
In situ confined synthesis of molybdenum oxide decorated nickel–iron alloy nanosheets from MoO42− intercalated layered double hydroxides for the oxygen evolution reaction†
Chao
Xie
a,
Yanyong
Wang
a,
Kui
Hu
a,
Li
Tao
a,
Xiaobing
Huang
b,
Jia
Huo
a and
Shuangyin
Wang
*a
aState Key Laboratory of Chem/Bio-sensing and Chemometrics, Provincial Hunan Key Laboratory for Graphene Materials and Devices, College of Chemistry and Chemical Engineering, Hunan University, Changsha, 410082, P. R. China. E-mail: shuangyinwang@hnu.edu.cn
bCollege of Chemistry and Chemical Engineering, Hunan University of Arts and Science, Changde, People's Republic of China
First published on 21st November 2016
Abstract
This work reports molybdenum oxide decorated NiFe alloy nanosheets with high OER activity by reducing MoO42− intercalated nickel–iron layered double hydroxides (LDHs). The presence of MoO42− successfully led to structural integrity, increase of active sites, and modification of the surface electronic properties of the NiFe alloy.
Water splitting has attracted significant attention of the scientific community because of the increasing demand for clean energy.1 The oxygen evolution reaction (OER) is the most sluggish reaction in water splitting.2,3 Iridium dioxide (IrO2) and ruthenium dioxide (RuO2) were found to be the best OER electrocatalysts with low overpotential.2,4 Nevertheless, these catalysts cannot be widely used due to their high cost and scarcity. Alternatively, researchers have found that non-precious first-row transition metals, metal oxides, and metal hydroxides have exhibited promising electro-catalytic activity towards the OER.5–8 Furthermore, in order to improve the electrocatalytic activity for the OER, bimetallic electrocatalysts based on Fe, Co, Ni, etc. have been developed, and their OER activity is often superior to the corresponding single-metal catalysts.9,10
Recently, layered double hydroxides (LDHs) have drawn much attention because of their two-dimensional layered structure and their attractive performance in electrocatalysis.11 LDHs have tunable cationic layers with guest charge-balancing anions intercalated between the layers and the large interlayer distance can increase the accessible surfaces to the electrolyte in electrochemical catalysis. In particular, LDHs based on transition metals like NiFe LDHs and CoFe LDHs have shown excellent performance for the OER.12,13 However, the bottleneck of LDHs as electrocatalysts for the OER is their poor conductivity and limited active sites. In order to improve the conductivity of catalysts, on one hand, researchers have prepared metal oxides, nitrides and sulfides derived from LDHs.14–16 On the other hand, carbon materials like graphene, carbon nanotubes and carbon dots were used as the support of LDHs.17–19 More recently, bimetallic alloys were found to be favorable OER electrocatalysts due to their higher conductivity even in the absence of any support.20,21 Generally, the main factors affecting the activity of OER electrocatalysts include the number of active sites, conductivity, mass transport and so on.22 Therefore, it is necessary to design OER electrocatalysts with more active sites, high conductivity and abundant mass transport channels.23
Because of the lattice confinement effect of LDHs and the atomic level highly dispersed forms of the tunable metal cations in cationic layers, it is an efficient way to obtain some well structured and active sites homogeneously distributed on mixed metal/metal oxide catalysts by calcination with LDHs as a single precursor. Besides, the interaction between the host cationic layers and changeable guest intercalated anions will influence various properties of LDHs.24 Researchers have studied whether increasing the host–guest interaction by exchanging different intercalated anions could enhance the thermal stability of LDHs compared to typical CO32− intercalated LDHs with the same metal cations. Wei et al.25 have used MoO42− intercalated Mg/Al/Fe LDHs as the precursor to get high density metal nanoparticles with good thermal stability. They have found that the presence of Mo could prevent the Fe nanoparticles from further sintering. Even more importantly, recent reports discovered that Mo and W with high valence states can modulate first-row transition metals surrounding them.26,27 In particular, the modulation of their electronic structure will significantly influence the electrocatalytic performance of metal catalysts.
Although LDHs have been demonstrated as efficient electrocatalysts for the OER, the electronic conductivity of LDHs limited their further improvement of activity for the OER. Alternatively, the corresponding metal alloys derived from LDHs could increase the conductivity. However, the transformation of LDHs into the corresponding metal alloys cannot preserve the layered structure due to the structural collapse during the high-temperature calcinations. In this work, we have successfully realized the in situ confinement synthesis of MoOx decorated NiFe alloy nanosheets (denoted as NiFe–MoOx NS) from MoO42− intercalated NiFe LDHs with high specific surface area.28 The NiFe–MoOx NS were synthesized by a simple calcination reduction process with the MoO42− intercalated NiFe LDHs (NiFe–MoO42− LDHs) as a single precursor. The presence of MoO42− confined within the LDH layers can effectively suppress the structural collapse during the high-temperature calcination to preserve the layered structure of the as-obtained NiFe alloy. Moreover, MoOx can also modify the electronic properties of the NiFe alloy to enhance the OER activity. Experimentally, the MoO42− intercalated NiFe LDH nanosheets were synthesized by a typical hydrothermal reaction. Then, this precursor was calcined at 500 °C under an Ar–H2 mixture atmosphere for 2 h to obtain NiFe–MoOx NS (scheme in Fig. 1A). With the unique structure and compositions, the NiFe–MoOx NS exhibit excellent performance for the OER in alkaline solutions. For comparison, a NiFe alloy directly derived from NiFe LDHs without MoO42− was also prepared.
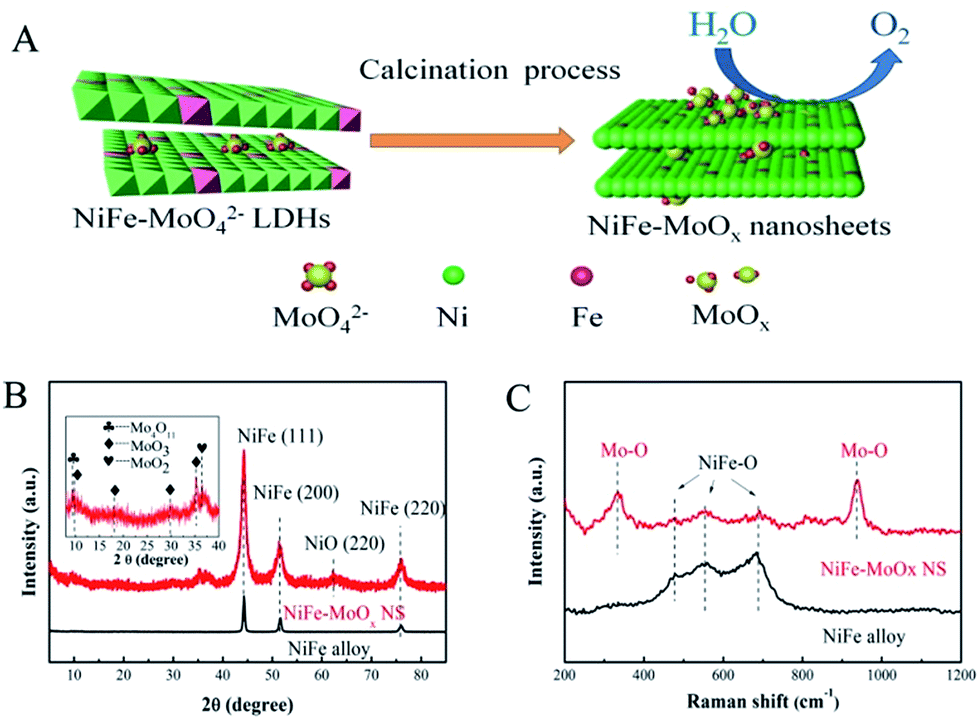 | ||
| Fig. 1 (A) The scheme of the synthesis of NiFe–MoOx NS. (B) The XRD pattern of NiFe–MoOx NS along with that of the NiFe alloy for comparison; the illustration shows a series of MoOx with a slight intensity. (C) Raman spectra of the NiFe–MoOx NS and NiFe alloy show the vibration of Mo–O and NiFe–O. | ||
The structural information of synthetic precursors NiFe LDHs and MoO42− intercalated NiFe LDHs (denoted as NiFe–MoO42− LDHs) was obtained by X-ray diffraction (XRD). As shown in Fig. S1,† the diffraction characteristic peaks of NiFe–MoO42− LDHs and NiFe LDHs could match well with those reported in the literature.28,29 The results demonstrate that the precursors NiFe–MoO42− LDHs and NiFe LDHs have been successfully synthesized. After the high-temperature annealing under Ar–H2, as shown in Fig. 1B, the XRD pattern of the NiFe alloy from the NiFe LDH is completely in conformity with the peaks of the NiFe alloy (JPDS card no. 38-0419). For NiFe–MoOx NS obtained by annealing MoO42− intercalated NiFe LDHs under Ar–H2, the main NiFe alloy characteristic peaks were observed. Apart from these peaks, there are several weak peaks corresponding to the NiO (220) (JPDS card no. 47-1049) and a few lattice planes of MoO3 (JPDS card no. 21-056-9), Mo4O11 (JPDS card no. 13-0142) and MoO2 (JPDS card no. 50-0739) (all of the molybdenum oxide species were denoted as MoOx) observed in the inset. This result indicates that the NiFe hydroxides could be reduced to a NiFe alloy under the reduction atmosphere at 500 °C. The presence of the weak peak of NiO indicates that metallic Ni was slightly oxidized to NiO while the dominant species are still the metallic NiFe alloy. In addition, it is obvious that the main peaks of the NiFe alloy in NiFeMoOx NS are wider than those of the pristine NiFe alloy, which indicates that NiFe–MoOx NS have a significantly smaller grain size than the NiFe alloy. The calculation results by applying the Scherrer formula indicate that the grain size of NiFe–MoOx is ∼10 nm and the size of the NiFe alloy is greater than 20 nm. The smaller grain size might be due to the presence of MoO42− ions during the calcination process, in which MoO42− ions could prevent the further growth of metal nanoparticles.
Raman spectroscopy is a surface-sensitive tool to investigate the surface properties of nanomaterials. The Raman spectra were collected between 200 cm−1 and 1200 cm−1 to investigate the electronic and surface properties. The Raman spectra of NiFe–MoO42− LDHs and NiFe LDHs (Fig. S1†) indicated that MoO42− ions were intercalated in LDHs nanosheets.28 As shown in Fig. 1C, the feature modes of NiFe–MoOx NS at 335 cm−1 and 938 cm−1 are respectively indexed to the bending modes and symmetric stretching modes of Mo–O,28 suggesting that molybdenum oxides exist in the production after the calcination process. Besides, the NiFe–MoOx NS and NiFe alloy show bands at 477 cm−1, 549 cm−1 and 684 cm−1, assigned to the vibration of NiFe–O,30,31 indicating that the surface of the NiFe alloy was partially oxidized while the bulk phases are dominated by the NiFe alloy phase. In addition, to estimate the proportion of NiFe and MoOx in NiFe–MoOx NS, TGA was carried out. According to the TGA test, the ratio of Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Fe:Mo:O was about 0.71:0.08:0.06:0.15.
:Fe:Mo:O was about 0.71:0.08:0.06:0.15.
Scanning electron microscopy (SEM) images of NiFe–MoO42− LDHs and NiFe LDHs (Fig. 2A and S2a†) indicate that the two precursors are composed of a number of nanosheets. It is obvious that the addition of MoO42− ions could make the nanosheets thinner. By annealing the corresponding precursors, we successfully obtained the NiFe alloy dominant phase, as evidenced by the XRD characterization. It is interesting to observe that, as shown in Fig. 2B, the reduction product NiFe–MoOx NS have inherited the layered structure of the LDHs at 500 °C. On the other hand, obviously, many NiFe alloy nanoparticles of around 10 nm were observed on the surface of the NiFe alloy nanosheets and some MoOx dispersed in the NiFe alloy, which is also confirmed by the high resolution transmission electron microscopy (HR-TEM) images in Fig. 2C, D, S5 and the EDX spectrum in Fig. S6.† In contrast, the morphological structure of NiFe LDHs has been completely destroyed and aggregated after the reduction process at 500 °C (Fig. S2b†). This comparison indicates that NiFe LDHs with MoO42− intercalated could retain the layered structure and inhibit the alloy sintering at 500 °C. We also annealed the NiFe–MoO42− LDHs at different temperatures (400, 600, and 700 °C, denoted as NiFe–MoOx-400, NiFe–MoOx-600 and NiFe–MoOx-700) to optimize the best reduction temperature (Fig. S4a–c†). When the temperature was 600 °C or 700 °C, the layer structure will be destroyed by the sintering of the alloy.
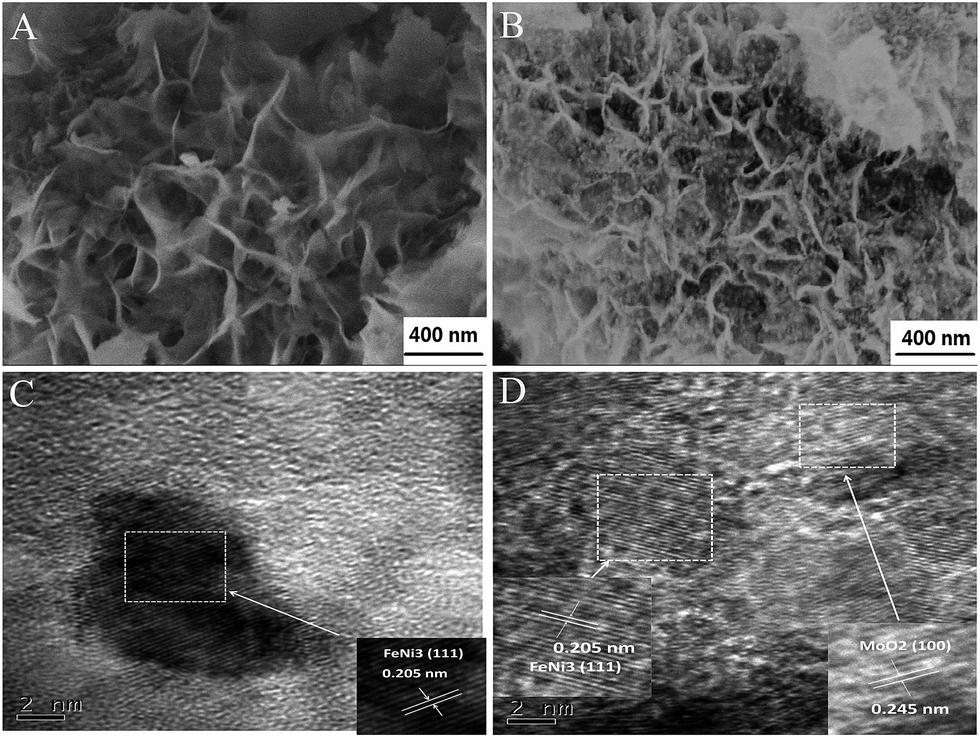 | ||
| Fig. 2 SEM images of NiFeMo LDHs (A), SEM images of NiFe–MoOx NS (B) and TEM images of NiFe–MoOx NS (C) and (D). | ||
An X-ray photoelectron spectroscope (XPS) was used for investigating the surface chemical states of the characteristic elements in the NiFe–MoOx NS and NiFe alloy. The Ni 2p3/2 XPS spectrum (Fig. 3A) shows that both the NiFe–MoOx NS and NiFe alloy have peaks of oxidized Ni2+/3+ and metallic Ni. Furthermore, it is obvious that the peak intensity of oxidized Ni2+/3+ at 855.54 eV is greater than that of metallic Ni (852.56 eV) in the XPS spectrum of NiFe–MoOx NS. The peak at 861.28 eV is the satellite peak of Ni2+/3+.32 For the pristine NiFe alloy, the peak of metallic Ni is the greatest peak, which manifests that metallic Ni is the leading component on the NiFe alloy surface. There is an apparent difference of ∼0.7 eV between the Ni2+/3+ binding energies of the NiFe alloy and NiFe–MoOx NS, which indicates that the introduction of MoOx could lead to a valence state rise of Ni species.33 As shown in Fig. 3B, the Fe 2p XPS spectrum was fitted using three components: a peak at 706.65 eV corresponding to metallic Fe and two peaks at 709.54 eV and 711.83 eV corresponding to Fe2+ and Fe3+, respectively.32 This result demonstrates that the chemical states of surface Fe have not changed distinctly with and without MoOx. The Fe on the surface is mostly in the form of oxidized Fe2+/3+ which might be due to the active chemical properties of Fe, so that the most surface metallic Fe could be easily oxidized to Fe2+/3+. The Mo 3d5/2 binding energies of NiFe–MoOx NS are shown in Fig. 3C, and it is clear that Mo exhibits Mo6+ (231.82 eV), Mo4+ (230.03 eV) and Mo3+ (228.84 eV) states,34 which confirms the presence of MoOx in NiFe–MoOx NS. The O 1s spectrum in Fig. 3D has three peaks corresponding to adsorbed H2O (l) (533.14 eV), M–OH (531.74 eV), and M–O (529.94 eV).35 Distinctly, the peak of M–O in NiFe–MoOx is much larger than that of the NiFe alloy, which demonstrates that the content of metal oxides (Ni oxides, MoOx and Fe oxides) on the surface of NiFe–MoOx is more than that on the surface of the NiFe alloy. All the above XPS results distinctly indicate that the presence of MoOx could alter the chemical state of surface Ni to higher oxidation states. Previous reports have reported that highly oxidized Ni could promote the formation and reaction of intermediate products in the OER in alkaline solutions. Ye et al. have used the plasmon-induced hot-electron excitation effect to increase the chemical state of Ni, which directly demonstrates that it is an effective approach to promote OER activity. In addition, the NiFe alloy transformed the surface metals into oxides before acting as a catalyst for the OER, and the higher content of surface oxides makes NiFe–MoOx NS more advantageous for OER catalysis than the NiFe alloy.
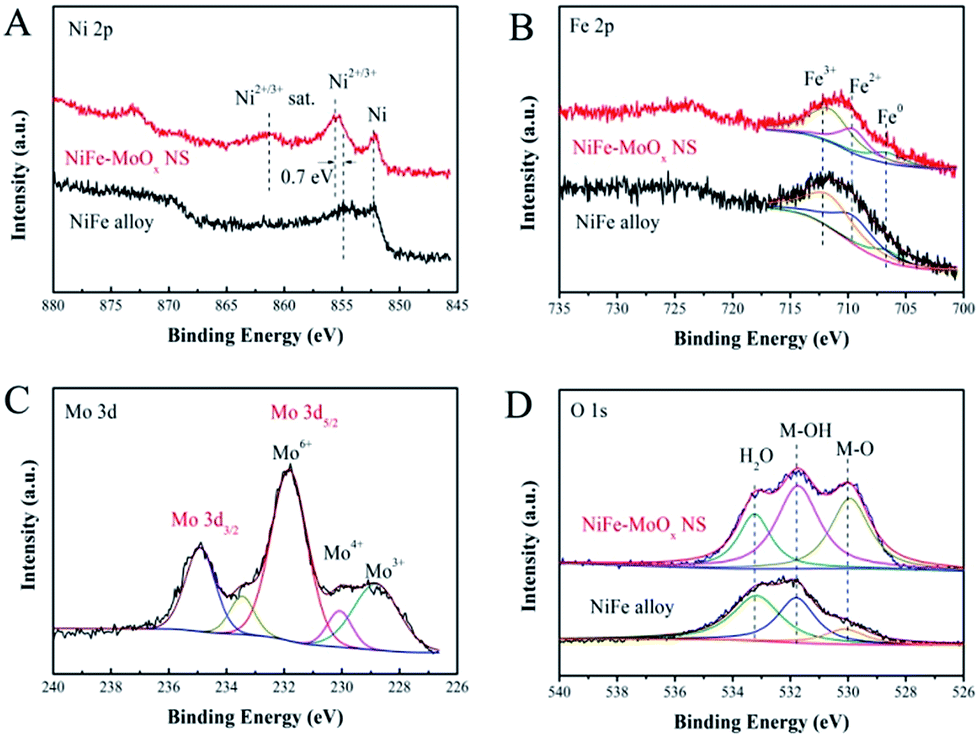 | ||
| Fig. 3 XPS spectra: (A) Ni 2p, (B) Fe 2p and (D) O 1s of NiFe–MoOx NS and the NiFe alloy; (C) Mo 3d of NiFe–MoOx NS. | ||
The electrocatalytic OER performance of NiFe–MoOx NS was investigated in 1 M KOH solution in a standard three-electrode system with a standard calomel electrode (SCE) as the reference electrode and a graphite rod as the counter electrode. The NiFe alloy and commercial RuO2 were tested under the same conditions for comparison. The catalysts were cast onto a glassy carbon electrode with a mass loading of ∼0.2 mg cm−2, and IR-corrected polarization curves were obtained at a scan rate of 5 mV s−1. As shown in Fig. 4A, the overpotential of NiFe–MoOx NS (276 mV) is much smaller than that of the NiFe alloy (370 mV) and equal to the overpotential of RuO2 (275 mV) at the current density of 10 mA cm−2. This overpotential is lower than some of the NiFe electrocatalysts in previous reports (Table S1†). In addition, it was profitable to coating on Ni foam for the OER test (Fig. S7d†). In order to investigate the influence of precursor LDHs on the structure and OER activity of the product, we prepared a sample by directly calcining the mixture of Ni, Fe and Mo sources (denoted as NiFe–MoOx Mix) for the OER (Fig. S7a†). It is obvious that the OER performance and the nanostructure of NiFe–MoOx NS are superior to those of the NiFe–MoOx mix. This result demonstrates that NiFe–MoOx NS catalysts with a structure derived from LDHs with species uniformly dispersed could improve the OER performance. Besides, samples calcined at different temperatures from NiFe–MoO42− LDHs were also tested under the same conditions for the OER (Fig. S7b†). The NiFe–MoOx NS obtained at 500 °C show the best performance in terms of the overpotential. Besides, the OER performance of the Ni foam substrate was also tested for comparison, which showed poor activity (Fig. S7c†).
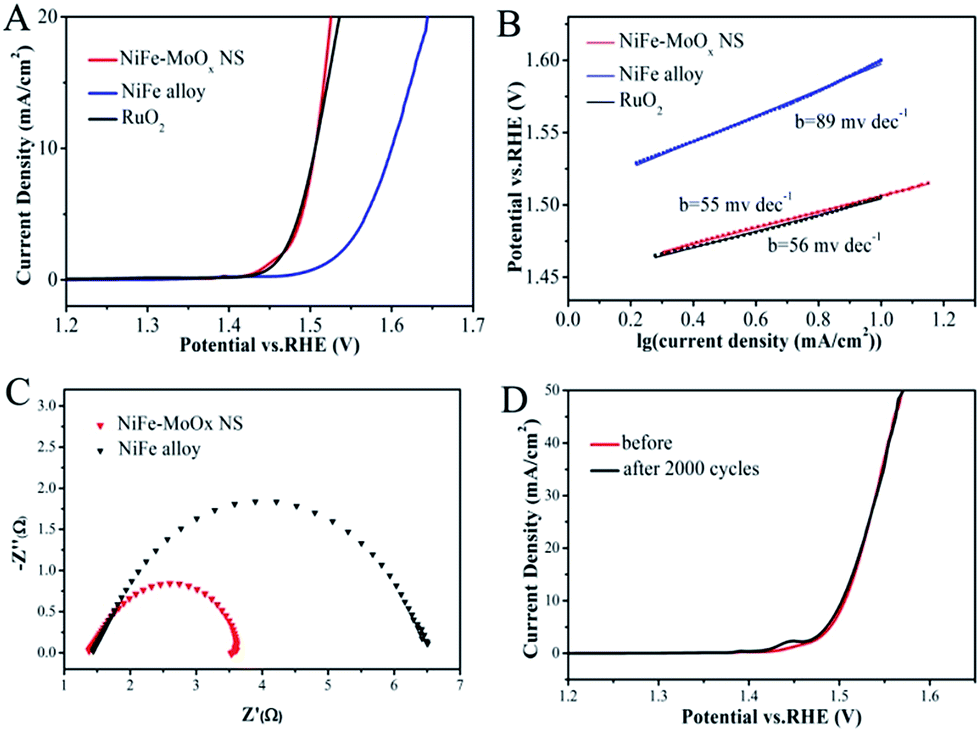 | ||
| Fig. 4 (A) LSV curves for the OER, (B) Tafel plots for the OER, (C) Nyquist plots obtained by EIS at 1.55 V (vs. RHE) for the OER of the NiFe–MoOx NS and NiFe alloy and (D) the stability testing of NiFe–MoOx NS for the OER. | ||
The OER kinetic limitation of the catalysts could be evaluated by calculating their Tafel slope. The Tafel slopes of the NiFe–MoOx NS, NiFe alloy and RuO2 measured in 1 M KOH solution are shown in Fig. 4B. The calculated Tafel slope of NiFe–MoOx NS is ∼55 mV dec−1, which is smaller than that of the NiFe alloy (∼56 mV dec−1) and RuO2 (∼89 mV dec−1). Furthermore, to investigate the reaction kinetics of the catalysts, electrochemical impedance spectroscopy (EIS) was performed in 1 M KOH solution. According to the EIS test (Fig. 4C), the NiFe–MoOx NS exhibit a smaller charge-transfer resistance than the NiFe alloy, which is highly accordant with the result of the Tafel slope. In addition, durability is another significant parameter to appraise an electrochemical catalyst. After cyclic voltammetry (CV) scanning between 1.25 V and 1.55 V for 2000 cycles in 1 M KOH solution, the linear sweeping voltammetry (LSV) curves of NiFe–MoOx NS nearly have no change, which indicates that the NiFe–MoOx catalyst exhibits a prominent durability (Fig. 4D). In order to investigate the changes of the material after the OER test, NiFe–MoOx NS were studied by XRD, XPS and TEM after the stability test. As shown in Fig. S8,† the XRD pattern indicates that the material maintains the NiFe alloy and MoOx composition. XPS spectra in Fig. S9† demonstrated that most of the metallic Ni on the surface has transformed into Ni2+/3+, and the valence state of Fe and Mo has nearly no change. As for O, the ratio of M–OH increased and that of M–OH decreased. These phenomena indicate that most of the superficial metallic Ni has transformed into Ni oxide and the metal oxides have transformed into hydroxides partially. In Fig. S10,† TEM images show some NiO crystal lattices on the surface of the material, which confirmed the results of XPS.
To investigate the specific surface and porosity of NiFe–MoOx NS and the corresponding comparisons, N2 adsorption measurement was used for testing the specific surface and pore size distribution. As shown in Fig. S11,† N2 isotherms were obtained for the NiFe–MoOx NS and NiFe alloy. Calculated from the N2 isotherms, the Brunauer–Emmett–Teller (BET) surface areas of the NiFe–MoOx NS and NiFe alloy were 109.8 m2 g−1 and 7.5 m2 g−1, respectively. The high specific surface area of NiFe–MoOx NS could be attributed to the well-reserved layered structure derived from the LDHs. Moreover, the pore size distribution of NiFe–MoOx NS is shown in Fig. S12.† There are both mesopores and micropores in NiFe–MoOx NS, which is more beneficial for the mass transfer in catalysis reactions. Besides, the LSV curves normalized by the BET surface are shown in Fig. S13.†
The intrinsic activity of NiFe–MoOx NS was further confirmed by determining the faradaic efficiency and turnover frequencies (TOFs) for this catalyst. Faradaic efficiency was measured and calculated to be about 95% according to Fig. S14† and the electrochemical methods in the ESI.† Furthermore, the TOFs were estimated to be 0.19 s−1 when the overpotential was 300 mV.
To investigate the possibility of extending the strategy, we also prepared MoO42− intercalated CoFe LDHs (denoted as CoFe–MoO42− LDHs) and calcined this precursor under the same conditions to form CoFe–MoOx nanosheets (CoFe–MoOx NS). CoFe LDHs for comparison were also prepared. As shown in Fig. S15,† the XRD pattern of CoFe–MoOx NS could coincide with that of the CoFe alloy and MoOx. The SEM images of CoFe–MoO42− LDHs and CoFe–MoOx NS demonstrate that the CoFe–MoOx could maintain the layered structure of the LDH nanosheets (Fig. S16†). The OER performance and Tafel slopes in Fig. S17† indicate that CoFe–MoOx has higher activity and better reaction kinetics than the CoFe alloy for OER electrocatalysis. This phenomenon proved that it is possible to prepare well-structured CoFe alloy nanosheets with high OER performance by the same method.
In summary, we have demonstrated a versatile method to produce molybdenum oxide modified NiFe alloy nanosheets with high OER activity. This electrocatalyst was derived from NiFe–MoO42− LDHs with high surface area and well-layered structure. The electrocatalytic OER performance of NiFe alloy materials is affected by the surface area, porosity and the surface electron structure. The excellent activity of the MoOx decorated NiFe alloy is not only contributed by the high surface area and porosity, but also improved by the valence state increase of surface Ni with the modification of MoOx. Therefore, this study successfully demonstrated an accessible strategy to prepare highly active bimetallic alloy electrocatalysts with high surface area and high porosity by reducing the MoO42− intercalated LDHs.
Acknowledgements
The authors acknowledge support from the National Natural Science Foundation of China (Grant No. 51402100 and 21573066) and the Provincial Natural Science Foundation of Hunan (Grant no. 2016JJ1006 and 2016TP1009).Notes and references
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110(11), 6446–6473 CrossRef CAS PubMed.
- F. Cheng and J. Chen, Chem. Soc. Rev., 2012, 41, 2172–2192 RSC.
- Y. Lee, S. Jin, K. J. May, E. E. Perry and S. H. Yang, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS PubMed.
- R. D. L. Smith, M. S. Prevot, R. D. Fagan, Z. Zhang, P. A. Sedach, K. J. S. Man, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60–63 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, W. Jian, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- L. Xu, Q. Jiang, Z. Xiao, X. Li, H. Jia, S. Wang and L. Dai, Angew. Chem., Int. Ed., 2016, 128, 5363–5367 CrossRef.
- S. Dou, L. Tao, J. Huo, S. Wang and L. Dai, Energy Environ. Sci., 2016, 9, 1320–1326 Search PubMed.
- D. Friebel, M. W. Louie, M. Bajdich, E. S. Kai, Y. Cai, A. M. Wise, M. J. Cheng, D. Sokaras, T. C. Weng and R. Alonso-Mori, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef PubMed.
- M. S. Burke, M. G. Kast, L. Trotochaud, A. M. Smith and S. W. Boettcher, J. Am. Chem. Soc., 2015, 137, 3638–3648 CrossRef CAS PubMed.
- Q. Wang and D. O'Hare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- J. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, H. J. Fan and M. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- H. Chen, L. Hu, M. Chen, Y. Yan and L. Wu, Adv. Funct. Mater., 2014, 24, 934–942 CrossRef CAS.
- Y. Li, H. He, W. Fu, C. Mu, X. Z. Tang, Z. Liu, D. Chi and X. Hu, Chem. Commun., 2015, 52, 1439–1442 RSC.
- Y. Wang, C. Xie, D. Liu, X. Huang, J. Huo and S. Wang, ACS Appl. Mater. Interfaces, 2016, 8(29), 18652–18657 Search PubMed.
- X. Long, G. Li, Z. Wang, H. Y. Zhu, T. Zhang, S. Xiao, W. Guo and S. Yang, J. Am. Chem. Soc., 2015, 137, 11900–11903 CrossRef CAS PubMed.
- M. Gong, Y. Li, H. Wang, Y. Liang, J. Z. Wu, J. Zhou, J. Wang, T. Regier, F. Wei and H. Dai, J. Am. Chem. Soc., 2013, 135, 8452–8455 CrossRef CAS PubMed.
- D. Tang, J. Liu, X. Wu, R. Liu, X. Han, Y. Han, H. Huang, Y. Liu and Z. Kang, ACS Appl. Mater. Interfaces, 2014, 6, 7918–7925 Search PubMed.
- X. Long, J. Li, S. Xiao, K. Yan, Z. Wang, H. Chen and S. Yang, Angew. Chem., Int. Ed., 2014, 53, 7584–7588 CrossRef CAS PubMed.
- M. Gong and H. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS.
- Y. Ullal and A. C. Hegde, Int. J. Hydrogen Energy, 2014, 39, 10485–10492 CrossRef CAS.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, ChemInform, 2014, 45, 102–121 CrossRef.
- E. Detsi, J. B. Cook, B. K. Lesel, C. L. Turner, Y. L. Liang, S. Robbennolt and S. H. Tolbert, Energy Environ. Sci., 2016, 9, 540–549 Search PubMed.
- C. Vaysse, L. Guerlou-Demourgues, A. Demourgues and C. Delmas, J. Solid State Chem., 2002, 167, 59–72 CrossRef CAS.
- M. Q. Zhao, Q. Zhang, W. Zhang, J. Q. Huang, Y. Zhang, D. S. Su and F. Wei, J. Am. Chem. Soc., 2010, 132, 14739–14741 CrossRef CAS PubMed.
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl and Y. M. Wang, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu and L. Zheng, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- N. Han, F. Zhao and Y. Li, J. Mater. Chem. A, 2015, 3, 16348–16353 RSC.
- A. Vaccari, Appl. Clay Sci., 1999, 14, 161–198 CrossRef CAS.
- A. Ahlawat, V. G. Sathe, V. R. Reddy and A. Gupta, J. Magn. Magn. Mater., 2011, 323, 2049–2054 CrossRef CAS.
- M. N. Iliev, D. Mazumdar, J. X. Ma, A. Gupta, F. Rigato and J. Fontcuberta, Phys. Rev. B, 2011, 83(1), 014108, DOI:10.1103/PhysRevB.83.014108.
- H. Alilöytty, M. W. Louie, M. R. Singh, L. Li, H. G. S. Casalongue, H. Ogasawara, E. J. Crumlin, Z. Liu, A. T. Bell and A. Nilsson, J. Phys. Chem. C, 2016, 120(4), 2247–2253 CrossRef.
- G. Liu, P. Li, G. Zhao, X. Wang, J. Kong, H. Liu, H. Zhang, K. Chang, X. Meng and T. Kako, J. Am. Chem. Soc., 2016, 138(29), 9128–9136 CrossRef CAS PubMed.
- P. A. Zosimova, A. V. Smirnov, S. N. Nesterenko, V. V. Yuschenko, W. Sinkler, J. Kocal, a. J. Holmgren and I. I. Ivanova, J. Phys. Chem. C, 2007, 111(40), 14790–14798 CrossRef CAS.
- K. Juodkazis, J. Juodkazytė, R. Vilkauskaitė and V. Jasulaitienė, J. Solid State Electrochem., 2008, 12, 1469–1479 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ta08149e |
| This journal is © The Royal Society of Chemistry 2017 |