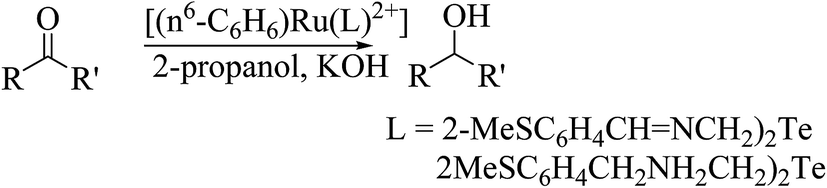DOI:
10.1039/C7RA09480A
(Review Article)
RSC Adv., 2017,
7, 55175-55198
Emerging trends in organotellurolate chemistry derived from platinoids
Received
26th August 2017
, Accepted 20th November 2017
First published on 5th December 2017
Abstract
This perspective begins with the discussion of various basic synthetic approaches applied for the synthesis of several organotellurium ligands, their chemistry derived from platinum group metals, and the reactivity difference among them. It also gives an overview on the development of various bi, tri, and high nuclearity complexes syntheses. Investigations targeting the organotellurium ligand systems revealed a remarkable reactivity due to the dynamic nature of the lone pair available on the tellurium metal, which has led to a serendipitous isolation of the complexes [Cp*Ir(ppy)(η1-Te2Ph2)]+, [Cp*IrOs3(μ-H)2(μ-Tetol)2(CO)7], [Pt{TeC5H3(3-R)N}2Te(PPh3)], [Pt{Ph2PCH(TeC5H3(3-R)NPPh2)}2] (R = H, Me), and various other high nuclearity heterometallic [Cp*Ir(CO)(μ-TeC6H4)2MCp*Cl]Cl (M = Rh, Ir) complexes. Studies of the various complexes investigated the various binding modes of coordination and the facile cleavage of the Te–C and Te–Te bonds of tellurium-based ligand systems. Attempts have been made to present a comprehensive account of the subject matter. Various promising aspects of these complexes, such as their synthesis, reactivity, structures, and applications, are covered in this review.
1. Introduction
Organosulphur and selenium derived metal complexes have been well documented for more than several decades.1–4 When one considers its homologues analog involving tellurium elements, it is clear that tellurium chemistry is still relatively uncharted. Due to their high reactivity, air sensitivity, and diffusive sets of orbitals, tellurium complexes are unstable and hence unexplored. Thus, the prompt reactivity and rich coordination of organotellurium ligand systems has drawn substantial interest over the last few years.5–10
The metalloid character of tellurium makes it amphoteric (acid as well as base) in nature depending upon the nature of the reactive substrate.6,11–13 Therefore, the reactions of organotellurium ligands with platinum group metal precursors represent an active area for further research.14–18 The noteworthy reactivity of platinoids has been utilized in synthetic chemistry to isolate products in quite good yields with great selectivity under normal conditions.19 These complexes are not only used as precursors but are also envisaged as an active species in various catalytic reactions. In particular, the superior stability of the platinum family complexes endow them with an opportunity to be utilized to comprehend the mechanistic details as well as the nature of complexes involved in particular catalytic cycles.20
Renewed interest in the field of coordination chemistry derived from organotellurium ligand systems has been stimulated by a number of recent publications7,8,21–23 dealing with the oxidative addition reactions of platinum group metal precursors with various diarylditelluride ligand systems. The outcomes of these reactions depend upon the nature of the solvent, especially in chlorinated solvents, which yield multinuclear complexes together with several unidentified products;21–23 whereas, the similar reaction in solvents like benzene and toluene affords different multinuclear complex. The formation of several products mainly results from the bond cleavage of Te–C bonds rather than Te–Te bonds.22,23 The comparable bond energies of Te–Te and Te–C bonds and the increased metallic character of Te may be the reason for the unexpected reactivity of organotellurium ligands.
Thus, this perspective aims to cover the various synthetic approaches for organotellurium ligands, different aspects and versatility of platinum group metal complexes derived from these ligand systems, with a particular emphasis on hemilabile tellurolate ligand systems since the subject was recently explored.
2. Synthetic strategy of various organo-tellurium ligands
2.1 Telluroethers
The synthetic approaches applied for the synthesis of various monodentate, bidentate, and hybrid telluroethers have been reviewed previously by various groups.6,24–28 However, there have been no significant recent developments reported. The common synthetic methods applied for the synthesis of telluroethers are highlighted in the following.
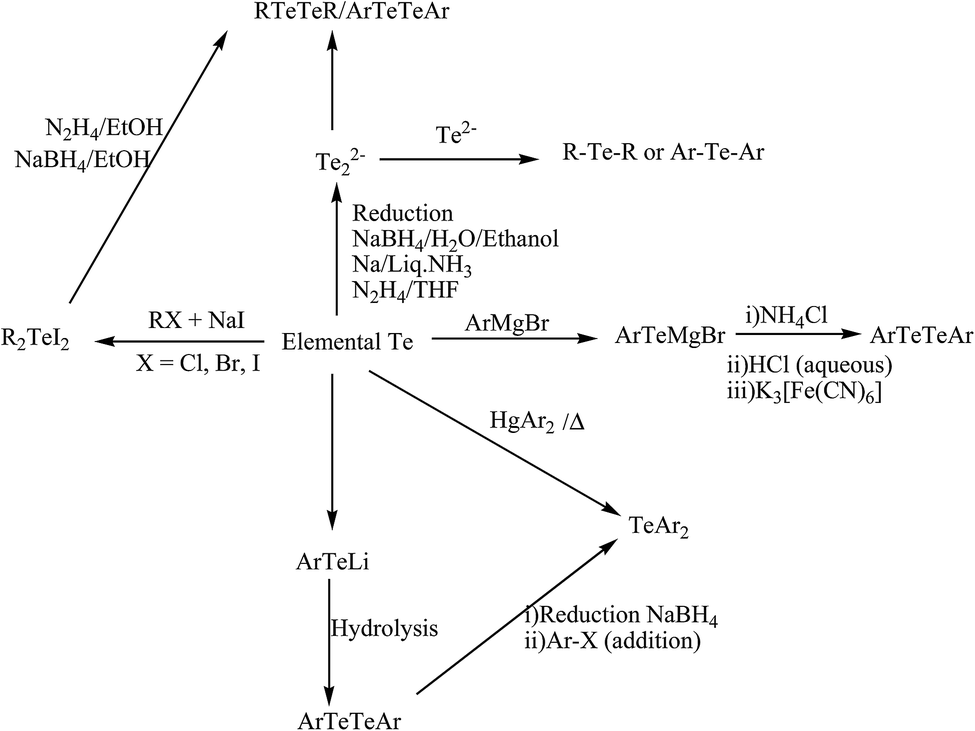
2.1.1. Reduction of ditellurides. This is one of the most effective methods used, in which the reduction of ditellurides takes place accompanied with the addition of organic halides compounds. The molar quantity of organic halides is usually double with respect to ditellurides. This method has also been used in the preparation of various asymmetrical telluroethers, but there are some issues around the poor yield due to the formation of symmetrical telluroethers. Most recently Prof. A. K. Singh et al. recently developed various telluroethers, such as (2-phenyltelluromethyl)tetrhydropyran/dioxane,24 (2-phenyltelluromethyl)tetrhydropyran,24 Te(CH2CH2R)2 (R = CH2NH2, C4H8N),25 (4-RC6H4)Te(CH2CH2){2-C5H4N} (R = H, Me, OMe),25 4-MeOC6H4Te(CH2)2N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(CH3)(2-HOC6H4),26 Te(CH2CH2NC(CH3)C6H4-2-OH)2,26 2-CH3SC6H4CNCH2CH2TeC6H4–OMe,27 4-MeOC6H4Te(CH2)nCN(2-HOC6H4)(C6H5),28 applying the same strategy.
C(CH3)(2-HOC6H4),26 Te(CH2CH2NC(CH3)C6H4-2-OH)2,26 2-CH3SC6H4CNCH2CH2TeC6H4–OMe,27 4-MeOC6H4Te(CH2)nCN(2-HOC6H4)(C6H5),28 applying the same strategy.
2.1.2. In situ generation of Na2Te. The second most efficient strategy for the formation of telluroethers is the in situ formation of Na2Te, followed by the addition of the corresponding alkyl halides to isolate the desired product. As an example, the class of cyclic ditelluroether [8]aneTe2 was synthesized in a similar fashion by the addition of a half equivalent of 1,3 dibromomethane in a THF solution of Na2Te, followed by the addition of NaBH4 and a further equivalent of 1,3 dibromomethane.29,30 The syntheses of mixed donor thia/tellura [9]aneS2Te2, [11]aneS2Te2, [12]aneS2Te2, [14]aneS3Te macrocyclic ligands and tripodal telluroether were also carried out based on the same strategy.31–35
2.1.3. Applying a Grignard reagent. Currently, Grignard reagent is also used as a promising methodology for the formation of telluroethers, especially for the isolation of heteroaromatic analogs and for unsymmetrical telluroether synthesis. In this case, the insertion of tellurium metal in to the Grignard reagent of the organic moiety corresponding to the expected telluroether is performed first. Subsequently, the addition of halo aryl or alkyl is followed in freezing conditions. Recently, an unsymmetrical 1-naphthyl-based telluroether and symmetrical pyridyl telluroethers have been isolated with the same strategy.36–38 In order to justify the mechanistic details, we performed the following process: (i) halopyridine underwent a complete metal–halogen exchange reaction with i-prMgCl to give pyridyl magnesium chloride, (ii) the latter compound was reacted with an equivalent of tellurium metal via an insertion mode, (iii) followed by the addition of alkyl/aryl halide to yield the desired telluroether with the elimination of MgCl2.
2.2 Diorganotellurides
The majority of synthetic approaches have been applied to the synthesis of various ditelluride ligand systems. Among these, the insertion of elemental tellurium in to reactive M–C bonds is a quite common methodology. Various approaches are described below.
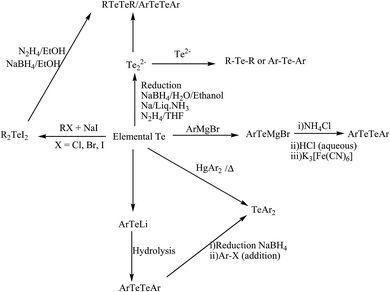
2.2.1. Insertion of chalcogen in to Li–Aryl bond. The insertion of elemental tellurium in to the Li–aryl bond followed by oxidation has been performed for the synthesis of various ditelluride ligand systems. The lithiation of bromoaryl compounds has been accomplished by the substitution of the bromo group through lithium metal with reagents like n-BuLi39,40 or ButLi41 in polar solvents like THF and petroleum ether at a temperature of −78 °C. The resulting lithiated aryl group reacts with active elemental tellurium metal to yield a lithiated chalcogenolate ion, which on hydrolysis gives the corresponding diaryl ditelluride ligand systems.
2.2.2. Reaction of E2− with haloaryl compounds. The reaction of heteroaromatic and aromatic halocompounds with ditellurido dianions in different solvents is another important synthetic pathway to synthesize a class of various ditelluride ligand systems. In most of the reactions, reducing reagents are generated through an in situ mode by a variety of reducing agents, like NaBH4, Li metal reaction, Na/NH3, and hydrazine.42–47 Sodium borohydride reduction of elemental tellurium in ethanol, water, and ethoxy ethanol has been applied for the synthesis for Na2Te2.45–48 The latter has been synthesized by an in situ and dropwise addition of haloaryl compounds, yielding the corresponding diaryl ditelluride; while hydrazine hydrate in the presence of NaOH in DMF has also been used to prepare Na2Te2, which on reaction with bromopyridine and chloropyrimidines, affords their corresponding ditellurides (Scheme 1).49
 |
| | Scheme 1 Synthetic approaches for the synthesis of various ditellurides and monotellurides. | |
2.2.3. Insertion of tellurium in to aryl magnesium halide. The lithiation of haloaryl compounds takes place at −78 °C, but this cryogenic condition and the instability of the lithiated products results in a poor yield, which makes this method really an inconvenient route for the synthesis of ditellurides. In contrast, the stability and ease of handling of aryl magnesium bromide compounds makes this synthetic strategy quite trendy. Normally for all cases, a Grignard reagent of the corresponding alkyl or arylhalo compounds is first synthesized,36,37,50,51 followed by the insertion of elemental tellurium, which on acid hydrolysis yields the corresponding ditellurides in a moderate yield.
3. Metal complexes with telluroether ligands
3.1 Ruthenium and osmium
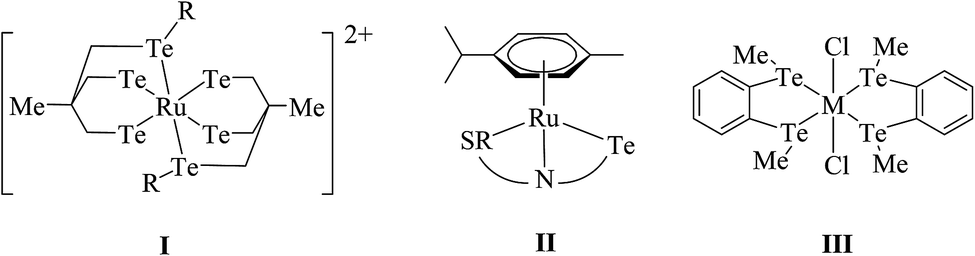
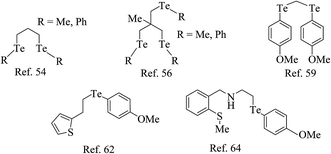
The reaction of RuCl3·nH2O, TePh2, and CO yields the complex [Ru(CO)2Cl2(TePh)2], which has also been obtained in moderate yield by the simple addition between [Ru{(CO)3Cl2}2] and telluroether.52,53 The reaction of chelated ditelluroether RTe(CH2)3TeR; (R = Me, Ph, or o-C6H4(TeMe)2) with RuCl3·nH2O, [Ru(dmso)4X2] gave a complex with the general composition trans-[RuX2(L∩L)2] (X = Cl, Br or I); L∩L = RTe(CH2)3TeR; (R = Me, Ph or o-C6H4(TeMe)2) (Scheme 2).54,55 All these products showed poor solubility in organic solvents. To overcome the solubility issues, a bulky phosphine-based ruthenium precursor, [RuCl2(PPh3)3], has been used, which on reaction with the same ditelluroether yielded a complex of [RuCl(PPh3)(L∩L)2]PF6 (Fig. 1) with good solubility in various organic solvents.55 In solution, the latter complex is converted to a product with the composition [RuCl2(PPh3){MeC(RTeCH2)}3], while a longer stay in the solvent results in its decomposition to unknown species;56 while this tripodal ligand on reaction with [Ru(dmf)6](SO3CF3)3 in methanol gave a homoleptic compound [Ru{MeC(CH2TeR)3}2](SO3CF3)2 (I).57,58
 |
| | Scheme 2 Homoleptic and heteroleptic telluroether ligands. | |
 |
| | Fig. 1 Crystal structure of [RuCl(PPh3){MeTeCH2}2]+ (redrawn from ref. 55). | |
A new class of carbon-backboned ligands (4-MeO–C6H4–Te)2CH2 and [(CH2)2C(CH2TeMe)2] on reaction with [RuCl4(dmso)4]/[RuCl2(p-cymene)]2 afforded the complex [RuCl2(dmso)2(L)]/[RuCl2(p-cymene)2(L)] (L= (4-MeO–C6H4–Te)2CH2 and [(CH2)2C(CH2TeMe)2]) which contains a telluroether in a chelating fashion with the formation of a strained four-membered ring. These complexes were configured in a distorted octahedral geometry around the Ru metal center.59
The reaction of [RuCl2(p-cymene)]2 with two equivalent of homoleptic telluroether Te{CH2CH2(Ar)}2 (Ar = 1,3 dioxane, thiophene)60 or a heteroleptic telluroether like [(thiophene)Te{CH2(R)}] (R = furan, pyrol),61 [{CH2CH2(R)}Te(C6H4-p-OMe)] (R = thiophene, phthalamide, benzotriazole)62,63 yielded [RuCl2(p-cymene)L] (L = Te{CH2CH2(Ar)}2, [{CH2CH2(R)}Te(C6H4-p-OMe)], [(thiophene)Te{CH2(R)}]) (Scheme 2). The same type of composition (II) has also been derived with various tridentate hybrid ligand systems.64–67
The chemistry of halo osmium precursors OsO4-HX or [OsX6] with telluroether has not had a successful outcome yet for dithio or diselenoether ligands. In this case, the reaction of [OsCl2(dmso)4] with carbon-backboned ditelluroether RTe(CH2)3TeR, CH2(TeMe)2, o-C6H4(CH2TeMe)2 led to a compound with the composition [OsCl2(L∩L)2] (III) (L∩L = RTe(CH2)3TeR, CH2(TeMe)2, o-C6H4(CH2TeMe)2).54,68 Unlike for thio and selenoether ligands, a very clean route to the final product has been achieved in the case of telluroethers. Compared to the ruthenium analogs, very few examples of telluroether-based osmium complexes are known so far.
3.2 Rhodium and iridium
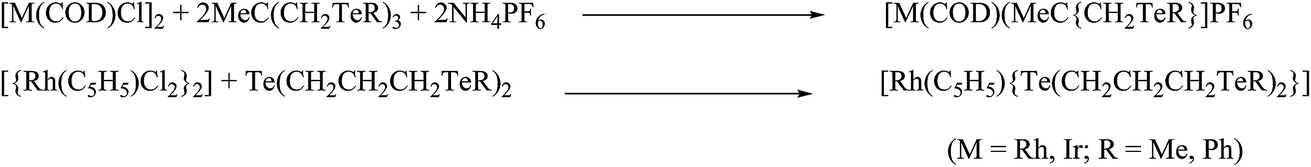
Very little work on rhodium and iridium complexes derived from organotellurium ligand systems, i.e., telluroethers, has been documented so far. Compared to ruthenium, the related references for rhodium and iridium complexes are less significant in number. The reaction of TeR2 (R = Me, Et) with metallocyclic [(C5H5)2Rh2(μ-CO)(CF3CCF3)] derived an adduct in which TeR2 was added to one of the rhodiums attached with bridging carbonyl, which was boned in a η1 fashion.69 Such a type of insertion followed by the addition of tellurium can be particularized due to diffusive sets of lone pairs and their ease of availability to coordination. While the reaction of RhCl3·3H2O and ditelluroethers Te(CH2)3TeR (R = Me, Ph) along with NH4PF6 in ethanol solution yielded the complex [RhCl2(L∩L)2]PF6 (Scheme 3). Similarly, various tripodal Rh(I) and Ir(I) complexes [M{MeC(CH2TeMe)3}(COD)]PF6 (M = Rh, Ir) were derived from umbrella-like telluroethers, namely with MeC(CH2TeR)3 (R = Me, Ph).55 However, Rh(III) complexes with the composition [Rh(η5-C5H5){MeC(CH2ER)3}][PF6]2 with a square pyramidal geometry were isolated by the reaction of [{RhCl2(η5-C5H5)}2] and the corresponding ligand systems.56 The fluxional behavior of the latter complex was established by its 1H NMR spectrum, which showed a single resonance for TeMe, CH2, and COD (Scheme 3). It was concluded, during a dynamic process, that a flip on and off movement takes place through the arm of the tripod around the metal center.
 |
| | Scheme 3 Reaction of rhodium and iridium precursors with tripodal telluroether. | |
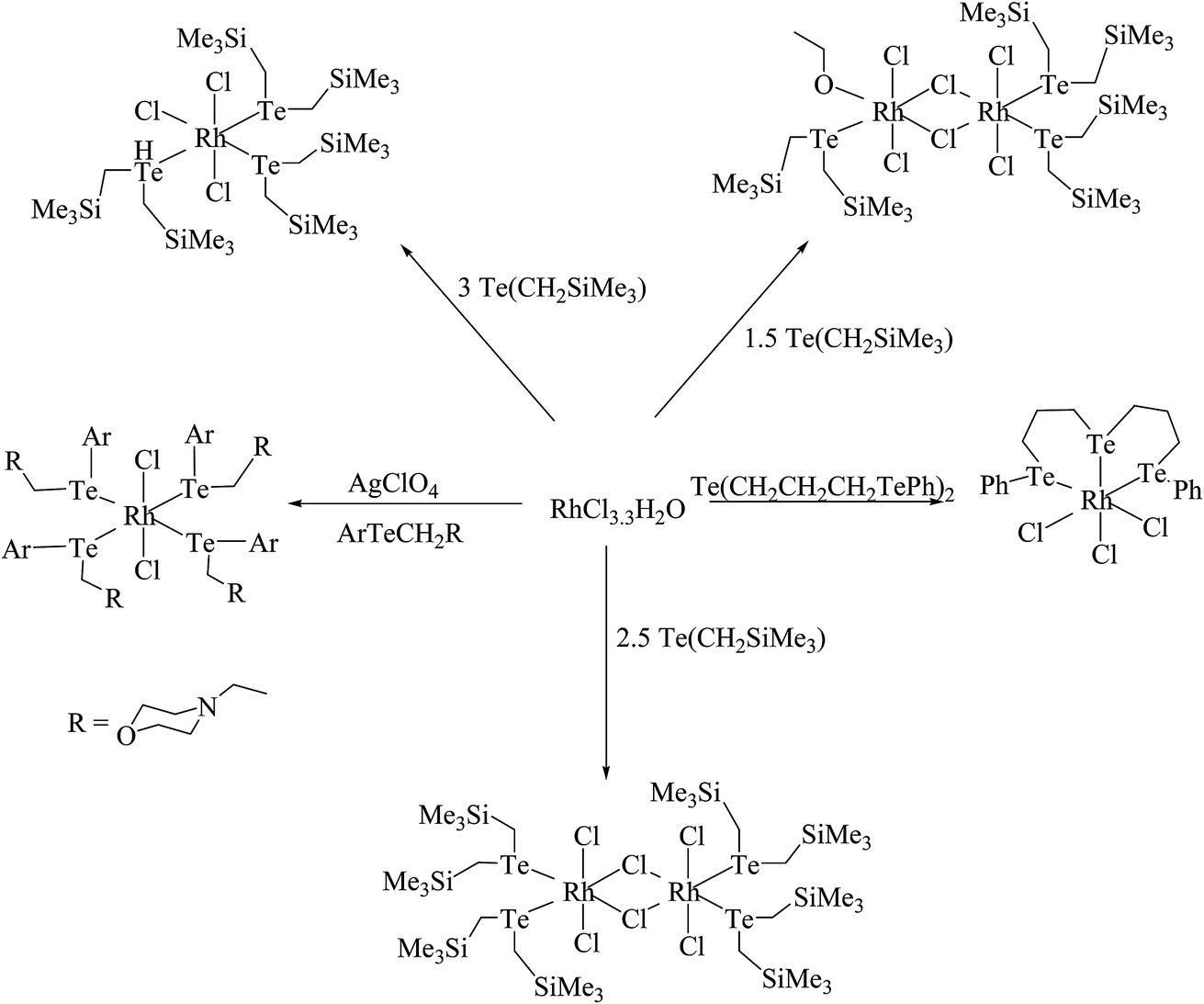
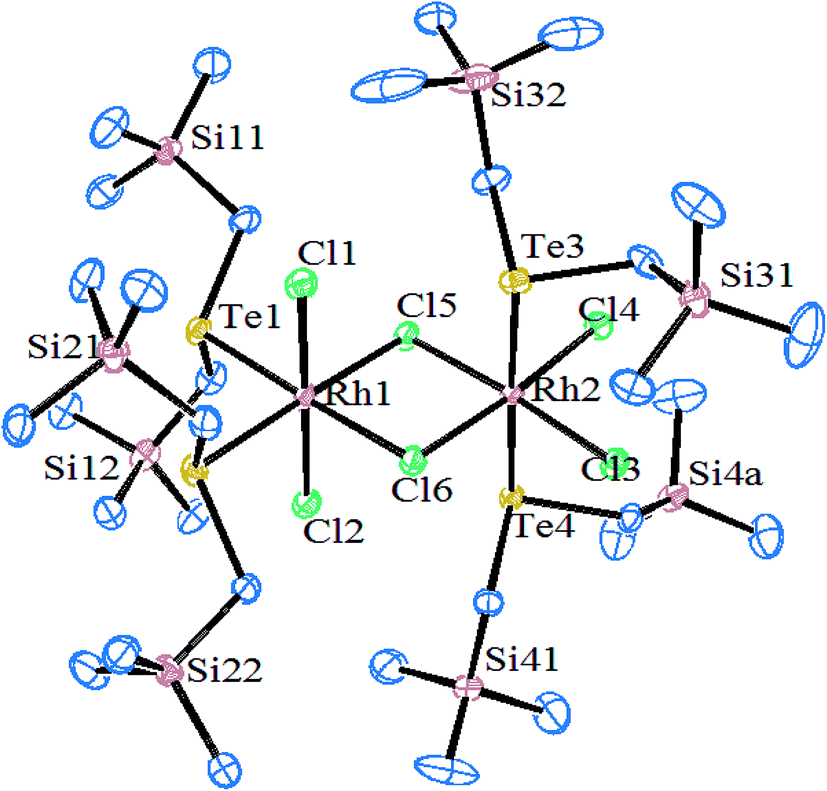
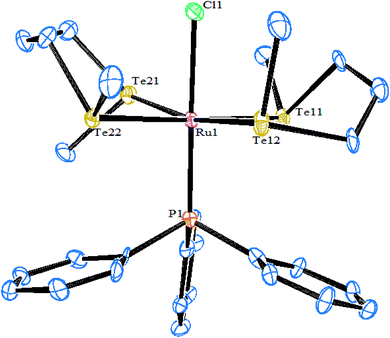
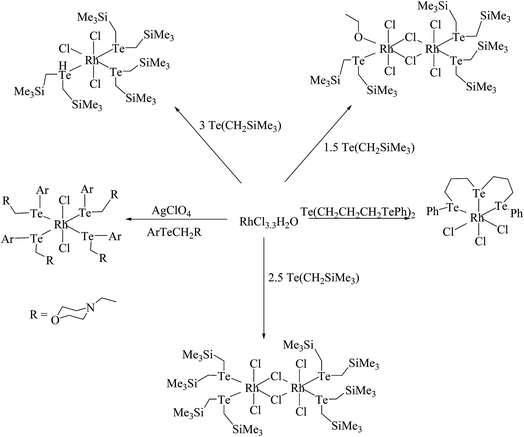
Similarly, when the reaction with RhCl3·3H2O is employed in various mole ratios with the ligand system Te(CH2SiMe3), a variety of products has been isolated (Scheme 4) depending upon the mole ratio of the ligand (Fig. 2).70 Most of these complexes have shown issues with their solubility in common organic solvents. The solubility issues have been overcome with the development of a new class of telluroether in which the backbone consists of a morpholine group. Rh(III) complexes derived from the same ligand N-{2aryltelluroethyl}morpholine (Scheme 4)71 have shown ready solubility in all organic solvents other than diethyl ether and hexane, in which the complexes are sparingly soluble. Their high solubility makes them promising catalysts in the hydrogenation reactions of ketones.
 |
| | Scheme 4 Schematic representation of the reaction of RhCl3·3H2O with various telluroethers. | |
 |
| | Fig. 2 Crystal structure of [Rh2Cl4(μ-Cl)2{Te(CH2SiMe3)2}4] (redrawn from ref. 70). | |
3.3 Palladium and platinum
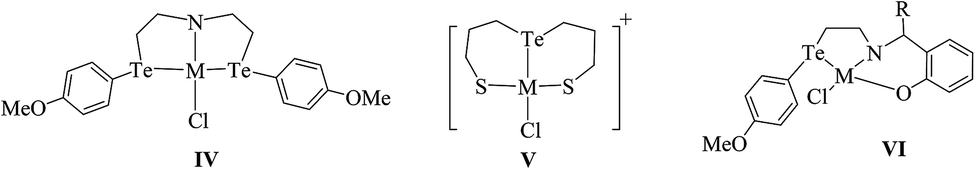
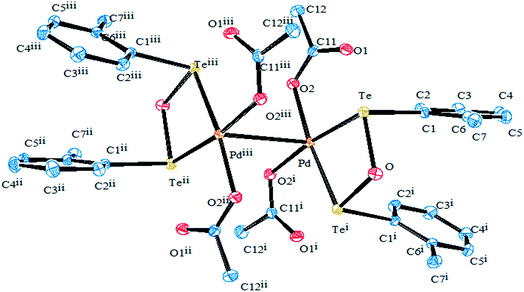
Compared to other platinoids, a significant amount of work has been documented on Pd(II) and Pt(II) complexes derived from telluroethers. The reaction of M2M′Cl4 (M = Na, K; M′ = Pd, Pt) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mole ratio with homoleptic telluroether Te{CH2CH2(Ar)}2 IV (Ar = 1,3 dioxane, thiophene)60,62 or a heteroleptic telluroether, like [(thiophene)Te{CH2(R)}] (R = furan, pyrol),61 [{CH2CH2(R)}Te(C6H4-p-OMe)] (R = thiophene, phthalamide, benzotriazole),62,63 led to a complex of the type [MCl2L2] L = Te{CH2CH2(Ar)}2, [{CH2CH2(R)}Te(C6H4-p-OMe)], [(thiophene)Te{CH2(R)}];63 while the reaction in 1:1 mole ratio with [{CH2CH2(R)}Te(C6H5-p-OMe)] (R = 2-CH3SC6H4CHN, 2-CH3SC6H4CHNH V, {C6H5}{2-HOC6H4}CN) VI and a variety of tridentate ligand yielded an expected additive product with the composition [MCl(L)]+.65–69 Among these cyclic complexes, [MX2{Te(CH2)4}2] (M = Pd, Pt; X = Cl, Br, I) has been isolated from the reaction with cyclic telluroethers, i.e., telluracyclopentane Te(CH2)4.72 In the case of the platinum metal system, both cis- and trans-configured complexes have been reported. However, only a trans isomer has been isolated in the case of palladium, which was also supported by its X-ray structure results (Fig. 3).72 The trans configuration was also obtained by ligand systems like 4 oxatellurane,73 1,3 dihydrobenzotellurophene, and 2,7-dihydro-1H-dibenzotellurophene ligands.74 A comparative study of bidentate ligand RECH2ER (R = Me, Ph and E = S, Se, Te)75 with both metals showed that the thio analogs led to a very stable compound, namely [MCl2(η1-PhSCH2SPh)2], while selenium complexes are slowly decomposed into their polymeric form with the composition [{MCl2(RECH2ER)2}n] and a free ligand; while tellurium analogs of the same ligand, i.e., RTeCH2TeR (R = Me, Ph), only gave the polymeric [{MCl2(RECH2ER)2}n] (M = Pd, Pt) compounds. The similar ligand system (4-MeOC6H4Te)2CH2 formed a monomeric structure [MCl2(L∩L)] compound comprised of a four-membered strained ring with an acute angle of 81° between Te–Pd–Te (Fig. 4).76,77
:2 mole ratio with homoleptic telluroether Te{CH2CH2(Ar)}2 IV (Ar = 1,3 dioxane, thiophene)60,62 or a heteroleptic telluroether, like [(thiophene)Te{CH2(R)}] (R = furan, pyrol),61 [{CH2CH2(R)}Te(C6H4-p-OMe)] (R = thiophene, phthalamide, benzotriazole),62,63 led to a complex of the type [MCl2L2] L = Te{CH2CH2(Ar)}2, [{CH2CH2(R)}Te(C6H4-p-OMe)], [(thiophene)Te{CH2(R)}];63 while the reaction in 1:1 mole ratio with [{CH2CH2(R)}Te(C6H5-p-OMe)] (R = 2-CH3SC6H4CHN, 2-CH3SC6H4CHNH V, {C6H5}{2-HOC6H4}CN) VI and a variety of tridentate ligand yielded an expected additive product with the composition [MCl(L)]+.65–69 Among these cyclic complexes, [MX2{Te(CH2)4}2] (M = Pd, Pt; X = Cl, Br, I) has been isolated from the reaction with cyclic telluroethers, i.e., telluracyclopentane Te(CH2)4.72 In the case of the platinum metal system, both cis- and trans-configured complexes have been reported. However, only a trans isomer has been isolated in the case of palladium, which was also supported by its X-ray structure results (Fig. 3).72 The trans configuration was also obtained by ligand systems like 4 oxatellurane,73 1,3 dihydrobenzotellurophene, and 2,7-dihydro-1H-dibenzotellurophene ligands.74 A comparative study of bidentate ligand RECH2ER (R = Me, Ph and E = S, Se, Te)75 with both metals showed that the thio analogs led to a very stable compound, namely [MCl2(η1-PhSCH2SPh)2], while selenium complexes are slowly decomposed into their polymeric form with the composition [{MCl2(RECH2ER)2}n] and a free ligand; while tellurium analogs of the same ligand, i.e., RTeCH2TeR (R = Me, Ph), only gave the polymeric [{MCl2(RECH2ER)2}n] (M = Pd, Pt) compounds. The similar ligand system (4-MeOC6H4Te)2CH2 formed a monomeric structure [MCl2(L∩L)] compound comprised of a four-membered strained ring with an acute angle of 81° between Te–Pd–Te (Fig. 4).76,77
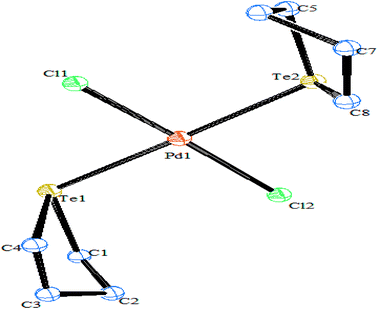 |
| | Fig. 3 Crystal structure of [PdCl{Te(CH2)4}2] (redrawn from ref. 72). | |
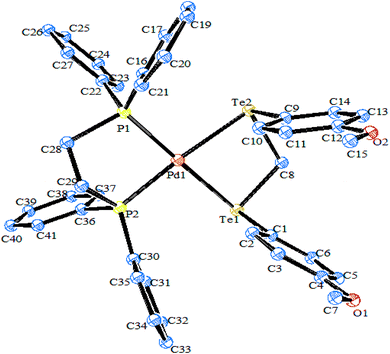 |
| | Fig. 4 Crystal structure of [Pd{(4MeO–C6H4Te2)CH2}(dppe)] (redrawn from ref. 77). | |
Platinum(IV) complexes with the composition [{PtMe3I}2{(4-RTe)2CH2}], (R = Ph, 4-MeOC6H4) and [{PtMe3I}2(PhTeCH2TePh)2], containing bridging, chelating, and monodentate ditelluroethers, respectively, have also been isolated.78 The homoleptic complexes [M(L∩L)2](PF6)2 (M = Pd, Pt); L∩L = RTe(CH2)3TeR, R = Me, Ph; o-CH2(TeMe)2, o-C6H4(CH2TeMe)2, MeC(CH2TeMe)3, and MeC(CH2TePh)3 have been prepared from [MCl2(MeCN)2], TlPF6, and the corresponding ligands in acetonitrile solution.54,55 The 1{H}NMR spectra of the above-mentioned complexes show broad resonance due to an inversion process. Prof. A. K. Singh et al. developed a series of telluroethers, namely 1,3-(4-MeOC6H4TeCH2)2CHOH,79 4-MeOC6H4TeCH2CH(OH)CH2OH, 4-MeOC6H4Te(CH2)nCN(2-HOC6H4)(C6H5),80 (2-phenyltelluromethyl)tetrhydropyran,81 (2-phenyltelluromethyl)tetrhydropyran,82 Te(CH2CH2R)2 (R = CH2NH2, C5H4N), (4-RC6H4)Te(CH2CH2{2-C5H4N}) (R = H, Me, OMe),83–88 N-{2-(4-MeOC6H4Te)CH2CH2}-phthalimide,89 with a possible one or more donor atoms, like oxygen and nitrogen, other than just the available tellurium atom (Scheme 5). Obviously, both metals (Pd, Pt) are directly bonded to tellurium with weak interactions through the donor atoms.
 |
| | Scheme 5 Tridentate hybrid telluroether ligands. | |
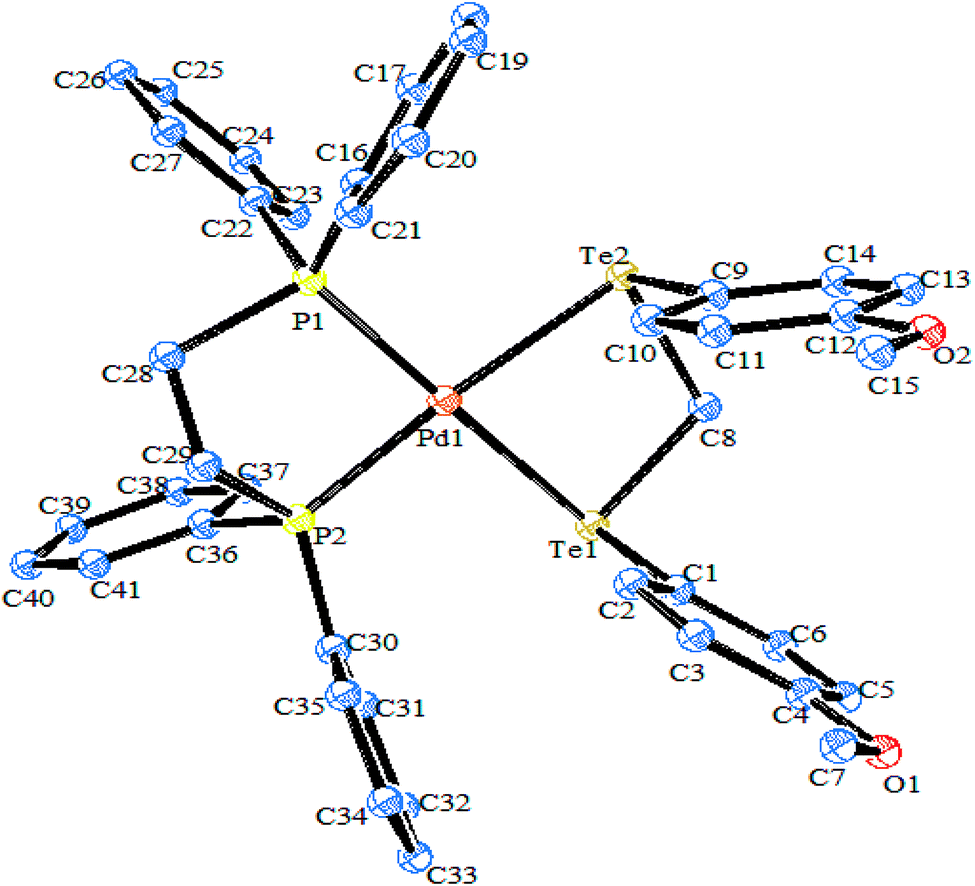
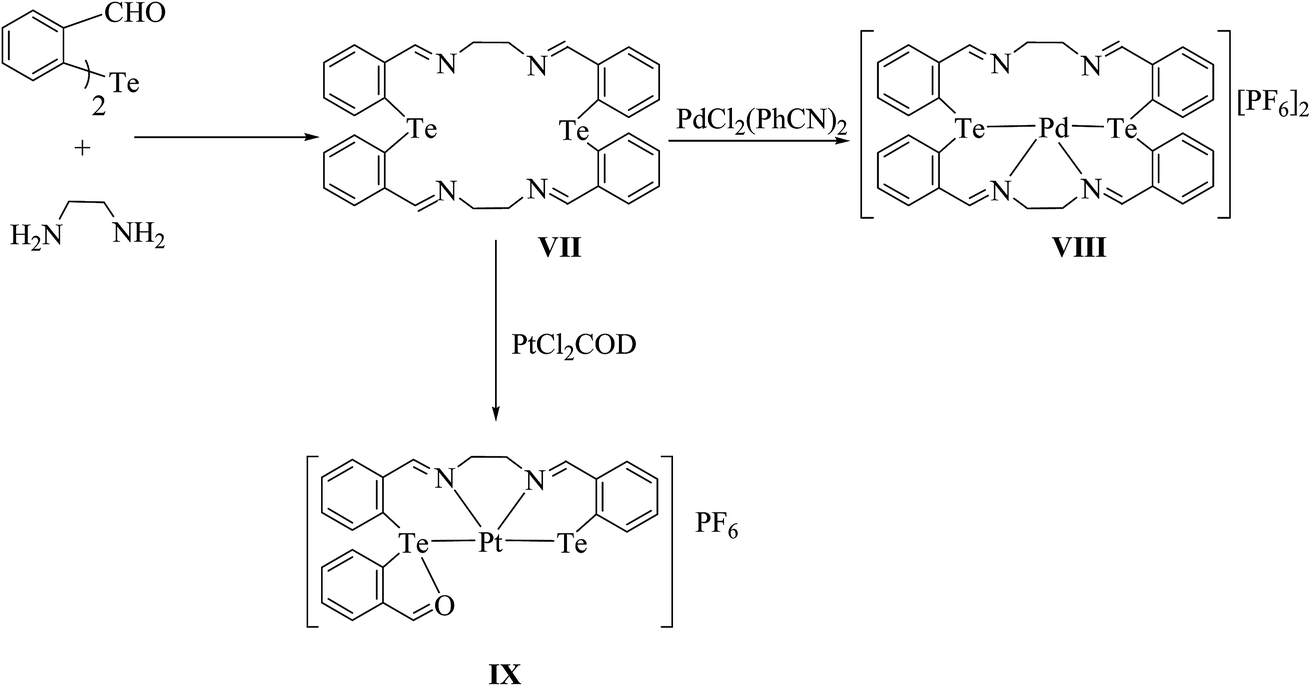
Tellurium has also been ligated into a macrocyclic Schiff base, and the resulting ligand VII upon reaction with [PdCl2(PhCN)2] yielded the complex [{PdCl2}2L]. In this complex, palladium is coordinated with each tellurium as well as the nitrogen of the Schiff base. However, the same reaction in a 1:1 ratio of palladium precursor to the ligand gave the product [PdL]+ VIII,90 in which palladium is coordinated to the Te2N2 core of the ligand, leaving the two nitrogen atoms uncoordinated (Scheme 6);91 while in the case of [PtCl2(COD)], a symmetrical ring opening of the ligand took place to give the product IX, where platinum is coordinated to the TeN2C core. Surprisingly, the complexation mode of the tellurium ligand system was totally different from the similar selenium analogs. In the case of the latter ligand system, palladium is directly coordinated to all four nitrogen atoms.92,93
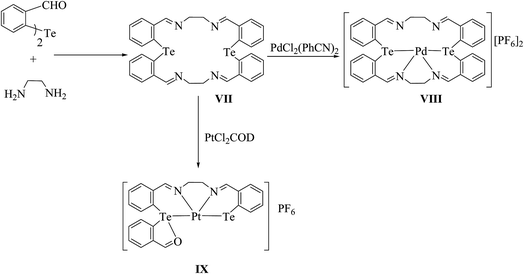 |
| | Scheme 6 Reaction of [MCl2(L)2] (M = Pd, Pt; L = PhCN or COD) with a tellurium-ligated macrocyclic Schiff base. | |
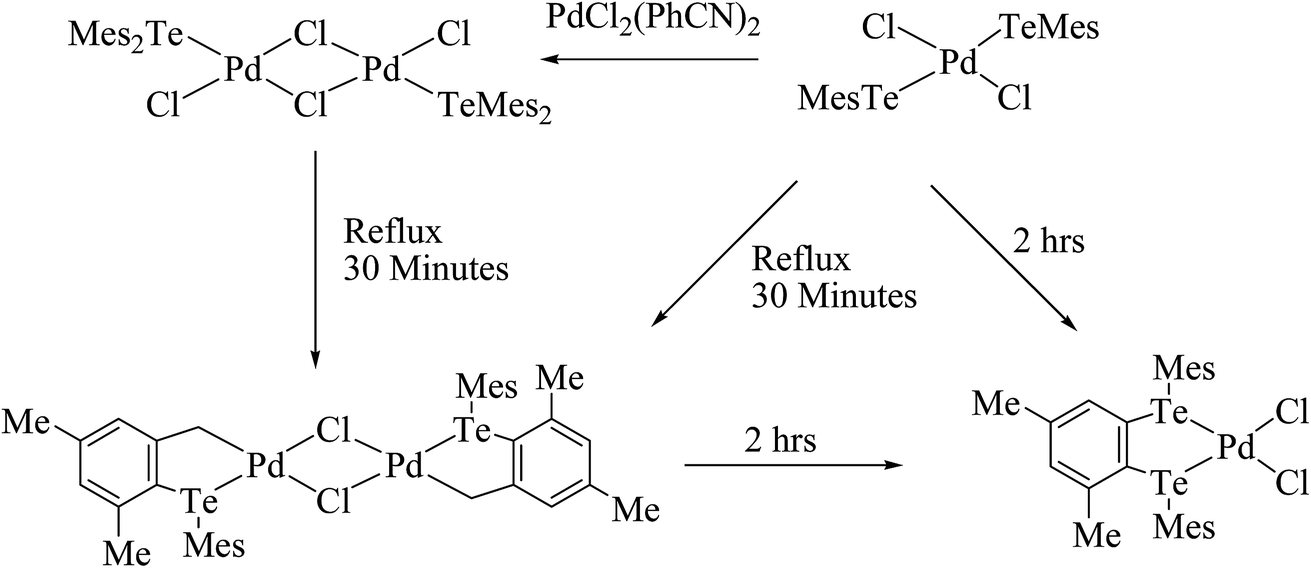
An interesting example of a halobridged complex, [M2X2(μ-X)2(TeR2)2] (M = Pd, Pt; X = Cl, Br; R = Mes, Ph, o-tol), was obtained by the treatment of [MX2(TeR2)2] (R = Mes, Ph, o-tol) with MX2 (M = Pd, Pt)/Na2PdCl4 (Scheme 7).93–95 These complexes were formed due to the chlorobridged cleavage reaction with the substitution of thio and seleoether through monotellurides. Usually, telluroether complexes are oriented in the cis form, which is slowly transformed to the trans isomer in solution. The conversion of the cis to the trans form was encountered in the 125TeNMR spectrum of the latter complexes, which exhibited a single resonance first, but in the longer acquisition results two prominent resonances were observed, attributed to the cis as well as trans forms.94 An agostic interaction between telluroethers and metal atoms has also been documented in these complexes, where a toluene–methanol solution of [Pd2Cl2(μ-Cl)2(TeMes2)2] or compound [PdCl2(TeMes)2] on refluxing for 30 min yielded a binuclear cyclopalladated complex [Pd2(μ-Cl)2{CH2C6H2(4,6-Me2)TeMes2}2] (Scheme 7). However, the latter binuclear compound was converted to the mononuclear compound [PdCl2{MesTeCH2C6H2(4,6-Me2)TeMes}2]. The formation of the mononuclear compound mainly arose due to nucleophilic attack of mesityl tellurolate at the Pd–C bond.93
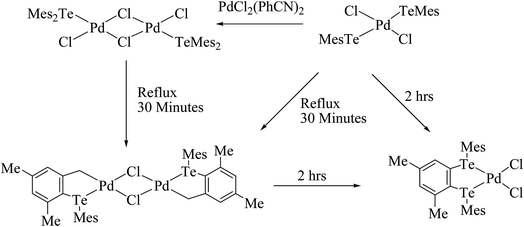 |
| | Scheme 7 Cyclometallation of [PdCl2(Mes)2] at different temperatures. | |
Recently, an intricate palladacycle was synthesized by the reaction between ditolyl telluride and palladium acetate in toluene solution, which yielded two complexes with the composition [(o-tolylTe)2OPd(OAc)2] (Fig. 5) and trinuclear [Pd(o-tolyl){di-o-tolyltelluride}]2Pd(μ-OAc)2. The former complex was a bidentate tellurinic acid anhydride, while the latter was coordinated to tolyl and telluride.96 The formation of both binuclear [(o-tolylTe)2OPd(OAc)2] and trinuclear [Pd(o-tolyl){di-o-tolyltelluride}]2Pd(μ-OAc)2 complexes can be encountered by intramolecular cyclopalladation via the bond formation between palladium metal centers and tolyl carbon.
 |
| | Fig. 5 Crystal structure of [(o-tolylTe)2OPd(OAc)2] (redrawn from ref. 96). | |
4. Metal tellurolates
4.1 Ruthenium and osmium
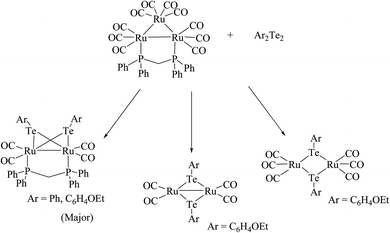
The coordination chemistry of ruthenium cluster complexes toward the highly reactive organotellurium ligand is very rich in the literature, but still various aspects of its reactivity pattern are uncultivated. With this prospect, the refluxing of Ru3(CO)12 with Na2Te2 and PPh4PBr at 80–130 °C for 8 h yielded the cluster complex [Ru4(Te)2(Te2)2(TeMe)2(CO)8](PPh4)2.97 The crystal structure of the latter complex consisted of four rectangles with a center of inversion in the midst. While the oxidative addition of diorganoditelluride to [Ru3(CO)12] yielded a variety of compounds. In particular, the reaction of diphenyl ditelluride resulted in the formation of a binuclear complex bridged by phenyl tellurolate along with the polymeric form of [Ru(CO)2(μ-TePh)]n.98,99 Performing the same reaction with the addition of halogen yielded the complex [Ru2X2(CO)6(μ-TePh)2] (X = Br, I), which was isolated by breaking of the Ru–Ru bond.99 Similarly, on refluxing a phosphine precursor of the ruthenium complex [Ru3(CO)10(μ-dppm)] with tetrahydrofuran solution of Ar2Te2 (Ar = C6H4OEt-4) in a 1:2 mole ratio for 6 h yielded a mixture of the products [Ru2(CO)4(μ-TeAr)2(μ-dppm)], [Ru2(CO)6(μ-TeAr)2] and [Ru(CO)4(TeAr)2] (Scheme 8).100 However in toluene solution, other than [Ru2(CO)4(μ-TeAr)2(μ-dppm)], several products101 have been afforded due to the competitive cleavage of Te–Te and Te–C bonds. At room temperature in CH2Cl2, reaction with a diphenyl ditelluride ligand system yielded the binuclear compound [Ru2(μ-TePh)2(CO)4(μ-dppm)] as well as the trinuclear unsaturated clusters [Ru3(μ3-Te)2(μ-TePh)2(CO)6(μ-dppm)] and [Ru2(μ3-Te)(μ-TePh)3(CO)6(η1-COPh)(μ-dppm)].102
 |
| | Scheme 8 Oxidative addition of [Ru3(CO)10(μ-dppm)] with diaryl ditelluride. | |
Heating a solution of [Ru3(μ-H){(μ3-η2-C4H2O)(μ-PC4H3O)2}(CO)7(μ-dppm)] with Ph2Te2 at 110 °C for 2 h resulted in various cluster complexes, mainly [Ru3(μ3-Te)2(μ-TePh)2(CO)6(μ-dppm)], [Ru3(μ3-η2-C4H3O){μ-PC4H3O2}(CO)5(μ-TePh)2(μ-dppm)] (Fig. 6), [Ru3(μ-H)(μ3-η2-C4H2O){μ-C4H3O2}(CO)5(μ-TePh)2(κ2-dppm)], and [Ru3(μ-H)(μ3-η2-C4H2O){(PC4H3O)3}{μ-P(C4H3O)2}(CO)4(μ-TePh)2(κ2-dppm)].103 However, the reaction of the tetranuclear ruthenium(II) complex with R2Te2 (R = Me, Et, Fc) yielded the diruthenium complex syn-[Cp*RuCl(μ-TeR)]2 in quite a good yield.104 Although the reaction of dialkylditellurides led to a clean pot synthesis of alkanechalcogenolate diruthenium complex, in the case of phenyl derivatives of the tellurium-ligated system, two products [Cp*RuCl(μ-TePh)]2 and [Cp*Ru(μ-TePh)3RuCp*]Cl were isolated.104
 |
| | Fig. 6 Crystal structure of [Ru3(μ3-η2-C4H3O){(μ-PC4H3O)2}(CO)5(μ-TePh)2(μ-dppm)] (redrawn from ref. 103). | |
Treatment of [(η6-cymene)Ru(μ-Cl)3Re(CO)3] with sodium salt phenyl telluride yielded a yellow heterometallic product [(η6-cymene)Ru(μ-TePh)3][Re2(CO)6(μ-TePh)3].105 Half sandwich complex [(Cp*Ru)2B2H6(μ-TePh)] has been obtained by the thermolysis of [(Cp*Ru)2B2H6S2] in the presence of Ph2Te2.106 The reactions of [Fe2(μ-Te)2(CO)6] with low valent precursor of ruthenium metal derive heteronuclear telluride bridged cluster compound by the insertion between the Te–Te bond.107–112 Therefore the treatment of [Ru4(CO)12] with [Fe2(μ-Te)2(CO)6] afforded a pentanuclear cluster complex of composition [Fe(CO)6Ru3(μ-Te)2(CO)11].107 Similar outcome has been obtained by the reaction of [Fe2(μ-EE′)2(CO)6] (E = S, Se, Te) with [M3(CO)12(NCMe)] (M = Ru, Os).109,110 These latter complexes are also isolated at room temperature by the precursor [M3(CO)10(MeCN)2] (M = Ru, Os).113 While the reaction with trinuclear compound [Fe3(μ3-Te)2(CO)9] results the substitution product [Ru4(μ4-Te)2(CO)11].114
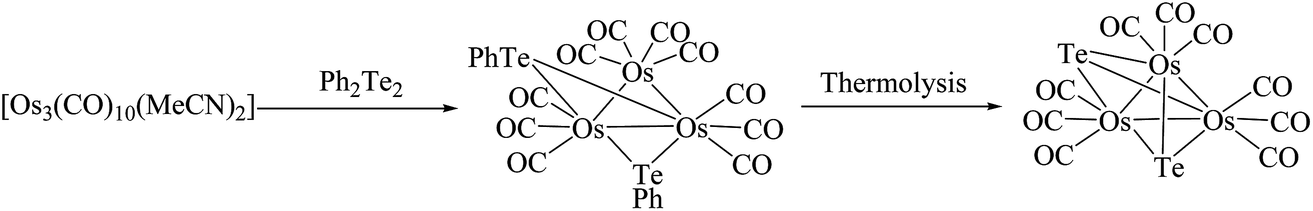
In contrast to ruthenium, the reaction of Ph2Te2 with the osmium precursor [Os3(CH3CN)2(CO)10] under normal conditions yielded the cluster [Os3(CO)10(μ-TePh)2], in which both tellurium linkages were at opposite Os–Os edges.115 Thermolysis of the latter complex resulted in the cleavage of Te–C bonds and yielded the product [Os3(CO)9(μ-Te)2] (Scheme 9).116 Different to the selenium analog, in this case there was no evidence of intact REER bonds in these complexes, but the conversion from one isomer to other isomer is much more facile; while the reaction with unsaturated osmium cluster compounds, like [Os3(μ-H)2(CO)10], are much more complicated. A variety of complexes were formed, out of which three cluster complexes, namely [Os3H2(μ-TePh)2(CO)10] (X), [Os3H(μ-TePh)(CO)10] (XI), and [OsH(CO)3(TePh)]2,115 were isolated. The same ligand upon reaction with phosphine-derived osmium precursors yielded the binuclear product [Os2(μ-TePh)2(CO)4(μ-dppm)] and two isomeric compounds with the composition [Os3(μ-TePh)2(CO)8(μ-dppm)] (Fig. 7), which mainly differ from the phenyl orientation attached to the telluride metal center.116
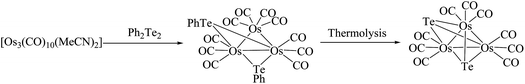 |
| | Scheme 9 Synthesis of a trinuclear osmium telluride bridged complex. | |
 |
| | Fig. 7 Crystal structure of [Os3(μ-TePh)2(CO)8(μ-dppm)] (redrawn from ref. 116). | |
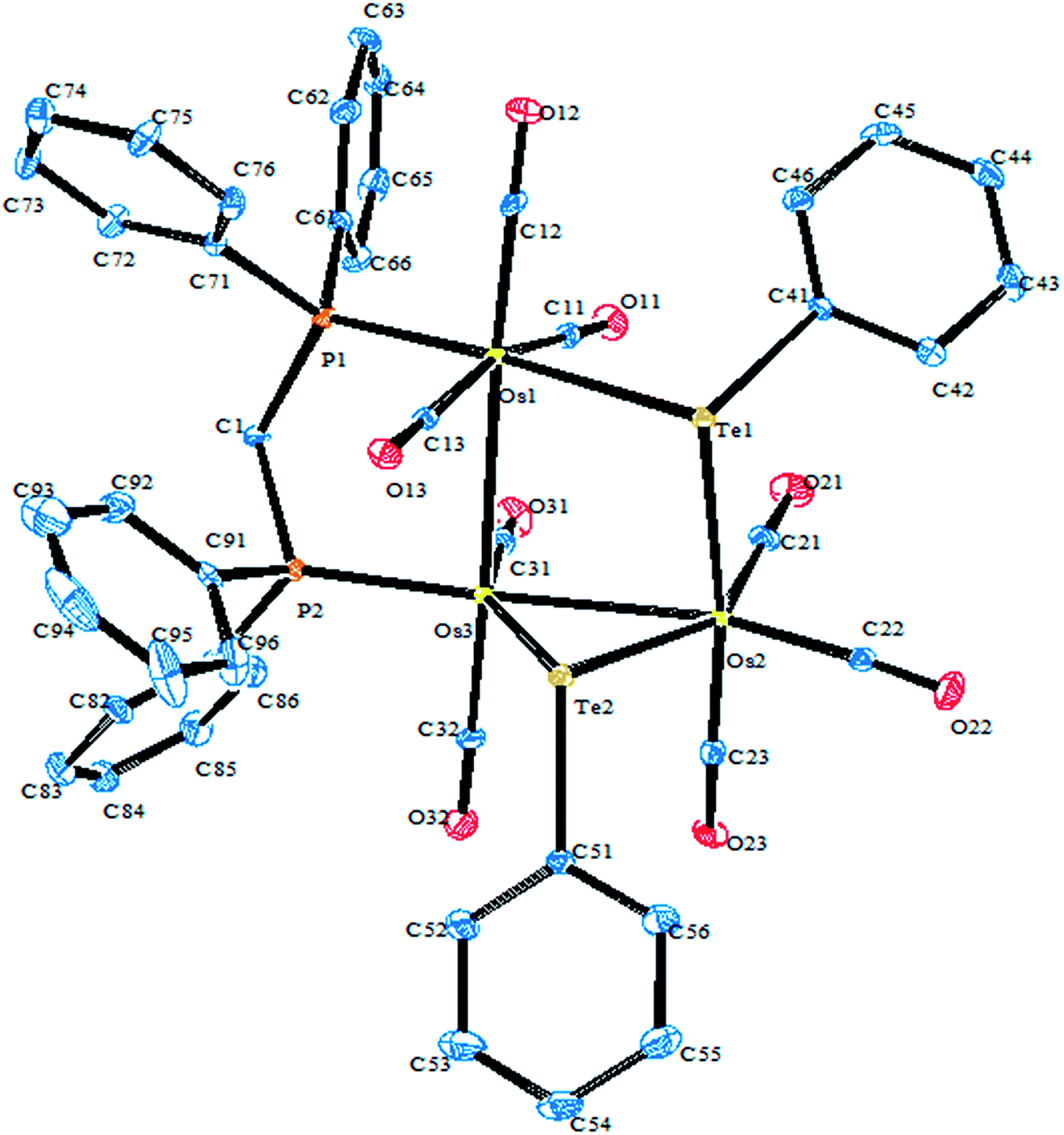
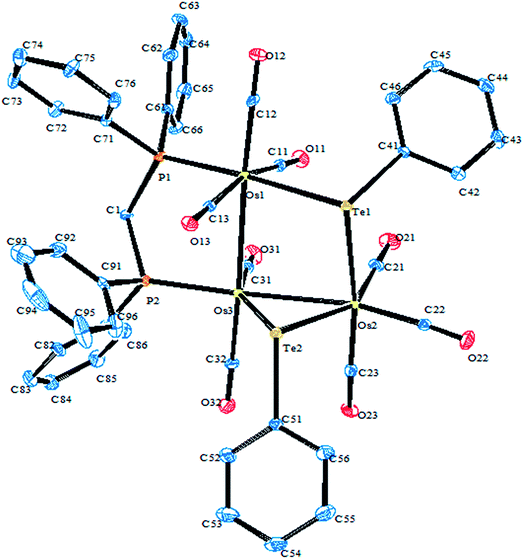
A very close output was obtained when performing the oxidative addition reaction of (TeTol)2 with the mixed cluster [Cp*IrOs3(μ-H)2(CO)10].117 In this case, three cluster complexes were isolated with the composition [Cp*IrOs3(μ-H)2(μ-Tetol)2(CO)7] (Fig. 8). These clusters had relatively different orientations of the tolyl group around the tellurium center, with two of them being stereoisomers having the tolyl group orientation away from the cluster core, i.e., exo, or inward toward the core, i.e., endo. These possibilities of obtaining various stereoisomers have only been reported in ditellurides systems, and can be encountered due to the dynamic nature of tellurium lone pairs.117
 |
| | Fig. 8 Crystal structure of [Cp*IrOs3(μ-H)2(μ-Tetol)2(CO)7] (redrawn from ref. 117). | |
4.2 Rhodium and iridium
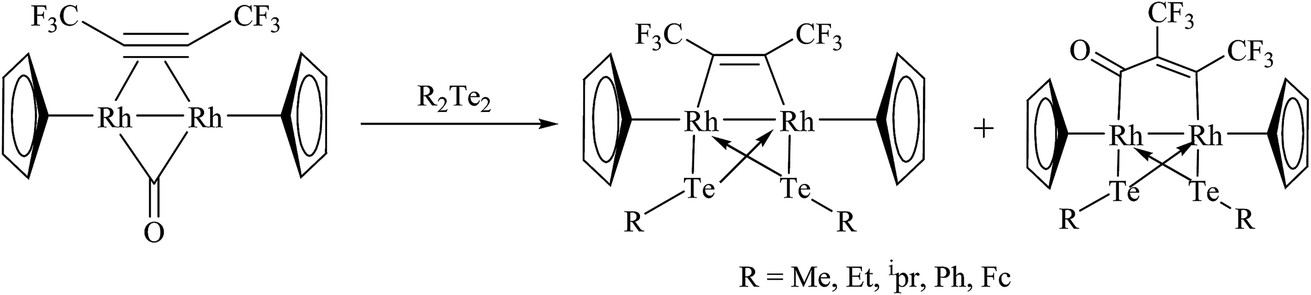
A new class of rhodium precursors, like [Tp*Rh(COE)(MeCN)] (Tp* = hydrotris(3,5 dimethylpyrazol-1-yl)borate; COE = cyclooctene), were explored to study the oxidative addition reaction with diphenyl ditelluride.118 The latter reaction mixture resulted in an oxidative additive product, namely [Tp*Rh(TePh)2(MeCN)], which was allowed to react further with a small amount of [(Cp*Ru)4(μ3-Cl)4] to yield the mixed binuclear compound [Tp*RhCl(μ-TePh)2RuCp*(CH3CN)]. However, on stirring the reaction mixture in an open atmosphere, it binds with oxygen to result in the complex [(Tp*RhCl(μ-TePh)2RuCp*O2)]. The reaction of [(Cp*)2Rh2(μ-CO)μ-η2:η2C2(CF3)2] with R2Te2 (R = Me, Et, ipr, Ph, Fc) yielded the product [(Cp*)2Rh(μ-TeR)2{μ-C2(CF3)2}]119,120 (Scheme 10) along with some of the product [(Cp*)2Rh(μ-TeR)2{μ-CO(CCF3)2}] formed via the insertion process. A new class of internally functionalized dianion ligands [Te(tBuN)P(μtBuN)2P(NtBu)Te]2− upon reaction with [Cp*Rh2(μ-Cl)2Cl2] afforded a cyclic compound with the composition [Cp*Rh{tBuN(Te)P(μ-NtBu)2P(Te)NBut}2(μ-Te)].121
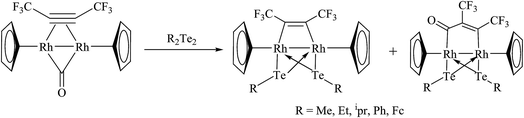 |
| | Scheme 10 Reaction of [(Cp*)2Rh2(μ-CO)μ-η2:η2C2(CF3)2] with R2Te2 (R = Me, Et, ipr, Ph, Fc). | |
The reaction of iridium carbonyl clusters with diphenyl ditelluride is very selective with a very sluggish rate, requiring 20 h of continuous stirring to complete the reaction. The refluxing of PhTeTePh and [Ir6(CO)15]2− gave the anionic cluster [Ir6(CO)14(μ-TePh)]− in tetrahydrofuran solution.122 By applying the same experimental conditions with a 2:1.5 mole ratio of PhTeTePh with [Ir6(CO)15]2− yielded exclusively the neutral product [Ir6(CO)13(μ-TePh)2]; however, similar reactions with other chalcogenides yielded a mixture of products in which a similar neutral product was isolated in very poor yield via a solvent extraction methodology. The best strategy applied to isolate the above neutral complex was the reaction of [Ir6(CO)16] with PhTeTePh in refluxing toluene. The same reaction with other chalcogenides was much less effective in terms of isolation of the cluster compound (Scheme 11).122
 |
| | Scheme 11 Oxidative addition reactions of diphenylditellurides with iridium carbonyl clusters. | |
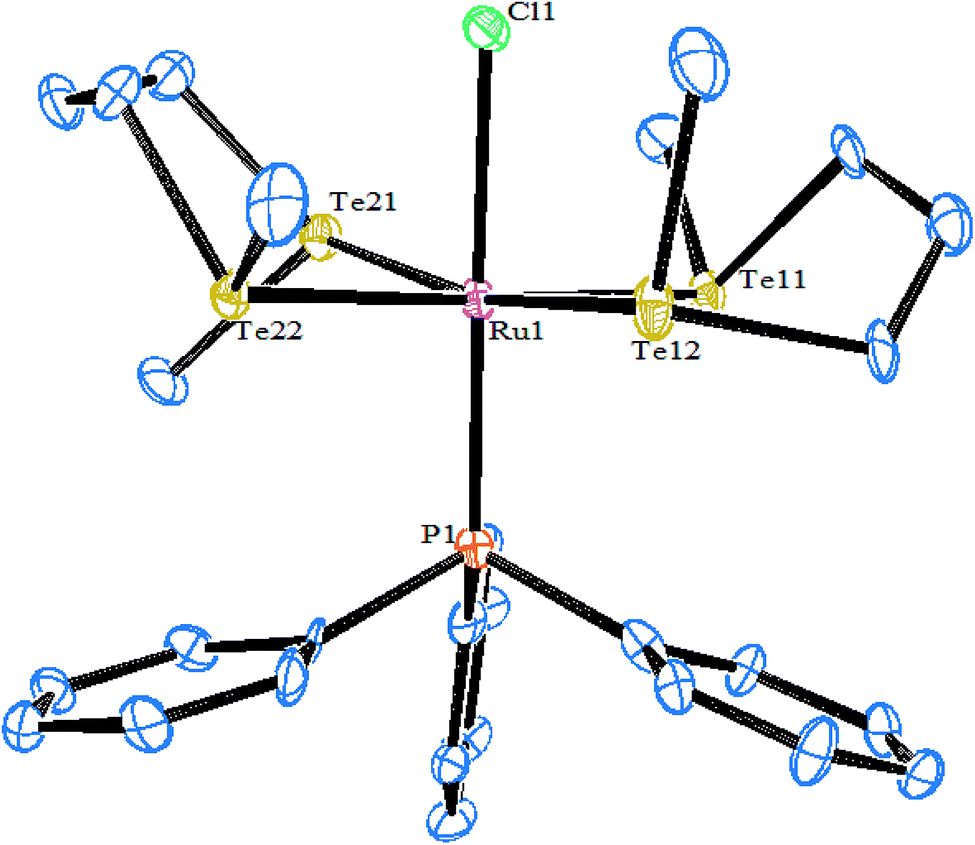
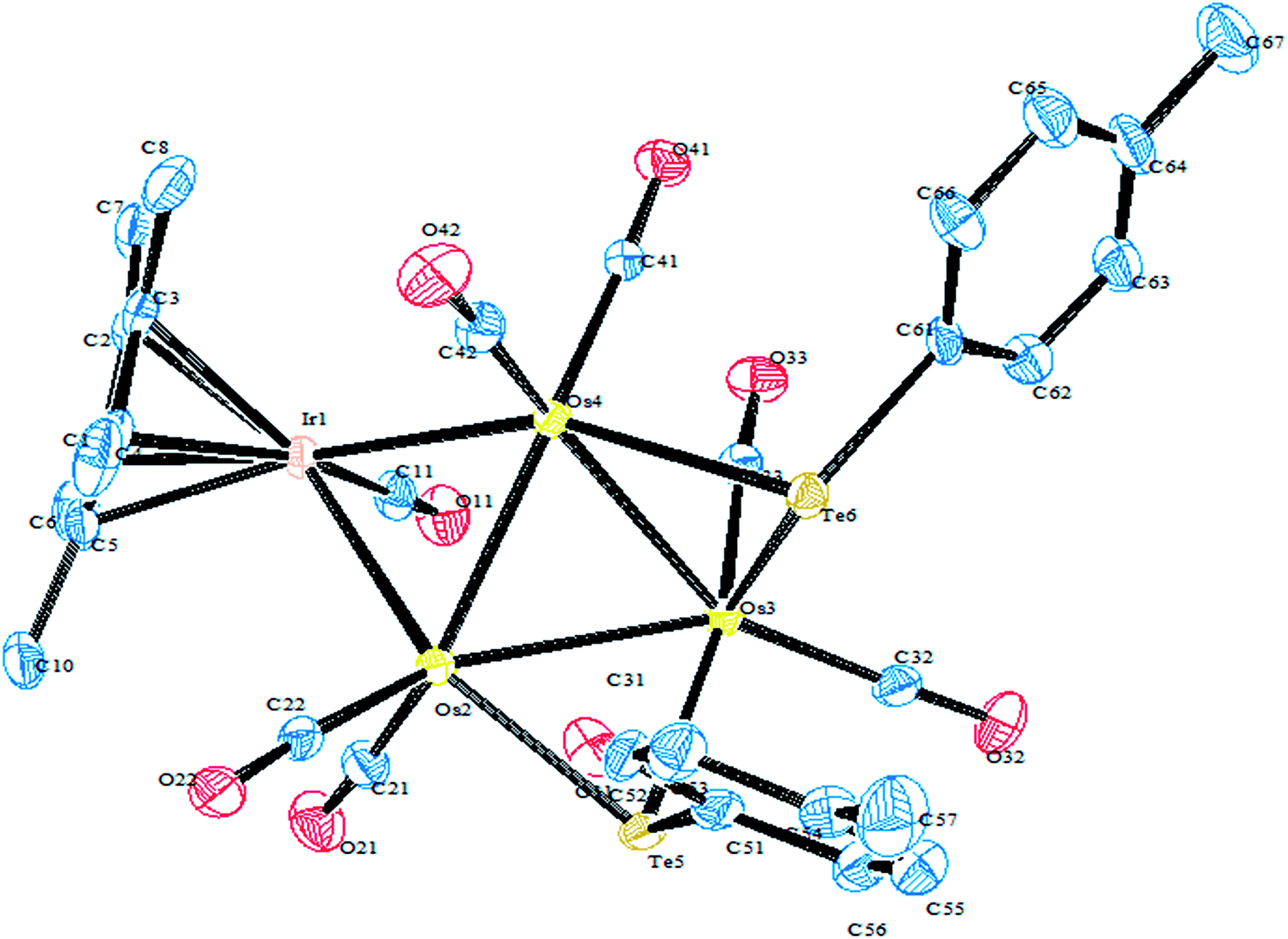
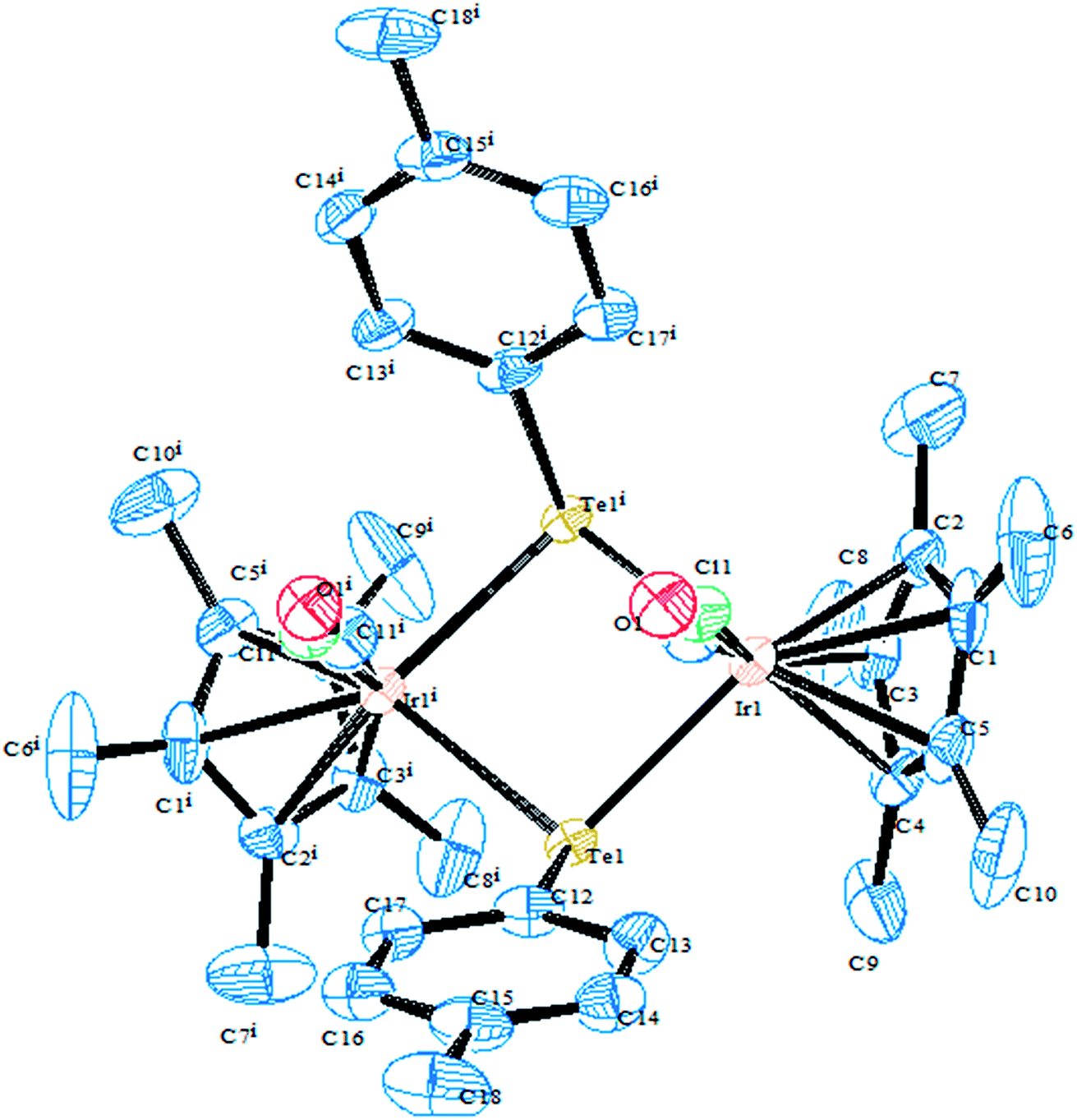
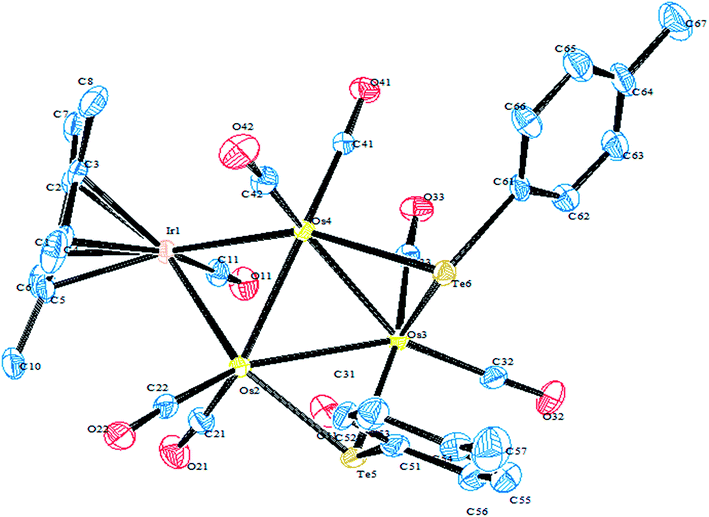
In an attempt to synthesize hyper-valent iridium complexes, organotellurium compounds have played a key role to isolate such complexes. In this context, an oxidative addition of Ph2Te2 with the Ir(I) compound [Cp*Ir(ppy)(solv)]+ was applied.123 Surprisingly the reaction led to the formation of the Ir(III) η1-ditelluride complex [Cp*Ir(ppy)(η1-Te2Ph2)](OTf). It is noteworthy that isolation of a complex with the coordination mode η1-REER is very much less common, e.g. [Cp*Mn(CO)2]2(μ-η1-REER),124 but it is strongly believed that these coordination modes derived compounds take part as an intermediate in the oxidative addition of R2E2 (E = S, Se, Te) to give the Pd(0) and Pt(0) precursors.125 The proven potency from the dynamic nature of tellurium metal can be encountered with the ease of synthesis of homo- and heterometallic complexes with iridium compounds derived from the tellurolate ligand system. As an example, the oxidative addition product [Cp*Ir(CO)(TeTol)2] upon reaction with [(Cp*MCl)2(μ-Cl)2] (M = Rh, Ir) yielded a stereoisomer of the binuclear complexes [Cp*Ir(CO)(μ-TeTol)2MCp*Cl]Cl (M = Rh, Ir) (Fig. 9) bridged with telluride systems (Scheme 12). Interestingly, a similar reaction of [Cp*Ir(CO)(TeTol)2] with [Cp*RuCl4(μ3-Cl)4] and [RuH(COD)(CH3CN)](BPh4) resulted in the formation of the tetranuclear [Cp*IrCl{μ-Te(η6-Tol)RuCp*]2RuCp*(CO)]Cl2 and binuclear [Cp*Ir(μ-H)(μ-TeTol)2Ru(CO)(COD)]BPh4 complexes, respectively (Scheme 12).126
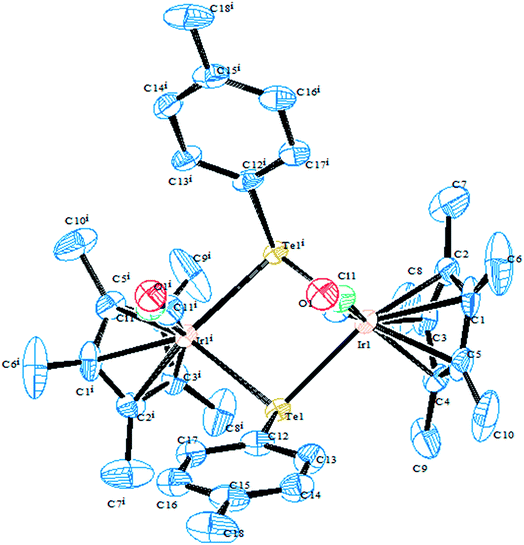 |
| | Fig. 9 Crystal structure of [Cp*Ir(CO)(μ-TeTol)2IrCp*Cl] (redrawn from ref. 126). | |
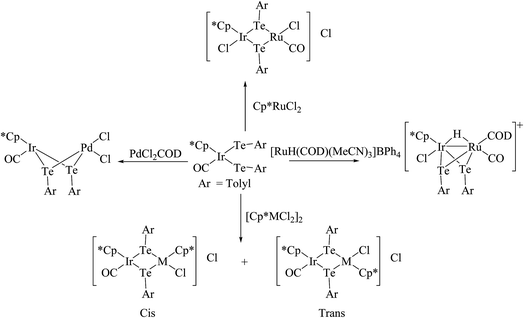 |
| | Scheme 12 Synthesis of various heterometallic complexes using [Cp*Ir(CO)(TeTol)2]. | |
4.3 Palladium and platinum
Tellurium complexes with palladium and platinum phosphine precursors are comparatively more stable than any other precursors. The main stability factor is the π bonding involving dxy orbitals of the palladium and platinum metals with the available empty orbitals of phosphine, which results in the extent of σ overlapping being stronger in the phosphine complexes.127,128 Hence, these complexes play a crucial role in a reduction of the electron density around the metal center, which is enhanced due to the ease of donation of the lone pair available on the tellurium center.
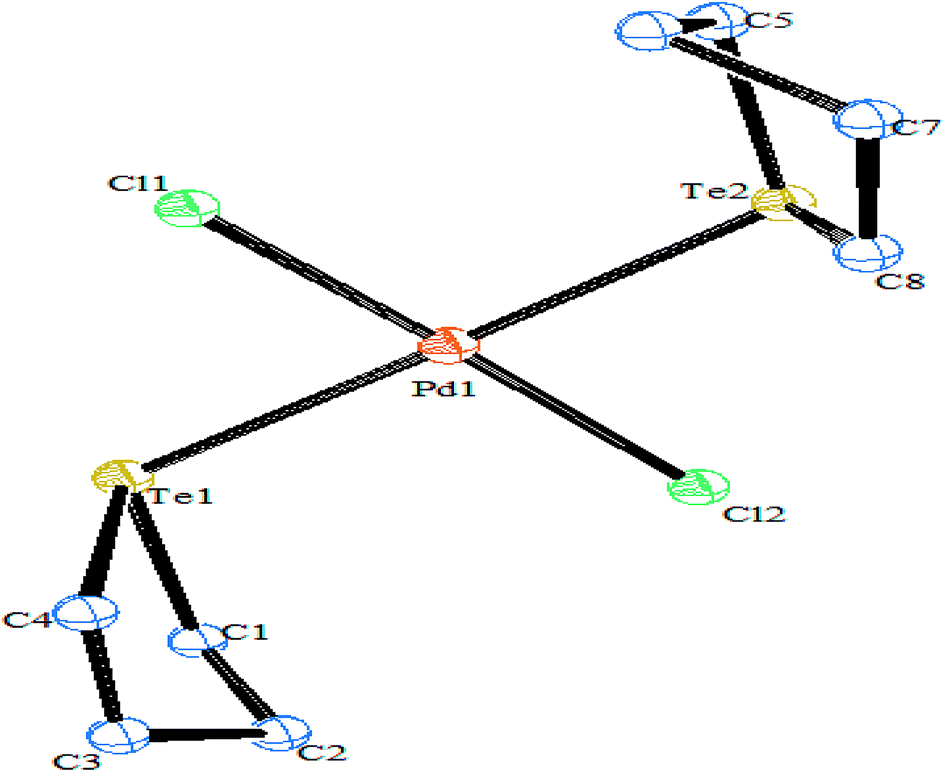
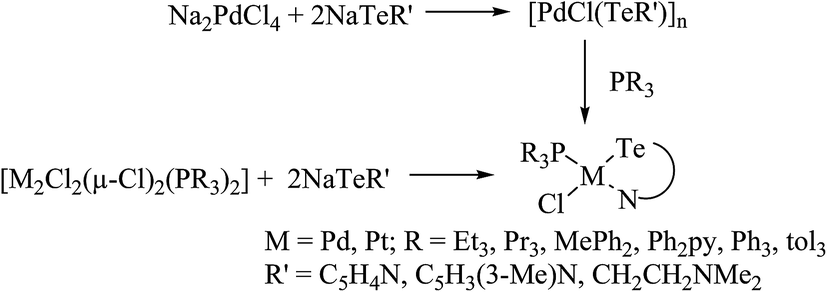
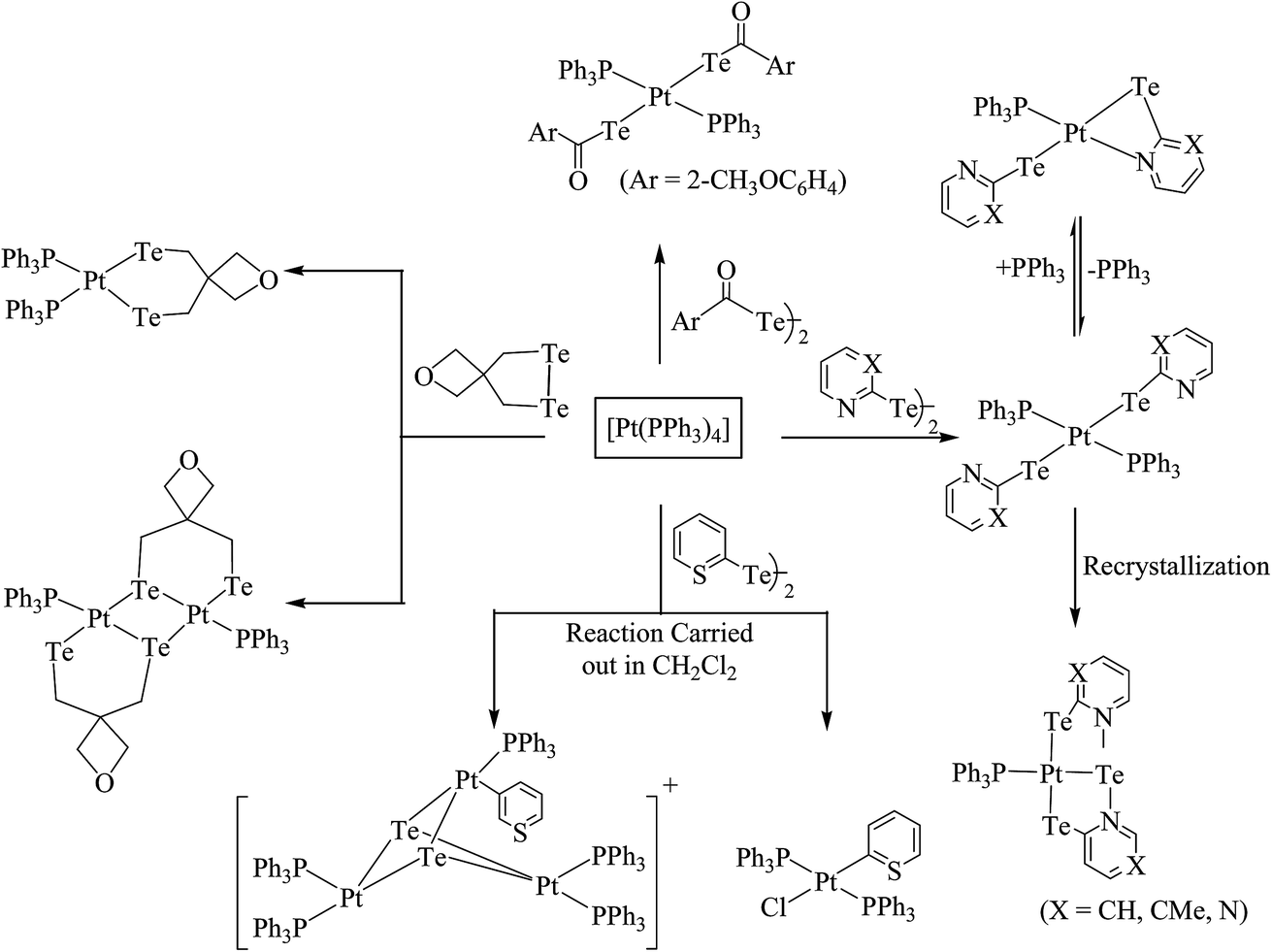
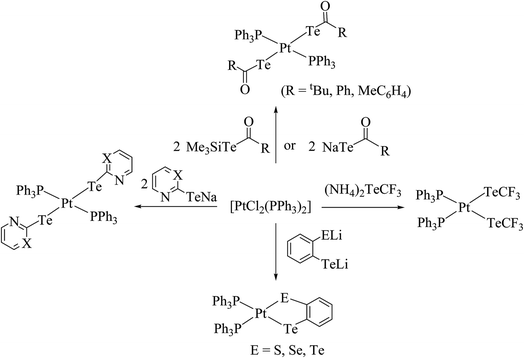
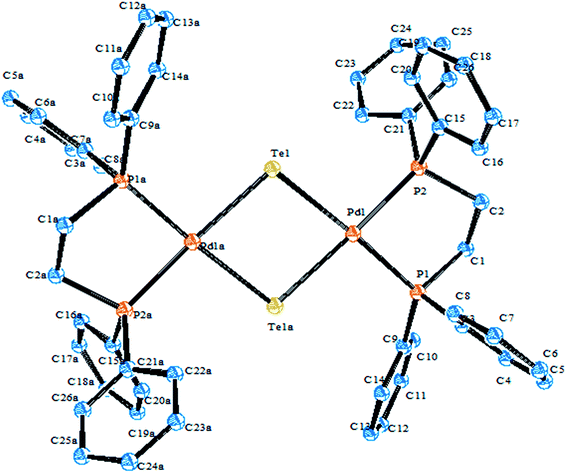
4.3.1. Reactivity of various tellurium systems with palladium and platinum nonchelated phosphine precursors. T. B. Rauchfuss and Rheingold synthesized a series of complexes of the type [Pt(ETeC6H4)(PPh3)2] (E = S; Se; Te) derived from ligand systems like alkeneditellurides and 1,2 benezeneditellurides,129 which represent the first examples of mixed chalcogenides ligand systems (Scheme 15). Various complexes of the type [MCl{TeC5H3(3-R′)N}(PR3)] (M = Pd, Pt; R = PEt3, Ph2Me) have been derived by cleaving of the bond between metal bridged chlorides, e.g., the reaction of [M2Cl2(μ-Cl)2(PR3)2] (M = Pd, Pt; R = PEt3, Ph2Me) with two equivalent of NaTe(R′C5H3N) (R′ = H, Me) (Scheme 13).130,131
 |
| | Scheme 13 Reactions of [M2Cl2(μ-Cl)2(PR3)2] with various sodium salts of tellurolate ligands. | |
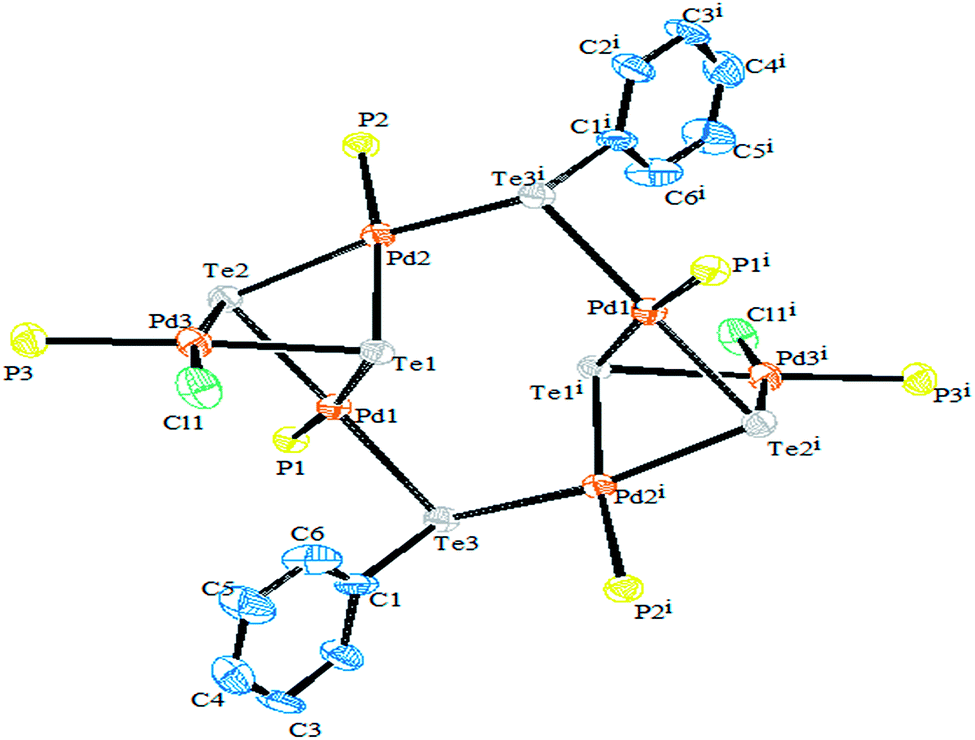
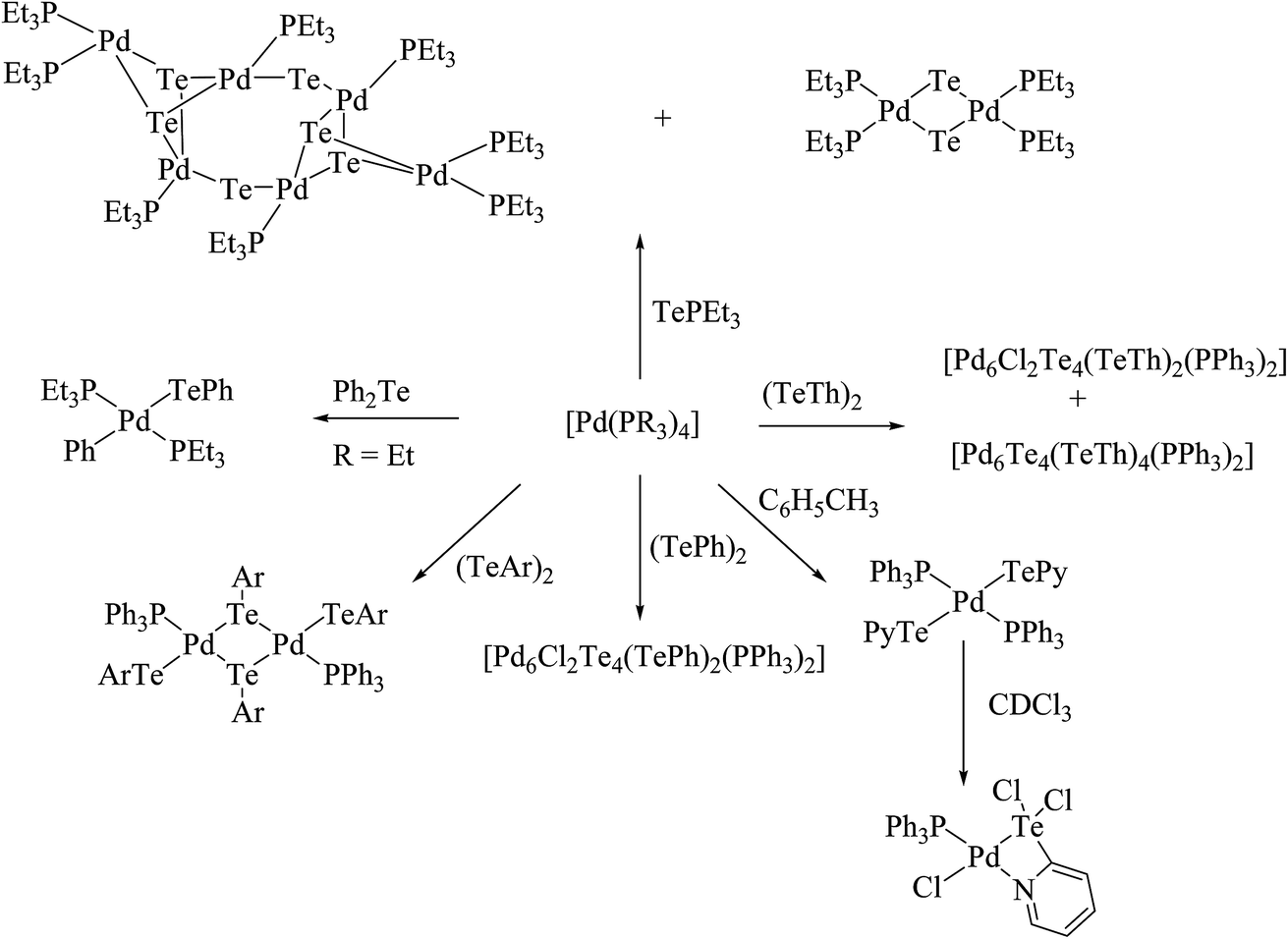
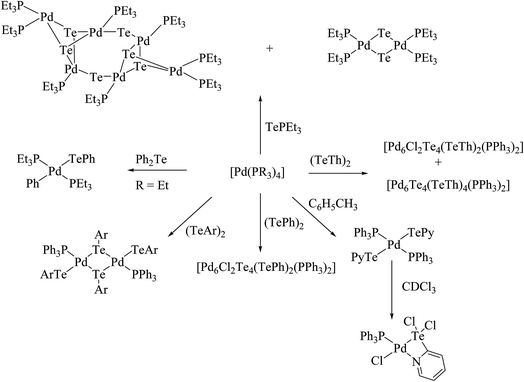
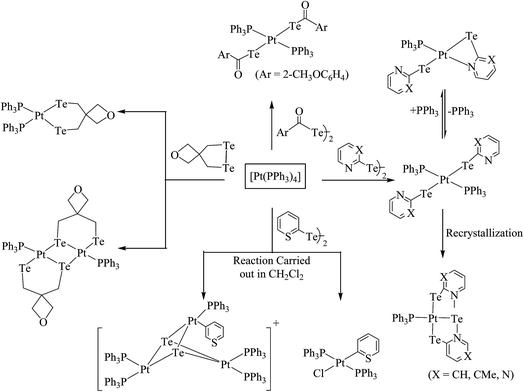
Performing an oxidative addition reaction between [Pd(PPh3)4] and various diaryl ditelluride ligand systems yielded binuclear products with the composition [Pd2(μ-TeAr)2(TeAr)2(PPh3)2].132 In the latter complex, both terminal and bridging positions are occupied with aryl tellurols (Scheme 13). On the other hand, an alike reaction upon performing with [M(PPh3)4] (M = Pd, Pt) with various ligand systems, such as CF3Te2 Th2Te2, Ph2Te2, afforded mono-, bi-, tri-, and hexanuclear complexes [Pd6Te6Cl2(PEt3)6] (Fig. 10) depending on the nature of the solvent (Schemes 14 and 16).21,133–138 It has been well established that the bonding energy between C–Te and Te–Te is quite comparable compared to other analogs of the chalcogen family, and therefore the reaction with tellurium ligands afforded polynuclear compounds. Tanaka et al. found that upon performing an oxidative addition between Pd(0) or Pt(0) species with various telluroethers, cleavage of the C–Te bonds take place, leading to isolation of a compound with the composition [M(Ar)(TeAr)(PEt3)2] (M = Pd, Pt) (Scheme 14).22
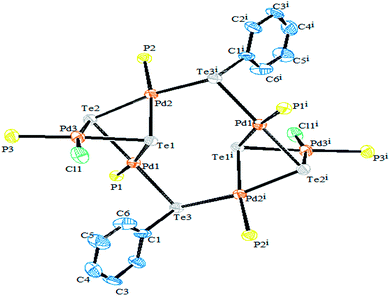 |
| | Fig. 10 Crystal structure of [Pd6Te6Cl2(PEt3)6] (redrawn from ref. 138). | |
 |
| | Scheme 14 Oxidative addition of [Pd(PR3)4] (R = Et, Ph) with various diorganoditellurides. | |
 |
| | Scheme 15 Reactions of [PtCl2(PPh3)2] with various sodium/lithium salts of aryl tellurolates. | |
 |
| | Scheme 16 Oxidative addition reactions of [Pt(PPh3)4] with various diaryl ditellurides. | |
Molecular orbital calculations also concluded that the activation energy barrier for the oxidative addition of dichlacogenides to [M(PH3)2] decreases in the order S > Se > Te (in terms of the addition of E–E bonds).125 Hence, the exothermicity of the reaction is also decreased with respect to the M–E–R bond strength. This statement quantifies that the oxidative addition of Te–Te bonds to low valent metal precursors is very simplistic compared to with the rest of the other dichalcogenides analogs and results in a complex (mononuclear) that is less stable compared to thiolato and selenolato complexes. However, the tendency for isolation of the dimerized product follows the reverse trend (S < Se < Te). Therefore, it can easily be concluded that binuclear complexes are higher in tellurium system compared to the other analogs.
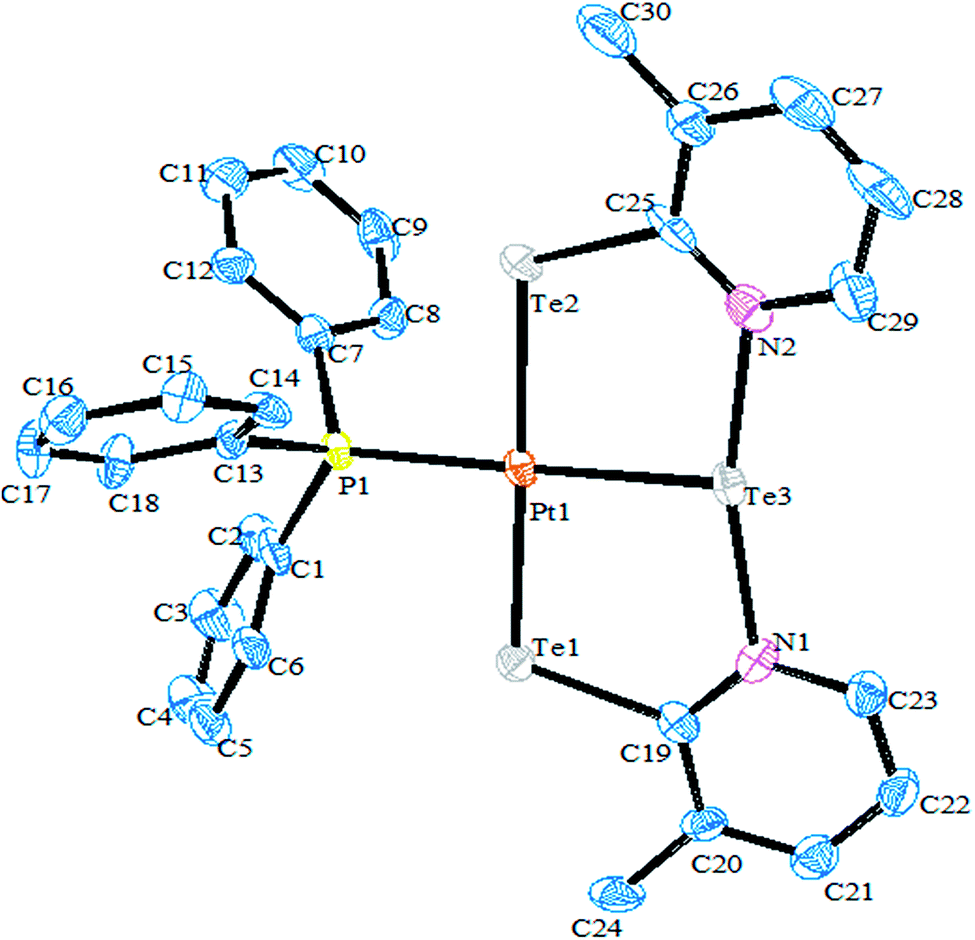
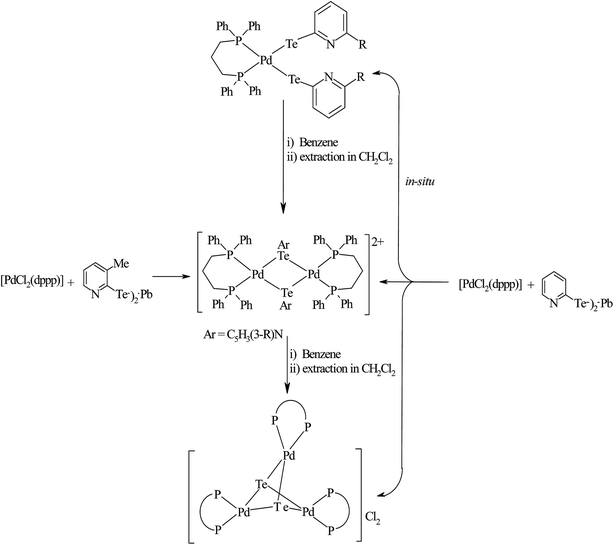
Recently, Jain et al. performed an oxidative addition among [Pt(PPh3)4] and hemilabile pyridyl ditelluride (C5H4N)2Te2 to isolate novel complexes with the composition [Pt{TeC5H3(3-R)N}2Te(PPh3)] (R = H, Me) (Scheme 16) along with an expected oxidative addition product.7 The former complex was the first example of its own type of tellurium (0) acting as a ligand system (Fig. 11). The serendipitous isolation of the latter one was also justified by the substitution reaction of [PtCl2(PPh3)2] with the 2NaTeC5H3(3-R)N (R = H, Me) (Scheme 15). The crystal structure of [Pt{TeC5H3(3-R)N}2Te(PPh3)] showed that the bare tellurium was directly coordinated to the metal center and flanked by the two pyridyl rings (Fig. 5). Density function calculations highlighted that the extra stability of this complex was due to the aromatic nature of the five-membered ring formed by Pt–Te–C–N–Te. While similar reactions in the case of palladium led to isolation of an expected oxidative addition product,8 which on keeping for a longer time period in CDCl3 solution, yielded a green product, [PdCl{Te(Cl)2C5H4N}(PPh3)], in which tellurium is oxidized from a +2 to a +4 oxidation state by the attachment of two chlorine atoms (Scheme 7).
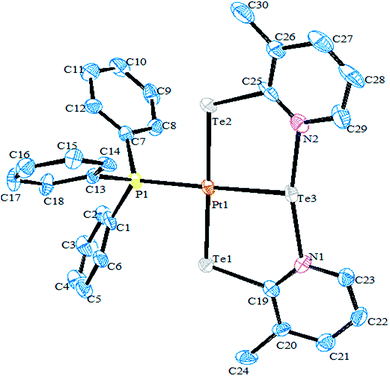 |
| | Fig. 11 Crystal structure of [Pt{TeC5H3(3-Me)N}2Te(PPh3)] (redrawn from ref. 7). | |
A new class of platinum(0) phosphine precursors [Pt(η2-norborene)(PPh3)2] has also been applied to study oxidative addition with bulky ditellurides of dibenzobarrelenyl and norborene. The reaction afforded the formation of four- and five-membered telluraplatinacycles compounds [Pt(Te∩C)(PPh3)3] in the presence of excess triphenyl phosphine.139
4.3.2. Reactivity of various tellurolates with palladium and platinum chelated phosphine precursors. The reactivity of the chelating phosphine diphenylphosphinomethane (dppm) ligand is comparatively higher than that of other chelating phosphines. The main striking factor of the reactivity is strain, caused by the projection of the four-membered ring. The cone angle drawn on P–M–P ranges from 70–72°, which means it is highly acute. To overcome the acuteness, the complementary angle is widened in space to provide the space to react with the incoming ligand. In the case of the substitution reaction between [PtCl2(dppm)] and the sodium salt of aryl tellurolates (aryl = Ph, Tol, Mes), cis configured mononuclear complexes were obtained.133,140,141 However, a similar substitution and an oxidative addition with hemilabile ligand systems, like derivatives of 2 and 4-dipyridylditelluride, yielded an expected mononuclear complex as well as C–H activated [Pt{Ph2PCH(TeC5H3(3-R)NPPh2}2)] (R = H, Me) (Scheme 17).8,131 The latter complexes had a distorted square planer geometry around the metal, with the angle around the activated carbon center varying from 100–126°, which shows an allylic configuration of the carbon center (Fig. 12). It can be concluded that the reactivity of the same palladium phosphine precursors with 4-pyridyl tellurolates to lead to the tetranuclear compound [Pd2(μ-Te)(μ2-TeC5H4N)(4-TeC5H4N)(μ-dppm)]2 (ref. 131) can be rationalized by the high reactivity of the palladium phosphine precursor and tellurium-based ligand.
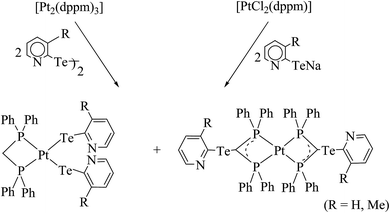 |
| | Scheme 17 Reactions of [Pt2(dppm)3]/[PtCl2(dppm)] with dipyridyl ditellurides/pyridyl tellurolates, respectively. | |
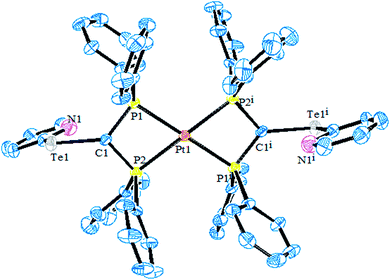 |
| | Fig. 12 Crystal structure of [Pt{PPh2CH(TeC5H4N)PPh2}2] (redrawn from ref. 8). | |
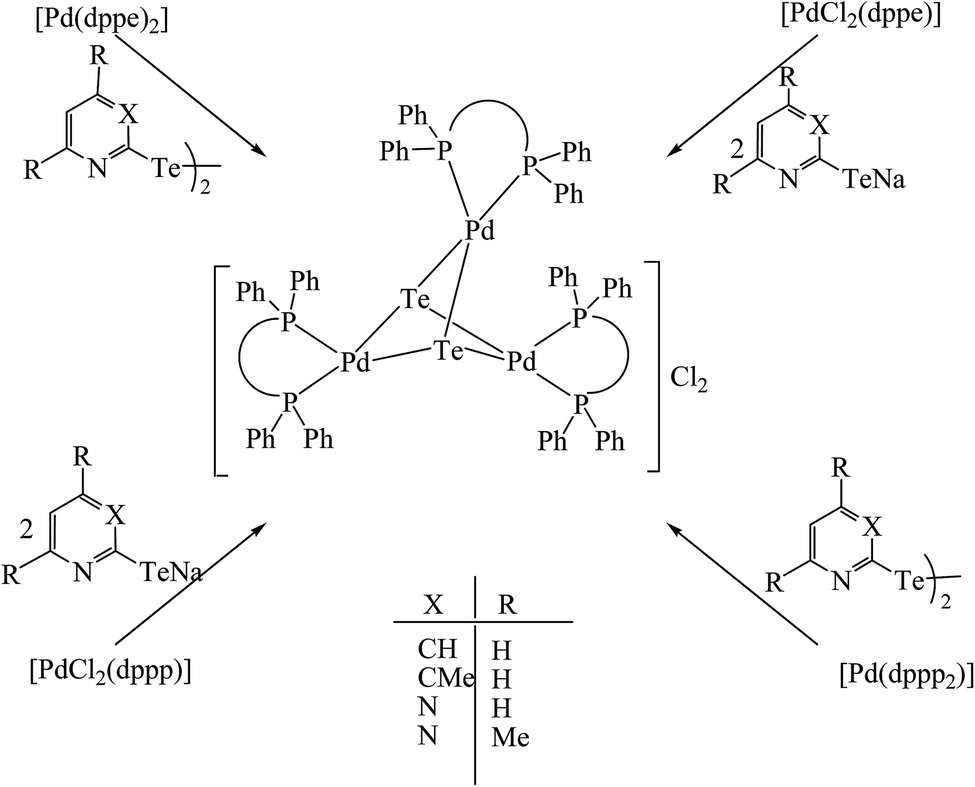
However, the chemistry related to diphenylphosphinoethane with tellurium systems seemed to be as expected. This may be due to the strainless five-membered ring project by “dppe” ligand. On performing an oxidative addition/substitution reaction between [Pt(dppe)2]/[PtCl2(dppe)] and the aryl ditellurides/sodium salt of aryl tellurolate (aryl = py, Ph, tol, Mes, Thienyl), a mononuclear product was isolated.8,140,141 Only in the case of the methyl-substituted telluropyridne ligand system was a complex with the composition [PtCl{TeC5H3(3-Me)N}(dppe)] isolated, which existed in equilibrium with the moiety [Pt{κ2-TeC5H3(3-Me)N}(dppe)]+. Conductometric measurements were performed to correlate the nature of the complex in solution and it was reported that in the case of highly polar solvents, like methanol and acetonitrile, the nature of the complex was a 1:1 electrolyte. Surprisingly, the palladium chemistry is totally different from their platinum analogs. On performing a similar reaction between palladium phosphine precursors [Pd(dppe)2]/[PdCl2(dppe)] with hemilabile ligand systems, like pyridyl (Fig. 13) and pyrimidyl ditelluride,8,48,141 resulted the rapid conversion of mono- to trinuclear complex in a chlorinated solvent (Scheme 18). This result shows the high susceptibility of tellurium ligands toward the chlorinated solvents.
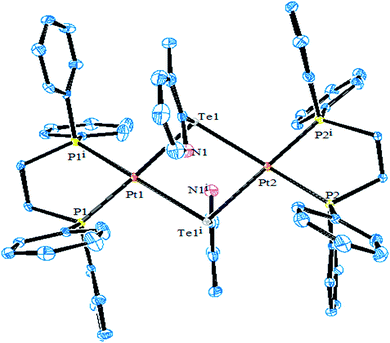 |
| | Fig. 13 Crystal structure of [Pd2{μTeC5H3(3-Me)N}2(dppe)] (redrawn from ref. 8). | |
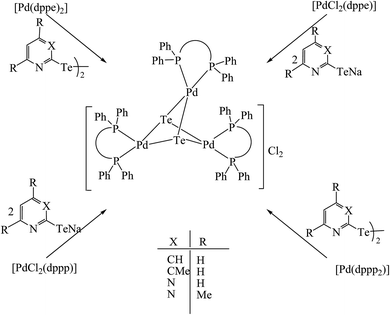 |
| | Scheme 18 Reactions of [Pd(P∩P)2] and [PdCl2(P∩P)] (P∩P = dppe, dppp) with Ar2Te2 (Ar = {C5H3(3-R)N}, {C5H2(4,6-R)N} (R = H, Me)) and their corresponding sodium salts. | |
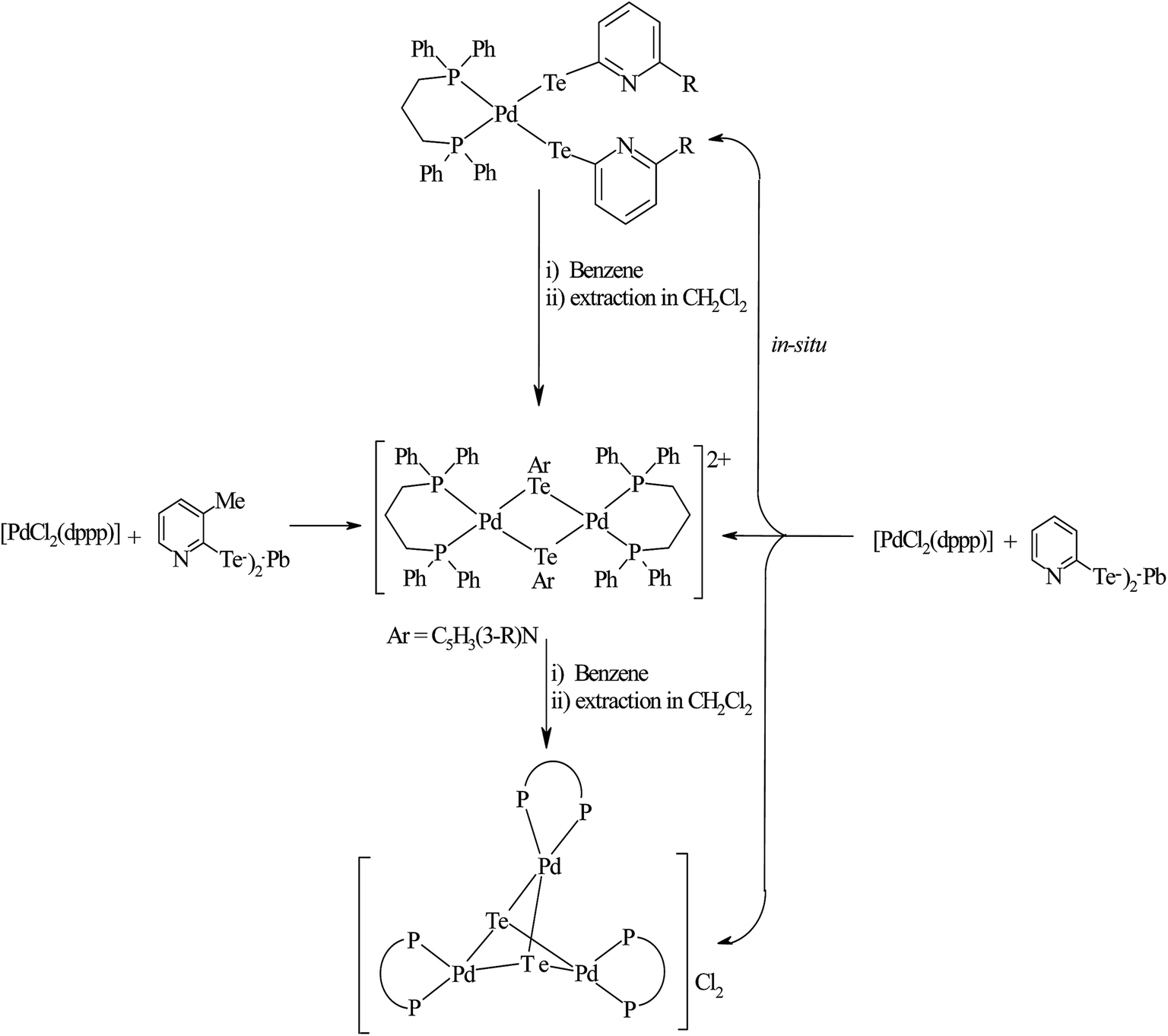
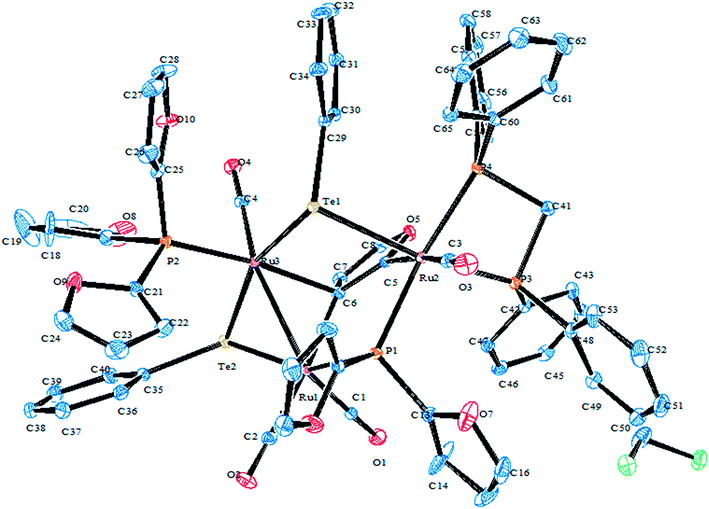
In the case of platinum precursors of diphenylphosphinopropane (dppp), like [Pt(dppp)2]/[PtCl2(dppp)], performing a similar reaction as mentioned above with various hemilabile ligand systems afforded mono- and trinuclear products.8,48 However, the reaction with palladium phosphine precursors yielded only trinuclear products in a moderately good yield. A mechanistic study was performed to isolate the trinuclear product by the mild reaction between [PdCl2(dppp)] and [Pb(TeC5H4N)2] (Scheme 19).8 Upon recording the 31P{1H} NMR within 2 h stirring of the reaction, there was a single resonance corresponding to mononuclear complexes, which again on further stirring for up to 4–5 h at the same temperature showed two quite shielded resonances, corresponding to bi- and trinuclear products. By the extraction of the product in dichloromethane, the trinuclear product was exclusively isolated. The formation of bi- and trinuclear products was also characterized with single crystal X-ray analysis.
 |
| | Scheme 19 Reactions of [PdCl2(dppp)] with the lead salts of 2-pyridine chalcogenolate. | |
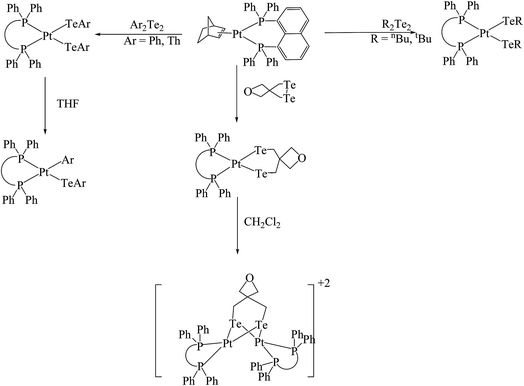
Weigand et al. had applied a new class of Pt(0) precursors [Pt(dppn)(nb)] derived from the chelating phosphine diphenylphosphineno naphthalene (Ph2P-napthyl-PPh2).136 These precursors showed a remarkable reactivity toward ditelluride systems. Upon reaction with various cyclic/saturated, acyclic/saturated, and cyclic/unsaturated ditellurides, the momonuclear [Pt(Te2C5H8O)(dppn)] and binuclear [Pt(Te2C5H8O)(dppn)] products were isolated, which on keeping for a prolonged time period in the solvent decomposed into several products (Scheme 20). It was noticed that alkyl tellurides result in the products by the cleavage of Te–Te bonds, however aryl telluride derives the products not only by the Te–Te bond cleavage but also by Te–C bond cleavage.136
 |
| | Scheme 20 Oxidative addition reaction between [Pt(dppn)(nb)] with various cyclic and acyclic diorgano ditellurides. | |
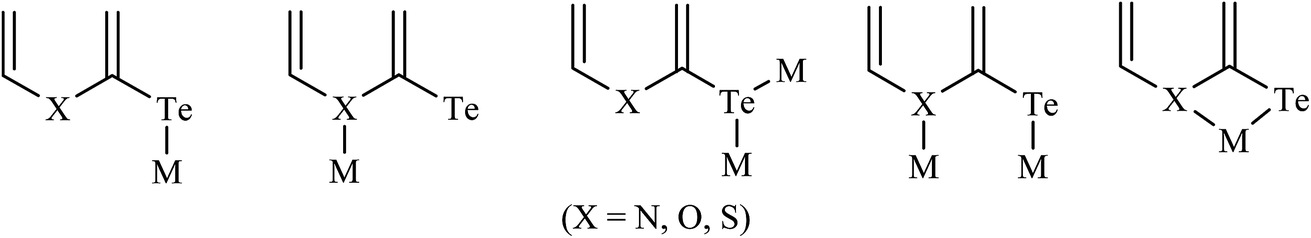
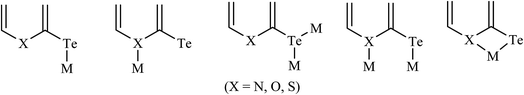
Notable attention7,8 has been drawn by the outcome of the above-mentioned document conclusion on hemilabile ligands. The discussion focused on complexes derived by hemilabile organochalcogens, which have been the subject of considerable interest due to their rich coordination chemistry. In fact, as a concluding remark, the coordination potential of hemilabile ligand systems, especially those that possess α heteroatoms as well as their corresponding anions, referred to as chalcogenolates, is immense.1,9,10 There is indeed a considerable versatility in the various coordination modes of these molecules, which may include monodentate binding through E or through heteroatoms, bridging through a single E, bridging through both E and N, or chelating via the E to N backbone.48,134 All these coordination modes, either alone or in combination, have been observed or assigned on the basis of spectroscopic and/or crystallographic evidence of both homo- and heteroleptic metal complexes (Scheme 21).
 |
| | Scheme 21 Various coordination modes in hemilabile tellurolate ligand systems. | |
5. Metal tellurides
The binuclear complexes [M2(μ-Te)2(dppe)2] (M = Pd, Pt) (Fig. 14) act as a powerful metallophilic ligand to provide a binding site for various transition metals, d10 systems, and lanthanide metal centers. The former complexes can be isolated by the reaction between M2+, dppe, and NaTeH in a N,N-dimethylformamide and acetonitrile mixture.142 The reaction of [Pt(CH3CN)4(NO3)2] with a similar composition to the former complexes led to the formation of pentanuclear complexes [Pt{Pd2(μ-Te)2(dppe)2}2]2+ (ref. 142) (Fig. 15) in which the platinum metal center was coordinated with all four bridging tellurido linkages. Similarly, various trinuclear products have been isolated by the reaction between M2+, P∩P, and NaTeH in N,N-dimethylformamide for more than 20 h stirring (Scheme 22).143 Undoubtedly, strong coordinating solvents, like DMF, CH3CN, may enable the coordination of tellurium to further platinum moieties with ease, resulting in tri- to pentanuclear products.143–146 Morley et al. also isolated the trinuclear product [Pt3{(μ-Te)2(dppe)2}2]2+ by the oxidative addition of [Pt(dppe)2] and vitreous tellurium under refluxing for 5 h.147 The isolation of the trinuclear product via an oxidative addition mode is unprecedented and could be rationalized by the fact that the higher nucleophilicity of tellurium makes it highly susceptible to the nucleophilic attack of chlorinated solvents.
 |
| | Fig. 14 Crystal structure of [Pd2(μ-Te)2(dppe)2]2+ (redrawn from ref. 142). | |
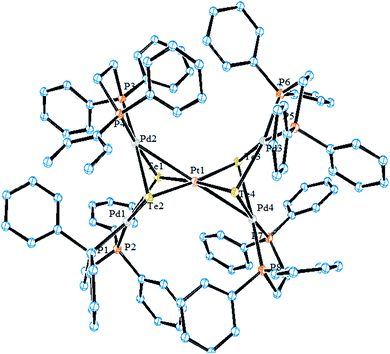 |
| | Fig. 15 Crystal structure of [Pt{Pd2(μ-Te)2(dppe)2}2]2+ (redrawn from ref. 142). | |
 |
| | Scheme 22 Various approached used for the synthesis of high nuclearity complexes. | |
Redox studies of these complexes were studied by cyclic voltammetry.142,144,145 In the case of telluride bridged cluster complexes, it all depends upon the nature of the phosphine and metal center. The cyclic voltammograms of dppe-derived complexes showed a reversible nature, whereas it was irreversible in other cases. The order of chemical reversibility followed the trend: dppe > dppp > dppm.
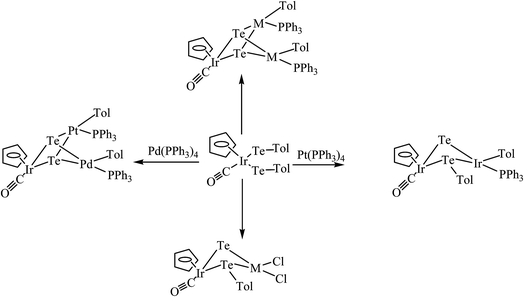
Cluster compounds like [PPh4]2[Pd(Te4)2] were synthesized by refluxing the palladium precursor with tellurium powder.148 This intriguing complex exists in two crystallographic forms: the α and β forms. The triclinic α and orthorhombic β forms were separated using ethylene diamine and ether solvents, respectively.148–150 Similarly, the complex [Li4(en)10][Pd6(μ3-Te)8] was formed by the reaction of [Pb2Te3]2− with [PdCl2(PPh3)2].151 Contrary to the palladium precursors, the platinum analog resulted in the cluster complex [Pt4Te4(Te3)6]4−, which was comprised of a Pt4Te4 cubane core with a distorted octahedral configuration around the platinum atom.152 Recently, several platinum group cluster complexes were isolated by refluxing TeCl4 with the corresponding metal precursors.153–156 The high nucleophilicity of the terminal telluride in mononuclear complexes makes them highly susceptible for further reaction with coordinative unsaturated complexes like the Pt(0) and Pd(0) precursors. Thus, when the oxidative addition product [CpIr(CO)(TeTol)2] was subjected to reaction with two equivalent of [M(PPh3)4] (M = Pd, Pt) (Scheme 23), a compound of the type [CpIr(CO)(μ3-Te)2{MTol(PPh3)}2] was isolated, while the similar reaction with one equivalent of [Pt(PPh3)4] yielded the intermediate product [CpIr(CO)(μ-Te)(μ-TeTol){PtTol(PPh3)}].157 The latter complexes upon reaction with [Pd(PPh3)4] derived the heterometallic complex [CpIr(CO)(μ-Te)2{PdTol(PPh3)}{PtTol(PPh3)}] (Fig. 16), which was formed by the facile inversion of the Pd(0) center in to a Te–Tol bond in the complex [CpIr(CO)(μ-Te)(μ-TeTol){PtTol(PPh3)}].126,157
 |
| | Scheme 23 Synthesis of various heterometallic complexes bridged with telluride systems. | |
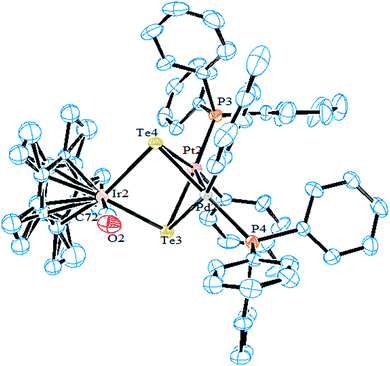 |
| | Fig. 16 Crystal structure of [CpIr(CO)(μ-Te)2{PdTol(PPh3)}{PtTol(PPh3)}] (redrawn from ref. 157). | |
Similarly, [Pt(TeR)2(dppe)] (R = Ph, Fc) upon reaction with a rhenium carbonyl cluster yielded heterometallic tellurolato bridged complexes [(dppe)Pt(μ-TeR)2ReCl(CO)3] (R = Ph, Fc).158,159
6. Applications
6.1 Catalysis
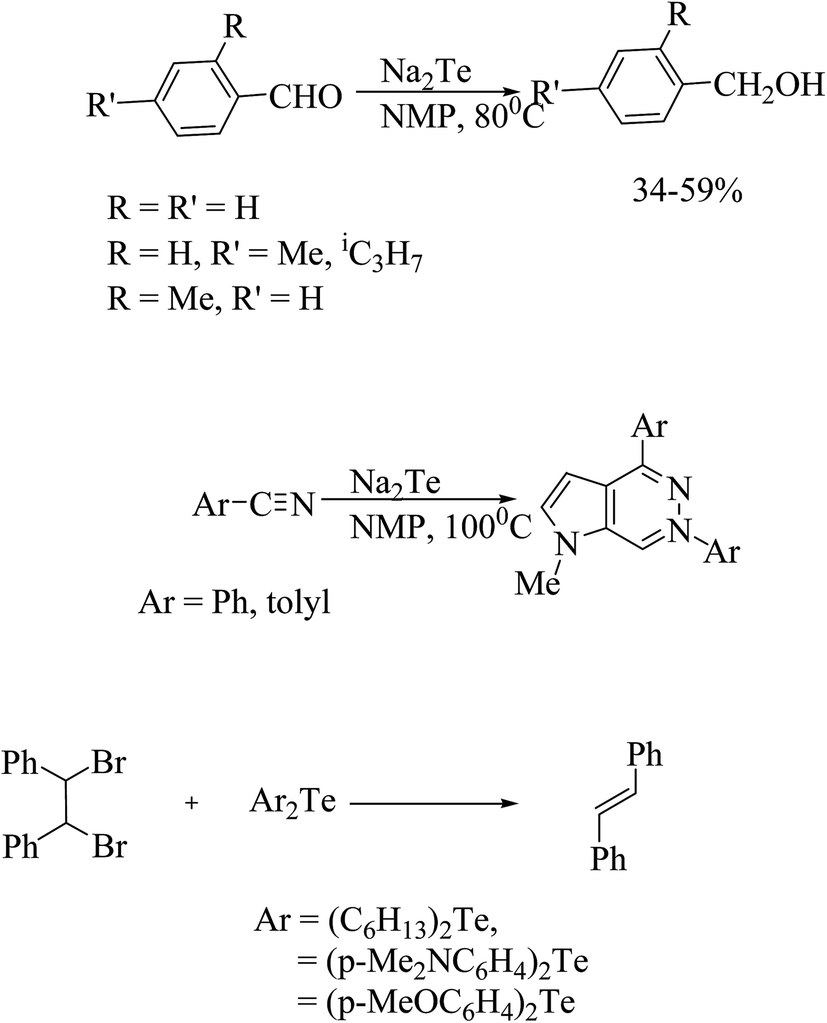
The efficient and selective transformation of various functional groups have taken place with sodium hydrogen telluride (NaHTe) and Na2Te, which have been reported in further applications in recent years.160–164 In particular, the reduction of aromatic aldehydes to alcohols is the most important transformation by Na2Te in NMP (N-methyl-2-pyrorolidone).165,166 Similarly, an attempted reduction of aromatic nitriles serendipitously led to the pharmacologically important product 7-diaza-9H purines in low yields (Scheme 24).166–168
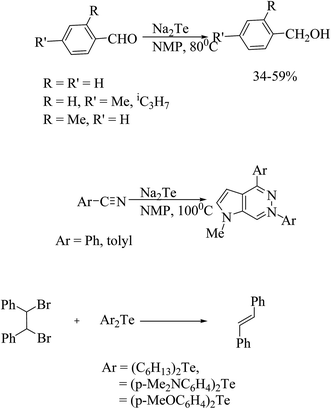 |
| | Scheme 24 Schematic representation of various reactions catalyzed by organotellurium analogs. | |
One more striking example of the conversion by debromination of vic-dibromides to alkenes is that catalyzed by p-methoxyphenyltelluride. Again on reinvestigation, it was found that the more electron-rich diorganoditellurides associated with reducing agents, like glutathione (GSH) or sodium ascorbate, are better debrominating agents than the previous reaction (Scheme 24).169–171
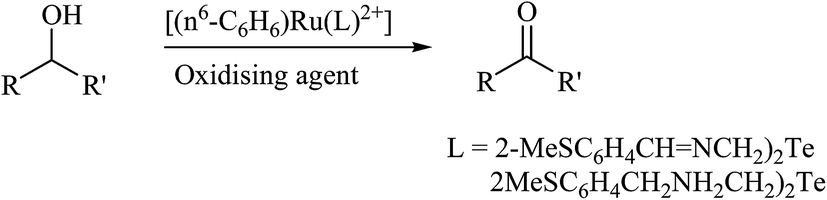
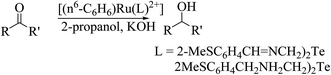
Recently, it was found that ruthenium complexes derived by various telluroethers, like [(η6-C6H6)Ru(L)](PF6) (L = 2-MeSC6H4CHNCH2CH2EC6H4-R; R = H, E = S, Se; 2-MeSC6H4CH2NHCH2CH2EC6H4-R; R = OMe, E = Te),63 have proven potential as an excellent catalyst in the oxidation of primary and secondary alcohols. The TON values for the oxidation of various alcohols, like cyclic, acyclic, and benzylic, ranged from 7.8–9.6 × 104. Strikingly, the complex (η6-C6H6)[Ru(2-MeSC6H4CH2NHCH2CH2TeC6H4–OMe)](PF6) appeared to be the most efficient in this process with the highest % conversion (98%). A comparative study of all the ruthenium complexes derived by various chalcogen analogs showed that the catalytic efficiency varies in the order of Te > Se > S. This fact can be rationalized by the softer ligand making it easier to form the intermediate oxy species RuO, which plays a pivotal role in any oxidation reaction (Scheme 25).
 |
| | Scheme 25 Oxidation of alcohols through ruthenium tellurolate complexes. | |
The second important class of reaction, i.e., transfer of hydrogen reaction, is also catalyzed by the complexes [(η6-C6H6)Ru(L)](PF6) (L = 2-MeSC6H4CHNCH2CH2EC6H4-R; R = H, E = S, Se; 2-MeSC6H4CH2NHCH2CH2EC6H4-R; R = OMe, E = Te) (Scheme 26). In the case of acetophenone, the conversion efficiency was up to 98%, while in various other aliphatic secondary ketones, it ranged up to 90%. Similarly as an oxidation reaction, the catalytic efficiency of these reactions also varies in the same order. An earlier well-established catalyst for this reaction was Ru(II) complexes of 2-(aminomethyl)pyridine phosphine with a TOF of 10−5 h−1 and up to 97% conversion in 2-propanol using NaOH as a base. Comparatively, a half sandwich ruthenium complex of the above-mentioned composition derived with various telluroethers of pyrrolidine, morpholine, and benzotriazole moieties62,172,173 also showed a similar efficiency rate with a short reaction time.
 |
| | Scheme 26 Transfer of hydrogen reaction catalyzed by ruthenium tellurolate complexes. | |
6.2 Suzuki and Heck reaction
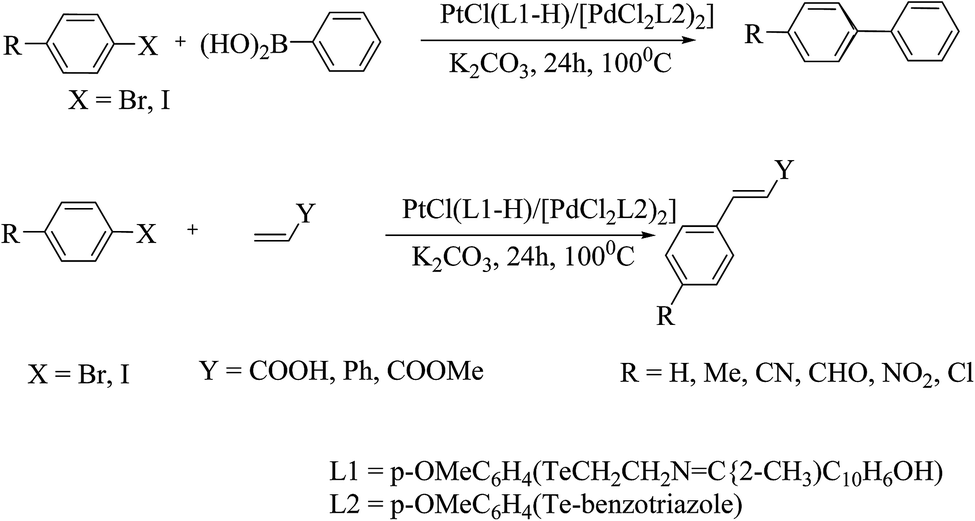
Due to the air and moisture sensitivity of phosphorus-derived complexes, there is increasing interest in phosphine-free ligands174–177 for the Suzuki–Miyaura reaction. With this prospect, palladium chalcogenolate178 complexes are considered as existing substituents for C–C coupling reactions. Various coupling reactions are primarily catalyzed by palladium selenated and tellurated compounds. An early study performed by Prof. A. K. Singh et al. showed that palladium tellurolate complexes are as efficient as their selenium analogs.179 The conversion was found to be up to 85%, particularly for activated 1-bromo-4-nitrobenzene (Scheme 27).
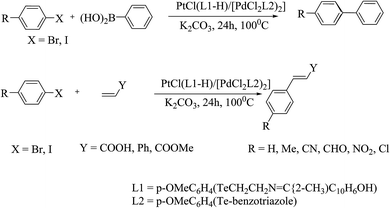 |
| | Scheme 27 Representation of palladium–platinum tellurolate catalyzed Suzuki and Heck reactions. | |
It is well documented that Pd–Se bonded compounds have promise for the Heck reaction.178 Within this continuation, a Schiff base-derived telluroether palladium complex showed good selectivity for the isolation of a trans-alkene product. The catalytic activity depended on the nature of the halide, while the electron-withdrawing groups on the aryl ring increased the reaction rate. For aryl bromides, a very small amount of complex (0.001 mmole) was sufficient to catalyze the Heck reaction (Scheme 27).179
6.3 Material science
Binary systems of Pt and Te (as a combination of metal and semiconductor) are important precursors for their application in the field of sensors and magnetic memories.180–182 In particular, the composites of PtTe2 are known to enhance the Seebeck coefficient of PbTe bulk particles.183 Similarly, multicomponent rod-shaped mixed composites FePt–PtTe2 have exhibited high coercivity (Hc > 500).184 Pt3Te4 also has shown a catalytic ability in the transformation of nitrophenol to aminophenol.185 Platinum group metal chalcogen materials are also prominently used for low resistance ohmic contacts. These thermodynamically stable contacts are very crucial for device durability. At the interface of Pt/CdTe diffusion couples, a non planner reaction layer of intermetallic CdPt and Pt–Te is formed.186
Platinum group metal chalcogenides comprise various binary and ternary chalcogenide materials. Undoubtedly, the preparatory methods dealing with platinum group metal sulfide and selenide materials applying single source molecular precursor's methods are greater in number with respect to their tellurium precursors.187 The complex [PdCl{Te(3-MeC5H3N)}(PR3)] (R = propyl, Ph) upon heating in a furnace at 340 °C under an argon atmosphere yielded a molecular precursor of the binary composition PdTe.130 The latter compositions supported on carbon have relevance in various catalytic reactions used at an industrial scale.188 However, complexes like [PdCl2(4-MeOC6H4TeCH2CH2N(CH2CH2)2O)] also result in the formation of Pd–Te nanoparticles of 5 nm diameter.189 A similar composition has also been documented by heating the hexanuclear complex at >250 °C.138 Thiolate- and selenolate-derived palladium complexes with the composition [Pd2(μ-ER)2(η3-C4H7)] (E = S, Se) upon refluxing in xylene solution afforded Pd4E (E = S, Se); however, the tellurolate analogs resulted in the formation of Pd3Te2 composites at room temperature.190 Recently, a composition of Pd7Te3 has been isolated upon refluxing the complex [PdCl2(TeMes2)2] in xylene solution.93
6.4 Biological importance
Upon evaluating the biological activity of tellurium-derived ruthenium complexes, it has been found that the complexes are quite potent as anticancer agents. Recently, a biological study of the complexes [(η6-p-Me C6H4Pri)2Ru2(μ-TeC6H5)3]PF6 and [(η5-C5H5)2Ru2(μ-TeC6H5)3]PF6 (ref. 96) showed their good biological activity on normal and human cancer cell lines. The former complexes are even cyto-toxic in nature, which revealed their strong selectivity to cancer cells compared to normal ones. These promising outcomes of ruthenium complexes as anticancer agents deserves more exploration toward various other biological activities.
7. Conclusions
The versatile reactivity of organotellurolate ligands is quite evident from the ongoing discussions and reports in the literature. Their remarkable reactivity is derived due to presence of soft (tellurium) and hard donor atoms (N, O, S) in various ligands derived from organic moieties, like thinyl, pyridyl, furan, and pyrimidyl. In the meantime, one in particular cannot be declined: that the notable reactivity may also arise due to the comparable bond energies of Te–Te and Te–C bonds, which facilitates a competitive cleavage among them. Furthermore, among all the other chalcogens, the easy availability of lone pairs and high polarizability makes them great building block synthons for various heterometallic and high nuclearity metal complexes. It is hoped that this perspective will support further enthusiasm in this field and provide momentum for further research in this field to help establish their potential application in various fields.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
One of the authors (RSC) is grateful to DST for the financial support under the DST young scientist scheme YSS/2014/000797. We thank Dr V. K. Jain for his encouragement with this work and for his valuable suggestions for the preparation of this article.
References
-
(a) P. D. Akrivos, H. J. Katsikis and A. Koumoutsi, Coord. Chem. Rev., 1997, 167, 95 CrossRef CAS;
(b) P. D. Akrivos, Coord. Chem. Rev., 2001, 213, 181 CrossRef CAS.
- P. J. Blower and J. R. Dilworth, Coord. Chem. Rev., 1987, 76, 121 CrossRef CAS.
- J. R. Dilworth and N. Wheatley, Coord. Chem. Rev., 2000, 199, 89 CrossRef CAS.
- W. Levason, S. D. Orchid and G. Reid, Coord. Chem. Rev., 2002, 225, 159 CrossRef CAS.
-
(a) S. Dey, V. K. Jain, A. Knoedler, A. Klein, W. Kaim and S. Zalis, Inorg. Chem., 2002, 41, 2864 CrossRef CAS PubMed;
(b) S. Dey, L. B. Kumbhare, V. K. Jain, T. Schurr, W. Kaim, A. Klein and F. Belaj, Eur. J. Inorg. Chem., 2004, 4510 CrossRef CAS.
- A. K. Singh and S. Sharma, Coord. Chem. Rev., 2000, 209, 49 CrossRef CAS.
-
(a) G. Kedarnath and V. K. Jain, Coord. Chem. Rev., 2013, 257, 1409 CrossRef CAS;
(b) R. S. Chauhan, G. Kedarnath, A. Wadawale, A. Munoz-Castro, R. Arratia-Perez, V. K. Jain and W. Kaim, Inorg. Chem., 2010, 49, 4179 CrossRef CAS PubMed.
-
(a) R. S. Chauhan, G. Kedarnath, A. Wadawale, A. L. Rheingold, A. Munoz-Castro, R. Arratia-Perez and V. K. Jain, Organometallics, 2012, 31, 1743 CrossRef CAS;
(b) R. S. Chauhan, G. Wadawale, A. M. Z. Slawin and V. K. Jain, Dalton Trans., 2013, 42, 259 RSC.
- T. Wirth, Organoselenium Chemistry: Modern Developments in Organic Synthesis, in Topics in Current Chemistry, Springer-Verlag, Berlin, 2000, vol. 208 Search PubMed.
- S. Narayan, V. K. Jain and B. Varghese, J. Chem. Soc., Dalton Trans., 1998, 2359 RSC.
-
(a) D. Dakternieks, R. D. Giacomo, R. W. Gable and B. F. Hoskins, J. Am. Chem. Soc., 1988, 110, 6541 CrossRef CAS;
(b) D. Dakternieks, R. D. Giacomo, R. W. Gable and B. F. Hoskins, J. Organomet. Chem., 1989, 377, 43 CrossRef.
-
(a) A. K. Singh, V. Srivastava, J. K. Basumatary, T. P. Singh and A. K. Saxena, Phosphorus, Sulfur Silicon Relat. Elem., 1994, 89, 31 CrossRef CAS;
(b) A. K. Singh, V. Srivastava, J. K. Basumatary, T. P. Singh and A. K. Saxena, Phosphorus, Sulfur Silicon Relat. Elem., 1993, 85, 175 CrossRef.
- J. E. Drake, R. J. Drake, L. N. Khasrou and R. Ratnani, Inorg. Chem., 1996, 35, 2831 CrossRef CAS.
- V. P. Ananikov and I. P. Beletskaya, Org. Biomol. Chem., 2004, 2, 284 CAS.
-
(a) V. P. Ananikov, M. A. Kabeshov, I. P. Beletskaya, V. N. Khrustalev and M. Y. Antipin, Organometallics, 2005, 24, 1275 CrossRef CAS;
(b) I. P. Beletskaya and V. P. Ananikov, Eur. J. Org. Chem., 2007, 3431 CrossRef CAS.
- L. B. Han, N. Choi and M. Tanaka, J. Am. Chem. Soc., 1997, 119, 1795 CrossRef CAS.
-
(a) N. Nakata, R. Uchiumi, T. Yoshino, T. Ikeda, H. Kamon and A. Ishii, Organometallics, 2009, 28, 1981 CrossRef CAS;
(b) A. Ishii, Y. Yamaguchi and N. Nakata, Dalton Trans., 2010, 39, 6181 RSC;
(c) A. Ishii, N. Nakata, R. Uchiumi and K. Murakami, Angew. Chem., Int. Ed., 2008, 47, 2661 CrossRef CAS PubMed.
- Comprehensive Organometallic Chemistry – II, ed. R. J. Puddephatt, E. W. Abel, F. G. A. Stone and G. Wilkinson, Pergamon, Oxford, 1st edn, 1995, vol. 9, p. 1 Search PubMed.
- Catalytic Heterofunctionalization, ed. A. Togni and H. Grützmacher, Wiley-VCH: Weinheim, 2001, p. 1 Search PubMed.
- M. E. Cucciolito, V. De Felice, G. Roviello and F. Ruffo, Eur. J. Inorg. Chem., 2011, 457 CrossRef CAS.
- R. Oilunkaniemi, R. S. Laitinen and M. J. Ahlgren, J. Organomet. Chem., 2001, 623, 168 CrossRef CAS.
- L. B. Han, N. Choi and M. Tanaka, J. Am. Chem. Soc., 1997, 119, 1795 CrossRef CAS.
- A. Khanna, B. L. Khandelwal, A. K. Saxena and T. P. Singh, Polyhedron, 1995, 14, 2705 CrossRef CAS.
- A. K. Singh, M. Kadarkaraisamy, M. Mishra, J. Sooriyakumar, J. E. Drake, M. B. Hursthouse, M. E. Light and J. P. Jasinski, Inorg. Chim. Acta, 2008, 361, 1426–1436 CrossRef.
- G. Singh, A. K. Singh, P. Sharma, J. E. Drake, M. B. Hursthous and M. E. Light, J. Organomet. Chem., 2003, 688, 20–26 CrossRef CAS.
- P. R. Kumar, A. K. Singh, R. J. Butcher, P. S. Shrama and R. A. Toscano, Eur. J. Inorg. Chem., 2004, 1107–1114 CrossRef.
- P. R. Kumar, S. Upreti and A. K. Singh, Inorg. Chim. Acta, 2008, 361, 1426–1436 CrossRef.
- A. Kumar, M. Agarwal, A. K. Singh and R. J. Butcher, Inorg. Chim. Acta, 2009, 362, 3208 CrossRef CAS; A. Kumar, M. Agarwal and A. K. Singh, J. Organomet. Chem., 2008, 693, 3533 CrossRef.
- Y. Takaguchi, E. Horn and N. Furukawa, Organometallics, 1996, 15, 5112 CrossRef CAS.
- W. Levason, G. Reid and V. A. Tolhurst, J. Chem. Soc., Dalton Trans., 1998, 3411 RSC.
- W. Levason, S. D. Orchard and G. Reid, Chem. Commun., 2001, 427 RSC.
- S. C. Menon, H. B. Singh, R. P. Patel and S. K. Kulshreshtha, J. Chem. Soc., Dalton Trans., 1996, 1203 RSC.
- E. G. Hope, T. Kemmit and W. Levason, Organometallics, 1988, 7, 78 CrossRef CAS.
- J. Conolly, A. R. J. Genge, W. Levason, S. D. Orchard, S. J. A. Pope and G. Reid, J. Chem. Soc., Dalton Trans., 1999, 2343 RSC.
- A. J. Barton, W. Levason, G. Reid and A. J. Ward, Organometallics, 2001, 20, 3644 CrossRef CAS.
- K. K. Bhasin, V. Arora, T. Klapotke and M. J. Crawford, Eur. J. Inorg. Chem., 2004, 4781 CrossRef CAS.
- K. K. Bhasin, N. Singh, R. Dhiman, G. Ram, Shivani, S. K. Mehta and R. J. Butcher, J. Organomet. Chem., 2006, 691, 628 CrossRef.
- K. K. Bhasin, Rishu, S. Singh, H. Kumar and S. K. Mehta, J. Organomet. Chem., 2010, 695, 648 CrossRef CAS.
- A. Rodriguez, J. Romero, J. A. Garcia-Vazuez, M. L. Duran, A. Sousa Pedrares, A. Sousa and J. Zubieta, Inorg. Chim. Acta, 1999, 284, 133 CrossRef CAS.
- K. K. Bhasin, P. Venugopalan and J. Singh, Phosphorus, Sulfur Silicon Relat. Elem., 2002, 177, 2579 CrossRef CAS.
- L. Engman and M. P. Cava, Organometallics, 1982, 1, 470 CrossRef CAS.
- F. P. Colonna, G. Distfano, V. Galasso, K. J. Irgolic, G. C. Pappalardo and L. Pope, J. Chem. Soc., Perkin Trans. 2, 1981, 281 RSC.
- L. A. Summers, J. Heterocycl. Chem., 1987, 24, 533 CrossRef CAS.
- L. Syper and J. Mlochowski, Tetrahedron, 1988, 44, 6119 CrossRef CAS.
- K. K. Bhasin, N. Singh, V. Trehan, P. K. Jain and J. Singh, Phosphorus, Sulfur Silicon Relat. Elem., 2003, 178, 753 CrossRef CAS.
- K. K. Bhasin, E. Arora, K. Kaur, S. K. Kang, M. Gobel, T. M. Klapoetke and S. K. Mehta, Tetrahedron, 2009, 65, 247 CrossRef CAS.
- K. K. Bhasin, E. Arora, S. K. Mehta and T. M. Klapoetke, J. Organomet. Chem., 2011, 696, 835 CrossRef CAS.
- R. S. Chauhan, R. K. Sahrma, G. Kedarnath, D. B. Cordes, A. M. Z. Slawin and V. K. Jain, J. Organomet. Chem., 2012, 717, 180 CrossRef CAS.
- K. K. Bhasin and J. Singh, J. Organomet. Chem., 2002, 658, 71 CrossRef CAS.
- K. K. Bhasin and V. Arora, Appl. Organomet. Chem., 2004, 18, 359 CrossRef CAS.
- K. K. Bhasin, V. Arora, S. K. Sharma and P. Venugopalan, Appl. Organomet. Chem., 2005, 19, 161 CrossRef CAS.
- R. Oilunkaniemi, R. S. Laitenen and M. Ahlgren, Inorg. Chem. Commun., 2000, 3, 8 CrossRef CAS.
- A. K. Singh, M. Misra and M. Kadarkaraisamy, Phosphorus, Sulfur Silicon Relat. Elem., 1998, 143, 101 CrossRef CAS.
- W. Levason, B. Patel, G. Reid and A. J. Ward, J. Organomet. Chem., 2001, 619, 218 CrossRef CAS.
- W. Levason, S. D. Orchard, G. Reid and V.-A. Tolhurst, J. Chem. Soc., Dalton Trans., 1999, 2071–2076 RSC.
- W. Levason, S. D. Orchard and G. Reid, J. Chem. Soc., Dalton Trans., 2000, 4551 Search PubMed.
- W. Levason, S. D. Orchard and G. Reid, J. Chem. Soc., Chem. Commun., 1999, 1071 RSC.
- W. Levason, S. D. Orchard and G. Reid, Inorg. Chem., 2000, 39, 3853 CrossRef CAS PubMed.
- A. K. Singh, M. Kadarkaraisamy, J. E. Drake and R. J. Butcher, Inorg. Chim. Acta, 2000, 304, 45 CrossRef CAS.
- A. K. Singh, J. Sooriyakumar, J. E. Drake, M. B. Hursthouse and M. E. Light, J. Organomet. Chem., 2000, 631, 244 CrossRef.
- S. Bali, A. K. Singh, J. E. Drake, M. B. Hursthouse and M. E. Light, J. Organomet. Chem., 2006, 3788 CrossRef CAS.
- S. Bali, A. K. Singh, P. Sharma, R. A. Toscaro, J. E. Drake, M. B. Hursthouse and M. E. Light, J. Organomet. Chem., 2004, 689, 2346 CrossRef CAS.
- D. Das, P. Singh and A. K. Singh, J. Organomet. Chem., 2010, 695, 955 CrossRef CAS.
- K. N. Sharma, H. Joshi, A. K. Sharma, O. Prakash and A. K. Singh, Chem. Commun., 2013, 49, 9344 RSC.
- S. Kumar, G. K. Rao, A. Kumar, M. P. Singh and A. K. Singh, Dalton Trans., 2013, 42, 16939 RSC.
- T. P. Lin and F. P. Gabbai, J. Am. Chem. Soc., 2012, 134, 12230 CrossRef CAS PubMed.
- P. Singh and A. K. Singh, Organometallics, 2010, 29, 6433 CrossRef CAS.
- A. J. Barton, W. Levason, G. Reid and V.-A. Tolhurst, Polyhedron, 2000, 19, 235 CrossRef CAS.
- M. Devery, R. S. Dickson, B. W. Skelton and A. H. White, Organometallics, 1999, 18, 5292 CrossRef CAS.
- L. Vigo, M. J. Poropudas, P. Salin, R. Oilunkaniemi and R. S. Laitinen, J. Organomet. Chem., 2009, 694, 2053 CrossRef CAS.
- P. Singh, D. Das, M. Singh and A. K. Singh, Inorg. Chem. Commun., 2010, 13, 988 CrossRef CAS.
- T. Kemmitt, W. Levason, R. D. Oldroyd and M. Webster, Polyhedron, 1992, 11, 2165 CrossRef CAS.
- A. K. Singh, M. Kadarkaraisamy, S. Husebye and K. W. Tornroos, J. Chem. Res., 2000, 64 CrossRef CAS.
- A. Z. Al-Rubaie, A. Al-Marzook and S. A. N. Al-Jadaan, Recl. Trav. Chim. Pays-Bas, 1996, 115, 427 CrossRef CAS.
- A. F. Chiffey, J. Evans, W. Levason and M. Webster, J. Chem. Soc., Dalton Trans., 1994, 2835 RSC.
- B. L. Khandelwal, A. Khalid, A. K. Singh, T. P. Singh and S. Karthikeyan, J. Organomet. Chem., 1996, 507, 65 CrossRef CAS.
- J. E. Drake, J. Yang, A. Khalid, V. Srivastava and A. K. Singh, Inorg. Chim. Acta, 1997, 254, 57 CrossRef CAS.
- H. B. Singh, A. Regini, J. P. Jasinski, E. S. Paight and R. J. Butcher, J. Organomet. Chem., 1994, 466, 283 CrossRef CAS.
- A. K. Singh and V. Singh, Phosphorus, Sulfur Silicon Relat. Elem., 1993, 80, 95 CrossRef CAS.
- R. Batheja, S. Katiyar, V. Singh and A. K. Singh, Polyhedron, 1994, 13, 777 CrossRef CAS.
- R. Batheja and A. K. Singh, Polyhedron, 1997, 16, 2509 CrossRef CAS.
- R. Batheja and A. K. Singh, Polyhedron, 1997, 16, 4337 CrossRef CAS.
- A. Khanna and B. L. Khandelwal, Indian J. Chem., 1996, 35A, 236 CAS.
- J. E. Drake, J. H. E. Bailey, A. K. Singh and V. Srivastava, Acta Crystallogr., 1993,(49), 684 Search PubMed; A. K. Singh, V. Srivastava and B. L. Khandelwal, Polyhedron, 1990, 9, 851–857 CrossRef CAS.
- A. Khalid, B. L. Khandelwal, A. K. Singh, T. P. Singh and B. Padmanabhan, J. Coord. Chem., 1994, 31, 19 CrossRef CAS.
- A. Khaled and A. K. Singh, Polyhedron, 1996, 16, 33 CrossRef.
- B. L. Khandelwal, A. Khalid and A. K. Singh, Indian J. Chem., 1996, 35A, 438 CAS.
- A. Regini, R. Kaur, N. Sudha, S. C. Menon and H. B. Singh, Proc. - Indian Acad. Sci., Math. Sci., 1995, 107, 431 CAS.
- R. Batheja, S. K. Dhingra and A. K. Singh, J. Organomet. Chem., 1995, 487, 173 CrossRef CAS.
- S. C. Menon, H. B. Singh, R. P. Patel and S. K. Kulshreshtha, J. Chem. Soc., Dalton Trans., 1996, 1203 RSC.
- S. C. Menon, A. Panda, H. B. Singh and R. J. Butcher, J. Chem. Soc., Chem. Commun., 2000, 143 RSC.
- A. Panda, S. S. Zade, H. B. Singh and R. J. Butcher, Eur. J. Inorg. Chem., 2006, 172 CrossRef.
- S. Kolay, M. Kumar, A. Wadawale, D. Das and V. K. Jain, Dalton Trans., 2014, 43, 16056 RSC.
- S. Kolay, M. Kumar, A. Wadawale, D. Das and V. K. Jain, J. Organomet. Chem., 2015, 794, 40 CrossRef CAS.
- S. Kolay, A. Wadawale, S. Nigam, M. Kumar, C. Majumder, D. Das and V. K. Jain, Inorg. Chem., 2015, 54, 11741 CrossRef CAS PubMed.
- T. Chakraborty, S. Sharma, H. B. Singh and R. J. Butcher, Organometallics, 2011, 30, 2525 CrossRef CAS.
- B. K. Das and M. G. Kanatzidis, Inorg. Chem., 1995, 34, 1011 CrossRef CAS.
- E. D. Schermer and W. H. Baddley, J. Organomet. Chem., 1971, 30, 67 CrossRef CAS.
- R. Nagarajaprakash, B. Ramakrishna, K. Mahesh, S. M. Mobin and B. Manimaran, Organometallics, 2013, 32, 7292 CrossRef CAS.
- N. Begum, M. I. Hyder, S. Islam, S. E. Kabir, K. Kundu and K. M. Abdul Malik, Indian J. Chem., 2005, 44A, 516 CAS.
- K. A. Azam, M. Bhowmick, G. M. G. Hossain, S. E. Kabir, K. Kundu, K. M. A. Malik and S. Pervin, J. Chem. Crystallogr., 2001, 31, 63 CrossRef CAS.
- N. Begun, M. I. Hyder, M. R. Hassan, S. E. Kabir, D. W. Bennet, D. T. Haworth, T. A. Sidhiquee, D. Rokhsana, A. Sharmin and E. Rosenberg, Organometallics, 2008, 27, 1550 CrossRef.
- S. Ghosh, G. Hogarth, S. E. Kabir, E. Nordlander, L. Salassa and D. A. Tocher, J. Organomet. Chem., 2011, 696, 1982 CrossRef CAS.
- Y. Nishibayashi, H. Imajima, G. Onodera, Y. Inade, M. Hidai and S. Uemura, Organometallics, 2004, 23, 5100 CrossRef CAS.
- A. A. Pasynskii, S. S. Shapovalov, I. V. Skabitsky, O. G. Tikhonova and T. A. Krishtop, Russ. J. Coord. Chem., 2015, 41, 234 Search PubMed.
- C. E. Rao, K. Yuvaraj and S. Ghosh, J. Organomet. Chem., 2015, 776, 123 CrossRef CAS.
- P. Mathur, I. J. Mavunkal and A. L. Rheingold, J. Chem. Soc., Chem. Commun., 1989, 382 RSC.
- P. Mathur and I. J. Mavunkal, J. Organomet. Chem., 1988, 350, 251 CrossRef CAS.
- S. Chatterjee, S. K. Patel and S. M. Mobin, J. Organomet. Chem., 2011, 696, 1782 CrossRef CAS.
- P. Mathur, P. Payra, S. Ghosh, M. M. Hossain, C. V. V. Satyanarayana, F. O. Chicote and R. K. Chadha, J. Organomet. Chem., 2000, 606, 176 CrossRef CAS.
- P. Mathur, P. Sekar, C. V. V. Satyanarayana and M. F. Mahon, J. Chem. Soc., Dalton Trans., 1996, 2173 RSC.
- P. Mathur, I. J. Mavunkal, V. Rugmini and M. F. Mahon, Inorg. Chem., 1990, 29, 4838 CrossRef CAS.
- P. Mathur, I. J. Mavunkal and V. Rugmini, Inorg. Chem., 1989, 28, 3616 CrossRef CAS.
- P. Mathur, B. H. S. Thimappa and A. L. Rheingold, Inorg. Chem., 1990, 29, 4658 CrossRef CAS.
- J. Zhang and W. K. Long, J. Chem. Soc., Dalton Trans., 2000, 1249–1253 RSC.
- S. E. Kabir, M. S. Saha, D. Tocher, G. M. G. Hossain and E. Rosenberg, J. Organomet. Chem., 2006, 691, 97 CrossRef CAS; E. D. Schermer and W. H. Baddley, J. Organomet. Chem., 1971, 30, 67 CrossRef.
- P. Srinivasan, M. E. Tai and W. K. Leong, J. Organomet. Chem., 2006, 691, 941–951 CrossRef CAS.
- S. Nagao, H. Seino, M. Hiddai and Y. Mizobe, Dalton Trans., 2005, 3166–3172 RSC.
- M. P. Devery, R. S. Dickson, G. D. Fallon, G. A. Koutsantonis, B. W. Skelton and A. H. White, Z. Anorg. Allg. Chem., 2008, 634, 675 CrossRef CAS.
- M. P. Devery, R. S. Dickson, B. W. Skelton and A. H. White, Organometallics, 1999, 18, 5292 CrossRef CAS.
- A. Nordheider, K. S. A. Arachchige, A. M. Z. Slawin, J. D. Woollins and T. Chivers, Dalton Trans., 2015, 44, 8781 RSC.
- R. D. Pergola, A. Ceriotti, A. F. de Biani, L. Garlaschelli, M. Manassero, R. Piacentini, M. Sansoni and P. Zanello, Organometallics, 1998, 17, 802 CrossRef.
- Y.-K. Sau, X. Yi Yi, K.-W. Chan, C.-S. Lai, I. D. Williams and W.-H. Leung, J. Organomet. Chem., 2010, 695, 1399 CrossRef CAS.
- R. D. Adams, J. W. Long and J. L. Perrin, J. Am. Chem. Soc., 1998, 120, 1922 CrossRef CAS.
- J. M. Gonzales, D. G. Musaev and K. Morokuma, Organometallics, 2005, 24, 4908 CrossRef CAS.
- T. Nakagawa, H. Seino and Y. Mizobe, J. Organomet. Chem., 2010, 695, 137 CrossRef CAS.
- L. M. Venanzi, Chem. Ber., 1968, 4, 162 CAS.
- E. Shustorovich and P. A. Dobosh, J. Am. Chem. Soc., 1979, 101, 4090 CrossRef CAS.
- D. M. Giolando, T. B. Rauchfuss and A. L. Rheingold, Inorg. Chem., 1987, 26, 1636 CrossRef CAS.
- S. Dey, V. K. Jain, J. Singh, V. Trehan, K. K. Bhasin and B. Varghese, Eur. J. Inorg. Chem., 2003, 744 CrossRef CAS.
- S. Dey, K. Vivekananda, A. P. Wadawale, V. K. Jain and N. Bhuvanesh, ChemistrySelect, 2017, 2, 5073 CrossRef CAS.
- L. Y. Chia and W. R. McWhinnie, J. Organomet. Chem., 1978, 148, 165 CrossRef CAS.
-
(a) M. Risto, E. M. Jahr, M. S. Hannu-Kurre, R. Oilunkaniemi and R. S. Laitinen, J. Organomet. Chem., 2007, 692, 2193 CrossRef CAS;
(b) R. Oilunkaniemi, R. S. Laitinen and M. Ahlgren, J. Organomet. Chem., 2000, 595, 232 CrossRef CAS.
- N. V. Kirji, W. Tyrra, I. Pantenburg, D. Naumann, H. Scherer and Y. L. Yagupolskii, J. Organomet. Chem., 2006, 691, 2679 CrossRef.
- S. Kato, O. Niyomura, Y. Kawahara and T. Kanda, J. Chem. Soc., Dalton Trans., 1999, 1677 RSC.
- A. Wagner, L. Vigo, R. Oilukaniemi, R. S. Laitinen and W. Weigand, Dalton Trans., 2008, 3535 RSC; M. M. Karjalainen, T. Wiegand, J. M. Rautiainen, A. Wagner, H. Görls, W. Weigand, R. Oilunkaniemi and R. S. Laitinen, J. Organomet. Chem., 2017, 836, 17 CrossRef CAS.
- A. Singhal, V. K. Jain, B. Varghese and E. R. T. Tiekink, Inorg. Chim. Acta, 1999, 285, 190 CrossRef CAS.
- J. G. Brennam, T. Siegrist, S. M. Stuczynski and M. L. Steigerwald, J. Am. Chem. Soc., 1990, 112, 9233 CrossRef.
- Y. Yamaguchi, N. Nakata and A. Ishii, Eur. J. Inorg. Chem., 2013, 5233 CrossRef CAS.
- R. S. Chauhan, G. Kedarnath, A. Wadawale, J. A. Golen, A. L. Rheingold and V. K. Jain, J. Organomet. Chem., 2013, 737, 40 CrossRef CAS.
- R. S. Chauhan, G. Kedarnath, J. A. Golen, A. L. Rheingold and V. K. Jain, Indian J. Chem., 2013, 52 A, 42 Search PubMed.
- C. Nishitani, T. Shizuka, K. Matsumoto, S. Okeya and H. Kioto, Inorg. Chem. Commun., 1998, 1, 325 CrossRef CAS.
- K. Matsumoto, K. Takahashi, M. Ikuzawa and H. Kimoto, Inorg. Chim. Acta, 1998, 281, 174 CrossRef CAS.
- A. L. Ma, J. B. Thoden and L. B. Dahl, J. Chem. Soc., Chem. Commun., 1992, 1516 RSC.
- K. Matsumoto, M. Ikuzawa, M. Kamibuko and S. Ooi, Inorg. Chim. Acta, 1994, 129 CrossRef CAS.
- K. Matsumoto, C. Nishitani, M. Tadokoro and S. Okey, J. Chem. Soc., Dalton Trans., 1996, 3335 RSC.
- M. R. Lewtas, C. P. Morley and M. Divaira, Polyhedron, 2000, 19, 751 CrossRef CAS.
- R. D. Adams, T. A. Wolfe, B. W. Eichhorn and R. C. Haushalter, Polyhedron, 1989, 8, 701 CrossRef CAS.
- H. Wolkers, K. Dehnicke, D. Fenske, J. Magull, C. Oliver and S. Weigerber, Z. Naturforsch., 1994, 64B, 4004 Search PubMed.
- M. G. Kanatzidis, Comments Inorg. Chem., 1990, 10, 161 (Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 1193) CrossRef CAS.
- G. Thiele, G. You and S. Dehnen, Inorg. Chem., 2015, 54, 2491 CrossRef CAS PubMed.
- J. M. McConnachie, J. Bollinger and J. A. Ibers, Inorg. Chem., 1993, 32, 3923 CrossRef CAS.
- A. Gunther, A. Isaeva, A. I. Baranov and M. Ruck, Chem.–Eur. J., 2011, 17, 6382 CrossRef PubMed.
- A. Gunther, M. Heise, F. R. Wagner and M. Ruck, Angew. Chem., Int. Ed., 2011, 50, 9987 CrossRef PubMed.
- A. Gunther, A. Isaeva and M. Ruck, Z. Anorg. Allg. Chem., 2013, 639, 254 CrossRef.
- A. Gunther and M. Ruck, Z. Anorg. Allg. Chem., 2012, 638, 317 CrossRef.
- T. Nakagawa, H. Seino, S. Nagao and Y. Mizobe, Angew. Chem., Int. Ed., 2006, 45, 7758 CrossRef CAS PubMed.
- A. A. Y. Pasynskii, V. Torubaev, A. Palova, I. V. Skanitsky, G. Denisov and V. Grinberg, J. Cluster Sci., 2015, 26, 247 CrossRef CAS.
- A. A. Pasynskii, Y. V. Torubaev, A. Pavlova, S. S. Shapovalov, I. V. Skabitskii and G. Denisov, Russ. J. Coord. Chem., 2014, 40, 611 CrossRef CAS.
- N. Petragnani and J. V. Comasseto, Synthesis, 1986, 1 CrossRef CAS.
- N. Petragnani and J. V. Comasseto, Synthesis, 1991, 793, 897 CrossRef.
- J. V. Comasseto, L. W. Ling, N. Petragnani and H. A. Stefani, Synthesis, 1997, 373 CrossRef CAS.
- N. Petragnani, in Tellurium in Organic Synthesis—Best Synthetic Methods, ed. O. Meth-Cohn, Academic, 1994 Search PubMed.
- N. Petragnani and A. McKillop, Tellurium, in Comprehensive Organometallic Chemistry, Pergamon, NewYork, 1995, vol. 11 Search PubMed.
- H. Suzuki and T. Nakamura, J. Org. Chem., 1993, 58, 241 CrossRef CAS.
- N. Petragani and H. A. Stefani, Tetrahedron, 2005, 61, 1613 CrossRef.
- G. Blay, L. Cardona, B. Garcia, L. Lahoz and J. R. Pedro, Tetrahedron, 1996, 52, 8611 CrossRef CAS.
- A. S. Pepito and D. C. Dittmer, J. Org. Chem., 1997, 62, 7920 CrossRef CAS PubMed.
- W. Li, X. J. Zhou and Q. Ma, Synth. Commun., 1995, 25, 553 CrossRef CAS.
- C. C. Silveira, E. J. Lenardao and J. V. Comasseto, Synth. Commun., 1994, 24, 575 CrossRef CAS.
- V. Bargues, G. Blay, J. Fernandez and J. R. Pedro, Synlett, 1996, 655 CrossRef CAS.
- P. Singh, M. Singh and A. K. Singh, J. Organomet. Chem., 2009, 694, 3872 CrossRef CAS.
- P. Singh and A. K. Singh, Eur. J. Inorg. Chem., 2010, 4187 CrossRef CAS.
- I. G. Jung, S. U. Son, K. H. Park, K.-C. Chung, J. W. Lee and Y. K. Chung, Organometallics, 2003, 22, 4715 CrossRef CAS.
- E. Dıez-Barra, J. Guerra, V. Hornillos, S. Merino and J. Tejeda, J. Organomet. Chem., 2003, 22, 4610 CrossRef; C. S. Consorti, M. L. Zanini, S. Leal, G. Ebeling and J. Dupont, Org. Lett., 2003, 5, 83 CrossRef PubMed.
- J. Masllorens, M. Moreno-Manas, A. Pla-Quintana and A. Roglans, Org. Lett., 2003, 5, 1559 CrossRef CAS PubMed.
- S. B. Park and H. Alper, Org. Lett., 2003, 5, 3209 CrossRef CAS PubMed; C. Mazet, L. H. Gade, Eur. J. Inorg. Chem., 2003, 1161 Search PubMed.
- Q. Yao, E. P. Kinney and C. Zheng, Org. Lett., 2004, 6, 2999 Search PubMed.
- A. Kumar, M. Agarwal and A. K. Singh, J. Organomet. Chem., 2008, 693, 3533 CrossRef CAS.
- D. H. Cobden, Nature, 2001, 409, 32 CrossRef CAS PubMed.
- Y. Cui, Q. Wei, H. Park and C. M. Lieber, Science, 2001, 293, 1289 CrossRef CAS PubMed.
- G. A. Prinz, Science, 1998, 282, 1660 CrossRef CAS PubMed.
- W. Zhou, J. Zhu, D. Li, H. H. Hng, F. Y. C. Boey, J. Ma, H. Zhang and Q. Yan, Adv. Mater., 2009, 21, 3196 CrossRef CAS.
- Q. Yan, M. S. Raghuveer, H. Li, B. Singh, T. Kim, M. Shima, A. Bose and G. Ramanath, Adv. Mater., 2007, 19, 3 CrossRef.
- A. K. Samal and T. Pradeep, Langmuir, 2010, 26, 19136 CrossRef CAS PubMed.
- H. Cordes and R. Schmid-Fetzer, Semicond. Sci. Technol., 1994, 9, 2085 CrossRef CAS.
- S. Dey and V. K. Jain, Platinum Met. Rev., 2004, 48, 16 CAS.
-
(a) L. Y. Chiang, J. W. Swirczewski, R. Kastrup, C. S. Hsu and R. B. Upasani, J. Am. Chem. Soc., 1991, 113, 6574 CrossRef CAS;
(b) M. Misono and N. Nojiri, Appl. Catal., 1990, 64, 1 CrossRef CAS.
- P. Singh, D. Das, M. Singh and A. K. Singh, Inorg. Chem. Commun., 2010, 13, 988 CrossRef CAS.
- A. Singhal, V. K. Jain, R. Mishra and B. Varghese, J. Mater. Chem., 2000, 10, 1121 RSC.
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a and
Neelam Shivranb
*a and
Neelam Shivranb