
Preparation of iodonium ylides: probing the fluorination of 1,3-dicarbonyl compounds with a fluoroiodane†
Gemma C. Geary,
Eric G. Hope,
Kuldip Singh and
Alison M. Stuart*
Department of Chemistry, University of Leicester, Leicester, LE1 7RH, UK. E-mail: Alison.Stuart@le.ac.uk
First published on 22nd January 2015
Abstract
The isolation of iodonium ylide 8, from the reaction of fluoroiodane 1 with ethyl 3-oxo-3-phenylpropanoate 5 in the presence of potassium fluoride, provides strong evidence that 1,3-dicarbonyl compounds undergo an addition reaction with fluoroiodane 1 to form an iodonium intermediate which can be deprotonated to generate an iodonium ylide. In the presence of TREAT-HF, however, the iodonium intermediate reacts to form the 2-fluoro-1,3-dicarbonyl product and we propose that fluoroiodane 1 simulates electrophilic fluorination via an addition/substitution mechanism. Further evidence to support this mechanism was obtained by successfully reacting the isolated iodonium ylide 8 with TREAT-HF, hydrochloric acid, acetic acid and p-toluenesulfonic acid to form the 2-fluoro-, 2-chloro-, 2-acetyl- and 2-tosyl-1,3-ketoesters respectively.
Introduction
The development of mild and efficient methods for introducing fluorine into organic molecules has always been a highly desirable target for the pharmaceutical and agrochemical industries because of the beneficial properties imparted by fluorine. Recently, hypervalent iodine reagents, in combination with nucleophilic sources of fluoride, have provided remarkable progress in the development of new fluorination synthetic strategies.1–6,8 Sanford has reported a mild Cu-catalysed nucleophilic fluorination of unsymmetrical diaryliodonium salts in order to access 18F-labelled aryl fluorides,1 whilst in 2014 two independent research groups introduced aryl iodonium ylides as alternatives to diaryliodonium salts for the direct preparation of electron rich and non-activated [18F]-fluoroaromatics.2Since Kitamura demonstrated that (difluoroiodo)arenes can be generated and used in situ for the fluorination of 1,3-dicarbonyl compounds and acetophenone derivatives, there has been renewed interest in the applications of these and related hypervalent iodine reagents.3,4 Both Kitamura and Shibata have now improved this process and shown that ArIF2 can be generated in situ using a catalytic amount of iodoarene in the presence of m-CPBA and hydrogen fluoride.3c,3d Shibata has extended this fluorination protocol to the intramolecular aminofluorination of alkenes and alternative procedures using PhI(OPiv)2/HF-pyridine and PhIO/BF3·Et2O have also been reported.4 A chiral (difluoroiodo)arene was first reported in 2013 and good enantioselectivities (61–81% ee) were obtained in the enantioselective aminofluorinations of a series of alkenes.4c
Togni first published two cyclic hypervalent iodine(III) reagents for electrophilic trifluoromethylation in 2006 and, since then, there has been an explosion in the number of publications worldwide on trifluoromethylation chemistry.5 Inspired by Togni's work, we reported the synthesis of an alternative electrophilic fluorinating reagent based on the cyclic hypervalent iodine(III) skeleton using a formal umpolung of fluoride. The air and moisture stable fluoroiodane 1 was prepared by the nucleophilic fluorination of hydroxyiodane 2 with triethylamine tris(hydrogen fluoride) (TREAT-HF) and by the nucleophilic substitution of trifluoroacetoxyiodane 3 and tosyliodane 4 with tetrabutylammonium fluoride (TBAF) (Scheme 1).6 Fluoroiodane 1 can also be synthesised by an electrophilic fluorination using Selectfluor, as well as by a nucleophilic substitution of the chloroiodane with spray-dried potassium fluoride.7 We, and Szabó, have demonstrated that fluoroiodane 1 can act like an electrophilic fluorinating reagent with 1,3-dicarbonyl compounds and styrenes respectively to create new C–F bonds.6,8 In this paper, however, we propose that the fluoroiodane actually simulates an electrophilic fluorination via an addition/substitution mechanism and evidence to support this mechanism will be presented.
 | ||
| Scheme 1 Synthesis of fluoroiodane 1. | ||
Results and discussion
The optimum reaction conditions for the fluorination of ethyl 3-oxo-3-phenylpropanoate 5 had been developed previously and used 2 equivalents of fluoroiodane 1 with 2.7 equivalents of TREAT-HF in dichloromethane at 40 °C for 24 hours (entry 1, Table 1).6 The air and moisture stability of fluoroiodane 1 is illustrated in entry 2 where it still performed well after being stored in air at room temperature for 7 weeks. Entry 3 demonstrates clearly that the addition of the acid, TREAT-HF, is essential for this fluorination since only 10% of the monofluorinated product 6 was isolated in the absence of TREAT-HF. However, when ethyl 3-oxo-3-phenylpropanoate 5 was reacted with fluoroiodane 1 under basic conditions using 1.2 equivalents of potassium fluoride in acetonitrile, the conversion to the monofluorinated product 6 decreased dramatically to only 18% and a new product 8 was observed for the first time (entry 4). On increasing the amount of potassium fluoride to 2.2 equivalents, conversion to 8 was much higher and, with 5.6 equivalents of potassium fluoride at 60 °C (entry 6), the conversion reached 100%. After purification by column chromatography, the spectroscopic and structural data indicated that the new product formed under basic conditions is the iodonium ylide 8.
|
||||||
|---|---|---|---|---|---|---|
| Entry | KF (no. equiv.) | TREAT-HF (no. equiv.) | Yield of 5a,b (%) | Yield of 6a,b (%) | Yield of 7a,b (%) | Yield of 8a,b (%) |
| a Determined by 1H and 19F NMR spectroscopy.b Isolated yield in parenthesis.c Fluoroiodane 1 stored in air at room temperature for 7 weeks.d Reaction at 60 °C. | ||||||
| 1 | 0 | 2.7 | 5 | 89 (63) | 6 | 0 |
| 2c | 0 | 2.7 | 11 | 85 | 3 | 0 |
| 3 | 0 | 0 | 80 | 20 (10) | 0 | 0 |
| 4 | 1.2 | 0 | 34 | 18 | 0 | 48 |
| 5 | 2.2 | 0 | 8 | 4 | 0 | 88 |
| 6d | 5.6 | 0 | 0 | 0 | 0 | 100 (42) |
The solid-state structure of iodonium ylide 8 is presented in Fig. 1. As expected, the C(10)–I(1) bond length is slightly shorter than the C(1)–I(1) bond length (Table 2) and is similar to those reported for other iodonium ylides.9 The negative charge of the ylide is delocalised into the carbonyl group of the ketone and consequently, the C(10)–C(11) bond length of 1.406(8) Å is slightly shorter than a normal C–C single bond, whilst the C(11)–O(2) bond length of 1.254(6) Å is slightly longer than a normal C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond.10 The C(10)–I(1)–C(1) bond angle of 98.7(2)° is typical for the pseudo T-shape of an iodonium ylide, where this bond angle is normally between 97 and 99°.9 Furthermore, there is a strong intramolecular interaction (2.729(3) Å) between I(1) and O(1) and the C(10)–I(1)⋯O(1) angle of 167.82(18)° is very similar to the F(1)–I(1)–O(1) bond angle in fluoroiodane 1 (166.4°).7a There are two further weak I(1)–O(2) and I(1)–O(4) intramolecular interactions (3.028(5) and 2.953(5) Å respectively), which are both shorter than the sum of the van der Waals radii (3.5 Å),11 and all of these intramolecular interactions contribute to the unusual stability of this acyclic iodonium ylide. In Fig. 2 the extended structure reveals that iodonium ylide 8 forms dimers in the solid-state through intermolecular hydrogen bonding between O(1) and O(2)′ (2.654(5) Å) and the solid-state packing diagram shows that there are also weak I(1)–O(3)′′ intermolecular interactions (2.975(4) Å) which causes long chains of these dimers to form (see Fig. 4 in the ESI†).
O double bond.10 The C(10)–I(1)–C(1) bond angle of 98.7(2)° is typical for the pseudo T-shape of an iodonium ylide, where this bond angle is normally between 97 and 99°.9 Furthermore, there is a strong intramolecular interaction (2.729(3) Å) between I(1) and O(1) and the C(10)–I(1)⋯O(1) angle of 167.82(18)° is very similar to the F(1)–I(1)–O(1) bond angle in fluoroiodane 1 (166.4°).7a There are two further weak I(1)–O(2) and I(1)–O(4) intramolecular interactions (3.028(5) and 2.953(5) Å respectively), which are both shorter than the sum of the van der Waals radii (3.5 Å),11 and all of these intramolecular interactions contribute to the unusual stability of this acyclic iodonium ylide. In Fig. 2 the extended structure reveals that iodonium ylide 8 forms dimers in the solid-state through intermolecular hydrogen bonding between O(1) and O(2)′ (2.654(5) Å) and the solid-state packing diagram shows that there are also weak I(1)–O(3)′′ intermolecular interactions (2.975(4) Å) which causes long chains of these dimers to form (see Fig. 4 in the ESI†).
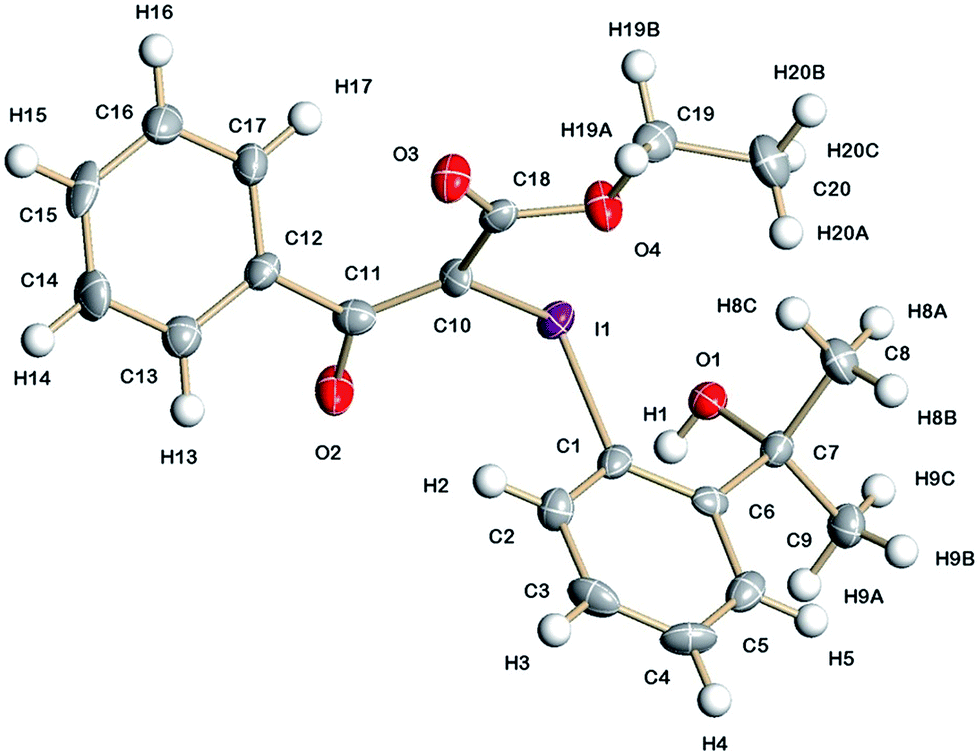 | ||
| Fig. 1 Molecular structure of iodonium ylide 8 showing 50% displacement ellipsoids. | ||
| Bond lengths | Iodonium ylide 8 | Bond lengths & angles | Iodonium ylide 8 |
|---|---|---|---|
| C(10)–I(1) | 2.068(5) | I(1)⋯O(1) | 2.729(3) |
| C(1)–I(1) | 2.132(6) | I(1)⋯O(2) | 3.028(5) |
| C(11)–C(10) | 1.406(8) | I(1)⋯O(4) | 2.953(5) |
| C(10)–C(18) | 1.445(7) | I(1)⋯O(3)′′ | 2.975(4) |
| C(11)–O(2) | 1.254(6) | O(1)⋯O(2)′ | 2.654(5) |
| C(18)–O(3) | 1.210(6) | C(10)–I(1)–C(1) | 98.7(2) |
| C(10)–I(1)⋯O(1) | 167.82(18) |
 | ||
| Fig. 2 Molecular structure of iodonium ylide 8 showing 30% displacement ellipsoids and intermolecular hydrogen bonding (2.654(5) Å). The hydrogen atoms have been omitted for clarity. | ||
The reaction between 1,3-dicarbonyl compounds and hypervalent iodine(III) reagents has been proposed to proceed via an iodonium intermediate3a,3d,12 and we propose a similar mechanism for the fluorination of 1,3-dicarbonyl compounds using fluoroiodane 1. The isolation of iodonium ylide 8, from the reaction of fluoroiodane 1 with ethyl 3-oxo-3-phenylpropanoate 5 in the presence of potassium fluoride (Table 1, entry 6), provides strong evidence that 5 underwent an addition reaction with fluoroiodane 1 to form an iodonium intermediate 9 which was then deprotonated to generate 8 (Scheme 2). The enol, which is the reactive tautomer of the ketoester, reacted with the electropositive iodine to form an iodinated intermediate which on ring opening the chelate sidearm, followed by proton transfer to the alkoxide, generated the iodonium intermediate 9. In the presence of base (potassium fluoride), 9 was deprotonated to form iodonium ylide 8 which can be stabilised by resonance. However, in the presence of TREAT-HF the iodonium intermediate 9 remained protonated, facilitating the formation of ethyl 2-fluoro-3-oxo-3-phenyl-propanoate 6 either by a reductive elimination or by a nucleophilic substitution.
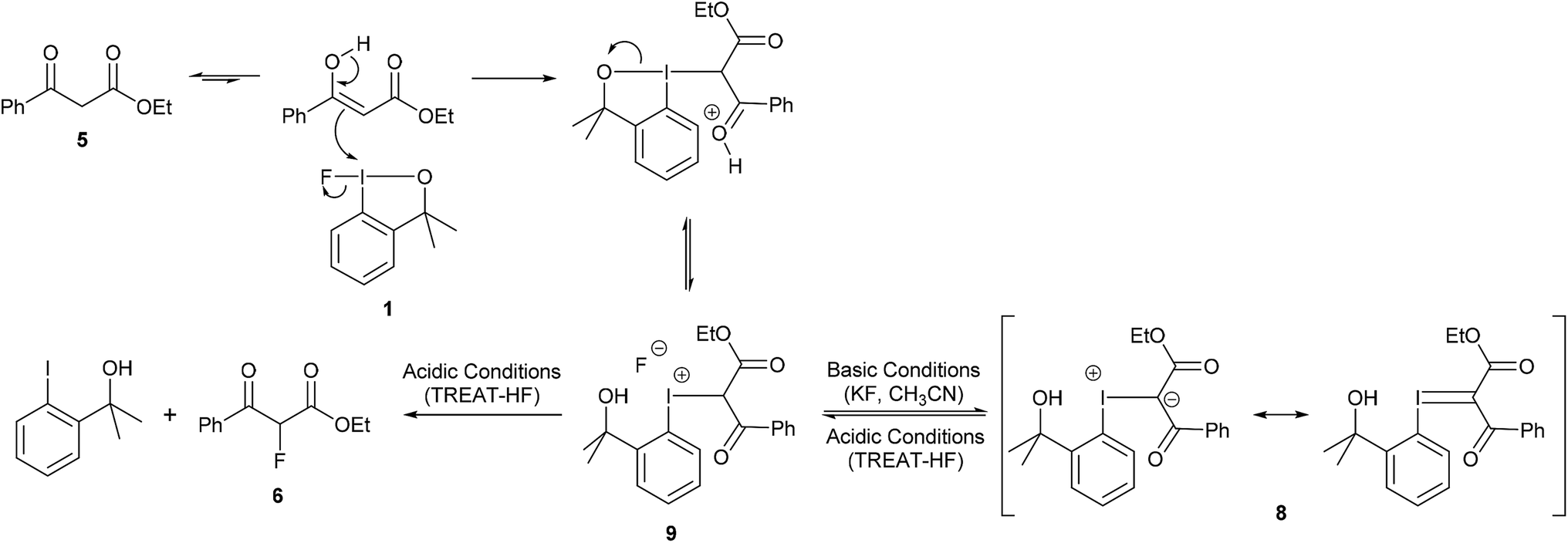 | ||
| Scheme 2 Proposed reaction mechanism. | ||
Since iodonium ylide 8 was synthesised under basic conditions, the reaction between ethyl 3-oxo-3-phenylpropanoate 5 and hydroxyiodane 2 was investigated because the reaction would be expected to generate hydroxide anion that could act directly as the base. Under these reaction conditions, iodonium ylide 8 was isolated in an excellent 84% yield (Scheme 3). Using the same approach, two new iodonium ylides 10 and 11 were prepared in good yields by reacting hydroxyiodane 2 with ethyl 3-(4-methoxyphenyl)-3-oxo-propanoate and 1,3-diphenyl-1,3-propanedione respectively (Scheme 3). Unfortunately, there was no reaction with diethyl malonate presumably because it exists exclusively as the keto tautomer and the reaction was only successful if the 1,3-dicarbonyl compound existed, at least to some extent, in the enol form.
 | ||
| Scheme 3 Synthesis of iodonium ylides from hydroxyiodane 2. | ||
Non-functionalised acyclic phenyliodonium ylides are normally prepared by reacting (diacetoxyiodo)benzene with 1,3-dicarbonyl compounds under basic conditions at 0 °C, but these iodonium ylides possess low thermal stability and can often only be prepared and used in situ.13,14 For example, the phenyliodonium ylide 12 derived from dimethyl malonate (Fig. 3) is unstable in organic solvents at room temperature and decomposes to iodobenzene and the dimethyl malonate dimer.14b Cyclic iodonium ylides (e.g. 13 which is derived from Meldrum's acid), however, are more stable and the increased stability is thought to be due to secondary C–I⋯O bonding which has been observed in the solid-state structures.9c Recently, the solubility and stability of iodonium ylides have been improved by the introduction of an ortho-alkoxy group in the phenyl ring of the phenyliodonium moiety (e.g. 14).9a,9b The new iodonium ylides (8, 10 and 11) reported here are stabilised by an ortho-propan-2-ol group making them surprisingly stable for acyclic iodonium ylides and three different intramolecular interactions between iodine and oxygen were observed in the molecular structure of iodonium ylide 8. Not only are these new iodonium ylides stable at room temperature, but they were all purified by column chromatography on silica gel.
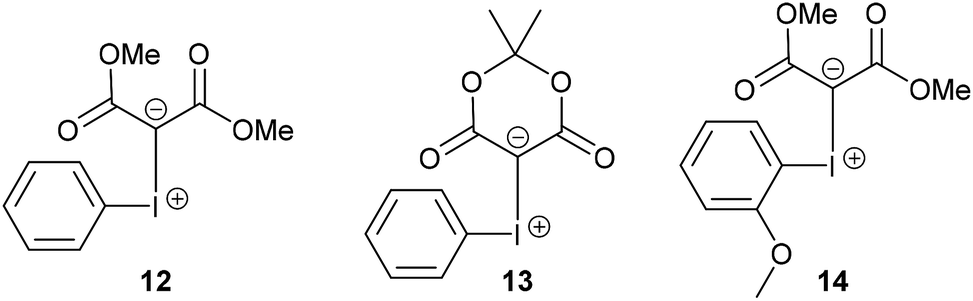 | ||
| Fig. 3 Examples of an acyclic iodonium ylide 12, a cyclic iodonium ylide 13 and an ortho-functionalised iodonium ylide 14. | ||
We decided to investigate the fluorinations of the iodonium ylides (Table 3) with TREAT-HF in order to test them as alternative starting materials in the proposed reaction mechanism in Scheme 2. When iodonium ylide 8 was reacted with 2.7 equivalents of TREAT-HF, the same monofluorinated product 6 was isolated in 36% yield (entry 1) presumably via a protonation and reductive elimination/nucleophilic substitution sequence. Crucially, no fluorinated products were obtained when 8 was reacted with a non-protic source of fluoride, TBAF (entry 2). A very similar result was also obtained with iodonium ylide 10, which on reaction with TREAT-HF formed the monofluorinated product in 28% yield. In contrast, the reaction between iodonium ylide 11 and TREAT-HF proceeded much better to give 2-fluoro-1,3-diphenylpropane-1,3-dione in 64% isolated yield. For the first time the difluorinated product (9%) was also produced along with 1,3-diphenylpropane-1,3-dione (15%). The difference in this reaction is that 2-fluoro-1,3-diphenylpropane-1,3-dione contains both keto and enol tautomers, whilst the monofluorinated products of the ketoesters only exist in the keto form. Consequently, 2-fluoro-1,3-diphenylpropane-1,3-dione could react with the ylide and then undergo a second fluorination.
|
|||||
|---|---|---|---|---|---|
| Entry | Ylide | Fluorinating reagent (no. equiv.) | Monofluoro producta,b (%) | Difluoro producta,b (%) | Dicarbonyl producta,b (%) |
| a Calculated by 1H and 19F NMR spectroscopy.b Isolated yields in parenthesis. | |||||
| 1 | 8 | Et3N·3HF (2.7 equiv.) | 100 (36) | Trace | Trace |
| 2 | 8 | TBAF (2.7 equiv.) | 0 | 0 | 0 |
| 3 | 10 | Et3N·3HF (2.7 equiv.) | 100 (28) | 0 | 0 |
| 4 | 11 | Et3N·3HF (2.7 equiv.) | 79 (64) | 11 (9) | 10 (15) |

We have extended this methodology to other protic acids (Table 4). When iodonium ylide 8 was reacted with 2.7 equivalents of hydrochloric acid, the monochlorinated product was isolated in 83% yield. Excellent isolated yields (72–93%) were also achieved by reacting 8 with acetic acid and p-toluenesulphonic acid in order to form the monoacetylated and monotosylated products respectively. The monotosylated product can also be prepared in 77% yield by the direct reaction between ethyl 3-oxo-3-phenyl-propanoate 5 and tosyliodane 4.

Conclusions
In conclusion, we propose that fluoroiodane 1 simulates electrophilic fluorination with 1,3-dicarbonyl compounds via an addition/substitution mechanism. The isolation of iodonium ylide 8, from the reaction of fluoroiodane 1 with ethyl 3-oxo-3-phenylpropanoate 5 in the presence of potassium fluoride, provided strong evidence that 5 underwent an initial addition reaction with fluoroiodane 1 to form an iodonium fluoride 9. Under basic conditions iodonium fluoride 9 was deprotonated to form iodonium ylide 8, whereas under acidic conditions it reacted with TREAT-HF to produce ethyl 2-fluoro-3-oxo-3-phenylpropanoate 6 either by a reductive elimination or by a nucleophilic substitution. The ortho-propan-2-ol group increased the stability of iodonium ylide 8 and the solid-state structure showed that it was stabilised by three different intramolecular iodine–oxygen interactions. Iodonium ylide 8 was also reacted successfully with a series of protic acids (TREAT-HF, hydrochloric acid, acetic acid and p-toluenesulfonic acid) to form the 2-fluoro-, 2-chloro-, 2-acetyl- and 2-tosyl-1,3-ketoesters respectively giving further evidence to support the proposed protonation and reductive elimination/nucleophilic substitution sequence.References
- (a) N. Ichiishi, A. F. Brooks, J. J. Topczewski, M. E. Rodnick, M. S. Sanford and P. J. H. Scott, Org. Lett., 2014, 16, 3224 CrossRef CAS PubMed; (b) N. Ichiishi, A. J. Canty, B. F. Yates and M. S. Sanford, Org. Lett., 2013, 15, 5134 CrossRef CAS PubMed.
- (a) B. H. Rotstein, N. A. Stephenson, N. Vasdev and S. H. Liang, Nat. Commun., 2014, 5, 5365 CrossRef PubMed; (b) J. Cardinale, J. Ermert, S. Humpert and H. H. Coenen, RSC Adv., 2014, 4, 17293 RSC.
- (a) T. Kitamura, S. Kuriki, M. H. Morshed and Y. Hori, Org. Lett., 2011, 13, 2392 CrossRef CAS PubMed; (b) T. Kitamura, K. Muta and K. Muta, J. Org. Chem., 2014, 79, 5842 CrossRef CAS PubMed; (c) T. Kitamura, K. Muta and S. Kuriki, Tetrahedron Lett., 2013, 54, 6118 CrossRef CAS PubMed; (d) S. Suzuki, T. Kamo, K. Fukushi, T. Hiramatsu, E. Tokunaga, T. Dohi, Y. Kita and N. Shibata, Chem. Sci., 2014, 5, 2754 RSC.
- (a) Q. Wang, W. Zhong, X. Wei, M. Ning, X. Meng and Z. Li, Org. Biomol. Chem., 2012, 10, 8566 RSC; (b) J. Cui, Q. Jia, R.-Z. Feng, S.-S. Liu, T. He and C. Zhang, Org. Lett., 2014, 16, 1442 CrossRef CAS PubMed; (c) W. Kong, P. Feige, T. de Haro and C. Nevada, Angew. Chem., Int. Ed., 2013, 52, 2469 CrossRef CAS PubMed.
- J. Charpentier, N. Früh and A. Togni, Chem. Rev., 2014, 114 DOI:10.1021/cr500223h.
- G. C. Geary, E. G. Hope, K. Singh and A. M. Stuart, Chem. Commun., 2013, 49, 9263 RSC.
- (a) C. Y. Legault and J. Prevost, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, o1238 CAS; (b) V. Matoušek, E. Pietrasiak, R. Schwenk and A. Togni, J. Org. Chem., 2013, 78, 6763 CrossRef PubMed.
- N. O. Ilchenko, B. O. A. Tasch and K. J. Szabó, Angew. Chem., Int. Ed., 2014, 53, 12897 CrossRef CAS PubMed.
- (a) C. Zhu, A. Yoshimura, P. Solntsev, L. Ji, Y. Wei, V. N. Nemykin and V. V. Zhdankin, Chem. Commun., 2012, 48, 10108 RSC; (b) C. Zhu, A. Yoshimura, L. Ji, Y. Wei, V. N. Nemykin and V. V. Zhdankin, Org. Lett., 2012, 14, 3170 CrossRef CAS PubMed; (c) N. W. Alcock, A. P. Bozopoulos, E. Hatzigrigoriou and A. Varvoglis, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 1300 CrossRef.
- (a) F. S. Stephens, J. Chem. Soc., 1965, 5640 RSC; (b) G. Berthier and J. Serre, The Chemistry of the Carbonyl Group, ed S. Patai, Wiley, New York, 1966, ch. 1, pp. 1–78 Search PubMed.
- R.-Y. Yang, L.-X. Dai and C.-G. Chen, J. Chem. Soc., Chem. Commun., 1992, 1487 RSC.
- K. Gondo and T. Kitamura, Molecules, 2012, 17, 6625 CrossRef CAS PubMed.
- V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299 CrossRef CAS PubMed.
- (a) J. Cardinale and J. Ermert, Tetrahedron Lett., 2013, 54, 2067 CrossRef CAS PubMed; (b) S. R. Goudreau, D. Marcoux and A. B. Charette, Org. Synth., 2010, 87, 115 CrossRef CAS; (c) S. R. Goudreau, D. Marcoux and A. B. Charette, J. Org. Chem., 2009, 74, 470 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1036695. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra15733h |
| This journal is © The Royal Society of Chemistry 2015 |