
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the effects of synthesis parameters on the properties and photoactivity of WO3–graphene oxide synthesized via a microwave route†
Bárbara S.
Rodrigues
a,
Markus
Niederberger
 b and
Juliana S.
Souza
*a
b and
Juliana S.
Souza
*a
aCentro de Ciências Naturais e Humanas, Universidade Federal do ABC, 09210-580, Santo André, SP, Brazil. E-mail: juliana.souza@ufabc.edu.br
bLaboratory for Multifunctional Materials, Department of Materials, ETH Züurich, 8093, Zurich, Switzerland
First published on 25th April 2024
Abstract
Tungsten oxide (WO3) is a promising material for photocatalysis. Coupling it with graphene-based materials can enhance its electronic conductivity. One effective technique for synthesizing WO3 nanomaterials is microwave-assisted solvothermal synthesis, which selectively heats the reaction species and reduces the reaction time. We report a straightforward route for preparing a WO3 nanomaterial modified with graphene oxide (WO3GO) using microwave-assisted solvothermal synthesis. We investigated the effect of various synthesis parameters, such as the irradiation time and reaction temperature. WO3 nanoplatelets were obtained under all conditions investigated; also, adding GO to the reaction did not change the WO3 morphology. It was observed that the crystal phase related to tungsten oxide can be modulated by temperature or time. Hexagonal WO3·H2O was obtained at temperatures of 160 and 180 °C, whereas at 200 °C, monoclinic WO3 was formed. All WO3GO materials were active for methylene blue, rhodamine B and methyl orange photodegradation. Also, incorporating GO increased the photoactivity of the materials.
1. Introduction
Tungsten oxide (WO3) has been widely reported as a promising material for photocatalysis and photoelectrocatalysis.1–4 Coupling WO3 with graphene-based materials such as graphene, graphene oxide (GO)5 or reduced graphene oxide (rGO)6 is very attractive. Some authors stated that adding around 1–4 wt% of graphene oxide is ideal for enhancing electron conduction without reducing photoactivity.5,7–9 This value is related to the volume of the filler (in this case, graphene materials) necessary to create an electron pathway, increasing the conductivity of a ceramic material. The incorporation of graphene-based materials also enhances the chemical stability10 of the surface area.11Several strategies have been reported for the synthesis of WO3/graphene, such as hydrothermal synthesis,12–14 sol–gel method,15,16 electrodeposition,17 spray pyrolysis,18 electrospinning,19 pulsed laser ablation in liquids,20 sonochemical approaches,21,22 and microwave-assisted synthesis.23–30
Microwave-assisted routes have been considered as a powerful technique to reduce the reaction time dramatically.31,32 This effect is attributed to the selective heating of species with high dielectric loss (ability to convert electromagnetic energy into heat), such as ionic species and polar solvents.33 The heating process involves two phenomena: dipolar polarization and ionic conduction. These species interact with the oscillating electric field of the microwave and try to reorientate, leading to increased collision and friction among the reactants, which enhances the temperature uniformly.32 Besides these thermal effects, non-thermal effects like superheating, selective microwave interactions, and the formation of molecular radiators (through the coupling of the dielectric energy and specific species of solution) can be determined to shorten the lifetime of the activated complex, boosting, even more, the reaction rate.34,35 Microwaves also have an essential effect on the interfacial process during particle growth. In this process, the nuclei adsorb species on their surfaces. Changing the interfacial properties can affect the reactivity in the early stages of the reaction.34,35 These interfacial effects can be even more critical concerning composites such as WO3 and graphene.
Even though there are some reports on WO3/graphene synthesis via microwave routes (Table 1), more information must be provided regarding the influence of microwave parameters on the product structure and morphology. For instance, the microwave field distribution depends on the cavity design (microwave oven), power delivery, vessel size, and volume of the precursor solution. These parameters directly influence the rate of nucleation and crystallization.31 Therefore, many procedures are not reproducible because the microwave parameters are usually not fully described. As a result, it is not easy to associate the observed materials' properties with the synthesis conditions.
| Material | Synthesis | Power (W) | Temperature (°C) | Heating rate/ramping time | Reaction time (min) | Volume of solution | Solvent | Microwave oven | Annealing (°C) | Morphology | Crystalline phase | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| - Not provided, * microwave solid-state treatment at 800 W. | ||||||||||||
| WO3/reduced graphene oxide | Microwave open vessel | — | — | — | 20 | — | Water | — | No | Disc-shaped onto nanosheets | Monoclinic WO3 and rGO | 23 |
| WO3/reduced graphene oxide | Microwave-assisted solvothermal and microwave solid-state | — | 200 | 10 min | 30 | — | Ethanol | Ultrawave – milestone | * | Nanowires | Hexagonal and rGO | 24 |
| WO3/reduced graphene oxide/g-C3N4 | Microwave-assisted hydrothermal | 600 | 190 | 5 °C min−1 | 10 | — | Water | Microwave reactor (CEM) | No | 2D/2D/2D sheet-on-sheet architecture | Monoclinic WO3 and no structural evidence of rGO | 25 |
| WO3·0.33H2O/reduced graphene oxide | Microwave-assisted hydrothermal | — | 160 | — | 60 | — | Water | — | No | Nanoneedles | Orthorhombic WO3 and rGO | 26 |
| WO3/graphene sheets | not specified | 850 | — | — | 10 | — | Water | — | 500 | Nanoplates on 2D graphene | Monoclinic and hexagonal WO3 and graphene | 27 |
| WO3/graphene nanocomposite | Microwave-assisted solvothermal | — | 180 | — | 25 | — | Ethanol | MWave-5000 | 105 | Flower-like | Hexagonal WO3 and graphene | 28 |
| WO3·0.33H2O/reduced graphene oxide | Microwave-assisted hydrothermal | — | 140 | — | 10 | — | Water | Microwave aided device BR200815393-A2 | 400 | Nanocolumn bundle-like | Hexagonal and orthorhombic WO3 and no structural evidence of rGO | 29 |
| WO3/graphene nanocomposite | Microwave-assisted solvothermal | — | 180 | — | 18 | — | Ethanol | MWave-5000 | 500 | Hemispherical | Monoclinic WO3 and graphene | 30 |
| WO3/graphene oxide micro composites | Microwave-assisted solvothermal | 300 W maximum | 1st step 180 °C, 2nd step 200 °C | Heat as fast as possible | 2 minutes | 7 mL (70% of the vial capacity) | Ethylene glycol | Monowave 400R (Anton paar) | No | Nanosheets | B-W3O and graphene graphitized | 36 |
| WO3/graphene oxide micro composites | Microwave-assisted solvothermal | 300 W maximum | 1st step 180 °C, 2nd step 200 °C | Heat as fast as possible | 2 minutes | 7 mL (70% of the vial capacity) | Ethylene glycol | Discover (CEM) | No | Nano agglomerates of spheres | Hexagonal and WO3·H2O, and graphene graphitized | 36 |
| WO3/graphene oxide micro composites | Microwave-assisted solvothermal | 300 W maximum | 200 °C | 10 min | 11 minutes | 7 mL (70% of the vial capacity) | Ethylene glycol | Monowave 400R (Anton paar) | No | Nanoflowers | B-W3O and graphene graphitized | 36 |
| WO3/graphene oxide micro composites | Microwave-assisted solvothermal | 300 W maximum | 200 °C | 10 min | 11 minutes | 7 mL (70% of the vial capacity) | Ethylene glycol | Discover (CEM) | No | Nano agglomerates of spheres | Hexagonal and graphene graphitized | 36 |
| WO3/graphene oxide composite | Microwave-assisted solvothermal | 1st step 50 W, 2nd step 90 W | 5 minutes | 11 minutes | 4 mL (40% of the vial capacity) | Benzyl alcohol | Discover (CEM) | No | Nanoplatelets | Monoclinic WO3 and graphene | This work | |
Furthermore, several methods of WO3 synthesis require a calcination step despite the hydrothermal synthesis to get the monoclinic phase of tungsten oxide, the most active phase for photocatalysis.37–39 However, annealing WO3/graphene can induce weight loss of graphene40 and its re-oxidation.41
Thus, we report a straightforward route to prepare a WO3 nanomaterial modified with graphene oxide by a microwave-assisted solvothermal route. This methodology allows the production of highly homogeneous monoclinic WO3-graphene oxide composite materials without the need for post-annealing treatment, which could change the morphology of WO3 and oxidize the graphene. Another interesting feature of this route is that the material can be obtained within ten minutes. The method provides advantages regarding reproducibility since, unlike the others reported in Table 1, all the synthesis conditions critical to its reproducibility are reported. In addition, our method is among those that employ simpler and milder reaction conditions in terms of the complexity of the reaction medium, reaction time, temperature, and irradiation power (which is the lowest among those reported in Table 1). This development contributes to optimizing synthesis processes and designing advanced nanocomposites for various applications.
2. Materials and methods
2.1. Reactants
Tungsten hexachloride (99.9% trace metal basis), benzyl alcohol (99.8%), and graphene oxide (4–10% edge oxidized) were purchased from Sigma Aldrich.2.2. Synthesis
The synthesis of tungsten oxide modified with graphene oxide was performed based on a previously reported modified synthesis.42 First, 0.5 mmol of tungsten hexachloride (WCl6) was dissolved in 200 μL of methanol in a microwave vial of 8 mL; then, the solution was stirred for 2 h. Second, 100 mg of graphene oxide was suspended in 20 mL of benzyl alcohol and sonicated for 2 h, forming a black suspension. Third, the GO suspension was added to the microwave vial, keeping the GO ratio at 3 wt%. Finally, the volume was adjusted with benzyl alcohol to 4 ml (in a vial of 10 mL capacity). The system was placed in the microwave cavity of a discover-CEM reactor. The microwave heating protocol followed two steps: (i) pre-heating for 1 minute at 100 °C, under constant stirring, using an irradiation power of 50 W; and (ii) a heating step at 160, 180, and 200 °C for 10 min, using an irradiation power of 80, 90, and 98 W, respectively. As a result, three different materials were obtained, and WO3 was synthesized following the same procedure without the addition of GO.Note that the weight ratio of GO was optimized to 3% after a study involving 0.5, 3 and 10% of GO. This study is reported in the ESI† (Fig. S1–S4).
The influence of time on the materials' crystal structure and morphology was also investigated. The materials synthesized at 180 °C and 200 °C were selected for this study based on their ability to form monoclinic WO3, aiming to correlate the synthesis parameters with crystal growth. It was investigated at two different reaction times: 1 and 10 min. All the mentioned conditions are reported in Table 2.
| Sample name | Investigation | Temperature (°C) | Time (min) | GO proportion (%) |
|---|---|---|---|---|
| WO3 | Graphene loading | 200 | 10 | 0 |
| WO30.5%GO | 200 | 10 | 0.5 | |
| WO33%GO | 200 | 10 | 3 | |
| WO310%GO | 200 | 10 | 10 | |
| WO3_160 °C-0 min | Synthesis stages | 160 | 0 | 0 |
| WO3_180 °C-0 min | 180 | 0 | 0 | |
| WO3_200 °C-0 min | 200 | 0 | 0 | |
| WO3GO_160 °C-0 min | 160 | 0 | 3 | |
| WO3GO _180 °C-0 min | 180 | 0 | 3 | |
| WO3 GO_200 °C-0 min | 200 | 0 | 3 | |
| WO3_180 °C-1 min | Temperature and time | 180 | 1 | 0 |
| WO3_180 °C-10 min | 180 | 10 | 0 | |
| WO3GO_180 °C-1 min | 180 | 1 | 3 | |
| WO3GO_180 °C-10 min | 180 | 10 | 3 | |
| WO3_200 °C-1 min | 200 | 1 | 0 | |
| WO3_200 °C-10 min | 200 | 10 | 0 | |
| WO3GO_200 °C-1 min | 200 | 1 | 3 | |
| WO3GO_200 °C-10 min | 200 | 10 | 3 |
All the related samples were washed 3 times with acetone, centrifuged and dried in an oven at 60 °C for 2 h.
2.3. Characterization
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were used to investigate the morphological aspects of the synthesized materials. The sample powders were suspended in isopropanol, dropped onto silicon wafer slides fixed on conductive carbon tapes (Ted Pella) and coated with 3 nm platinum on a sputter coater (CCU-010, Safematic). High-resolution transmission electron microscopy (HRTEM) and scanning transmission electron microscopy (STEM) images were collected on a FEI Talos F200X microscope and were analyzed at 200 kV. The samples were suspended in isopropanol and placed on gold grids. Scanning transmission electron microscopy was performed on a JEOL JEM-2100F microscope. For X-ray diffraction (XRD), a PANalytical Empyrean with a PIXcel 1D detector and Cu Kα radiation was used to investigate the crystal phases. Additionally, Raman spectroscopy was applied to evaluate the crystal structure using a MicroRaman Renishaw inVia spectrometer coupled to a He–Ne laser (532 nm). The surface area was investigated by nitrogen gas sorption using the Brunauer–Emmett–Teller (BET) method from data collected using a Quantachrome Autosorb iQ analyzer after outgassing the samples at 100 °C for 24 h.2.4. Dye adsorption and degradation
Photocatalytic degradation of methyl blue (MB), rhodamine B (RhB) and methyl orange (MO) was used to evaluate the activity of WO3_180 °C-0 min, WO3GO_180 °C-0 min, WO3_200 °C-0 min, WO3GO_200 °C-0 min, WO3_180 °C-10 min, WO3GO_180 °C-10 min, WO3_200 °C-10 min and WO3GO_200 °C-10 min.Adsorption assays were performed to rule out any adsorption effects. First, 6 mg of the photocatalyst was suspended in 10 mL of an aqueous solution of the dye (60 mg L−1) and sonicated for 10 s. Then, the suspension was stirred in the dark. Aiming to investigate the adsorption over time, aliquots of 1 mL of the suspension were extracted at 0 min, 1 min, 5 min, 10 min, 30 min, 60 min, 120 min, 480 min and 1440 min intervals, using a syringe coupled to a Millipore filter with 0.2 μm pore size (Merck). After that, the filtered portion was centrifuged for 10 min at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. The electronic spectra of the supernatant were analyzed using UV-vis spectroscopy (Cary 50Scan-Varian). The characteristic bands of methylene blue, rhodamine B and methyl orange at 664 nm, 555 nm and 489 nm, respectively, were monitored.
000 rpm. The electronic spectra of the supernatant were analyzed using UV-vis spectroscopy (Cary 50Scan-Varian). The characteristic bands of methylene blue, rhodamine B and methyl orange at 664 nm, 555 nm and 489 nm, respectively, were monitored.
For the photocatalytic assay, 0.3 mg mL−1 of the catalysts were suspended in the dye solution (60 mg L−1) and stirred in the dark for 24 h. After that, the mixture was placed in a cuvette at a known distance from the sunlight simulator with a filter A.M. 1.5G spectrum (power of 100 mW cm−2) for 1 h. Aliquots of the mixture were collected every 15 min for 1 h, centrifuged, and the absorbance of the supernatant was measured using a UV-vis spectrophotometer. The characteristic absorption band of each dye as a function of time was monitored. The weight of the degraded dye was calculated using eqn (1).
| Dye degradation (mg) = mi − m | (1) |
3. Results and discussion
During microwave irradiation, a direct coupling occurs between the microwaves and the reactant species. As a result, some effects, including superheating and forming “molecular radiators,” can occur.43 From the point of view of inorganic nanoparticle synthesis, these effects directly affect the interface events, impacting nuclei formation in the initial synthesis steps.44 As a result, the growth process can occur in an inhomogeneous way.45 Therefore, it is essential to guarantee a “slow” heating process to avoid it. Thus, to produce highly crystalline WO3GO materials, we developed a microwave-assisted synthesis protocol in two steps. First, all samples were subjected to a pre-heating step for 1 minute at 100 °C under constant stirring (Fig. 1). Then, the irradiation power was set to 50 W, the minimum required to achieve the desired temperature. The main goal of this step is to complete the dissolution of the precursors in the solvent, control the reaction medium heating rate, and avoid any inhomogeneity of the temperature in the reaction media, which can affect the crystal growth and induce the formation of agglomerates.46,47 In the second step, the temperature is increased to 180 °C or 200 °C (Fig. 1) using the minimum irradiation power necessary, as described in the methodology. Again, the main goal is to avoid overheating.48,49 Unfortunately, studies involving higher heating temperatures could not be performed due to the pressure increase beyond the equipment limitation.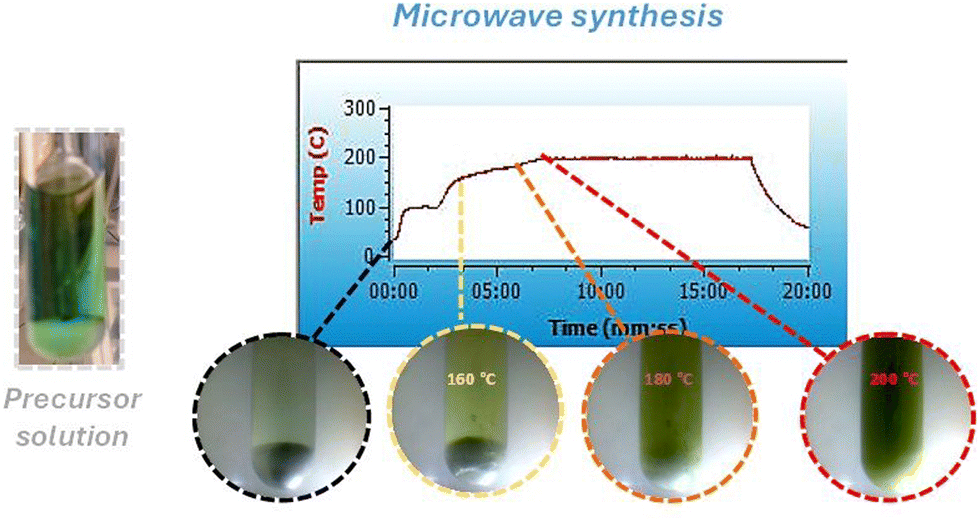 | ||
| Fig. 1 Temperature vs. reaction time graph of the microwave synthesis of WO3, along with images of the reaction solutions. | ||
Since the microwave oven is connected to a camera, color changes were observed during the heating process at 160 °C, 180 °C and 200 °C (Fig. 1). Initially, tungsten is coordinated to six chloride anions (WCl6), a purple powder. Upon dissolution in methanol and the addition of benzyl alcohol, the color changes to greenish-blue. After a few seconds of microwave heating, the color changes to pale yellow. At 160 °C, the solution turns yellow with occasional green spots, which precipitate upon cooling. At 180 °C, a precipitate forms even before cooling, and at 200 °C, the color of the precipitate shifts to yellow-green.
These color variations can be correlated with changes in the chemical environment surrounding the tungsten (W) atoms. Initially, W is coordinated with six chlorides; introducing water molecules through the dissolution of the solid salt alters this coordination, promoting the partial substitution of some coordinated chloride to water molecules. Furthermore, heating the precursor can also modify the composition of the coordination compound, resulting in different solution colors.
However, depending on synthesis conditions, the samples may contain coordinated water in the crystal lattice, as evidenced by XRD analysis (Fig. 2) of WO3 at 180 °C for 0 minutes, which reveals the crystal structure of WO3·H2O. Also, tungsten can present different oxidation numbers (W6+ or Wx+) depending on the chemical environment surrounding it. In both cases, it can affect the color of the precipitate.
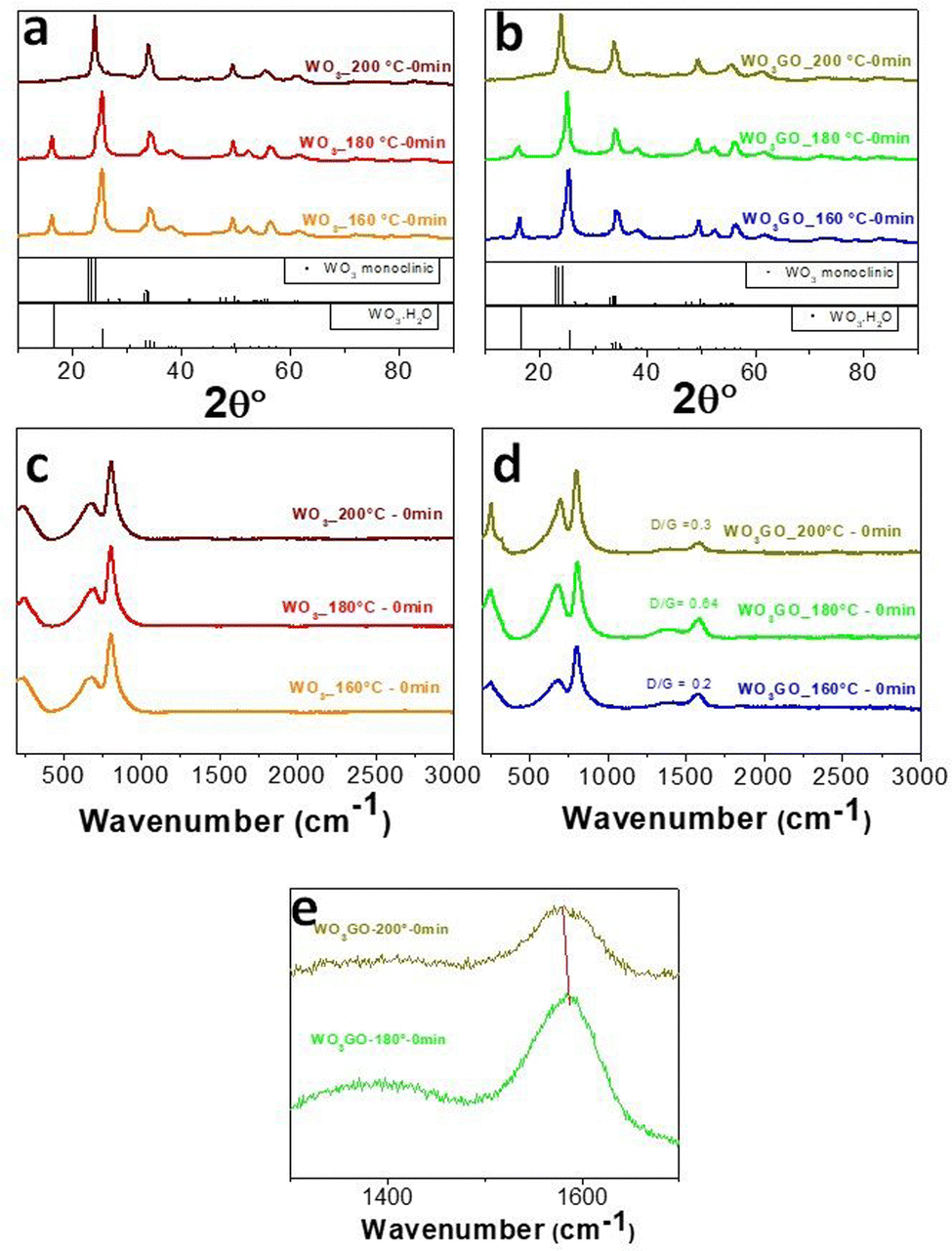 | ||
| Fig. 2 XRD of pristine WO3 samples (a) and WO3 modified with GO (b). Raman spectra of pristine (c) and GO-modified samples (d). Enlarged view of the graph (d), emphasizing the shift of the G band between WO3GO-180 °C-0 min and WO3GO-200 °C-0 min (e). | ||
3.1. Study of synthesis stages
Aiming to understand the growth pathway of WO3 and the influence of graphene on the crystal growth, the precipitate was collected after different reaction times following each change of color that we could observe during the reaction between WCl6 and BnOH (at 160 °C, 180 °C and 200 °C). The same procedure was performed for the sample WO3GO.The crystal structure was analyzed through XRD (Fig. 2a and b). The results show that only when the temperature reaches 200 °C, we can obtain the monoclinic phase of WO3 (Fig. 2a, PDF 83-0951). The broadening and overlapping of the XRD peaks are associated with oxygen deficiency.50 At temperatures lower than 200 °C, the only product observed was the hydrated form of tungsten oxide – WO3·H2O (PDF 18-1418).51
The hypothesis is that, in the first step of the synthesis, the alcoholysis of the precursor occurs due to the interaction of metal chloride with benzyl alcohol.52 Niederberger and coworkers have studied the mechanism of synthesis for WCl6 and BnOH.53 In this paper, they proposed the following overall reaction for the formation of WO3·H2O at 100 °C through the characterization of the synthesis intermediates (eqn (2)):
| WCl6 + 8.7BnOH → WO3·H2O + 3.3BnCl + 2.7BnOBn + 2.7HCl + 2H2O | (2) |
The synthesis mechanism leads to the formation of HCl and water. However, the pathway can be different in our work here since the previous study did not perform a predilution step in methanol. Also, graphene has been added to the reaction medium in this work. Furthermore, increasing the temperature to 200 °C leads to the formation of the monoclinic phase of WO3 instead of WO3·H2O (Fig. 2).
The same trends were observed for WO3GO. The XRD patterns of the WO3GO samples (Fig. 2b) did not show changes in crystal structures compared to the pristine WO3.
Raman spectroscopy data of WO3 (Fig. 2c) show bands around 806 cm−1 and 701 cm−1 ascribed to the O–W–O antisymmetric stretching and the W2O6 and W3O8 stretching, respectively.50,54 As expected, samples modified with GO (Fig. 2d) have the same bands associated with WO3. The spectra also show two additional bands around 1350 and 1604 cm −1,55 corresponding to the D and G bands of the graphitic structure, respectively. The G band is related to the C–C vibrations of carbon with sp2 orbitals; also, it is a characteristic peak of graphitic sheets. The D band around 1351 cm−1 is related to the vibrational defects of the C–C bond. Intensity ratios between D/G bands higher than 1 are associated with more significant structural defects. This feature is usually associated with the transition between GO and rGO. WO3GO samples (Fig. 2d) exhibited graphene oxide bands with low structural defects, which means low content of reduced graphene oxide. Another aspect related to the formation of rGO is the shift of the bands towards lower wavenumbers, as observed for WO3GO-200 °C-0 min compared to WO3GO-180 °C-0 min (Fig. 2e).55
SEM images were used to evaluate the morphology of the samples (Fig. 3). For the synthesis performed at 180 °C, WO3 exhibited a platelet-like shape with roundish edges of 120–150 nm in length (Fig. 3a). WO3 prepared at 200 °C comprises irregular nanoplatelets (Fig. 3b), mostly ranging from 80–100 nm with sharp edges. The addition of graphene did not affect the morphology of the samples (Fig. 3c and d). Thus, it is not possible to identify which particles could be graphene. However, it was not possible to identify the presence of GO in the SEM images of WO3GO_180 °C-0 min and WO3GO_200 °C-0 min samples. STEM images of WO3GO_180 °C-0 min (Fig. S5, ESI†) show that WO3 and GO are in different regions on the samples. This result indicates that longer irradiation times are required to obtain a homogeneous mixture of WO3 and GO, as seen for WO3GO_180 °C-10 min and WO3GO_200 °C-10 min samples, discussed later in the paper.
 | ||
| Fig. 3 SEM images of (a) WO3_180 °C-0 min, (b) WO3_200 °C-0 min, (c) WO3GO_180 °C-0 min and (d) WO3GO_200 °C-0 min. | ||
3.2. Effect of time and temperature
The effect of irradiation time on the structure and morphology of the products was also tracked for WO3 and WO33%GO synthesis. In this study, the temperature was maintained at 180° or 200 °C varying the reaction time from 1 to 10 min. The XRD results (Fig. 4) show that keeping the temperature at 180 °C for 10 min is enough to form the monoclinic phase of WO3 (Fig. 4a); below 10 minutes of synthesis, only the pattern associated with WO3·H2O is observed. In addition, at 200 °C, the time does not influence the crystal phase.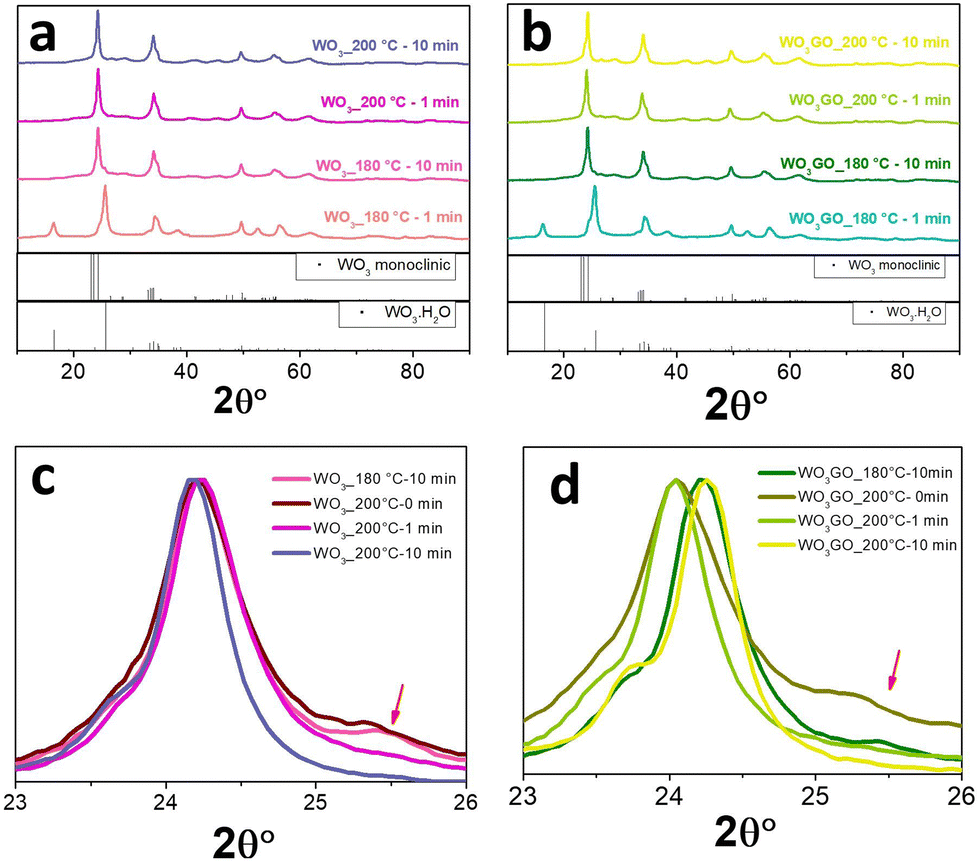 | ||
| Fig. 4 XRD of (a) WO3_180 °C-1 min, WO3_180 °C-10 min, WO3_200 °C-1 min, WO3_200 °C-10 min and (b) WO3GO_180 °C-1 min, WO3GO_180 °C-10 min, WO3GO_200 °C-1 min and WO3GO_200 °C-10 min. Magnified XRD region between 23 and 26° of (c) WO3_180 °C-10 min, WO3_200 °C-0 min, WO3_200 °C-1 min, WO3_200 °C-10 min and (d) WO3GO_180 °C-10 min, WO3GO_200 °C-0 min, WO3GO_200 °C-1 min and WO3GO_200 °C-10 min. | ||
The presence of GO in the sample did not change the diffraction pattern observed for WO3 (Fig. 4b). The most intense peak of monoclinic WO3, around 24° (Fig. 4c), remains at the same position independently of the reaction conditions. However, WO3_200 °C-10 min exhibited a narrower peak that can be associated with higher crystallinity (Fig. 4c). Also, WO3_180 °C-10 min and WO3_200 °C-0 min samples show a small peak at 25.5°, which may be ascribed to the reminiscent of the hydrated phase of tungsten oxide. On the other hand, samples of WO3GO show a shift towards higher 2θ values of the most intense peak of monoclinic WO3. This displacement increases with the synthesis time for both WO3_180 °C-10 min and WO3_200 °C-10 min. According to the literature, this property is associated with the narrowing of crystallographic planes related to the constriction of the lattice.56 In addition, again, WO3GO_200 °C-10 min exhibited a narrower peak compared to the other conditions. XRD was also used to evaluate the reproducibility of the method (Fig. S6, ESI†).
Thermal gravimetric analysis (TGA) and the first derivative curves obtained in the air atmosphere were used to investigate the thermal behavior of the samples obtained at 180 °C and 200 °C (Fig. S7, ESI†). Generally, the weight loss observed for all samples (Fig. S7a–i, ESI†) below 300 °C is commonly associated with the loss of surface adsorbed water and residual solvent.57 Also, the weight loss percentage is higher for the samples exhibiting or containing residues of WO3·H2O, such as WO3_180 °C-10 min and WO3GO_180 °C-10 min.
Furthermore, the weight loss was significantly higher for all samples modified with graphene, probably due to GO structural instability at higher temperatures that induced a continuous weight loss, which has also been observed for GO_200 °C-10 min (ESI† file). Additionally, WO3_200 °C-10 min and WO3GO_200 °C-10 min had a lower weight loss value than WO3_180 °C-10 min and WO3GO_180 °C-10 min, probably due to the formation of a stable monoclinic crystal phase and an efficient removal of the residual solvent after the synthesis.
The crystal structure of WO3GO samples was confirmed by Raman spectroscopy (Fig. S7, ESI†). The two bands associated with WO3 are observed for WO3 (Fig. S8a, ESI†) and WO3GO (Fig. S8b, ESI†), as we also observed in the spectra presented in Fig. 2c and d. The spectra of samples modified with GO (Fig. S8b, ESI†) showed the peaks characteristic of graphene around 1350 and 1604 cm −1 with low intensity. Therefore, the spectra were zoomed in between 1100 and 1800 cm−1 in Fig. S7c (ESI†). In this case, it is possible to notice an intense G band associated with multilayer graphene with low structural defects and asymmetry content.58 The D/G ratio (Fig. S7d, ESI†) decreases with increasing temperature and time; this may be associated with the restacking of graphene that can be induced by microwave interaction.59 The reduction of GO promoted by removing oxygen-containing groups induces the formation of π–π bonds between the graphene layers.60,61
The incorporation of GO in the sample WO3GO_200 °C-10 min could be observed by STEM in the high-angle annular dark-field (HAADF) mode images (Fig. 5 and Fig. S10 (ESI†) for WO3GO_180 °C-10 min). WO3 exhibits a nanoplatelet shape, while graphene also forms plates that tend to be less homogeneous in size (Fig. S10, ESI†). HAADF-STEM coupled with energy-dispersive X-ray (EDX) images allowed us to characterize the spatial distribution of WLα and OKα, reinforcing that the homogeneous nanoplatelets are composed of W and O (Fig. 5a). Tungsten is marked in red whereas oxygen in blue, the combination of both colors at exactly the same region results in the observation of the color magenta. In contrast, the more significant portion mainly comprises carbon that can be associated with graphene oxide (Fig. 5a). In the image obtained in the HAADF-STEM mode, the heavier elements appear with brighter contrast (Fig. 5b), and the circular area highlighted in green was selected to observe the HRTEM image of GO (Fig. 5c). The purple circle was chosen for collecting the HRTEM image of WO3 (Fig. 5d).
 | ||
| Fig. 5 (a) HAADF-STEM micrograph of WO3GO_200 °C-10 min with the elemental map obtained from WLα, OKα, and CKα. (b) HAADF image with selected regions of WO3 and GO and (c) HRTEM images of GO and (d) WO3 with a fast-Fourier-transform pattern (FFT – inset). (e) FFT pattern fitted with the schematic diagram of monoclinic WO3 (301) and (f) calculated diffraction pattern (301). | ||
HRTEM was used to confirm the crystal phase of WO3 nanoparticles (Fig. 5d – inset WO3). The lattice fringes observed are associated with the FFT pattern (Fig. 5e) that corresponds to the (301) plane of the WO3-monoclinic phase (Fig. 5f).62 On the other hand, the region previously associated with graphene (Fig. 5c) exhibited “wavy” fringes that cannot be related to an FFT pattern. In other words, it does not show a defined crystal structure. This result is expected since the exfoliation of multilayer graphene or graphite and partial reduction typically lead to the partial loss of the crystalline structure and structural defects.63,64
HAADF-STEM images of WO3GO_180 °C-10 min (Fig. S10, ESI†) also pointed toward the presence of GO and WO3 in the sample. However, delimiting graphene as a more extensive particle was impossible. They were homogeneously dispersed in this case, and GO is comparable in size to the WO3 nanoparticles. In addition, the HRTEM image of WO3GO_180 °C-10 min (Fig. S9, ESI†) also showed fringes that can be associated with the FFT of the monoclinic phase of WO3.
The zeta potential of the materials was measured in an aqueous dispersion with pH 5 (Table 3), which is approximately the pH of the aqueous solution of methyl blue used in the photocatalytic tests. The surface charges of a semiconductor normally occur due to the surface hydration and dissociation of hydroxyl groups, where the negative charges originate from the acidic dissociation of hydroxyl groups, and the positive charges arise from the addition of a proton to the neutral surface hydroxyl groups.65 All related materials showed large negative values of zeta potential, meaning that the surfaces of the samples are negatively charged.66 For the samples synthesized by holding the maximum temperature for 0 min, the zeta potential did not change upon incorporating GO into the WO3.
| Sample | Zeta potential (mV) |
|---|---|
| WO3_180 °C-0 min | −22.8 ± 2.1 |
| WO3GO_180 °C-0 min | −24.9 ± 1.8 |
| WO3_200 °C-0 min | −16.7 ± 1.3 |
| WO3GO_200 °C-0 min | −16.7 ± 1.2 |
| WO3_180 °C-10 min | −26.8 ± 2.0 |
| WO3GO_180 °C-10 min | −20.3 ± 1.6 |
| WO3_200 °C-10 min | −16.5 ± 1.3 |
| WO3GO_200 °C-10 min | −10.5 ± 0.9 |
This result can be ascribed to a low interaction between the GO and hydroxyl groups at the surface of WO3. These hydroxyl groups are responsible for changing the zeta potential in dependence of the pH.67 Therefore, we hypothesize that the reaction time was insufficient to promote a bond between these two species.
On the other hand, for the samples synthesized holding the maximum temperature for 10 min, the insertion of GO caused a decrease in the zeta potential value, which can result from the bond of the –OH surface groups of WO3 and the –OH surface groups of GO.68,69
The samples produced using the maximum temperature of 180 °C exhibit zeta potentials that are more negative than those made at 200 °C, which can be ascribed to the higher surface area of the first set of materials, as described below.
The surface area of WO3GO and WO3 samples synthesized under 10 min of microwave irradiation was determined by BET analysis (Fig. 6). WO3_200 °C-10 min (Fig. 6a) exhibited a surface area of 38.46 m2 g−1, while WO3GO_200 °C-10 min has 36.65 m2 g−1 (Fig. 6b). Thus, the surface area did not show drastic changes upon the incorporation of GO. On the other hand, the surface area of WO3_180 °C-10 min (Fig. 6c) was calculated as 40.88 m2 g−1, whereas WO3GO_180 °C-10 min was 44.09 m2 g−1. Thus, modification with GO promoted a slight surface area increase for these materials. The central hypothesis to explain why graphene incorporated into WO3 did not increase the surface area at 200 °C can be due to a very homogeneous distribution of WO3 nanoparticles partially blocking the surface of GO or to restacking of graphene layers. The restacking phenomenon was previously reported by other authors60,61 who discussed that microwave heating onto graphene oxide sheets (as we also discussed earlier in the Raman section) could be thermally induced (Fig. S8, ESI†).
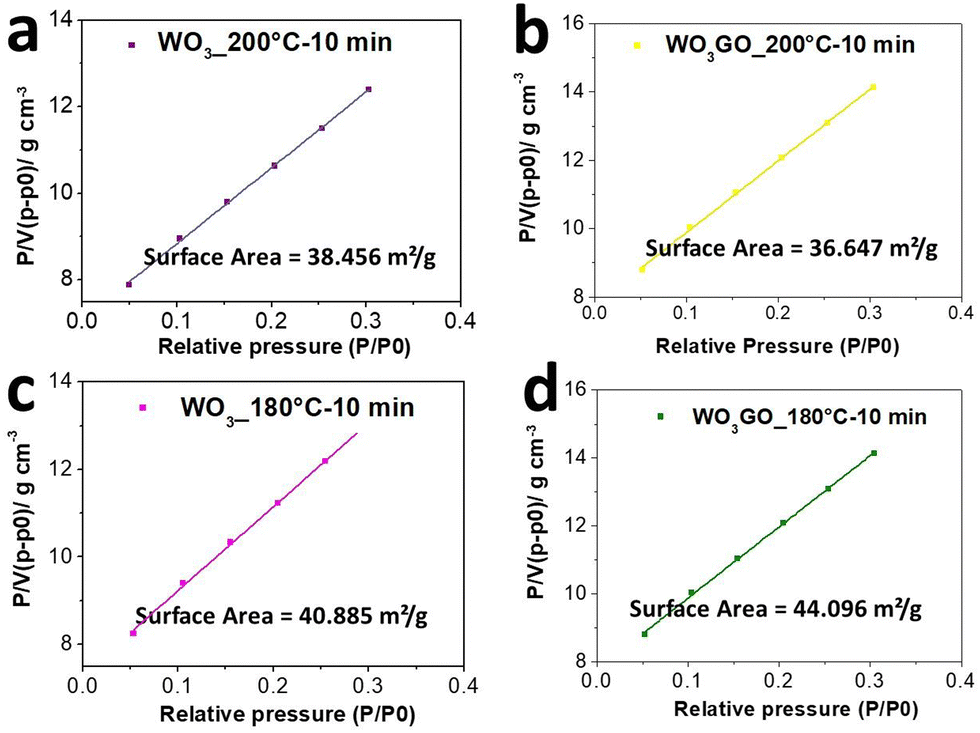 | ||
| Fig. 6 BET curves of (a) WO3_200 °C-10 min, (b) WO3GO_200 °C-10 min, (c) WO3_180 °C-10 min and (d) WO3GO_180 °C-10 min. | ||
3.3. Dye adsorption and degradation
The mass of MB, RhB and MO adsorbed onto the WO3_180 °C-0 min, WO3GO_180 °C-0 min, WO3_200 °C-0 min and WO3GO_200 °C-0 min photocatalysts as a function of time is shown in Fig. 7a, e and i. It is observed that there is no clear tendency between the mass of the dye adsorbed and the photocatalyst or the charge of the dye. This result may be ascribed to the fact that the adsorption ability of a species onto an adsorbent is multifactorial. Therefore, the crystalline structure, surface area, zeta potential and chemisorption must be considered. Fig. 4a shows that WO3GO_180 °C-0 min, WO3_200 °C-0 min and WO3GO_200 °C-0 min exhibit a monoclinic structure, whereas the structure of WO3GO_180 °C-0 min is ascribed to WO3·H2O. Fig. 6 shows that the temperature also plays a significant role in the surface area of the studied catalyst; it also affects the zeta potential (Table 3). Finally, differences in the shape of the graphs of the mass of dye adsorbed as a function of time (Fig. 7a, e and i) show that the adsorption model probably changes upon changing the adsorbed dye. As a result, it was not possible to find a single explanation for the results obtained.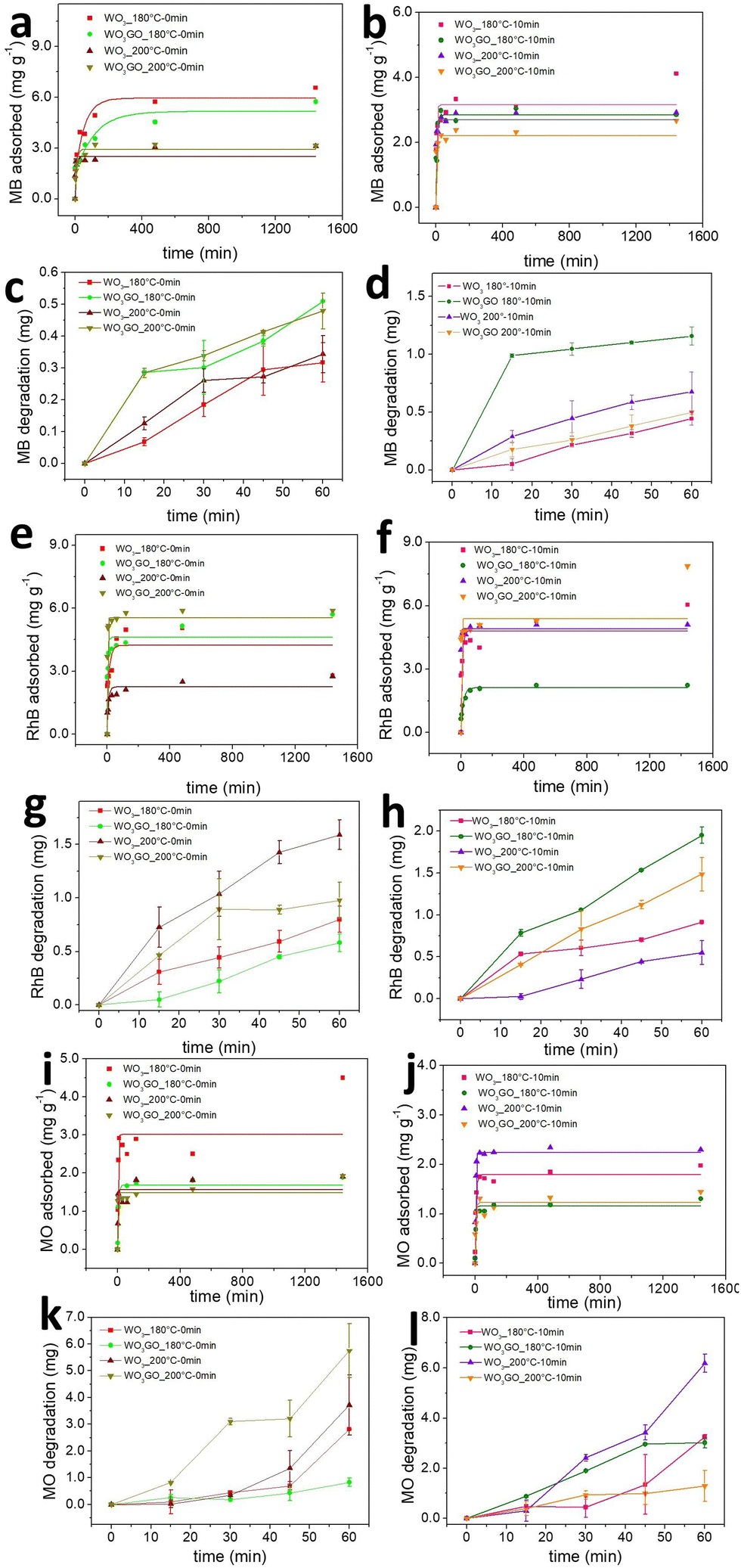 | ||
| Fig. 7 Mass of MB (a), RhB (e) and MO (i) adsorbed onto WO3_180 °C-0 min, WO3GO_180 °C-0 min, WO3_200 °C-0 min and WO3GO_200 °C-0 min. Mass of MB (b), RhB (f) and MO (j) adsorbed onto WO3_180 °C-10 min, WO3GO_180 °C-10 min, WO3_200 °C-10 min and WO3GO_200 °C-10 min. Mass of MB (c), RhB (g) and MO (k) degraded by WO3_180 °C-0 min, WO3GO_180 °C-0 min, WO3_200 °C-0 min and WO3GO_200 °C-0 min photocatalysts. Mass of MB (d), RhB (h) and MO (l) degraded by WO3_180 °C-10 min, WO3GO_180 °C-10 min, WO3_200 °C-10 min and WO3GO_200 °C-10 min photocatalysts. | ||
Fig. 7b, f and j show the adsorbed mass of MB, RhB and MO, respectively, onto the surface of WO3_180 °C-10 min, WO3GO_180 °C-10 min, WO3_200 °C-10 min and WO3GO_200 °C-10 min. It is possible to notice that increasing the reaction time of the synthesis of the photocatalysts did not increase the capacity of the resultant material to adsorb the dyes (considering the order of magnitude of the mass of dye adsorbed). On the other hand, it changed the trend of the amount of dye adsorbed as a function of the photocatalyst used as an adsorbent; it also changed the shape of the graphs of the mass of dye adsorbed as a function of time. This observation may be ascribed to the changes in the crystal structure of materials upon increasing the reaction time (more specifically, considering the WO3_180 °C-0 min and WO3_180 °C-10 min materials). Additionally, the surface area, zeta potential and dispersion of GO onto WO3 have changed upon increasing the reaction time. Again, since the capacity of the dyes to be adsorbed onto the photocatalyst surface is a multifactorial property, it is impossible to rationalize a simple trend in the obtained results.
In general, it has been observed that for all dyes and catalysts studied, equilibrium is reached after 500 minutes. Therefore, the photocatalytic experiments were conducted only after this minimum time of adsorption.
The adsorption kinetics of the dye adsorption onto all photocatalysts are well fitted with the pseudo-second-order model (Fig. S11–S13 (ESI†), eqn (3)).70
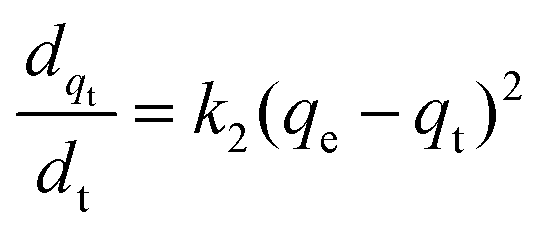 | (3) |
| Dye | Sample | k 2 | R 2 |
|---|---|---|---|
| Methyl blue | WO3_180 °C-0 min | 0.0006 | 0.9972 |
| WO3GO_180 °C-0 min | 0.0004 | 0.9905 | |
| WO3_180 °C-10 min | 0.0013 | 0.9968 | |
| WO3GO_180 °C-10 min | 0.0033 | 0.9996 | |
| WO3_200 °C-0 min | 0.0025 | 0.9990 | |
| WO3GO_200 °C-0 min | 0.0070 | 0.9998 | |
| WO3_200 °C-10 min | 0.0158 | 0.9999 | |
| WO3GO_200 °C-10 min | 0.0294 | 0.9974 | |
| Rhodamine B | WO3_180 °C-0 min | 0.0010 | 0.9997 |
| WO3GO_180 °C-0 min | 0.0004 | 0.9979 | |
| WO3_180 °C-10 min | 0.0006 | 0.9960 | |
| WO3GO_180 °C-10 min | 0.4913 | 0.9996 | |
| WO3_200 °C-0 min | 0.0033 | 0.9979 | |
| WO3GO_200 °C-0 min | 0.0020 | 0.9999 | |
| WO3_200 °C-10 min | 0.0052 | 0.9999 | |
| WO3GO_200 °C-10 min | 0.0037 | 0.9767 | |
| Methyl orange | WO3_180 °C-0 min | 0.0014 | 0.9465 |
| WO3GO_180 °C-0 min | 0.0040 | 0.99959 | |
| WO3_180 °C-10 min | 0.0057 | 0.99916 | |
| WO3GO_180 °C-10 min | 1.4716 | 0.9988 | |
| WO3_200 °C-0 min | 0.0067 | 0.99915 | |
| WO3GO_200 °C-0 min | 0.0197 | 0.9944 | |
| WO3_200 °C-10 min | 0.0256 | 0.9999 | |
| WO3GO_200 °C-10 min | 0.1012 | 0.9983 |
Fig. 7c, d, g, h, k and l show the MB, RhB and MO mass adsorbed by each studied photocatalyst. From the graphs, it is possible to observe different activities depending on the catalyst and the dye without any noticeable trend. Fig. 8a shows a correlation between the mass of the dyes adsorbed onto each catalyst after 1440 min of adsorption in the dark. Fig. 8b shows the mass of dye degraded after 60 min of simulated sunlight irradiation. First, it is possible to observe that most catalysts exhibited higher photoactivity for MO degradation. Coincidently, MO is the dye that adsorbs less in all catalysts, probably due to its negative charge. This result suggests that strong dye adsorption onto the photocatalyst's surface may create a layer of photosensitive material that absorbs the light. As a result, a smaller portion of light activates the photocatalyst. RhB was the second dye most degraded by all catalysts, and the second dye most adsorbed by WO3_180 °C-0 min and WO3GO_180 °C-0 min, which can be ascribed to the WO3·H2O structure exhibited only for these two samples. This feature is also reflected in the slower adsorption kinetics of these samples (Fig. 7a). For WO3G_200 °C-0 min and WO3GO_180–10 min, the amount of RhB adsorbed is also lower than that of MB, which can be a combination of surface area, zeta potential and chemical interaction. RhB was degraded more efficiently for the other catalysts, although it was more intensely adsorbed than MB. This result cannot be explained by blocking the light effect caused by a layer of dye on the catalyst surface. These results suggest different chemical interactions with the cationic dyes.
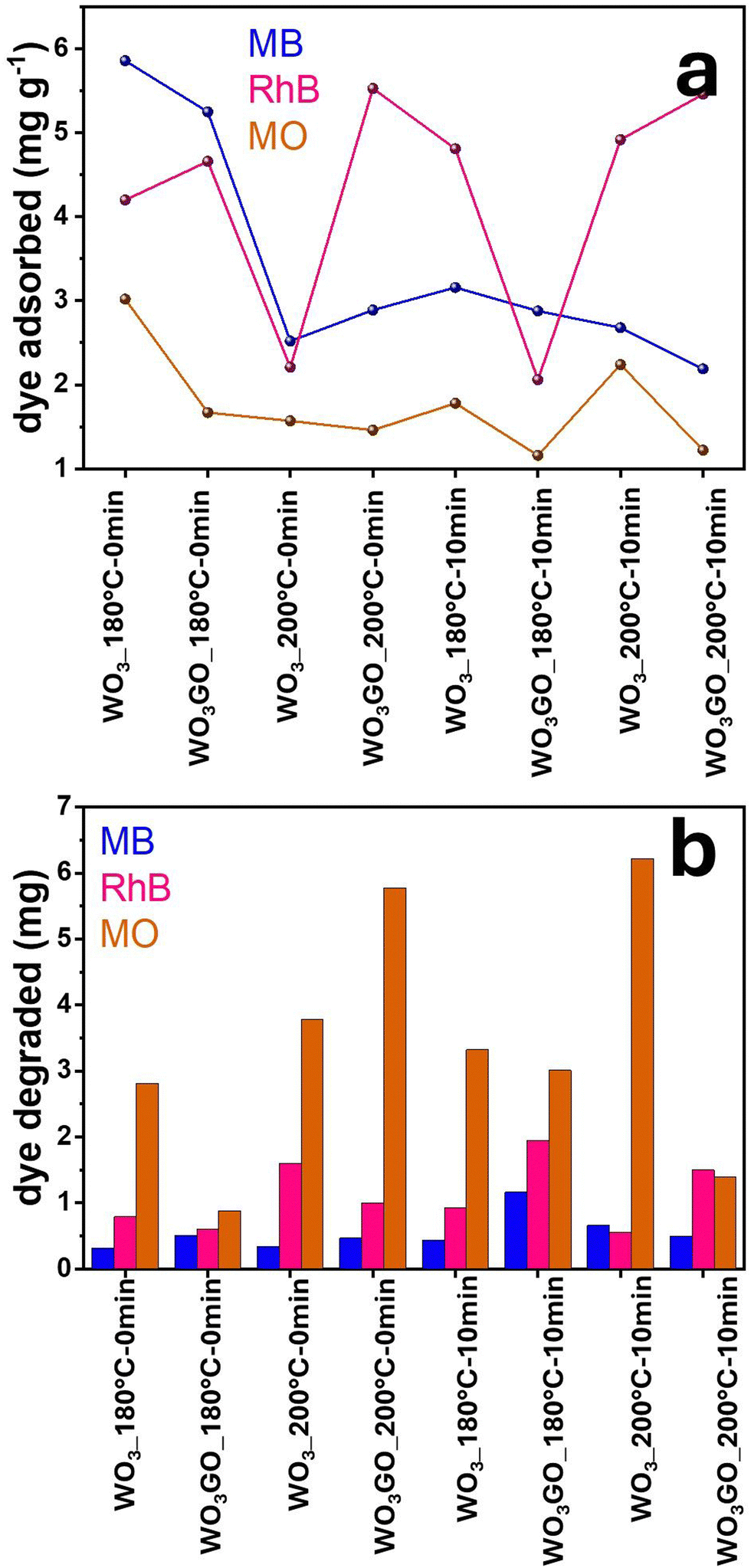 | ||
| Fig. 8 Correlation between the MB, RhB and MO mass adsorbed onto each catalyst after 1440 min of adsorption in the dark (a) and the mass of dye degraded after 60 min of simulated sunlight irradiation (b). | ||
In this sense, we infer that the photoactivity cannot be explained by one single property of WO3GO. Instead, several variables can hinder or increase the activity. Therefore, further optimizations must be done to create highly efficient materials.
4. Conclusions
The present study reports the successful synthesis of WO3 and WO3GO samples via a rapid microwave route. Different synthesis parameters, such as temperature and time, influence the properties of the materials, particularly their morphology and crystal structure. Hence, we observed that rounded platelets with the crystal structure of WO3·H2O form WO3GO materials synthesized at lower temperatures for shorter periods. However, the monoclinic phase is formed by increasing the time to 10 min at 180 °C or a temperature of 200 °C. Although the presence of GO in the synthesis medium does not seem to interfere with particle growth, the interaction between GO and microwaves at high temperatures may promote the restacking of graphene sheets. WO3GO_200 °C samples exhibited an intense and sharp G band at Raman spectra compared to WO3GO_180 °C samples, which is associated with re-stacking. In addition, microwave treatment at 200 °C did not increase the degree of reduction of graphene or improve the surface area of WO3GO. Finally, we observed that a longer reaction time is required to improve the binding between GO and WO3.We found that the highest photoactivities were observed when MO was used as a model dye, which has been ascribed to its lower adsorption onto the catalyst's surfaces. Therefore, the dye does not block the interaction of light with the semiconductor. For the cationic dyes RhB and MB, the crystal structure of the catalyst as well as its surface area, zeta potential, and chemical interaction play more critical roles. Therefore, we infer that a single property of WO3GO cannot explain the photoactivity, and several variables must be considered to increase the photoactivity. Further investigations focused on synthetic parameters can be done to modulate the properties of WO3GO materials.
Author contributions
JSS designed and supervised the research. BSR performed the synthesis, characterization and photocatalysis assays. MN and BSR discussed and analyzed the microwave synthesis and characterization data. BSR has written the manuscript based on inputs from all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by FAPESP (grants 2021/05958-4, 2019/26010-9 and 2021/12018-8). This study was also financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001 and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). The authors also acknowledge the Multifunctional Materials group at ETH Zürich, especially Elena Tervoort-Gorokhova, for collecting the images of SEM and TEM—the National Laboratory of Nanotechnology (LNNano) for the STEM images (Proposal #20233865). Finally, we thank the Multi-users platform (CEM) at UFABC for instrumental facilities.References
- P. Shandilya, S. Sambyal, R. Sharma, P. Mandyal and B. Fang, Properties, optimized morphologies, and advanced strategies for photocatalytic applications of WO3 based photocatalysts, J. Hazard. Mater., 2022, 428, 128218, DOI:10.1016/j.jhazmat.2022.128218.
- S. Adhikari, K. Sarath Chandra, D.-H. Kim, G. Madras and D. Sarkar, Understanding the morphological effects of WO3 photocatalysts for the degradation of organic pollutants, Adv. Powder Technol., 2018, 29, 1591–1600, DOI:10.1016/j.apt.2018.03.024.
- P. Jineesh, T. C. Bhagya, R. Remya and S. M. A. Shibli, Photocatalytic hydrogen generation by WO3 in synergism with hematite-anatase heterojunction, Int. J. Hydrogen Energy, 2020, 45, 18946–18960, DOI:10.1016/j.ijhydene.2020.05.043.
- G. Mineo, M. Scuderi, E. Bruno and S. Mirabella, Engineering Hexagonal/Monoclinic WO3 Phase Junctions for Improved Electrochemical Hydrogen Evolution Reaction, ACS Appl. Energy Mater., 2022, 5, 9702–9710, DOI:10.1021/acsaem.2c01383.
- G. Jeevitha, R. Abhinayaa, D. Mangalaraj and N. Ponpandian, Tungsten oxide-graphene oxide (WO3-GO) nanocomposite as an efficient photocatalyst, antibacterial and anticancer agent, J. Phys. Chem. Solids, 2018, 116, 137–147, DOI:10.1016/j.jpcs.2018.01.021.
- L. Tie, C. Yu, Y. Zhao, H. Chen, S. Yang, J. Sun, S. Dong and J. Sun, Fabrication of WO3 nanorods on reduced graphene oxide sheets with augmented visible light photocatalytic activity for efficient mineralization of dye, J. Alloys Compd., 2018, 769, 83–91, DOI:10.1016/j.jallcom.2018.07.176.
- M. G. Hosseini, P. Y. Sefidi, Z. Aydin and S. Kinayyigit, Toward enhancing the photoelectrochemical water splitting efficiency of organic acid doped polyaniline-WO3 photoanode by photo-assisted electrochemically reduced graphene oxide, Electrochim. Acta, 2020, 333, 135475, DOI:10.1016/j.electacta.2019.135475.
- N. Lu, P. Wang, Y. Su, H. Yu, N. Liu and X. Quan, Construction of Z-Scheme g-C3N4/RGO/WO3 with in situ photoreduced graphene oxide as electron mediator for efficient photocatalytic degradation of ciprofloxacin, Chemosphere, 2019, 215, 444–453, DOI:10.1016/j.chemosphere.2018.10.065.
- P.-G. Su and S.-L. Peng, Fabrication and NO2 gas-sensing properties of reduced graphene oxide/WO3 nanocomposite films, Talanta, 2015, 132, 398–405, DOI:10.1016/j.talanta.2014.09.034.
- A. Khan, N. Y. Bhosale, S. S. Mali, C. K. Hong and A. V. Kadam, Reduced graphene oxide layered WO3 thin film with enhanced electrochromic properties, J. Colloid Interface Sci., 2020, 571, 185–193, DOI:10.1016/j.jcis.2020.03.029.
- B. Ahmed, A. K. Ojha, A. Singh, F. Hirsch, I. Fischer, D. Patrice and A. Materny, Well-controlled in-situ growth of 2D WO3 rectangular sheets on reduced graphene oxide with strong photocatalytic and antibacterial properties, J. Hazard. Mater., 2018, 347, 266–278, DOI:10.1016/j.jhazmat.2017.12.069.
- W. Mu, Q. Yu, R. Hu, X. Li, H. Wei and Y. Jian, Porous three-dimensional reduced graphene oxide merged with WO3 for efficient removal of radioactive strontium, Appl. Surf. Sci., 2017, 423, 1203–1211, DOI:10.1016/j.apsusc.2017.06.206.
- R. Samal, B. Chakraborty, M. Saxena, D. J. Late and C. S. Rout, Facile Production of Mesoporous WO3-rGO Hybrids for High-Performance Supercapacitor Electrodes: An Experimental and Computational Study, ACS Sustainable Chem. Eng., 2019, 7, 2350–2359, DOI:10.1021/acssuschemeng.8b05132.
- W. Dang, W. Wang, Y. Yang, Y. Wang, J. Huang, X. Fang, L. Wu, Z. Rong, X. Chen, X. Li, L. Huang and X. Tang, One-step hydrothermal synthesis of 2D WO3 nanoplates@ graphene nanocomposite with superior anode performance for lithium ion battery, Electrochim. Acta, 2019, 313, 99–108, DOI:10.1016/j.electacta.2019.04.184.
- M. Covei, C. Bogatu, D. Perniu, A. Duta and I. Visa, Self-cleaning thin films with controlled optical properties based on WO3-rGO, Ceram. Int., 2019, 45, 9157–9163, DOI:10.1016/j.ceramint.2019.01.256.
- M. Zhi, W. Huang, Q. Shi, M. Wang and Q. Wang, Sol–gel fabrication of WO3/RGO nanocomposite film with enhanced electrochromic performance, RSC Adv., 2016, 6, 67488–67494, 10.1039/C6RA13947G.
- Y. E. Firat, Pseudocapacitive energy storage properties of rGO-WO3 electrode synthesized by electrodeposition, Mater. Sci. Semicond. Process., 2021, 133, 105938, DOI:10.1016/j.mssp.2021.105938.
- J. M. Won, M. Y. Son, J.-H. Seo and Y. C. Kang, Electrochemical properties of WO3-reduced graphene oxide composite powders prepared by one-pot spray pyrolysis process, J. Alloys Compd., 2016, 688, 647–653, DOI:10.1016/j.jallcom.2016.07.238.
- L. Mei, H. Zhao and B. Lu, Ultra-Efficient Photocatalytic Properties in Porous Tungsten Oxide/Graphene Film under Visible Light Irradiation, Adv. Sci., 2015, 2, 1500116, DOI:10.1002/advs.201500116.
- Y. O. Ibrahim, M. A. Gondal, A. Alaswad, R. A. Moqbel, M. Hassan, E. Cevik, T. F. Qahtan, M. A. Dastageer and A. Bozkurt, Laser-induced anchoring of WO3 nanoparticles on reduced graphene oxide sheets for photocatalytic water decontamination and energy storage, Ceram. Int., 2020, 46, 444–451, DOI:10.1016/j.ceramint.2019.08.281.
- J. Guo, Y. Li, S. Zhu, Z. Chen, Q. Liu, D. Zhang, W.-J. Moon and D.-M. Song, Synthesis of WO3@Graphene composite for enhanced photocatalytic oxygen evolution from water, RSC Adv., 2012, 2, 1356–1363, 10.1039/C1RA00621E.
- G. Jeevitha, R. Abhinayaa, D. Mangalaraj, N. Ponpandian, P. Meena, V. Mounasamy and S. Madanagurusamy, Porous reduced graphene oxide (rGO)/WO3 nanocomposites for the enhanced detection of NH3 at room temperature, Nanoscale Adv., 2019, 1, 1799–1811, 10.1039/C9NA00048H.
- R. Bhargava and S. Khan, Fabrication of WO3–reduced graphene oxide (WO3–G) nanocomposite for enhanced optical and electrical properties, J. Mater. Sci.: Mater. Electron., 2020, 31, 8370–8384, DOI:10.1007/s10854-020-03372-0.
- S. Xiao, C. Zhou, X. Ye, Z. Lian, N. Zhang, J. Yang, W. Chen and H. Li, Solid-Phase Microwave Reduction of WO3 by GO for Enhanced Synergistic Photo-Fenton Catalytic Degradation of Bisphenol A, ACS Appl. Mater. Interfaces, 2020, 12, 32604–32614, DOI:10.1021/acsami.0c06373.
- Y. Bao, H. Guo, L. Jiang, Z. Liu, J. Qu, C. Zhang, X. Jia and K. Chen, Heterostructured WO3/RGO/protonated g-C3N4 three-layer nanosheets for enhanced visible-light photocatalytic activity, Appl. Surf. Sci., 2019, 496, 143639, DOI:10.1016/j.apsusc.2019.143639.
- T. M. Perfecto, C. A. Zito, T. Mazon and D. P. Volanti, Flexible room-temperature volatile organic compound sensors based on reduced graphene oxide–WO3·0.33H2O nano-needles, J. Mater. Chem. C, 2018, 6, 2822–2829, 10.1039/C8TC00324F.
- P. Nagaraju, A. Alsalme, A. M. Alkathiri and R. Jayavel, Rapid synthesis of WO3/graphene nanocomposite via in-situ microwave method with improved electrochemical properties, J. Phys. Chem. Solids, 2018, 120, 250–260, DOI:10.1016/j.jpcs.2018.04.046.
- Y. Gui, Z. Liu, S. Fang, J. Tian and F. Gong, Synthesis of flower-like WO 3/graphene nanocomposite by microwave-assisted hydrothermal method and the enhanced gas-sensing properties to aniline, J. Mater. Sci.: Mater. Electron., 2016, 27, 2890–2895 CrossRef CAS.
- T. M. Perfecto, C. A. Zito and D. P. Volanti, Room-temperature volatile organic compounds sensing based on WO3·0.33H2O, hexagonal-WO3, and their reduced graphene oxide composites, RSC Adv., 2016, 6, 105171–105179, 10.1039/C6RA16892B.
- Y. Gui, J. Zhao, W. Wang, J. Tian and M. Zhao, Synthesis of hemispherical WO3/graphene nanocomposite by a microwave-assisted hydrothermal method and the gas-sensing properties to triethylamine, Mater. Lett., 2015, 155, 4–7, DOI:10.1016/j.matlet.2015.04.100.
- I. Bilecka and M. Niederberger, Microwave chemistry for inorganic nanomaterials synthesis, Nanoscale, 2010, 2, 1269–1528, 10.1039/b9nr00377k.
- M. B. Schütz, L. Xiao, T. Lehnen, T. Fischer and S. Mathur, Microwave-assisted synthesis of nanocrystalline binary and ternary metal oxides, Int. Mater. Rev., 2018, 63, 341–374, DOI:10.1080/09506608.2017.1402158.
- B. Reeja-Jayan, K. L. Harrison, K. Yang, C.-L. Wang, A. E. Yilmaz and A. Manthiram, Microwave-assisted Low-temperature Growth of Thin Films in Solution, Sci. Rep., 2012, 2, 1003, DOI:10.1038/srep01003.
- M. M. Maitani, T. Yamada, H. Mashiko, K. Yoshimatsu, T. Oshima, A. Ohtomo and Y. Wada, Microwave Effects on Co–Pi Cocatalysts Deposited on α-Fe2O3 for Application to Photocatalytic Oxygen Evolution, ACS Appl. Mater. Interfaces, 2017, 9, 10349–10354, DOI:10.1021/acsami.6b16319.
- Y. Tsukahara, A. Higashi, T. Yamauchi, T. Nakamura, M. Yasuda, A. Baba and Y. Wada, In Situ Observation of Nonequilibrium Local Heating as an Origin of Special Effect of Microwave on Chemistry, J. Phys. Chem. C, 2010, 114, 8965–8970, DOI:10.1021/jp100509h.
- B. S. Rodrigues, M. R. S. Vicente and J. S. Souza, Investigating the role of microwave thermal and non-thermal effects on WO3-graphene oxide composite synthesis, RSC Adv., 2023, 13, 26794–26803, 10.1039/D3RA04113A.
- A. Apolinário, T. Lopes, C. Costa, J. P. Araújo and A. M. Mendes, Multilayered WO3 Nanoplatelets for Efficient Photoelectrochemical Water Splitting: The Role of the Annealing Ramp, ACS Appl. Energy Mater., 2019, 2, 1040–1050, DOI:10.1021/acsaem.8b01530.
- B. Scola Rodrigues, C. M. Branco, M. R. S. Vicente, J. Rodríguez-López and J. dos Santos de Souza, Influence of the Solvent Used for Microwave-Assisted Synthesis of W−BiVO4 on Properties and Photoelectroactivity of W−BiVO4/WO3, ChemElectroChem, 2022, 9, e202200098, DOI:10.1002/celc.202200098.
- J. Yang, X. Chen, X. Liu, Y. Cao, J. Huang, Y. Li and F. Liu, From Hexagonal to Monoclinic: Engineering Crystalline Phase to Boost the Intrinsic Catalytic Activity of Tungsten Oxides for the Hydrogen Evolution Reaction, ACS Sustainable Chem. Eng., 2021, 9, 5642–5650, DOI:10.1021/acssuschemeng.1c00485.
- T. Kavinkumar, D. Sastikumar and S. Manivannan, Effect of functional groups on dielectric, optical gas sensing properties of graphene oxide and reduced graphene oxide at room temperature, RSC Adv., 2015, 5, 10816–10825, 10.1039/C4RA12766H.
- M. D. Sandeepa Lakshad Wimalananda, J.-K. Kim, S. W. Cho and J.-M. Lee, Highly efficient two-step nitrogen doping of graphene oxide-based materials in oxygen presence atmosphere for high-performance transistors and electrochemical applications, J. Sci.: Adv. Mater. Devices, 2022, 7, 100481, DOI:10.1016/j.jsamd.2022.100481.
- S. Hilaire, M. J. Süess, N. Kränzlin, K. Bieńkowski, R. Solarska, J. Augustyński and M. Niederberger, Microwave-assisted nonaqueous synthesis of WO3 nanoparticles for crystallographically oriented photoanodes for water splitting, J. Mater. Chem. A, 2014, 2, 20530–20537, 10.1039/C4TA04793A.
- Y.-J. Zhu and F. Chen, Microwave-Assisted Preparation of Inorganic Nanostructures in Liquid Phase, Chem. Rev., 2014, 114, 6462–6555, DOI:10.1021/cr400366s.
- Wm. C. Conner and G. A. Tompsett, How Could and Do Microwaves Influence Chemistry at Interfaces, J. Phys. Chem. B, 2008, 112, 2110–2118, DOI:10.1021/jp0775247.
- Q. Zhang, S.-J. Liu and S.-H. Yu, Recent advances in oriented attachment growth and synthesis of functional materials: concept, evidence, mechanism, and future, J. Mater. Chem., 2009, 19, 191–207, 10.1039/B807760F.
- S. Xu, G. Zhong, C. Chen, M. Zhou, D. J. Kline, R. J. Jacob, H. Xie, S. He, Z. Huang, J. Dai, A. H. Brozena, R. Shahbazian-Yassar, M. R. Zachariah, S. M. Anlage and L. Hu, Uniform, Scalable, High-Temperature Microwave Shock for Nanoparticle Synthesis through Defect Engineering, Matter, 2019, 1, 759–769, DOI:10.1016/j.matt.2019.05.022.
- A. E. Souza, G. T. A. Santos, B. C. Barra, W. D. Macedo Jr., S. R. Teixeira, C. M. Santos, A. M. O. R. Senos, L. Amaral and E. Longo, Photoluminescence of SrTiO3: Influence of Particle Size and Morphology, Cryst. Growth Des., 2012, 12, 5671–5679, DOI:10.1021/cg301168k.
- S. H. Jhung, T. Jin, Y. K. Hwang and J.-S. Chang, Microwave Effect in the Fast Synthesis of Microporous Materials: Which Stage Between Nucleation and Crystal Growth is Accelerated by Microwave Irradiation, Chem. – Eur. J., 2007, 13, 4410–4417, DOI:10.1002/chem.200700098.
- M. Kang and H.-S. Kim, Microwave-assisted facile and ultrafast growth of ZnO nanostructures and proposition of alternative microwave-assisted methods to address growth stoppage, Sci. Rep., 2016, 6, 1–13 CrossRef PubMed.
- I. M. Szilágyi, B. Fórizs, O. Rosseler, Á. Szegedi, P. Németh, P. Király, G. Tárkányi, B. Vajna, K. Varga-Josepovits, K. László, A. L. Tóth, P. Baranyai and M. Leskelä, WO3 photocatalysts: Influence of structure and composition, J. Catal., 2012, 294, 119–127, DOI:10.1016/j.jcat.2012.07.013.
- J. T. Szymanski and A. C. Roberts, The crystal structure of tungstite, WO3. H2O, Can. Mineral., 1984, 22, 681–688 CAS.
- N. Le Houx, G. Pourroy, F. Camerel, M. Comet and D. Spitzer, WO3 Nanoparticles in the 5–30 nm Range by Solvothermal Synthesis under Microwave or Resistive Heating, J. Phys. Chem. C, 2010, 114, 155–161, DOI:10.1021/jp908669u.
- I. Olliges-Stadler, J. Stötzel, D. Koziej, M. D. Rossell, J.-D. Grunwaldt, M. Nachtegaal, R. Frahm and M. Niederberger, Study of the Chemical Mechanism Involved in the Formation of Tungstite in Benzyl Alcohol by the Advanced QEXAFS Technique, Chem. – Eur. J., 2012, 18, 2305–2312, DOI:10.1002/chem.201101514.
- H. Yoon, M. Kim, S. S. Al-deyab and S. S. Yoon, Electrostatic spray deposition of transparent tungsten oxide thin-film photoanodes for solar water splitting Electrostatic spray deposition of transparent tungsten oxide thin-film photoanodes for solar water splitting, Catal. Today, 2015, 260, 89–94, DOI:10.1016/j.cattod.2015.03.037.
- J. Kaur, K. Anand, K. Anand and R. C. Singh, WO3 nanolamellae/reduced graphene oxide nanocomposites for highly sensitive and selective acetone sensing, J. Mater. Sci., 2018, 53, 12894–12907, DOI:10.1007/s10853-018-2558-z.
- X. Wen, J. Luo, K. Xiang, W. Zhou, C. Zhang and H. Chen, High-performance monoclinic WO3 nanospheres with the novel NH4+ diffusion behaviors for aqueous ammonium-ion batteries, Chem. Eng. J., 2023, 458, 141381, DOI:10.1016/j.cej.2023.141381.
- I. Szilágyi, J. Pfeifer, C. Balázsi, A. Tóth, K. Varga-Josepovits, J. Madarász and G. Pokol, Thermal stability of hexagonal tungsten trioxide in air, J. Therm. Anal. Calorim., 2008, 94, 499–505 CrossRef.
- A. Niilisk, J. Kozlova, H. Alles, J. Aarik and V. Sammelselg, Raman characterization of stacking in multi-layer graphene grown on Ni, Carbon, 2016, 98, 658–665, DOI:10.1016/j.carbon.2015.11.050.
- S.-H. Bae, K. Karthikeyan, Y.-S. Lee and I.-K. Oh, Microwave self-assembly of 3D graphene-carbon nanotube-nickel nanostructure for high capacity anode material in lithium ion battery, Carbon, 2013, 64, 527–536, DOI:10.1016/j.carbon.2013.08.003.
- J. Liang, C. Li, W. Li, J. Qi and C. Liang, Microwave-assisted polyol preparation of reduced graphene oxide nanoribbons supported platinum as a highly active electrocatalyst for oxygen reduction reaction, J. Appl. Electrochem., 2018, 48, 1069–1080, DOI:10.1007/s10800-018-1235-x.
- H. Chen, F. Guo, Y. Liu, T. Huang, B. Zheng, N. Ananth, Z. Xu, W. Gao and C. Gao, A defect-free principle for advanced graphene cathode of aluminum-ion battery, Adv. Mater., 2017, 29, 1605958 CrossRef PubMed.
- X. Hu, P. Xu, H. Gong and G. Yin, Synthesis and Characterization of WO3/Graphene Nanocomposites for Enhanced Photocatalytic Activities by One-Step In-Situ Hydrothermal Reaction, Materials, 2018, 11, 147–163, DOI:10.3390/ma11010147.
- V. Harnchana, S. Chaiyachad, S. Pimanpang, C. Saiyasombat, P. Srepusharawoot and V. Amornkitbamrung, Hierarchical Fe3O4-reduced graphene oxide nanocomposite grown on NaCl crystals for triiodide reduction in dye-sensitized solar cells, Sci. Rep., 2019, 9, 1494, DOI:10.1038/s41598-018-38050-z.
- Q. Lu, L. Liu, X. Zhang, Y. Cheng, Y. Huang, Y. Shan, Q. Zhao, G. Zhang and D. Li, Graphene transparent conductive films directly grown on quartz substrates by assisted catalysis of Cu nanoparticles, J. Mater. Sci., 2019, 54, 10312–10324, DOI:10.1007/s10853-019-03621-6.
- P. Jayaweera, S. Hettiarachchi and H. Ocken, Determination of the high temperature zeta potential and pH of zero charge of some transition metal oxides, Colloids Surf., A, 1994, 85, 19–27, DOI:10.1016/0927-7757(93)02737-Y.
- X. Liu, A. Jin, Y. Jia, T. Xia, C. Deng, M. Zhu, C. Chen and X. Chen, Synergy of adsorption and visible-light photocatalytic degradation of methylene blue by a bifunctional Z-scheme heterojunction of WO3/g-C3N4, Appl. Surf. Sci., 2017, 405, 359–371, DOI:10.1016/j.apsusc.2017.02.025.
- N. Wang, C. Hsu, L. Zhu, S. Tseng and J.-P. Hsu, Influence of metal oxide nanoparticles concentration on their zeta potential, J. Colloid Interface Sci., 2013, 407, 22–28, DOI:10.1016/j.jcis.2013.05.058.
- J. Fu, Q. Xu, J. Low, C. Jiang and J. Yu, Ultrathin 2D/2D WO3/g-C3N4 step-scheme H2-production photocatalyst, Appl. Catal., B, 2019, 243, 556–565, DOI:10.1016/j.apcatb.2018.11.011.
- B. I. Kharisov, O. V. Kharissova, A. Vázquez Dimas, I. Gómez De La Fuente and Y. Peña Méndez, Review: Graphene-supported coordination complexes and organometallics: properties and applications, J. Coord. Chem., 2016, 69, 1125–1151, DOI:10.1080/00958972.2016.1170817.
- T. R. Sahoo and B. Prelot, Nanomaterials for the Detection and Removal of Wastewater Pollutants, in Adsorption processes for the removal of contaminants from wastewater: the perspective role of nanomaterials and nanotechnology, ed. B. Bonelli, F. S. Freyria, I. Rossetti and R. Sethi, Elsevier, 2020, ch. 7, pp. 161–222 DOI:10.1016/B978-0-12-818489-9.00007-4.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ma00972f |
| This journal is © The Royal Society of Chemistry 2024 |