
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCombination of cowpea mosaic virus (CPMV) intratumoral therapy and oxaliplatin chemotherapy†
Miguel A.
Moreno-Gonzalez
 adefg,
Zhongchao
Zhao
adefg,
Adam A.
Caparco
adeh and
Nicole F.
Steinmetz
*abcdefgh
adefg,
Zhongchao
Zhao
adefg,
Adam A.
Caparco
adeh and
Nicole F.
Steinmetz
*abcdefgh
aDepartment of NanoEngineering, University of California San Diego, 9500 Gilman Dr, La Jolla, California 92093, USA. E-mail: nsteinmetz@ucsd.edu
bDepartment of Bioengineering, University of California San Diego, 9500 Gilman Dr, La Jolla, California 92093, USA
cDepartment of Radiology, University of California San Diego, 9500 Gilman Dr, La Jolla, California 92093, USA
dCenter for Nano-ImmunoEngineering, University of California San Diego, 9500 Gilman Dr, La Jolla, California 92093, USA
eShu and K.C. Chien and Peter Farrell Collaboratory, University of California San Diego, 9500 Gilman Dr, La Jolla, California 92093, USA
fCenter for Engineering in Cancer, Institute of Engineering in Medicine, University of California San Diego, 9500 Gilman Dr, La Jolla, California 92093, USA
gMoores Cancer Center, University of California, University of California San Diego, 9500 Gilman Dr, La Jolla, California 92093, USA
hInstitute for Materials Discovery and Design, University of California San Diego, 9500 Gilman Dr, La Jolla, California 92093, USA
First published on 7th May 2024
Abstract
Cowpea mosaic virus is a potent intratumoral immunotherapy agent that has shown promise in preclinical studies and canine cancer trials with tumor- and tissue-agnostic efficacy. As we move towards the clinic, it is imperative to investigate combination strategies that synergize to further improve the potency of the approach. Here, we combined CPMV with the clinically approved chemotherapeutic agent oxaliplatin. CPMV's ability to recruit and activate naive immune cells synergized with oxaliplatin's ability to induce immunogenic cell death in the ID8-Defb29/Vegf-A ovarian and B16F10 melanoma murine cancer models with an increase of median survival of 57.7% and 162.2%, respectively. The combination therapy outperformed the CPMV or oxaliplatin monotherapy, and achieved a percent difference in tumor burden of 26.1% and 170.6% in the ID8-Defb29/Vegf-A ovarian and B16F10 melanoma models, respectively. Immunofluorescence staining of treated tumor sections elucidated the role of damage associated molecular patterns (calreticulin and HMGB1), innate immune cells (myeloid cells – likely neutrophils, NK cells, and macrophages), and regulatory T cells (Tregs) as a function of the treatment regimen. Overall, our proposed combination therapy modulated the dormant tumor microenvironment which resulted in effective tumor cell death. This study demonstrates the potential for clinical combination of chemotherapy and CPMV intratumoral immunotherapy.
Introduction
Cowpea mosaic virus (CPMV) is a plant virus and drug candidate under development as an intratumoral immunotherapy. Intratumorally administered CPMV is immunostimulatory and repolarizes the immunosuppressive tumor microenvironment (TME) from a pro-tumor to an anti-tumor phenotype. Mechanism studies have elucidated the role of the innate immune system in CPMV cancer immunotherapy.1–4 The repeating nature of the nucleoprotein assembly is recognized as a pathogen-associated molecular pattern (PAMP) and agonizes pattern recognition receptors (PRRs), which are highly conserved innate immune receptors.5 Gene knockout studies identified Toll-like receptors (TLRs) 2, 4, and 7 as the PRRs responsible for CPMV recognition and the resulting anti-tumor efficacy.6 TLR signaling initiates the activation of transcription factors which leads to the upregulation of proinflammatory cytokines (IFN-α, IFN-β, IFN-γ, IL-6, TNF-α, and GM-CSF) and downregulation of immunosuppressive cytokines (TGF-β and IL-10).1–4 This shift in the TME cytokine profile not only recruits and activates innate immune cells, it also promotes the switch from immunosuppressive to cytotoxic macrophage and neutrophil phenotypes (M2 → M1, and N2 → N1 switch).1 GM-CSF in particular has the ability to differentiate immunosuppressive G-MDSCs and M-MDSCs into activated neutrophils, macrophages, and dendritic cells.3 In doing so, CPMV turns the “cold” tumor into a “hot” tumor that is recognized and eliminated by the immune system. Immune cell killing and antigen processing by the innate immune cells, which then become antigen presenting cells (APCs), ultimately primes activation of tumor-specific cytotoxic CD8+ T cells and CD4+ helper T cells.3 It is the adaptive response that leads to abscopal effect and immune memory that treats and prevents the formation of metastatic spreading and recurrence.7 Although CD4+ T cells seem to have no bearing in the treatment of primary tumors, they are essential for the maturation of CD8+ memory T cells that remain in circulation to prevent recurrence,8 highlighting the beauty and complexity of cancer immunology.Cancer is a complex disease, and the most successful therapeutic interventions are synergistic combinations. Given the mechanism of CPMV, combination with agents with tumor cell killing functions, e.g. radiation9 or chemotherapy10 are thought to act in synergy. Tumor cell death will release neoantigens and tumor-associated antigens to be processed by the innate immune cells recruited and activated by CPMV, therefore expanding the pool of anti-tumor CD8 T cells. In this work, we set out to test the efficacy of CPMV + oxaliplatin chemotherapy. Oxaliplatin is an FDA-approved chemotherapy that is used for the treatment for several tumor types in the clinic. Compared to other platinum drugs such as cisplatin, oxaliplatin is considered less toxic and less prone to resistance.11 This alkylating agent disrupts DNA replication and transcription by binding platinum to the GC-rich regions of DNA which leads to intra- and inter-strand crosslinking.11,12 As cancer cells lose their ability to replicate, they undergo immunogenic cell death (ICD) which exposes damage-associated molecular patterns (DAMPs) on the cell surface.13 These DAMPs are recognized by dendritic cells of the innate immune system which promote tumor cell phagocytosis and cytotoxic T cell activation.13,14
We hypothesized that the combination of PAMPs from CPMV and exposure of DAMPs as a result of oxaliplatin-induced ICD would lead to a synergistic effect with improved anti-tumor efficacy. We tested this hypothesis in murine tumor mouse models of ovarian cancer and melanoma. Immunofluorescence staining of tumor sections was performed to gain insights into the mechanism of the CPMV solo and combination therapy.
Materials and methods
CPMV production
We obtained CPMV by infecting Vigna unguiculata (black-eyed pea No. 5) plants using previously described methods.15 Briefly, primary leaves were mechanically inoculated using a carborundum abrasive and harvested ∼14 days post-infection when the leaves showed the typical mosaic pattern. The infected leaves were subjected to a series of homogenization, filtration, extraction, PEG precipitation, and isopycnic centrifugation steps to extract CPMV from the infected leaf tissue. Pure CPMV was resuspended in 0.1 M potassium phosphate buffer pH 7; in the following referred to as KP.CPMV characterization
CPMV integrity and concentration was assessed using a NanoDrop 2000 spectrophotometer. We measured the absorbance at 260 nm and 280 nm, and confirmed that the A260/A280 was ∼1.7 for an intact CPMV. CPMV concentration was calculated using Beer–Lambert Law (A260 = εcl), where ε is the extinction coefficient of CPMV (8.1 mL × cm−1 × mg−1), c is the concentration (mg mL−1), and l is the path length of the spectrophotometer (0.1 cm).NuPAGE was used to confirm the presence of the small (24 kDa) and large (42 kDa) coat proteins of native CPMV and absence of protein contaminants. A sample of 5 μg of CPMV in KP, 1× NuPAGE™ LDS Sample Buffer (Invitrogen), and 1× NuPAGE™ Sample Reducing Agent (Invitrogen) was prepared and then denatured at 100 °C for 5 minutes and loaded on a pre-cast NuPAGE™ 4–12% bis-Tris protein gel (Invitrogen). SeeBlue™ Plus2 Prestained Standard was used to confirm the molecular weight of the CPMV proteins. We performed electrophoresis at 200 V and 120 mA for 35 minutes in 1 × MOPS SDS Running Buffer (Invitrogen). The gel was then stained using GelCode™ Blue Safe Protein Stain (Thermo Scientific). Images were obtained with a ProteinSimple FluorChem R imaging system.
Agarose gel electrophoresis was used to confirm colocalization of RNA and coat protein indicative of structural integrity. A sample of 15 μg of CPMV in KP and 1× Gel Loading Dye Purple (New England BioLabs) was prepared and analyzed on a 0.8% (w/v) agarose gel with 1× GelRed® Nucleic Acid Stain in 10 mM KP (10 mM KP was also used as the running buffer). Electrophoresis was carried out at 120 V and 400 mA for 30 minutes. The gel was imaged under UV light for RNA visualization, and after addition of GelCode™ Blue Safe Protein Stain (Thermo Scientific) under white light for coat protein visualization. Images were obtained with a ProteinSimple FluorChem R imaging system.
CPMV particle size was measured using dynamic light scattering (DLS) using a Malvern Instruments Zetasizer Nano at 25 °C. CPMV at 1 mg mL−1 in KP was loaded in a disposable single sealed cuvette (Eppendorf).
Particle size and integrity was further corroborated using fast protein liquid chromatography (FPLC) on the ÄKTA pure™ chromatography system (Cytiva). CPMV was prepared at 0.5 mg mL−1 in KP and analyzed using a Superose 6 Increase column at a flow rate of 0.5 mL min−1; RNA was detected at 260 nm and protein at 280 nm.
CPMV was visualized using transmission electron microscopy (TEM). 4 μL 0.5 mg mL−1 CPMV in PBS was applied to a glow-discharged carbon film with a 300-mesh Cu grid for 30 s; after blotting by filter paper, 4 μL Milli-Q water was applied to the grid for 30 s followed by filter paper blotting; lastly 4 μL 1% (w/v) uranyl acetate (Electron Microscopy Sciences) was applied onto the grid for 30 s; after another filter paper blotting, grid was air dried and imaged using a ThermoFisher Talos Transmission Electron Microscope at a nominal magnification of 120![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×.
000×.
Cell culture
B16F10 cells were cultured in DMEM with 4.5 g L−1 glucose and L-glutamine without sodium pyruvate (corning), heat-inactivated 10% (v/v) FBS, and 1% (v/v) Pen/Strep. ID8-Defb29/Vegf-A ovarian cancer cells were cultured in RPMI 1640 media with 2 mM L-glutamine, heat-inactivated 10% (v/v) FBS (VWR), 1% (v/v) Pen/Strep (Cytiva), 1 mM sodium pyruvate (ThermoFisher Scientific), and 0.05 mM β-mercaptoethanol (ThermoFisher Scientific).16 All cells were passaged and washed with trypsin-EDTA (Gibco) and phosphate-buffered saline (PBS) (corning), respectively. All cells were maintained in an incubator at 37 °C with 5% CO2.Mice
All animal studies were approved by the Animal Ethics Committee of the University of California, San Diego (UCSD) and in accordance with the Institutional Animal Care and Use Committee (IACUC) of UCSD. Female C57BL/6 mice (6–8 weeks old) from The Jackson Laboratory were used for the ID8-Defb29/Vegf-A and B16F10 studies. Animals were housed in groups of four with unlimited food and water.In vivo studies
Adherent ID8-Defb29/Vegf-A cells were harvested using Trypsin-EDTA and centrifuged at 400 × g/4 minutes/4 °C. Cells were then diluted in 1× PBS at a concentration of 10 × 106 cells per mL, and cells were injected in the peritoneal cavity (200 μL injection volume using a 28G1/2 insulin syringe; Exel International). Intraperitoneal (i.p.) treatment with CPMV, oxaliplatin, or a combination thereof was repeated 8 times weekly beginning on day 7 after tumor implantation. CPMV monotherapy was injected at a 1 mg mL−1 concentration and an injection volume of 200 μL. Oxaliplatin monotherapy was injected at a 0.5 mg mL−1 concentration and an injection volume of 200 μL. High dose CPMV combination treatment consisted of 100 μL CPMV at 2 mg mL−1 and 100 μL of oxaliplatin at 1 mg mL−1 administered i.p. simultaneously. Low dose CPMV combination treatment consisted of 100 μL CPMV at 1 mg mL−1 and 100 μL of oxaliplatin at 1 mg mL−1 administered i.p. simultaneously. Tumor growth was monitored by circumference (using a measuring tape around the abdomen) and body weight, and animals were euthanized once the circumference reached 9 cm, or the body weight reached 30 g.Adherent B16F10 cells were harvested using Trypsin-EDTA and centrifuged at 400× g/4 minutes/4 °C. Cells were then diluted in 1× PBS at a concentration of 6.7 × 106 cells per mL, and the mice received a 30 μL intradermal injection on the right flank using a 31G insulin syringe (Exel International). CPMV monotherapy was injected intratumorally (i.t.) at a 5 mg mL−1 concentration and an injection volume of 20 μL. High dose oxaliplatin monotherapy was injected i.p. at a 0.25 mg mL−1 concentration and an injection volume of 200 μL. Low dose oxaliplatin monotherapy was injected i.p. at a 0.125 mg mL−1 concentration and an injection volume of 200 μL. High dose oxaliplatin combination treatment consisted of 20 μL CPMV at 5 mg mL−1 i.t. and 200 μL of oxaliplatin at 0.25 mg mL−1 i.p. Low dose oxaliplatin combination treatment consisted of 20 μL CPMV at 5 mg mL−1 i.t. and 200 μL of oxaliplatin at 0.125 mg mL−1 i.p. CPMV was administered on days 7, 14, and 21, whereas oxaliplatin was administered on days 7, 10, 13, 16, 19, and 22. Tumor growth was monitored using calipers and the volume was calculated using V = (L × W2)/2, where V is tumor volume, L is tumor length (longer dimension), W is tumor width (shorter dimension). Animals were euthanized once the tumor volume exceeded 1000 mm3.
Immunofluorescence staining
B16F10 tumors were inoculated using methods described above. Animals received a single treatment of 100 μg CPMV i.t., 50 μg oxaliplatin i.p., combination, or PBS 7 days post tumor challenge; and tumors were collected 2 days post treatment. Tumors were snap frozen in liquid nitrogen and stored at −80 °C. The tumors were then submerged in Tissue-Tek™ O.C.T. Compound (Sakura), and cryosectioned using the Leica CM1860 cryostat. 10 μm sections were collected on Superfrost™ Plus Microscope Slides (Fisherbrand) and placed at −80 °C until further use.For immunofluorescence staining, O.C.T. was washed off by submerging the slides in 1× PBS for 5 minutes. Tumors were isolated using a hydrophobic ImmEdge™ Pen (Vector Laboratories) and fixed with 4% paraformaldehyde (PFA) diluted in 1× PBS for 10 minutes at room temperature (RT). To remove excess PFA, slides were then washed using 1× PBS for 5 minutes. The tumors that required permeabilization (i.e. for calreticulin, Ki67, and FoxP3 staining) were treated with 0.1% (v/v) Triton X-100 in 1× PBS for 20 minutes at RT. Then slides were washed 3 times followed by blocking using 1% (w/v) BSA in 1× PBS with 0.05% (v/v) Tween 20 (PBST) for 1 hour at RT. Primary antibodies were prepared in 1% (w/v) BSA in 1× PBST to block non-specific binding; staining was allowed to proceed overnight at 4 °C in a humidified staining chamber following 3 washing steps using 1× PBST. Primary antibodies dilutions were as follows: rat anti-mouse CD8α – 1:100 (Invitrogen Cat# 14-0081-85), rabbit anti-mouse CD4 – 1:200 (Abcam Cat# ab183685), rat anti-mouse FoxP3 – 1:200 (Invitrogen Cat# 14-5773-82), rabbit anti-mouse Ki67 – 1:250 (Invitrogen Cat# MA5-14520), rabbit anti-mouse HMGB1 – 1:100 (Invitrogen Cat# PA1-16926), rabbit anti-mouse calreticulin – 1:100 (Cell Signaling Cat# 62304S), rat anti-mouse F4/80 – 1:100 (Invitrogen Cat# MA5-16624), rabbit anti-mouse CD161c/NK1.1 – 1:100 (Bioss Cat# BS-4682R), and rat anti-mouse Ly-6G/Ly-6C – 1:100 (Invitrogen Cat#14-5931-82). Finally, secondary antibodies (diluted in 1× PBST with 1% BSA) were added for 1 hour at RT in the dark. Secondary antibody dilutions were as follows: Alexa Fluor™ 555 goat anti-rabbit IgG – 1:500 (Invitrogen Cat# A-21428) and Alexa Fluor™ 555 goat anti-rat IgG – 1:1000 (Invitrogen Cat# A-21434). Tumors were washed 3 times using 1× PBST in the dark; slides were dried and mounted using Fluoroshield™ with DAPI histology mounting media (Sigma-Aldrich) by incubation for 5–10 minutes at RT. The tissues were then covered with 12 mm circular cover glass (Electron Microscopy Sciences) and sealed with nail polish (Electron Microscopy Sciences). The slides were stored at 4 °C and imaged on the Nikon A1R Confocal/TIRF STORM confocal microscope.
TUNEL assay
Tumor inoculation, treatment, and collection was the same as for immunofluorescence staining established above. To visualize and quantify apoptotic cells in our tumor sections, we used the TUNEL (terminal deoxynucleotidyl transferase dUTP nick end labeling) Assay Kit - HRP-DAB (Abcam Cat#ab206386). We utilized reagents and protocols as per manufacturer's instructions. Sections were imaged using an ECHO Rebel microscope.Image analysis
Confocal fluorescence images were analyzed using the General Analysis module in the Nikon Elements software for image segmentation and quantification of intensity. Brightfield images were analyzed in MATLAB R2020b (Natick, Massachusetts USA). In brief, images were loaded into the software and transformed from the RGB to CIELAB color space. Thresholding was done in the lightness channel to remove background. In the green and magenta channel, values were thresholded to isolate contributions from green and brown pixels. No thresholding was done on the blue and yellow channel. The number of nonzero elements of the green and brown signals were determined.MATLAB
Image analysis MATLAB code:function [ratio, nnz_green, nnz_red, green_int, red_int] = cellrat(A)
% A must be a char string of the name and directory of the image file
% load image
I = imread(A);
% transform into color space LAB
I_color = rgb2lab(I);
% Look at first channel of color
I1 = I_color(:,:,1);
% Find indices that filter out yellow background
I1_prime = I1 < 85;
% Look at second channel of color
I2 = I_color(:,:,2);
% Apply indices that filter yellow background
I2_prime = I2(I1_prime);
% Split into green and red/brown sections
green = I2_prime < 0;
red = I2_prime ≥ 0;
% Find number of colored pixels and average intensity of each
nnz_green = nnz(green);
nnz_red = nnz(red);
end
Statistical analysis
All graphs and statistical analyses were made using GraphPad Prism version 10.0.0 for Windows (GraphPad Software, Boston, Massachusetts USA). For in vivo studies groups were randomly assigned with n = 8 for efficacy studies and n = 3 for imaging studies. Tumor growth curves were analyzed using two-way analysis of variance (ANOVA) followed by Tukey's multiple comparisons test. Survival curves were analyzed using the log-rank (Mantel–Cox) test. Violin plots were analyzed using the one-way ANOVA followed by Kruskal–Wallis multiple comparisons test. Asterisks on figures represent the following: **** = P ≤ 0.0001, *** = P ≤ 0.001, ** = P ≤ 0.01, * = P ≤ 0.05, ns = P > 0.05.Results and discussion
CPMV production and characterization
CPMV (Fig. 1A) was isolated from Vigna unguiculata (black-eyed pea No. 5) plant using our established methods. The purity and structural integrity of the viral nanoparticles were evaluated using a combination of gel electrophoresis, size exclusion chromatography using fast protein liquid chromatography (FPLC), and dynamic light scattering (DLS), and transmission electron microscopy (TEM). NuPAGE of the denatured CPMV samples confirmed the presence of a small and large coat protein corresponding to bands at 24 kDa and 42 kDa, respectively, Fig. 1B. Native electrophoresis using agarose gels confirmed that the RNA and protein co-migrated which indicates that intact CPMV was isolated, Fig. 1C and Fig. S1 (ESI†). FPLC analysis of CPMV confirmed elution from the Superose 6 Increase column at the expected volume (∼12–15 mL) with no signs of aggregation or broken particles; again RNA (at 260 nm) and protein (at 280 nm) co-eluted, Fig. 1D. Particle size was assessed using DLS which showed a monodisperse profile with an average hydrodynamic radius of ∼31 nm and a polydispersity index (PDI) of 0.035; consistent with the size of CPMV, Fig. 1E. Structural integrity of CPMV was visualized using TEM imaging, which confirmed the icosahedral symmetry of CPMV, Fig. 1F. All methods were consistent and did not indicate any detectable impurities.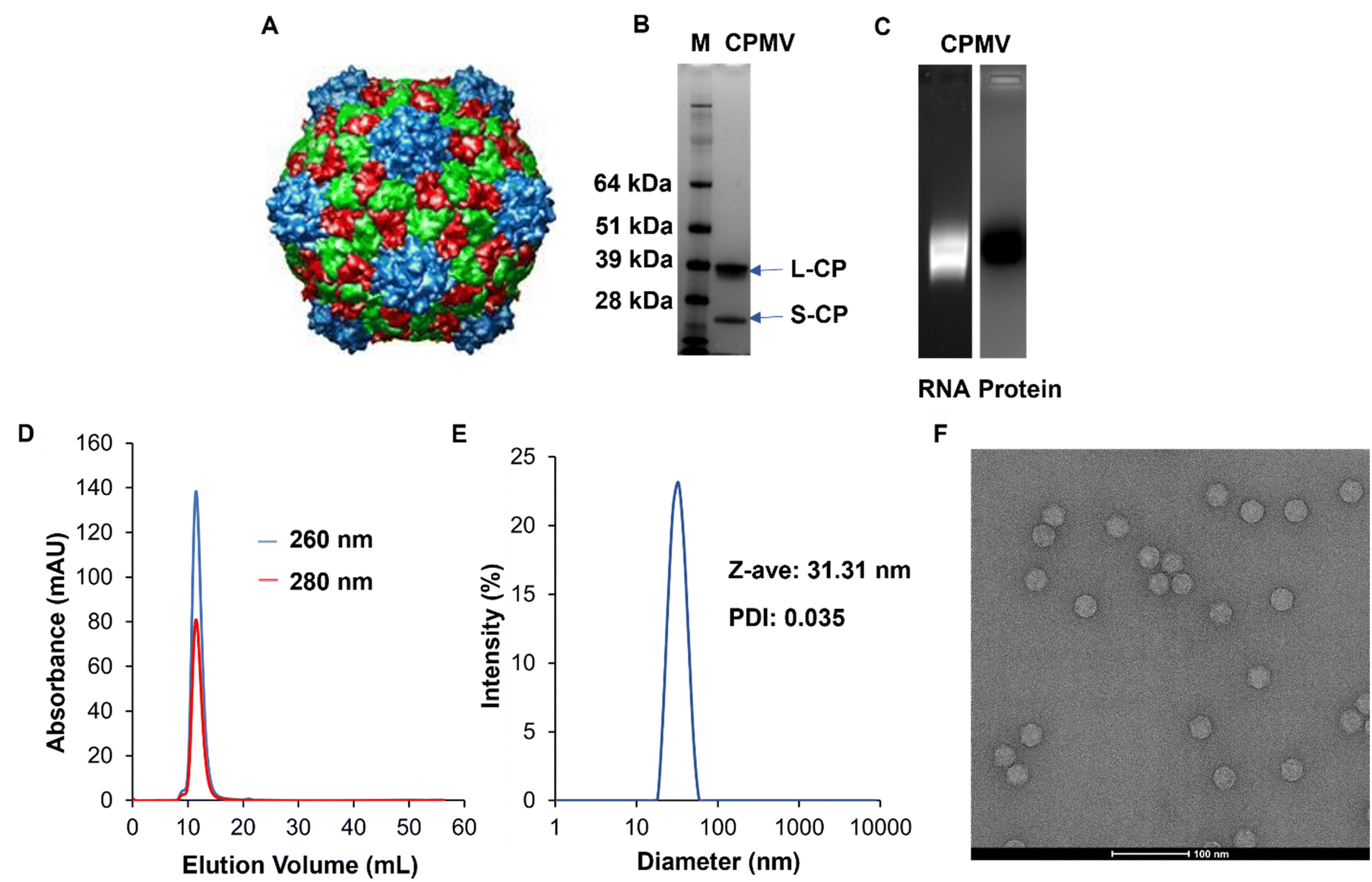 | ||
| Fig. 1 Characterization of CPMV nanoparticles. (A) UCSF Chimera image of CPMV viral capsid structure. The image was obtained from VIPERdb (https://viperdb.org) (B) NuPAGE showing small (24 kDa) and large (42 kDa) coat proteins of CPMV. Lane M: SeeBlue™ Plus2 Prestained Standard, lane 1: CPMV. (C) Agarose gel electrophoresis showing co-localization of CPMV RNA (lane 1, under UV light and with GelRed staining) and coat protein (lane 2, under white light after GelCode™ Blue Safe Protein staining). Complete gels are shown in Fig. S1 (ESI†). (D) FPLC of CPMV using Superose 6 Increase size exclusion column and Äkta purifier showing the typical elution profile of intact CPMV. (E) DLS of CPMV showing monodisperse distribution of particles, Z-ave = diameter, PDI = polydispersity index. (F) TEM of CPMV showing structural integrity, scale bar = 100 nm. | ||
CPMV + oxaliplatin against ID8-Defb29/Vegf-A ovarian cancer
Prior research from our lab showed efficacy of CPMV against intraperitoneal (i.p.) disseminated ovarian tumors from the ID8-Defb29/Vegf-A epithelial mouse cancer cell line,17,18 so we opted to implement the combination approach using this highly aggressive, metastatic tumor model. ID8-Defb29/Vegf-A cells are engineered to express vascular endothelial growth factor-A (Vegf-A) which promotes tumor angiogenesis by suppressing dendritic cells (DCs) and β-defensin-29 (Defb29) which recruits immature dendritic cells which further promote immune suppression.19 Co-expression of Defb29 and Vegf-A accelerates ID8 tumor development by polarizing DCs towards an endothelial cell-like phenotype.19 This suggests that the recruited DCs no longer operate as immune cells, but as endothelial cells capable of developing new blood vessels, resulting in aggressive disease.C57BL/6 mice received an i.p. injection of 2 × 106 ID8-Defb29/Vegf-A cells and 8 weekly i.p. treatments beginning on day 7 post tumor inoculation. While chemotherapeutic drugs are typically infused intravenously (i.v.) into human patients, repeated i.v. administration in mice can lead to collapsed veins. Therefore, we injected the oxaliplatin chemotherapy and CPMV immunotherapy i.p., which is considered safer in mice.20 Of note, CPMV was also administered i.p. as an “intratumoral” agent against the i.p. disseminated tumors (in contrast, intratumoral injections directly into the tumors were performed when studying the mice with dermal melanomas). The mice were randomly assigned to groups that received doses of 200 μg CPMV, 100 μg oxaliplatin (OxPt), a combination of 200 μg CPMV and 100 μg oxaliplatin (high dose CPMV combination), a combination of 100 μg CPMV and 100 μg oxaliplatin (low dose CPMV combination), or PBS (vehicle only), Fig. 2A. Tumor burden was monitored by circumference and body weight (not shown) measurements (increased circumference and body weight is a result of tumor growth and ascites development). As expected, CPMV delayed tumor growth; but free OxPt did not have significant impact on tumor burden when compared to PBS – it is noted that likely efficacy of solo OxPt therapy could be achieved by increased dosing. Nevertheless, the CPMV + OxPt combination therapy – independent of CPMV dose – showed increased potency compared to CPMV therapy alone. Differences became apparent 60 days post tumor challenge. The CPMV group started to reach endpoint on day 69 with an average circumference of 7.10 cm (PBS and OxPt started to reach endpoint on days 52 and 59, respectively), Fig. 2B and Fig. S2 (ESI†). In contrast, the combination therapy controlled tumor growth, and circumference was measured at ∼5.5 cm at this timepoint (for low and high dose CPMV + OxPt) – thus the circumference was up to 26.11% (P < 0.0001) reduced comparing the CPMV + OxPt combination vs. CPMV solo therapy. The combination therapy also resulted in increased median survival: the low and high dose CPMV + OxPt combination resulted in a median survival of 87.5 days or 83.5 days, respectively, while CPMV alone reached 74 days (OxPt reached only 59 days, and PBS 55.5 days median survival). Compared to the PBS control group, the CPMV + OxPt treatment increased median survival by up to 57.7% (P ≤ 0.05). It is of note, there was one animal in the PBS group in which tumor growth was not indicated for 90+ days; if this animal was considered an outlier, the statistical analysis results in an increased median survival of 59.1% with an overall significance of P < 0.0001 comparing the combination vs. PBS group, Fig. 2C.
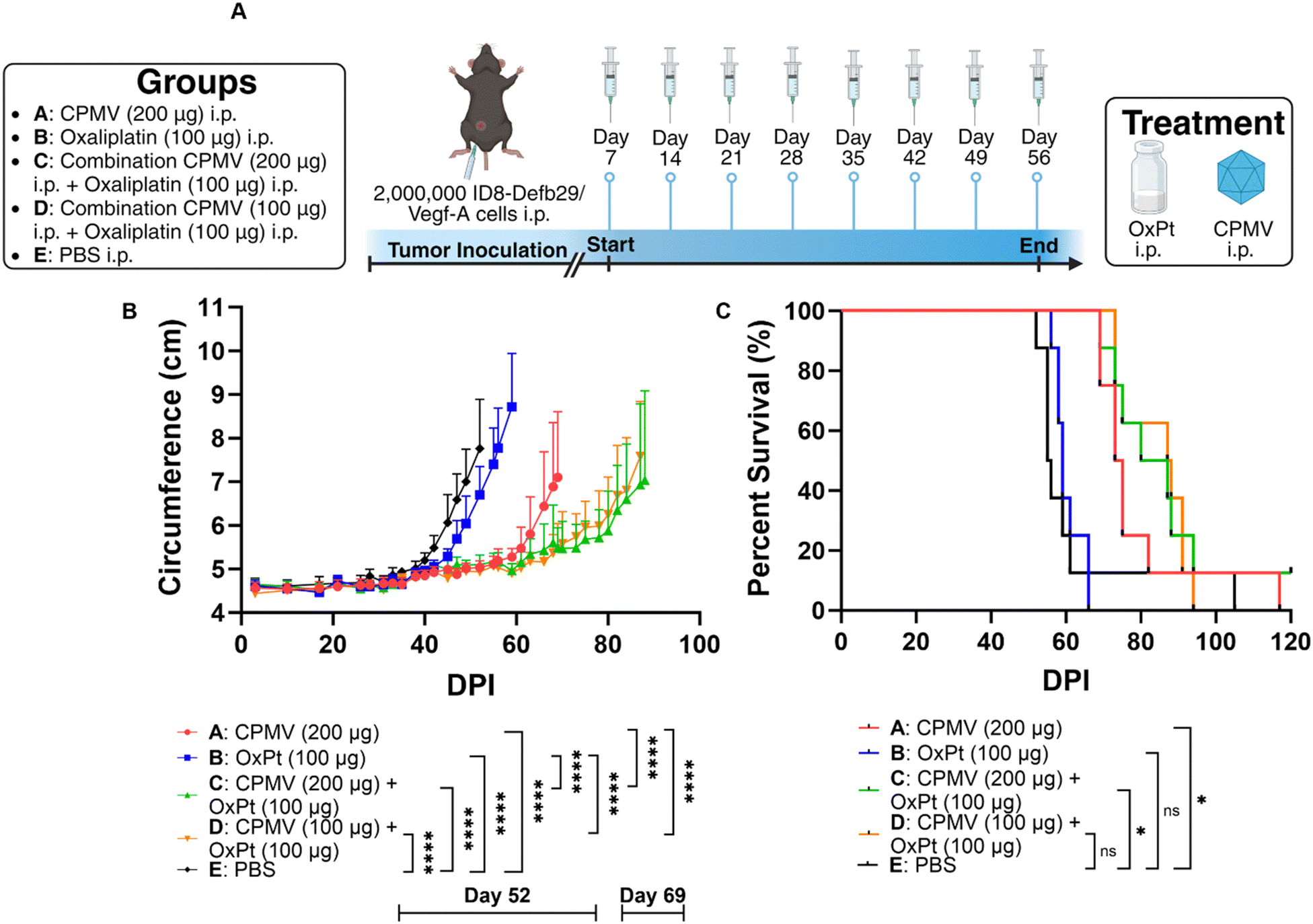 | ||
| Fig. 2 CPMV + OxPt combination therapy against i.p. disseminated ID8-Defb29/Vegf-A ovarian cancer in C57BL/6 mice (n = 8). (A) Treatment schedule: animals received 8 weekly i.p. treatments starting on day 7; solo CPMV and OxPt and CPMV + OxPt combination at two dose levels were compared; animals were monitored until they reached endpoint (defined as abdominal circumference > 9 cm; or body weight > 30 g). Created with BioRender.com. (B) Tumor burden as measured by abdominal circumference. Curves were terminated when the first animal of the corresponding group reached the endpoint. Statistical significance data on day 52 when the first animal in Group E reached endpoint, and day 69 when the first animal in Group A reached endpoint. Statistical analysis for growth curves using two-way ANOVA (**** = P ≤ 0.0001). (C) Survival data. Statistical analysis of survival curves using log-rank (Mantel–Cox) test (* = P ≤ 0.05, ns = P > 0.05). | ||
CPMV + oxaliplatin against B16F10 melanoma
Next, we tested efficacy against B16F10 melanoma – this model is considered aggressive and poorly immunogenic because the murine cell line does not express costimulatory molecules or major histocompatibility complex (MHC) making it highly adept at immune evasion.21 We have reported efficacy of CPMV against this tumor model.22,23We injected 200000 B16F10 cells in C57BL/6 mice intradermally and began treatment after 7 days. The subjects for this study were separated into groups that received 100 μg of CPMV i.t., 50 μg of oxaliplatin i.p., a combination of 100 μg of CPMV i.t. and 50 μg of oxaliplatin i.p. (high dose OxPt combination), 25 μg of oxaliplatin i.p., a combination of 100 μg of CPMV i.t. and 25 μg of oxaliplatin i.p. (low dose OxPt combination), or PBS i.t (vehicle only). CPMV and PBS were administered on days 7, 14, and 21, whereas oxaliplatin was administered on days 7, 10, 13, 16, 19, and 22, Fig. 3A. Preliminary data (not shown) using the B16F10 melanoma model showed that weekly oxaliplatin doses of 100 μg i.p. were poorly tolerated by the mice. Therefore, we optimized the treatment schedule by injecting lower doses more frequently to give the subjects time to adjust to the strong cytotoxic drug. Tumor burden was assessed as tumor volume using calipers. CPMV delayed tumor growth while OxPt had virtually no effect – again likely because we underdosed. While the high dose combination therapy was more potent than the CPMV monotherapy alone, the low dose combination therapy did not result in synergy. On day 30 post tumor inoculation, the CPMV group started to reach endpoint with a mean tumor volume of 323.38 mm3 (PBS, high dose OxPt, low dose OxPt, and low dose OxPt combination started to reach endpoint on days 16, 19, 19, and 26, respectively), Fig. 3B. At this timepoint, the high dose combination therapy had a much more pronounced anti-tumor effect with a mean tumor volume of 25.68 mm3, equivalent to a percent difference of 170.6% (P = 0.0031). The potency of the CPMV + high dose OxPt combination therapy was also reflected by significantly increased median survival of 48.5 days, while CPMV monotherapy reached 41 days (PBS, high dose OxPt, low dose OxPt, and low dose OxPt combination had a median survival of 18.5, 19.5, 22.5, and 29.5 days, respectively). In conclusion, the high dose oxaliplatin combination drastically increased median survival by 162.2%. with 50% of the animals achieving complete tumor clearance, Fig. 3C and Fig. S3 (ESI†).
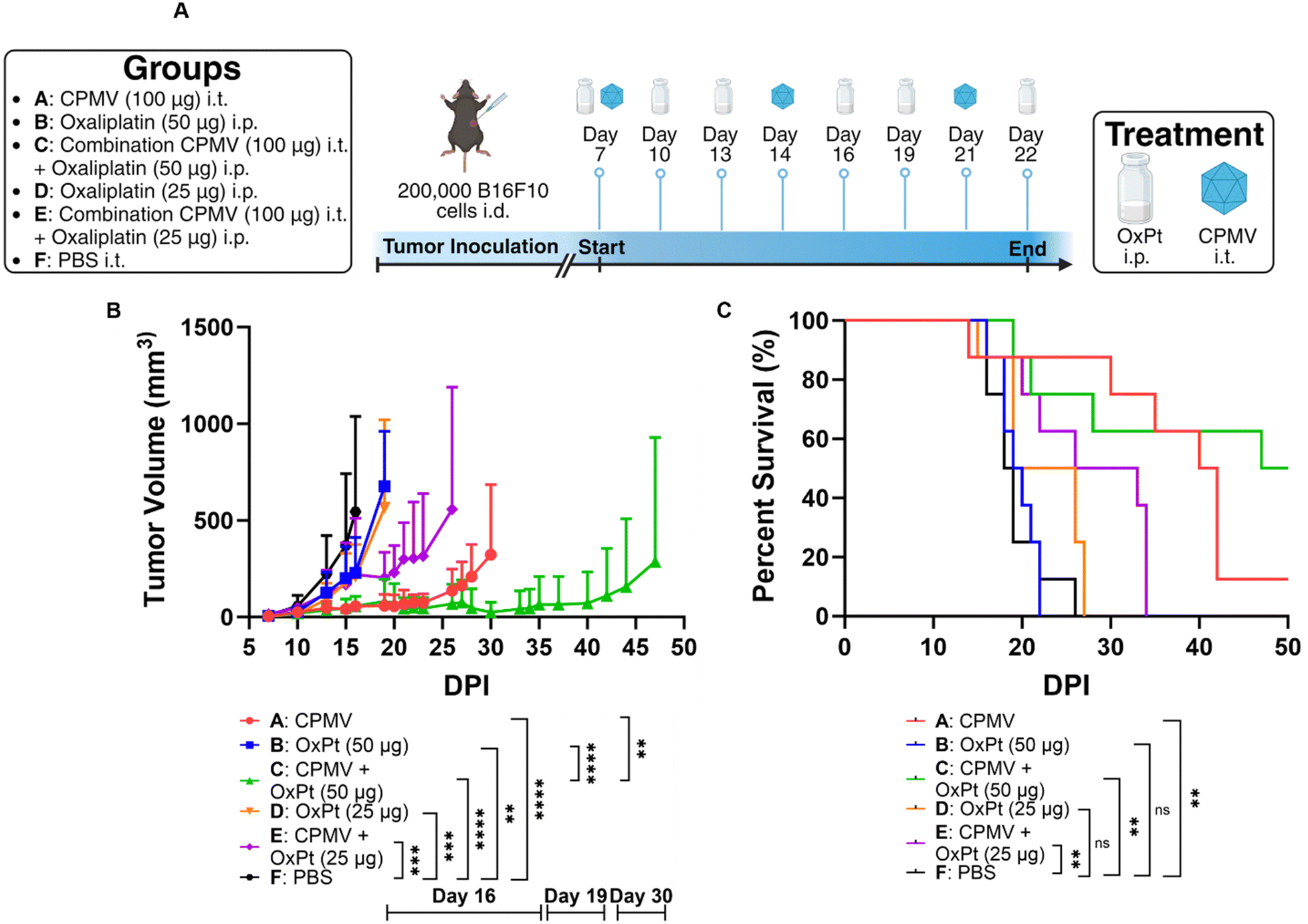 | ||
| Fig. 3 CPMV + OxPt combination therapy using the intradermal B16F10 melanoma model and C57BL/6 mice (n = 8). (A) Treatment schedule: animals received 3 weekly CPMV i.t. injections, OxPt was given i.p. every 3 days for 15 days; mono- and combination therapy was evaluated using two dose levels and treatment began on day 7 post tumor challenge. Tumor growth was monitored until the animals reached endpoint (defined as tumor volume > 1000 mm3). Created with BioRender.com. (B) Tumor burden as measured by tumor volume. Curves were terminated when the first animal of the corresponding group reached the endpoint. Statistical significance data on day 16 when the first animal in Group F reached endpoint, day 19 when the first animal in Group B reached endpoint, and day 30 when the first animal in Group A reached endpoint. Statistical analysis for growth curves using two-way ANOVA (**** = P ≤ 0.0001, *** = P ≤ 0.001, ** = P ≤ 0.01). (C) Survival data. Statistical analysis of survival curves using log-rank (Mantel–Cox) test (** = P ≤ 0.01, ns = P > 0.05). | ||
CPMV + oxaliplatin combination therapy stimulates the innate immune system
Given the potent efficacy observed using the B16F10 model, we utilized this tumor model for the subsequent immune mechanism studies using immunofluorescence staining and confocal microscopy of tumor sections. B16F10 dermal tumors were established and treated on day 7 post tumor inoculation; to analyze innate immune cell changes within the TME, tumors were collected 48 hours following a single treatment of 100 μg CPMV, 50 μg oxaliplatin, CPMV + OxPt combination, or PBS. For the innate immune cell panel, myeloid cells (Ly-6G/Ly-6C), natural killer (NK1.1) cells, and macrophages (F4/80) were stained – these leukocytes were previously indicated to play a role in CPMV-induced anti-tumor immunity.4 We analyzed 6–11 images per staining; quantitative data were generated by analyzing violin plots where each data point corresponds to the total intensity from a single image section per staining; Fig. 4 shows representative images and additional slides are shown in Fig. S4–S10 (ESI†).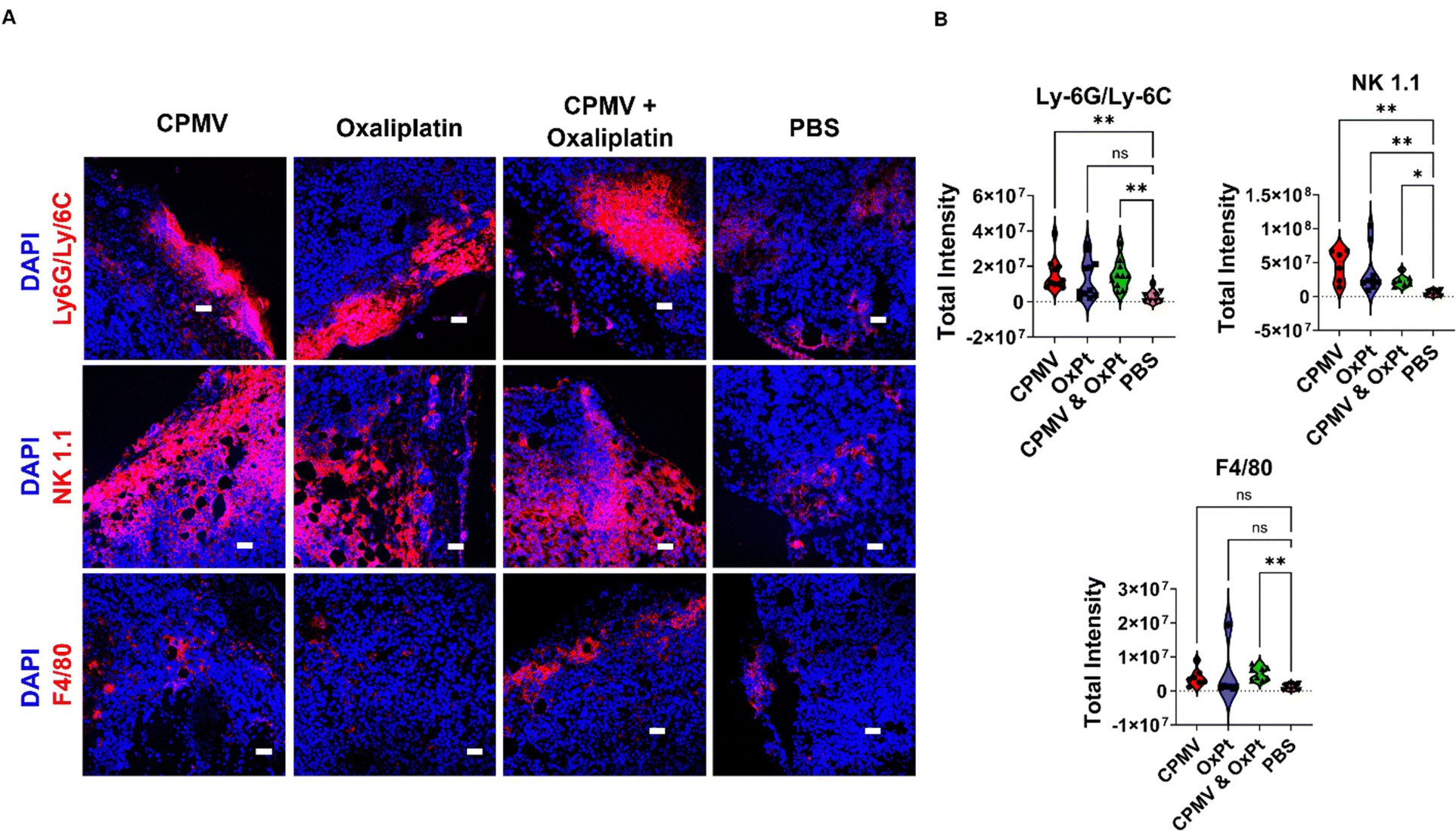 | ||
| Fig. 4 (A) Immunofluorescence imaging of innate immune cells in B16F10 tumor sections after single treatment with solo- or CPMV + OxPt combination therapy. Animals received one treatment on day 7 post tumor inoculation, and the tumors were collected 48 hours post treatment. The tumors were cryosectioned and stained with DAPI, anti-mouse Ly-6G/Ly-6C (myeloid cells), anti-mouse CD161c/NK1.1 (NK cells), and anti-mouse F4/80 (macrophages). The scale bar is 50 μm. (B) Quantification of confocal images from innate immunity panel (analysis was based on 6–11 slides per staining, each data point in the violin plots corresponds to the total intensity from an individual image). | ||
First, we used the anti-mouse Ly-6G/Ly-6C primary antibody to stain for myeloid cells and found that CPMV, OxPt, and the combination treatment, each led to a 6.3, 5.8, and 6.2-fold increase in myeloid cell infiltration into the tumor, respectively, Fig. 4 and Fig. S4 (ESI†). Ly-6G/Ly-6C is a broad differentiation marker present in Ly6G+ granulocytes, and Ly-6C+ monocytes/macrophages. The most abundant Ly-6G+ granulocyte in mice are neutrophils making up 20–30% of all leukocytes.24 Neutrophils are phagocytic cells that can either have pro-tumor or anti-tumor properties.25 Our prior research indicates that CPMV repolarizes tumor associated neutrophils (TANs) toward a cytotoxic N1 phenotype.1,22 These N1 neutrophils can kill tumor cells by secreting H2O2, upregulating pro-inflammatory/immunostimulatory cytokines, inducing antibody-dependent cellular cytotoxicity (ADCC), activating DCs, and operating as antigen presenting cells (APCs).26,27 Neutrophils thus are implicated in the mechanism of action of the CPMV intratumoral therapy.
Staining with the NK1.1 primary antibody revealed that the treatments also led to increased presence of NK cells with a 4.8-fold increase by the CPMV + OxPt combination treatment, however the combination led to a lesser degree of NK infiltration compared to the monotherapies (8.0-fold for CPMV and 9.7-fold for OxPt), Fig. 4 and Fig. S5 (ESI†). It is noted that while the median for CPMV and OxPt lies within the same range, NK infiltration for tumors treated with OxPt chemotherapy was more variable, with a few slides staining for a high degree of NK cells, while more frequent events showed a lesser degree of NK cell infiltration. Thus, data indicates that while CPMV is an effective NK cell recruiter, OxPt is less so and this may explain the reduced abundance, albeit increased presence vs. PBS, in the CPMV + OxPt combination group. In fact, we have previously shown that CPMV monotherapy leads to prominent recruitment of NK cells;28 and CPMV and NK agonist combination therapy leads to synergistic efficacy in preclinical tumor models. NK cells recognize aberrant cells following the downregulation of surface MHC-I or DNA damage from cellular stress.25 Upon recognition of cancerous cells, NK cells secrete cytotoxic granules, induce death receptor-mediated apoptosis, secrete cytokines and chemokines, recruit DCs and macrophages, and induce ADCC.13,29
Staining with the F4/80 macrophage marker indicated that the combination treatment significantly improved macrophage infiltration into the tumor with a 5.0-fold increase over PBS, Fig. 4 and Fig. S6 (ESI†). A similar trend was noted for the monotherapy groups (3.4-fold increase for CPMV and 5.6-fold increase for OxPt). Macrophages and DCs are APCs that interface with the adaptive immune system. Similar to neutrophils, macrophages can have an M1 or M2 phenotype depending on the TME, and prior research indicates CPMV induces a switch towards the M1 anti-tumor population.1 M1 macrophages can kill tumor cells by secreting reactive oxygen species (ROS) and nitric oxide (NO), or by participating in ADCC.30 They can also prime naive CD4+ T cells through MHC-II and costimulatory molecule surface expression.1
Overall, the combination as well as monotherapies led to innate immune cell infiltration, and given that the innate cells can become antigen presenting cells to prime anti-tumor T cells, we also stained slides for T cells. Fig. S7 and S8 (ESI†) indicate that CPMV monotherapy and CPMV + OxPt combination treatment, but less so the OxPt monotherapy, led to reduced abundance of immunosuppressive regulatory FoxP3+ Tregs (with a 7.6-fold decrease in the FoxP3 Treg marker for CPMV + OxPt, 5.3-fold decrease of CPMV, and 2.2-fold decrease for OxPt). Changes in CD4 helper T cells or CD8 cytotoxic T cells were not apparent, likely reflecting the early time point – tumors were collected and stained 48 hours post treatment, Fig. S7, S9 and S10 (ESI†).
The imaging data is consistent with CPMV as well as CPMV + OxPt intratumoral therapy being immunostimulatory which leads to priming a ‘hot’ TME characterized by infiltration of neutrophils, macrophages and NK cells. Similar to our approach, it has been shown that papaya mosaic virus (PapMV) induces antitumor immunity mainly through endocytosis by plasmacytoid dendritic cells (pDCs) which stimulates the production of interferon-alpha (IFN-α), likely through recognition of the ssRNA cargo by TLR7/8.31 Other viral nanoparticle treatments such as TLR9 agonist-loaded Qβ bacteriophage also owe their success to the stimulation of macrophages and DCs.32 Beyond the use of TLR agonists, other pathways are the STING pathway and nanoparticles and other biologics conjugated with STING have shown promise as anti-cancer immunotherapy as well.33,34 The remodeling of the TME by innate immune cells leads to cancer cell death and processing of tumor-associated antigens to prime systemic anti-tumor immunity.
CPMV + oxaliplatin combination efficiently kills cancer cells and induces immunogenic cell death
Next, we used imaging to analyze the tumor cell proliferative vs. apoptotic indices. Ki67 staining indicates a reduced proliferation of tumor cells in slices collected from animals receiving OxPt (8.3-fold) and CPMV + OxPt (4.1-fold), Fig. 5 and Fig. S11 (ESI†). TUNEL assay was used to visualize apoptotic cells highlighting that the treatments induced cell death with CPMV + OxPt > OxPt > CPMV. A 2.3-fold increase in the number of apoptotic cells over the PBS control was noted for the CPMV + OxPt groups (vs. 1.4-fold and 1.2-fold for the OxPt and CPMV groups) Fig. 5 and Fig. S12 (ESI†). Taken together, the imaging studies confirm that the combination of CPMV and oxaliplatin is a cytotoxic treatment regimen, and the cytotoxic activity can be attributed to OxPt activity. Of note, we do see a trend of increased number of apoptotic cells in tumor sections from the CPMV only treatment group (differences were not statistically significant). CPMV is not an oncolytic virus and there is no evidence that it induces cell death – however we have previously shown that CPMV recruits and activates myeloid cells that induce tumor cell death.1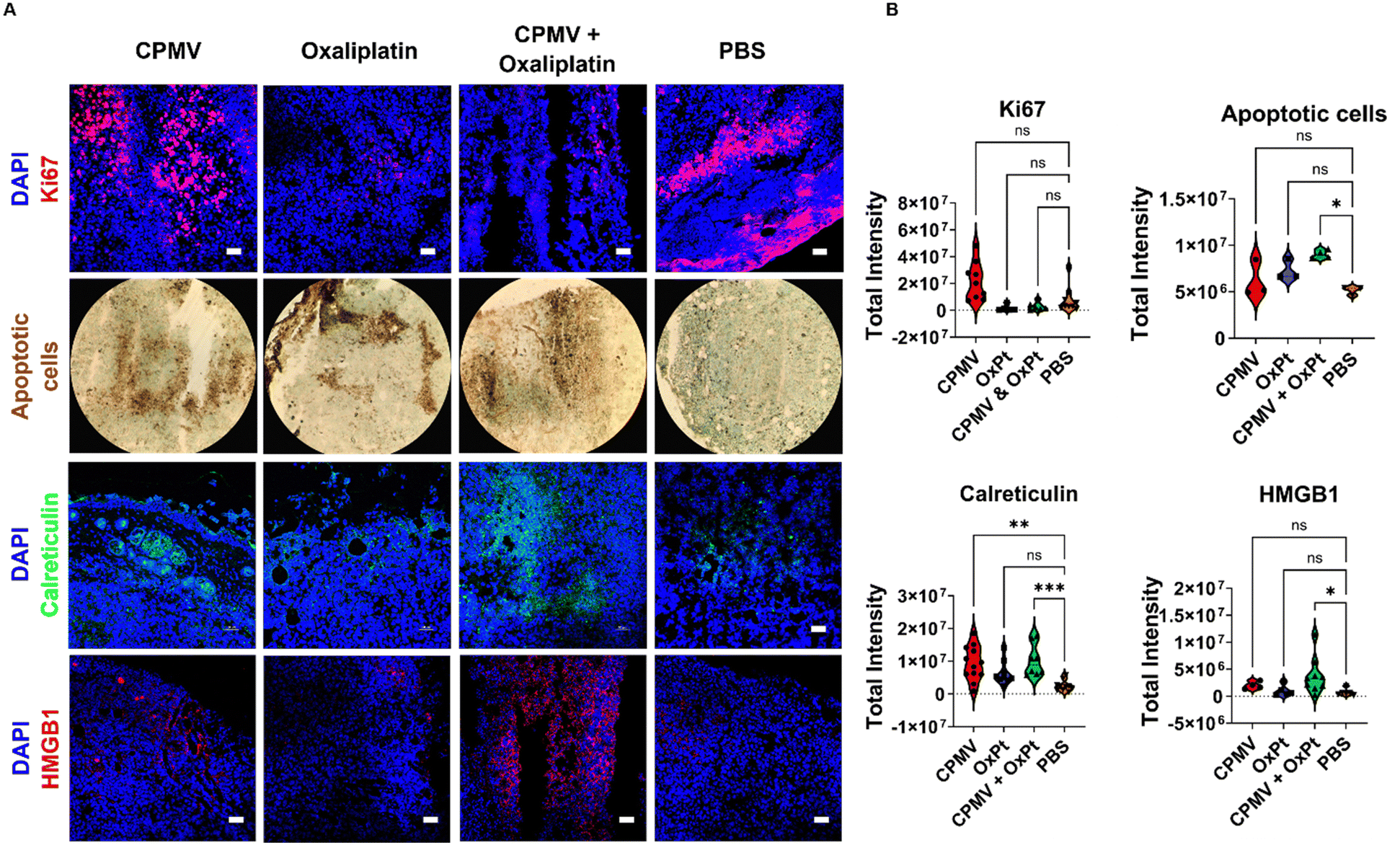 | ||
| Fig. 5 (A) Imaging cancer cell survival and immunogenic cell death. Animals received one treatment on day 7 post tumor inoculation, and the tumors were collected 48 hours post treatment. Tumor cryosections were stained with anti-mouse Ki67 (proliferation marker), anti-mouse calreticulin (DAMP), and anti-mouse HMGB1 (DAMP). The scale bar is 50 μm. Second row shows TUNEL assay of apoptotic nuclei imaged with a 10× magnification. (B) Quantification of confocal images with 3–12 images per staining, each data point in the violin plots corresponds to the total intensity from an individual image. | ||
Cell death can either be homeostatic or immunogenic.14 In the case of homeostatic cell death, the cell simply undergoes apoptosis with no immune stimulation. The more interesting scenario occurs when a cell undergoes immunogenic cell death (ICD) which triggers an innate immune response against the antigens that are released from the dying cell. In this process, DAMPs, i.e. expression of high-mobility group box 1 (HMGB1) and calreticulin, become beacons for innate immune cells. Calreticulin is an endoplasmic reticulin (ER) protein essential for cellular homeostasis and antigen presentation.35 Under regular physiological conditions, calreticulin has functions ranging from protein folding to MHC-I expression.35 In the case of ICD, calreticulin gets transported from the ER to the cell surface where it promotes phagocytosis by APCs.14,35 HMGB1 is a nuclear protein that regulates nucleosome structure, gene transcription and recombination, DNA repair, and telomere homeostasis.36 In the context of cancer development, HMGB1 promotes inflammation, accelerates cell metabolism, promotes metastasis, promotes angiogenesis, and inhibits immune cells.36 However, the passive secretion of HMGB1 as a result of ICD has proven to aid in immune cell recognition of dying cancerous cells. Extracellular HMGB1 gets recognized by TLR4 in DCs thus activating the cytotoxic adaptive immune response.13,14,36
The CPMV + OxPt led to significant staining of calreticulin and HMGB1 with a 3.3-fold and 8.1-fold increase, respectively; of note also the increase in calreticulin but to a lesser degree HMGB1 (4.6 and 1.9-fold, respectively) in tumor sections from animals receiving CPMV solo therapy. Changes for the OxPt monotherapy group were rather moderate, in line with the observed lack of efficacy in vivo, which may be explained by the low dose treatment regimen and analysis after single treatment14 (observed changes were a 2.3-fold increase in calreticulin and 1.2-fold increase in HMGB1 expression). Data are shown in Fig. 5 and Fig. S13, S14 (ESI†). Overall, this data is consistent with the combination therapy inducing ICD and may indicate the potential of CPMV to induce ICD. The analysis was performed at 48 hours post single treatment; while at this timepoint tumor cell killing was not evident by the CPMV monotherapy, the upregulation of calreticulin may be indicative of onset of apoptosis mediated by innate immune cells. We would expect to observe tumor cell death in tumors treated by CPMV after repeated treatments or later time points, mediated through immune cells (myeloid cells, NK cells, and macrophages) stimulated by CPMV.
Conclusion
The research delineates a combination therapy leveraging the immunogenic properties of plant virus-based nanoparticles, specifically cowpea mosaic virus (CPMV), and the immunostimulatory effects of immunogenic cell death (ICD) inducers, such as oxaliplatin. The synergistic interaction between pathogen-associated molecular patterns (PAMPs) from CPMV and damage-associated molecular patterns (DAMPs) from oxaliplatin-induced ICD was observed to markedly enhance anti-tumor efficacy. This dual-therapy regimen manifested pronounced therapeutic outcomes against multiple cancer models, indicating its potential as a broadly applicable treatment strategy in oncology.The regimen was first evaluated using the ID8-Defb29/Vegf-A murine ovarian cancer model, where CPMV alone had previously shown therapeutic promise. Oxaliplatin alone did not yield significant therapeutic benefit, whereas the CPMV and oxaliplatin combination significantly impeded tumor progression and extended median survival, illustrating a superior therapeutic index. The combination treatment achieved a substantial 44.4% reduction in tumor growth compared to controls (P < 0.0001) and a 57.7% increase in median survival. The scope of the combination therapy's efficacy was further assessed on B16F10 melanoma, a cutaneous tumor model. The results were consistent, with the combination therapy outperforming CPMV monotherapy in tumor volume reduction by 170.6% (P = 0.0031) and significantly elevating median survival by 162.2% relative to the control group. Moreover, the combination treatment culminated in complete tumor remission in 50% of the subjects. These findings underscore the clinical potential of this combination therapy as a robust, tumor-type-independent treatment option in cancer therapeutics.
Immunofluorescence staining revealed that CPMV and CPMV + OxPt therapy led to a significant augmentation in the infiltration of myeloid cells (likely neutrophils), NK cells, and macrophages – consistent with priming a ‘hot’ TME. When analyzing T cells, we also noted a reduction in CD4+ regulatory T cells (Tregs) post-treatment, further indicating a reversal of the immunosuppressive TME. This immunological shift is postulated to contribute to the enhanced anti-tumor response, promoting more efficient tumor cell eradication and clearance. CPMV + OxPt therapy and OxPt induced apoptosis which was consistent with reduced proliferative indices in these tumor sections. Lastly, DAMP staining provided further evidence, showing elevated expression levels of calreticulin and HMGB1, hallmarks of immunogenic cell death – this was observed in particular for the combination treatment. These immunological insights corroborated the observed in vivo efficacy highlighting the opportunity and potential of CPMV intratumoral immunotherapy combined with chemotherapy, such as oxaliplatin.
Data availability:
Data will be available from the authors upon reasonable request.Conflicts of interest
The authors declare the following competing financial interest(s): Dr Steinmetz is a co-founder of, has equity in, and has a financial interest with Mosaic ImmunoEnginering Inc. Dr Steinmetz is a co-founder of, and serves as manager of Pokometz Scientific LLC, under which she is a paid consultant to Flagship Labs 95 Inc. and Arana Biosciences Inc. The other authors declare no potential COI.Acknowledgements
This work was supported by NIH grants R01-CA253615 and R01-CA253615-02S1 Diversity Supplement as well the Shaughnessy Family Fund for Nano-ImmunoEngineering (nanoIE) at UCSD. A. A. C. was supported by NIFA 2022-67012-36698. The authors thank the UCSD Cancer Center Microscopy Shared Facility (Specialized Support Grant P30 CA23100-28) for providing equipment, services, and expertise to complete this work.References
- C. Wang, S. N. Fiering and N. F. Steinmetz, Cowpea Mosaic Virus Promotes Anti-Tumor Activity and Immune Memory in a Mouse Ovarian Tumor Model, Adv. Ther., 2019, 2, 1900003 CrossRef PubMed.
- S. Shukla, et al., The unique potency of Cowpea mosaic virus (CPMV) in situ cancer vaccine, Biomater. Sci., 2020, 8, 5489–5503 RSC.
- A. A. Murray, C. Wang, S. Fiering and N. F. Steinmetz, In Situ Vaccination with Cowpea vs. Tobacco Mosaic Virus against Melanoma, Mol. Pharm., 2018, 15, 3700–3716 CrossRef CAS PubMed.
- C. Wang, V. Beiss and N. F. Steinmetz, Cowpea Mosaic Virus Nanoparticles and Empty Virus-Like Particles Show Distinct but Overlapping Immunostimulatory Properties, J. Virol., 2019, 93(21) DOI:10.1128/jvi.00129-19.
- S. N. Lester and K. Li, Toll-Like Receptors in Antiviral Innate Immunity, J. Mol. Biol., 2014, 426, 1246–1264 CrossRef CAS PubMed.
- C. Mao, V. Beiss, J. Fields, N. F. Steinmetz and S. Fiering, Cowpea mosaic virus stimulates antitumor immunity through recognition by multiple MYD88-dependent toll-like receptors, Biomaterials, 2021, 275, 120914 CrossRef CAS PubMed.
- C. Mao, et al., In situ vaccination with cowpea mosaic virus elicits systemic antitumor immunity and potentiates immune checkpoint blockade, J. Immunother. Cancer, 2022, 10, e005834 CrossRef PubMed.
- B. J. Laidlaw, J. E. Craft and S. M. Kaech, The multifaceted role of CD4+ T cells in CD8+ T cell memory, Nat. Rev. Immunol., 2016, 16, 102–111 CrossRef CAS PubMed.
- K. E. A. Duval, et al., Cowpea Mosaic Virus Nanoparticle Enhancement of Hypofractionated Radiation in a B16 Murine Melanoma Model, Front. Oncol., 2020, 10, 594614 CrossRef PubMed.
- H. Cai, C. Wang, S. Shukla and N. F. Steinmetz, Cowpea Mosaic Virus Immunotherapy Combined with Cyclophosphamide Reduces Breast Cancer Tumor Burden and Inhibits Lung Metastasis, Adv. Sci., 2019, 6, 1802281 CrossRef PubMed.
- S. Faivre, D. Chan, R. Salinas, B. Woynarowska and J. M. Woynarowski, DNA strand breaks and apoptosis induced by oxaliplatin in cancer cells, Biochem. Pharmacol., 2003, 66, 225–237 CrossRef CAS PubMed.
- T. Alcindor and N. Beauger, Oxaliplatin: a review in the era of molecularly targeted therapy, Curr. Oncol., 2011, 18, 18–25 CrossRef CAS PubMed.
- S.-R. Woo, L. Corrales and T. F. Gajewski, Innate Immune Recognition of Cancer, Annu. Rev. Immunol., 2015, 33, 445–474 CrossRef CAS PubMed.
- G. Kroemer, L. Galluzzi, O. Kepp and L. Zitvogel, Immunogenic Cell Death in Cancer Therapy, Annu. Rev. Immunol., 2013, 31, 51–72 CrossRef CAS PubMed.
- A. A. Murray, M. R. Sheen, F. A. Veliz, S. N. Fiering and N. F. Steinmetz, In Situ Vaccination of Tumors Using Plant Viral Nanoparticles, Methods Mol. Biol., 2019, 2000, 111–124 CrossRef CAS PubMed.
- R. Patel, A. E. Czapar, S. Fiering, N. L. Oleinick and N. F. Steinmetz, Radiation Therapy Combined with Cowpea Mosaic Virus Nanoparticle in Situ Vaccination Initiates Immune-Mediated Tumor Regression, ACS Omega, 2018, 3, 3702–3707 CrossRef CAS PubMed.
- C. Wang and N. F. Steinmetz, A Combination of Cowpea Mosaic Virus and Immune Checkpoint Therapy Synergistically Improves Therapeutic Efficacy in Three Tumor Models, Adv. Funct. Mater., 2020, 30, 2002299 CrossRef CAS PubMed.
- S. Shukla, C. Wang, V. Beiss and N. F. Steinmetz, Antibody Response against Cowpea Mosaic Viral Nanoparticles Improves In Situ Vaccine Efficacy in Ovarian Cancer, ACS Nano, 2020, 14, 2994–3003 CrossRef CAS PubMed.
- J. R. Conejo-Garcia, et al., Tumor-infiltrating dendritic cell precursors recruited by a β-defensin contribute to vasculogenesis under the influence of Vegf-A, Nat. Med., 2004, 10, 950–958 CrossRef CAS PubMed.
- J. N. Davis, C. L. Courtney, H. Superak and D. K. Taylor, Behavioral, clinical and pathological effects of multiple daily intraperitoneal injections on female mice, Lab. Anim., 2014, 43, 131–139 CrossRef PubMed.
- J. Wang, S. Saffold, X. Cao, J. Krauss and W. Chen, Eliciting T Cell Immunity Against Poorly Immunogenic Tumors by Immunization with Dendritic Cell-Tumor Fusion Vaccines1, J. Immunol., 1998, 161, 5516–5524 CrossRef CAS.
- P. H. Lizotte, et al., In situ vaccination with cowpea mosaic virus nanoparticles suppresses metastatic cancer, Nat. Nanotechnol., 2016, 11, 295–303 CrossRef CAS PubMed.
- P. L. Chariou, V. Beiss, Y. Ma and N. F. Steinmetz, In situ vaccine application of inactivated CPMV nanoparticles for cancer immunotherapy, Mater. Adv., 2021, 2, 1644–1656 RSC.
- K. E. O’Connell, et al., Practical Murine Hematopathology: A Comparative Review and Implications for Research, Comp. Med., 2015, 65, 96–113 Search PubMed.
- L. Maiorino, J. Daßler-Plenker, L. Sun and M. Egeblad, Innate Immunity and Cancer Pathophysiology, Annu. Rev. Pathol.: Mech. Dis., 2022, 17, 425–457 CrossRef CAS PubMed.
- C. Furumaya, P. Martinez-Sanz, P. Bouti, T. W. Kuijpers and H. L. Matlung, Plasticity in Pro- and Anti-tumor Activity of Neutrophils: Shifting the Balance, Front. Immunol., 2020, 11, 2100 CrossRef CAS PubMed.
- X. Wang, L. Qiu, Z. Li, X.-Y. Wang and H. Yi, Understanding the Multifaceted Role of Neutrophils in Cancer and Autoimmune Diseases, Front. Immunol., 2018, 9, 2456 CrossRef PubMed.
- E. C. Koellhoffer and N. F. Steinmetz, Cowpea Mosaic Virus and Natural Killer Cell Agonism for In Situ Cancer Vaccination, Nano Lett., 2022, 22, 5348–5356 CrossRef CAS PubMed.
- M. Cheng, Y. Chen, W. Xiao, R. Sun and Z. Tian, NK cell-based immunotherapy for malignant diseases, Cell. Mol. Immunol., 2013, 10, 230–252 CrossRef CAS PubMed.
- Y. Pan, Y. Yu, X. Wang and T. Zhang, Tumor-Associated Macrophages in Tumor Immunity, Front. Immunol., 2020, 11, 583084 CrossRef CAS PubMed.
- M.-È. Lebel, et al., Potentiating Cancer Immunotherapy Using Papaya Mosaic Virus-Derived Nanoparticles, Nano Lett., 2016, 16, 1826–1832 CrossRef CAS PubMed.
- C. D. Lemke-Miltner, et al., Antibody opsonization of a TLR9-agonist-containing virus-like particle enhances in situ immunization, J. Immunol., 2020, 204, 1386–1394 CrossRef CAS PubMed.
- Y. Wu, et al., Tumor-targeted delivery of a STING agonist improves cancer immunotherapy, Proc. Natl. Acad. Sci., 2022, 119, e2214278119 CrossRef CAS PubMed.
- P. A. Bielecki, et al., Immunostimulatory silica nanoparticle boosts innate immunity in brain tumors, Nanoscale Horiz., 2021, 6, 156–167 RSC.
- J. Fucikova, R. Spisek, G. Kroemer and L. Galluzzi, Calreticulin and cancer, Cell Res., 2021, 31, 5–16 CrossRef CAS PubMed.
- R. Kang, Q. Zhang, H. J. Zeh, M. T. Lotze and D. Tang, HMGB1 in Cancer: Good, Bad, or Both?, Clin. Cancer Res., 2013, 19, 4046–4057 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Complete gel images, tumor growth curves of individual mice, confocal image panels. See DOI: https://doi.org/10.1039/d4ma00427b |
| This journal is © The Royal Society of Chemistry 2024 |