
Improved lithium-ion battery cathode rate performance via carbon black functionalization†
Donghyuck
Park
 a,
Peter C.
Sherrell
ab,
Fangxi
Xie
ac and
Amanda V.
Ellis
*a
a,
Peter C.
Sherrell
ab,
Fangxi
Xie
ac and
Amanda V.
Ellis
*a
aDepartment of Chemical Engineering, The University of Melbourne, Grattan Street, Parkville, Victoria 3010, Australia. E-mail: amanda.ellis@unimelb.edu.au
bSchool of Science, RMIT University, Melbourne, Victoria 3001, Australia
cSchool of Chemical Engineering and Technology, Sun Yat-Sen University, Zhuhai, 519082, P. R. China
First published on 24th January 2024
Abstract
Carbon black (CB) creates essential electron transport pathways in lithium-ion battery (LiB) cathodes. Here, we show that by modifying the surface of CB via mild hydrogen peroxide or nitric acid treatment, the rate performance of a LiB cathode can be increased up to 350% at 0.75 C-rate charging. We demonstrate that this improvement is predominately due to the presence of introduced carbonyl groups on the surface of the CB which increases the surface redox reaction of the nickel manganese cobalt oxide (NMC) cathode active material. As a result, there is both a decrease in the overpotential (∼37% during 0.25 C-rate charging) and electrochemical impedance. A mechanism is proposed which describes how the cathode performance is influenced by electrostatic interactions between the CB surface and solvated lithium ions. An improved rate effect was demonstrated across various cathode active materials, clearly highlighting the versatility of this simple approach.
1. Introduction
Lithium-ion batteries (LiBs), as a bridge towards a green energy transformation, have a rapidly expanding role in society, ranging from powering personal electronics,1 to hybrid or fully electric vehicles,2–4 as well as managing intermittent power supply from renewable energy on the national grid.5,6 Amongst the components within LiBs, the cathode plays a critical role in the cell-level performance metrics such as accessible capacity and rate performance.7–9 The most commonly researched cathode is nickel manganese cobalt oxide (NMC), with an electrical resistance of ∼10−7 to ∼10−2 S m−1.10 Most LIB research has focused on the active material including its chemistry,8,11 coating,12 cathode–electrolyte interface,13–15 and electrode design.16 In particular, focus has been on increasing the cathode capacity and rate performance by enhancing the Li+ ion transport at the cathode–electrolyte interface.In order to boost a cathodes electronic conductivity, carbon black (CB) is added as a conductive additive in the vast majority of LiBs.17,18 In addition, CB aids in the formation of ionic pathways for lithium-ion (Li+) diffusion where the electrolyte fills the pores in the cathode.19 In 2019, Park et al.20 used conductive single-walled carbon nanotubes (SWCNTs) in high energy density LiBs, finding that the high aspect ratio, conductivity and mechanical strength of the SWCNTs provided exceptional performance, up to 29 mA h cm−2. However, CB remains the conductive additive of choice in LiBs due to its simple synthesis and low cost. However, while the electronic properties of CBs are well understood, how CB influences ionic transport within the cathode requires further study.
One path to improve CBs ionic transport properties is via chemical functionalization of its surface. The chemical modification of CB, in general, has been studied extensively, particularly for the introduction of oxygen-containing functionalities onto its surface.21 The majority of approaches have used strong chemical oxidizing agents such as sulfuric acid (H2SO4),22–24 potassium permanganate (KMnO4),25,26 or high concentration nitric acid (>60% HNO3),27,28 all of which oxidize the target carbon material while simultaneously generating defects within the CB structure.
In LiB anodes, the role of defects and surface functionalities of the graphite active material has been well studied and shown to contribute to an increased Li+ ion storage capacity and facilitate Li+ ion diffusion.29–33 In particular, oxygen groups have been shown to exhibit strong Li+ ion absorption, leading to enhanced capacity and rate performance.34 Furthermore, hydroxyl (–C–OH) and carboxyl (–COOH) groups on the graphite have potential to reduce the energy barrier of Li+ ion intercalation by generating a chemical interaction with the electronegative oxygen atoms in the carbonate molecules within the electrolyte.35 Despite these detailed studies on graphite anodes, the role of oxygen-containing groups on CBs within a cathode have not been well explored.
In this work, the role of oxygen functional groups (hydroxyl, carbonyl (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and carboxyl) on CB, in LiB cathodes, is studied. Commercial CB, SuperP, was treated under various mild oxidation conditions. Raman spectroscopy and X-ray photoelectron spectroscopy (XPS) were used to show defect generation and oxygen surface functionalities, respectively. NMC cathodes doped with functionalized CBs were investigated for surface redox behaviour, electrical conductivity, rate performance and electrochemical impedances. Finally, the functionalized CB was applied with other cathode active materials, namely, Ni0.8Mn0.2Co0.2O2 (NMC811), lithium manganese oxide (LiMn2O4) (LMO), and nickel cobalt aluminium oxide (Ni0.8Co0.15Al0.05O2) (NCA) to evaluate the rate performances compare to pristine SuperP.
O) and carboxyl) on CB, in LiB cathodes, is studied. Commercial CB, SuperP, was treated under various mild oxidation conditions. Raman spectroscopy and X-ray photoelectron spectroscopy (XPS) were used to show defect generation and oxygen surface functionalities, respectively. NMC cathodes doped with functionalized CBs were investigated for surface redox behaviour, electrical conductivity, rate performance and electrochemical impedances. Finally, the functionalized CB was applied with other cathode active materials, namely, Ni0.8Mn0.2Co0.2O2 (NMC811), lithium manganese oxide (LiMn2O4) (LMO), and nickel cobalt aluminium oxide (Ni0.8Co0.15Al0.05O2) (NCA) to evaluate the rate performances compare to pristine SuperP.
2. Results and discussion
2.1 Chemical modification of carbon black
Elucidating the role that CB plays in ion transport is critical to minimizing the rapid charging and discharging in LiBs. Modifying CB with various surface functional groups without sacrificing electronic conductivity will enable the study of such ion transport effects. To avoid the formation of a significant number of defects in the CB, and associated loss of electronic conductivity, mild oxidizing agents were used to enable control over surface functional group formation (Fig. 1a). To this end, CB was treated with H2O2 at 70 °C for 24, 48 or 72 h, as well as 20% v/v or 70% v/v HNO3 for 1.5 h. Raman spectroscopy was performed on the pristine and modified CBs to measure defect generation. The Raman spectra of CB is dominated by the D-band (∼1350 cm−1, disordered phase, out-of-plane vibration attributed to the defects36) and the G-band (∼1582 cm−1, graphitic phase, in-plane vibration of sp2 bonding37), with the ID/IG ratio is proportional to the degree of sp3 defects.38Fig. 1b and c displays the ID/IG ratios for the H2O2- and HNO3-treated CBs, respectively. The intensity of the ID/IG ratio for the H2O2-treated CBs (Fig. 1b) remained constant from over the course of 72 h treatment at 1.63 ± 0.03 (pristine) to 1.61 ± 0.10 (72 h), indicating minimal defect generation.39,40 Mild HNO3 (20% v/v) treated CB showed a similar defect level (ID/IG = 1.60 ± 0.19) to both untreated CB (1.63 ± 0.03) and 72 h H2O2 (1.61 ± 0.10) treated CB (Fig. 1c). This lack of defect generation is important as it avoids changes in chemical and electrical properties arising from defects that can interfere with the evaluation of oxygen groups. However, stronger HNO3 (70% v/v) treatment generated significant new defects (ID/IG = 2.03 ± 0.21) (Fig. 1d).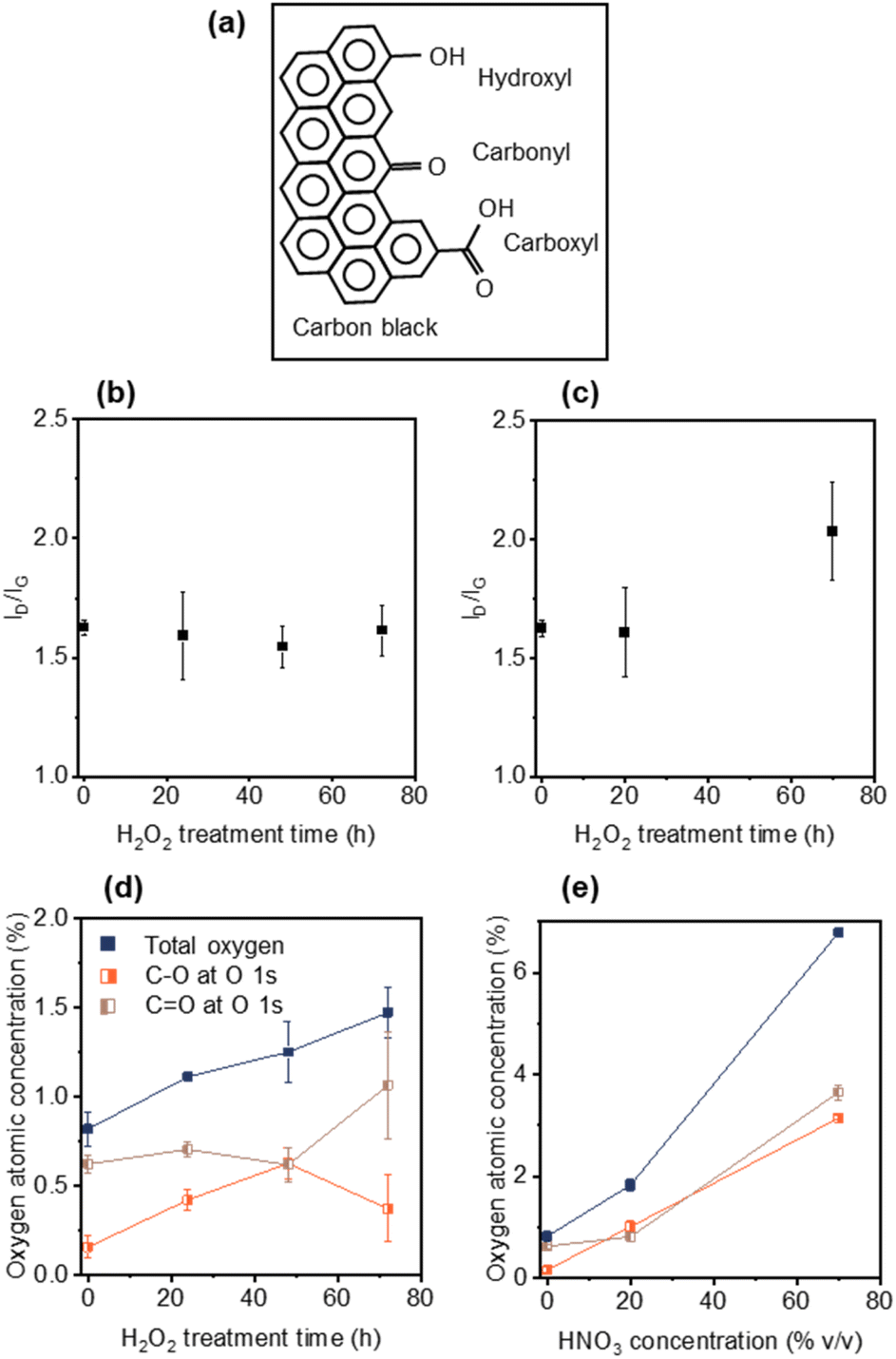 | ||
| Fig. 1 (a) Schematic of oxygen-containing functional groups on CB, namely –C–OH (hydroxyl), –CO (carbonyl) and –COOH (carboxyl). Oxygen atomic concentrations of analysed XPS spectra for (b) H2O2-treated CB for 0, 24, 48 and 72 h and (c) HNO3-treated CB with 20% v/v and 70% v/v. Raman intensity ratios of D- to G-band (ID/IG) for (d) H2O2-treated CB for 0, 24, 48 and 72 h and (e) HNO3-treated CB with 20% v/v and 70% v/v. The error bars represent standard error. | ||
Oxygen functionalization of the CBs was monitored using XPS (Fig. S1–S4†). XPS peaks in the O 1s region were fitted using CASA XPS software (see Fig. S3 and Table S1†). The peak centred at ∼533.0 eV was assigned to C–O bonding, which mainly corresponds to C–OH and/or COOH.41 The peak centred at ∼531.5 eV was assigned to CO bonding which mainly corresponds to –CO or –COOH functional groups42 (Fig. 1a). Peng et al.39 and Vincente-Santiago et al.40 have demonstrated that the atomic concentration ratios of the C–O to CO can be used to identify the dominant oxygen functional group species (C–OH, CO, or COOH) on a carbon material.
Treatment with H2O2 for 24 h, 48 h and 72 h showed an increase in the minimal total oxygen content from 0.82 ± 0.09 at% (pristine) to 1.47 ± 0.14 at% (72 h) (Fig. 1d and Table 1). After 24 h H2O2 treatment the C–OH content appeared to dominate (noting that the CO at% remains higher than the C–O at% due to the initial functional groups on pristine CB), with the C–O content increasing from 0.16 ± 0.06 at% to 0.42 ± 0.06 at%. From 24 h to 48 h the C–O content increased from 0.42 ± 0.06 at% to 0.63 ± 0.09 at% with a constant level of CO. However, between 48 h and 72 h a marked change in C–O content was observed, decreasing from 0.63 ± 0.09 at% to 0.37 ± 0.19 at% with a corresponding increase in CO content from 0.62 ± 0.10 at% to 1.07 ± 0.30 at%, resulting in CO groups dominating the modified CB. The tendency of H2O2 treatment to preferentially oxidize C–OH groups to CO groups, over generating new C–OH groups on the surface of CB, is in good agreement with reported literature.39,40
| Treatment | Sample name | Atomic concentration (at%) | I D/IG | ||
|---|---|---|---|---|---|
| Total O | O 1s C–O | O 1s CO |
|||
| Pristine | CB | 0.82 ± 0.09 | 0.16 ± 0.06 | 0.62 ± 0.05 | 2.95 ± 0.10 |
| H2O2 24 h | CB–OH (24 h) | 1.11 ± 0.01 | 0.42 ± 0.06 | 0.71 ± 0.04 | 3.08 ± 0.15 |
| H2O2 48 h | CB–OH (48 h) | 1.25 ± 0.17 | 0.63 ± 0.09 | 0.62 ± 0.10 | 3.11 ± 0.20 |
| H2O2 72 h | CBO (72 h) |
1.47 ± 0.14 | 0.37 ± 0.19 | 1.07 ± 0.30 | 3.24 ± 0.22 |
| HNO3 20% v/v | CB–COOH (20%) | 1.83 ± 0.11 | 1.01 ± 0.11 | 0.81 ± 0.09 | 3.18 ± 0.27 |
| HNO3 70% v/v | CB–COOH (70%) | 6.79 ± 0.10 | 3.15 ± 0.04 | 3.65 ± 0.14 | 3.69 ± 0.32 |
In contrast, for both 20% v/v and 70% v/v HNO3 treatments both C–O and CO content are shown to increase (Fig. 1e and Table 1), consistent with the reported literature for the formation of COOH groups via HNO3 treatment (where 2CH2 + 5HNO3 → 2COOH + 5HNO2 + H2O).24,28,39,43–45 The 20% v/v HNO3 treated CB showed a higher oxygen content than H2O2 72 h CB (1.82 vs. 1.47 at%) despite similar defect levels. This result is explained by the tendency of HNO3 treatment to form carboxyl-functional groups, which have 2 oxygen atoms per sp3 defect site (compared to 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for hydroxyl- or carbonyl- functional groups in Fig. 1a). Note that no nitrogen signal was observed in the XPS spectrum (Fig. S4†) and that epoxy groups (C–O–C bending at ∼850 cm−2)46 were not observed in any of the Raman spectra (Fig. S5†).
:1 for hydroxyl- or carbonyl- functional groups in Fig. 1a). Note that no nitrogen signal was observed in the XPS spectrum (Fig. S4†) and that epoxy groups (C–O–C bending at ∼850 cm−2)46 were not observed in any of the Raman spectra (Fig. S5†).
Thus, from here on each sample is denoted by its oxygen-functionality dominance, as CB; CB–OH (24 h); CB–OH (48 h); CBO (72 h); CB–COOH (20%); and CB–COOH (70%).
2.2 Electrochemical characterisation
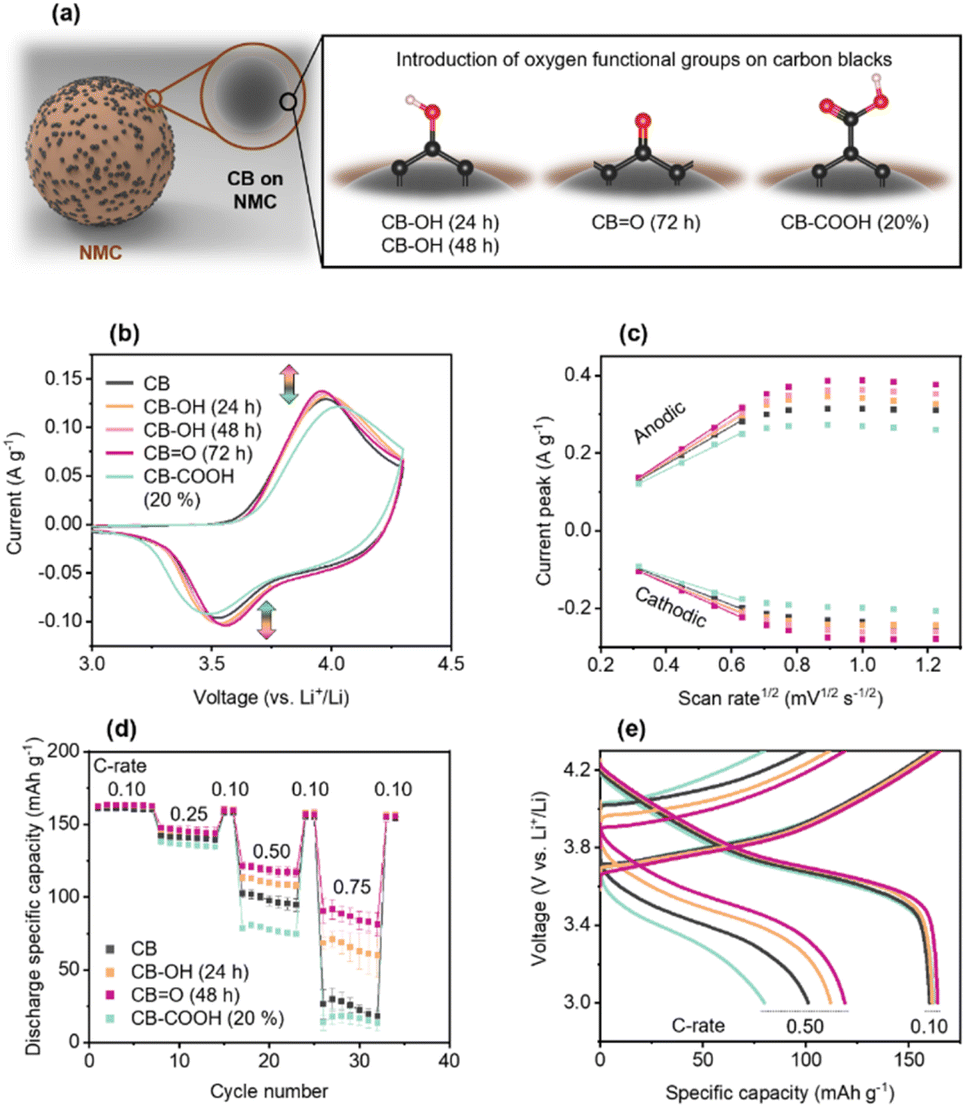 | ||
| Fig. 2 Electrochemical tests for NMC622 cathodes fabricated using modified CBs. (a) Schematic of oxygen-containing functional groups on CB. (b) CV with a scan rate of 0.1 mV s−1. (c) Anodic and cathodic current peaks at a scan rate of 0.1–1.5 mV s−1versus the square root of the scan rate (mV1/2 s−1/2), a linear interval of 0.1–0.4 mV s−1 was fitted. (d) Rate performance of the NMC622 cathodes at 0.10, 0.25, 0.50, and 0.75 C-rates, error bars represent standard deviations. (e) GCD curves at 0.1 and 0.5 C-rates. | ||
The electrochemical performance of the pristine and modified CBs was investigated using cyclic voltammetry (CV) at a scan rate of 0.1 mV s−1 (Fig. 2b). In the CV, the anodic peak corresponds to Li+ ion de-insertion, and the anodic peak corresponds to Li+ ion insertion. The data shows an increase in anodic and cathodic peaks in the order of: CBO (72 h) 0.138 A g−1, −0.104 A g−1 > CB–OH (48 h) 0.135 A g−1, −0.103 A g−1 > CB–OH (24 h) 0.133 A g−1, −0.102 A g−1 > CB 0.129 A g−1, −0.096 A g−1 > CB–COOH (20%) 0.122 A g−1, −0.092 A g−1. The largest current peaks for CBO (72 h) indicate that this cathode has the fastest reaction rate of deintercalation and intercalation of Li+ ions. According to the Nernst equation, the gap between the specific voltages at which the anodic and cathodic peaks appear, is inversely proportional to the electron transfer rate. Thus, as CBO (72 h) has the lowest peak separation potential (Table S1†) as it undergoes the most efficient electrochemical reaction for charging/discharging compared to the other cathodes.
CVs at various scan rates (0.1, 0.2, 0.3, 0.4, 0.5, 0.6, 0.8, 1.0, 1.2 and 1.5 mV s−1) were performed on the CB, CB–OH (24 h), CB–OH (48 h), CBO (72 h), and CB–COOH (20%) cathodes (Fig. S7†). The current response could be distinguished into faradaic current (iF) and non-faradaic electrochemical double-layer current (EDLC), with iF arising from both the surface redox reaction at the electrode–electrolyte interface (iS) and bulk diffusion reactions (iB) on the cathode material.50,51 Note, that the current originating from the EDLC is commonly ignored because of its small overall contribution;52 thus iF can be derived from the Randles–Sevcik equation, eqn (1).53,54
| iF = iS + iB = k1v + k2v1/2 | (1) |
O (72 h) (anodic slope: 0.57) have advantageous (faster) surface redox reactions compared to pristine CB in both the anodic and cathodic reactions. In contrast, the lower slopes of the CB–COOH (20%) (anodic slope: 0.42) indicate that the surface redox reaction is hindered by COOH groups. The same trends were observed in the peak separation potential analysis (Fig. S8 and Table S5†).
The practical performance of the cathodes was evaluated via GCD in the voltage range of 3.0–4.3 V vs. Li+/Li, as shown in Fig. 2d and e. The potential window of 3.0–4.3 V vs. Li+/Li was chosen as standard cut-off voltage of NMC cathode.55–57 All samples showed similar GCD curves, delivering a discharge capacity of ∼160 mA h g−1 at 0.10C (C-rate is calculated based on the theoretical capacity of ∼277.4 mA h g−1). NMC622 typically has an accessibly capacity of 158–174 mA h g−1 at 0.10C depending on the active material mass loading and electrode structure.58
As the C-rate is increased to 0.10, 0.25, 0.50, and 0.75C, differences in the cathode rate performance of NMC622 cathodes made with modified CBs are observed. The pristine CB cathode showed a significant decrease in capacity at 0.75C down to 24.4 mA h g−1, around 15% of the capacity at 0.10C. Similarly, the CB–COOH (20%) cathode showed a lower performance at 16.1 mA h g−1 at 0.75C. However, both CB–OH (24 h) and CBO (72 h) cathodes showed a dramatically improved performance at 0.75C, at 65.4 mA h g−1 and 86.5 mA h g−1, respectively.
For all samples, switching from a fast (0.25, 0.50, 0.75C) to a slow 0.10 C-rate demonstrated full capacity recovery. This recovery confirms that the performance deterioration of CB–COOH (20%) at high C-rate may originate from electrostatic interactions of Li+ ions with carboxyl groups. Komaba et al.59 have previously shown that Li+ ions can be trapped by electronegative carboxyl groups. Since Li+ trapping is an electrostatic attraction, and reversible, full capacity recover of the CB–COOH (20%) should be expected. A modified CB/polyvinylidene difluoride composite in the cathode, operating between 3 and 4.3 V vs. Li+/Li using CV, confirmed this reversibility (Fig. S7†). Polarization of the electrical double layer is observed up to 300 mV, with no oxidation (anodic) peak. Shuichi et al.60 have observed that oxygen groups in a carbonate medium do not decompose in the range of 1.9–4.9 V vs. Li+/Li (symmetric activated carbon electrodes with 1 M triethylmethylammonium (TEMA) tetrafluoroborate (BF4) in propylene carbonate (PC)). Thus, the lack of an oxidation peak precludes any irreversible phase change from occurring.
O (72 h) the resistance to different processes occurring within the cathode were studied. For electrochemical reactions at the LiBs cathode, the resistance experienced by Li+ ions during diffusion occur in 3 locations: (1) inside the NMC (ZWarburg for solid-state diffusion), (2) within the porous structure (Rion for ionic transportation) and (3) on the surface of NMC (RCEI for cathode–electrolyte interphase and Rct for charge-transfer).61–64 In the case of solid-state diffusion a known deterioration mechanism is the acidic attack of the active material which leads to transition metal dissolution, hydrofluoric acid (HF) production, and associated crystal structure changes.65 In this work, the oxygen groups are not involved in the deterioration mechanism. The HF can be produced when lithium salt (LiPF6) and water react,66,67 but the oxygen groups were not decomposed in the 3–4.3 V vs. Li+/Li operating range (Fig. S9†). This means that there is no evidence oxygen groups are involved in the acidic attack. In addition, recovery of the capacity at 0.10C after a high current rate (Fig. 2c) showed NMC crystal structure is not influenced by the oxygen groups. Thus, the introduction of oxygen groups on CB does not appears to affect the solid-state diffusion of Li+ ions within the NMC622 active material.
To identify electronic transfer and Li+ ion diffusion within the porous NMC622 cathode structure, out-of-plane electrical conductivity was investigated as a function of CB content (0.7–5 wt%) (Fig. 3a and Note S1, ESI†). The out-of-plane conductivity indirectly demonstrates not only electronic transportation from a current collector to active sites,20,68,69 but also the passively formed porous structure upon CB dispersion.19,70Fig. 3a shows an exponential increase in electrical conductivity with CB content, explained by percolation theory.71,72 The percolation threshold for each CB in the NMC622 cathode was calculated as follows: CB (ϕc = 0.74 ± 0.05 wt%); CB–OH (24 h) (ϕc = 0.76 ± 0.09 wt%); CBO (72 h) (ϕc = 0.74 ± 0.05 wt%); and CB–COOH (20%) (ϕc = 0.71 ± 0.07 wt%). This data clearly shows that there is no significant difference on the distribution of the CBs in the cathode. The electrical resistances of the NMC622 cathode with 5 wt% CB showed CO (72 h) to have the highest value (CB = 63.2 ± 7.9 Ω m, CB–OH (24 h) = 78.3 ± 5.1 Ω m, CBO (72 h) = 83.5 ± 7.2 Ω m, and CB–COOH (20%) = 58.0 ± 9.4 Ω m) (Fig. 3b). Despite the highest electrical resistivity, the CBO (72 h) shows the best electrochemical rate performance in Fig. 2b. Therefore, the enhancement in rate performance does not derive from electrical resistivity of the cathode, rather it may come from chemical and interfacial effects.
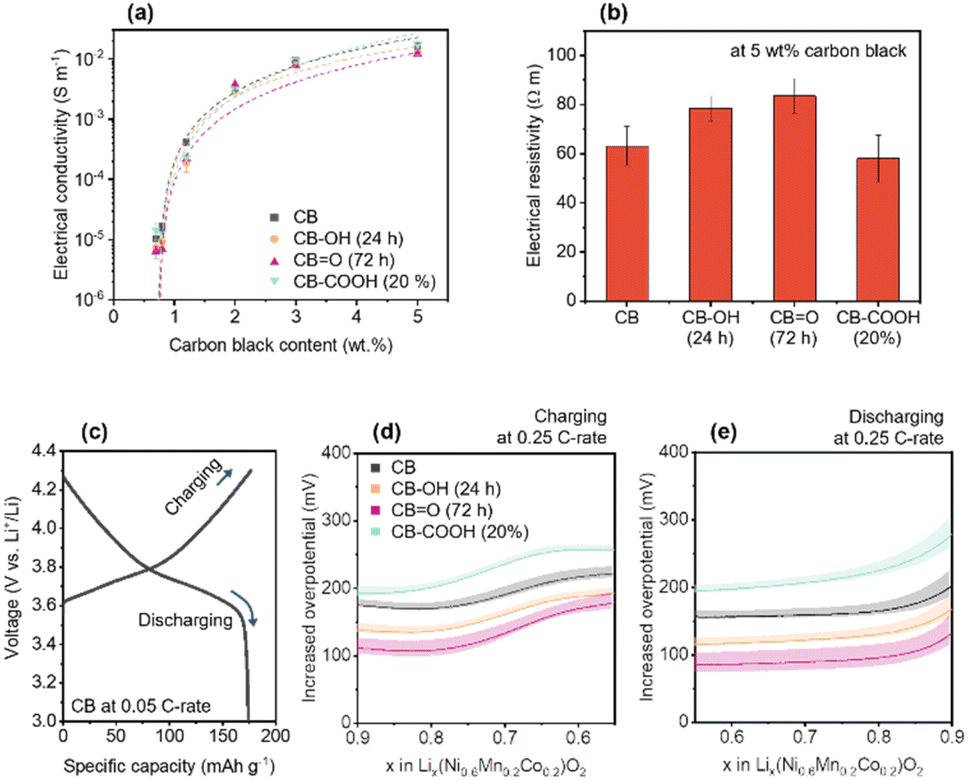 | ||
| Fig. 3 (a) Out-of-plane electrical conductivity of NMC622 cathodes with various CB content. The electrical conductivity is fitted with percolation equation (see Note S1, ESI†). (b) Electrical resistivity of the cathodes with 5 wt% CB content. (c) GCD curve for CB at 0.05 C-rate. Calculated overpotentials of the cathodes at 0.25 C-rate versus CB at 0.05 C-rate during (d) charging and (e) discharging. The error bars and bands represent standard deviation. | ||
To understand how oxygen groups on the surface of CB affect the Li+ ions at the surface of NMC, experiments comparing the potential difference between a pristine CB sample at slow rate of 0.05C (Fig. 3c), and at moderate rate of 0.25C (Fig. 3d and e) was investigated for CB, CB–OH (24 h), CBO (72 h), and CB–COOH (20%). The potential difference (Fig. 3d and e) is estimated as the voltage difference between NMC622 samples (CB, CB–OH (24 h), CBO (72 h), and CB–COOH (20%) at 0.25C) with CB (at 0.05C), and thus is termed overpotential.73 In Fig. 3c, the GCD curve of NMC622 with CB at 0.05C (discharge capacity ∼175 mA h g−1) is shown as reference. The x value in Lix(Ni0.6Mn0.2Co0.2)O2 (lithiated NMC622) is calculated based on the theoretical capacity of ∼277.4 mA h g1.
The measured overpotential at 0.25C (Fig. 3d and e) corresponds to the extra voltage that needs to be applied to the system to charge at the faster rate. As the C-rate increases, extra voltage or overpotential is required due to a departure from equilibrium charging/discharging processes, and the subsequent formation of a concentration gradient of Li+ ions in the electrode.74 In the range of x = 0.9 to 0.55 during charging, the NMC is undergoing a phase transition of H1 → H2 which corresponds to the expansion of a axis and the contraction of c axis of the NMC lattice simultaneously.75 As expected, within this x = 0.9 to 0.55 range, all NMC cathodes (CB, CB–OH (24 h), CBO (72 h), and CB–COOH (20%)) cathodes show an increased overpotential at 0.25C compared to a 0.05C CB cathode (Fig. 3d). The CBO (72 h) cathode demonstrates the lowest overpotential (109 mV at x = 0.9) compared to CB 173 mV, CB–OH (24 h) 136 mV, and CB–COOH (20%) 193 mV, indicating that the CBO (72 h) cathode requires the least energy to charge at faster rates. This trend is also consistent with overpotential on discharging. In Fig. 3e, in the range of x = 0.55 to 0.9, the overpotentials are low in order of: CBO (72 h) 101 mV, CB–OH (24 h) 136 mV, CB 171 mV, and CB–COOH (20%) 235 mV at x = 0.55. The CBO (72 h) reduced the overpotentials, energy barrier for electrochemical reaction, of 64 and 70 mV compared to pristine CB at 0.25C charging/discharging, respectively.
O (72 h) compared to CB within the NMC622 cathode (Fig. 2c and d; 3d and e), electrochemical impedance spectroscopy (EIS) was performed (Fig. 4).
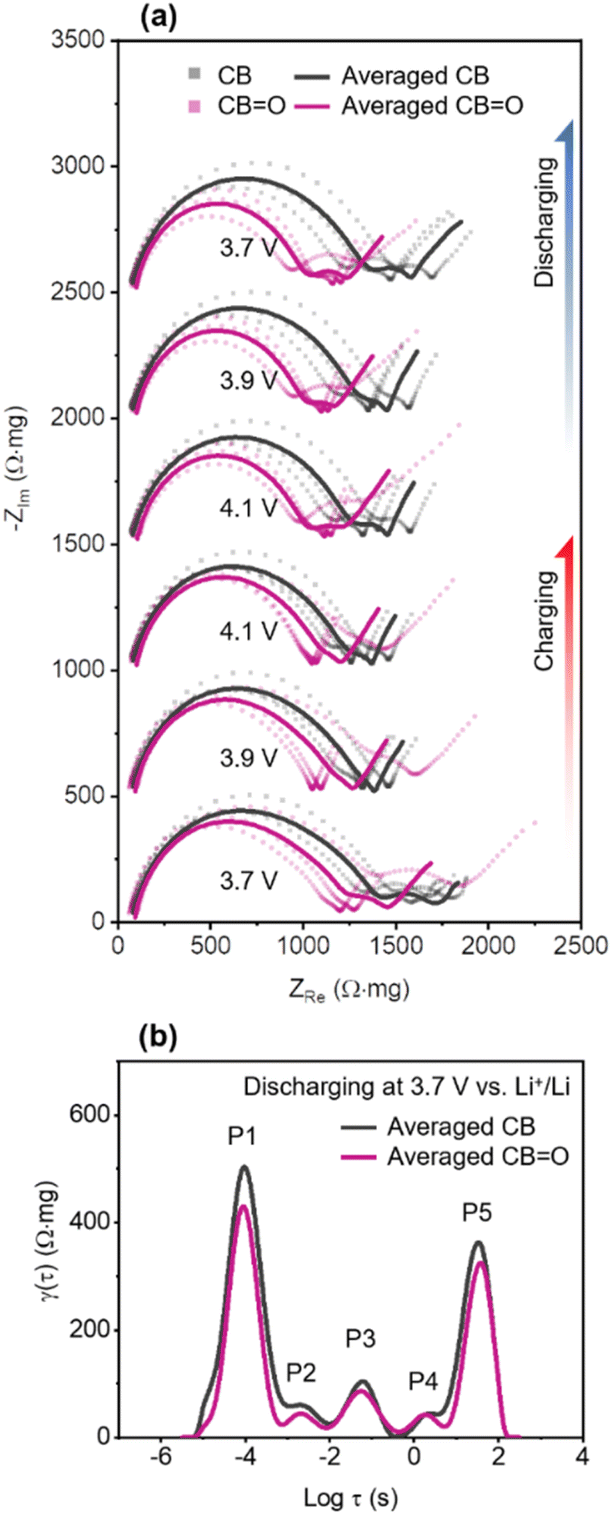 | ||
| Fig. 4 (a) Nyquist plots of pristine CB and CBO (72 h) at 3.7, 3.9, and 4.1 V vs. Li+/Li during charging and discharging. The dotted lines denote measurement of individual cells and solid lines are averages. (b) Distribution of relaxation times (DRT) of averaged pristine CB and CBO (72 h) at 3.7 V vs. Li+/Li discharging. | ||
The measured EIS spectra were averaged from three cells after normalisation.73,77 In Fig. 4a, Nyquist plots of CB and CBO (72 h) cathodes are shown at different applied potentials during charging and discharging. These Nyquist plots comprise of three key regions: (1) a semi-circle at high frequency (100 kHz to 20 Hz) describing interfacial resistance (e.g., cathode–electrolyte interphase); (2) a relatively small semi-circle at intermediate frequency (20 Hz to 300 mHz) related to charge-transfer resistance; and (3) a Warburg tail at low frequency (<300 mHz) for restricted solid-state diffusion.78,79 During both charging and discharging processes, CBO (72 h) NMC622 cathodes exhibited a lower real impedance (ZRe) at high frequencies compared to CB NMC622 cathodes at all studied voltages, denoted by the first semi-circle, indicating lower resistance at cathode–electrolyte interface.
To quantify the impedance of the three different electrochemical processes described above in CB and CBO (72 h) cathodes, the distribution of relaxation times technique (DRT) was applied to represent discrete impedance data onto continuous time functions (Fig. S10†).80–84 The DRT curves of CB/NMC622 cathodes and CBO (72 h)/NMC622 cathodes at 3.7 V vs. Li+/Li discharging are illustrated in Fig. 4b, showing 5 distinct polarisation contributions (peaks), labelled P1–P5. The five polarisation contributions can be distinguished by their relaxation time: cathode–electrolyte interphase (P1 at logτ = −4.0), charge-transfer reaction (P2–P4 at logτ = −2.6, −1.3, and 0.7), and solid-state diffusion (P5 at logτ = 1.5).
Firstly, in the case of interfacial resistance at the cathode–electrolyte interphase (CEI), CB/NMC622 cathodes show a higher peak amplitude (494 Ω mg) than CBO (72 h)/NMC622 cathodes (431 Ω mg). This higher peak amplitude indicates that CB/NMC622 cathode has thicker CEI than the CBO/NMC622 (72 h) cathode. The CB/NMC622 cathode exhibits inferior surface redox behaviour, in comparison to the CBO/NMC622 (72 h) cathode, which results in the concentration of Li+ ions at the surface of NMC622. The higher concentration of Li+ ions lead to an increase local polarisation and current,85 and subsequently the formation of a thicker CEI.65 In the case of charge-transfer reaction, CB/NMC622 and CBO (72 h)/NMC622 cathodes show differences in P2 (59 vs. 45 Ω mg) and P3 (106 vs. 86 Ω mg) with a decreased improvement at slower charge transfer rates at P4 (44 vs. 41 Ω mg). To date, factors contributing to the resistance of each electrochemical reaction are not fully understood.86 However, improved wetting of the CBO with the electrolyte (EC/EMC) (Fig. S11†), which may arise from hydrogen bonding interactions between EC/DMC and the carbonyl functional group will increase electrochemically active surface area compared to pristine CB. The improved wetting was demonstrated by dropping 20 μL of 1 M LiPF6 EC:EMC onto a film of CB and CBO (72 h) in an argon atmosphere. On CB, the 1 M LiPF6 EC:EMC showed the distinct formation of a droplet, in contrast it rapidly fully wet the CBO with no surface droplet visible (Fig. S11†). An increased electrochemically active surface area, particularly creating a higher triple boundary area between NMC622|CB|electrolyte, can enhance the efficiency of a solvated Li+ ion migrating into NMC622 by Li accepting an electron. For P5, which represents solid-state diffusion in the DRT curve (Fig. 4b),87 CBO/NMC622 was observed to have lower impedance compared to CB/NMC622 (372 vs. 333 Ω mg). This can be explained by the increased wetting ability of the electrolyte on the CBO/NMC622 and, consequently an increase in the electrochemically active triple boundary area (Fig. 5). This increased electrochemically active boundary area in CBO/NMC622 means that although the NMC622 is identical in both samples, CBO/NMC622 has a shorter diffusion length than CB/NMC622. Thus, since the solid-state diffusion coefficient is proportional to the square of diffusion length,61 the CBO/NMC622 (with a relatively shorter diffusion length) presents a lower solid-state diffusion impedance (Fig. 4b).
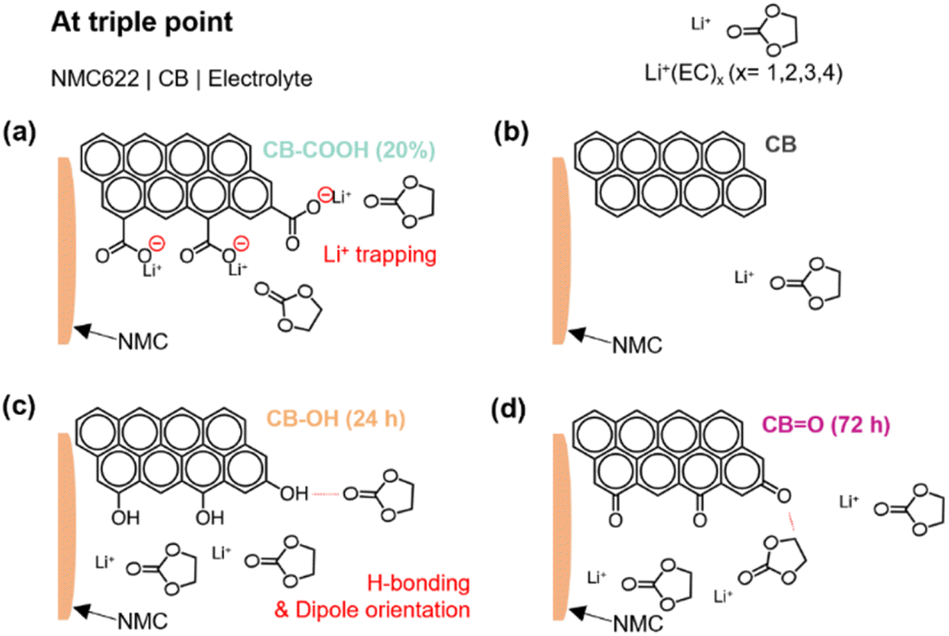 | ||
| Fig. 5 Schematic model for the role of oxygen-containing groups on the surface of CB at the triple boundary: (a) CB–COOH (20%), (b) CB, (c) CB–OH (24 h), and (d) CBO (72 h). | ||
2.3 Demonstration with other cathode materials
To demonstrate the broad applicability of this approach, a comparative rate performance study of CB and CBO (72 h) as a conductive additive for NMC811, LMO, and NCA cathodes were conducted (Table 2 and Fig. S12†). Compared to CB, the rate performance of all CBO (72 h) cathodes increased at 0.75C: 62.1 mA h g−1 for NMC622, 71.0 mA h g−1 for NMC811, 6.5 mA h g−1 for LMO and 98.5 mA h g−1 for NCA. Although the dominant cause of rate-limiting differs depending on the cathode materials or electrode structure, CBO (72 h) can be a general solution to enhance rate capability for LiB cathodes by improving the surface reaction.
| Active material | Carbon black | Active material mass [mg cm−2] | Averaged discharge capacity [mA h g−1] | ||||
|---|---|---|---|---|---|---|---|
| 0.1C | 0.25C | 0.5C | 0.75C | Δ at 0.75C | |||
| NMC622 | CB | 23.40 ± 0.64 | 160.8 | 140.7 | 98.4 | 24.4 | 0.0 |
| NMC622 | CB–OH (24 h) | 21.20 ± 1.89 | 162.7 | 144.9 | 110.4 | 65.4 | 41.0 |
| NMC622 | CBO (72 h) |
19.70 ± 0.89 | 162.9 | 145.5 | 119.0 | 86.5 | 62.1 |
| NMC622 | CB–COOH (20%) | 18.99 ± 0.54 | 162.2 | 136.1 | 77.7 | 16.1 | −8.3 |
| NMC811 | CB | 22.10 ± 0.90 | 181.3 | 152.5 | 111.1 | 65.2 | 0.0 |
| NMC811 | CBO (72 h) |
20.67 ± 1.88 | 193.6 | 180.7 | 159.9 | 136.2 | 71.0 |
| LMO | CB | 18.99 ± 0.33 | 116.8 | 111.7 | 103.0 | 91.9 | 0.0 |
| LMO | CBO (72 h) |
20.42 ± 1.03 | 117.3 | 113.2 | 106.5 | 98.4 | 6.5 |
| NCA | CB | 22.24 ± 0.12 | 165.1 | 123.9 | 45.5 | 0.0 | 0.0 |
| NCA | CBO (72 h) |
21.00 ± 0.42 | 153.8 | 140.2 | 120.8 | 98.5 | 98.5 |
2.4 Proposed mechanism of rate enhancement
From the CV, GCD and EIS data, it is clear that by modifying CB with CO functional groups increases the rate performance of NMC cathodes, while COOH groups demonstrate a negative effect. The improvement observed for CBO (72 h) arises from a decreased resistance to electrochemical reactions within the cathode active material, including cathode–electrolyte interphase resistance, solid-state diffusion, and charge transfer properties.
The suggested roles of various oxygen functionalities on the surface of CB, for electrochemical reactions at the triple boundary, is illustrated in Fig. 5. In the case of CB–COOH (20%), the negative charge on deprotonated carboxyl groups (COO−) can lead to a strong interaction with approaching Li+ ions (Fig. 5a). This interaction creates an energy barrier, meaning more energy (i.e., voltage/overpotential) is required to enable further Li+ ion motion (via diffusion, intercalation or deintercalation).
In contrast, CB–OH (24 h) (Fig. 5c) and CBO (72 h) (Fig. 5d) showed an increased rate performance compared to pristine CB due to the increased electrolyte wettability (Fig. S11†) arising from dipole interactions between the hydrogen atom (e.g., CBO⋯ in ethylene carbonate) and the oxygen atom (e.g., CB–OH⋯O in ethylene carbonate) of the carbonate solvent. Considering that Li+ ions are transported through the solvent phase, through an interaction with the oxygen group in the carbonate solvent, the proposed dipole attraction may limit Li+ ion transportation by acting as a competing mechanism.
3. Conclusions
Carbon black is a critical, yet understudied, component of the LiB cathode. Here, mild oxidation processes are used to tune the oxygen-containing functional groups on the surface of carbon blacks, which results in a marked improvement in NCM622 cathode rate capability (from 24.4 mA h g−1 to 86.5 mA h g−1 at 0.75C). Introducing carbonyl groups (CO) or hydroxyl groups (C–OH) onto the surface of carbon black resulted in a dramatic improvement in rate performance, while the formation of carboxyl groups (COOH) resulted in significantly lower cathode rate capability. This concept was extended, with CO (72 h) functionalised carbon blacks showing improved rate performance across commonly used cathode active materials, including NMC811, LMO, and NCA. This work provides a simple pathway for enhancing the rate capability of LiB cathodes in carbonate solvents.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Future Battery Industries Cooperative Research Centre (FBICRC). The authors are also grateful to the Graduate Research Scholarship, University of Melbourne. This work was performed in part at the Materials Characterization and Fabrication Platform (MCFP) at the University of Melbourne. P. C. S. acknowledges support from the Elizabeth and Vernon Puzey Foundation at the University of Melbourne, and from the Vice-Chancellor's Fellowship Scheme at RMIT University. The authors thank Dr Kimberley Callaghan for her warm advice.Notes and references
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- W. Cao, B. C. Mecrow, G. J. Atkinson, J. W. Bennett and D. J. Atkinson, IEEE Trans. Ind. Electron., 2011, 59, 3523–3531 Search PubMed.
- J. Rosero, J. Ortega, E. Aldabas and L. Romeral, IEEE Aero. Electron. Syst. Mag., 2007, 22, 3–9 Search PubMed.
- M. Gutsch and J. Leker, J. Energy Storage, 2022, 52, 105030 CrossRef.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS PubMed.
- A. Kwade, W. Haselrieder, R. Leithoff, A. Modlinger, F. Dietrich and K. Droeder, Nat. Energy, 2018, 3, 290–300 CrossRef.
- A. Manthiram, Nat. Commun., 2020, 11, 1550 CrossRef CAS PubMed.
- Z. Chen, W. Zhang and Z. Yang, Nanotechnology, 2020, 31, 012001 CrossRef CAS PubMed.
- R. Amin and Y.-M. Chiang, J. Electrochem. Soc., 2016, 163, A1512 CrossRef CAS.
- L.-X. Yuan, Z.-H. Wang, W.-X. Zhang, X.-L. Hu, J.-T. Chen, Y.-H. Huang and J. B. Goodenough, Energy Environ. Sci., 2011, 4, 269–284 RSC.
- X. Zhu, T. U. Schulli, X. Yang, T. Lin, Y. Hu, N. Cheng, H. Fujii, K. Ozawa, B. Cowie, Q. Gu, S. Zhou, Z. Cheng, Y. Du and L. Wang, Nat. Commun., 2022, 13, 1565 CrossRef CAS PubMed.
- X. Han, Y. Gong, K. K. Fu, X. He, G. T. Hitz, J. Dai, A. Pearse, B. Liu, H. Wang, G. Rubloff, Y. Mo, V. Thangadurai, E. D. Wachsman and L. Hu, Nat. Mater., 2017, 16, 572–579 CrossRef CAS PubMed.
- X. Yu and A. Manthiram, Energy Environ. Sci., 2018, 11, 527–543 RSC.
- W. Li, A. Dolocan, P. Oh, H. Celio, S. Park, J. Cho and A. Manthiram, Nat. Commun., 2017, 8, 14589 CrossRef PubMed.
- S. Choudhury, R. Mangal, A. Agrawal and L. A. Archer, Nat. Commun., 2015, 6, 10101 CrossRef CAS PubMed.
- T. Marks, S. Trussler, A. Smith, D. Xiong and J. Dahn, J. Electrochem. Soc., 2010, 158, A51 CrossRef.
- W. Bauer and D. Nötzel, Ceram. Int., 2014, 40, 4591–4598 CrossRef CAS.
- Y. Itou, N. Ogihara and S. Kawauchi, J. Phys. Chem. C, 2020, 124, 5559–5564 CrossRef CAS.
- S.-H. Park, P. J. King, R. Tian, C. S. Boland, J. Coelho, C. Zhang, P. McBean, N. McEvoy, M. P. Kremer, D. Daly, J. N. Coleman and V. Nicolosi, Nat. Energy, 2019, 4, 560–567 CrossRef CAS.
- M. Jerigova, M. Odziomek and N. Lopez-Salas, ACS Omega, 2022, 7, 11544–11554 CrossRef CAS PubMed.
- J. Zhang, H. Zou, Q. Qing, Y. Yang, Q. Li, Z. Liu, X. Guo and Z. Du, J. Phys. Chem. B, 2003, 107, 3712–3718 CrossRef CAS.
- Y.-C. Chiang, W.-H. Lin and Y.-C. Chang, Appl. Surf. Sci., 2011, 257, 2401–2410 CrossRef CAS.
- T. A. Saleh, Appl. Surf. Sci., 2011, 257, 7746–7751 CrossRef CAS.
- T. J. Aitchison, M. Ginic-Markovic, J. G. Matisons, G. P. Simon and P. M. Fredericks, J. Phys. Chem. C, 2007, 111, 2440–2446 CrossRef CAS.
- A. Rasheed, J. Y. Howe, M. D. Dadmun and P. F. Britt, Carbon, 2007, 45, 1072–1080 CrossRef CAS.
- V. Datsyuk, M. Kalyva, K. Papagelis, J. Parthenios, D. Tasis, A. Siokou, I. Kallitsis and C. Galiotis, Carbon, 2008, 46, 833–840 CrossRef CAS.
- K. A. Wepasnick, B. A. Smith, K. E. Schrote, H. K. Wilson, S. R. Diegelmann and D. H. Fairbrother, Carbon, 2011, 49, 24–36 CrossRef CAS.
- D. Pan, S. Wang, B. Zhao, M. Wu, H. Zhang, Y. Wang and Z. Jiao, Chem. Mater., 2009, 21, 3136–3142 CrossRef CAS.
- E. Lee and K. A. Persson, Nano Lett., 2012, 12, 4624–4628 CrossRef CAS PubMed.
- Y. Okamoto, J. Phys. Chem. C, 2016, 120, 14009–14014 CrossRef CAS.
- K. Nishidate and M. Hasegawa, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 245418 CrossRef.
- V. Meunier, J. Kephart, C. Roland and J. Bernholc, Phys. Rev. Lett., 2002, 88, 075506 CrossRef PubMed.
- C. B. Robledo, J. E. Thomas, G. Luque, E. P. M. Leiva, O. Cámara, D. Barraco and A. Visintin, Electrochim. Acta, 2014, 140, 160–167 CrossRef CAS.
- T.-H. Kim, E. K. Jeon, Y. Ko, B. Y. Jang, B.-S. Kim and H.-K. Song, J. Mater. Chem. A, 2014, 2, 7600–7605 RSC.
- M. J. Matthews, M. A. Pimenta, G. Dresselhaus, M. Dresselhaus and M. Endo, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, R6585 CrossRef CAS.
- T. Jawhari, A. Roid and J. Casado, Carbon, 1995, 33, 1561–1565 CrossRef CAS.
- S. Eigler, C. Dotzer and A. Hirsch, Carbon, 2012, 50, 3666–3673 CrossRef CAS.
- Y. Peng and H. Liu, Ind. Eng. Chem. Res., 2006, 45, 6483–6488 CrossRef CAS.
- J. Vicente-Santiago, J. Cornejo-Jacob, D. Valdez-Perez, J. Ruiz-Garcia and R. A. Guirado-Lopez, Phys. Chem. Chem. Phys., 2019, 21, 24077–24091 RSC.
- S. Wang, Z.-H. Chen, W.-J. Ma and Q.-S. Ma, Ceram. Int., 2006, 32, 291–295 CrossRef CAS.
- E. Desimoni, G. Casella, A. Morone and A. M. Salvi, Surf. Interface Anal., 1990, 15, 627–634 CrossRef CAS.
- J. J. Ternero-Hidalgo, J. M. Rosas, J. Palomo, M. J. Valero-Romero, J. Rodríguez-Mirasol and T. Cordero, Carbon, 2016, 101, 409–419 CrossRef CAS.
- J.-H. Kim, S. Y. Hwang, J. E. Park, G. B. Lee, H. Kim, S. Kim and B. U. Hong, Carbon Lett., 2019, 29, 281–287 CrossRef.
- G. Gao, M. Pan and C. D. Vecitis, J. Mater. Chem. A, 2015, 3, 7575–7582 RSC.
- M. Acik, G. Lee, C. Mattevi, M. Chhowalla, K. Cho and Y. Chabal, Nat. Mater., 2010, 9, 840–845 CrossRef CAS PubMed.
- C. D. Quilty, G. P. Wheeler, L. Wang, A. H. McCarthy, S. Yan, K. R. Tallman, M. R. Dunkin, X. Tong, S. Ehrlich and L. Ma, ACS Appl. Mater. Interfaces, 2021, 13, 50920–50935 CrossRef CAS PubMed.
- S. Li, T. Zhao, K. Wang, C. Sun, W. Jia, M. Zhang, H. Wang, A. Shao and Y. Ma, Adv. Funct. Mater., 2022, 32, 2203010 CrossRef CAS.
- Y. Bai, R. Essehli, C. J. Jafta, K. M. Livingston and I. Belharouak, ACS Sustainable Chem. Eng., 2021, 9, 6048–6055 CrossRef CAS.
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater., 2010, 9, 146–151 CrossRef CAS PubMed.
- A. J. Bard, L. R. Faulkner and H. S. White, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons, 2022 Search PubMed.
- G. Lai, P. Ruan, X. Hu, B. Lu, S. Liang, Y. Tang and J. Zhou, J. Mater. Chem. A, 2023, 11, 15211–15218 RSC.
- X. Yang and A. L. Rogach, Adv. Energy Mater., 2019, 9, 1900747 CrossRef.
- T. Kim, W. Choi, H.-C. Shin, J.-Y. Choi, J. M. Kim, M.-S. Park and W.-S. Yoon, J. Electrochem. Sci. Technol., 2020, 11, 14–25 CrossRef CAS.
- M. Evertz, F. Horsthemke, J. Kasnatscheew, M. Börner, M. Winter and S. Nowak, J. Power Sources, 2016, 329, 364–371 CrossRef CAS.
- I. Buchberger, S. Seidlmayer, A. Pokharel, M. Piana, J. Hattendorff, P. Kudejova, R. Gilles and H. A. Gasteiger, J. Electrochem. Soc., 2015, 162, A2737 CrossRef CAS.
- R. Jung, F. Linsenmann, R. Thomas, J. Wandt, S. Solchenbach, F. Maglia, C. Stinner, M. Tromp and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, A378 CrossRef CAS.
- P. Zhu, V. Trouillet, S. Heißler and W. Pfleging, J. Energy Storage, 2023, 66, 107401 CrossRef.
- S. Komaba, N. Yabuuchi, T. Ozeki, K. Okushi, H. Yui, K. Konno, Y. Katayama and T. Miura, J. Power Sources, 2010, 195, 6069–6074 CrossRef CAS.
- S. Ishimoto, Y. Asakawa, M. Shinya and K. Naoi, J. Electrochem. Soc., 2009, 156, A563 CrossRef CAS.
- C. Zhu, R. E. Usiskin, Y. Yu and J. Maier, Science, 2017, 358, 1400 CrossRef CAS PubMed.
- H. Gao, Q. Wu, Y. Hu, J. P. Zheng, K. Amine and Z. Chen, J. Phys. Chem. Lett., 2018, 9, 5100–5104 CrossRef CAS PubMed.
- N. Ogihara, S. Kawauchi, C. Okuda, Y. Itou, Y. Takeuchi and Y. Ukyo, J. Electrochem. Soc., 2012, 159, A1034 CrossRef CAS.
- W. Choi, H.-C. Shin, J. M. Kim, J.-Y. Choi and W.-S. Yoon, J. Electrochem. Sci. Technol., 2020, 11, 1–13 CrossRef CAS.
- K. Guo, S. Qi, H. Wang, J. Huang, M. Wu, Y. Yang, X. Li, Y. Ren and J. Ma, Small Sci., 2022, 2, 2100107 CrossRef CAS.
- Y. Tesfamhret, R. Younesi and E. J. Berg, J. Electrochem. Soc., 2022, 169, 010530 CrossRef CAS.
- Y. Tesfamhret, H. Liu, Z. Chai, E. Berg and R. Younesi, ChemElectroChem, 2021, 8, 1516–1523 CrossRef CAS.
- R. Tian, N. Alcala, S. J. K. O'Neill, D. V. Horvath, J. Coelho, A. J. Griffin, Y. Zhang, V. Nicolosi, C. O'Dwyer and J. N. Coleman, ACS Appl. Energy Mater., 2020, 3, 2966–2974 CrossRef CAS.
- X. Zhang, Z. Ju, Y. Zhu, K. J. Takeuchi, E. S. Takeuchi, A. C. Marschilok and G. Yu, Adv. Energy Mater., 2020, 11, 2000808 CrossRef.
- H. Bockholt, W. Haselrieder and A. Kwade, Powder Technol., 2016, 297, 266–274 CrossRef CAS.
- Z. Ju, X. Zhang, S. T. King, C. D. Quilty, Y. Zhu, K. J. Takeuchi, E. S. Takeuchi, D. C. Bock, L. Wang, A. C. Marschilok and G. Yu, Appl. Phys. Rev., 2020, 7, 041405 CAS.
- D. Park and K. Kim, J. Appl. Phys., 2021, 130, 204101 CrossRef CAS.
- J. J. Kuo, S. D. Kang and W. C. Chueh, Adv. Energy Mater., 2022, 12, 2201114 CrossRef CAS.
- S. Müller, J. Eller, M. Ebner, C. Burns, J. Dahn and V. Wood, J. Electrochem. Soc., 2018, 165, A339 CrossRef.
- C. D. Quilty, D. C. Bock, S. Yan, K. J. Takeuchi, E. S. Takeuchi and A. C. Marschilok, J. Phys. Chem. C, 2020, 124, 8119–8128 CrossRef CAS.
- S. Wang, J. Zhang, O. Gharbi, V. Vivier, M. Gao and M. E. Orazem, Nat. Rev. Methods Primers, 2021, 1, 41 CrossRef CAS.
- M. A. Kamenskii, S. N. Eliseeva, E. G. Tolstopjatova, A. I. Volkov, D. V. Zhuzhelskii and V. V. Kondratiev, Electrochim. Acta, 2019, 326, 134969 CrossRef CAS.
- G. Assat, D. Foix, C. Delacourt, A. Iadecola, R. Dedryvere and J. M. Tarascon, Nat. Commun., 2017, 8, 2219 CrossRef PubMed.
- L. Wang, J. Zhao, X. He, J. Gao, J. Li, C. Wan and C. Jiang, Int. J. Electrochem. Sci., 2012, 7, 345–353 CrossRef CAS.
- Q. Meyer, S. Liu, Y. Li and C. Zhao, J. Power Sources, 2022, 533, 231058 CrossRef CAS.
- R. Soni, J. B. Robinson, P. R. Shearing, D. J. L. Brett, A. J. E. Rettie and T. S. Miller, Energy Storage Mater., 2022, 51, 97–107 CrossRef.
- H. Jia, L. Zou, P. Gao, X. Cao, W. Zhao, Y. He, M. H. Engelhard, S. D. Burton, H. Wang, X. Ren, Q. Li, R. Yi, X. Zhang, C. Wang, Z. Xu, X. Li, J. G. Zhang and W. Xu, Adv. Energy Mater., 2019, 9, 1900784 CrossRef.
- F. Ciucci and C. Chen, Electrochim. Acta, 2015, 167, 439–454 CrossRef CAS.
- T. H. Wan, M. Saccoccio, C. Chen and F. Ciucci, Electrochim. Acta, 2015, 184, 483–499 CrossRef CAS.
- A. Jana, S. I. Woo, K. S. N. Vikrant and R. E. García, Energy Environ. Sci., 2019, 12, 3595–3607 RSC.
- R. He, Y. He, W. Xie, B. Guo and S. Yang, Energy, 2023, 263, 125972 CrossRef CAS.
- X. Zhu, L. Cheng, H. Yu, F. Xu, W. Wei and L.-Z. Fan, J. Materiomics, 2022, 8, 649 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental information, XPS analysis, Raman spectroscopy analysis, SEM images, cyclic voltammograms at various scan rates, peak separation potential analysis, electrochemical stability test, distribution of relaxation times analysis details, contact angle measurements, rate performance and GCD curves analysis for various cathode materials, schematic diagram of chemical treatment, XRD analysis, note for electronic conductivity and percolation effect. See DOI: https://doi.org/10.1039/d3ta05093a |
| This journal is © The Royal Society of Chemistry 2024 |