
Potential in vitro and in vivo colon specific anticancer activity in a HCT-116 xenograft nude mice model: targeted delivery using enteric coated folate modified nanoparticles
Renuka Khatika,
Pankaj Dwivedia,
Vijayabhaskar Reddy Junnuthulaa,
Komal Sharmaa,
Krishna Chuttanib,
Anil Kumar Mishrab and
Anil Kumar Dwivedi*a
aPharmaceutics Division, Central Drug Research Institute, Sector 10, Janki Puram Extention, Sitapur Road, Lucknow 226031, India. E-mail: anilcdri@gmail.com; renukadops@gmail.com; Tel: +91 9415910144
bDivision of Cyclotron & Radiopharmaceutical Sciences, Institute of Nuclear Medicine and Allied Sciences (INMAS), DRDO, Delhi-110054, India
First published on 12th January 2015
Abstract
The aim of this study was to develop a drug delivery system for specific targeting in colon cancer treatment. We have developed a Eudragit S-100 (ES) coated folic acid (FA) conjugated gliadin (Gd) delivery system for the effective targeting of overexpressed folate receptors (FRs) in colon cancer. The FA conjugate with Gd (FA–Gd) was synthesized and characterized using FTIR and 1H NMR, and this developed conjugate was used to prepare curcumin (CU) loaded nanoparticles (NPs) by a desolvation method. FA–CU–GdNPs were further coated with ES and ES–FA–CU–GdNPs were obtained. The ES–FA–CU–GdNPs were also capable of inducing cell caspase dependent apoptosis in Caco-2 cell lines and exhibited DNA intercalating activity. In therapeutic experiments the ES–FA–CU–GdNPs were administered orally to HCT-116 tumor-bearing nude mice. In vivo bio-distribution data showed that ES–FA–CU–GdNPs had delivered the maximum amount of NPs to the colon and tumor after 12 hours, reflecting its targeting potential for the colon and tumor. A gamma scintigraphy study suggested that ES–FA–CU–GdNPs remain intact at low pH and released NPs slowly at pH 7.4 in the colon. This study provides evidence that ES–FA–CU–GdNPs hold the promise to address overexpressed FRs in colorectal cancer and were found to be safe for oral administration for a prolonged duration.
1. Introduction
Cancer is a principal cause of distress and death, with more than 10 million people being diagnosed with the disease every year.1,2 The most commonly used treatments of cancer are chemotherapy or surgical procedures, which are associated with a number of disadvantages, i.e. they damage healthy tissues leading to systemic toxicity, multi-drug resistance, nonselective distribution of drugs and adverse side effects.3,4 Nanotechnology, one of the efficacy technologies which have been adapted by medical experts can pave the way to overcoming the problems related to safe drug delivery. Sustained release of drugs might overcome problems associated with high dose related toxicity and drug resistance, which can also be used as a new concept in chemotherapy.2 The increased efficacy of traditional medicines may open up a new opportunity for future cancer therapies.5 Taking this into consideration, the upcoming anticancer drug from a natural herbal extract, curcumin (CU), offers a solution to the obstacles involved in chemotherapy, by showing safety and a chemopreventive effect which can be loaded into nanoparticles (NPs) that deliver the drug into a malignant cell.6CU, a natural hydrophobic phenolic compound, isolated from the rhizome of Curcuma longa (turmeric), has a wide spectrum of healthy functions and pharmacological activities.7,8 It also exhibits anticarcinogenic properties9 and sensitizes tumor cells for chemotherapy and radiation therapies, with a low intrinsic toxicity.10,11 Despite all of these advantages, CU has a limited usage due to its poor aqueous solubility, and thus, minimal systemic bioavailability. Various formulations of CU were investigated in an effort to improve its oral bioavailability, including encapsulation in liposomes12,13 and CU complexed with phospholipids14 and cyclodextrin.15 NPs have also been reported to improve its bioavailability.16
In view of this, one of the approaches to achieve enhanced bioavailability of CU may be to encapsulate it with natural polymers (e.g., polysaccharides, glycoproteins, polypeptides, and lipids). Natural polymers are less toxic and safer than synthetic polymers.17,18 In addition, natural polymers or glycoproteins are promising carriers for the delivery and controlled release of drugs, because they are biodegradable, nontoxic, and stable.19 Gliadin (Gd), a plant protein, contains neutral amino acids which enhance hydrogen bonding within the mucus layer, and lipophilic amino acids that support hydrophilic interactions.20,21 Gliadin (Gd) has been reported to serve as an enzyme carrier of superoxide dismutase, protecting the acid degradation of the enzyme and allowing it to release in the intestine. This property can be used to deliver the maximum amount of drug to the colon. Gd has previously been used for the preparation of NPs for drug delivery and controlled release applications.22 The stability of gliadin nanoparticles (GdNPs) increased when they were chemically cross-linked with glutaraldehyde. Although various drugs have been loaded into GdNPs for delivery and controlled release, GdNPs loaded with anticancer drugs have not yet been fully explored.
In order to improve the effectiveness and uptake of the NPs into specific targeted cells, target-specific ligands, such as FA can be used. FA is a stable compound that has a high affinity for FRs23,24 which are overexpressed on many human epithelial cancer cell surfaces, especially in the colon. The targeting ability of a drug can be improved by conjugating the drug delivery carrier with FA.25,26 FA conjugate delivery systems, which are covalently derivatized via the folate’s γ-carboxyl moiety, can maintain a high affinity for the FRs.27 Later, the unligated FRs may recycle to the cell surface to move more FA conjugates into the cell cytoplasm.24 The mechanism of endocytosis is mediated by FRs.
In this scenario, the choice of a receptor mediated targeted delivery has more advantages for efficient intracellular delivery over the one that relies on cell membrane markers. Colon cancer cells have a FA deficiency hence they arrest more FA conjugated NPs than normal cells.1,28–30 To date, NPs have been widely functionalized with ligands and antibodies to target FRs on cancer cells.31,32
In the present investigation, FA rich FA–CU–GdNPs, have been prepared and found to be more effective in targeting the overexpressed FRs in colorectal cancer when compared to CU–GdNPs. We can conclude that the FA–CU–GdNPs have a higher cancer targeting potential in tumor-bearing nude mice. We also determined the pharmacokinetics, biodistribution and tumor growth inhibition. We then evaluated the potential of these FA–CU–GdNPs coated with Eudragit S-100 (ES) in inhibiting angiogenesis and suppressing the growth of colon tumors in vitro and in vivo.
2. Results
2.1. Conjugated FA–Gd characterization by FTIR
FA conjugation to Gd was accomplished by a new application for the Mitsunobu reaction (Fig. 1). The FTIR spectra of the FA modified Gd showed typical peaks (Fig. 2). The O–H broadened stretching band in the region of 3281 cm−1 was found. IR spectra of the FA conjugated Gd, show characteristic absorption bands at 1063 cm−1 which were due to C–O stretching vibrations of the ester formed. The IR spectra of FA conjugated Gd shows C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at nearly 1630 cm−1.
O stretching at nearly 1630 cm−1.
 | ||
| Fig. 1 Schematic diagram showing conjugation of FA with Gd. | ||
 | ||
| Fig. 2 FT-IR spectrum of FA conjugated Gd. | ||
2.2. NMR spectroscopic characterization of FA conjugation to Gd
Chemical conjugation of FA with Gd was also confirmed using 1H NMR spectroscopy. 1H NMR spectra showed that the FA was successfully conjugated to Gd. In the 1H NMR (300 MHz) spectrum of FA conjugated Gd (Fig. 3), the appearance of aromatic protons (Ar–H) from δ 6.7 to δ 7.9 indicates the presence of a FA moiety in our product while C–H and –CH2 protons were found from δ 0.8 to δ 2.2. Some –CH2 protons present near –N– were shifted and were present from δ 3.7 to δ 4.5 and –OH protons of FA conjugated Gd appeared from δ 5.2 to δ 5.3.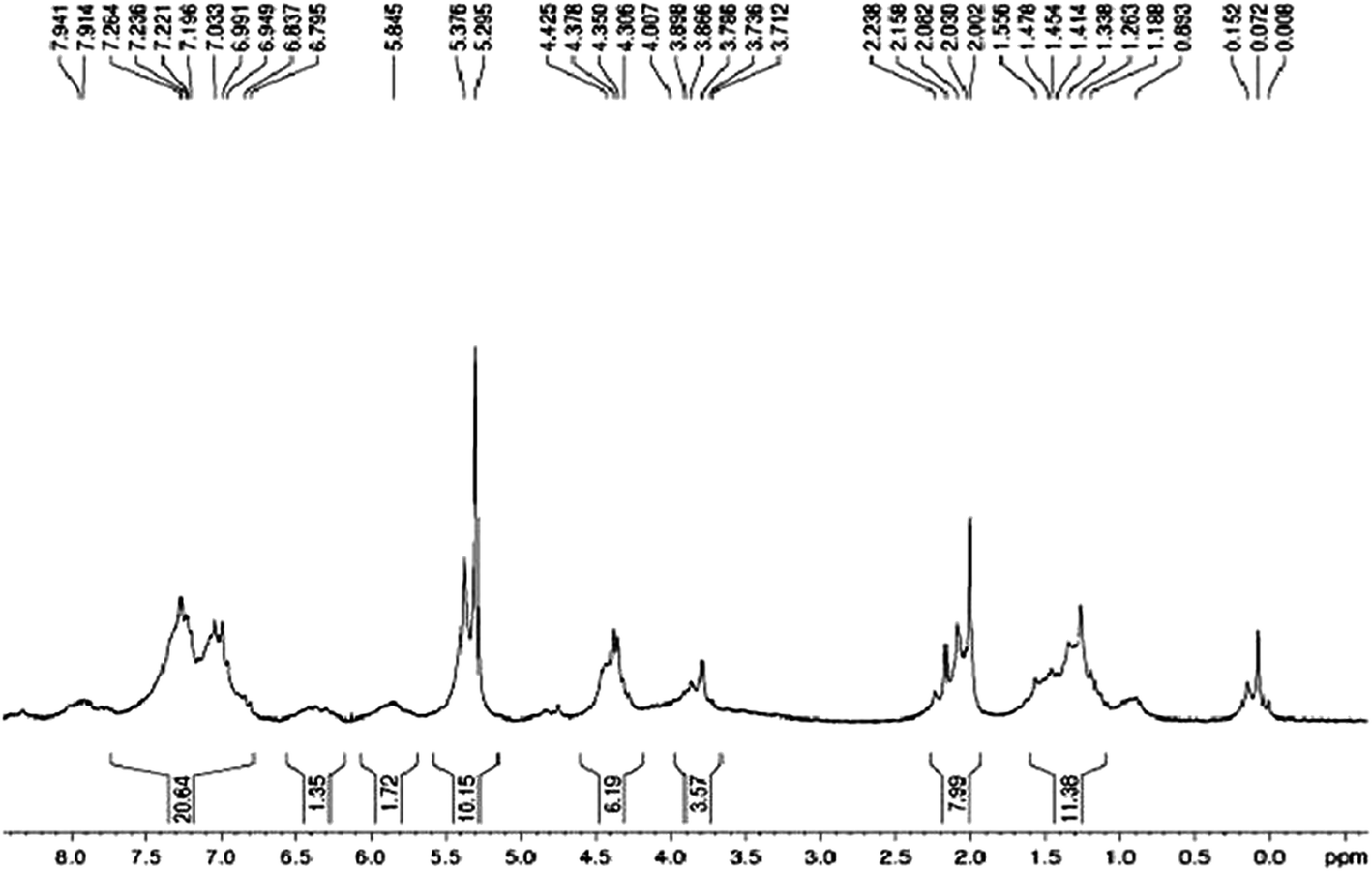 | ||
| Fig. 3 1H NMR spectrum of FA conjugated Gd. | ||
2.3. Differential scanning calorimetry (DSC)
Interaction of CU with Gd was also confirmed by DSC, which gives information about the compatibility of CU with Gd. DSC thermograms of CU, Gd and a physical mixture of CU–Gd are shown in (Fig. 4) in which a sharp endothermic peak at 183 °C has been observed during the DSC of CU due to the dehydration of the crystalline network, whereas in the thermogram of Gd, a broad hump near 70–75 °C emerged. However, in the case of the physical mixture of CU–Gd the thermogram shows no difference in their physical characteristics and peaks emerged in the regular places, at 70–75 °C for Gd and 183 °C for CU revealing no interaction between these two.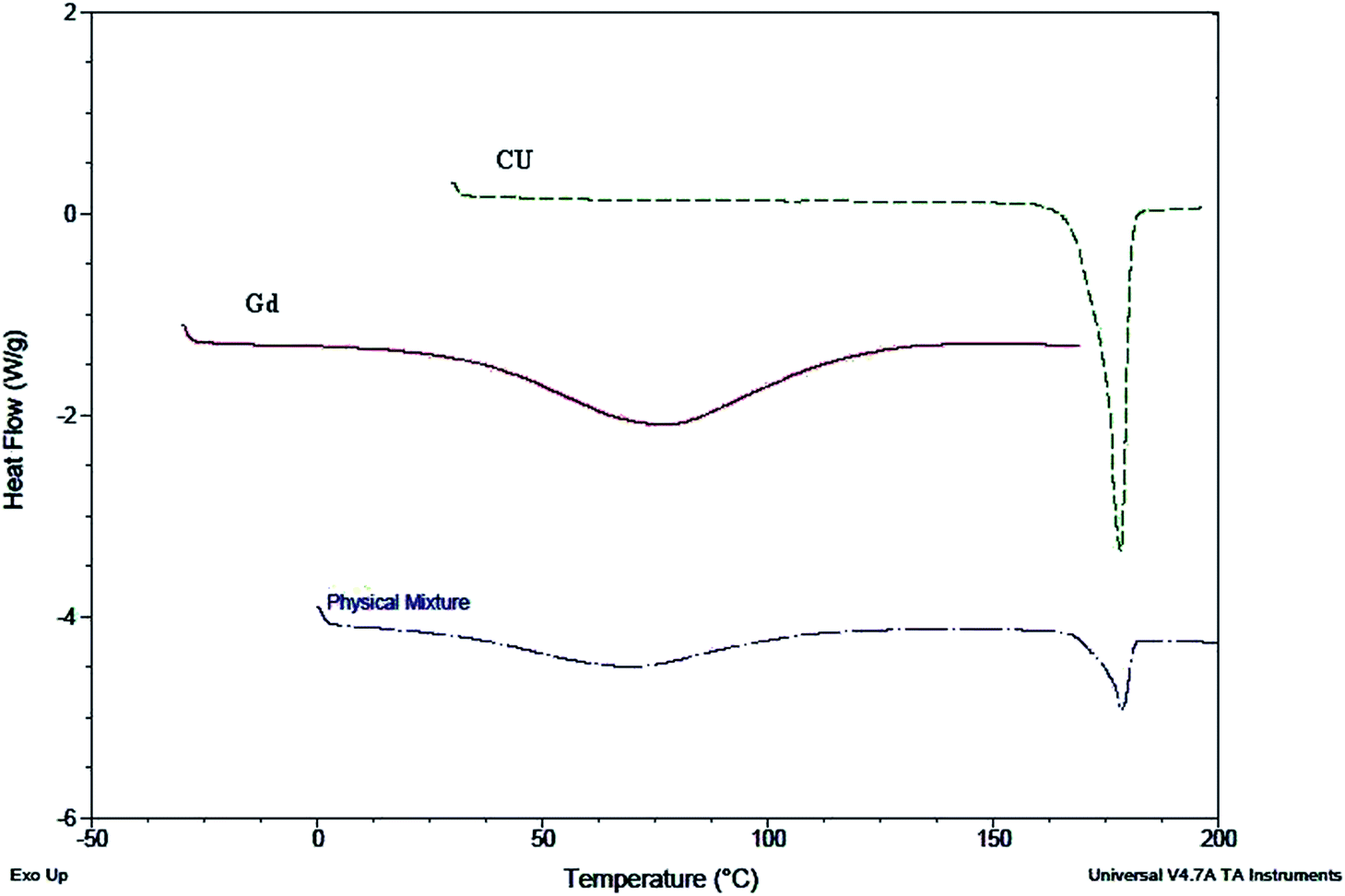 | ||
| Fig. 4 The compatibility of Gd and CU measured using DSC. | ||
2.4. Characterization of ES–CU–GdNPs and ES–FA–CU–GdNPs
The mean particle sizes of ES–GdNPs, ES–FA–GdNPs, ES–CU–GdNPs and ES–FA–CU–GdNPs were found to be 98.3 ± 3.5 nm, 112.4 ± 8.6 nm, 137.2 ± 4.1 nm and 249.3 ± 7.2 nm with PDI values of 0.131, 0.127, 0.191 and 0.175 respectively. The zeta potentials of ES–GdNPs, ES–FA–GdNPs, ES–CU–GdNPs and ES–FA–CU–GdNPs were found to be −8.3 ± 1.2 mV, −21.3 ± 1.6 mV, −10.7 ± 2.0 mV and −27.1 ± 1.8 mV respectively, with narrow size distributions measured by DLS. These data have been illustrated in Table 1. The %EE for ES–CU–GdNPs was found to be 49.4 ± 3.8%, whereas for ES–FA–CU–GdNPs there was increase in the %EE (53.2 ± 4.1%). Fig. 5 shows TEM images of the morphological characteristics of FA–CU–GdNPs. Surface modification is signified by the incessant opaque and irregular layer of the FA–CU–GdNPs.| Formulation | Particle size (nm)± SD | PDI | Zeta potential (mV) ± ZD | %Entrapment efficacy |
|---|---|---|---|---|
| ES–FA–CU–GdNPs | 249.3 (7.2) | 0.175 | −27.1 (1.8) | 53.2 (4.1) |
| ES–CU–GdNPs | 137.2 (4.1) | 0.191 | −10.7 (2.0) | 49.4 (3.8) |
| ES–GdNPs | 98.3 (3.5) | 0.131 | −8.3 (1.2) | — |
| ES–FA–GdNPs | 112.4 (8.6) | 0.127 | −21.3 (1.6) | — |
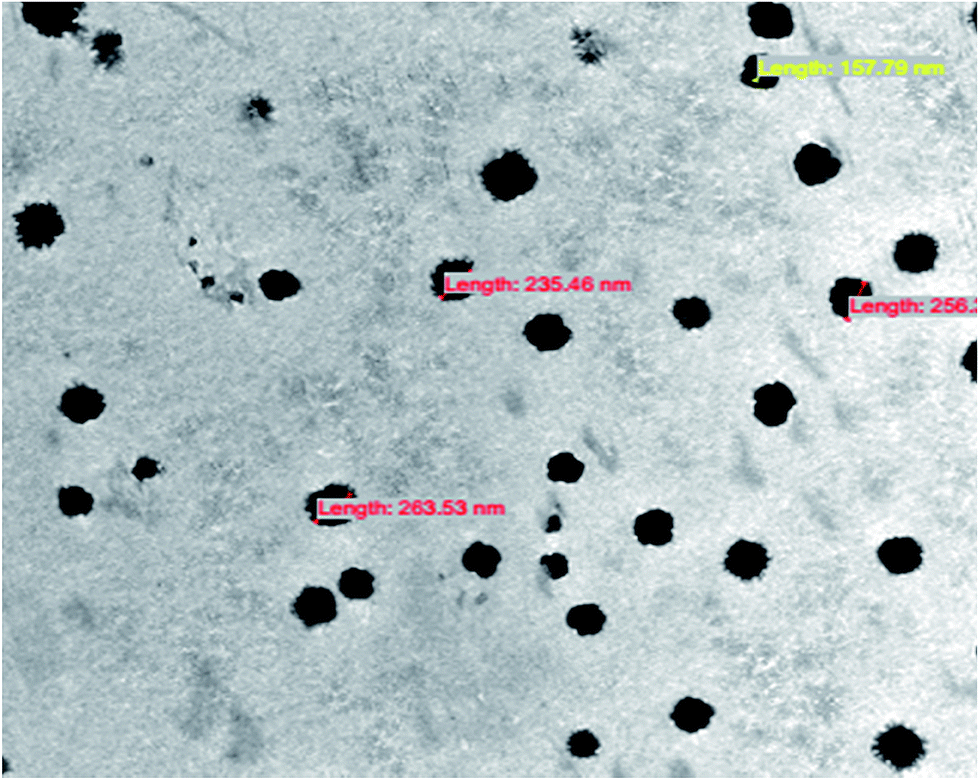 | ||
| Fig. 5 TEM image of ES–FA–CU–GdNPs. | ||
2.5. In vitro release study
The in vitro release profiles of CU from ES–CU–GdNPs and ES–FA–CU–GdNPs were studied by using a pre-activated dialysis bag (Sigma; cut off MW 12 kD) at pH 1.2, 7.4 and 6.8, while the temperature of the medium was kept at 37 °C. The in vitro release of CU began instantly and was complete within 6 h. It was observed that there was no drug release up to 2–3 hours in the case of both the ES–CU–GdNPs and ES–FA–CU–GdNPs, and begins release only after 3 hours in the SIF at pH 7.5. This can be explained by the fact that the ES polymer contains carboxyl groups that ionize from neutral in alkaline media. As the ionization takes place, the integrity of the coating is disturbed and the NPs are released. Furthermore, the release conducted in the presence of rat caecal and colonic contents, which have a pH of 6.8, from the cumulative amount of CU released from ES–CU–GdNPs, the drug release was found to be 52.23% ± 2.16%, 64.09 ± 2.32% and 68.3% ± 2.9% at the end of 8, 12 and 24 hours, respectively. In the case of the ES–FA–CU–GdNPs, drug release was found to be 46.44% ± 1.39%, 58.16% ± 2.07% and 63.23 ± 2.17% at the end of 8, 12 and 24 hours, respectively. The results shown in Fig. 6 indicate that ES–CU–GdNPs and ES–FA–CU–GdNPs release the maximum amount of CU in the colon environment due to the sustained release from the matrix system. | ||
| Fig. 6 In vitro drug release profile of CU, ES–CU–GdNPs and ES–FA–CU–GdNPs in different gastrointestinal simulated fluids (SGF, SIF, and SCF). | ||
2.6. In vitro uptake studies
An in vitro uptake study has been conducted to scrutinize the significance of prepared ES–FA–CU–GdNPs over ES–CU–GdNPs. The study was conducted on Caco-2 cells using flow cytometry and the uptake intensity of ES–CU–GdNPs and ES–FA–CU–GdNPs were measured against a blank. Fig. 7 shows the results of the macrophage uptake of ES–CU–GdNPs and ES–FA–CU–GdNPs in Caco-2 cells from the flow cytometric uptake study. The mean fluorescence intensity shows an almost two fold (>2.127) enhanced uptake of ES–FA–CU–GdNPs over ES–CU–GdNPs in Caco-2 cell lines revealing the significantly higher (P < 0.05) uptake of ES–FA–CU–GdNPs in comparison with ES–CU–GdNPs, as the presence of folate receptors might be mediating the uptake.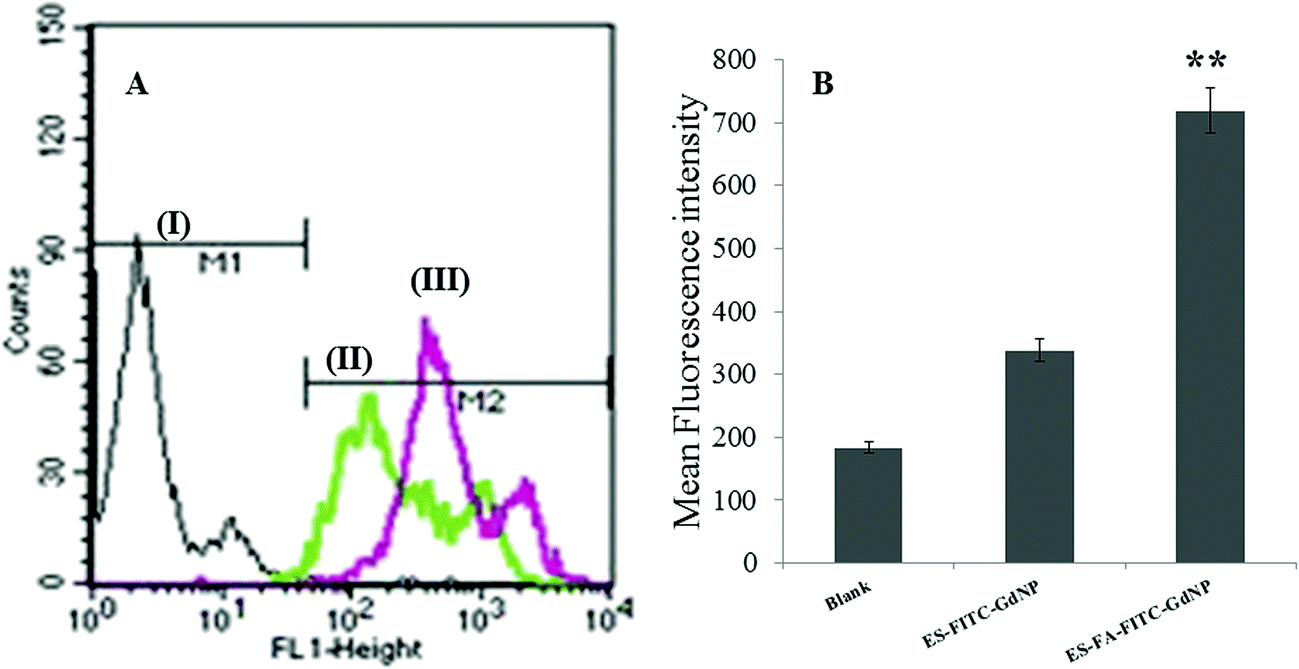 | ||
| Fig. 7 Flow cytometric uptake studies in Caco-2 cells. (A) X-axis represents fluorescence inside the cells of (i) control cells (ii) ES–FITC–GdNPs and, (iii) ES–FA–FITC–GdNPs. (B) Bar graph represents the mean fluorescent intensity in (i) control cells (ii) ES–FITC–GdNPs and, (iii) ES–FA–FITC–GdNPs. **p<0.001. | ||
2.7. Apoptosis studies
Toward finding a possible reason for how ES–CU–GdNPs and ES–FA–CU–GdNPs are able to kill cancer cells, we have analyzed the apoptosis induction in ES–CU–GdNPs and ES–FA–CU–GdNPs treated colon cancer cells such as Caco-2. Apoptosis is one of the major cell death pathways induced by anticancer agents and we have evaluated the apoptosis inducing effects of ES–CU–GdNPs and ES–FA–CU–GdNPs by Annexin V/propidium iodide (PI) binding. As can be seen in Fig. 8, The fraction of apoptotic cells was higher in the case of cells treated with ES–FA–CU–GdNPs than with ES–CU–GdNPs or with Plain CU, the ES–FA–CU–GdNPs increased the percentage of apoptotic cells in Caco-2 cells from 0.40% in the control, while CU had very few cells i.e. 9.69% in the apoptotic phase. The values for ES–CU–GdNPs and ES–FA–CU–GdNPs were shown to be 16.82% and 54.70% in treated cells. From the above data it is evident that ES–FA–CU–GdNPs truly induced apoptosis specifically in colon cancer cells irrespective of ER status and did not induce apoptosis in normal cells. | ||
| Fig. 8 Apoptotic cells detected using flow cytometry with Annexin V conjugated with PI. The stained Caco-2 cells were treated with different formulations; (A) control (B) Plain CU (C) ES–CU–GdNPs and (D) ES–FA–CU–GdNPs. | ||
2.8. Cytotoxicity study
The dose dependent cytotoxicity of the NPs was determined by MTT assay.33 Caco-2 cells were plated with plain CU, ES–CU–GdNPs and ES–FA–CU–GdNPs at different concentrations and cultured for 24 h with 5% CO2 at 37 °C and the cytotoxicity for cells was determined by MTT assay. The percentage cell viability for different NPs with differing concentrations was found to be significantly less in comparison to that of plain CU. The blank formulations ES–GdNPs and ES–FA–GdNPs showed slight cytotoxicity (less than 10%) revealing the safety of the excipients used. The cell viability at concentrations of 5 μM, 10 μM, 20 μM and 40 μM was estimated and the results revealed that the viability of the cells exposed to ES–FA–CU–GdNPs was less than that of the cells exposed to ES–CU–GdNPs. At a higher concentration (40 μM), ES–FA–CU–GdNPs and ES–CU–GdNPs showed 16.4 ± 0.92% and 22.7 ± 1.13% cell viability respectively. At 20μM, ES–FA–CU–GdNPs and ES–CU–GdNPs showed less than 40% cell viability, whereas at a lower concentration (10 μM) ES–FA–CU–GdNPs showed 33.5 ± 1.67% and ES–CU–GdNPs showed more than 45% cell viability. The data have been represented in (Fig. 9). | ||
| Fig. 9 %Cell cytoxicity of Caco-2 cells exposed to 5 μM, 10 μM, 20 μM and 40 μM equivalent concentrations of CU in plain CU, ES–CU–GdNPs and ES–FA–CU–GdNPs. The values shown are means and standard deviations (n = 3), **p < 0.001. | ||
2.9. Analysis of pharmacokinetic parameters after oral administration
The overall pharmaceutical targeting efficacy of the CU loaded surface engineered ES–FA–CU–GdNPs was evaluated in HCT-116 tumor-bearing nude mice. The plasma concentration profiles of a CU dose equivalent to 25.0 mg per kg body weight after oral administration of the free CU, ES–CU–GdNPs and ES–FA–CU–GdNPs were measured and the pharmacokinetic parameters were determined using non-compartment modeling as summarized in Table 2 (Fig. 10). The MRT and AUC0–∞ values for the ES–FA–CU–GdNPs (14.65571 ± 3.52 h, 108![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 37.5 ± 102.34 ng h−1 mL) were approximately 3.8 fold higher, compared to those for free CU and those for the ES–CU–GdNPs (11.4301 ± 2.14 h, 8123.57 ± 85.1 ng h−1 mL) were approximately 2.9 fold higher, compared to those for free CU (5.655 ± 2.57 h, 2794.44 ± 61.13 ng h−1 mL−1). The ES–CU–GdNPs exhibited a rapid increase in the plasma level, which reached a maximal plasma concentration of Cmax = 847.067 ± 29.67 ng mL−1 compared with that of ES–FA–CU–GdNPs (Cmax = 706.7 ± 24.89 ng mL−1) and CU (Cmax = 537.47 ± 15.05 ng mL−1). This clearly evinces the enhanced pharmacokinetics, better bioavailability and a more prolonged retention in systemic circulation of the ES–FA–CU–GdNPs compared with the properties observed upon administration of ES–CU–GdNPs and plain CU.
37.5 ± 102.34 ng h−1 mL) were approximately 3.8 fold higher, compared to those for free CU and those for the ES–CU–GdNPs (11.4301 ± 2.14 h, 8123.57 ± 85.1 ng h−1 mL) were approximately 2.9 fold higher, compared to those for free CU (5.655 ± 2.57 h, 2794.44 ± 61.13 ng h−1 mL−1). The ES–CU–GdNPs exhibited a rapid increase in the plasma level, which reached a maximal plasma concentration of Cmax = 847.067 ± 29.67 ng mL−1 compared with that of ES–FA–CU–GdNPs (Cmax = 706.7 ± 24.89 ng mL−1) and CU (Cmax = 537.47 ± 15.05 ng mL−1). This clearly evinces the enhanced pharmacokinetics, better bioavailability and a more prolonged retention in systemic circulation of the ES–FA–CU–GdNPs compared with the properties observed upon administration of ES–CU–GdNPs and plain CU.
| Formulation | Cmax (ng ml−1) | Tmax (h) | AUC0–∞(ng h−1 ml−1) | MRT (h) |
|---|---|---|---|---|
| a Cmax: maximal plasma concentration, Tmax: time to reach maximal plasma concentration, AUC: area under plasma concentration versus time curve, MRT: mean residual time. **p < 0.01 Significances vs. CU, ##p < 0.01 denotes ES–FA–CU–GdNPs vs. ES–CU–GdNPs. | ||||
| CU | 537.47 ± 15.05 | 3 ± 0.14 | 2794.44 ± 61.13 | 5.655 ± 2.57 |
| ES–CU–GdNPs | 706.7 ± 24.89 | 6 ± 0.09 | 8123.57 ± 85.1** | 11.4301 ± 2.14 |
| ES–FA–CU–GdNPs | 847.067 ± 29.67 | 8 ± 1.28 | 10837.5 ± 102.34** ## | 14.65571 ± 3.52 |
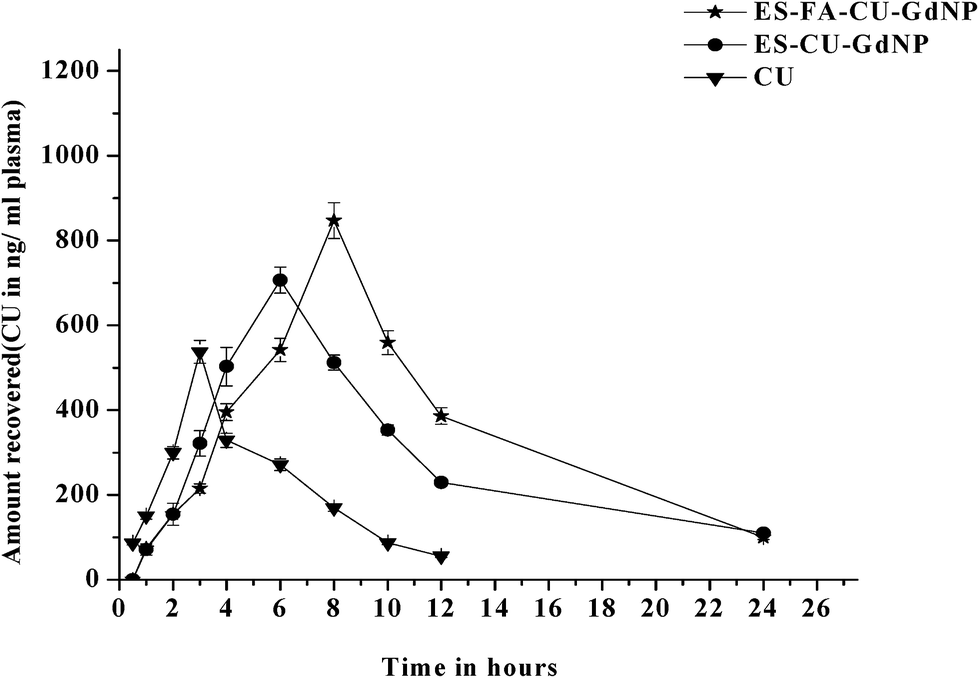 | ||
| Fig. 10 The in vivo bioavailability of CU, ES–CU–GdNPs and ES–FA–CU–GdNPs. The values are mean ± SD (n = 3). | ||
2.10. Tissue distribution study
A tissue distribution study was carried out to evaluate the in vivo CU release in different parts of the GIT from enteric coated NPs. The results of the drug distribution from the ES–CU–GdNPs and ES–FA–CU–GdNPs in the upper GIT after oral administration are displayed in Fig. 11. Entire GIT parts were homogenized, and the content of CU was quantitatively evaluated using an HPLC assay method. The free CU accumulates progressively in the stomach, where up to 72.9% of the dose is localized after 2 h post administration whereas only 1.91% of CU was found after 6 h. In all of the cases, the CU concentrations were considerably higher for the ES–FA–CU–GdNPs than for the ES–CU–GdNPs. The CU concentrations in the colon and tumor tissue after 12 hours were observed to be 9.34 and 11.6 (ES–CU–GdNPs) and 8.26 and 30.44 μg CU per g tissue (ES–FA–CU–GdNPs), respectively.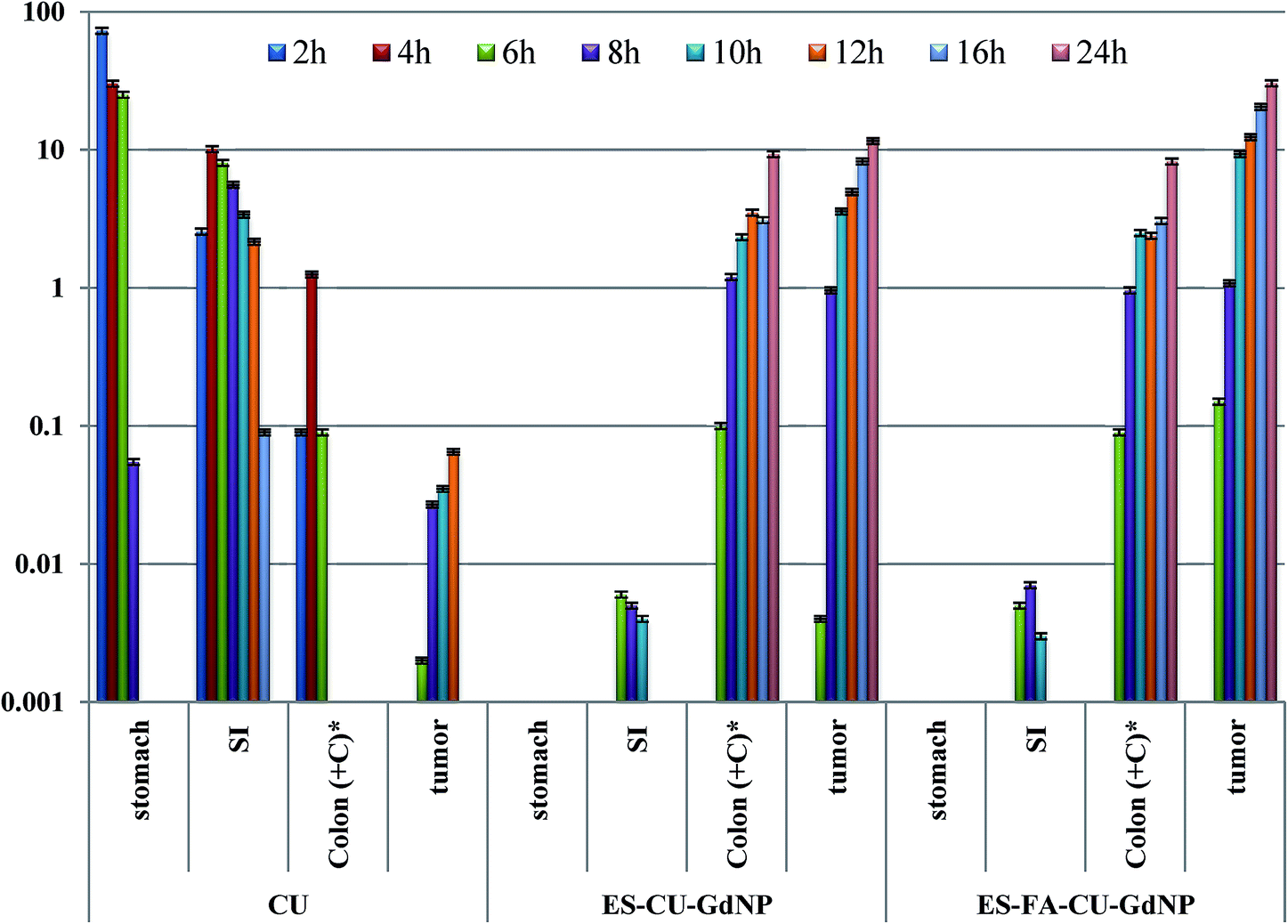 | ||
| Fig. 11 Distribution profiles of CU, ES–CU–GdNPs and ES–FA–CU–GdNPs in various tissues as a function of time after oral dose (25 mg kg−1) to mice represented by mean ± SD values obtained from three animals per time point. Colon (+C)* includes colon contents and tissue, caecal contents, and tissue; CU; curcumin, SI; small intestine. | ||
2.11. γ-Scintigraphy study
The gastric transit and colon arrival times of the designed NPs, bearing 99mTc-DTPA, in nude mice were recorded using γ-scintigraphy. The mean gastric emptying time of the formulation was found to be 1.50 ± 0.17 h. The mean intestinal transit time was calculated to be 3.20 ± 0.22 h and the mean colon arrival time was 6.60 ± 0.27 h (Fig. 12) after the administration of the NPs.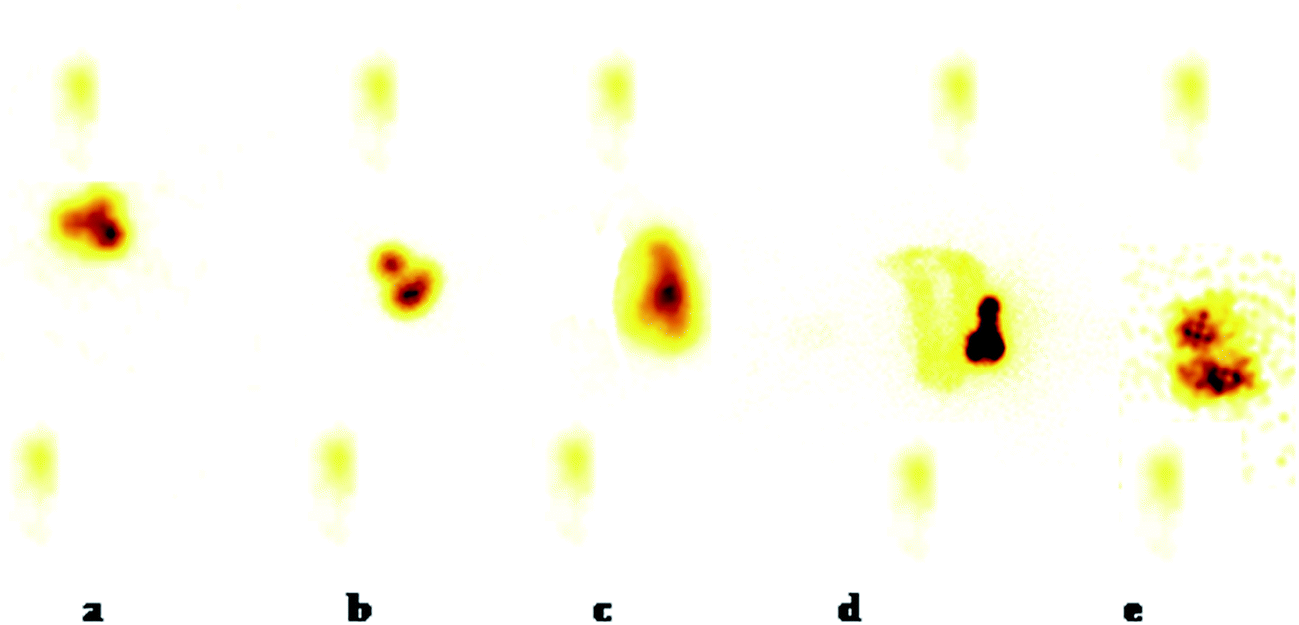 | ||
| Fig. 12 γ-Scintigraphs captured at different time intervals following oral administration of NPs: (a) after 1 h showing the release of a negligible amount of tracer in the stomach from enteric coated NPs, (b) after 3 h showing the release of tracer in the ascending colon from enteric coated NPs, (c) after 6 h showing the release of tracer in the ascending colon and the entry of the NPs into the transverse colon, (d) after 12 h showing the distribution of liberated radioactivity through the whole colon from the enteric coated NPs and (e) after 24 h showing the radioactivity in the distal part of the colon from the enteric coated NPs. | ||
2.12. Assessment of anti-tumor cancer targeting efficiency
The percentage body weight changes of the HCT-116 tumor-bearing nude mice model after oral administration of the CU loaded formulations was calculated up to 35 days as shown in Fig. 13(A). It clearly suggests that the developed formulations do not affect the body weight of the nude mice, while in case of the normal saline treated group the loss of body weight was observed These results further confirmed the higher tumor treatment potential demonstrated by the ES–FA–CU–GdNPs.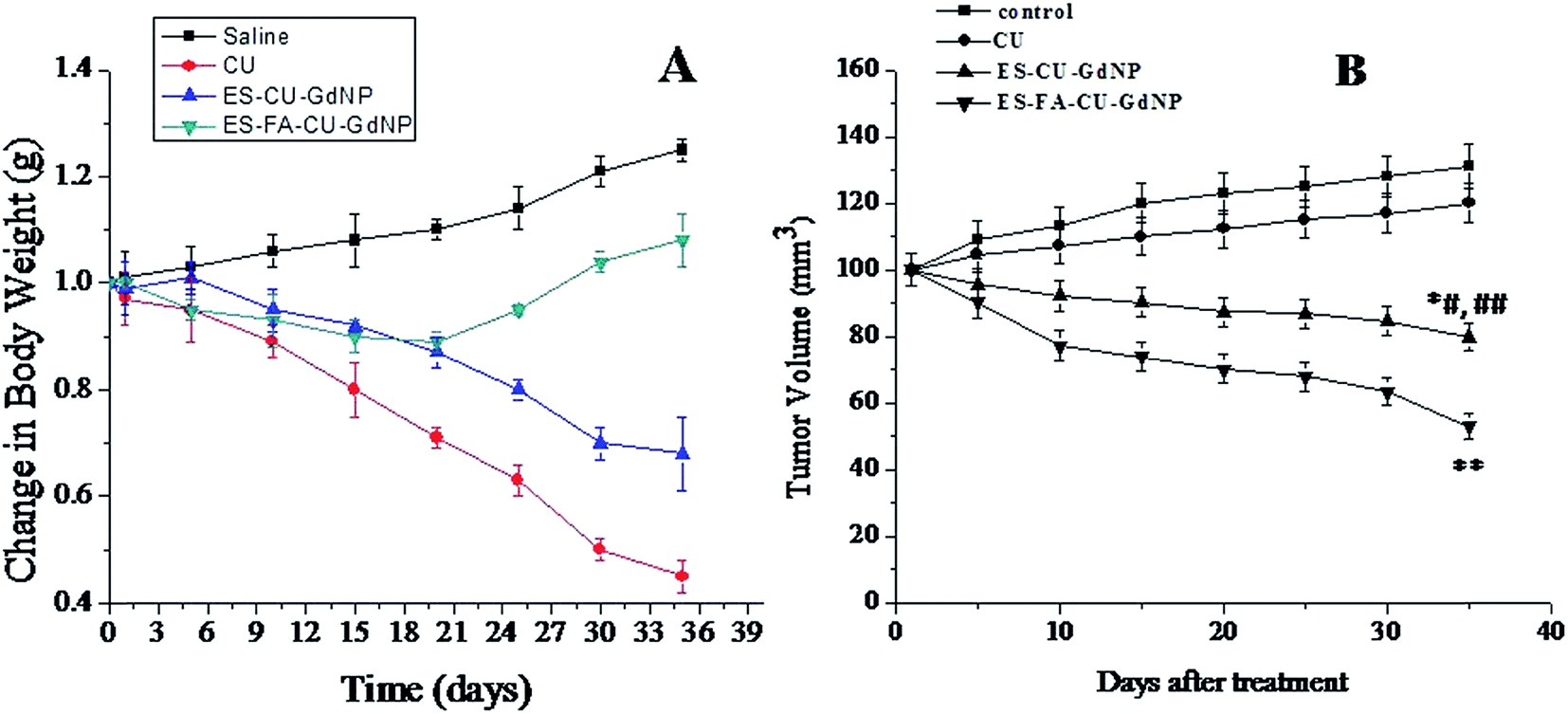 | ||
| Fig. 13 (A) Body weight changes in tumor-bearing nude mice after administration of CU solution and CU loaded NPs formulations and (B) tumor regression analysis after the oral administration of the control, CU, ES–CU–GdNPs and ES–FA–CU–GdNPs (25 mg per kg body weight dose). The ES–FA–CU–GdNPs treated group showed suppression of tumor growth compared with the other groups (lower) (n = 3). **p < 0.001 is for ES–FA–CU–GdNPs versus the control and CU, *#p < 0.01 is for ES–CU–GdNPs versus the control and ##p < 0.05 depicts the comparison of ES–CU–GdNPs versus CU. | ||
The in vivo tumor targeting efficacy was assayed in the HCT-116 tumor-bearing nude model and the starting tumor size was approximately 100 mm3 for all dose receiving groups, for the developed ES–CU–GdNPs and ES–FA–CU–GdNPs as well as normal saline and plain CU. The tumor volume (mm3) was 129.01, 118.7, 102.5 and 90.5 in the case of normal saline, free CU, ES–CU–GdNPs and ES–FA–CU–GdNPs after 5 days post treatment. The size of the tumor volume (mm3) was reduced to 79.8 and 43.89 after 35 days post treatment with the ES–CU–GdNP and ES–FA–CU–GdNP formulations, respectively [Fig. 13(B)]. (**p < 0.001 is for ES–FA–CU–GdNPs versus the control and CU, *#p < 0.01 is for ES–CU–GdNPs versus the control and ##p < 0.05 depicts the comparison of ES–CU–GdNPs versus CU.
3. Discussion
NPs derived from natural sources have been established to be of enormous profit for drug delivery and cancer therapy.34,35 The aim of this present investigation was to target CU to the colon, delivered by encapsulating it in ES–FA–CU–GdNPs. The motive of the specific delivery of CU to the colon for targeting the colon cancer was not achievable by delivering the CU by unmodified, simple CU–GdNPs, due to the unspecific delivery and loss of CU in undesired areas. A specific targeting moiety which can be exploited to deliver CU to the colon is required. To fulfill our requirements, FA, which has been reported previously26 to bind to the FRs present on the colon, has been exploited. The binding ability of the FA to the FRs overexpressed in malignancies in the colon allows for the specific delivery of CU. FA, a stable, inexpensive, and generally poorly immunogenic chemical was conjugated with Gd. Gd was conjugated to FA molecules using a new application for the Mitsunobu reaction. The formation of FA–Gd was confirmed through FT-IR and NMR spectroscopic studies, which clearly indicated conjugate formation. The FA–Gd conjugate was further used for preparing FA–CU–GdNPs, an FA rich delivery system, by a desolvation method, this system has been further coated with ES in order to specifically target CU to the colon.26,36 The FA binds to cell surface FRs with nanomolar affinity. After binding, the cell membrane introverts and pinches off to form an endosome. As the endosomal partition acidifies, the folate conjugate and CU discharge from the receptor into the cytosol. Although the reduced folate carrier is thought to be present in virtually all cells, in contrast, membrane bound FRs largely reprocess back to the cell surface, allowing for the delivery of further folate-linked drugs into the cell.37 To examine the targeting specificity of ES–FA–CU–GdNPs, ES–CU–GdNPs, and blank ES–GdNPs have also been prepared by the same desolvation method. TEM micrographs confirmed the nanosize range and show clearly coated spherical shaped ES–FA–CU–GdNPs and the change in the amount of surface charge alters the dielectric state of the ES–FA–CU–GdNPs, consequently resulting in increases in the zeta potential values. The %EE of ES–FA–CU–GdNPs was found to be slightly higher than that of ES–CU–GdNPs. The compatibility of Gd with CU was confirmed by DSC and was found to be attuned. The in vitro CU release profiles for the ES–FA–CU–GdNPs and ES–CU–GdNPs over 24 h are shown in Fig. 6. For the ES–FA–CU–GdNPs and ES–CU–GdNPs, there was no release of CU at pH 1.2 as anticipated; however, the release was observed when the pH of the medium was adjusted to 7.4 or 6.8. This clearly exhibits that the maximum fraction of CU was available in the colon and that this pH-dependent solubility of ES was exploited to avoid the rapid dissolution of CU during the initial transit of the ES–FA–CU–GdNPs through the gastric cavity. The drug release from the ES–FA–CU–GdNPs shows a similar behavior to that of the ES–CU–GdNPs, apart from it having a slightly slower rate due to FA conjugated on the surface of the CU–GdNPs which provides the possibility to often fight against cancer cells, resulting in a decreased cancer cell viability (shown in the in vitro cytotoxicity section).38 Conventional dissolution testing is less likely to allow accurate calculation of the in vivo performance of the colon specific delivery systems triggered by bacteria residing in the colon, due to factors related to the colon environment such as insufficiency of fluid, decreased motility, and presence of micro flora.39 Hence, release studies were performed in an alternate release medium (PBS 6.8 with rat caecal contents). After 12 h the percentage of CU released in the SCF (pH 6.8) containing 4% rat caecal contents with enzyme initiation at 37 °C, was found to be 68% and 63% for the ES–CU–GdNP and ES–FA–CU–GdNP formulations respectively. Thus, the step release of CU from the matrix at pH 6.8 in the colonic fluid was due to diffusion through nanopores of the polymer network or degradation of the polymer matrix by colonic bacteria. Significantly higher (p < 0.001) uptake of the ES–FA–CU–GdNPs over the ES–CU–GdNPs in Caco-2 cell lines generates interest that there is still scope for the enhanced localization of ES–FA–CU–GdNPs in colon macrophages. The ES–FA–CU–GdNPs were able to localize a higher intensity of fluorescence inside the cells compared to the ES–CU–GdNPs because of better interaction with the FRs on the macrophages, confirmed by apoptosis.Apoptosis is a key regulator of physiological growth control and for regulation of tissue homeostasis, but it is highly deregulated in cancer. Implementation of apoptosis is regulated by an excess of cell signaling pathways and recent chemotherapeutic agents solely exhibit their action by targeting these signaling pathways. The enhanced therapeutic ability of the dual effect of formulations with distinct cellular targets is able to hit signaling cascades simultaneously, thus ultimately causing enhanced apoptosis.
The cytotoxicity of ES–FA–CU–GdNPs was found to be significantly (p < 0.001) higher than both that of ES–CU–GdNPs and CU. This effect could have been attributed to the preferential binding of FA with the FRs in the Caco-2 cells, thus inhibiting up-regulated cell growth. The results illuminate the low toxicity of the blank NPs as well as the ability of the ES–FA–CU–GdNPs to preferentially target CU to colon cancer cells. The cell cytotoxicity studies reveal a negligible cytotoxicity of both the blank ES–FA–GdNPs and GdNPs in the Caco-2 cells lines indicating that the FA is not adding any kind of further cytotoxicity to the formulation, instead it facilitates the uptake without any damage. Having the dual advantage of the surface modified NPs for the release of CU in the colon, as well as the receptor mediated uptake by the cancer cells, its application for the targeted delivery of bioactive is in the offing.
Our experimental conclusions that FA causes ligand specific internalization and enhanced endocytosis of the NPs which leads to superior cytotoxicity also are supported by the related findings of various research groups. Folate decorated poly(lactide)–vitamin E TPGS nanoparticles for the targeted delivery of paclitaxel were synthesized by the group of Pan et al., and they have shown the improved advantages of the NPs versus the pure drug in achieving an enhanced therapeutic effect.40 Further, the behavior of the NPs in vivo correlated well with their in vitro interaction with tumor cells. Indeed, the in vitro anti-tumor activity of the NPs was tested by using the MTT method and was performed after the cells directly interacted with free CU or the NP formulation. In this process, the cellular uptake and the resulting intracellular release play important roles. But in vivo, the NPs with an anti-tumor effect undergo a complex biological process, in which the CU should first be targeted to the tumor site and then operate an inhibitory effect on the tumor cells, determined by factors that are greatly different from those in vitro.
One of the major interests in using ES–FA–CU–GdNPs is to improve the in vivo bioavailability of CU. Interestingly, our results showed that ES–FA–CU–GdNPs and ES–CU–GdNPs have a 3.8 and 2.9 fold greater AUC0–∞ compared to plain CU, respectively, which improved bioavailability.
Biodistribution studies indicated that the ES–FA–CU–GdNPs reached the tumor and were more effective in treating colon tumors as compared to ES–CU–GdNPs and the free CU. This may be due to the ligand mediated uptake of ES–FA–CU–GdNPs in the tumor and release of the drug in the tumor instead of the colon. The reason for this amount of drug in the colon may be due to the larger number of NPs uptaken from the tumor surface via receptor mediated endocytosis and a few of the NPs are also expected to penetrate through tumor leaky vasculature at the FRs; in a human colon adenocarcinoma the gaps could be as large as 400 nm and the NP size is well below this pore size i.e. in the size range of 150 to 335 nm, hence it could be a reason for successfully achieving the targeting strategy.
The most useful technique, to date, to evaluate in vivo behavior of dosage forms in animals and humans is external scintigraphy or γ-scintigraphy, After testing the in vitro colon targeting behavior of the ES–FA–CU–GdNPs, it was worthwhile to evaluate their in vivo behavior in terms of the residence time of the NPs in different parts of the GI tract. An interesting fact about the transit of the material is that during the initial hours of the study, the NPs remain firm which may cause fast transit and as time passed, the rigidity of the NPs became weaker due to penetration of solvent, and that led to slow transit through the rest of the GI tract.41 Also, gastric emptying values from 0.4 to 4.4 h were observed when multi-particulate radiolabeled NPs were administered to human volunteers.
It was clearly seen from the captured scintigraphs that a small amount of the radiolabeling was released into the stomach (Fig. 12a). This corresponds to the effective enteric coating of the NPs. It is evident from the scintigraph taken after 3 h (Fig. 12b) that a small quantity of tracer was released into the small intestine during a 1.85 to 3.20 h time period. The NPs remain intact as they descend down through the small intestine. The radiolabeled tracer activity, which was released in the small intestinal environment, is visible around the NPs. After 3.0 h, as the formulation entered into the ascending colon, the release of the tracer increased (Fig. 12c). This is due to a change in the pH of the lower GIT leading to the dissolution of the enteric coating and thus, release of the tracer. After 5 and 6 h, the formulation remained in the ascending colon but the release of radioactivity from the NPs was considerably improved as the coating dissolved with time in the colon, and its shape was distorted with the liberation of a considerably higher amount of tracer. After 11–12 h (Fig. 12d) the formulation entered into the transverse colon leaving the ascending colon filled with the tracer released. The NPs completely disintegrated after 10 h (Fig. 12e) and after 24 h, the movement of intact radiolabelled NPs from the stomach and the small intestine to the colon, without the tracer being released, and complete degradation of the network in the colon reveals the efficiency of this pH dependent system.
The in vivo anticancer study once again indicated that compared with the CU, the ES–CU–GdNPs and ES–FA–CU–GdNPs exhibited a notably enhanced anticancer efficacy. There was a significant difference observed between mice treated with saline and CU with the NP treated mice. Fig. 13A and B show that the relative body weight and the tumor volume in the tumor-bearing nude mice were obviously different compared with the various experimental groups. Both saline and CU showed similar anti-cancer activities within 30–35 days. Neither saline nor CU had any measurable influences on tumor growth, in both groups the tumor volumes and weights increased rapidly. For nude mice treated with ES–FA–CU–GdNPs, the decrease in body weight was limited and a complete tumor regression was observed for over 50% of the mice compared with the saline or CU groups. The increase in the body weights of saline-treated mice and CU-treated mice has been observed due to the increase in the tumor volume. It can be concluded that preparation of ES–FA–CU–GdNPs and ES–CU–GdNPs were beneficial against colon cancer.
4. Materials and methods
4.1. Materials
Curcumin (CU) and gliadin (Gd) were purchased from Merck India Ltd., Mumbai (India). Sodium azide-fluorescein isothiocyanate (FITC), fetal bovine serum (FBS), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), Tween 80 and Sorbitan trioleate (Span85), folic acid (FA),1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), sodium chloride, N,N′-dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP) were purchased from Sigma Aldrich (St. Louis, USA). Triethyl citrate, and the dialysis bag (cut off mol. wt. 12000 Dalton) were purchased from Himedia (Mumbai, India). HPLC grade acetonitrile and methanol were purchased from Spectrochem India. Well plates for cytotoxicity and uptake studies were purchased from Greiner Bio One 110 (Frickenhausen, Germany). All other chemicals used were of analytical grade. Triple distilled water was used throughout the study prepared by a Milli-Q plus 185 purification system (Bedford, Massachusetts).
4.2. Cell culture
Caco-2 cells were maintained in Roswell Park Memorial Institute medium (RPMI 1640, Merck), supplemented with 10% FBS (Merck) and a 1% antibiotic–antimycotic solution (Merck) at 37 °C in a humidified incubator with 5% CO2. All of the stock solutions of the compound were prepared in cell culture grade DMSO and stored at −20 °C. The human colorectal carcinoma cell line HCT-116 was kindly gifted by Dr Jayanta Sarkar, Scientist, Biochemistry Division CSIR-Central Drug Research Institute (CDRI). The cells were grown in McCoy’s 5A media (Sigma Aldrich) supplemented with antibiotics, L-glutamine and 10% fetal bovine serum (Gibco BRL) at 37 °C. All of the NPs were diluted in culture media prior to their use in experiments.4.3. Synthesis of FA conjugated Gd
Gd was esterified by FA using the Mitsunobu reaction, to give the FA conjugated Gd.42 FA and Gd were refluxed in the presence of tetrahydrofuran (THF), diethyl azodicarboxylate (DEAD) and triphenylphosphine (Ph3P). The product obtained was dialyzed for purification in two steps; firstly against a phosphate buffered saline solution (PBS pH 7.4) for 3 days and then against water for 4 days. The FA–Gd conjugate was collected by lyophilization and kept for further use. A schematic representation of the reaction has been illustrated in Fig. 1.4.4. Development of CU–GdNP and FA–CU–GdNP formulations
CU–GdNPs were prepared using a desolvation method as previously reported with slight modifications.36 Briefly, Gd and CU were dissolved in a 20 mL mixture of ethanol:water (7:3 v/v) and this solution was added dropwise into a stirred saline phase (0.9% NaCl) containing 0.5% Pluronic F-68 as a stabilizer. After the addition, the ethanol was removed by evaporation under reduced pressure, centrifuged at 40000g for 30 min, and then the supernatant was removed and the pellets were re-suspended in water, centrifuged again and finally, the precipitated CU–GdNPs were freeze-dried to obtain purified CU–GdNPs. Batches of CU–GdNPs were hardened by the addition of 2 mg glutaraldehyde per mg of CU–GdNPs by stirring for 2 h at room temperature. The same method was followed for the preparation of FA–CU–GdNPs in which FA conjugated Gd was used instead of Gd. Blank NPs (FA–GdNPs and GdNPs) were also prepared using the same method described above by omitting the addition of CU in the procedure.
4.5. ES coating of the CU–GdNPs and FA–CU–GdNPs
The above prepared CU–GdNPs were further coated with ES using an oil-in-oil solvent evaporation method using the coat:core ratio (2:1).43 CU–GdNPs were dispersed in 10 mL of coating solution prepared from the dissolution of ES in ethanol:acetone (2:1). This dispersion was then poured in 50 mL of light liquid paraffin containing 1% w/v Span 85. This dispersion was maintained under an agitation speed of 600 rpm at room temperature for 3 hours, after which the organic phase was removed under vacuum. The resultant solution was washed with 3 × 50 ml of n-hexane to remove the liquid paraffin and then dried to obtain the ES–CU–GdNPs. ES–FA–CU–GdNPs have also been prepared using the same method; mentioned above.
Radiolabeled ES–FA–GdNPs bearing technetium 99 m–labeled diethylenetriamine pentaacetate (99mTc-DTPA) were prepared using a similar method to that discussed above, and all of the components were used in the same quantity, except that CU was replaced by sodium chloride with a radioactive (99mTc-DTPA) tracer adsorbed onto its surface.
5. Characterization
5.1. Fourier transform infrared spectroscopy (FTIR)
The conjugation of FA with Gd was confirmed from FTIR spectra which were recorded using an FTIR multiscope spectrophotometer (Perkin-Elmer, Seer Green, Beaconsfield, and Buckinghamshire, United Kingdom) equipped with spectrum v3.02 software.5.2. NMR spectroscopy
The FA conjugation with Gd was further confirmed from NMR spectra recorded using a 1H NMR spectrometer (Bruker 400 ultrashield, Switzerland) and the Topspin software.5.3. Differential scanning calorimetry (DSC)
The compatibility of CU with Gd has been established by performing DSC with a Perkin-Elmer DSC apparatus (Perkin-Elmer, Wellesley, MA). The samples were weighed and placed in a 30 μL hermetic aluminum pan and sealed. The sample was scanned over a temperature range of 10 °C to 200 °C using a heating rate of 5°C min−1 in a nitrogen atmosphere (flow rate, 10 mL min−1).5.4. Particle sizes, zeta potentials and polydispersity indices
The mean particle sizes, size distributions and zeta potentials of ES–GdNPs, ES–FA–GdNPs, ES–CU–GdNPs and ES–FA–CU–GdNPs were determined by a Malvern Zetasizer NanoZS (Malvern 3000HS, France). Each sample was measured in triplicate.5.5. Entrapment efficiency (%EE)
The amounts of the CU entrapped within ES–CU–GdNPs and ES–FA–CU–GdNPs were determined by an ultracentrifugation method. 10 mg of lyophilized ES–CU–GdNPs or ES–FA–CU–GdNPs were suspended separately in 10 mL of PBS and centrifuged at 60000 rpm for 3 hours at 4 °C. The ES–CU–GdNPs and ES–FA–CU–GdNPs settled as a pellet and the supernatant was analyzed for free CU using RP-HPLC at 425 nm.44 The %EE was calculated from the amount of CU in the supernatant and the total amount of CU taken for loading. The %EE was determined as follows:| %EE = [(Wi − Wf)/Wi] × 100 |
5.6. Transmission electron microscopy (TEM)
TEM characterization of ES–FA–CU–GdNPs was carried out. A drop of the NPs suspended in triple distilled water, was placed onto a carbon-coated copper grid, forming a thin liquid film. The film on the grid was negatively stained with a drop of 1% (w/v) phosphotungstic acid. The excess staining solution was removed using filter paper and the sample was then air-dried. These samples were viewed and imaged using TEM.5.7. HPLC analysis
A validated RP-HPLC method was developed for estimating the CU content in ES–CU–GdNPs and ES–FA–CU–GdNPs. The HPLC system was equipped with two 10 ATVP binary gradient pumps (Shimadzu), a Rheodyne (Cotati, CA, USA) model 7125 injector fitted with a 20 μl loop and a SPD-M10 AVP UV detector (Shimadzu). HPLC separation was achieved on a RP-C18 column (250 mm, 4.6 mm, 5 μm, Merck). Column effluents were monitored at 425 nm. Data was acquired and processed using the Shimadzu class VP software. The mobile phase was a mixture of acetonitrile: water: glacial acetic acid (650:340:10) v/v. The solution was filtered and degassed before use. Chromatography was performed at 25 °C with a flow rate of 1.0 ml min−1. The calibration curve of CU was in the range of 1–10 μg mL−1. Under these conditions the drug shows a retention time of about 4 min.
5.8. In vitro release study
In order to evaluate the difference in the release pattern of the prepared NPs, an in vitro release study of CU, ES–CU–GdNPs and ES–FA–CU–GdNPs was performed in simulated gastric fluid (SGF) at pH 1.2 (2 h), in simulated intestinal fluid (SIF) at pH 7.4 (6 h) and simulated colonic fluids (SCF) at pH 6.8 (24 h), at 37 °C.39 The SGF (pH 1.2) contained sodium chloride (1.0 g), pepsin (1.6 g) and hydrochloric acid (3.5 ml) and the volume was made up to 500 ml with triple distilled water. The SIF (pH 7.4) consisted of monobasic potassium phosphate (3.4 g), 0.2 N sodium hydroxide (90 ml) and pancreatin (5 g) and the volume was made up to 500 ml with triple distilled water. SCF was prepared by a method reported previously.45,46The release studies were carried out in a USP dissolution test apparatus (Apparatus II, 100 rpm, 37 ± 0.5 °C). Briefly, 500 ml of dissolution medium was taken into the container and immersed into the water bath of the apparatus. The activated dialysis bag containing CU, ES–CU–GdNPs and ES–FA–CU–GdNPs were first immersed in the dissolution media separately; first into the SGF solution, then into the SIF and finally into the SCF. For the release study, Tween 80 (1%, v/v) was added to the dissolution medium to facilitate the CU released from ES–CU–GdNPs and ES–FA–CU–GdNPs. Samples (2 ml) were withdrawn from each dissolution medium at different time intervals (1, 2, 3, 4, 5, 6, 8, 12 and 24 h). The perfect sink condition was maintained by replacing the solution with the same volume of the respective dissolution medium after each sampling. The sample was centrifuged at 10000 rpm for 15 min, and then the supernatant was filtered through a 0.4 μm membrane filter and the filtrate was subjected to HPLC analysis after appropriate dilution using the method reported above.
5.9. In vitro uptake study
The Caco-2 cell line was used for uptake studies of ES–FA–CU–GdNPs and ES–CU–GdNPs and was evaluated using a fluorescence activated cell sorter (FACS) instrument (BD Biosciences, FACS Aria, and Germany). Aliquots (100 μL) containing Caco-2 (1 × 105) cells suspended in 0.9 ml of fresh RPMI-1640 medium supplemented with penicillin 10 U ml−1, 10% FBS, 100 μg ml−1 streptomycin, 1 mM sodium pyruvate, and 10 mM HEPES medium. These were transferred into 24 well plates containing fresh medium and suspended in a 37 °C humidified incubator with a 5% CO2 atmosphere. After 24 h the culture medium was replaced with a fresh culture medium. Fluorescein isothiocyanate (FITC) loaded ES–CU–GdNPs (ES–FITC–GdNPs) and FITC loaded ES–FA–CU–GdNPs (ES–FA–FITC–GdNPs), were prepared using the same method used for the preparation of ES–CU–GdNPs and ES–FA–CU–GdNPs, and FITC was used instead of CU. All of the protocols were performed in dark conditions. ES–FITC–GdNPs and ES–FA–FITC–GdNPs containing equivalent amounts of FITC were added to cells and incubated for 4 h at room temperature in the dark. The cells were scraped and the extent of the uptake of the ES–FITC–GdNPs and ES–FA–FITC–GdNPs was examined using flow cytometry. The cell-associated fluorescence was measured using FACS at an excitation wavelength of 480 nm and an emission wavelength of 550 nm.475.10. Annexin V/propidium iodide staining for apoptotic cells
The quantitation of apoptotic cells using Annexin V staining was performed according to the manufacturer’s instructions (Calbiochem). Briefly, Caco-2 cells (5 × 105 cells per well) were seeded into 6 well plates and treated with 5 mM of the formulations i.e. the control, the plain drug, the ES–CU–GdNPs and the ES–FA–CU–GdNPs for 24 h. After the incubations, the cells were suspended in 500 mL of cold PBS, centrifuged for 5 min at 1000×g and then resuspended in 500 mL cold × binding buffer. 1.25 mL of Annexin V-FITC was added and then the suspension was incubated for 15 min at RT in the dark, and centrifuged at 1000×g for 5 min at RT. The supernatent was discarded, the cells were then gently resuspended in 500 mL cold × binding buffer and 10 mL of PI was added. Flow cytometry was performed using a FACScan (Becton Dickinson, Mountain View, CA) flow cytometer, equipped with a single 488 nm argon laser. Annexin V-FITC was analyzed using excitation and emission settings of 488 nm and 535 nm (FL-1 channel); PI, 488 nm and 610 nm (FL-2 channel). Debris and clumps were gated out using forward and orthogonal light scattering. The experiment was repeated three times independently. The values were expressed as mean ± SEM.5.11. Cytotoxicity study
In vitro cytotoxicity of CU, ES–CU–GdNPs and ES–FA–CU–GdNPs was analyzed in Caco-2 cell lines by (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) MTT assay. The caco-2 cell line was maintained as described above. The Caco-2 cells were seeded into 96 well plates at a density of 4 × 105 cells per well for 24 h and CU, ES–CU–GdNPs and ES–FA–CU–GdNPs at equivalent concentrations of CU i.e. 1 μM, 10 μM, 25 μM and 50 μM were incubated for 24 h.48 Blank formulations of ES–Gd–NPs and ES–FA–GdNPs at all of the equivalent concentrations were also incubated.49 The cytotoxicity was estimated by staining live cells with 0.5 mg mL−1 MTT for 3 h (formazan crystal formation), and optical density values were determined at 540 nm using an ELISA plate reader. Triplicates of each sample were analyzed.5.12. In vivo studies
Organs and luminal contents were weighed and homogenized with 2 mL PBS (pH 7.4) using tissue homogenizer (MAC Micro Tissue Homogenizer; New Delhi, India) and vortexed after the addition of a chloroform (CHCl3) and methanol (CH3OH) mixture and centrifuged for 5 min at 2000×g on Sigma 3–16 K (Frankfurt, Germany). After centrifugation, the obtained supernatant was decanted into another eppendorf and evaporated to dryness under vacuum using a speed vacuum concentrator (Savant Instrument, Farmingdale, USA). The residue was reconstituted in 50 μL of the mobile phase, vortexed and 20 μL of it was injected onto an analytical column. The concentration of CU in the tissues was determined using HPLC, using the method reported previously. Similarly, the blood samples were prepared and analyzed for their drug content using the HPLC method.
6. Statistical analysis
All data are expressed as mean ± standard deviation. Statistical analysis was done by one-way analysis of variance (ANOVA) followed by the Turkey–Kramer multiple comparison test, using Graph Pad Instat software (Graph Pad Software Inc., San Diego, California). A probability of p < 0.05 was considered as significant, and p < 0.01 and p < 0.001 were considered as extremely significant.7. Conclusions
It is observed that there is a regression in tumor size after the administration of ES–FA–CU–GdNPs. Experimental observations also suggest that ES–FA–CU–GdNPs are more effective against HCT-116 tumor-bearing nude mice than free CU and ES–CU–GdNPs. The results from plasma and tissue distribution studies demonstrate that the maximum amount of CU is recovered from the colon and tumors indicating the efficacy of colon specific drug delivery. In addition, since the surface of the ES–CU–GdNPs could be similarly functionalized with other targeting ligands besides FA, they should serve as an effective carrier for targeted drug delivery to different types of cancer. ES–FA–CU–GdNPs might have potential in cancer diagnosis and therapies which are more affected by FRs expressed on the tumor surface, offering the dual advantage of localizing tumors by non-invasive imaging and also treating them.8. Ethical statement
Eight-ten week old nude mice (male, 30–35 g) were obtained from INMAS New Delhi, India and were acclimatized in a climate- and light cycle-controlled environment for 2 days before investigation. The animals were maintained at a controlled temperature of 23 ± 1 °C and a humidity of 55 ± 5% in a 14 h light/10 h dark cycle. Throughout the study, the animals were provided with soy-free food and filtered drinking water. The animal experiments have been carried out by following the experimental protocol guidelines of the Council for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Ministry of Social Justice and Empowerment, Government of India. The experiments were conducted after ethical clearance by the Institutional Animal Ethics Committee of the institute (INM/DASQA/IAEC/09/015).Conflict of interest statement
The authors declare they have no competing financial interest.Acknowledgements
The authors are particularly grateful to SAIF, CDRI, Lucknow, India, for spectral analysis, AIIMS, New Delhi, India for TEM analysis. The author is thankful to ICMR, New Delhi India for providing SRF. This work was supported by a grant from the Indian Council of Medical Research, New Delhi, India. The CDRI communication number is 8892.References
- L. Brannon-Peppas and J. O. Blanchette, Adv. Drug Delivery Rev., 2004, 56, 1649–1659 CrossRef CAS PubMed.
- S. S. Feng, Expert Rev. Med. Devices, 2004, 1, 115–125 CrossRef CAS PubMed.
- M. Das, C. Mohanty and S. K. Sahoo, Expert Opin. Drug Delivery, 2009, 6, 285–304 CrossRef CAS PubMed.
- S. K. Sahoo and V. Labhasetwar, Drug Discovery Today, 2003, 8, 1112–1120 CrossRef CAS.
- P. Anand, C. Sundaram, S. Jhurani, A. B. Kunnumakkara and B. B. Aggarwal, Cancer Lett., 2008, 267, 133–164 CrossRef CAS PubMed.
- B. B. Aggarwal, A. Kumar and A. C. Bharti, Anticancer Res., 2003, 23, 363–398 CAS.
- E. Balogun, M. Hoque, P. Gong, E. Killeen, C. J. Green, R. Foresti, J. Alam and R. Motterlini, Biochem. J., 2003, 371, 887–895 CrossRef CAS PubMed.
- A. Mazumder, K. Raghavan, J. Weinstein, K. W. Kohn and Y. Pommier, Biochem. Pharmacol., 1995, 49, 1165–1170 CrossRef CAS.
- A. Duvoix, R. Blasius, S. Delhalle, M. Schnekenburger, F. Morceau, E. Henry, M. Dicato and M. Diederich, Cancer Lett., 2005, 223, 181–190 CrossRef CAS PubMed.
- W. M. Abuzeid, S. Davis, A. L. Tang, L. Saunders, J. C. Brenner, J. Lin, J. R. Fuchs, E. Light, C. R. Bradford, M. E. Prince and T. E. Carey, Arch. Otolaryngol., Head Neck Surg., 2011, 137, 499–507 CrossRef PubMed.
- S. V. Bava, V. T. Puliappadamba, A. Deepti, A. Nair, D. Karunagaran and R. J. Anto, J. Biol. Chem., 2005, 280, 6301–6308 CrossRef CAS PubMed.
- L. Li, B. Ahmed, K. Mehta and R. Kurzrock, Mol. Cancer Ther., 2007, 6, 1276–1282 CrossRef CAS PubMed.
- N. K. Narayanan, D. Nargi, C. Randolph and B. A. Narayanan, Int. J. Cancer, 2009, 125, 1–8 CrossRef CAS PubMed.
- K. Maiti, K. Mukherjee, A. Gantait, B. P. Saha and P. K. Mukherjee, Int. J. Pharm., 2007, 330, 155–163 CrossRef CAS PubMed.
- M. M. Yallapu, M. Jaggi and S. C. Chauhan, Macromol. Biosci., 2010, 10, 1141–1151 CrossRef CAS PubMed.
- S. Bisht, G. Feldmann, S. Soni, R. Ravi, C. Karikar and A. Maitra, J. Nanobiotechnol., 2007, 5, 3 CrossRef PubMed.
- A. O. Elzoghby, W. S. El-Fotoh and N. A. Elgindy, J. Controlled Release, 2011, 153, 206–216 CrossRef CAS PubMed.
- Y. Lu and S. C. Chen, Adv. Drug Delivery Rev., 2004, 56, 1621–1633 CrossRef CAS PubMed.
- C. Coester, P. Nayyar and J. Samuel, Eur. J. Pharm. Biopharm., 2006, 62, 306–314 CrossRef CAS PubMed.
- I. Ezpeleta, M. A. Arangoa, J. M. Irache, S. Stainmesse, C. Chabenat, Y. Popineau and A. M. Orecchioni, Int. J. Pharm., 1999, 191, 25–32 CrossRef CAS.
- C. Duclairoir, A. M. Orecchioni, P. Depraetere, F. Osterstock and E. Nakache, Int. J. Pharm., 2003, 253, 133–144 CrossRef CAS.
- R. B. Umamaheshwari, S. Ramteke and N. K. Jain, AAPS PharmSciTech, 2004, 5, e32 CrossRef CAS PubMed.
- Y. Zhang, N. Kohler and M. Zhang, Biomaterials, 2002, 23, 1553–1561 CrossRef CAS.
- E. K. Park, S. B. Lee and Y. M. Lee, Biomaterials, 2005, 26, 1053–1061 CrossRef CAS PubMed.
- M. O. Oyewumi, R. A. Yokel, M. Jay, T. Coakley and R. J. Mumper, J. Controlled Release, 2004, 95, 613–626 CrossRef CAS PubMed.
- S. Mansouri, Y. Cuie, F. Winnik, Q. Shi, P. Lavigne, M. Benderdour, E. Beaumont and J. C. Fernandes, Biomaterials, 2006, 27, 2060–2065 CrossRef CAS PubMed.
- P. Chan, M. Kurisawa, J. E. Chung and Y. Y. Yang, Biomaterials, 2007, 28, 540–549 CrossRef CAS PubMed.
- Y. Lu and P. S. Low, Adv. Drug Delivery Rev., 2002, 54, 675–693 CrossRef CAS.
- J. Sudimack and R. J. Lee, Adv. Drug Delivery Rev., 2000, 41, 147–162 CrossRef CAS.
- S. Wang and P. S. Low, J. Controlled Release, 1998, 53, 39–48 CrossRef CAS.
- J. F. Kukowska-Latallo, K. A. Candido, Z. Cao, S. S. Nigavekar, I. J. Majoros, T. P. Thomas, L. P. Balogh, M. K. Khan and J. R. Baker Jr, Cancer Res., 2005, 65, 5317–5324 CrossRef CAS PubMed.
- Y. Liu, K. Li, J. Pan, B. Liu and S. S. Feng, Biomaterials, 2010, 31, 330–338 CrossRef CAS PubMed.
- M. He, Z. Zhao, L. Yin, C. Tang and C. Yin, Int. J. Pharm., 2009, 373, 165–173 CrossRef CAS PubMed.
- M. Gulfam, J.-e. Kim, J. M. Lee, B. Ku, B. H. Chung and B. G. Chung, Langmuir, 2012, 28, 8216–8223 CrossRef CAS PubMed.
- C. V. Rao and B. S. Reddy, Curr. Cancer Drug Targets, 2004, 4, 29–42 CrossRef CAS.
- M. A. Arangoa, G. Ponchel, A. M. Orecchioni, M. J. Renedo, D. Duchene and J. M. Irache, Eur. J. Pharm. Sci., 2000, 11, 333–341 CrossRef CAS.
- C. M. Paulos, J. A. Reddy, C. P. Leamon, M. J. Turk and P. S. Low, Mol. Pharmacol., 2004, 66, 1406–1414 CrossRef CAS PubMed.
- Y. Liu, K. Li, J. Pan, B. Liu and S.-S. Feng, Biomaterials, 2010, 31, 330–338 CrossRef CAS PubMed.
- M. Sharma, R. L. Salisbury, E. I. Maurer, S. M. Hussain and C. E. Sulentic, Nanoscale, 2013, 5, 3747–3756 RSC.
- J. Pan and S.-S. Feng, Biomaterials, 2008, 29, 2663–2672 CrossRef CAS PubMed.
- J. G. Hardy, S. S. Davis, R. Khosla and C. S. Robertson, Int. J. Pharm., 1988, 48, 79–82 CrossRef CAS.
- V. P. Fitzjarrald and R. Pongdee, Tetrahedron Lett., 2007, 48, 3553–3557 CrossRef CAS PubMed.
- T. Oosegi, H. Onishi and Y. Machida, Eur. J. Pharm. Biopharm., 2008, 68, 260–266 CrossRef CAS PubMed.
- T. Sartori, F. Seigi Murakami, A. Pinheiro Cruz and A. Machado de Campos, J. Chromatogr. Sci., 2008, 46, 505–509 CAS.
- Y. S. Krishnaiah, V. Satyanarayana, B. Dinesh Kumar and R. S. Karthikeyan, Eur. J. Pharm. Sci., 2002, 16, 185–192 CrossRef CAS.
- V. R. Sinha, B. R. Mittal, K. K. Bhutani and R. Kumria, Int. J. Pharm., 2004, 269, 101–108 CrossRef CAS PubMed.
- R. Khatik, R. Mishra, A. Verma, P. Dwivedi, V. Kumar, V. Gupta, S. Paliwal, P. Mishra and A. Dwivedi, J. Nanopart. Res., 2013, 15, 1–15 CrossRef.
- P. Dwivedi, S. Kansal, M. Sharma, R. Shukla, A. Verma, P. Shukla, P. Tripathi, P. Gupta, D. Saini, K. Khandelwal, R. Verma, A. K. Dwivedi and P. R. Mishra, J. Drug Targeting, 2012, 20, 883–896 CrossRef CAS PubMed.
- S. K. Sahoo, W. Ma and V. Labhasetwar, Int. J. Cancer, 2004, 112, 335–340 CrossRef CAS PubMed.
- K. Flatmark, G. M. Mælandsmo, M. Martinsen, H. Rasmussen and Ø. Fodstad, Eur. J. Cancer, 2004, 40, 1593–1598 CrossRef PubMed.
- E. E. Ibrahim, R. Babaei-Jadidi, A. Saadeddin, B. Spencer-Dene, S. Hossaini, M. Abuzinadah, N. Li, W. Fadhil, M. Ilyas, D. Bonnet and A. S. Nateri, Stem Cell, 2012, 30, 2076–2087 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |