
Thermo-responsive phase-transition polymer grafted magnetic FePt nanoparticles with tunable critical temperature for controlled drug release†
 *ac
*ac
Abstract
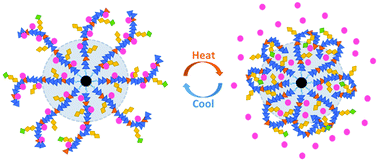
A temperature stimuli-responsive drug release system is presented in this work. Polymer (poly(N-isopropylacrylamide), PNIPAM based polymer) grafted FePt nanoclusters were fabricated, tethered with folic acid (FA) on their surfaces for cancer-cell specific targeting. The reversible thermal response of the prepared nanoclusters was successfully verified, and the threshold temperature for drug release is tunable as the lower critical solution temperature (LCST) of the polymer could be modulated from 32 to 45 °C by adjusting comonomers and their relative composition ratios. The resultant nanoclusters realized both amphipathic and hydrophobic cargo release resulting from the shrinkage of the polymer responding to temperature rise. Fluorescence spectroscopic analysis indicates that amphipathic molecules can be released out of the nanoclusters more efficiently than hydrophobic molecules. Additionally, superparamagnetic FePt NPs have the potential to serve as the heat source of the system through the magnetocaloric effect. And the nanoclusters can respond to pH as well, which holds promise for the therapy of various tissues with different pH values. With excellent cytocompatibility and flexible composition design, these thermal responsive nanoclusters have great prospects for controlled drug release to address a series of clinical indications.
 Please wait while we load your content...
Please wait while we load your content...

