera, a Zeiss Ultra Plus Field Emission Scanning Electron Microscope (FE-SEM) and an atomic force microscope (AFM) respectively. Fourier transform infrared spectrophotometry (FTIR) was performed on a Perkin-Elmer Model 1600 Series FTIR Spectro-photometer. The magnetic moment of each sample was investigated over a range of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 G of applied magnetic fields using 30 min sweep time. Dynamic light scattering (DLS) measurements were performed on a Zetasizer Nano ZS90 (Malvern, USA) equipped with a 4.0 mW He–Ne laser operating at 633 nm and an avalanche photodiode detector.
000 G of applied magnetic fields using 30 min sweep time. Dynamic light scattering (DLS) measurements were performed on a Zetasizer Nano ZS90 (Malvern, USA) equipped with a 4.0 mW He–Ne laser operating at 633 nm and an avalanche photodiode detector.
Synthesis of FePt nanoparticles
Fe–Pt nanoparticles were synthesized with an average diameter of 5 nm using the procedure described as follows. 0.197 g (0.50 mmol) of Pt(acac)2 and 0.176 g (0.50 mmol) of Fe(acac)3 were added to 10 ml of oleylamine in a 100 ml three-neck flask and stirred (500 rpm) under a N2 atmosphere. The solution was then purified under vacuum at room temperature (RT) for 0.5 h and then heated to 110 °C under a N2 atmosphere. 1.35 ml (4.25 mmol) of oleic acid was sequentially injected through a rubber septum into the reactant solution and held for 0.5 h. The reactant mixture was heated to 290 °C and stirred for 0.5 h. Thereafter, the mixture was cooled to RT and the reactants were purified in air. The suspension was centrifuged at 9000 rpm for 20 min to precipitate large aggregates and discard the aqueous supernatant liquid. The aggregate products obtained as oleic acid-coated FePt nanoparticles were washed with ethanol three times and stored in hexane.Preparation of acrylamide-grafted FePt
2-Aminoethanethiol (0.06 g) was added to the re-dispersed oleic acid-coated FePt nanoparticles (0.03 g) in a round bottom flask, followed by addition of ethanol (10 ml) and stirring for 24 h at RT under a N2 atmosphere. Then the prepared amino-grafted FePt (FePt–NH2) nanoparticles were re-dispersed in NaOH solution (1.5 g in 10 ml of deionized water) after magnetic separation and cleaning with ethanol and deionized water. Acryloyl chloride (2.0 ml) was added dropwise to the dispersion in an ice-water bath and the solution was continuously stirred at RT for 24 h. After separation and cleaning of the above mixture, the final product FePt–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 was dispersed and stored in water (0.01 g of FePt–CHCH2 in 1 ml of water).
CH2 was dispersed and stored in water (0.01 g of FePt–CHCH2 in 1 ml of water).
Synthesis of polymer grafted FePt nanoclusters
Polymer grafted FePt nanoclusters were synthesized via a free radical polymerization using APS as an initiator. In a typical polymerization, 10 ml of deionized water was added to a solution of FePt–CHCH2 nanoparticles (3 ml) in a 100 ml three-neck flask, followed by addition of NIPAM (0.1 g) and APS (0.02 g in 0.05 ml of deionized water). The mixture was set at 70 °C and stirred for 0.5 h under a N2 atmosphere. After that, different amounts of PEGMA and AA were added dropwise into the mixture for 30 min to obtain samples with different polymer compositions. The product was washed with deionized water to remove ungrafted copolymers or unreacted monomers and centrifuged at 5000 rpm for 20 min to precipitate large aggregates and discard the aqueous supernatant liquid. In the end, polymer-grafted FePt nanoclusters were re-dispersed in water (0.001 g ml−1) after removing the water using a freezing machine.
Preparation of folic acid tethered polymer grafted FePt nanoclusters
Polymer grafted FePt nanoclusters with FA were activated via EDC/NHS chemistry using the following procedure. In brief, 0.16 mmol of FA (70.6 mg) was dissolved in 10 ml of a DMSO–Milli-Q water mixture (1:1 v/v), followed by the addition of EDC (0.32 mmol, 65.92 mg) and NHS (0.32 mmol, 36.83 mg), maintaining the pH at 7.0–8.0 with sodium hydroxide (NaOH) solution. After 6 h FA activation, 5 ml of an aqueous dispersion of polymer grafted FePt nanoclusters was added dropwise to the solution, and the mixture was stirred overnight in the dark at RT. Finally, the FA-modified polymer grafted FePt nanoclusters were magnetically precipitated and washed with water and DMSO several times.
Loading and release of fluorescent molecules
A mixture of a polymer-grafted FePt nanocluster solution (1 mg) and a fluorescent molecule solution (0.1 mg ml−1, 10 ml) was kept in a shaker for 24 h in the dark to achieve uniform cargo-loaded nanocarriers. Then, the prepared nanocarrier solution was transferred into a dialysis bag (Mw = 10 kDa) suspended in distilled water at RT to remove the unencapsulated drugs. And the release medium was renewed every 8 hours 3 times. To analyse how the cargo release behaviour related to temperature, the separated nanocarrier solution was incubated in a water bath and the fluorescence intensities of the experimental cellular solutions were examined and recorded at selected time intervals from 5 to 60 min at different temperatures.Results and discussion
Characterization of polymer grafted FePt nanoclusters
In this work, an illustration of the three steps involved in the preparation of polymer grafted FePt nanoclusters is given in Fig. 1. The FePt NPs were synthesized with an average diameter of 5 nm using the procedure mentioned earlier. Then the functionalization of the FePt surface with the amino group was followed by covalent conjunction with thermo-responsive (PNIPAM, AA, PEGMA) polymers successively via a free radical polymerization using APS as an initiator. Finally, FA-polymer grafted FePt nanoclusters were successfully obtained after tethering FA to the surfaces of the nanoclusters.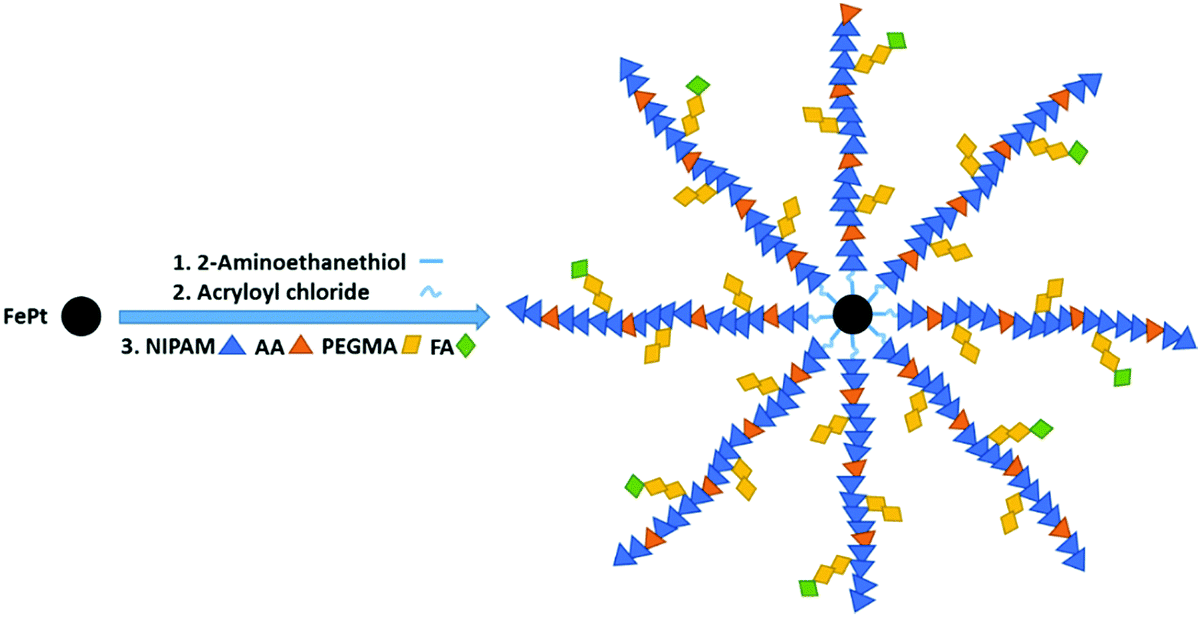 | ||
| Fig. 1 An illustration of three steps involved in the preparation of polymer grafted FePt nanoclusters. | ||
Morphological and structural characterization
It can be seen from the TEM image of FePt NPs as well as the FE-SEM and AFM images of the PNIPAM grafted FePt nanoclusters (Fig. 2a, d and e) that a large quantity of the prepared samples was uniformly dispersed on the substrates. The starting FePt NPs have a diameter distribution ranging from 2 to 10 nm. Fig. 2b and c show that the FePt NPs were well grafted with polymers. And the final nanoclusters have diameters of 120 ± 20 nm, which are well within the preferred size range of the NPs for effective drug delivery. Moreover, the 3D-image of the nanocluster (Fig. 2f) demonstrates that the nanocluster has a capacity for loading some small molecules like dyes or drugs because of its non-solid structure.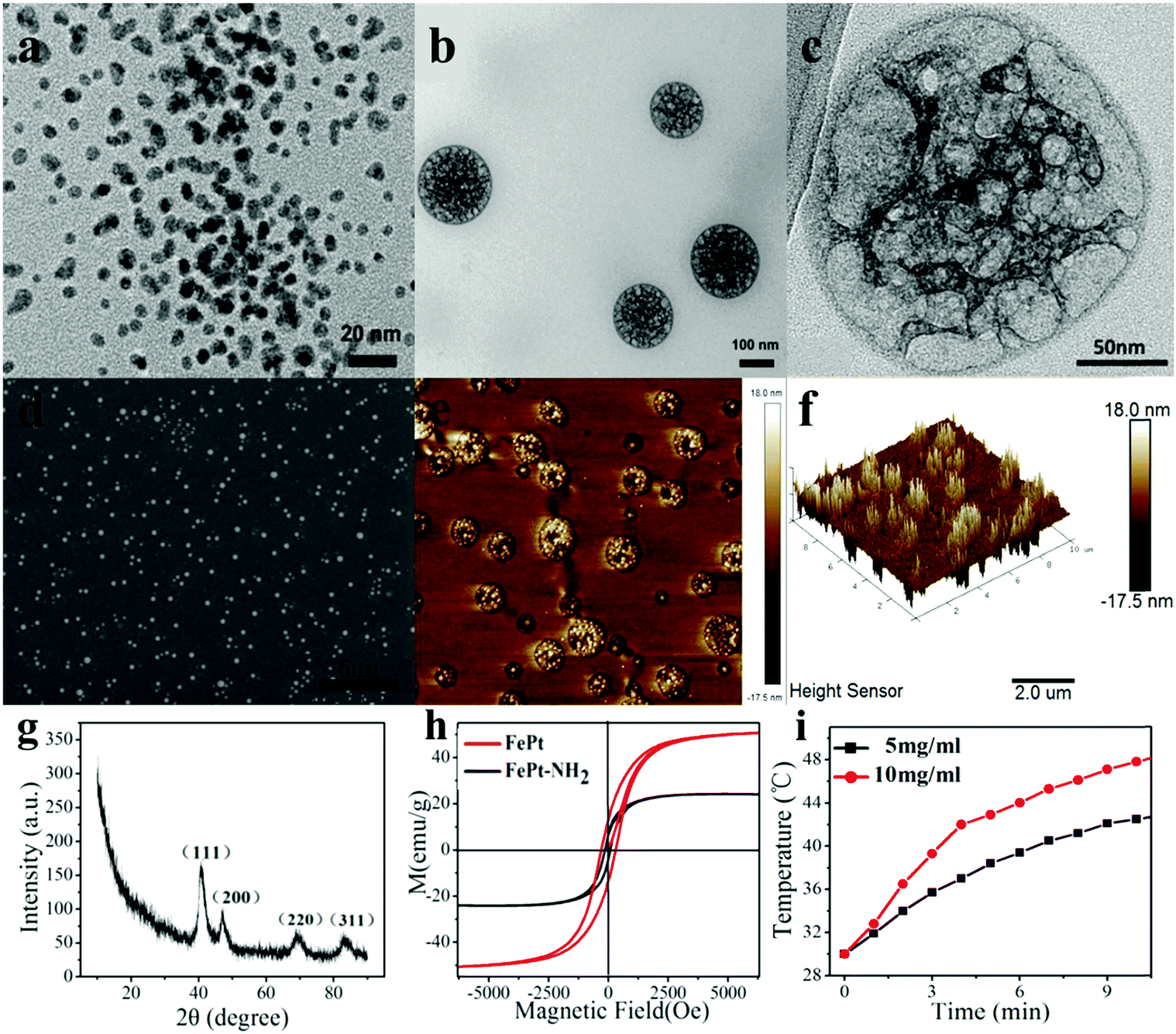 | ||
| Fig. 2 (a) TEM image of FePt NPs. (b and c) High magnification TEM images of PNIPAM grafted FePt nanoclusters. (d) FE-SEM image of PNIPAM grafted FePt nanoclusters. (e) AFM topographical image of PNIPAM grafted FePt nanoclusters. (f) 3D-Image of a PNIPAM grafted FePt nanocluster. (g) XRD analysis image of FePt NPs. (h) M–H curves for FePt and FePt–NH2 NPs. (i) High-frequency induction heating curves of FePt NPs. | ||
X-ray diffraction studies and magnetic measurements
The crystalline nature and the phase purity of the prepared FePt NPs were studied via XRD analysis. Fig. 2g shows the characteristic peak of FePt NPs on the (111) surface at 40 degrees, suggesting their disordered face-centred cubic structures, in which Fe or Pt atoms randomly occupy any position of the crystal lattice rather than a certain point of the cubic cell. All of the diffraction peaks in the diffraction pattern are well consistent with a standard XRD-PDF pattern.The magnetic properties of FePt NPs were ascertained using a superconducting quantum interference device (SQUID) under external magnetic fields from −8000 G to 8000 G at RT. The saturated magnetization values for FePt NPs and FePt–NH2 NPs were calculated to be 50.601 emu g−1 and 24.22 emu g−1, respectively, from the magnetic hysteresis loops as shown in Fig. 2h. Although there is a gradual decrease in the Ms values with surface modification due to a decrease in the magnetic dipolar interaction with diamagnetic coating, the NPs still showed high enough Ms values and exhibited superparamagnetic behaviour at RT. Fig. 2i displays the results of high-frequency induction heating experiments of FePt NPs (5 mg ml−1 and 10 mg ml−1), verifying that the FePt NPs are able to regulate the temperature near 37 °C by the magnetocaloric effect.
FT-IR spectral analysis
The presence of surface functional groups on NPs was analysed by the FTIR method (Fig. 3). The FTIR spectrum of FePt shows no characteristic signals. The spectrum of FePt–CHCH2 shows peaks at 3400, 2974, 1600, 1450, 1383, and 1050 cm−1, which correspond to O–H stretching, C–H bond vibration, N–H bending, and C–N stretching, demonstrating that FePt–CHCH2 was obtained through coupling between amino-grafted FePt and acryloyl chloride. After decoration with polymers, characteristic peaks of the nanoclusters were observed at 1050, 1087, and 1273 cm−1, confirming the polymer linkage via an acrylamide bond.
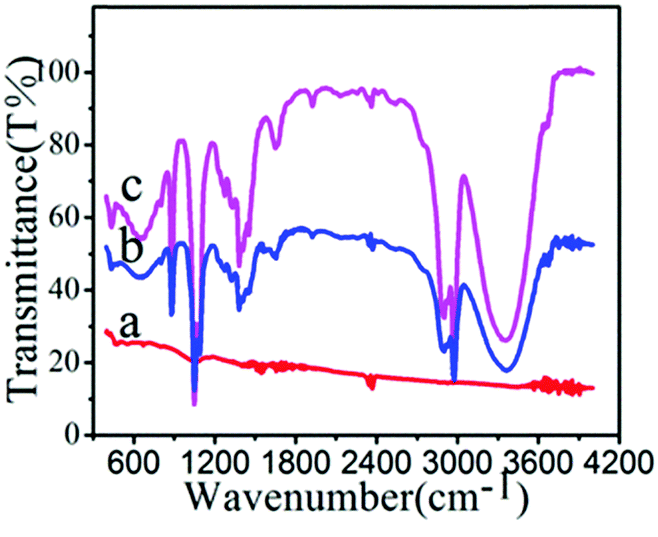 | ||
| Fig. 3 FTIR spectra of (a) FePt, (b) FePt–CHCH2 and (c) NIPAM grafted FePt nanoclusters. | ||
Thermal response test of polymer grafted FePt nanoclusters
The nanocluster solution was dialyzed using a microporous filtering film (450 nm) to remove impurities before the thermal response test. The aqueous solution of PNIPAM grafted FePt nanoclusters (PNIPAM–FePt) became turbid upon heating to 50 °C and became clear again after cooling to RT as shown in Fig. 4a, indicating that the phase transition of the nanoclusters with temperature change is reversible. The UV-vis spectra of the PNIPAM–FePt solution at different temperatures between 20 and 50 °C are shown in Fig. 4b, from which the temperature-dependent optical transmittance (λ = 380 nm) of the PNIPAM–FePt solution is plotted in Fig. 4c, revealing that the solution became opaque above 32 °C. Similarly, the transmittance change of the FePt nanoclusters grafted with polymers (NIPAM:AA = 5:1) decreased as the temperature decreased and the nanoclusters showed a LCST increase to about 36 °C (Fig. S2, ESI†). The hydrodynamic volume of the PNIPAM–FePt solution at pH 7.4 decreased near the LCST, resulting from the dissociation of hydrogen bonds between the hydrophilic groups of the polymers and water (Fig. 4d). It has been proved that the thermal responsive volume changes of the nanoclusters are irrelevant to the FePt NPs (Fig. S3, ESI†). Moreover, pH could also affect the formation of the hydrogen bonds, thereby affecting the volumes of the nanoclusters, which can be a regulation strategy for drug release systems (Fig. S4, ESI†).
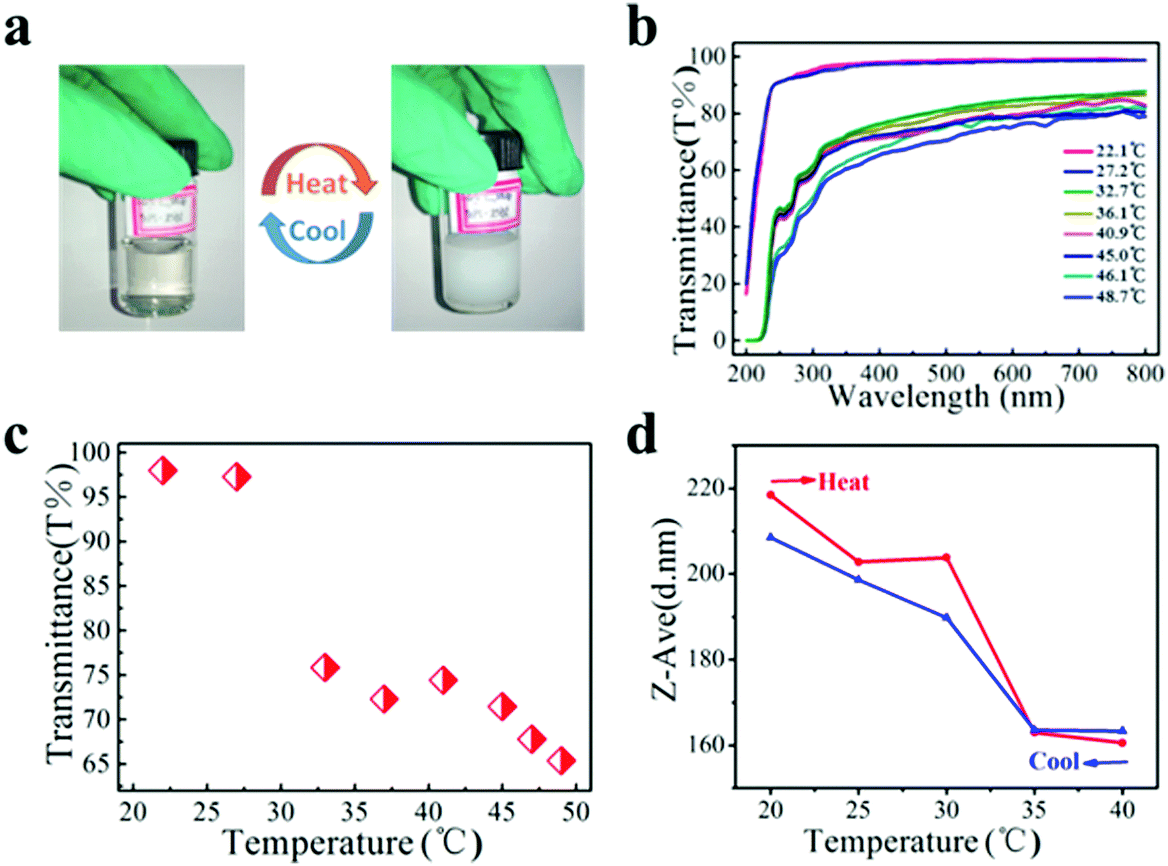 | ||
| Fig. 4 The results of the thermal response test for PNIPAM grafted FePt nanocluster. (a) Photograph of a PNIPAM–FePt solution in vials at 25 °C (below the LCST, left) and 45 °C (above the LCST, right). (b) UV-vis spectra of the temperature-responsive optical transmittance of PNIPAM–FePt in water. (c) Temperature-dependent optical transmittance of PNIPAM–FePt (λ = 380 nm) in water. (d) DLS measurements of PNIPAM–FePt in water. | ||
The modulation of the phase transition temperature
Initially, we obtained PNIPAM–FePt with a LCST of 32 °C in water via growing a PNIPAM shell at the FePt NPs’ surfaces. This phase transition temperature makes the NPs unsuitable for clinical applications, as it is lower than body temperature. In chemical synthesis, the copolymerization of NIPAM with different monomers can shift the LCSTs of the resulting polymers from 32 °C to higher temperature. In this work, the LCSTs of the polymers increased up to 33–36 °C, 35–38 °C, 37–40 °C, 40–45 °C, respectively, with different mole ratios of NIPAM to allylamine (AA) of 10:1, 8:1, 5:1, 3:1, which is promising for clinical application with various temperature demands (Fig. 5).
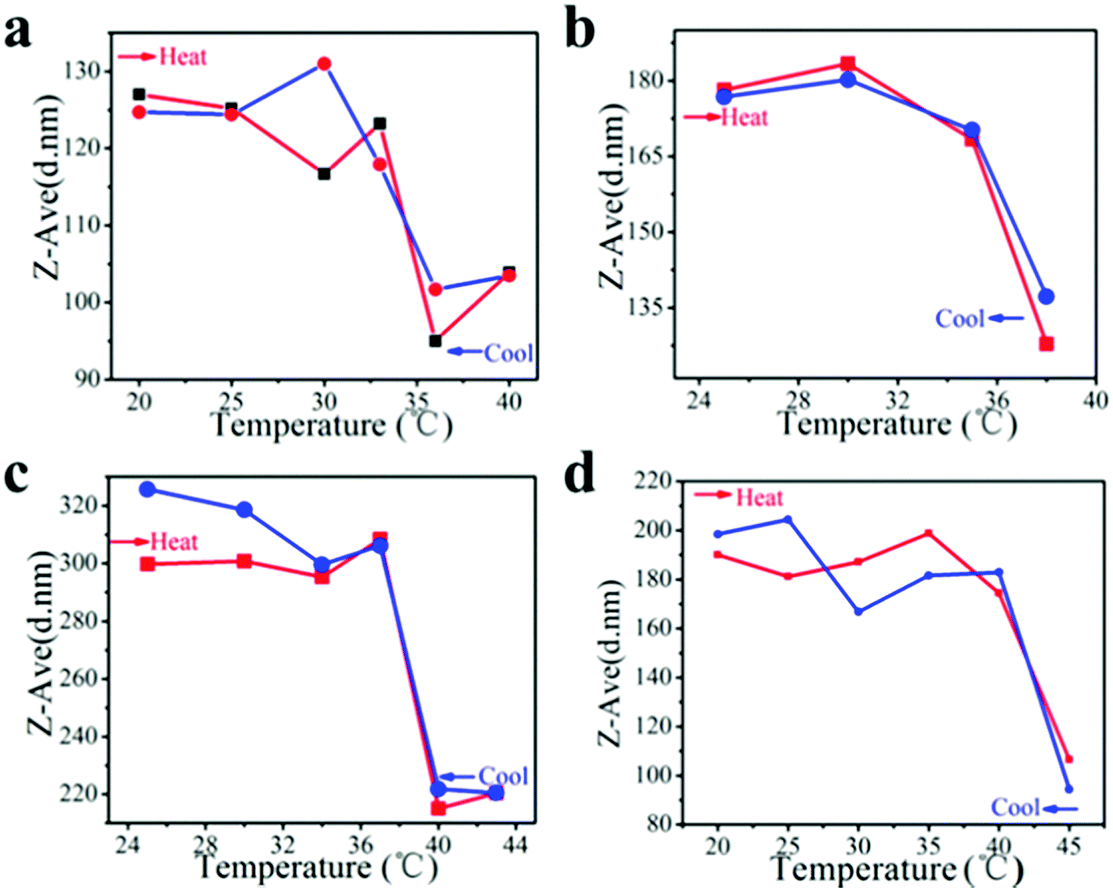 | ||
| Fig. 5 DLS measurements of polymer grafted FePt nanoclusters with NIPAM and AA as copolymerization monomers at different mole ratios: (a) NIPAM:AA = 10:1; (b) NIPAM:AA = 8:1; (c) NIPAM:AA = 5:1; and (d) NIPAM:AA = 3:1. | ||
Drug release experiment
Small molecule therapeutic agents inevitably cause adverse reactions and systemic toxicity. There exist certain issues like poor aqueous solubility, rapid clearance and metabolism, and nonspecific tissue accumulation. Nano-therapeutic delivery strategies aim to overcome certain problems and show great prospects particularly in anticancer therapeutics including DOX, paclitaxel (PTX), 10-hydroxycamptothecin, 5-fluorouracil (5-FU), tamoxifen and others. Clinical drug molecules can be divided into two broad categories: hydrophobic and amphipathic. Therefore, hydrophobic Nile red42 and amphipathic Rhodamine B, two kinds of fluorescent molecules, were treated as substitutes for therapeutic agents to track drug release, taking advantage of their fluorescence in hydrophobic environments but non-fluorescence in polar aqueous environments.According to the phase transition forms of the polymers, the following drug release mechanism is proposed. As illustrated in Fig. 6a and b, the shrinkage of the polymers caused by hydrophobic transition with increasing temperature is accompanied by the squeezing of the loaded molecules out of the nanocluster to aqueous solution. But hydrophobic and amphipathic loading molecules differ slightly in specific release kinetics.
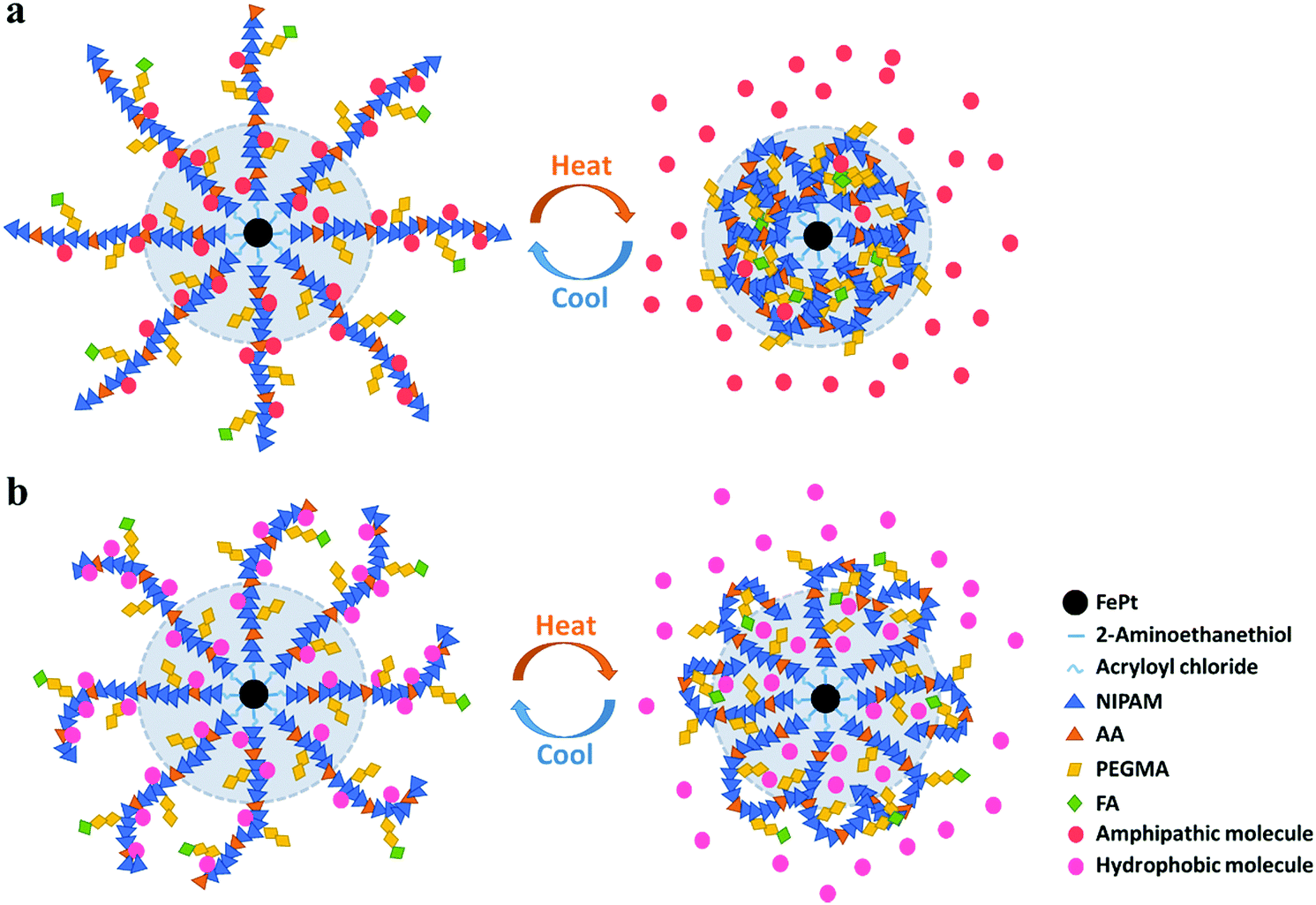 | ||
| Fig. 6 Drug release mechanism of the polymer grafted nanocluster loaded with (a) amphipathic molecules and (b) hydrophobic molecules. | ||
For amphipathic molecules, the outer and even inner molecules (inside the light blue shaded area in Fig. 6a) would distribute into the solution due to the intense squeezing effect between hydrophobic polymers and amphipathic cargos. For hydrophobic molecules (Fig. 6b), however, the inner molecules remain in situ, and some outer molecules would release into the solution, while the rest would be absorbed and wrapped by the polymers owing to the hydrophobic interaction.
Rhodamine B, as a model amphipathic drug, was encapsulated in PNIPAM–FePt to carry out the drug release test (Fig. 7). The fluorescence intensity of the Rhodamine B-loaded nanoclusters decreased with higher temperature, especially for the temperature above the NIPAM's LCST (32 °C) as shown in Fig. 7a and b. Meanwhile, as Fig. 7c depicts, the stable and reversible cargo release behaviour of the nanoclusters was proved using alternate heating and cooling experiments. When the temperature is above the LCST, the molecular chains of the polymers would contract, leading to a decrease in the nanocluster diameter (Fig. 7d). The amphipathic Rhodamine B almost realized complete release into water as is mentioned earlier, which inhibited the movement of the conjugated electron cloud of Rhodamine B, consistent with its lower fluorescence intensity at higher temperatures.
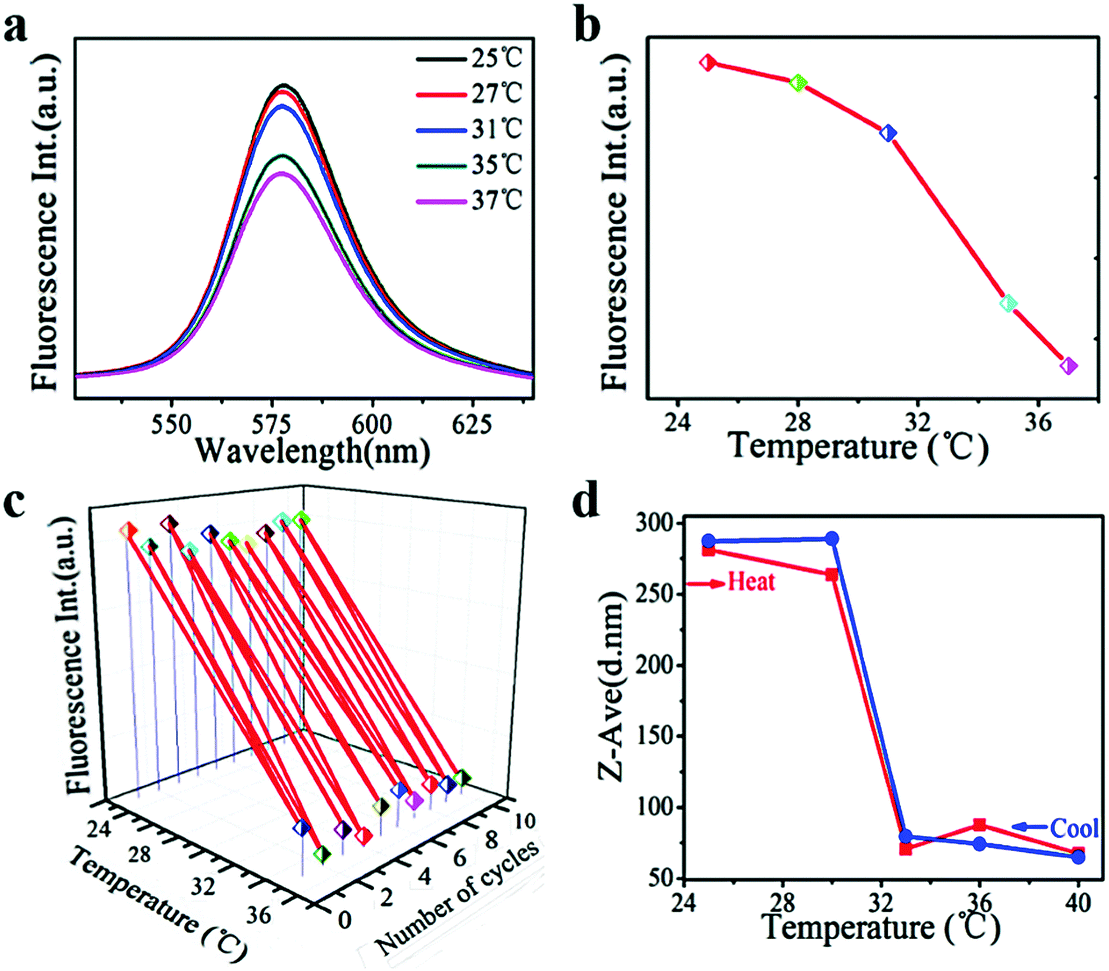 | ||
| Fig. 7 (a) The fluorescence spectra of Rhodamine B-loaded PNIPAM–FePt at different temperatures. (b) The fluorescence intensity of Rhodamine B-loaded PNIPAM–FePt in water. (c) Alternate heating and cooling experiments of Rhodamine B-loaded PNIPAM–FePt in water between 25 °C (below the LCST) and 37 °C (above the LCST). (d) DLS measurements of Rhodamine B-loaded PNIPAM–FePt in water. | ||
Nile red (used as a model hydrophobic drug) loaded PNIPAM–FePt was treated almost in the same way as Rhodamine B-loaded PNIPAM–FePt. As illustrated in Fig. 8a and b, the fluorescence intensity of the Nile red-loaded nanoclusters increased with higher temperature, especially after the temperature reached the polymer's LCST (32 °C) and vice versa. Also, the result remained the same for several heating cycles, again confirming the stability of the nanoclusters (Fig. 8c). The curve of the sizes of the Nile red-loaded nanoclusters versus temperature displays the same trend as those of the Rhodamine B-loaded nanoclusters, but presents smaller initial diameters and larger final diameters (Fig. 8d), which is attributed to the hydrophobic properties and incomplete release of Nile red. Based on the hydrophobic drug release mechanism, more Nile red molecules were located in the hydrophobic area compared to the starting circumstance, leading to the reduction of the fluorescence quenching effect, which explains the fluorescence enhancement with temperature increase. In addition, FePt nanoclusters decorated with different polymers (NIPAM:AA = 5:1) were heated to a series of temperatures to accomplish the Nile red release test. The results show a sharp increase in fluorescence intensity above 36 °C (Fig. S5, ESI†), suggesting the possibility of drug release at various temperatures by adjusting the polymers’ compositions.
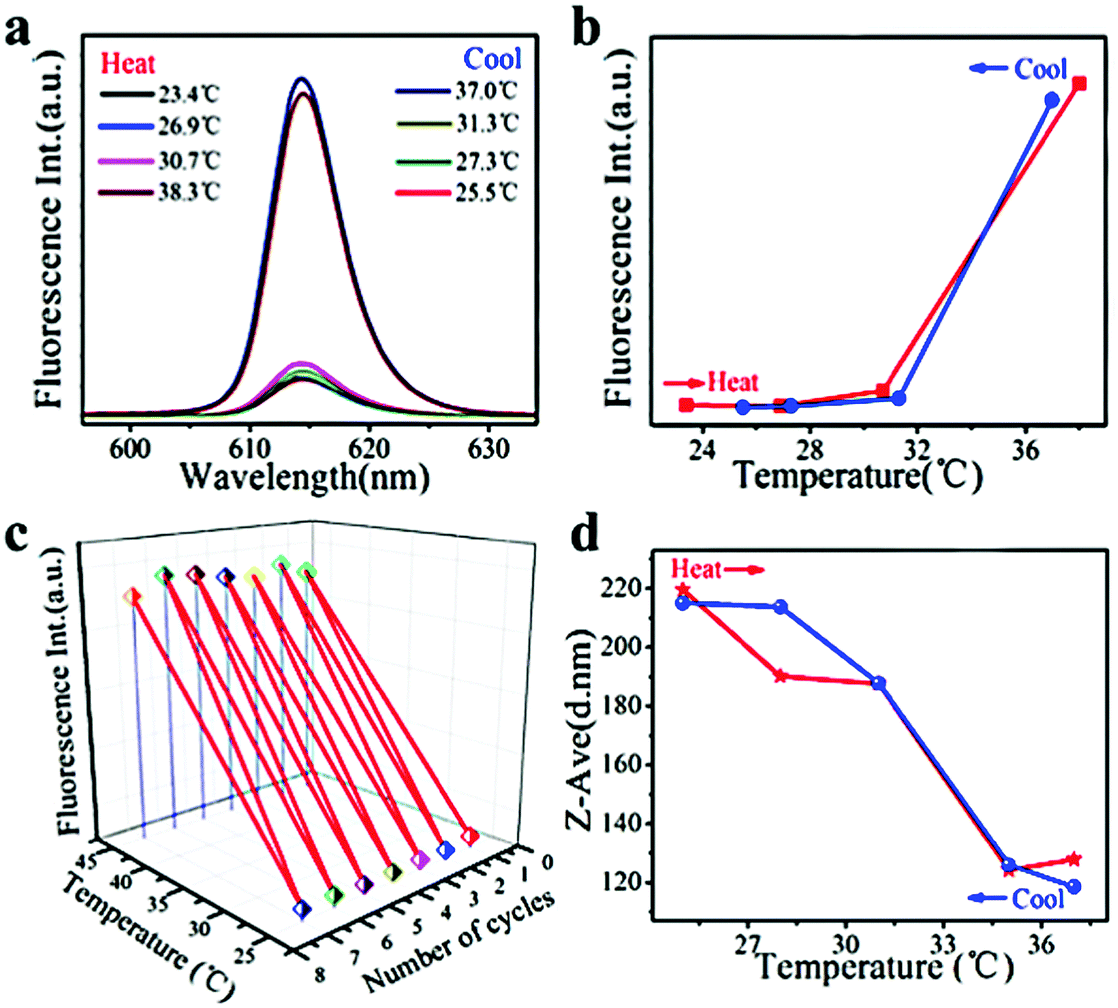 | ||
| Fig. 8 (a) The fluorescence spectra of Nile red-loaded PNIPAM–FePt. (b) The fluorescence intensity of Nile red-loaded PNIPAM–FePt in water. (c) Alternate heating and cooling experiments of Nile red-loaded PNIPAM–FePt in water between 25 °C (below the LCST) and 45 °C (above the LCST). (d) DLS measurements of Nile red-loaded PNIPAM–FePt in water. | ||
Conclusions
In summary, polymer grafted FePt nanoclusters tethered with FA, possessing different LCSTs, have been designed and developed as nanocarriers for targeted drug delivery systems. Superparamagnetic FePt NPs are expected to serve as the heat source of the system through the magnetocaloric effect, and to meet the demand of thermoregulation near body temperature. The phase transition temperature of the nanoclusters could be tuned from 32 °C to 45 °C by modifying the monomers and their relative ratios in the synthesis of thermo-responsive polymers. As is investigated, the cargo release of the nanoclusters is induced by the shrinkage of the polymers responding to the temperature increase. It has also been confirmed using fluorescent model drug release experiments that the nanoclusters are more suitable for the release of amphipathic molecules than hydrophobic molecules. Furthermore, FA, with targeting characteristics towards tumour cells, has been tethered at the nanocluster surfaces, indicating the availability of connection with other cell-specific ligands for the targeted therapy of various diseases. The clear pH-responsiveness of the nanoclusters also provides evidence for their application in the treatment for various tissues with different pH values. Accordingly, these magnetic and thermal responsive nanoclusters appear to be promising for specific targeting and therapeutics in nanomedicine.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Major Research Program of China (No. 2013CB932602), the National Key Research and Development Program of China (2016YFA0202701), the Program of Introducing Talents of Discipline to Universities (B14003), the National Natural Science Foundation of China (No. 51527802, 51232001, 51372020 and 51602020), the Beijing Municipal Science and Technology Commission (Z151100003315021), the China Postdoctoral Science Foundation (No. 2016M600039) and the Fundamental Research Funds for the Central Universities (FRF-TP-16-031A1).References
- H. Maeda and Y. Matsumura, Crit. Rev. Ther. Drug Carrier Syst., 1989, 6, 193–210 Search PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef PubMed.
- H. Maeda, J. Fang, T. Inutsuka and Y. Kitamoto, Int. Immunopharmacol., 2003, 3, 319–328 CrossRef PubMed.
- H. Maeda, Adv. Enzyme Regul., 2001, 41, 189–207 CrossRef PubMed.
- P. T. Wong and S. K. Choi, Chem. Rev., 2015, 115, 3388–3432 CrossRef PubMed.
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef PubMed.
- B. Sahoo, K. S. Devi, R. Banerjee, T. K. Maiti, P. Pramanik and D. Dhara, ACS Appl. Mater. Interfaces, 2013, 5, 3884–3893 Search PubMed.
- R. J. Lee and P. S. Low, Biochim. Biophys. Acta, Biomembr., 1995, 1233, 134–144 CrossRef.
- A. R. Hilgenbrink and P. S. Low, J. Pharm. Sci., 2005, 94, 2135–2146 CrossRef PubMed.
- P. S. Low and S. A. Kularatne, Curr. Opin. Chem. Biol., 2009, 13, 256–262 CrossRef PubMed.
- Y. K. Huang, C. H. Su, J. J. Chen, C. T. Chang, Y. H. Tsai, S. F. Syu, T. T. Tseng and C. S. Yeh, ACS Appl. Mater. Interfaces, 2016, 8, 14470–14480 Search PubMed.
- T. Li, X. Shen, Y. Geng, Z. Chen, L. Li, S. Li, H. Yang, C. Wu, H. Zeng and Y. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 13748–13758 Search PubMed.
- N. Parvin, Q. Jin, Y. Z. Wei, R. B. Yu, B. Zheng, L. Huang, Y. Zhang, L. H. Wang, H. Zhang, M. Y. Gao, H. J. Zhao, W. P. Hu, Y. L. Li and D. Wang, Adv. Mater., 2017, 29, 1606755 CrossRef PubMed.
- T. Ishida, Y. Okada, T. Kobayashi and H. Kiwada, Int. J. Pharm., 2006, 309, 94–100 CrossRef PubMed.
- X. Wu, Z. Wang, D. Zhu, S. Zong, L. Yang, Y. Zhong and Y. Cui, ACS Appl. Mater. Interfaces, 2013, 5, 10895–10903 Search PubMed.
- D. Li, L. Y. Sun, Y. T. Zhang, M. Yu, J. Guo and C. C. Wang, Mater. Chem. Front., 2017, 1, 521–529 RSC.
- L. Zhu, T. Wang, F. Perche, A. Taigind and V. P. Torchilin, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17047–17052 CrossRef PubMed.
- C. J. Chen, J. C. Wang, E. Y. Zhao, L. Y. Gao, Q. Feng, X. Y. Liu, Z. X. Zhao, X. F. Ma, W. J. Hou and L. R. Zhang, Biomaterials, 2013, 34, 5303–5316 CrossRef PubMed.
- S. Kashyap, N. Singh, B. Surnar and M. Jayakannan, Biomacromolecules, 2016, 17, 384–398 CrossRef PubMed.
- J. Wang, X. Sun, W. Mao, W. Sun, J. Tang, M. Sui, Y. Shen and Z. Gu, Adv. Mater., 2013, 25, 3670–3676 CrossRef PubMed.
- K. Kono, M. Taishi, T. Yoshida, Y. Haba, S. Kanaoka, T. Takagishi and S. Aoshima, Bioconjugate Chem., 2005, 16, 1367–1374 CrossRef PubMed.
- X. Y. Zhang, L. Han, M. Y. Liu, K. Wang, L. Tao, Q. Wan and Y. Wei, Mater. Chem. Front., 2017, 1, 807–822 RSC.
- G. J. Czarnota, R. Karshafian, P. N. Burns, S. Wong, A. Al Mahrouki, J. W. Lee, A. Caissie, W. Tran, C. Kim, M. Furukawa, E. Wong and A. Giles, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2033–2041 CrossRef PubMed.
- X. J. Zhou, Q. Yang, J. Y. Li, J. J. Nie, G. P. Tang and B. Y. Du, Mater. Chem. Front., 2017, 1, 369–379 RSC.
- A. Baeza, E. Guisasola, E. Ruizhernández and M. Valletregí, Chem. Mater., 2012, 24, 517–524 CrossRef.
- J. L. Viveroescoto, I. I. Slowing, C. W. Wu and V. S. Y. Lin, J. Am. Chem. Soc., 2009, 131, 3462–3463 CrossRef PubMed.
- C. S. Kumar and F. Mohammad, Adv. Drug Delivery Rev., 2011, 63, 789–808 CrossRef PubMed.
- E. Guisasola, A. Baeza, M. Talelli, D. Arcos, M. Moros, J. M. de la Fuente and M. Vallet-Regi, Langmuir, 2015, 31, 12777–12782 CrossRef PubMed.
- S. R. Deka, A. Quarta, R. Di Corato, A. Riedinger, R. Cingolani and T. Pellegrino, Nanoscale, 2011, 3, 619–629 RSC.
- Q. Yan, J. Yuan, Z. Cai, Y. Xin, Y. Kang and Y. Yin, J. Am. Chem. Soc., 2010, 132, 9268–9270 CrossRef PubMed.
- J. Rao and A. Khan, J. Am. Chem. Soc., 2013, 135, 14056–14059 CrossRef PubMed.
- T.-Y. Liu, S.-H. Hu, D.-M. Liu, S.-Y. Chen and I. W. Chen, Nano Today, 2009, 4, 52–65 CrossRef.
- S. Purushotham and R. V. Ramanujan, Acta Biomater., 2010, 6, 502–510 CrossRef PubMed.
- M. M. Rahman and A. Elaissari, J. Colloid Sci. Biotechnol., 2012, 1, 3–15 CrossRef.
- T. Shang, C. D. Wang, L. Ren, X. H. Tian, D. H. Li, X. B. Ke, M. Chen and A. Q. Yang, Nanoscale Res. Lett., 2013, 8, 1–4 CrossRef PubMed.
- H. Wei, C. Cheng, C. Chang, W. Q. Chen, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, Langmuir, 2008, 24, 4564–4570 CrossRef PubMed.
- N. Shamim, L. Hong, K. Hidajat and M. S. Uddin, Colloids Surf., B, 2007, 55, 51–58 CrossRef PubMed.
- S. Meerod, B. Rutnakornpituk, U. Wichai and M. Rutnakornpituk, J. Magn. Magn. Mater., 2015, 392, 83–90 CrossRef.
- N. I. Taib, V. Agarwal, N. M. Smith, R. C. Woodward, T. G. S. Pierre and K. S. Iyer, Mater. Chem. Front., 2017, 1, 2335–2340 RSC.
- M. Pernia Leal, A. Torti, A. Riedinger, R. La Fleur, D. Petti, R. Cingolani, R. Bertacco and T. Pellegrino, ACS Nano, 2012, 6, 10535–10545 CrossRef PubMed.
- X. Guo, D. Li, G. Yang, C. Shi, Z. Tang, J. Wang and S. Zhou, ACS Appl. Mater. Interfaces, 2014, 6, 8549–8559 Search PubMed.
- M. K. Gupta, J. R. Martin, T. A. Werfel, T. Shen, J. M. Page and C. L. Duvall, J. Am. Chem. Soc., 2014, 136, 14896–14902 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00154e |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2018 |