
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Decoupling multiscale morphological effects in templated porous Ag electrodes for electrochemical CO2 reduction†
Maaike E. T.
Vink-van Ittersum‡
 a,
Karen
van den Akker‡
ab,
Peter
Ngene
a and
Petra E.
de Jongh
*a
a,
Karen
van den Akker‡
ab,
Peter
Ngene
a and
Petra E.
de Jongh
*a
aMaterials Chemistry & Catalysis, Debye Institute for Nanomaterials Science, Utrecht University, Universiteitsweg 99, 3584CG Utrecht, The Netherlands. E-mail: P.E.deJongh@uu.nl
bLeiden Institute of Chemistry, Leiden University, PO Box 9502, 2300 RA Leiden, The Netherlands
First published on 14th March 2025
Abstract
Electrochemical reduction of CO2 using renewable electricity is a promising strategy to produce sustainable fuels and chemical feedstocks. The use of porous electrodes is a promising approach to increase the activity of electrocatalysts such as Ag which exhibit high CO selectivity. However, it is challenging to fully understand the impact of their complex morphologies. We varied electrodeposition conditions to obtain different micrometer-scale morphologies: flat catalysts and more dendritic (“coral”) catalysts. Performing this electrodeposition in either the absence or the presence of a template, allowed to independently introduce additional porosity of 180 nm cages connected via smaller windows. The structures were relatively stable in catalysis, with some changes on the 10 nm scale at the most negative potentials. The templated Ag catalysts consistently reached higher CO partial current densities than non-templated equivalents. Interestingly, where CO production scaled with the internal electrode surface area, simultaneous H2 evolution was impeded in the mesoscale pore network. Therefore, our work shows a promising assembly strategy to deconvolute morphology effects on different length scales, and demonstrates the importance of porosity specifically at the 100 nm scale to enhance CO2 conversion to CO in porous Ag electrodes.
Introduction
The anthropogenic rise in atmospheric CO2 levels induces global warming.1 To limit climate change, CO2 concentrations need to be extensively reduced. Most urgent is to stop further emission of CO2 by replacing fossil fuels by renewable energy. On top of that, the current CO2 levels have to be lowered. Converting captured CO2 into fuels and chemical feedstocks with electrochemical CO2 reduction using renewable energy is a promising approach to close the carbon cycle. However, the effectiveness is contingent upon the source of energy and conversion efficiency.2 For electrochemical CO2 conversion, Hori et al. showed the importance of the metal composition of the electrocatalyst for product selectivity and activity.3–7 Bagger et al. elaborated on this by relating the selectivity to the binding energies of reaction intermediates.8 Specifically, they demonstrated that Ag, Au, and Zn are the most promising metals to obtain CO during electrochemical CO2 reduction.3,8 CO, if combined with hydrogen, can be converted into fuels and chemical feedstocks with existing processes (e.g. Fischer–Tropsch).9 However, it is challenging to achieve the operating conditions, in particular the current density, required for industrials applications (>300 mA cm−2 at <0.5 V overpotential at >70% faradaic efficiency10). Generally, higher current densities can be achieved using porous metals due to their higher surface area, which makes them promising candidates to investigate.Templating is a useful tool to obtain ordered porous model systems, as this synthesis method offers high control over the formed porosity. These model systems can be helpful in fundamental understanding of the processes influencing catalysis, as they allow the variation of one single parameter at a time. On top of that, they are very useful to study the stability of the catalyst material. Although templating has been widely used to create inverse opals of various metal(oxides),11–16 only a few examples of application in electrochemical CO2 reduction are known. Yoon et al. were the first to use Ag invers opals in the electrochemical reduction of CO2. They prepared porous Ag catalysts via electrodeposition of Ag in the voids of a polystyrene (PS) template, showing the effect of the layer thickness on the selectivity during electrochemical CO2 reduction.17 This inspired Sutter et al. to develop a mathematical model based on local concentrations, which was then used to verify the effect of layer thickness and to predict the effect of pore size on the selectivity and activity.18
Interestingly, these papers only looked at the effect of templated ordered porosity, a change in the morphology in the 100 nm range. Previous research on CO2 reduction catalysts has shown that changes in the morphology on the 1 or 10 nm scale are important too. For Ag, it is known that the (110) facet performs best in catalysis.19 Also, low-coordinated surface Ag atoms in porous structures are known to produce more CO.20 At the same time, the diffusion layer thickness of CO2 in these systems is often in μm range,21 so also corrugation on this scale is important. Changes in the morphology on the μm scales can be obtained by varying the electrodeposition solution and conditions.22–24
The aim of this paper is to investigate how the morphology of porous Ag obtained via templating varies on different scales with changing electrodeposition conditions and to find the dominant scale for the catalytic performance during electrocatalytic CO2 reduction. To better visualize and understand the changes on the μm scale, we compare the templated porous Ag electrodes with non-templated equivalents. The results show that porosity on the 100 nm scale plays a more important role in the catalytic performance than the morphology of porous Ag electrode on the μm scale, and that the porous electrodes exhibit high stability, despite slight changes in the edge smoothness. On top of that, we discuss a simple but versatile model that describes the increase in surface area when using both electrodeposition and templating as synthesis methods for porous structures.
Experimental
Electrode preparation
Template-based porous Ag catalysts were synthesized in five steps, which are schematically depicted in Fig. 1. First, PMMA spheres were prepared by polymerizing 100 mL MMA (Sigma Aldrich, >99%) in 400 mL milliQ using 69 mg potassium persulfate (Emsure, 99%) as inhibitor in a 2L round bottom flask heated to 70 °C with an overhead stirrer at 350 rpm, following the procedure described by van den Reijen et al.11 based on the work of Zhou et al.25 and Schroden et al.26 This led to a 2.6% w/w PMMA sphere suspension with a diameter of ∼180 nm after filtration. | ||
| Fig. 1 Schematic representation of the synthesis of template-based porous Ag by electrodeposition of Ag on a PMMA sphere template, involving (1) the synthesis of PMMA spheres; (2) the formation of a template on a substrate; (3) electrodeposition of a metal M and (4) removal of the template. | ||
In the second step, this suspension was mixed with water and Nafion®D-520 solution in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.015 volume ratio. To form the sacrificial templates, the resulting mixture was dropcasted on wrinkled Ag foil (Alfa Aesar, 99.95%) and dried in air overnight. The Nafion®D-520 solution was added to facilitate adhesion of the PMMA spheres to the Ag.
:1:0.015 volume ratio. To form the sacrificial templates, the resulting mixture was dropcasted on wrinkled Ag foil (Alfa Aesar, 99.95%) and dried in air overnight. The Nafion®D-520 solution was added to facilitate adhesion of the PMMA spheres to the Ag.
Then, Ag was electrodeposited in the voids of the PMMA sphere templates using a three electrode set-up consisting of a Pt wire anode, a 3 M Ag/AgCl reference electrode, and a glassy carbon disc (SIGRADUR K disc) with the PMMA-modified Ag electrode on top of it as the cathode. The default conditions consisted of potentiostatic deposition at −0.1 V vs. Ag/AgCl until a total charge of 2C cm−2 had passed in a solution of 0.05 M AgNO3 (Thermofischer 99.9 + % and Alfa Aesar, 99.9 + %), 0.5 M NH4OH (Emsure, 28–30% and Sigma Aldrich, 28–30%), 1.0 M NaNO3 (Thermo Fischer, 99.0% and Alfa Aesar, 99.0%) and 0.01 M EDTA (Sigma Aldrich, 98–103%), leading to sample 1, p-flat-default, in Table 1. Based on the cyclic voltammogram of the solution (Fig. S1, ESI†) only Ag deposition is expected to take place at this potential. Five other samples were prepared with slightly altered reaction conditions (see samples 2 to 6 in Table 1). Specifically, these changes were the use of galvanostatic deposition instead of potentiostatic deposition, a higher NH4OH concentration, a lower NaNO3 concentration, the absence of EDTA as dendrite suppressing agent, and the application of a more negative potential. All samples were made using a charge of 2C cm−2, ensuring equal catalyst loadings for all samples. After the Ag deposition, the electrodes were rinsed with milliQ and left to dry in the air.
| Sample | Name | Templated | Parameter varied | Potential (Vvs.Ag/AgCl) |
AgNO3 (M) | NH4OH (M) | NaNO3 (M) | EDTA (M) |
|---|---|---|---|---|---|---|---|---|
| 1 | t-flat-default | Yes | — | −0.1 | 0.05 | 0.5 | 1.0 | 0.01 |
| 2 | t-flat-5mA | Yes | Deposition type | Galvanostatic deposition (−5 mA) | 0.05 | 0.5 | 1.0 | 0.01 |
| 3 | t-flat- 1MNH4OH | Yes | Ammonia concentration | −0.1 | 0.05 | 1 | 1.0 | 0.01 |
| 4 | t-flat-0.5MNaNO3 | Yes | Electrolyte concentration | −0.1 | 0.05 | 0.5 | 0.5 | 0.01 |
| 5 | t-coral-0MEDTA | Yes | Presence of dendrite-suppressing additive | −0.1 | 0.05 | 0.5 | 1.0 | 0 |
| 6 | t-coral-0.4V | Yes | Deposition potential | −0.4 | 0.05 | 0.5 | 1.0 | 0.01 |
| 7 | nt-flat-default | No | — | −0.1 | 0.05 | 0.5 | 1.0 | 0.01 |
| 8 | nt-flat-5mA | No | Deposition type | Galvanostatic deposition (−5 mA) | 0.05 | 0.5 | 1.0 | 0.01 |
| 9 | nt-flat-1MNH4OH | No | Ammonia concentration | −0.1 | 0.05 | 1 | 1.0 | 0.01 |
| 10 | nt-flat-0.5MNaNO3 | No | Electrolyte concentration | −0.1 | 0.05 | 0.5 | 0.5 | 0.01 |
| 11 | nt-coral-0MEDTA | No | Presence of dendrite-suppressing additive | −0.1 | 0.05 | 0.5 | 1.0 | 0 |
| 12 | nt-coral-0.4V | No | Deposition potential | −0.4 | 0.05 | 0.5 | 1.0 | 0.01 |
Lastly, in the fourth step, the PMMA sphere templates were removed by soaking the sample in acetone (VWR chemicals) for at least 1 h. In addition to these porous samples, non-templated Ag equivalents were prepared. These were made by depositing Ag directly onto Ag foil using the same six electrodeposition conditions as for the porous Ag electrodes (see samples 7 to 12 in Table 1).
Characterisation
To visualize the sphere size of the PMMA template and the morphology of the Ag electrodes, scanning electron microscopy (SEM) was used. These measurements were performed on a Thermo Fisher Scientific (TFS) Helios G3 UC microscope which was operated at 2 kV and 50 pA or 5 kV and 20 nA. Prior to the SEM measurements on the PMMA template, a 10 nm Pt layer was first deposited onto the spheres. The sphere size distribution of the PMMA spheres was determined by counting the sphere sizes in image J. Scanning electron microscopy (SEM) energy-dispersive X-ray spectroscopy was performed at 15 kV and 5 pA using a Oxford Instruments X-MaxN 150 mm2 detector. X-Ray diffraction (XRD) was measured on a Bruker D2 Phaser with a Cu Kα X-ray source (1.54187 Å) between 35° and 85°. X-ray photoelectron spectroscopy (XPS) was measured on a SPECS XPS using a Al Kα source and Epass of 100 eV, which has been described in more detail in literature.27For all samples, the double layer capacitance and catalytic performance were measured by a series of experiments, performed in a custom-built three-electrode H-type cell described by Mattarozzi et al.28 This cell consisted of an anodic and a cathodic compartment of 18 mL each, and contained a Pt disc as counter electrode, a 3 M Ag/AgCl reference electrode, and a Nafion 117 proton exchange membrane. The samples were placed in the cathodic compartment on top of a glassy carbon disk that had previously been polished with diamond suspension (MetaDi Supreme; 1 μm, 0,25 μm and 0,05 μm). To each compartment, 15 mL of a 0.1 M KHCO3 (Honeywell Fluka, 99.7%) electrolyte solution, pretreated with Chelex® (100 sodium form, 50–100 mesh), was added. The cathodic compartment was stirred with a stirring bar at 400 rpm. Prior to any measurement, the cell was purged with a CO2 flow of 10 mL min−1 for 1 h. Both the stirring and purging were applied to prevent concentration gradients in the bulk during the electrochemical measurements, as is schematically depicted in Fig. S2 (ESI†). In addition, a cyclic voltammetry (CV) was performed between 0.1 V and −1.4 V vs. RHE -as catalysis was performed between these potentials- with a scan rate of 0.01 V s−1 for 5 cycles to reduce any Ag2O present.
Next, the double-layer capacitances (DLC) were measured to calculate the electrochemical surface area (ECSA). For this, CVs were performed between 0.6 and 0.4 V vs. RHE or 0.3 and 0.1 vs. RHE with scanning rates between 0.01 and 0.08 V s−1 or between 0.6 and 0.2 V vs. RHE with scanning rates between 0.01 and 1.0 V s−1. Then, the current at respectively 0.5 V vs. RHE or 0.4 V vs. RHE was plotted against the scan rate. The slope of a linear fit in the linear regime of these current vs. scan rate curves gave the capacitance of the sample. Using the capacitance of a flat Ag foil as a reference, which was measured in the same cell under the same conditions, the ECSA of the electrodes was determined.
Electrocatalytic performance
After that, the catalytic measurements were performed. During these measurements, a fixed potential of subsequently −0.7, −0.9, −1.2, and −1.4 V vs. RHE was applied to the cathode, and the current response was measured. Afterwards, the potentials were iR compensated using respectively Ohmic resistances of 25, 24, 22 and 18 Ω. Gaseous products were detected by an on-line Gas Analyzer Solution Compact GC 4.0 gas chromatogram (GC) using a Rt-QBond (10 m × 0.32 mm, Agilent), a Molecular Sieve 5A (10 m × 0.53 mm, Restek) and a Carboxen 1010 (8 m × 0.32 mm, Agilent) column, connected with respectively a FID detector, a FID detector (together with methanizer to increase the CO sensitivity) and a TCD detector to measure the presence of CH4, C2H4 and C2H6 (first column), CO and CH4 (second column) and H2 and CO2 (third column). These measurements were performed in duplo. For one electrode of a duplo couple, a second cycle of catalysis (−0.7, −0.9, −1.2, and −1.4 V vs. RHE) was measured.For the duplo measurement, electrical impedance spectroscopy (EIS) was measured after each potential between 0.01 and 105 Hz at the potential (−0.7, −0.9, −1.2, and −1.4 V vs. RHE) from catalysis with a voltage amplitude of 10 mV. Fitting the Nyquist plot with a R1 + Q2/R2 circuit gave the resistances (R1 and R2) and the constant phase element (Q2) which was considered to be the capacitance of the system. The resistance R2 is the electron transfer resistance, the capacitance Q2 was used to calculate the electron transfer rate via the formula ω = 1/RC.
The stability of the catalysts was tested in a 250 mL beaker filled with ∼150 mL 0.1 M KHCO3. Then, the same procedure was followed as for normal catalysis, except for the fact, that after each step, a small piece was cut out of the electrode for SEM analysis. All pretreatment steps were performed whilst applying a 10 mL min−1 CO2 flow. The catalytic part was performed without CO2 flow.
Results and discussion
Morphology of porous Ag electrodes
All templated Ag samples were synthesized using the same batch of PMMA spheres (diameter 182 ± 22 nm see Fig. S3, ESI†) as sacrificial template. Firstly, porous Ag was prepared under so-called default conditions (−0.1 V vs. Ag/AgCl, 0.05 M AgNO3, 1 M NaNO3, 0.01 M EDTA, 0.5 M NH4OH). Scanning electron microscopy energy-dispersive X-ray spectroscopy showed Ag was the only metal deposited, there was no Pt contamination from the anode (Fig. S4, ESI†). The X-ray diffractogram (Fig. S5, ESI†) is in line with this result. The X-ray photoelectron spectroscopy image (Fig. S6, ESI†) shows the additional presence of F, C and O on Ag, which can be explained by the presence of Nafion binder and partial oxidation of the Ag. Consequently, 5 different morphologies were obtained by varying one of these reaction parameters per synthesis, as summarized in Table 1. Also for these electrodes, the X-ray diffractograms showed only the presence of crystalline Ag (Fig. S5, ESI†).In Fig. 2 scanning electron microscopy (SEM) images for the six different templated Ag electrodes are shown. Clearly, all samples have an ordered porosity that is the inverse of the 100 nm scale template, resulting in a porosity consisting of ∼180 nm cages connected by smaller (∼70 nm) windows. Additionally, on the ∼5 μm scale, two types of porous structures are found. Specifically, 4 samples are rather flat (t-flat-default, t-flat-5mA, t-flat-1MNH4OH and t-flat-0.5MNaNO3 in respectively Fig. 2(a), (b), (d) and (e)) and the remaining 2 samples demonstrate a coral-like structure (t-coral-0.4V and t-coral-0MEDTA in respectively Fig. 2(c) and (f)). These two coral-like structures can be explained by the dendritic growth conditions due to their electrodeposition conditions: a rather cathodic potential and the absence of EDTA. Regarding the rather catholic potential, Guo et al. previously described how the applied potential influences the growth regime changing from kinetic control to mass transport control. The growth regime determines the morphology formed. At high overpotentials, the growth turns into a mass transport-limited process, which results in dendrite formation.29 Regarding the absence of EDTA, EDTA is known to be an additive that suppresses dendrite formation.22,30 So, its absence will cause dendrite formation at less negative potentials.
 | ||
| Fig. 2 SEM images of porous Ag electrodes prepared via Ag electrodeposition in voids of a PMMA sphere with different synthesis conditions: (A) default (−0.1 V vs. Ag/AgCl, 0.05 M AgNO3, 1 M NaNO3, 0.01 M EDTA, 0.5 M NH4OH); (B) −5 mA; (C) −0.4 V; (D) 1 M NH4OH; (E) 0.5 M NaNO3; (F) 0 M EDTA. | ||
Also on a slightly smaller scale, ∼1 μm, differences were found. These are most probably caused by the synthesis conditions, but harder to see in Fig. 2 because of the ordered pores. Hence, to deconvolute the influence of templating and synthesis conditions, non-templated analogues were prepared as well under the exact same electrodeposition conditions, but without PMMA spheres. Fig. 3 shows the SEM images of the non-templated electrodes. Interestingly, the electrodes have similar overall morphologies as their non-templated equivalents. To be more precise, the default samples (2a and 3a) consist of a round needle-like structure. The −5 mA samples (2b and 3b) consist of a sharp-edged block structure, which can be explained by the fact that the potential can change, hence over time there is a mixture of diffusion and kinetically controlled growth.31 The −0.4 V samples (2c and 3c) consist of a dendrite-like structure due to the large overpotential leading to diffusion-limited growth.29 Then, the 1 M NH4OH samples look like flakes (2d and 3d), which is the result of an increased pH and formation of diaminesilver(I) complexes.22,30 The 0.5 M NaNO3 samples look like flakes (2e and 3e) as well, implying that although the ionic strength decreased because of the decreasing NaNO3 concentration,32,33 these samples were still in the same growth regime as the 1 M NH4OH samples. Finally, the 0 M EDTA samples are open structures of large spheres (2f and 3f), which can be explained by the absence of EDTA.22,30
 | ||
| Fig. 3 SEM images of non-templated Ag electrodes prepared via Ag electrodeposition with different synthesis conditions: (A) default (−0.1 V vs. Ag/AgCl, 0.05 M AgNO3, 1 M NaNO3, 0.01 M EDTA, 0.5 M NH4OH); (B) −5 mA; (C) −0.4 V; (D) 1 M NH4OH; (E) 0.5 M NaNO3; (F) 0 M EDTA. | ||
These findings highlight that the presence of a template does not significantly alter the deposition process and, consequently, the morphology of the resulting electrode. Hence, we have demonstrated that templated electrodeposition can be a versatile tool to introduce nanoscale (∼200 nm) porosity while maintaining the micrometer scale (1–10 μm) structure. To the best of our knowledge, this has not been described in literature before.
Increase in surface area
Since the templated Ag electrodes have different morphologies, it is important to determine their surface areas before moving on to the catalytic performance. These electrochemically active surface areas (ECSA) were determined using double-layer capacitance (DLC) measurements performed in 0.1 M KHCO3. Plots of the current vs. the scan rate can be found in Fig. S7 (ESI†). The plots show that when too large currents are drawn, the current reaches a plateau, which can be explained by limitations in ion diffusion.34,35 Fig. S8 (ESI†) shows that the limiting current differs for the samples ranging between 1.3 mA and 2.6 mA. It is important to realize that these diffusion limitations are present, as these might affect the catalytic performance: due to changes in the local environment, e.g. pH, H2 formation can be suppressed whilst CO formation is increased.17,36To determine the ECSA, only the linear parts of the current vs. scan rate curves were used.34,35Table 2 shows the ECSA for both the porous Ag electrodes and their non-templated equivalents prepared with the different synthesis conditions. Next to the absolute surface areas, Table 2 gives the roughness factor (RF) of the electrodes. The roughness factor is defined as:
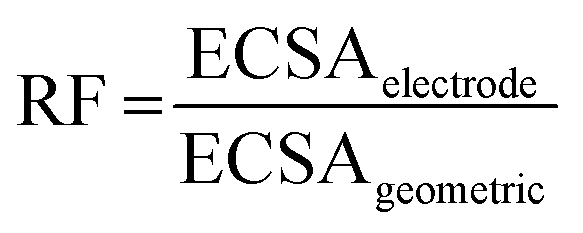 | (1) |
| Electrodeposition conditions | ECSA templated (cm2) | RF templated | ECSA non-templated (cm2) | RF non-templated | S |
|---|---|---|---|---|---|
| Default | 57 | 15 | 9 | 2.4 | 6.4 |
| −5 mA | 64 | 17 | 11 | 2.9 | 5.8 |
| 1 M NH4OH | 58 | 15 | 7 | 1.8 | 8.8 |
| 0.5 M NaNO3 | 96 | 25 | 7 | 1.8 | 14.3 |
| 0 M EDTA | 47 | 12 | 18 | 4.7 | 2.6 |
| −0.4 V | 61 | 16 | 74 | 19.4 | 0.8 |
Except for the t-flat-0.5MNaNO3 sample, all templated porous electrodes have a roughness factor of 12 to 17. For the non-templated samples, the nt-coral-0MEDTA and in particular the nt-coral-0.4V samples show higher roughness factors (respectively 4.7 and 19.4) than the other non-templated samples (with roughness factors between 1.8 and 2.9), which is in line with their coral-like structure as previously observed by SEM.
To understand if the increase in surface area found is realistic, it is good to compare the experimental data with a theoretical surface area increase. Although Suter et al. show data simulating the roughness factor based on the thickness, to our knowledge, there is no literature on a model including inhomogeneity in the FCC structure. Therefore, we built a mathematical model describing the factor of increase in surface area, S. Briefly, the model is based on stacking layers of spherical cages, while correcting for surface loss due to windows in the porous structure and imperfections in the cage connectivity causing inaccessible of some pores. The full derivation, which can be found in the ESI† including the underlying assumptions, leads to the following description of S
 | (2) |
This derivation of S assumes perfectly smooth surfaces. However, our electrodes already have some roughness caused by the different μm scale deposition morphology, as evidenced by the roughness factor of the non-templated samples of around 2 (see Table 2). Assuming that this extra roughness is equal for both the templated and non-templated electrodes, the increase of surface area for the templated porous electrodeposited electrodes becomes
| Rtemplated,deposition = S × Rdeposition | (3) |
 | (4) |
To prove that our model is generally applicable, the model was applied to literature data on template-based porous Ag and Au electrodes as published by respectively Yoon et al.17 and Hall et al.37 Based on the SEM images of Yoon et al., the a value is 0.4 and the b value is close to 1.0. In Fig. 4, the green dotted line indicates the S values for varying numbers of layers for a = 0.40 and b = 0.98. The green data points are the samples from Yoon et al. with RF 43, 78, and 110 (using their non-templated electrodes with RF 4 to calculate S). Fig. 4 shows that our model nicely fits the data of Yoon et al. The same holds for the electrodes of Hall et al., where the purple dotted line (for S using a = 0.30 and b = 0.98 based on the SEM images) in Fig. 4 fits the purple data points (for electrodes with RF 4, 10,27 assuming an Rdeposition of 2).
 | ||
| Fig. 4 Simulations of the surface area increase factor S as a function of the number of porous layers for a = 0.40 and b = 0.98 (green dotted line) as fit for the data of Yoon et al.,17a = 30 and b = 0.98 (purple dotted line) as fit for the data of Hall et al.37 and a = 0.7 and b = 0.7 (grey dotted line) as fit for the sample t-flat-default, t-flat-5mA, t-flat-1MNH4OH and t-flat-0.5MNaNO3. | ||
Secondly, our ordered porous catalysts were analyzed to see how these fit into the model. Only the flat samples were included, as the model assumes continuous porous structures. Based on very rough estimations from the SEM images in Fig. 2, both a and b values should be around 0.7. The number of layers is unknown, but the same number of layers as Yoon et al. found for their 2C cm−2 catalyst was assumed, as we used spheres of a similar diameter. Compared to the electrodes of Yoon et al. and Hall et al. a higher a value and a lower b value was found, implying that our structures have smaller windows and worse pore connection. Apart from the t-flat-0.5MNaNO3 sample, the model nicely matches our data. Also, it shows that for these values of a and b, S reaches a plateau. Hence, the assumption made on the number of layers will not influence our conclusions. So, this simple model is a versatile tool to describe the increase in surface area by templating.
Catalytic performance
Regarding the catalytic performances, the main gaseous products found during catalytic testing (−0.7, −0.9, −1.2, and −1.4 V vs. RHE, iR compensation was done during data analysis) were H2 and CO. The results shown are the average of the duplo measurements, of which the raw data is given in Table S1 (ESI†). In Fig. 5 the CO partial current densities, normalized to the geometric surface area, are provided. When comparing the porous electrodes (blue) to their non-templated equivalents (red), a clear difference between the partial current densities of the templated porous catalysts and their non-templated equivalents is observed. The porous catalysts have higher CO partial current densities than all their non-templated equivalents (except nt-coral-0.4V), in particular at more negative potential. So, clearly, the introduction of porosity by templating is useful to obtain higher current densities. However, it should be noted that these porous catalysts also have higher electrochemical surface areas.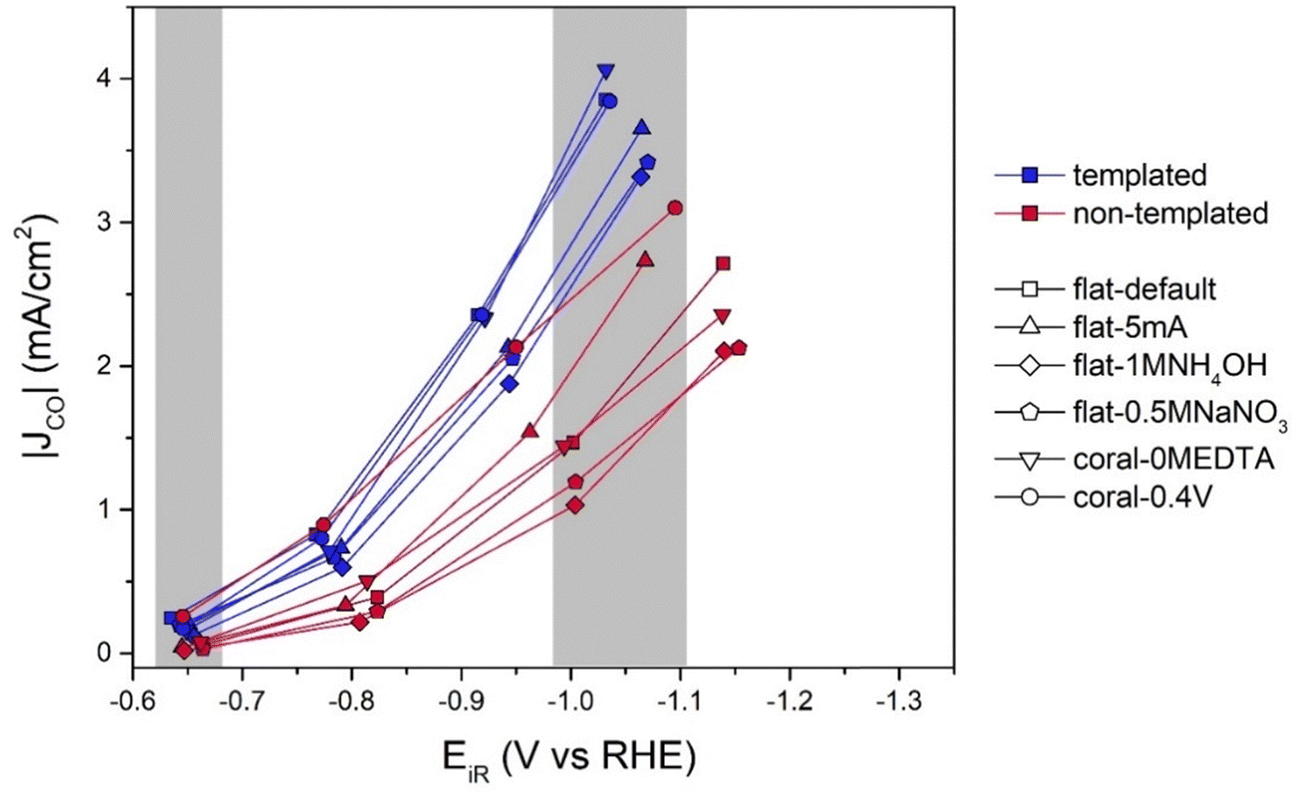 | ||
| Fig. 5 partial current densities to CO for both the template-based porous (blue) and non-templated equivalent (red) samples (n)t-flat-default, (n)t-flat-5mA, (n)t-flat-1MNH4OH, (n)t-flat-0.5MNaNO3, (n)t-coral-0MEDTA and (n)t-coral-0.4V. The areas in gray indicate the datapoints included in Fig. 6(a) and (b). | ||
Therefore, it is important to analyze surface area effects. To this end, in Fig. 6(a) and (b) the geometric CO partial current densities at respectively −0.65 V vs. RHE and −1.05 V vs. RHE (indicated by the grey areas in Fig. 5) are plotted against the ECSA. At −0.65 V vs. RHE (Fig. 6(a)), a linear correlation between the ECSA and |JCO| is observed. However, at higher overpotentials, such as −1.05 V vs. RHE (Fig. 6(b)), this linear correlation was not found. The templated samples only perform slightly better than the non-templated samples. This is an indication that at −1.05 V vs. RHE internal mass transport limitations hamper the CO2RR activity. This is not surprising, as the DLC measurements already indicated the presence of diffusion limitations. However, it is interesting to see if the specific limitation around ∼4 mA cm−2 can be explained. Usually, rotating disc electrodes (RDEs) can be used to verify the presence of mass transport limitations and explain the trend in selectivity.38,39 However, this does not work for porous systems because the limiting diffusion is in the pores, as the diffusion inside pores is always lower (Deff = p/τ × Dbulk, were p is the porosity and τ is the tortuosity of the catalyst).36 Working with an RDE only changes diffusion towards the surface of an electrode, not inside the pores.
 | ||
| Fig. 6 Partial current densities to CO for both the template-based porous (blue) and non-templated equivalent (red) samples (n)t-flat-default, (n)t-flat-5mA, (n)t-flat-1MNH4OH, (n)t-flat-0.5MNaNO3, (n)t-coral-0MEDTA and (n)t-coral-0.4V at (A) −0.65 V vs. RHE and (B) −1.05 V vs. RHE plotted versus the ECSA. A dashed gray line is provided as a guide for the eye. | ||
Next to the CO partial current densities, it is interesting to investigate the H2 partial current densities, which can be found in Fig. 7 (and Table S1, ESI†). Remarkably, all the Ag samples, both templated porous and non-templated produce similar amounts of H2, which cannot be correlated to their different ECSA and porosity. It might seem that the non-templated samples produce more H2, but given the much smaller scale of H2 production compared to CO production, these differences are negligible.
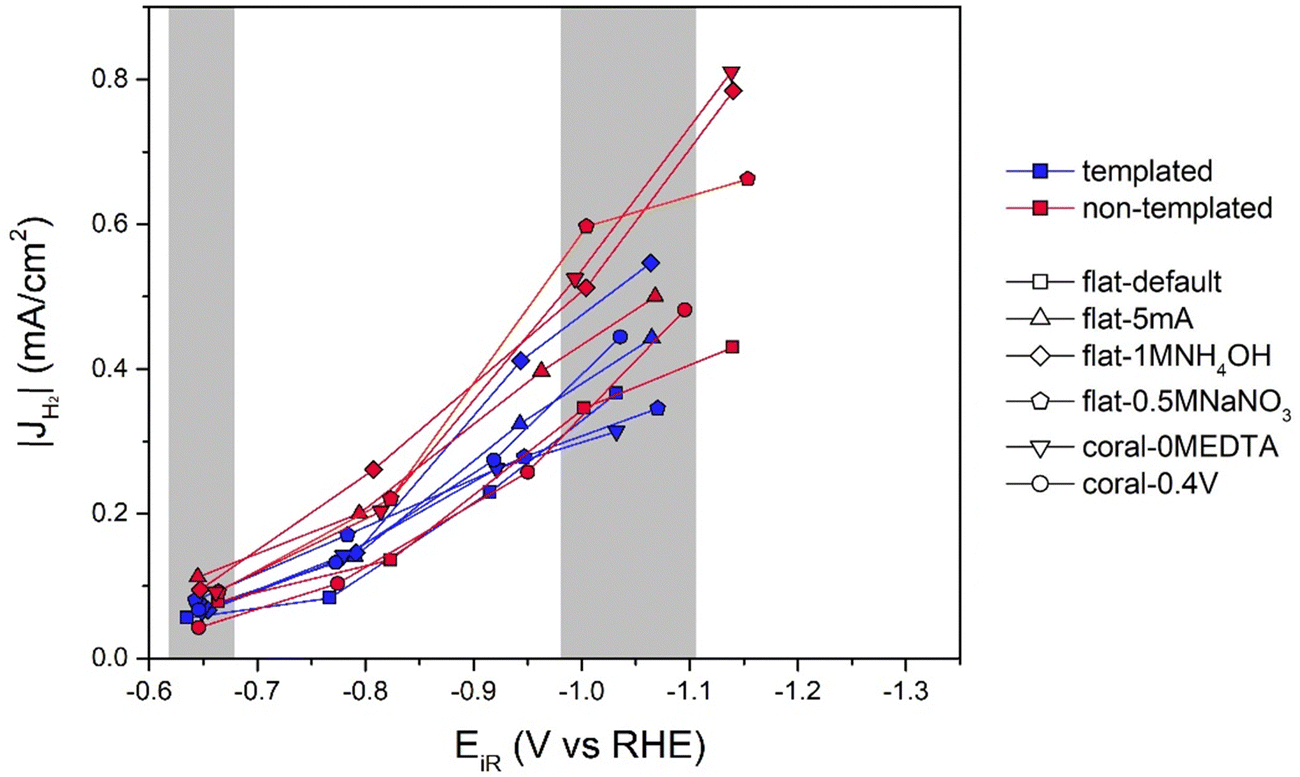 | ||
| Fig. 7 Partial current densities to H2 for both the template-based porous (blue) and non-templated equivalent (red) samples (n)t-flat-default, (n)t-flat-5mA, (n)t-flat-1MNH4OH, (n)t-flat-0.5MNaNO3, (n)t-coral-0MEDTA and (n)t-coral-0.4V. The areas in gray indicate the datapoints included in Fig. 8(a) and (b). | ||
The data in Fig. 8 confirm that neither at low overpotentials (Fig. 8(a)) nor at high overpotentials (Fig. 8(b)) the H2 partial current density scales with the ECSA. To understand this better, Fig. 8(a) and (b) show the partial current density of H2 produced on bare Ag foil as well. Interestingly, the |JH2| is in the same range as all the other catalysts. A possible explanation is that the HER is dominated by the electrode surface directly in contact with the bulk electrolyte and not inside the pores. However, it would be unlikely that the diffusion of protons or H2 would be slower than that of CO2, CO and/or associated carbonate ions. Another possibility is that the formation of H2 inside porous systems might be influenced by a difference in local pH insides the pores due to diffusion limitations, as postulated by Goyal et al.36 Also it cannot be excluded that more complex effects, such as an Ohmic drop over the porous structure influence the results.36
 | ||
| Fig. 8 Partial current densities to H2 for both the template-based porous (blue) and non-templated equivalent (red) samples (n)t-flat-default, (n)t-flat-5mA, (n)t-flat-1MNH4OH, (n)t-flat-0.5MNaNO3, (n)t-coral-0MEDTA and (n)t-coral-0.4V at (A) −0.65 V vs. RHE and (B) −1.05 V vs. RHE plotted versus the ECSA. | ||
In line with the trend in partial current density for CO and H2, the data in Table S2 (ESI†) shows the electron transfer resistance goes down and the electron transfer rate goes up for more negative potentials. Fig. S13 (ESI†) shows the electron transfer rate versus the ECSA at −1.05 V vs. RHE. Interestingly, this trend is exactly opposite to the trend found in |JCO| at this potential (see Fig. 6(b)). This result is in line with the hypothesis that internal mass transport limitations play an important role (see Fig. S2, ESI†).
So far, we have focused on the differences between porous and non-templated structures, but not on the effect of the different morphologies that have been discussed previously (Fig. 2). Comparing the flat templated structures (t-flat-default, t-flat-5mA, t-flat-1MNH4OH and t-flat-0.5MNaNO3) with the coral templated structures (t-coral-0.4V and t-coral-0MEDTA), one could expect faster diffusion in the coral structures -as they are more open- and, hence, differences in catalytic performance. This was not found: the only parameter that really defined the catalytic performance, was the ECSA of the catalyst. This implies that the nanoscale morphology is dominant for the catalytic performance, and not the 1–10 μm scale. So, the catalytic performance is robust, making the system more attractive for research application.
Catalyst stability
Porous Ag catalysts are reported to have an enhanced stability compared to non-templated structures.40 After catalysis, no significant Ag detachment was observed. However, the SEM images in Fig. 9 – showing t-flat-default before (Fig. 9(a)) and after (Fig. 9(b)) catalysis – illustrate that the catalyst morphology changes during catalysis. Although the porosity remains, the local fine structure is softened. Similar results were found for the other 5 porous Ag catalysts (Fig. S14, ESI†). | ||
| Fig. 9 HR-SEM images of p-flat-default (A) before catalysis and (B) after 1 cycle of catalysis at potentials between −0.65 V vs. RHE and −1.05 V vs. RHE. | ||
The question arises when exactly these changes in morphology take place. As the template-based porous electrodes have very well-defined structures, it is easy to follow changes over time. Therefore, the reaction was performed in a glass beaker (see the experimental section for more details). After each step of either the pretreatment or the catalysis, a small piece was cut out of the electrode and analyzed using SEM. To our knowledge, this has not been described in literature before. In the results that will follow, the potentials will be given without iR compensation.
Fig. 10 shows SEM images before catalysis (Fig. 10(a)) after each step of the pretreatment (Fig. 10(b)–(i)) and after each potential of catalysis (Fig. 10(j)–(l)). In Fig. 10(a), apart from the porous structure, the original grain boundaries from electrodeposition are visible. After saturating the electrolyte with CO2 (no potential applied, Fig. 10(b)), these boundaries have smoothened, but are still present. In addition, small spheres (10 nm) and large cubes (μm) are present on the porous structure. Thus, the morphology already started to change at this point. Then, after the first half cycle of the CV (from 0.1 to −1.4 V vs. RHE, Fig. 10(c)) these boundaries are almost fully smoothened. So, apparently, the negative potential applied drives this reconstruction. During the second half of the CV (Fig. 10(d)) the structure does not change much. Then, during the rest of the CV (the second, third, fourth and fifth cycle in respectively Fig. 10(e), (f), (g) and (h)) also the edges of the structure slowly begin to soften, probably fueled by the negative potential applied. During the DLC, the morphology does not change further, which is logical as the DLC was performed in the non-faradaic regime. The images after catalysis at −0.7 V (Fig. 10(j)), −0.9 V (Fig. 10(k)), −1.2 V (Fig. 10(l)) and −1.4 V (Fig. 10(m)), show that during catalysis, the morphology changes further. For all the images, small spheres (10 nm) are present. The more cathodic the potential, the softer the edge becomes. This is most clearly visible after catalysis at −1.2 V vs. RHE (Fig. 10(l)). These results agree with the assumption that negative potentials are responsible for the morphological changes of the catalyst.
 | ||
| Fig. 10 HR-SEM Images of the porous p-flat-default electrode (A) before catalysis; (B) after saturation (10 mL CO2 per min for 1 h); (C) after the first half CV cycle (0.1 → −1.4 vs. RHE); (D) after the second half CV cycle (−1.4 → 0.1 vs. RHE); (E) after the second CV cycle; (F) after the third CV cycle; (G) after the fourth CV cycle; (H) after the fifth CV cycle; (I) after DLC; (J) after catalysis at −0.7 V vs. RHE; (K) after catalysis at −0.9 V vs. RHE; (L) after catalysis at −1.2 V vs. RHE and (M) after catalysis at −1.4 V vs. RHE. | ||
It is important to realize that the morphology already changed before catalysis, implying that the structures in the SEM images of Fig. 2 are not exactly the same as the active phase during catalysis. This might explain why there was no significant difference in the catalytic performances of the four flat porous structures (t-flat-default, t-flat-5mA, t-flat-1MNH4OH, and t-flat-0.5MNaNO3). Next to that, the surface areas obtained via DLC might deviate from the values during catalysis. Here, the differences will be smaller, as DLC was measured shortly before the catalytic testing. Hence, at the least cathodic potentials, the structure will be almost the same. Only at the more cathodic potential the structure, and therefore the surface area, will differ. So, to summarize, these results show the importance of verifying the stability of the catalyst and checking the morphology of the electrode during and/or after catalysis.
In Fig. S15 (ESI†) the partial current densities to CO and H2 in the first and second cycle of catalysis are compared for all the templated electrodes to see if the catalytic performance is stable over time. The second cycle involves slightly altered morphologies discussed above. The electrodes produce more CO and similar amounts of H2. This can be explained by the local pH gradually increasing over time in the pores.36 This is known to lead to an increased CO production. These results hence indicate that diffusion limitations are more important than changes in the structure.
Conclusions
In this work we unraveled the effects of the morphology at different scales of templated porous Ag electrodes on their performance in electrocatalytic CO2 reduction. Clearly, the morphology is defined by the template used, and varying the electrodeposition parameters enabled us to tune the morphology of the porous electrodes successfully. The obtained structures were divided into two categories based on their shape on the μm scale: open, coral structures and closed, flat structures. Interestingly, the morphology on the μm scale obtained by varying the deposition conditions is the same as for the non-templated Ag equivalents on this scale, despite changes on the 100 nm scale due to templating. This enabled, for the first time, to discriminate between the effects of porosity on the 100 nm scale and the morphology on other scales on the catalytic properties of porous Ag electrodes. Additionally, we developed a mathematical model to estimate the roughness factor of the template-based porous Ag catalysts. Regarding the catalytic performance, we demonstrated that for all the porous Ag electrodes, the partial current density to CO was increased compared to their non-templated equivalents. At lower overpotentials, this is predominantly an effect of the higher surface area, as at −0.65 V vs. RHE a linear correlation between |JCO| and the ECSA was found. At higher overpotentials, the increase in |JCO| seems to be hampered by the limited diffusion of CO2 and/or other ions into the pores. Surprisingly, the partial H2 current density is independent of the catalyst's structure: where the CO production scaled with the internal electrode surface area, the H2 production was severely impeded at the internal electrode surface. Interestingly, the morphology on the μm scale (i.e. for coral, flat) did not influence the catalytic performance. This implies that the effect of templating on the nm scale (porosity) is the dominant factor for the catalyst's performance. Finally, it is noteworthy that although the boundaries and edges on the 10 nm scale are smoothened, the porosity remains after a long catalysis cycle, demonstrating the relatively good stability of the porous electrocatalysts for CO2 reduction.Data availability
The data supporting this article have been included as part of the ESI.† The data included in Fig. 4 that were produced by Yoon et al. and Hall et al. are available at respectively DOI: 10.1002/anie.201607942 and DOI: 10.1021/jacs.5b08259.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the reversible large-scale energy storage (RELEASE) consortium, a NWO program with project number 17621. Claudia Keijzer is acknowledged for measuring SEM in early stages of the project. Martijn Korporaal, Thom Heijnen and Alex Klaver are acknowledged for their help in sample preparation. Jan Willem de Rijk is acknowledged for his help with the catalytic set-up during this project and Eric Hellebrand for his technical support with SEM. Bert Weckhuysen and Ramon Oord from the Inorganic Chemistry and Catalysis group in Utrecht are acknowledge for the XRD measurements. Rik Mom and Alfred Larsson from Leiden University are acknowledge for the XPS measurements. Matt Peerlings, Francesco Mattarozzi and Valerio Gulino and Marc Koper from Leiden University are acknowledged for their useful discussions on the results.References
- Intergovernmental Panel on Climate Change, “Climate Change 2021 The Physical Science Basis Summary for Policymakers Working Group I Contribution to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change,” 2021. [Online]. Available: https://www.ipcc.ch/report/ar6/wg1/.
- M. Jouny, W. Luc and F. Jiao, General Techno-Economic Analysis of CO2 Electrolysis Systems, Ind. Eng. Chem. Res., 2018, 57(6), 2165–2177, DOI:10.1021/acs.iecr.7b03514
.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrocatalytic Process of CO Selectivity in Electrochemical Reduction of CO2 at Metal Electrodes in Aqueous Media, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS
- Y. Hori, A. Murata and R. Takahashi, Formation of Hydrocarbons in the Electrochemical Reduction of Carbon Dioxide at a Copper Electrode in Aqueous Solution, J. Chem. Soc., Faraday Trans. 1, 1989, 85(8), 2309–2326 RSC
- A. Murata and Y. Hori, Formation of Hydrocarbons in Electrochemical Reduction of Carbon Monoxide at an Fe Electrode in Connection with Electrochemical Reduction of Carbon Dioxide, Denki Kagaku, 1991, 6, 499–503 CrossRef
- Y.-C. Hsieh, S. D. Senanayake, Y. Zhang, W. Xu and D. E. Polyansky, Effect of Chloride Anions on the Synthesis and Enhanced Catalytic Activity of Silver Nanocoral Electrodes for CO2 Electroreduction, ACS Catal., 2015, 5(9), 5349–5356, DOI:10.1021/acscatal.5b01235
- Y. Hori and S. Suzuki, Electrolytic Reduction of Carbon Dioxide at Mercury Electrode in Aqueous Solution, Bull. Chem. Soc. Jpn., 1982, 55(3), 660–665, DOI:10.1246/BCSJ.55.660
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, Electrochemical CO2 Reduction: A Classification Problem, ChemPhysChem, 2017, 18(22), 3266–3273, DOI:10.1002/cphc.201700736
- X. Li, P. Anderson, H. R. M. Jhong, M. Paster, J. F. Stubbins and P. J. A. Kenis, Greenhouse Gas Emissions, Energy Efficiency, and Cost of Synthetic Fuel Production Using Electrochemical CO2 Conversion and the Fischer-Tropsch Process, Energy Fuels, 2016, 30(7), 5980–5989, DOI:10.1021/acs.energyfuels.6b00665
- M. Jouny, W. Luc and F. Jiao, General Techno-Economic Analysis of CO2 Electrolysis Systems, Ind. Eng. Chem. Res., 2018, 57(6), 2165–2177, DOI:10.1021/acs.iecr.7b03514
- J. E. van den Reijen, P. H. Keijzer and P. E. de Jongh, Pore structure stabilization during the preparation of single phase ordered macroporous α-alumina, Materialia, 2018, 4, 423–430, DOI:10.1016/j.mtla.2018.10.016
- H. Yan, C. F. Blanford, W. H. Smyrl and A. Stein, Preparation and structure of 3D ordered macroporous alloys by PMMA colloidal crystal templating, Chem. Commun., 2000, 49, 10.1039/b003147j
- A. Stein, Sphere templating methods for periodic porous solids, Microporous Mesoporous Mater., 2001, 44–45, 227–239, DOI:10.1016/S1387-1811(01)00189-5
- K. Zhang, X. Tan, J. Zhang, W. Wu and Y. Tang, Template-dealloying synthesis of ultralow density Au foams with bimodal porous structure, RSC Adv., 2014, 4, 7196–7201, 10.1039/c3ra47195k
- H. Yan,
et al., Fabrication of 2D and 3D ordered porous ZnO films using 3D opal templates by electrodeposition, Electrochem. Commun., 2005, 7(11), 1117–1121, DOI:10.1016/j.elecom.2005.08.011
- A. Stein and R. C. Schroden, Colloidal crystal templating of three-dimensionally ordered macroporous solids: Materials for photonics and beyond, Curr. Opin. Solid State Mater. Sci., 2001, 5(6), 553–564, DOI:10.1016/S1359-0286(01)00022-5
- Y. Yoon, A. S. Hall and Y. Surendranath, Tuning of Silver Catalyst Mesostructure Promotes Selective Carbon Dioxide Conversion into Fuels, Angew. Chem., Int. Ed., 2016, 55(49), 15282–15286, DOI:10.1002/anie.201607942
- S. Suter and S. Haussener, Optimizing mesostructured silver catalysts for selective carbon dioxide conversion into fuels, Energy Environ. Sci., 2019, 12(5), 1668–1678, 10.1039/c9ee00656g
- N. Hoshi, M. Kato and Y. Hori, Electrochemical reduction of CO2 on single crystal electrodes of silver Ag(111), Ag(100) and Ag(110), J. Electroanal. Chem., 1997, 440(1–2), 283–286, DOI:10.1016/S0022-0728(97)00447-6
- J. Rosen,
et al., Mechanistic Insights into the Electrochemical Reduction of CO2 to CO on Nanostructured Ag Surfaces, ACS Catal., 2015, 5(7), 4293–4299, DOI:10.1021/acscatal.5b00840
-
A. C. Fisher, Electrode Dynamics, Oxford University Press, 1996 Search PubMed
- G. M. De Oliveira, L. L. Barbosa, R. L. Broggi and I. A. Carlos, Voltammetric study of the influence of EDTA on the silver electrodeposition and morphological and structural characterization of silver films, J. Electroanal. Chem., 2005, 578(1), 151–158, DOI:10.1016/j.jelechem.2004.12.033
- A. Liu,
et al., A Combined Theoretical and Experimental Study for Silver Electroplating, Sci. Rep., 2015, 4, 35–43, DOI:10.1038/srep03837
- D. Kang, T. W. Kim, S. R. Kubota, A. C. Cardiel, H. G. Cha and K. S. Choi, Electrochemical Synthesis of Photoelectrodes and Catalysts for Use in Solar Water Splitting, Chem. Rev., 2015, 115(23), 12839–12887, DOI:10.1021/acs.chemrev.5b00498
- R. S. D. Zou, S. Ma, R. Guan, M. Park, L. Sun and J. J. Aklonis, Model filled polymers. V. Synthesis of crosslinked monodisperse polymethacrylate beads, J. Polym. Sci., Part A: Polym. Chem., 1992, 30(1), 137–144, DOI:10.1007/BF00419975
- R. C. Schroden, N. Balakrishnan, A. Stein and M. D. Ward, Inverse Opal Photonic Crystals A Laboratory Guide, Univ. Minnesota, Mater. Res. Sci. Eng. Center, Minnesota, 1–42, 2001, [Online]. Available: http://scholar.google.com/scholar?hl=en&btnG=Search&q=intitle:Inverse+Opal+Photonic+Crystals+A+Laboratory+Guide+Authors#0.
- H. Javed, K. Kolmeijer, N. Klein, J. A. Trindell, G. Schneider and R. V. Mom, A laboratory-based electrochemical NAP-XPS system for operando electrocatalysis studies, Vacuum, 2025, 231, 113755, DOI:10.1016/j.vacuum.2024.113755
- F. Mattarozzi,
et al., Alkylamine-Functionalized Carbon Supports to Enhance the Silver Nanoparticles Electrocatalytic Reduction of CO2 to CO, ChemElectroChem, 2023, 10(19), 1–9, DOI:10.1002/celc.202300295
- L. Guo and P. C. Searson, On the influence of the nucleation overpotential on island growth in electrodeposition, Electrochim. Acta, 2010, 55(13), 4086–4091, DOI:10.1016/j.electacta.2010.02.038
- G. M. De Oliveira and I. A. Carlos, Silver-copper electrodeposition from ammonium hydroxide solution: Influence of EDTA and HEDTA, J. Appl. Electrochem., 2009, 39(8), 1217–1227, DOI:10.1007/s10800-009-9777-6
- S. S. V. Tatiparti and F. Ebrahimi, Potentiostatic versus galvanostatic electrodeposition of nanocrystalline Al–Mg alloy powders, J. Solid State Electrochem., 2012, 16(3), 1255–1262, DOI:10.1007/s10008-011-1522-5
-
F. Nasirpouri, Fundamentals and principles of electrode-position, 2017, vol. 62 Search PubMed
- Y. Jeon, S. Choe, H. C. Kim, M. J. Kim and J. J. Kim, Electrodeposition of Cu-Ag films in ammonia-based electrolyte, J. Alloys Compd., 2019, 775, 639–646, DOI:10.1016/j.jallcom.2018.10.023
- H. Wang and L. Pilon, Physical interpretation of cyclic voltammetry for measuring electric double layer capacitances, Electrochim. Acta, 2012, 64, 130–139, DOI:10.1016/j.electacta.2011.12.118
- D. M. Morales and M. Risch, Seven steps to reliable cyclic voltammetry measurements for the determination of double layer capacitance, JPhys Energy, 2021, 3(3), 1–18, DOI:10.1088/2515-7655/abee33
- A. Goyal, C. J. Bondue, M. Graf and M. T. M. Koper, Effect of pore diameter and length on electrochemical CO2 reduction reaction at nanoporous gold catalysts†, Chem. Sci., 2022, 13(11), 3288–3298, 10.1039/d1sc05743j
- A. S. Hall, Y. Yoon, A. Wuttig and Y. Surendranath, Mesostructure-Induced Selectivity in CO2 Reduction Catalysis, J. Am. Chem. Soc., 2015, 137(47), 14834–14837, DOI:10.1021/jacs.5b08259
- A. Goyal and M. T. M. Koper, Understanding the role of mass transport in tuning the hydrogen evolution kinetics on gold in alkaline media, J. Chem. Phys., 2021, 155(13), 134705, DOI:10.1063/5.0064330
- A. Goyal, G. Marcandalli, V. A. Mints and M. T. M. Koper, Competition between CO2 Reduction and Hydrogen Evolution on a Gold Electrode under Well-Defined Mass Transport Conditions, J. Am. Chem. Soc., 2020, 142(9), 4154–4161, DOI:10.1021/jacs.9b10061
- H. Wang,
et al., Enhanced CO selectivity and stability for electrocatalytic reduction of CO2 on electrodeposited nanostructured porous Ag electrode, J. CO2 Util., 2016, 15, 41–49, DOI:10.1016/j.jcou.2016.04.013
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ma00939h |
| ‡ Authors contributed equally to this research. |
| This journal is © The Royal Society of Chemistry 2025 |