
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNaphthoquinone based chemosensors for transition metal ions: experiment and theory†
Prajkta Gosavi-Mirkute‡
 a,
Amit Patil‡a,
Dipali N. Landea,
Debamitra Chakravartyb,
Shridhar P. Gejjia,
Surekha Satputec and
Sunita Salunke-Gawali*a
a,
Amit Patil‡a,
Dipali N. Landea,
Debamitra Chakravartyb,
Shridhar P. Gejjia,
Surekha Satputec and
Sunita Salunke-Gawali*a
aDepartment of Chemistry, Savitribai Phule Pune University, Pune 411007, India. E-mail: sunitas@chem.unipune.ac.in
bCentral Instrumentation Facility, Department of Chemistry, Savitribai Phule Pune University, Pune 411007, India
cDepartment of Microbiology, Savitribai Phule Pune University, Pune 411007, India
First published on 4th December 2017
Abstract
The synthesis and characterization of 2-((pyridine-2-yl)methylamino)naphthalene-1,4-dione (H-1), 2-((thiophen-2-yl)methylamino)naphthalene-1,4-dione (H-2) and 2-((pyridine/thiophen-2-yl)ethylamino)naphthalene-1,4-dione (H-3 and H-4) have been carried out. Molecular recognition abilities of these ligands toward transition metal ions in methanol, methanol–water, methanol–triethylamine or methanol–water–triethylamine mixtures, stoichiometries and association constants of H-1 and H-3 have been determined. It has been shown that H-1 and H-3 coordinate to metal ions via two nitrogen atoms and oxygen and exhibit remarkable selectivity towards Cu2+ ions in methanol or methanol–water mixtures, the complexation being accompanied by a color change from orange to intense blue. LOD (Limit of Detection) of Cu2+ with H-1, H-3 are 1.48 × 10−8 mol L−1 and 1.59 × 10−8 mol L−1 respectively. The vibrational spectra, 1H NMR chemical shifts and optical properties of H-1 to H-4 derived from density functional theory are also presented.
Introduction
The α,β-unsaturated carbonyl derivatives of 1,4-naphthoquinones1 are of growing interest owing to their anticancer,2–7 antimicrobial,8–10 antifungal,11–13 antitumor14–18 and antiviral19–21 activities. Their molecular attributes, namely conjugation and electrophilicity, govern metabolic pathways and the key processes in chemotherapy that require redox cycling drugs.22 The underlying redox switching of such naphthoquinones, which on the acceptance of one or two electrons yield a radical anion or dianion, are thus interesting. It is further discernible to note that the intra-molecular charge transfer (ICT) transition is shown to be strongly dependent on the amine substituent and in particular, the charge density on the nitrogen center.23 These ligands, which serve as chemosensors, have shown remarkable molecular recognition and gave easily detectable, sensitive signals. A colorimetric and ratiometric fluorescent chemosensor combined with attributes such as high fluorescence sensitivity and aesthetic appeal of colorimetric assay makes these napthoquinone sensors very fascinating.24Naphthoquinone based chemosensors viz., 2-((pyridin-2-yl)methylamino)naphthalene-1,4-dione (H-1), 2-((thiophen-2-yl)methylamino)naphthalene-1,4-dione (H-2) 2-((pyridine/thiophen-2-yl)ethylamino)naphthalene-1,4-dione (H-3 and H-4) (Fig. 1) are synthesized and characterized using FT-IR, 1H and 13C NMR and single-crystal X-ray diffraction studies. The chemosensing ability of ligands with metal ions; La3+, Hg2+, Cd2+, Zn2+, Cu2+, Cu+, Ni2+, Co2+, Mn2+, Cr3+ and Ca2+ in methanol, methanol–water mixture and in the presence of a mild base such as triethylamine has been evaluated. Moreover, the chemosensors H-1 and H-3 are shown to be useful for detection of different metal ions viz., Cu2+, Ni2+ and Co2+ revealing a color change that can be observed with the naked eye and further can be studied through the UV-visible and fluorescence experiments. On the other hand, H-2 and H-4 do not show any chemosensing ability toward the metal ions studied herein.
 | ||
| Fig. 1 Molecular structure of H-1, H-2, H-3 and H-4. | ||
Experimental
Materials and methods
1,4-napthoquinone, 2-methoxy-1,4-napthoquinone, 2-picolyl amine, 2-thiophenemethylamine, 2-(2′-aminoethyl)pyridine, 2-thiopheneethylamine from Sigma-Aldrich; CuCl2·2H2O, CuCl, NiCl2·6H2O, ZnCl2, HgCl2, CoCl2, CaCl2, triethylamine, methanol and dichloromethane from Merck chemicals; FeCl3·6H2O from Qualigens Chemicals, CrCl3·6H2O, CdSO4, MnCl2·4H2O from Fluka; LaCl3·7H2O from Thomas and Baker were obtained. Milli-Q water was used for preparation of aqueous solutions. Anhydrous methanol was prepared by literature reported procedure.25 FT-IR spectra (Fig. S1 in ESI†) were recorded between 4000 and 400 cm−1 as KBr pellets on SHIMADZU FT 8400 and Bruker FT-IR Spectrophotometers. The elemental analysis was performed on Thermo Finnigan EA 1112 Flash series Elemental Analyzer. 1H and 13C NMR (Fig. S2–S5 in ESI†) were obtained in DMSO-d6, with the help of Varian Mercury NMR spectrometer using the TMS (tetramethylsilane) as a reference. UV-vis spectra were measured on the SHIMADZU UV 1650 in DMSO between 200 nm to 800 nm. The fluorescence spectra were recorded on JASCO spectrofluorometer FP-8300. Melting points were observed using the DSC experiments on TA waters model Q2000 instrument (Fig. S6 in ESI†) equipped with Tzero aluminium pan as a sample holder.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol) with a color change from yellow to orange. The mixture after refluxing was further kept for 24 hours for evaporation of solvent which yield the red colored crystals.:methanol (9:1) as eluent. A red colored product was obtained subsequent to a slow evaporation of eluent.:9) solvent system. Red coloured solid products were obtained after evaporation of pure fraction from the column.
:methanol) with a color change from yellow to orange. The mixture after refluxing was further kept for 24 hours for evaporation of solvent which yield the red colored crystals.:methanol (9:1) as eluent. A red colored product was obtained subsequent to a slow evaporation of eluent.:9) solvent system. Red coloured solid products were obtained after evaporation of pure fraction from the column.Characterization
X-ray crystal structures
X-ray data for H-1, H-3 and H-4 were collected on D8 Venture PHOTON 100 CMOS diffractometer using graphite monochromatized Mo-Kα radiation (λ = 0.7107 Å) with exposure/frame = 10 s. The X-ray generator was operated at 50 kV and 30 mA. An initial set of cell constants and an orientation matrix were calculated from total 24 frames. The optimized strategy used for data collection included different sets of ϕ and ω scans with 0.5° steps in ϕ/ω. Crystal to detector distance was 5.00 cm with 512 × 512 pixels/frame, oscillation/frame −0.5°, maximum detector swing angle = −30.0°, beam centre = (260.2, 252.5), in plane spot width = 1.24. Data integration was carried out by Bruker SAINT program and empirical absorption correction for intensity data were carried out using the Bruker SADABS. The programs are integrated in APEX II package.26 The data were corrected for Lorentz and polarization effect. The structure was solved by direct method using the SHELX-97 (ref. 27) with the final refinement of the structure performed by a full-matrix least-squares technique with anisotropic thermal data for non-hydrogen atoms on F2. The non-hydrogen atoms were refined anisotropically where the hydrogen atoms were refined at the calculated positions as riding atoms with isotropic displacement parameters.27 Molecular diagrams were generated using the ORTEP-3 (ref. 28) and Mercury programs.29 Structural calculations were performed using SHELXTL30 and PLATON.31Metal ion binding studies, Job plot, competitive binding studies, pH studies and limit of detection (LOD) experiments
Metal ion binding studies were evaluated by following methods, (i) in methanol, (ii) in 1:1 methanol–water, (iii) in presence of triethylamine in (i) and (ii). In typical experiments 2 ml of H-1 and H-3 was mixed with 2 ml of metal ion solutions at room temperature (26 °C). 1 ml of triethylamine was added in chemosensor solution in case of (i) and (ii) prior to addition of metal ion solutions. UV-visible and fluorescence spectra of all solutions were measured; the concentration of ∼2.5 × 10−4 M was used.
Micromolar concentration (10–100 μM) of H-1, H-3 and metal ions was used for Jobs plot experiment. Solutions were prepared by varying concentration of ligands and metal ions. Binding constant of Cu2+ and Ni2+ was determined by Stern–Volmer plot. 5 × 10−4 M solution of chemosensor and 1 × 10−3 M metal ion solutions in methanol were used to obtained the same.
For competitive binding of metal ions (5 × 10−3 M) with H-1 and H-3 (5 × 10−3 M) equal volumes were mixed and fluorescence spectra of all solutions were measured.
For quantum yield experiment fluorescien is used as standard. Fluorescien (1 × 10−6 M), H-1 and H-3 (1 × 10−6 M) concentration is used. 0.1 M NaOH was used for the deprotonation of fluorescien.
In pH dependent metal ion binding studies experiments, 2 ml of chemosensor H-1 and H-3 was mixed with 2 ml of metal ion solutions (Cu2+, Ni2+, Zn2+ and Co2+) at room temperature (26 °C). 2 ml of buffer solution at pH 4, pH 7 and pH 9 was added respectively prior to addition of metal ion solutions. UV-visible spectra of all solutions were measured.
Micromolar concentrations (10 to 100 μM) of H-1, H-3 and Cu2+ metal ions were used for LOD experiments. Solutions were prepared by varying concentrations of H-1, H-3 and Cu2+ metal ions.
Computational methodology
Optimizations of naphthoquinone derivatives were carried out using the GAUSSIAN-09 program.32 Global hybrid meta GGA (M06-2X) functional was used in conjuction with the internally stored 6-311++G(d,p) basis set. Stationary point structures thus obtained were confirmed to be the local minima on the potential energy surfaces through vibrational frequency calculations. Frontier orbitals were derived in the same framework of theory. Electronic spectra were computed using the time dependent density functional theory (TD-DFT).Results and discussion
Synthesis and characterization of H-1, H-2, H-3 and H-4
Chemosensor H-1, H-2, H-3 and H-4 were synthesized and obtained in high yields at the room temperature (26 °C) without using any exhaustive synthetic procedures. H-1, H-2, H-3 and H-4 were characterized by FT-IR (Fig. S1 ESI†), 1H and 13C NMR (Fig. S2 through Fig. S5 in ESI†), elemental analysis, UV-visible spectroscopy and single crystal X-ray diffraction studies. Melting point of chemosensor ligands were obtained by Differential Scanning Calorimetry (DSC) (Fig. S6 in ESI†). FT-IR spectra of the ligands reveals characteristic N–H stretching observed at the 3355 cm−1 (H-1), 3354 cm−1 (H-2), 3336 cm−1 (H-3) and 3338 cm−1 (H-4). The carbonyl frequency was assigned to the ∼1672 cm−1 in H-1 and H-3, 1670 cm−1 in H-2 and ∼1669 cm−1 in H-4. The C–S frequency was observed in the range ∼1467 cm−1 (in H-3) to 1432 cm−1 (in H-1).ORTEP of H-1, H-3 and H-4 are shown in Fig. 2 and crystallography data have been summarized in Table 1. H-1 and H-3 shows the monoclinic space group while H-4 shows triclinic space group. The carbonyl bond distances (1.21 Å and 1.23 Å) in these compounds resemble to those observed in the oxidized form of compounds. The bond distances represent quinonoid distortion with two short and four long C–C bonds. The carbonyl C(1)–O(1) and C(4)–O(2) bond distances are found to be 1.220 Å and 1.234 Å, respectively. The C(2)–N(1) bond distances (1.335 Å) match well with those of amino derivatives of 1,4-naphthoquinone.33–38
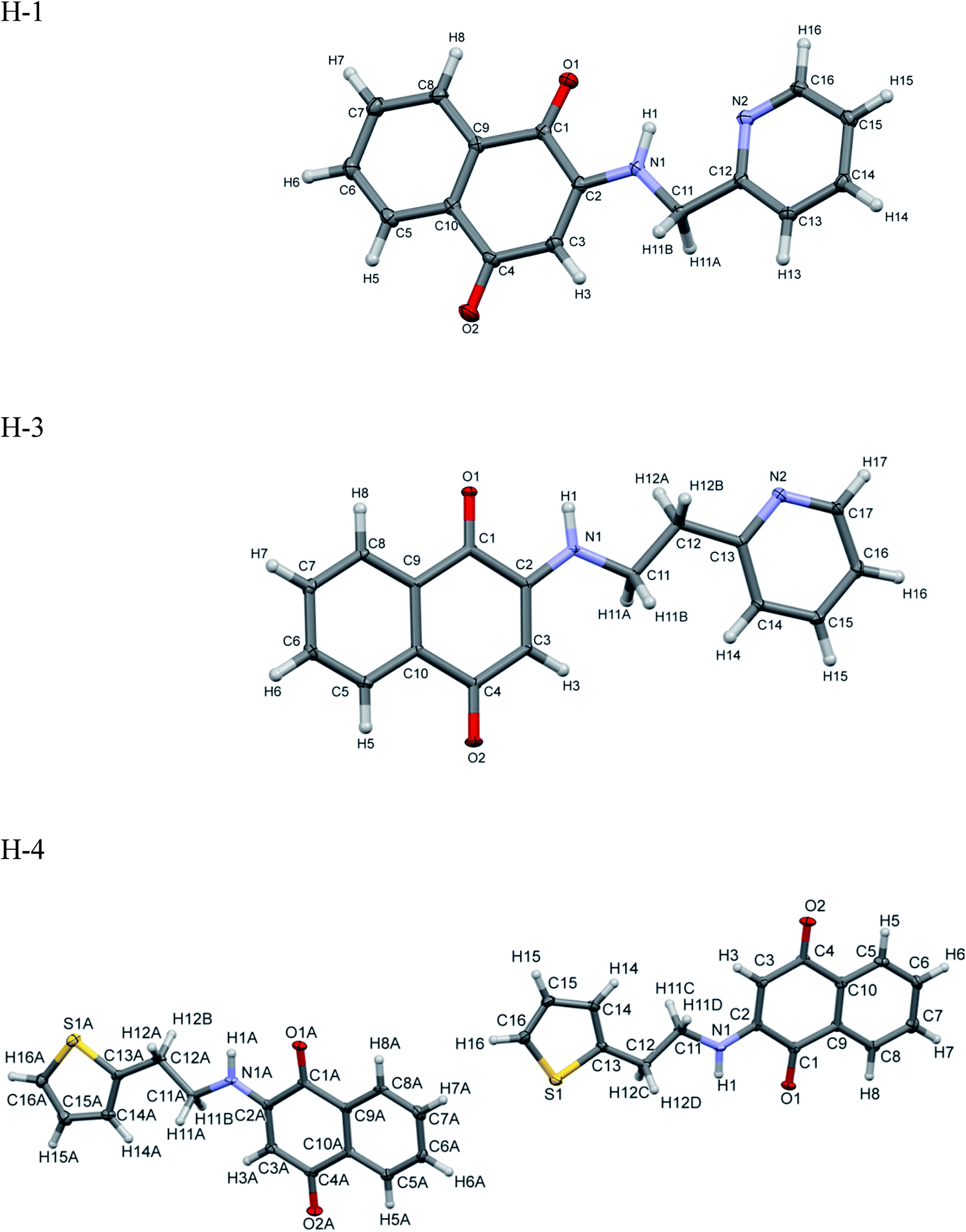 | ||
| Fig. 2 ORTEP of H-1, H-2, H-3 and H-4. The ellipsoid was drawn with 50% probabilities. | ||
| Identification code | H-1 | H-3 | H-4 |
| Empirical formula | C16H12N2O2 | C17H14N2O2 | C16H13NO2S |
| Formula weight | 264.28 | 278.3 | 283.33 |
| Temperature | 100(2) K | 100(2) K | 100(2) K |
| Wavelength | 0.71073 Å | 0.71073 Å | 0.71073 Å |
| Crystal system | Monoclinic | Monoclinic | Triclinic |
| Space group | P21/n | P21/n | P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| Unit cell dimensions | a = 5.7063(4) Å | a = 5.4000(4) Å | a = 5.4069(5) Å, α = 92.338(4)° |
| b = 8.4245(6) Å, β = 93.229(2)° | b = 30.026(2) Å, β = 99.278(2)° | b = 8.0971(7) Å, β = 91.024(4)° | |
| c = 25.6881(19) Å | c = 8.0578(6) Å | c = 29.735(3) Å, γ = 98.992(4)° | |
| Volume | 1232.94(15) Å3 | 1289.39(17) Å3 | 1284.3(2) Å3 |
| Z | 4 | 4 | 4 |
| Density (calculated) | 1.424 Mg m−3 | 1.434 Mg m−3 | 1.465 Mg m−3 |
| Absorption coefficient | 0.096 mm−1 | 0.096 mm−1 | 0.252 mm−1 |
| F(000) | 552 | 584 | 592 |
| Crystal size | 0.34 × 0.26 × 0.24 mm3 | 13.000 × 0.210 × 0.070 mm3 | 0.41 × 0.21 × 0.04 mm3 |
| Theta range for data collection | 2.893 to 28.388° | 2.713 to 28.345° | 2.841 to 28.611° |
| Index ranges | −7<=h<=7, −11<=k<=11, −34<=l<=34 | −7<=h<=7, −40<=k<=40, −10<=l<=10 | −7<=h<=7, −10<=k<=10, −39<=l<=37 |
| Reflections collected | 48104 |
51719 |
24810 |
| Independent reflections | 3076 [R(int) = 0.0306] | 3213 [R(int) = 0.0643] | 6062 [R(int) = 0.0555] |
| Completeness to theta = 25.242° | 99.90% | 99.90% | 97.60% |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 3076/0/181 | 3213/0/191 | 6062/0/361 |
| Goodness-of-fit on F2 | 1.032 | 1.054 | 1.155 |
| Final R indices [I > 2sigma(I)] | R1 = 0.0393, wR2 = 0.1039 | R1 = 0.0610, wR2 = 0.1054 | R1 = 0.0926, wR2 = 0.2044 |
| R Indices (all data) | R1 = 0.0445, wR2 = 0.1079 | R1 = 0.0784, wR2 = 0.1114 | R1 = 0.1233, wR2 = 0.2187 |
| Extinction coefficient | n/a | 0.0039(6) | n/a |
| Largest diff. peak and hole | 0.344 and −0.273 e Å−3 | 0.440 and −0.361 e Å−3 | 0.756 and −0.593 e Å−3 |
As may readily be noticed, H-1 shows naphthoquinone and pyridyl rings to be coplanar (Fig. S7 in ESI†) in and render inter-as well as the intra-molecular N–H⋯O hydrogen bonding which arise from quinonoid oxygen (Table 2).
| Compound | D–H⋯A | D⋯H (Å) | H⋯A (Å) | D⋯A (Å) | D–H⋯A (°) | Symmetry |
|---|---|---|---|---|---|---|
| H-1 | N(1)–H(1)⋯O(1) | 0.8797 | 2.3087(8) | 2.672(1) | 104.79 | Intra |
| C(15)–H(15)⋯O(1) | 0.949(1) | 2.5782(8) | 3.363(1) | 140.26(7) | 2 − x, 1 − y, −z | |
| C(16)–H(16)⋯N(2) | 0.950(1) | 2.559(1) | 3.391(1) | 146.41(7) | 2 − x, 1 − y, −z | |
| C(13)–H(13)⋯O(2) | 0.950(1) | 2.3391(8) | 3.166(1) | 145.14(7) | −x, −y, −z | |
| C(14)⋯C(5) | 3.398(2) | (−x, −y, −z) | ||||
| C(4)⋯O(1) | 3.216(1) | (1 − x, 1 − y, −z) | ||||
| C(1)⋯O(2) | 3.065(1) | (−x, 1 − y, −z) | ||||
| H-3 | N(1)–H(1)⋯O(1) | 0.88 | 2.252 | 3.099(1) | 161.65 | −x, −y, 1 − z |
| N(1)–H(1)⋯O(1) | 0.88 | 2.302 | 2.667(2) | 104.91 | Intra | |
| C(3)–H(3)⋯O(2) | 0.951 | 2.511 | 3.446(2) | 168.1(3) | 2 − x, −y, 2 − z | |
| C(14)–H(14)⋯O(2) | 0.949 | 2.633 | 3.564 (2) | 167 | 2 − x, −y, 2 − z | |
| C(11)–H(11B)⋯O(2) | 0.989 | 2.717 | 3.346 (2) | 121.84 | 2 − x, −y, 2 − z | |
| C(12)–H(12A)⋯O(1) | 0.99 | 2.658 | 3.211(2) | 115.5 | −x, −y, 1 − z | |
| C(12)–H(12B)⋯O(2) | 0.99 | 2.624 | 3.421 (2) | 137.64 | 1 − x, −y, 2 − z | |
| C(11)–H(11A)⋯C(8) | 0.99 | 2.807 | 3.476(2) | 125.4 | 1 − x, −y, 2 − z | |
| C(11)–H(11B)⋯C(3) | 0.989 | 2.789 | 3.650 (2) | 145.7 | 1 − x, −y, 2 − z | |
| C(4)⋯O(1) | 3.167(2) | −1 + x, y, z | ||||
| C(1)⋯O(2) | 3.141(7) | −1 + x, y, z | ||||
| C(1)⋯N(1) | 3.223(2) | 1 − x, −y, 1 − z | ||||
| C(13)⋯C(7) | 3.394(2) | 1 − x, −y, 1 − z | ||||
| H-4 | N(1)–H(1)⋯O(1) | 0.880(4) | 2.258(3) | 3.104(5) | 161.4(3) | 2 − x, 1 − y, 2 − z |
| N(1A)–H(1A)⋯O(1A) | 0.880(4) | 2.280(3) | 3.121(5) | 159.7(3) | 1 − x, −y, 1 − z | |
| N(1)–H(1)⋯O(1) | 0.88 | 2.307 | 2.670(5) | 104.8 | Intra | |
| N(1A)–H(1A)⋯O(1A) | 0.88 | 2.301 | 2.664(5) | 104.7 | Intra | |
| C(3)–H(3)⋯O(2) | 0.950(5) | 2.649(3) | 3.584(6) | 168.1(3) | −x, −y, 2 − z | |
| C(3A)–H(3A)⋯O(2A) | 0.951(5) | 2.607(3) | 3.545(6) | 168.9(3) | 3 − x, 1 − y, 1 − z | |
| C(14)–H(14)⋯O(2) | 0.950(5) | 2.504(3) | 3.440(6) | 168.2(3) | −x, −y, −z | |
| C(16)–H(16)⋯S(1A) | 0.949(5) | 2.951(3) | 3.681(5) | 134.7(3) | 1 − x, −y, 1 − z | |
| C(14A)–H(14A)⋯O(2A) | 0.950(5) | 2.508(3) | 3.445(6) | 168.5(3) | 3 − x, −y, 1 − z | |
| C(7)⋯C(13) | 3.387(7) | 1 − x, −y, 2 − z | ||||
| N(1)⋯C(1) | 3.244(7) | 1 − x, −y, 2 − z | ||||
| N(1A)⋯C(1A) | 3.240(7) | 2 − x, −y, 2 − z |
Two molecules of H-1 oriented in the opposite directions orientations which facilitate further intermolecular C–H⋯N (C(16)–H(16)⋯N(2)) and C–H⋯O(C(15)–H(15)⋯O(1)) hydrogen bonding. Subsequently the dimeric units are linked by the C–H⋯O (C(13)–H(13)⋯O(2)) interactions along a polymeric chain down a-axis (cf. Fig. 3 and 4) facilitate C–H⋯π interactions between the alkyl proton C(11)–H(11B) and quinonoid ring; with the corresponding distance being 3.231 Å. Benzenoid carbon C(5) in close contact with neighbouring molecules pyridine ring carbon C(14) (3.398(2) Å, (1 − x, −y, −z) (cf. Fig. 5); the C(4)⋯O(1) (−1 + x, y, z) separation of neighbouring molecules was 3.216 Å.
 | ||
| Fig. 3 Polymeric chains of H-1 molecules down a-axis. | ||
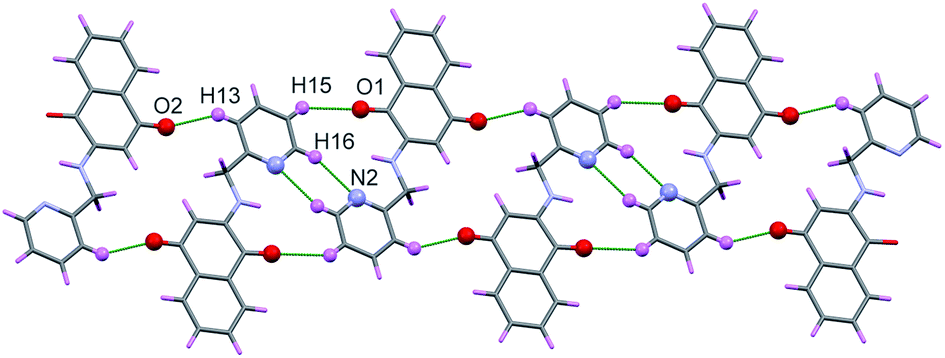 | ||
| Fig. 4 Polymeric chain of H-1 showing C–H⋯O and C–H⋯N interaction. | ||
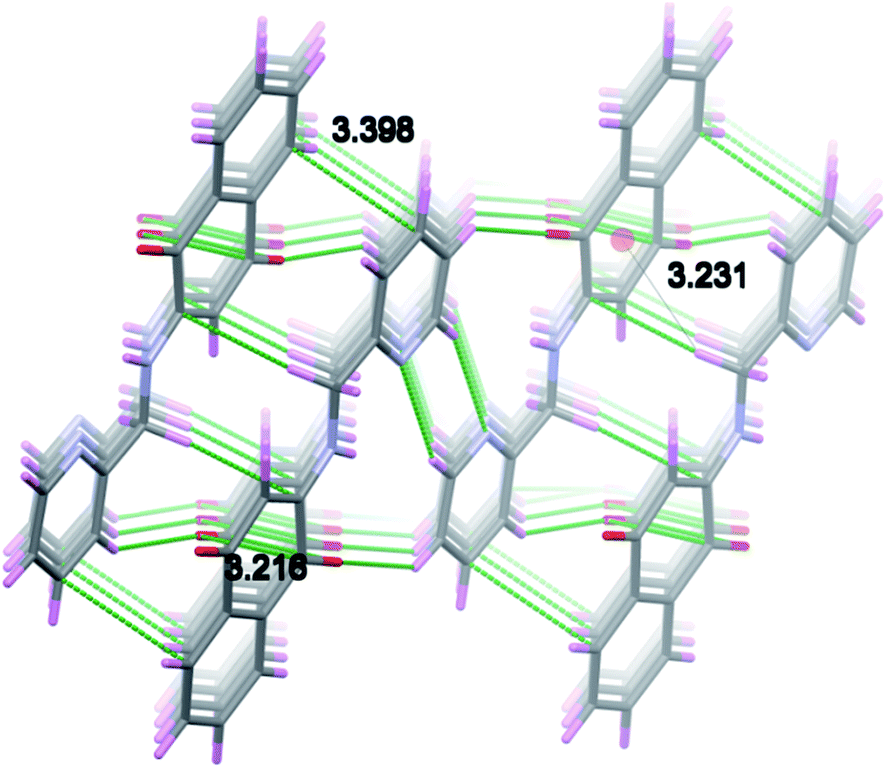 | ||
| Fig. 5 Figure showing π–π stacking and C–H⋯π interactions to H-1 molecules. | ||
The mutual orientation of pyridyl ring with naphthoquinone show a marginal deviation (Fig. S8 in ESI†) from the planarity as adjudged from the C(11)–N(1)–C(2)–C(1) dihedral angle parameters. A bidentate coordination of H-3 ligand with the metal ions is favoured; as inferred earlier in ref. 39–41.
As far as H-3 is concerned the opposite orientation was noticed (with respect pyridine ring) with a polymeric chain therein extending from one end by N(1)–H(1)⋯O(1) and C(12)–H(12A)⋯O(1), C(3)–H(3)⋯O(2), C(12)–H(12B)⋯O(2) and C(14)–H(14)⋯O(2) hydrogen bonding interactions, which are depicted in Fig. 6 and 7. Furthermore the polymeric chain brings about C–H⋯π, C⋯C(C(7)⋯C(13)), C⋯N (C(1)⋯N(1)) and C⋯O(C(4)⋯O(4)) stacking interactions as shown in Fig. 8. Besides the C–H⋯π interactions from ethyl protons and C(11)–(11A) of the benzenoid ring and those between C(12)–H(12B) and quinonoid can readily be noticed.
 | ||
| Fig. 6 Polymeric chains of H-3 molecules down a-axis. | ||
 | ||
| Fig. 7 Polymer chain of H-3 showing N–H⋯O and C–H⋯O interaction. | ||
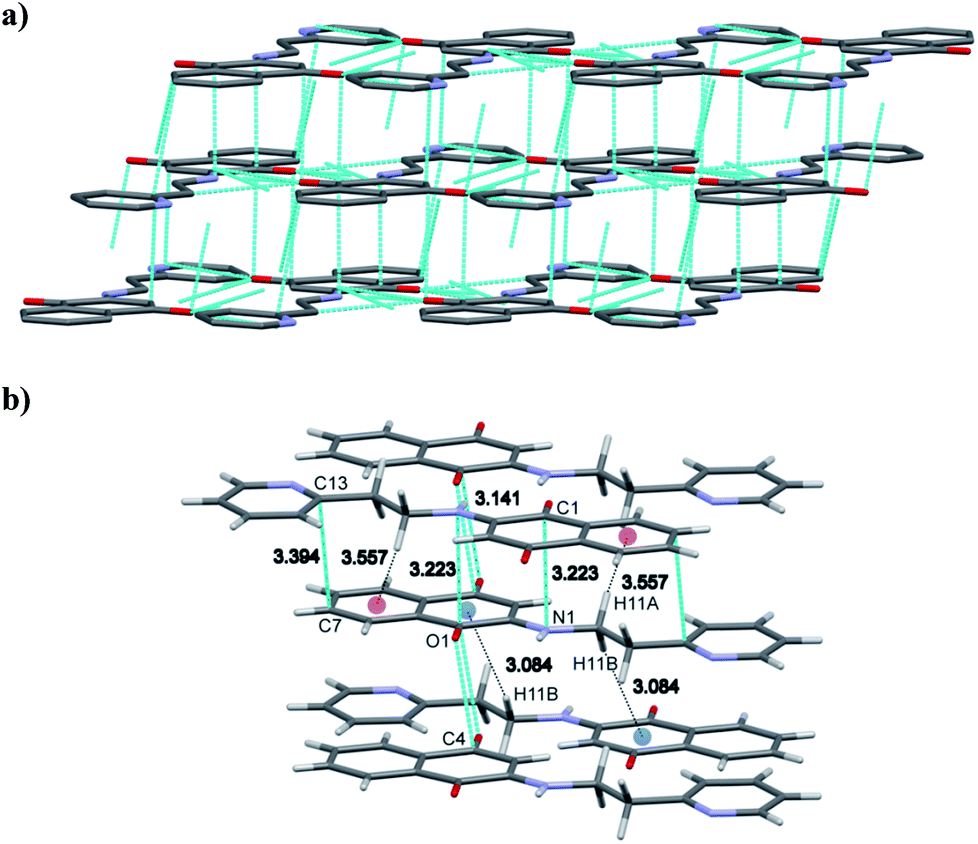 | ||
| Fig. 8 (a) and (b) showing π–π stacking and C–H⋯π interaction in H-3 molecule. | ||
Unlike H-1 and H-3 the polymeric chains of H-4 reveal two asymmetric molecules linked by the C(16)–H(16)⋯S(1A) (1 − x, −y, 1 − z) interactions which are depicted in Fig. 9. In addition to these C(11)–H(11A)⋯π and N(1)–H(1A)⋯π interactions from the quinonoid ring are transparent. On the other hand the C(12)–H(12B)⋯π interactions bring forth the binding with benzenoid moiety as can be deciphered from Fig. 10. Subsequently the interactions between asymmetric molecules engender marginal variations in their corresponding proton distances.
 | ||
| Fig. 9 Figure showing two asymmetric molecules of H-4 are joined by C–H⋯S interaction. | ||
 | ||
| Fig. 10 C–H⋯π and N–H⋯π interaction of H-4 molecules. | ||
Sensing metal ion binding of H-1 and H-3
No significant colour change was observed in metal ions binding studies experiments of H-2 and H-4. The metal ion binding studies thus focuses on H-1 and H-3 ligands. Metal ion binding studies35 were carried out (i) in methanol (ii) in methanol–water, (iii) in presence of mild base triethylamine (TEA) in (i) and (ii) conditions. TEA was used for deprotonation of ligand for detection of metal ions.Equal volumes of (2.5 × 10−4 M) of different metal ion solutions were mixed with H-1 and H-3 in methanol at room temperature (26 °C). A color change from orange in H-1 on addition of Cu2+, Cu+, Ni2+, Zn2+ and Co2+ ions can be attributed to deprotonation of N–H of the ligand which can be visualized with the naked eye (Fig. 11). A new band appeared in the range of 500–700 nm for Zn2+, Cu2+, Cu+, Ni2+ and Co2+ in the UV-visible spectra portrayed in Fig. 13a. The addition of triethylamine prior to addition of the metal ion solution, show a color change in the H-1 (shown in Fig. 11b) only for Zn2+, Cu2+, Cu+, Ni2+ and Co2+ which is accompanied by an emergence of a new band in ∼500–700 nm region (Fig. 13b).
 | ||
| Fig. 11 (a) Color changes observed to chemosensor H-1 (2.5 × 10−4 M) with metal ions (2.5 × 10−4 M) in methanol. (b) Color changes observed to chemosensor H-1 (2.5 × 10−4 M) with metal ions (2.5 × 10−4 M) in methanol–triethylamine. (c) Color changes observed to chemosensor H-1 (2.5 × 10−4 M) with metal ions (2.5 × 10−4 M) in methanol–water media. (d) Color changes observed to chemosensor H-1 (2.5 × 10−4 M) with metal ions (2.5 × 10−4 M) in methanol–triethylamine–water. | ||
Interestingly H-3 only shows color change with Cu+ and Cu2+ in methanol as can readily be noticed from Fig. 12a, subsequent addition of triethylamine led to binding with Cu+, Cu2+, Ni2+ and Co2+ ions concomitantly bringing about the color change for H-3 as evident from Fig. 12b (and also Fig. S9 shown in ESI†).
 | ||
| Fig. 12 (a) Color changes observed to chemosensor H-3 (2.5 × 10−4 M) with metal ions (2.5 × 10−4 M) in methanol. (b) Color changes observed to chemosensor H-3 (2.5 × 10−4 M) with metal ions (2.5 × 10−4 M) in methanol–triethylamine. (c) Color changes observed to chemosensor H-3 (2.5 × 10−4 M) with metal ions (2.5 × 10−4 M) in methanol–water. (d) Color changes observed to chemosensor H-3 (2.5 × 10−4 M) with metal ions (2.5 × 10−4 M) in methanol–triethylamine–water. | ||
Fig. 11c displays H-1 in the 1:1 mixture of methanol–water. Here the color change from yellow to grey was noticed only for the Cu2+. No color change was observed for the rest of the metal ions studied. In other words, H-1 selectively detects Cu2+ ion in methanol–water solvent mixture with the naked eye. Similar inferences are drawn for the H-3 ligand shown in Fig. 12c.
As inferred from Fig. 11d and 12d; H-1 and H-3 do not show any noticeable color change for triethylamine and methanol–water solvent mixtures. On addition of Ni2+ and Zn2+ solution the UV-visible spectra emerge with new bands near λmax 340 nm and 360 nm displayed in Fig. 13d and Fig. S9d in ESI.†
 | ||
| Fig. 13 (a) UV-visible spectra of H-1 in presence of various metal ions in methanol. (b) UV-visible spectra of H-1 in presence of various metal ions in methanol–triethylamine. (c) UV-visible spectra of H-1 in presence of various metal ions in methanol:water media. (d) UV-visual spectra of H-1 in presence of various metal ions in methanol–TEA–water. | ||
Competitive binding, pH dependent binding studies and detection limit of H-1 and H-3
The stoichiometry of H-1 (Fig. S10 through Fig. S12 in ESI†) and H-3 (Fig. S14 through Fig. S16 in ESI†) for Cu2+, Ni2+ was determined by the Job's plot method. As illustrated H-1 with Cu2+ showed 1:1 stoichiometry in methanol as well as methanol–water mixtures, whereas the 2:1 stoichiometry was suggested for the Ni2+ in methanol (Fig. S10 in ESI†). Besides Cu2+ in methanol–water–triethylamine mixture (Fig. S11 in ESI†) revealed 2:1 stoichiometry. Likewise the H-3 on combining with Cu2+ in methanol, methanol–triethylamine and methanol–water solvent mixture the stoichiometry was shown to be in 1:1 proportions. H-1 and H-3 ligands coordinating in tridentate manner with two nitrogen and a quinonoid oxygen bound to metal ions. The tetrahedral 1:1 complex Cu2+ and 1:2 octahedral complex of Ni2+ was proposed for such type of ligands.40,41
Association constant of Cu2+ ion was determined using the Stern–Volmer plots from fluorescence experiments (Fig. S13 (for H-1) and Fig. S17 (for H-3) in ESI†). The concentrations of H-1 or H-3 used were 5 × 10−4 M and that for the metal being 1 × 10−3 M. The methanol solutions were used. The association constants (Ka) for H-1 turned out to be 4932 M−1 for Cu2+ whereas that for the Ni2+ was 1542 M−1 (Fig. S13a and b in ESI†). Likewise the association constants (Ka) for H-3 were observed to be 938.9 M−1 and 790.7 M−1 respectively, for Cu2+ and Ni2+.
A competitive binding of metal ions was further studied through experiments in which equal volumes of H-1 and H-3 (1 × 10−4 M) were mixed with those of Cu2+, Ni2+ and Co2+ (1 × 10−3 M). Noticeable colour changes can be noticed in solution mixture of metal ions (Fig. 14 and S18 in ESI†) for H-1 and H-3. UV-visible (Fig. S19 in ESI†) and fluorescence spectra of selected metal ion are displayed in Fig. 15. Ligands H-1 and H-3 are fluorescent and showed low quantum yields of 0.99% and 1.89%, respectively. Noteworthy enough, Cu2+ demonstrate significant fluorescence quenching for H-1 and H-3. The experiments further showed Ni2+ and Co2+ bring about competitive binding to H-1 and H-3 however Cu2+ prevailing over other metal ions which is in accordance with its large binding constant.
 | ||
| Fig. 14 Color changes observed to H-1 and metal ions (1 × 10−4 M) in methanol. | ||
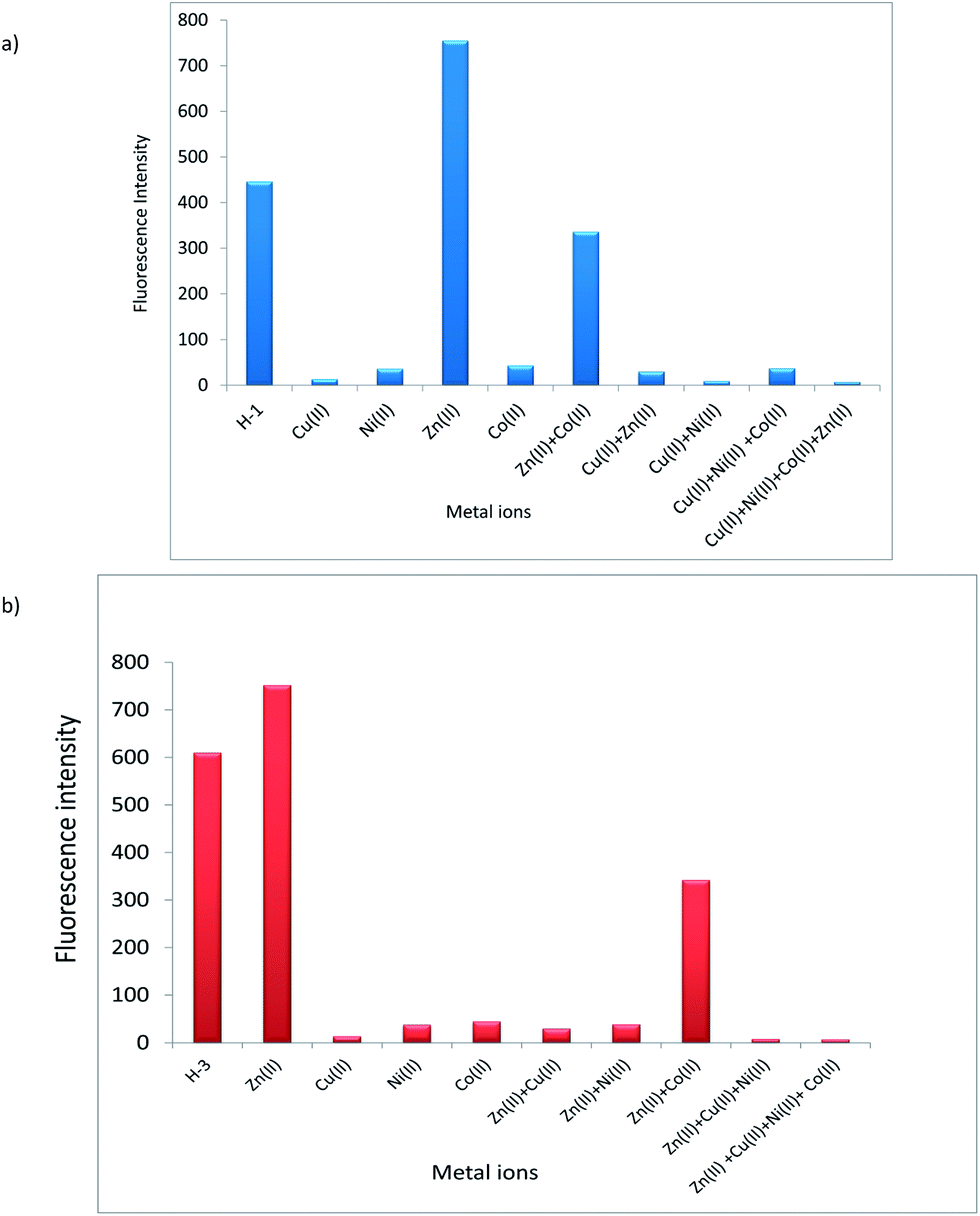 | ||
| Fig. 15 (a) Fluorescence spectra of competitive binding of metal ions with chemosensor H-1. (b) Fluorescence spectra of competitive binding of metal ions with chemosensor H-3. | ||
pH dependent metal ion binding experiments of H-1 and H-3 were carried out on metal ions Zn2+, Cu2+, Ni2+ and Co2+ in buffer solution of pH 4, 7 and 9. The metal ion binding was observed only in the basic pH (Fig. S20 in ESI†) and not for pH = 4.
The limit of detection (LOD) from the fluorescence experiments42,43 with Cu2+ and H-1 or H-3 ligands are observed to be 1.48 × 10−8 mol L−1 for H-1 (Fig. S21 in ESI†) and 1.59 × 10−8 mol L−1 (Fig. S22 in ESI†) for H-3.
DFT investigations
Optimized structures of aminonaphthoquinone derivatives obtained from the M06-2X based density functional theory are depicted in Fig. 16. Selected bond distances and angles are compared with X-ray crystal data in Table 3. As shown the overall structural parameters agree well with the experiments. A comparison of H-1 and H-2 shows that carbonyl C(1)O(1) bond distances are marginally elongated on substitution of thiophene instead of pyridine. | ||
| Fig. 16 Optimized geometries of (a) H-1, (b) H-2, (c) H-3 and (d) H-4. | ||
| H-1 | H-3 | H-4 | ||||
|---|---|---|---|---|---|---|
| Theo. | Obs. | Theo. | Obs. | Theo. | Obs. | |
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O1 O1 |
1.209 | 1.219 | 1.210 | 1.220 | 1.211 | 1.219 |
| CO2 |
1.217 | 1.234 | 1.216 | 1.237 | 1.216 | 1.246 |
| C3–H3 | 1.083 | 0.950 | 1.083 | 0.950 | 1.083 | 0.950 |
| N1–H1 | 1.015 | 0.880 | 1.012 | 0.880 | 1.012 | 0.880 |
Structural ramifications in vibration frequencies are analyzed. Calculated vibrational frequencies of H-1, H-2, H-3 and H-4 are compared with the experiment in Table 4. As may be inferred the N–H stretching assigned to 3247 cm−1 of H-1 engenders the frequency up shift of ∼42 cm−1 for the ethyl substituted H-3. As oppose to this in case of thiophene on substitution of the ethyl functionality engender the lowering of frequency from 3311 cm−1 (H-2) to 3285 cm−1 (H-4). The inferences borne out from DFT are thus in consonance with the experiment.
| H-1 | H-2 | H-3 | H-4 | |||||
|---|---|---|---|---|---|---|---|---|
| Theo. | Obs. | Theo. | Obs. | Theo. | Obs. | Theo. | Obs. | |
| ν(N1–H1) | 3247 | 3355 | 3311 | 3354 | 3289 | 3336 | 3285 | 3338 |
| ν(C3–H3) | 2936 | 3061 | 2926 | 3061 | 2921 | 2960 | 2911 | 2915 |
| ν(CO1) |
1650 | 1672 | 1645 | 1670 | 1649 | 1672 | 1645 | 1669 |
| ν(CO2) |
1610 | 1632 | 1620 | — | 1617 | 1600 | 1618 | — |
| ν(CC) |
1541 | 1593 | 1541 | 1599 | 1539 | 1570 | 1538 | 1593 |
1H-NMR chemical shifts (δH) in H-1, H-2, H-3 and H-4 in DMSO (as solvent) were simulated through the SCRF-PCM theory. A comparison with the experiment is given in Table 5. The H(1) proton participating in N–H⋯O and N–H⋯N interactions in H-1 emerge with relatively large deshielding (8.1 ppm) in the spectra. The signals of the corresponding proton in the rest of aminonaphthoquinone derivatives facilitate the N–H⋯O interactions and appear at ∼6.5 ppm. The alkyl protons emerge with up-field δH signals in the calculated 1H NMR. These inferences on the calculated δH values concur with the experiment.
| H-1 | H-2 | H-3 | H-4 | |||||
|---|---|---|---|---|---|---|---|---|
| Theo. | Obs. | Theo. | Obs. | Theo. | Obs. | Theo. | Obs. | |
| H1 | 8.1 | 8.08 | 6.5 | 8.11 | 5.6 | 7.62 | 5.6 | 7.55 |
| H3 | 6.2 | 5.59 | 6.1 | 5.74 | 6.2 | 5.71 | 6.2 | 5.72 |
| H5 | 8.4 | 7.90 | 9.0 | 7.90 | 8.4 | 7.92 | 8.4 | 7.33 |
| H6 | 9.0 | 7.81 | 8.6 | 7.80 | 9.0 | 7.81 | 8.9 | 7.72 |
| H7 | 8.6 | 7.77 | 8.5 | 7.71 | 9.0 | 7.70 | 9.1 | 7.82 |
| H8 | 9.4 | 8.00 | 9.1 | 7.97 | 9.3 | 7.95 | 9.3 | 7.93 |
| H11 | 4.6 | 4.52 | 4.9 | 4.61 | 3.6 | 3.54 | 3.6 | 3.45 |
| H13 | 8.3 | 7.37 | 7.7 | 7.10 | 7.7 | 7.32 | 7.3 | 6.94 |
| H14 | 8.5 | 7.73 | 7.7 | 6.97 | 8.5 | 7.72 | 7.6 | 6.96 |
| H15 | 8.0 | 7.30 | 8.1 | 7.40 | 8.1 | 7.23 | 8.2 | 7.97 |
| H16 | 9.7 | 8.55 | 9.6 | 8.51 | 3.4 | 3.12 | ||
| H17 | 3.6 | 3.04 | ||||||
The molecular Frontier orbital's HOMO and LUMO (isosurface of ±0.04 au) in aminonaphthoquinone ligands are portrayed in Fig. 17. The complimentarily of charge distributions between HOMO and LUMO is thus transparent. The electronic absorption spectra except H-4 revealed the band near ∼336 nm assigned to the HOMO to LUMO transition. The HOMO–LUMO energies, global indices of reactivity, namely, chemical potential (μ), hardness (η), and electrophillicity index (ω), are summarized in Table 6.
 | ||
| Fig. 17 Fronteir molecular orbitals of (a) H-1, (b) H-2, (c) H-3 and (d) H-4. | ||
| Molecular properties | H-1 | H-2 | H-3 | H-4 |
| ΔEHOMO–LUMO | 5.6 | 5.6 | 5.7 | 5.6 |
| Global hardness (η) | 2.8 | 2.8 | 2.9 | 2.8 |
| Electronic chemical potential (μ) | 4.7 | 4.9 | 4.9 | 4.9 |
| Electronegativity (χ) | 4.7 | 4.9 | 4.9 | 4.9 |
| Global electrophilicity index (ω) | 4.0 | 4.3 | 4.3 | 4.3 |
Conclusions
Naphthoquinone based chemosensors are characterized from the X-ray diffraction and spectroscopy experiments in conjunction with the density functional theory. X-ray diffraction experiments revealed the C–H⋯π interactions in molecular packing those bring about a variation in their optical properties. The metal ion binding of H-1 and H-3 in methanol, methanol–water mixture and in the presence of mild base triethylamine were examined. The binding of H-1 and H-3 with Cu2+ revealed 1:1 stoichiometry in methanol water mixture compared to that of Ni2+ which was 1:2 proportions when the methanol or methanol–triethylamine mixture were used. The association constant of Cu2+ with H-1 was observed to be ∼5.2 times as larger than that for the H-3. Secondly, the ligands H-1 and H-3 display remarkable recognition ability toward the Cu2+, Ni2+, Co2+ ions in methanol. Both ligands showed remarkable selectivity in the detection of Cu2+ in methanol/H2O solution. The experimental investigations further were corroborated through the inferences drawn based on the 1H NMR and infrared spectra derived from the density functional theory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SSG and SS grateful to DST-SERB (EMR/2016/007912). SPG acknowledges support from the Research Project (37(2)/14/11/2015-BRNS) from the Board of Research in Nuclear Sciences (BRNS), India. PM, AP and DNL thankful to Savitribai Phule Pune University for the award of research fellowship through the University of Potential excellence scheme from the University Grants Commission, New Delhi, India. SPG thanks Centre for Development of Advanced Computing (C-DAC), Pune for computer time on National Param Supercomputing Facility.References
- L. Li, E. Leyva and R. Las Garcia, Rev. Mex. Cienc. Farm., 2011, 42, 6–17 CAS.
- R. P. Varma, Med. Chem., 2006, 6, 489–499 Search PubMed.
- H. R. Lawrence, A. Kazi, Y. Luo, R. Kendig, Y. Ge, S. Jain, K. Daniel, D. Santiago, W. C. Guida and S. M. Sebti, Bioorg. Med. Chem., 2007, 18, 5576–5592 CrossRef PubMed.
- V. K. Tandon, H. K. Maurya, A. Tripathi, G. B. Shivakeshava, P. V. Shukla, P. Srivastava and D. Panda, Eur. J. Med. Chem., 2009, 44, 1088–1092 Search PubMed.
- E. H. da Cruz, C. M. Hussene, G. G. Dias, E. B. Diogo, I. M. de Melo, B. L. Rodrigues, M. G. da Goukrt, B. C. Cavalcanti, C. Pessoa and E. N. da Silva Junior, Bioorg. Med. Chem., 2014, 22, 1609–1619 CrossRef PubMed.
- H. R. Nasiri, M. G. Madej, R. Panisch, M. Lafontaine, J. W. Bats, C. R. Lancaster and H. Schwalbe, J. Med. Chem., 2013, 56, 9530–9541 CrossRef CAS PubMed.
- D. Bhasin, S. N. Chettiar, J. P. Etter, M. Mok and P. K. Li, Bioorg. Med. Chem., 2013, 21, 4662–4669 CrossRef CAS PubMed.
- C. V. Ryu and D. H. Kim, Arch. Pharmacal Res., 1992, 15, 263–268 CrossRef CAS.
- V. K. Tandon, H. K. Maurya, N. N. Mishra and P. K. Shukla, Eur. J. Med. Chem., 2009, 44, 3130–3137 CrossRef CAS PubMed.
- J. J. Inbaraj and C. F. Chignell, Chem. Res. Toxicol., 2004, 17, 55–62 CrossRef CAS PubMed.
- V. K. Tandon, R. B. Chhor, R. V. Singh, S. Raj and D. B. Yadav, Bioorg. Med. Chem. Lett., 2004, 14, 1079–1083 CrossRef CAS PubMed.
- K. Sasaki, H. Abe and F. Yoshizaki, Biol. Pharm. Bull.l, 2002, 25, 669–670 CrossRef CAS.
- V. K. Tandon, D. B. Yadav, H. K. Maurya, A. K. Chaturvedi and P. K. Shukla, Bioorg. Med. Chem., 2006, 14, 6120–6126 CrossRef CAS PubMed.
- T. Kayashima, M. Mori, H. Yoshida, Y. Mizushina and K. Matsubara, Cancer Lett., 2009, 278, 34–40 CrossRef CAS PubMed.
- E. N. Da Silva Jr, D. F. de Deus, B. C. Cavalcanti, C. Pessoa, L. V. Costa-Lotufo, R. C. Motenegro, M. O. de Moraes, M. C. E. R. Pinto, C. A. de Simone, V. F. Fereira, M. O. F. Goulart, C. K. Z. Andrade and A. V. Pinto, J. Med. Chem., 2010, 53, 504–508 CrossRef PubMed.
- O. D. Zakharova, L. P. Ovchinnikova, L. I. Goryunov, N. M. Troshkova, V. D. Shteingarts and G. A. Nevinsky, Eur. J. Med. Chem., 2010, 45, 2321–2326 CrossRef CAS PubMed.
- J. Benites, J. A. Valderrama, K. Bettega, R. C. Pedrosa, P. B. Calderon and J. Verrax, Eur. J. Med. Chem., 2010, 45, 6052–6057 CrossRef CAS PubMed.
- N. Pradidphol, N. Kongakathip, P. Sittikul, N. Boonyalai and B. Kongkathip, Eur. J. Med. Chem., 2012, 49, 253–270 CrossRef CAS PubMed.
- V. K. Tandon, R. V. Singh, S. Raj, R. B. Chhor and Z. K. Khan, Boll. Chim. Farm., 2002, 141, 304–310 CAS.
- T. V. Ilina, E. A. Semenova, T. R. Pronvaeva, A. G. Pokrovskii, I. N. Nechepurenko, E. E. Shults, O. I. Anderrva, S. N. Kochetkov and G. A. Tolstikov, Dokl. Biochem. Biophys., 2002, 382, 56–59 CrossRef CAS PubMed.
- A. J. M. da Silva, C. D. Buarque, F. V. Brito, L. Aurelian, L. F. Macedo, L. H. Malkas, R. J. Hickey, D. V. S. Lopes, Y. L. B. Murakami, N. M. V. Silva, P. A. Melo, R. R. B. Laruso, N. G. Castro and P. R. R. Costa, Bioorg. Med. Chem., 2002, 10, 2731–2738 CrossRef CAS PubMed.
- E. N. da Silva, M. Cecillia, B. V. de Souza, A. V. Pinto, F. R. Pinto, M. O. F. Goulart, F. W. A. Barros, C. Pessoa, L. V. Costa-Lotufa, R. C. Montenegro, M. Moraesel and V. F. Ferreiraa, Bioorg. Med. Chem., 2007, 15, 7035–7042 CrossRef PubMed.
- M. A. Maetinez, G. Uevas, M. J. Estrada, I. Gonzalez, B. L. Hennasen and N. M. Ruvalcaba, J. Org. Chem., 1999, 64, 3684–3694 CrossRef.
- (a) Z. Xu, Y. Xiao, X. Qian, J. Cui and D. Cui, Org. Lett., 2005, 7, 889–892 CrossRef CAS PubMed; (b) Y. Kubo, M. Ymamoto, M. Ikeda, M. Takeuchi, S. Shinkai, S. Yamaguchi and K. Tamao, Angew. Chem., Int. Ed., 2003, 42, 2036–2040 CrossRef CAS PubMed.
- D. D. Perrin, W. L. Armarego and D. R. Perrin, Purification of Laboratory Chemicals, Pergamon Press, London, 1988, p. 260 Search PubMed.
- Bruker, APEX2, SAINT and SADABS, Bruker AXS Inc, Madison, Wisconsin, USA, 2007 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- L. R. Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef.
- L. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Akajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian03, revision, Gaussian, Inc., Pittsburgh, PA, 2003 Search PubMed.
- R. Patil, D. Chadar, D. Chaudhari, J. Peter, M. Nikalje, T. Weyhermüller and S. Salunke-Gawali, J. Mol. Struct., 2014, 1075, 345–351 CrossRef CAS.
- S. Salunke-Gawali, L. Kathawate, Y. Shinde, V. G. Puranik and T. Weyhermüller, J. Mol. Struct., 2012, 1010, 38–45 CrossRef CAS.
- S. Salunke-Gawali, O. Pawar, M. Nikalje, R. Patil, T. Weyhermüller, V. G. Puranik and V. B. Konkimalla, J. Mol. Struct., 2014, 1056–1057, 97–103 CrossRef CAS.
- S. Salunke-Gawali, L. Kathawate and V. G. Puranik, J. Mol. Struct., 2012, 1022, 189–196 CrossRef CAS.
- S. Salunke-Gawali, S. Y. Rane, V. G. Puranik, C. Guyard-Duhayan and F. Varret, Polyhedron, 2004, 23, 2541–2547 CrossRef CAS.
- S. Pal, M. Jadhav, T. Weyhermüller, Y. Patil, M. Nethaji, U. Kasabe, L. Kathawate, V. Badireenath Konkimalla and S. Salunke-Gawali, J. Mol. Struct., 2013, 1049, 355–361 CrossRef CAS.
- A. P. Ware, A. Patil, S. Khomane, T. Weyhermüller, S. S. Pingale and S. Salunke-Gawali, J. Mol. Struct., 2015, 1093, 39–48 CrossRef CAS.
- A. Patil, D. Lande, A. Nalkar, S. P. Gejji, D. Chakravarty, R. Gonnade, T. Moniz, M. Rangel, E. Periera and S. Salunke-Gawali, J. Mol. Struct., 2017, 1143, 495–514 CrossRef CAS.
- G. Agarwal, D. N. Lande, D. Chakravarty, S. P. Gejji, P. Gosavi-Mirkute, A. Patil and S. Salunke-Gawali, RSC Adv., 2016, 6, 88010–88029 RSC.
- Q. Lin, T. Lu, X. Zhu, T. Wei, H. Li and Y. Zhang, Chem. Sci., 2016, 7, 5341–5346 RSC.
- Q. Lin, P. Mao, Y. Fan, L. Liu, J. Liu, Y. Zhang, H. Yao and T. Wei, Soft Matter, 2017, 13, 7085–7089 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: FT-IR, 1H and 13C NMR, DSC are presented in Fig. S1 through Fig. S6. crystallography figures Fig. S7 and S8. Metal ion binding studies on H-3. Job plot for H-1 Fig. S10 through Fig. S12. Binding constant for H-1 Fig. S13. Job plot for H-3 Fig. S14 through Fig. S16. Binding constant for H-1 Fig. S17. Competative metal ion binding studies for H-3, Fig. S18 through Fig. S19. pH dependent binding (Fig. S20). Limit of detetion (LOD) in Fig. S21 and S22. Crystallographic Tables S1–S12. CCDC 1506931, 1575689 and 1506932 for H-1, H-3 and H-4 respectively. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ra10490a |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |