
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoactivatable aggregation-induced emission probes for lipid droplets-specific live cell imaging†
Meng
Gao‡
 a,
Huifang
Su‡
b,
Yuhan
Lin
a,
Xia
Ling
a,
Shiwu
Li
a,
Anjun
Qin
*a and
Ben Zhong
Tang
*ab
a,
Huifang
Su‡
b,
Yuhan
Lin
a,
Xia
Ling
a,
Shiwu
Li
a,
Anjun
Qin
*a and
Ben Zhong
Tang
*ab
aGuangdong Innovative Research Team, State Key Laboratory of Luminescent Materials & Devices, South China University of Technology, Guangzhou 510640, China. E-mail: msqinaj@scut.edu.cn
bDepartment of Chemistry, Hong Kong Branch of Chinese National Engineering Research Center for Tissue Restoration and Reconstruction, The Hong Kong University of Science & Technology, Clear Water Bay, Kowloon, Hong Kong, China. E-mail: tangbenz@ust.hk
First published on 21st December 2016
Abstract
Photoactivatable probes for lipid droplets (LDs)-specific live-cell imaging are powerful tools for investigating their biological functions through precise spatial and temporal control. Ideal photoactivatable probes for LDs imaging require high concentration accumulation of fluorophores in LDs, simple synthetic procedures, and excellent photoactivation efficiency. However, it is difficult to overcome these challenges by conventional fluorophores due to aggregation-caused quenching (ACQ). In this study, a new class of photoactivatable and LDs-specific fluorescent probes was developed based on dihydro-2-azafluorenones, which can easily undergo photooxidative dehydrogenation reaction to afford 2-azafluorenones with aggregation-induced emission (AIE) properties. Dihydro-2-azafluorenones as photoactivatable and LDs-specific probes display significant advantages of excellent photoactivation efficiency and lack of self-quenching in the aggregated state, and are expected to have broad applications in study of biological functions of LDs' through light-controlled spatiotemporal imaging.
Introduction
Lipid droplets (LDs) as reservoirs of lipids and proteins are dynamic organelles and vary in number, morphology, and size in different cells.1 Due to the multifunctions of LDs in energy sources, membrane synthesis, and protein degradation, LDs are linked to various diseases, such as inflammation, virus infection, and obesity.2 Recently, the elevated expression level of LDs has been proposed as a biomarker of cancer due to the strong requirement for fatty acids and phospholipids in the rapidly growing cancer cells.3 Photoactivatable fluorescent probes are powerful tools for cell biology studies through light-controlled imaging at high spatial and temporal resolution.4 It is thus highly desirable to develop LDs-specific photoactivatable probes to investigate their various biological functions.5 However, to the best of our knowledge, to date, LDs-specific photoactivatable probes based on small organic molecules, which are less disruptive to the native biology and more convenient for operation than fluorescent proteins, have not been reported in the literature.To achieve the photoactivatable fluorescent imaging of LDs, two major challenges need to be overcome. First, fluorophores should selectively accumulate in LDs at high concentration to emit bright fluorescence with a high signal-to-noise ratio; however, this is difficult for conventional fluorophores due to aggregation-caused quenching (ACQ).6 Second, photoactivatable probes are required to achieve light-controlled spatiotemporal imaging of LDs, but previously reported photoactivatable probes suffer from limited light-up mechanisms, complicated synthetic procedures, generation of toxic byproducts, and difficulty to specifically accumulate in LDs.7 Although several LDs-specific fluorescent dyes, such as Nile Red, BODIPY493/503 green, monodansylpentane, AFN, NPBDP, LipidTOX red, and LD540, have been developed, the lack of photoactivatable ability and self-quenching at high concentration have severely limited their applications.8
Recently, aggregation-induced emission (AIE) has been proposed as a fundamental solution to solve the fluorescence self-quenching problem in the aggregated state.9 The AIE light-up mechanism has been proven to arise from the restriction of intramolecular motion.10 AIE-based bioprobes have unique advantages in terms of superior brightness, long-term in situ retention ability, high photostability, and low cytotoxicity.11 We have recently developed several AIEgens, including TPE-AmAl, TPE-AC, FAS, and DPAS, for LDs-specific imaging with the advantages of absence of self-quenching and easily adjustable emission spectra.12 However, it is difficult to introduce photoresponsive groups into these classic AIEgens. Therefore, LDs-specific probes based on a new class of AIEgens with easy availability and excellent photoactivation efficiency are highly required to explore the biological functions of LDs.
2-Azafluorenones are traditionally studied as core structures in many biologically active molecules; however, their photophysical properties and bioimaging applications have been rarely investigated.13 Unexpectedly, we found that 2-azafluorenone 2 can emit strong fluorescence in the aggregated state with typical AIE properties via the restriction of intramolecular motion and twisted intramolecular charge transfer (TICT) mechanisms (Scheme 1). Moreover, we found that dihydro-2-azafluorenone 1 can be efficiently transformed into 2-azafluorenone 2via photooxidative dehydrogenation reaction and can be used for LDs-specific imaging with an excellent photoactivation efficiency.
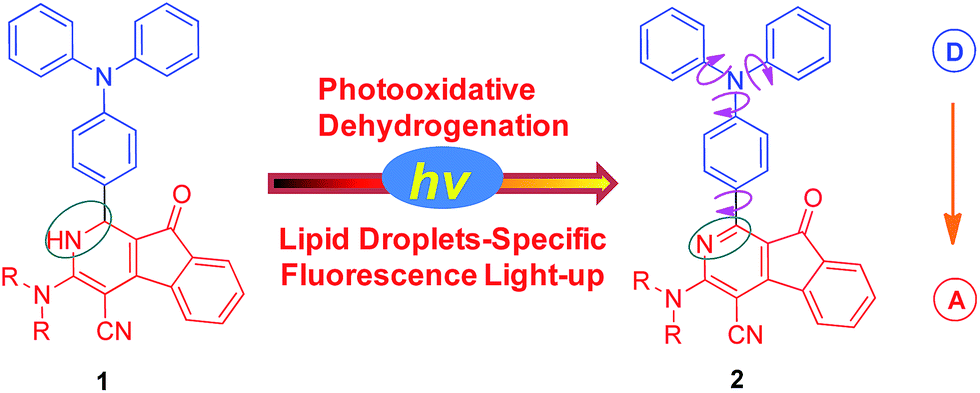 | ||
| Scheme 1 Photooxidative dehydrogenation of dihydro-2-azafluorenone 1 to afford 2-azafluorenone 2 with AIE properties. | ||
Experimental section
General procedures for the synthesis of compounds 1 and 2 (1a and 2a are presented as an example)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate = 5:1) to give 1a as a red solid (126 mg, 47% yield).
:ethyl acetate = 8:1) to give 1a as an orange red solid (162 mg, 61% yield).
:ethyl acetate = 5:1) to give 1a as a red solid (126 mg, 47% yield).
:ethyl acetate = 8:1) to give 1a as an orange red solid (162 mg, 61% yield).
Cell cultures
HCC827, A549, and HLF cell lines were purchased from ATCC. HCC827 cells were cultured in RPMI-1640 with 1% penicillin–streptomycin and 10% FBS at 37 °C in a humidified incubator with 5% CO2. A549 and HLF cells were cultured in DMEM with 1% penicillin–streptomycin and 10% FBS at 37 °C in a humidified incubator with 5% CO2. The culture medium was changed every other day and the cells were collected by treating with 0.25% trypsin–EDTA solution after they reached confluence.Cell viability
HCC827, A549, and HLF cells were respectively seeded in 96-well plates at a density of 5 × 104 cells per mL. After 24 h of culture, different concentrations of 1 or 2 were added and further incubated for 24 h. The sample and control wells were washed twice with PBS buffer and added with freshly prepared MTT medium solution (0.5 mg mL−1, 100 μL). After 3 h of incubation at 37 °C, the MTT medium solution was carefully removed and washed twice with PBS buffer. DMSO (100 μL) was then added into each well and the plate was gently stirred for 10 min at room temperature to dissolve all the precipitates that were formed. The absorbance of sample and control wells at 570 nm was then measured by a microplate reader. Cell viability was then calculated by the ratio of the absorbance of the sample wells to control cells.Cell imaging
Cells were grown in a 35 mm Petri dish with a coverslip at 37 °C. After incubation with 1 (20 μM) for 15 min, the cells were washed with PBS for three times. The photoactivated images were obtained using a confocal microscope via increasing scans at 405 nm with 1% laser power (the scanning rate was 22.4 s per frame). For 1a, the emission filter was 438–604 nm; for 1b, the emission filter was 515–690 nm; and for 1c, the emission filter was 551–675 nm.Confocal co-localization
For co-staining with lipid dye BODIPY493/503 green, cells were first incubated with probe 1 (20 μM) and BODIPY493/503 green (100 nM) at 37 °C for 15 min. The medium was then removed and the cells were rinsed with PBS for three times, and then imaged using a confocal microscope. For probe 1, the fluorescence was first photoactivated by irradiation at 405 nm (1% laser power) for designed time intervals, and then the fluorescence images were obtained. For 1a, the emission filter was 572–607 nm; for 1b, the emission filter was 572–689 nm; and for 1c, the emission filter was 572–675 nm; for BODIPY493/503 green, the excitation was 488 nm and the emission filter was 510–553 nm.Spatially and temporally controlled cell imaging
The observation window containing multicells was first imaged under irradiation at 405 nm (0.2% power). The selected cells were then irradiated in a bleach model with 405 nm laser (0.2% power) for 20 scans (the scanning rate was 22.4 s per frame). Subsequently, the whole observation window was imaged under irradiation at 405 nm (0.2% power). This process was repeated until all the selected cells in the observation window were lit-up.Results and discussion
We first established an efficient and direct method for preparing dihydro-2-azafluorenone 1 through a one-step reaction from easily available 2-((Z)-2-(4-(diphenylamino)benzylidene)-1,2-dihydro-1-oxoinden-3-ylidene)malononitrile (BDOYM) and amines, including morpholine, diethylamine, and ammonia solution (Fig. 1A). We also found that 2-azafluorenone 2 can be directly obtained from BDOYM and amines under light irradiation (7 W fluorescent bulb) and using air as the oxidant (for proposed reaction mechanism, see Scheme S1 in the ESI†). Structures of all obtained compounds were elucidated by NMR spectroscopy and high-resolution mass spectrometry (Fig. S1–S6†). The structure of compound 2b was also verified by single crystal X-ray diffraction (Fig. 1B and C).14 In the crystal of 2b, the dihedral angle between the planar 2-azafluorenone ring and the 1-linked phenyl ring was 41.9(6)°, whereas the triphenylamine moiety exhibited a propeller conformation with dihedral angles of 61.3(2)°, 67.9(1)°, and 73.3(6)° between the aryl rings, respectively. This kind of conformation can contribute to the non-irradiative release of energy in solution via intramolecular rotation. In the crystal packing structure, various intermolecular C–H⋯π, C–H⋯O, and van der Waals interactions were observed, which can help to restrict the intramolecular motion and block the non-radiative release of energy in the aggregated state.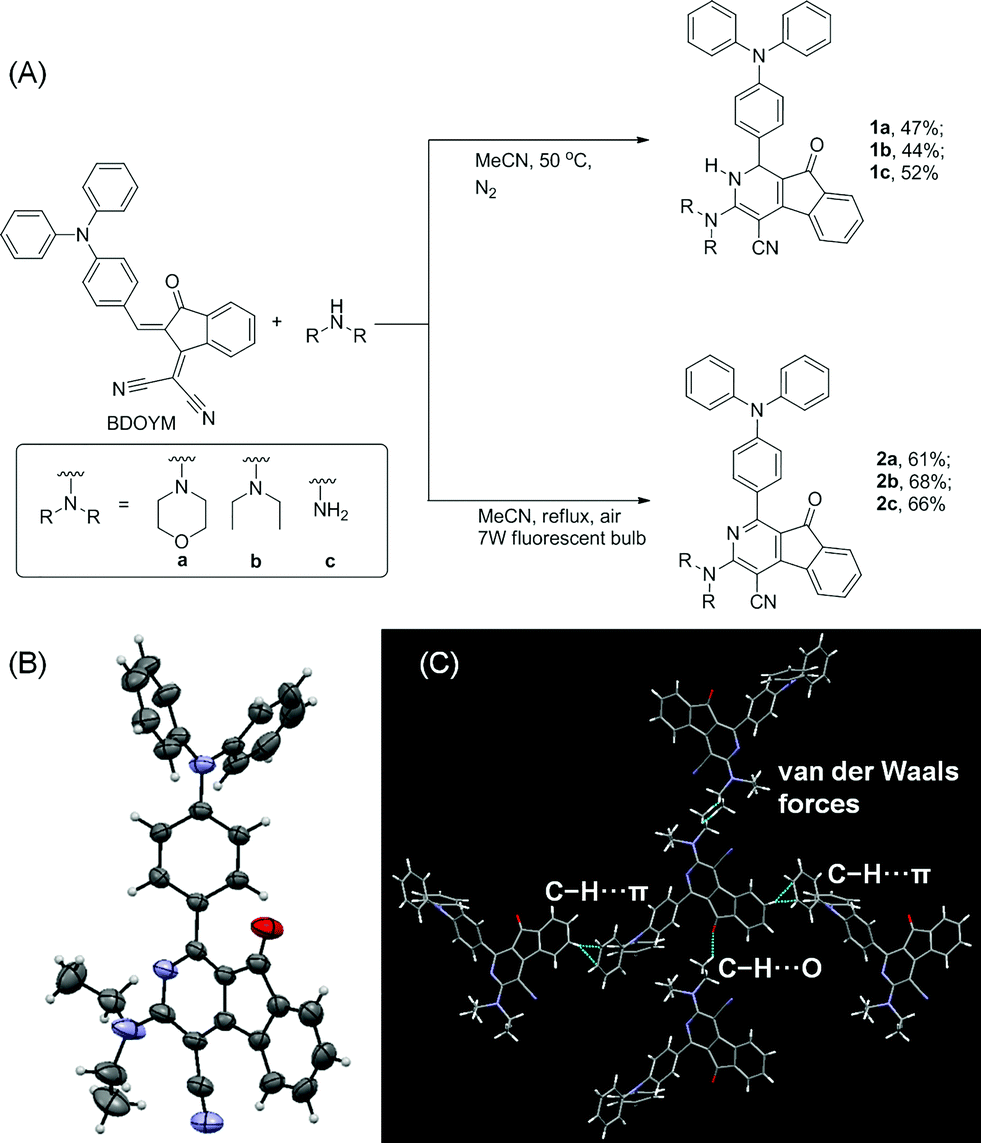 | ||
| Fig. 1 (A) Synthetic routes for dihydro-2-azafluorenone 1 and 2-azafluorenone 2. (B) Oak ridge thermal ellipsoid plot program (ORTEP) drawing of 2b. (C) Crystal packing structure of 2b. Intermolecular C–H⋯π, C–H⋯O, and van der Waals interactions are shown in green dashed lines. | ||
Intrigued by the increased fluorescence intensity of 2 during drying on thin-layer chromatography (TLC) plates, we first examined their photophysical properties. For compound 2a, an absorption peak at 409 nm and a weak emission peak at 624 nm were observed in dilute THF solution (Fig. 2). In the THF/water mixture, its emission first underwent quenching from water fraction (fw) = 10 to 70%, and this phenomenon probably originated from the twisted internal charge transfer (TICT) effect,15 which was verified by the red-shifted and decreased emission of 2a with the increasing solvent polarity (Fig. S7†). Density functional theory based calculation of 2a also verified its typical donor–acceptor structural feature (Fig. S8†). The highest occupied molecular orbital (HOMO) of 2a is localized on the triphenylamine moiety, whereas the lowest unoccupied molecular orbital (LUMO) is localized on the 2-azafluorenone moiety. Interestingly, further increase in the water fractions (fw) from 70 to 90% led to a significant emission enhancement, which can be due to the formation of aggregates of 2a caused by the poor solvating ability of THF/water mixture with high water content. Successively, the emission intensity of 2a showed a little decrease at 99% water fraction, which can be possibly due to the fast decrease in the solubility leading to the formation of amorphous aggregates with lower fluorescence efficiency.16 To verify that the AIE phenomenon is caused by the restriction of intramolecular motion, we measured the emission of 2a in polar solvents of ethanol and glycerol with low (η = 1.2 mPa S) and high viscosity (η = 945 mPa S), respectively.17 A 158-fold emission enhancement at 600 nm was observed in glycerol than that in ethanol, which clearly verified that the AIE phenomenon is caused by the restriction of intramolecular motion (Fig. S9†).18 Similar to 2a, both 2b and 2c first showed a decrease in the fluorescence intensity with the increase in the water fraction (fw) from 0 to 70%, and then exhibited significant emission enhancements as fw increased from 70 to 99% and 80%, respectively. We then measured the photophysical properties of 2a–c in the film state (Table S1†). Compared to those in the THF solution state, 2a, 2b, and 2c in the film state, respectively, exhibited 20, 18.4, and 12-fold increase in the fluorescence quantum yields (Φf), 3.4, 8.8, and 7.0-fold increase in the fluorescence lifetime (τ), 2.5, 2.1, and 1.7-fold increase in the radiative decay rates (kr), and 3.6, 9.2, and 7.6-fold decrease in the nonradiative decay rates (knr). They also exhibited large Stokes shifts of 8424, 8668, and 8607 cm−1 in the film state, respectively. The excellent emission efficiency enhancement and large Stokes shifts in the aggregated state can greatly favor their applications in bioimaging.
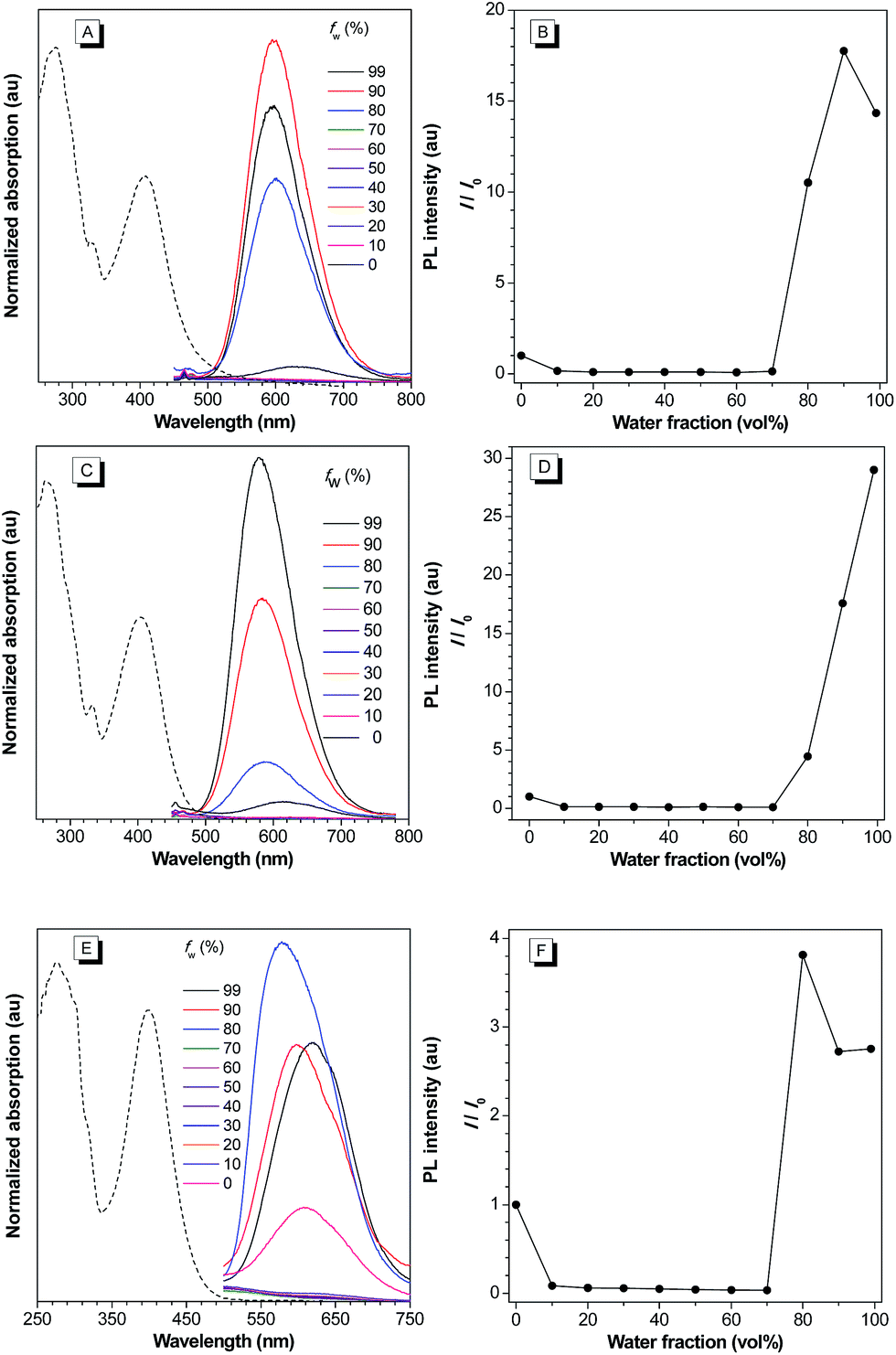 | ||
| Fig. 2 (A, C, and E) Normalized UV-vis absorption spectra of 2a, 2b, and 2c THF (dashed line); PL spectra of 2a, 2b, and 2c in THF and THF/water mixture with different water fractions (fw). (B, D, and F) Plots of relative maximum emission intensity (I/I0) of 2a, 2b, and 2cversus the solvent composition of THF/water mixture. [2a] = [2b] = [2c] = 10 μM. For 2a, λex = 409 nm. For 2b, λex = 402 nm. For 2c, λex = 400 nm. | ||
We next examined the photooxidative transformation of dihydro-2-azafluorenone 1 into 2-azafluorenone 2. Upon irradiation at 365 nm using a hand-held UV lamp, a gradually increased fluorescence was observed for 1a in aqueous solution (Fig. 3A and B), indicating that 1a can efficiently undergo photooxidative transformation to afford AIE-active 2a. Various water soluble oxidants, such as H2O2, t-BuOOH, NaOCl, sodium persulfate, and potassium peroxymonosulfate, have also been tested for the oxidation of 1a into 2a under dark conditions at room temperature; however, all of them can not promote this transformation (Fig. S10†). The photooxidative transformation was also verified by the UV-vis absorption spectra changes of 1a, in which the maximum absorption wavelength progressively shifted from 530 nm to a shorter wavelength at 413 nm (Fig. S11†). Moreover, the stacking 1H NMR spectra of 1a and 2a showed that the Ha and Hb in 1a (peaks at 6.25 and 5.61 ppm, respectively) completely disappeared in 2a (Fig. 3C), which clearly verified the proposed photooxidative dehydrogenation mechanism. Similar to 1a, compounds 1b and 1c also exhibited the fast light-up fluorescence and blue-shifted absorption under UV light irradiation (Fig. S12–S15†), which indicates their smooth transformation to 2b and 2cvia the photooxidative reaction.
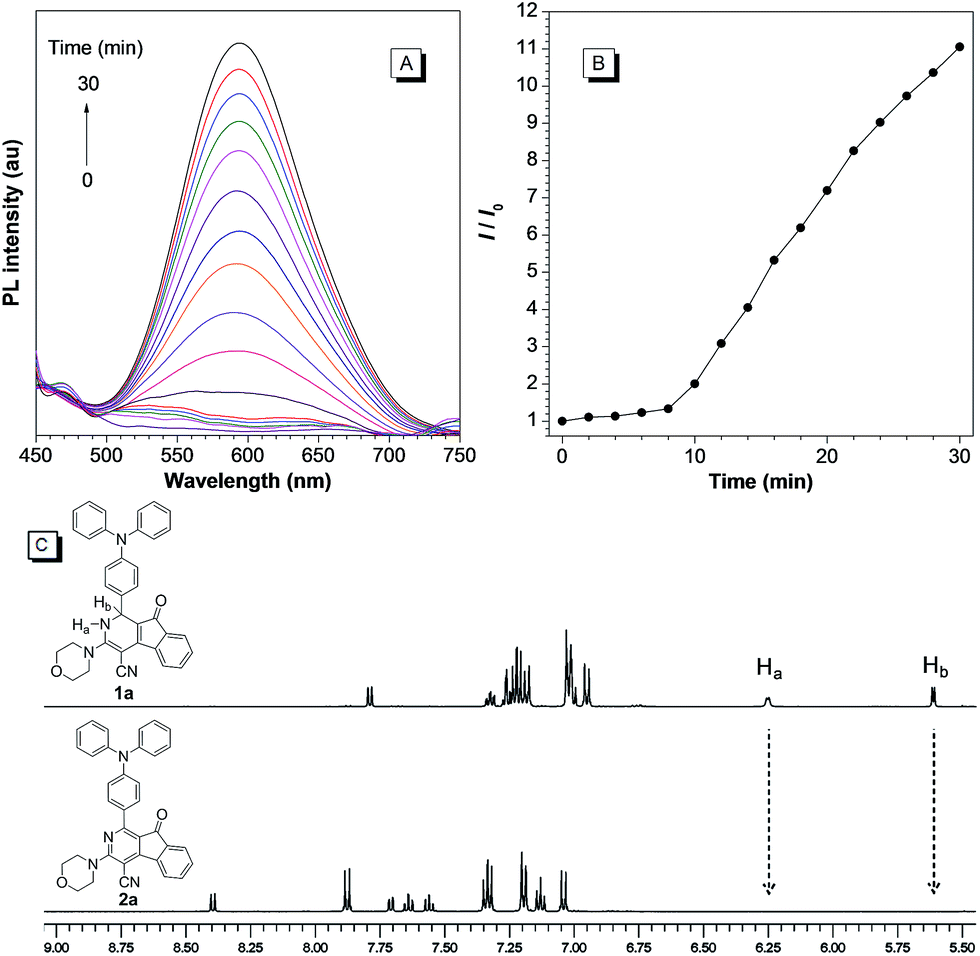 | ||
| Fig. 3 (A) The PL spectra of 1a in DMSO/water mixture with 99% water fraction under irradiation at 365 nm for 0–30 min. λex = 409 nm. (B) Plot of relative PL intensity (I/I0) of 1a at 600 nm versus the irradiation time. (C) The stacking 1H NMR spectra of 1a and 2a. | ||
We then used 1a as a photoactivatable probe for live-cell imaging. Based on MTT assay, the cytotoxicity of 1a and 2a was first evaluated for lung cancer HCC827, A549 cells, and normal lung HLF cells. As shown in Fig. S16,† no significant change in the cell viability was observed even when a high concentration of 100 μM of 1a or 2a was present in the culture medium, which suggests that 1a or 2a has very low cytotoxicity. Based on 1a, we then conducted the photoactivatable imaging experiment in the lung cancer HCC827 cells. After incubation at 37 °C for 15 min, 1a can be efficiently uptaken by HCC827 cells and a very fast light-up process (less than 2 min) was observed via irradiation at 405 nm with very mild irradiation of only 1% laser power (Fig. 4A). Through statistically analyzing the increased fluorescence intensity of 1a in HCC827 cells, an excellent light-up ratio of 265-fold was obtained (Fig. 4B). The co-localization experiment with lipid dye BODIPY493/503 green was then conducted to test the specificity of 1a for LDs (Fig. 4C). The results show that photoactivated 1a could co-localize well with BODIPY493/503 green with a high overlap coefficient of 0.99 (Fig. S17†). Through light irradiation at 405 nm, a fast light-up process of 1a was also observed in the A549 cells (Fig. 4D, E and S18†), and its specificity for LDs was also verified by co-staining with BODIPY493/503 green (Fig. S19†). Compared to lung cancer HCC827 and A549 cells, only few LDs were observed in the normal lung HLF cells by photoactivated 1a (Fig. 4F and G). The much larger number of LDs in the lung cancer HCC827 and A549 cells is probably due to their faster growing rate compared with normal lung HLF cells.3 This experiment suggests that 1a as a photoactivatable and LDs-specific probe can be used to discriminate between the cancer and normal cells through their different expression levels for LDs.
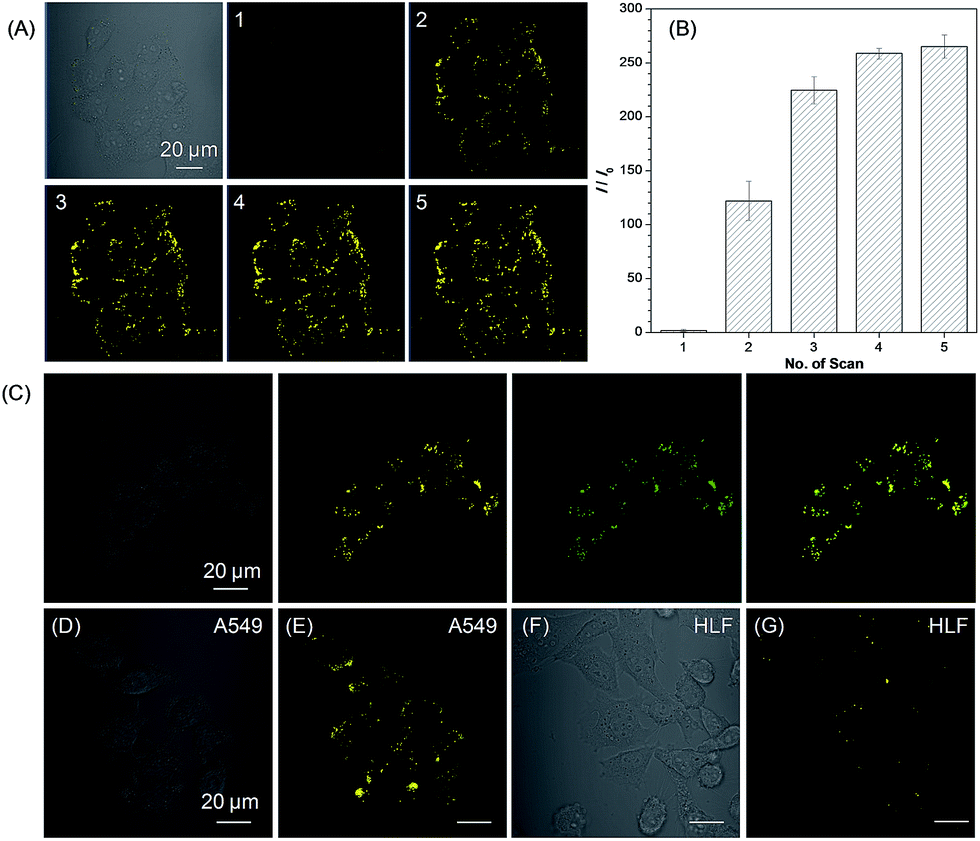 | ||
| Fig. 4 (A) Bright field and fluorescence images of live HCC827 cells obtained under white light illumination and increasing scans at 405 nm with 1% laser power (the number of scans are shown in the upper left corner and the scanning rate was 22.4 s per frame). (B) Plot of fluorescence enhancement (I/I0) of HCC827 cells with the increasing scans at 405 nm. (C) Bright field and fluorescence images of the HCC827 cells stained with 1a after photoactivation (yellow), BODIPY493/503 (green), and the merged image. (D) Bright field, and (E) fluorescence images of the live A549 cells stained with 1a after photoactivation. (F) Bright field, and (G) fluorescence images of live HLF cells stained with 1a after photoactivation by irradiation. [1a] = 20 μM. Scale bar = 20 μm. | ||
Light as an external trigger is a valuable and easily controllable tool with high spatial and temporal accuracy. As shown in Fig. 5, sequential photoactivation of 1a in selected HCC827 cells can be achieved in a multicellular environment. This experiment suggests that 1a as a photoactivatable probe is potential to monitor the dynamic events of LDs in a complex biological sample.
 | ||
| Fig. 5 (A) Bright field of HCC827 cells. (B–D) Sequential photoactivation of HCC827 cells (cells 1, 2, and 3) by irradiation at 405 nm (0.2% laser power). Dashed lines indicate the periphery of cells. [1a] = 20 μM. Scale bar = 20 μm. | ||
We also tested the targeting specificity and photoactivatable imaging ability of 1b–c for LDs in the lung cancer HCC827 and A549 cells. Through irradiation at 405 nm with 1% laser power, fast light-up processes were observed for both 1b and 1c in HCC827 and A549 cells, and maximum emission intensities could be achieved in 3 min (Fig. S20–S23†). The co-localization experiment with lipid dye BODIPY493/503 green clearly showed that 1b and 1c can also be used for LDs-specific photoactivatable imaging (Fig. 6). In HCC827 cells, the overlap coefficients for 1b and 1c with lipid dye BODIPY493/503 green are 0.99 and 0.98, respectively; whereas in A549 cells, the overlap coefficients for 1b and 1c with BODIPY493/503 green are 0.97 and 0.99, respectively.
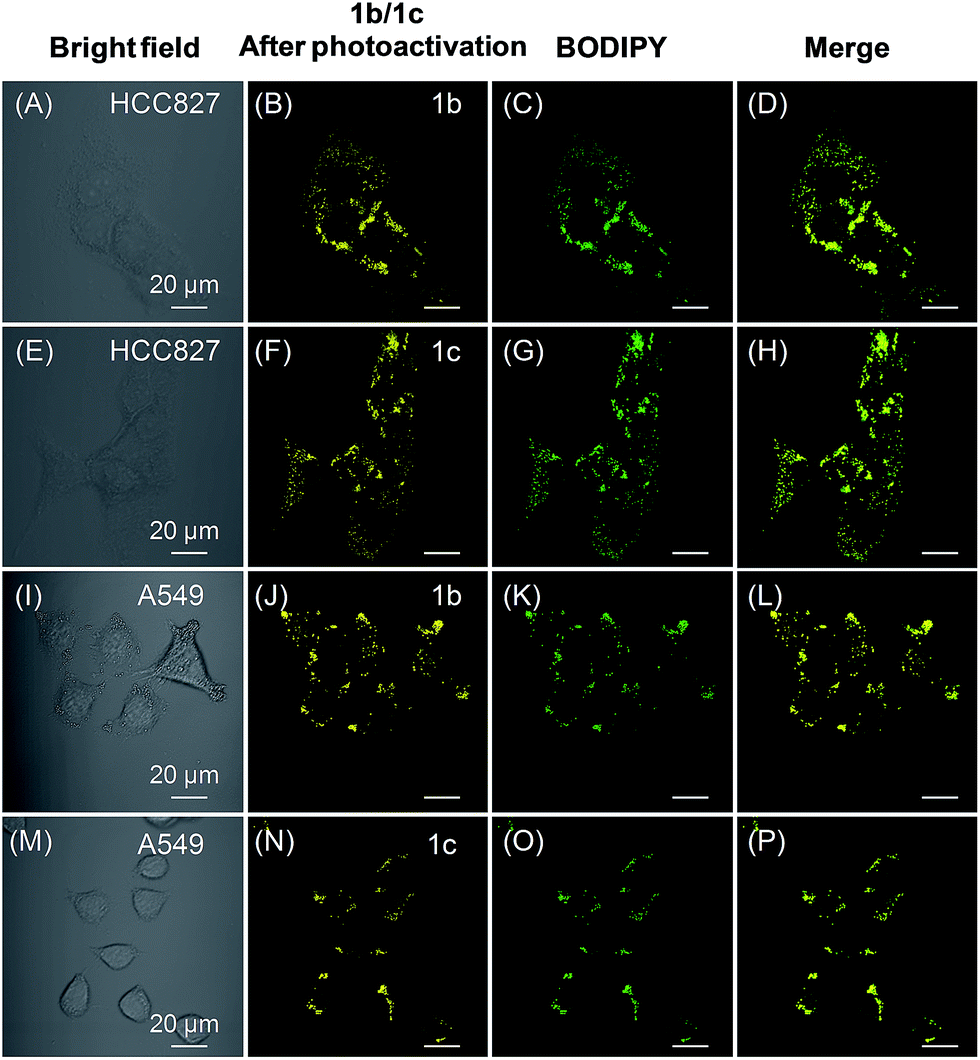 | ||
| Fig. 6 (A and E) Bright-field images of HCC827 cells. (B and F) Fluorescence images of HCC827 cells from 1b and 1c after photoactivation, respectively. (C and G) Fluorescence images of HCC827 cells from BODIPY493/503 green. (D and H) The merged images. (I and M) Bright-field images of A549 cells. (J and N) Fluorescence images of A549 cells from 1b and 1c after photoactivation, respectively. (K and O) Fluorescence images of A549 cells from BODIPY493/503 green. (L and P) The merged images. Both 1b and 1c were photoactivated through irradiation at 405 nm for 3 min (1% laser power). [1b] = [1c] = 20 μM. Scale bar = 20 μm. | ||
Conclusions
In conclusion, we have developed photoactivatable and LDs-specific probes based on dihydro-2-azafluorenones, which can easily undergo photooxidative dehydrogenation reaction to afford 2-azafluorenones with typical AIE properties. Even with different amine substituents, dihydro-2-azafluorenones are generally applicable for LDs-specific imaging in live cells with an excellent photoactivation efficiency and high signal-to-noise ratio. They can also be used for the sequential photoactivation of selected cells in a multicellular environment. Moreover, they can efficiently discriminate between lung cancer and normal lung cells through their different expression levels for LDs. Benefiting from the advantages of absence of self-quenching in the aggregated state, easy preparation, fast cell uptake, low cytotoxicity, and excellent photoactivation efficiency, the photoactivatable AIE probes developed in this study are expected to have broad applications in the biological studies of LDs.Acknowledgements
This work was financially supported by China Postdoctoral Science Foundation Grant (2015M580716 and 2016T90778), the Key Project of the Ministry of Science and Technology of China (2013CB834702), National Science Foundation of China (51620105009 and 21602063), Natural Science Foundation of Guangdong Province (2016A030313852 and 2016A030312002), Fundamental Research Funds for the Central Universities (2015ZY013 and 2015ZZ104), Innovation and Technology Commission of Hong Kong (ITC-CNERC14SC01), and Guangdong Innovative Research Team Program (201101C0105067115).Notes and references
- (a) S. O. Olofsson, P. Bostrom, L. Andersson, M. Rutberg, J. Perman and J. Boren, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2009, 1791, 448–458 CrossRef CAS PubMed; (b) S. Martin and R. G. Parton, Nat. Rev. Mol. Cell Biol., 2006, 7, 373–378 CrossRef CAS PubMed.
- (a) Y. Miyanari, K. Atsuzawa, N. Usuda, K. Watashi, T. Hishiki, M. Zayas, R. Bartenschlager, T. Wakita, M. Hijikata and K. Shimotohno, Nat. Cell Biol., 2007, 9, 1089–1097 CrossRef CAS PubMed; (b) P. T. Bozza and J. P. Viola, Prostaglandins, Leukotrienes Essent. Fatty Acids, 2010, 82, 243–250 CrossRef CAS PubMed; (c) Y. Guo, T. C. Walther, M. Rao, N. Stuurman, G. Goshima, K. Terayama, J. S. Wong, R. D. Vale, P. Walter and R. V. Farese, Nature, 2008, 453, 657–661 CrossRef CAS PubMed.
- (a) H. Abramczyk, J. Surmacki, M. Kopec, A. K. Olejnik, K. Lubecka-Pietruszewska and K. Fabianowska-Majewska, Analyst, 2015, 140, 2224–2235 RSC; (b) L. Tirinato, C. Liberale, S. Di Franco, P. Candeloro, A. Benfante, R. La Rocca, L. Potze, R. Marotta, R. Ruffilli, V. P. Rajamanickam, M. Malerba, F. De Angelis, A. Falqui, E. Carbone, M. Todaro, J. P. Medema, G. Stassi and E. Di Fabrizio, Stem Cells, 2015, 33, 35–44 CrossRef CAS PubMed; (c) C. R. Santos and A. Schulze, FEBS J., 2012, 279, 2610–2623 CrossRef CAS PubMed; (d) R. Chowdhury, B. Jana, A. Saha, S. Ghosh and K. Bhattacharyya, Med. Chem. Commun., 2014, 5, 536 RSC.
- (a) F. M. Raymo, Phys. Chem. Chem. Phys., 2013, 15, 14840–14850 RSC; (b) W. H. Li and G. Zheng, Photochem. Photobiol. Sci., 2012, 11, 460–471 RSC; (c) C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446–8476 CrossRef CAS PubMed; (d) Q. Shao and B. Xing, Chem. Soc. Rev., 2010, 39, 2835–2846 RSC; (e) M. J. Rust, M. Bates and X. Zhuang, Nat. Methods, 2006, 3, 793–796 CrossRef CAS PubMed.
- N. Kory, A. R. Thiam, R. V. Farese Jr and T. C. Walther, Dev. Cell, 2015, 34, 351–363 CrossRef CAS PubMed.
- (a) R. I. MacDonald, J. Biol. Chem., 1990, 265, 13533–13539 CAS; (b) A. Reisch and A. S. Klymchenko, Small, 2016, 12, 1968–1992 CrossRef CAS PubMed; (c) J. M. Swiecicki, F. Thiebaut, M. Di Pisa, S. Gourdin-Bertin, J. Tailhades, C. Mansuy, F. Burlina, S. Chwetzoff, G. Trugnan, G. Chassaing and S. Lavielle, Sci. Rep., 2016, 6, 20237 CrossRef CAS PubMed.
- (a) Y. Zhang, S. Swaminathan, S. Tang, J. Garcia-Amoros, M. Boulina, B. Captain, J. D. Baker and F. M. Raymo, J. Am. Chem. Soc., 2015, 137, 4709–4719 CrossRef CAS PubMed; (b) M. K. Lee, P. Rai, J. Williams, R. J. Twieg and W. E. Moerner, J. Am. Chem. Soc., 2014, 136, 14003–14006 CrossRef CAS PubMed; (c) T. Kobayashi, T. Komatsu, M. Kamiya, C. Campos, M. Gonzalez-Gaitan, T. Terai, K. Hanaoka, T. Nagano and Y. Urano, J. Am. Chem. Soc., 2012, 134, 11153–11160 CrossRef CAS PubMed; (d) Z. Yu, L. Y. Ho and Q. Lin, J. Am. Chem. Soc., 2011, 133, 11912–11915 CrossRef CAS PubMed; (e) S. J. Lord, N. R. Conley, H.-l. D. Lee, R. Samuel, N. Liu, R. J. Twieg and W. E. Moerner, J. Am. Chem. Soc., 2008, 130, 9204–9205 CrossRef CAS PubMed; (f) N. Gagey, P. Neveu, C. Benbrahim, B. Goetz, I. Aujard, J.-B. Baudin and L. Jullien, J. Am. Chem. Soc., 2007, 129, 9986–9998 CrossRef CAS PubMed.
- (a) S. Daemen, M. A. M. J. van Zandvoort, S. H. Parekh and M. K. C. Hesselink, Molecular Metabolism, 2016, 5, 153–163 CrossRef CAS PubMed; (b) J. Zhai, Y. Zhang, C. Yang, Y. Xu and Y. Qin, Analyst, 2014, 139, 52–54 RSC; (c) E. Kim, S. Lee and S. B. Park, Chem. Commun., 2012, 48, 2331–2333 RSC; (d) S. Rabhi, I. Rabhi, B. Trentin, D. Piquemal, B. Regnault, S. Goyard, T. Lang, A. Descoteaux, J. Enninga and L. Guizani-Tabbane, PLoS One, 2016, 11, e0148640 Search PubMed; (e) J. H. Lee, J. H. So, J. H. Jeon, E. B. Choi, Y. R. Lee, Y. T. Chang, C. H. Kim, M. A. Bae and J. H. Ahn, Chem. Commun., 2011, 47, 7500–7502 RSC; (f) A. Sharma, S. Umar, P. Kar, K. Singh, M. Sachdev and A. Goel, Analyst, 2016, 141, 137–143 RSC; (g) J. Spandl, D. J. White, J. Peychl and C. Thiele, Traffic, 2009, 10, 1579–1584 CrossRef CAS PubMed; (h) A. B. Neef and C. Schultz, Angew. Chem., Int. Ed., 2009, 48, 1498–1500 CrossRef CAS PubMed; (i) D. Eggert, K. Rosch, R. Reimer and E. Herker, PLoS One, 2014, 9, e102511 Search PubMed.
- X. Lim, Nature, 2016, 531, 26–28 CrossRef CAS PubMed.
- (a) J. Mei, Y. Hong, J. W. Lam, A. Qin, Y. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429–5479 CrossRef CAS PubMed; (b) J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed; (c) F. Bu, R. Duan, Y. Xie, Y. Yi, Q. Peng, R. Hu, A. Qin, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2015, 54, 14492–14497 CrossRef CAS PubMed; (d) N. L. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Lam and B. Z. Tang, Chem.–Eur. J., 2014, 20, 15349–15353 CrossRef CAS PubMed; (e) Q. Li and Z. Li, Sci. China: Chem., 2015, 58, 1800–1809 CrossRef CAS.
- (a) D. Ding, K. Li, B. Liu and B. Z. Tang, Acc. Chem. Res., 2013, 46, 2441–2453 CrossRef CAS PubMed; (b) R. T. Kwok, C. W. Leung, J. W. Lam and B. Z. Tang, Chem. Soc. Rev., 2015, 44, 4228–4238 RSC; (c) J. Liang, B. Z. Tang and B. Liu, Chem. Soc. Rev., 2015, 44, 2798–2811 RSC; (d) X. Gu, E. Zhao, T. Zhao, M. Kang, C. Gui, J. W. Lam, S. Du, M. M. Loy and B. Z. Tang, Adv. Mater., 2016, 28, 5064–5071 CrossRef CAS PubMed; (e) X. Gu, E. Zhao, J. W. Lam, Q. Peng, Y. Xie, Y. Zhang, K. S. Wong, H. H. Sung, I. D. Williams and B. Z. Tang, Adv. Mater., 2015, 27, 7093–7100 CrossRef CAS PubMed.
- (a) Z. Wang, C. Gui, E. Zhao, J. Wang, X. Li, A. Qin, Z. Zhao, Z. Yu and B. Z. Tang, ACS Appl. Mater. Interfaces, 2016, 8, 10193–10200 CrossRef CAS PubMed; (b) E. Wang, E. Zhao, Y. Hong, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem. B, 2014, 2, 2013 RSC; (c) M. Kang, X. Gu, R. T. Kwok, C. W. Leung, J. W. Lam, F. Li and B. Z. Tang, Chem. Commun., 2016, 52, 5957–5960 RSC.
- (a) G. R. Heintzelman, J. L. Bullington and K. C. Rupert, WO2005042500A1, 2005; (b) T. Landmesser, A. Linden and H.-J. Hansen, Helv. Chim. Acta, 2008, 91, 265–284 CrossRef CAS.
- CCDC 1497612 contains the supplementary crystallographic data for this paper.
- R. R. Hu, E. Lager, A. Aguilar-Aguilar, J. Z. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. C. Zhong, K. S. Wong, E. Pena-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845–15853 CAS.
- A. Shao, Y. Xie, S. Zhu, Z. Guo, S. Zhu, J. Guo, P. Shi, T. D. James, H. Tian and W. H. Zhu, Angew. Chem., Int. Ed., 2015, 54, 7275–7280 CrossRef CAS PubMed.
- M. A. Haidekker, T. P. Brady, D. Lichlyter and E. A. Theodorakis, Bioorg. Chem., 2005, 33, 415–425 CrossRef CAS PubMed.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Structural and photophysical characterization data for 1 and 2, UV-vis and PL spectra, DFT calculations, cell imaging, cell viability, and NMR spectra (PDF). Crystallographic data of 2b (CIF). CCDC 1497612. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6sc04842k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |