
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stimuli-responsive synthetic helical polymers
María
Lago-Silva
,
Manuel
Fernández-Míguez
 ,
Rafael
Rodríguez
,
Emilio
Quiñoá
and
Félix
Freire
*
,
Rafael
Rodríguez
,
Emilio
Quiñoá
and
Félix
Freire
*
Centro Singular de Investigación en Química Biolóxica e Materiais Moleculares (CiQUS) and Departamento de Química Orgánica, Universidade de Santiago de Compostela, E-15782 Santiago de Compostela, Spain. E-mail: felix.freire@usc.es
First published on 18th December 2023
Abstract
Synthetic dynamic helical polymers (supramolecular and covalent) and foldamers share the helix as a structural motif. Although the materials are different, these systems also share many structural properties, such as helix induction or conformational communication mechanisms. The introduction of stimuli responsive building blocks or monomer repeating units in these materials triggers conformational or structural changes, due to the presence/absence of the external stimulus, which are transmitted to the helix resulting in different effects, such as assymetry amplification, helix inversion or even changes in the helical scaffold (elongation, J/H helical aggregates). In this review, we show through selected examples how different stimuli (e.g., temperature, solvents, cations, anions, redox, chiral additives, pH or light) can alter the helical structures of dynamic helical polymers (covalent and supramolecular) and foldamers acting on the conformational composition or molecular structure of their components, which is also transmitted to the macromolecular helical structure.
 From left to right: María Lago, Rafael Rodríguez, Manuel Fernández-Míguez, Félix Freire, Emilio Quiñoá | María Lago studied a BSc in Chemistry and a MSc with a specialization in Organic Chemistry at the University of Vigo (Master's project with Professor M. M. Cid). She then joined the Group of Prof. Félix Freire and Emilio Quiñoá in 2019 as a PhD student at the CiQUS-University of Santiago de Compostela, working on the study of helical structure control mechanisms. In 2022, she enjoyed a short stay at the University of Southampton (UK) under the supervision of Professor Stephen M. Goldup, working on the synthesis of mechanically chiral interlocked structures. Manuel Fernández Míguez pursued his undergraduate studies in Chemistry (BSc) and Chemistry at the Interface with Biology and Materials Science (MSc) at the Universidad de Santiago de Compostela. He is currently a PhD candidate in the laboratory of Professors Félix Freire and Emilio Quiñoá at the CiQUS-Universidad de Santiago de Compostela. Rafael Rodríguez received his MSc and PhD at the University of Santiago de Compostela/CiQUS (Spain) working on the structural elucidation and applications of stimuli responsive helical polymers—awarded as best thesis in science in 2018—under the supervision of Prof. Félix Freire and Prof. Emilio Quiñoá. He performed two postdoctoral stays at Kanazawa University (Japan) and the Institut des Sciences Chimiques de Rennes (University of Rennes 1, France) in the Research groups of Prof. Katsuhiro Maeda and Dr Jeanne Crassous to work in chiral materials with CPL activity. In 2021, he was awarded with a Juan de la Cierva Incorporación fellowship to move back to University of Santiago de Compostela/CiQUS (Spain) to develop chiral materials with switchable CPL activity. Recently, he was awarded with a prestigious Ramón y Cajal contract. Emilio Quiñoá obtained his PhD in Chemistry from the University of Santiago de Compostela (USC) on the topic of bioactive marine natural products. After postdoctoral research at the University of California, Santa Cruz (UCSC), he became an Associate Professor (1989) and later a Full Professor of Organic Chemistry (1998) at USC. His research then addressed the development of methods for the assignment of absolute configurations by NMR spectroscopy among other interests. He has coauthored more than 160 scientific publications, several textbooks and patent applications and has received several research awards. In 2010 he joined the Center for Research in Biological Chemistry and Molecular Materials (CiQUS) at USC. His current research interests focus on the study of dynamic helical polymers and their applications in nanotechnology. Félix Freire obtained his BSc (2000), MSc (2002), and PhD (2005) in Chemistry from the University of Santiago de Compostela (USC). In 2005 he did a postdoctoral stay in the group of Prof. Jesús Jiménez Barbero at CSIC-Madrid, where he worked on NMR studies of biomolecules. During 2006–2008 he was a Fulbright Postdoctoral Fellow in the group of Samuel H. Gellman, studying the folding of parallel β-sheets in water. Since 2009 he has worked at the University of Santiago, first as a Ramón y Cajal Researcher and now as Full Professor of Organic Chemistry (2023). His research interests include stimuli-responsive polymers, molecular self-assembly, and chiral polymer particles. |
1. Introduction
Non-natural helical polymers including foldamers,1–17 supramolecular5,18–38 and covalent helical polymers5,39–60 have been extensively studied during the last decades due to the possibility of creating a large variability of helical scaffolds with different functionalities by rational design. These materials emerged as consequence of the structure/function relationship found in biomacromolecules such as DNA, peptides or polysaccharides, whose helical scaffolds are directly related to their important biological functions such as signalling, storage, duplication, amplification, and processing of information.61–63 However, while the building blocks used by nature are limited, chemistry offers a wide range of chemical structures with different functionalities limited mainly by the creativity of the scientist.64–70 In this line, synthetic helical polymers with stimuli-responsive properties have been extensively studied due to the possibility of using an external stimulus (input) to produce a structural change, such as helix inversion (output), in the polymer. In this review we will focus our attention on those synthetic helical structures (covalent polymers, supramolecular polymers and foldamers) whose helical scaffold (sense and elongation) can be altered if an external stimulus triggers a rational mechanism of information transmission through changes in molecular conformations.71To create a synthetic stimuli-responsive helix by design, the starting material must meet certain requirements, some of which are discussed below.
1.1 Foldamers
In helical foldamers1–17 (Fig. 1a), the building blocks must preferably adopt a twisted conformation that propagates towards the formation of a helical structure when an oligomer is formed.70–74 A problem found in the preparation of stimuli-responsive foldamers is that if the conformation adopted by the building block is too stable, the foldamer will behave as quasi-static, making it difficult to tune its helical sense. This effect is the most abundant when a foldamer is formed by chiral residues, such as biotic foldamers by β- and/or γ-amino acids. In these systems, a positive or negative torsion of the chiral residue is favoured due to its intrinsic chirality, being difficult to reorient it in the opposite direction. To avoid this problem, the foldamer must be constituted mainly by achiral residues,75–79 such as aminoisobutyric acid (Aib) (Fig. 1b), employed in the preparation of stimuli-responsive biotic foldamers where the two P (plus) and M (minus) twisted conformations are equally favoured. To induce a helical sense in the foldamer, two approaches are usually followed, such as supramolecular or covalent interactions with a chiral fragment at one end of the foldamer helix described by the groups of Clayden and Inai (Fig. 1c–e).80–85 In these foldamers, the conformational preference adopted by the chiral residue is transmitted to the adjacent achiral residue by adoption of a preferred P or M twisted conformer and propagated to the other achiral residues through a screw sense induction mechanism. Variations in the conformational composition of the chiral residue, or the replacement of the chiral residue by its enantiomer leads to the induction of the opposite helical sense, generating a dynamic helical foldamers.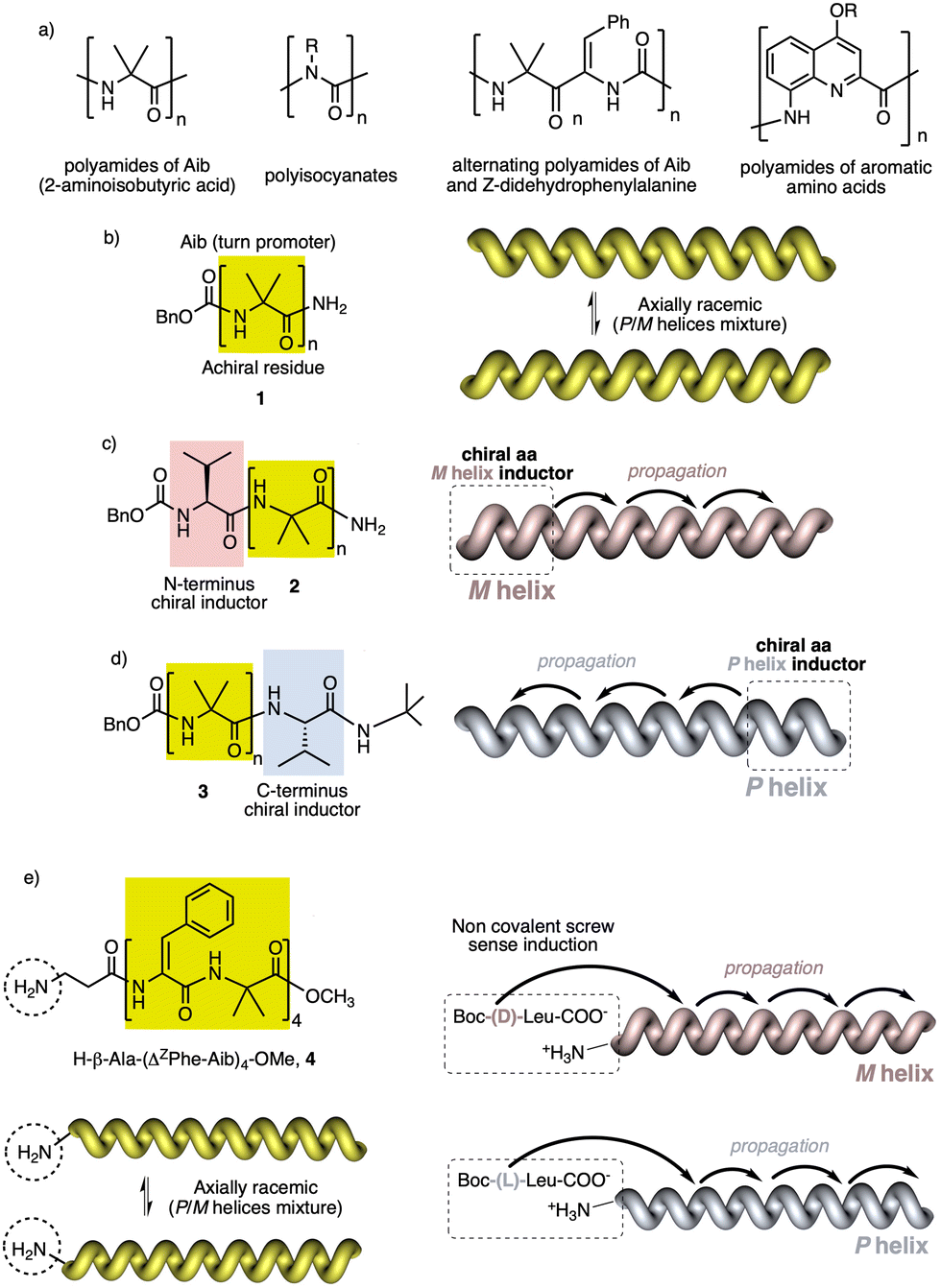 | ||
| Fig. 1 (a) Some achiral oligomer families forming axially racemic helices. Schematic illustration of (b) an axially racemic oligoamide of Aib and its (c) M or (d) P screw sense induction by introducing a chiral (L)-amino acid at the N- and C-terminus respectively. (e) Schematic illustration of the non-covalent screw sense induction of the axially racemic foldamer H-β-Ala-(ΔZPhe-Aib)4-OMe by interaction with chiral acids. | ||
In other cases, the chiral residue can be introduced in the middle of the foldamer sequence86 or encapsulated by the helical scaffold,13,87 although the mechanism of screw sense induction is produced mostly by fixation of a conformation in one of the residues of the foldamer backbone which is then transmitted to the rest of the oligomer that adopts a preferred P or M screw sense excess.
1.2 Dynamic helical polymers
In stimuli responsive covalent helical polymers, helix induction mechanisms are different from those observed in foldamers. In general, in these polymers, the main chain or polymer backbone is achiral, e.g., polyacetylenes and their derivatives,39–57 polyisocyanides,58,59 polysilanes88–90 or poly(quinoxaline-2,3-diyl)s (PQXs)53,91–97 (Fig. 2a), which adopt a preferred helical sense due to the conformational composition adopted by the pendant group. Thus, if the pendant is chiral, e.g., poly-5, a poly(phenylacetylene) (PPA) that bears the benzamide of (S)-phenylglycine methyl ester,98 the presence of a preferred conformer at the chiral centre results in an induced helical sense in the polymer main chain.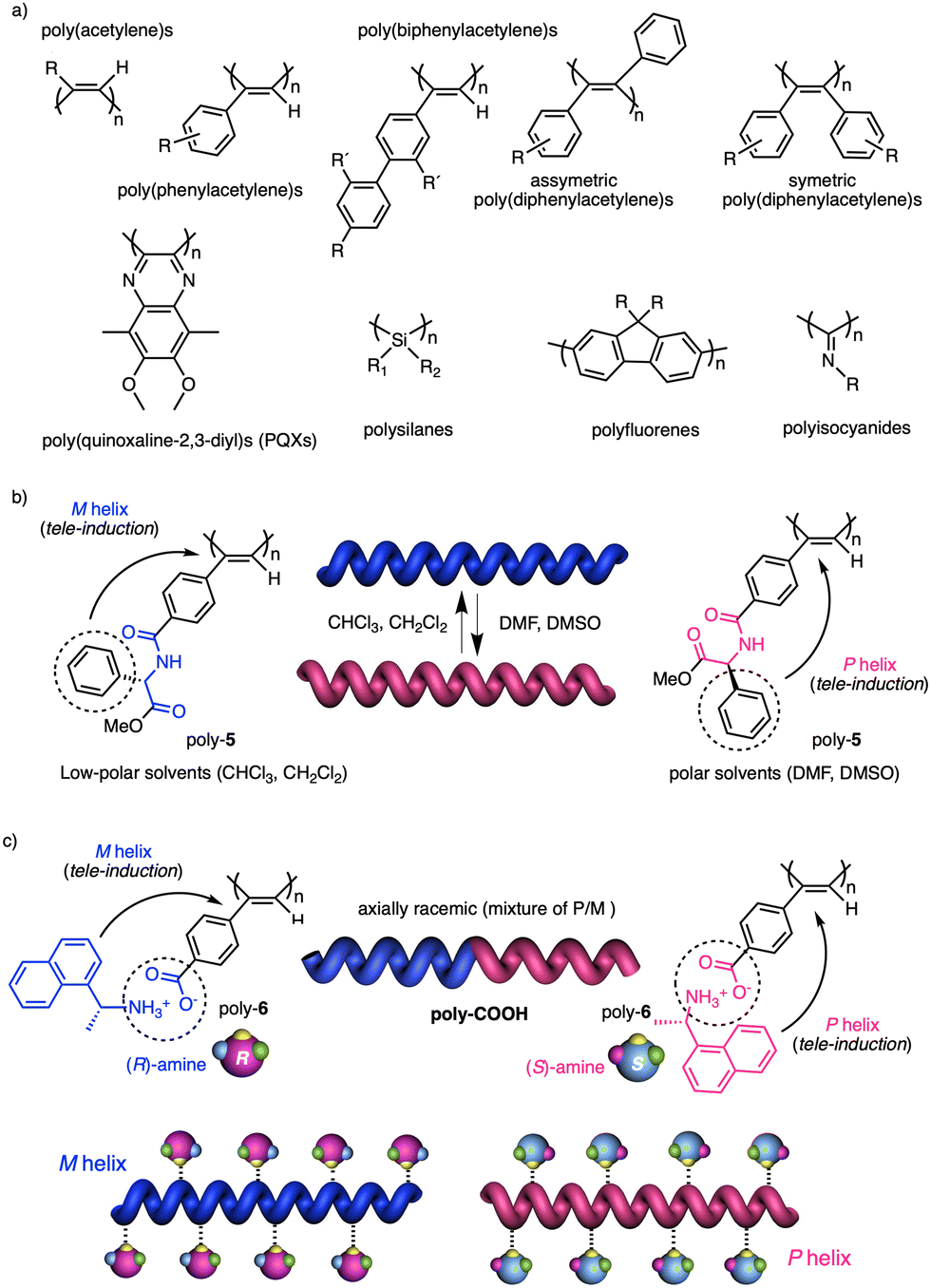 | ||
| Fig. 2 (a) Some families of dynamic helical polymers. (b) Helix inversion of a chiral poly(phenylacetylene) bearing 4-benzamide of (S)-phenylglycine methyl ester as pendant group by tuning its conformational composition using solvent polarity as external stimulus. (c) Chiral amplification of an achiral poly(phenylacetylene) bearing 4-benzoic acid as pendant using chiral amines as external stimuli. | ||
Variations in the conformational composition of the pendant group by the presence of external stimuli, such as solvent polarity or the addition of metal ions, can result in either helical sense enhancement or helical inversion effect (Fig. 2b). On the other hand, if the helical polymer is achiral, e.g., a PPA bearing a benzoic group as pendant (poly-6),99,100 a phenomenon of asymmetry amplification (helical induction) arises from interactions between the polymer and external chiral molecules, such as chiral amines used as external stimuli.
The most frequently helix induction mechanism that governs the screw sense excess adopted by chiral and achiral polymers is tele-induction.101 In this mechanism, changes in the spatial distribution of substituents in the chiral centres or in the species associated with the pendant groups result in stimuli being transmitted through space to the polymer backbones, which adopt specific conformations that generate P or M helical structures (Fig. 2b and c).
Interestingly, other helix induction mechanisms operating from the pendants to the main chains of polymers have been explored in recent years.
1.2.1.1 Chiral harvesting. It is based on the introduction of a chiral source at a remote position separated from the backbone by achiral spacers. In such a case, the information transmission mechanism can follow two different paths. The induction of: (i) a conformational change in the achiral spacer,102 or (ii) a tilting degree in the supramolecular arrangement of the achiral spacers along the helix.103 Both mechanisms are triggered by changes in the conformational composition of a chiral group placed in a remote position, which are transmitted to the achiral spacer and further harvested by the polymer backbone that adopts a P or an M screw sense excess.
Two illustrative examples are poly-7 and poly-8, PPAs that bear an achiral and flexible Aib residue102 and a planar and rigid oligo(phenylene-ethynylene) (OPE)103 as spacers (Fig. 3a and b, respectively).
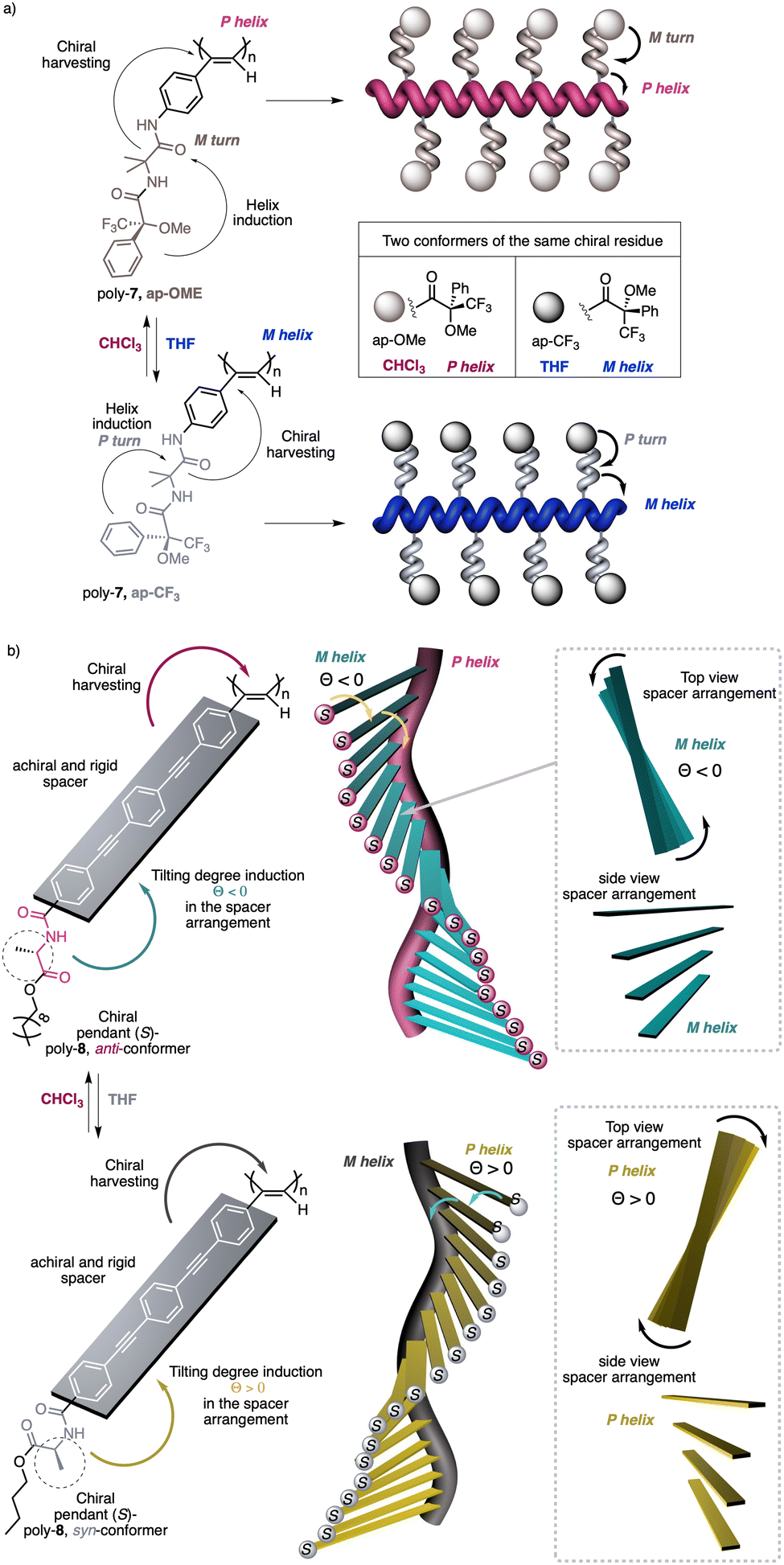 | ||
| Fig. 3 Screw sense induction of helical polymers through chiral harvesting using (a) flexible and (b) rigid achiral spacers. | ||
1.2.1.2 Chiral overpass. Usually, when the pendant of a helical polymer is composed by two or more chiral centres, the one closest to the backbone of the polymer is more likely to control its handedness, while the other centre has low-to-null impact on the structural features of the helical structure.104 Interestingly, the possibility of playing with the conformational composition of these multi-chiral side chains makes possible to activate a chiral-overpass effect.105,106 For example, in poly-9, which bears the anilide of (S)-alanine-(R)-methoxyphenylacetate—(S)-Ala-(R)-MPA dimer—as pendant,106 two different conformations, extended and bent, can occur (Fig. 4a). Thus, if the dimer adopts an extended conformation, (S)-alanine, located closer to the poly(phenylacetylene) main chain, commands a preferred macromolecular structure (screw sense and elongation) in the PPA (M helix, Fig. 4a). On the other hand, by fixing a bent structure at the dimeric pendant, it is possible to place the second chiral residue close to the backbone, which is now responsible for the screw sense preference adopted by the macromolecular helical polymer105,106 (P helix, Fig. 4a).
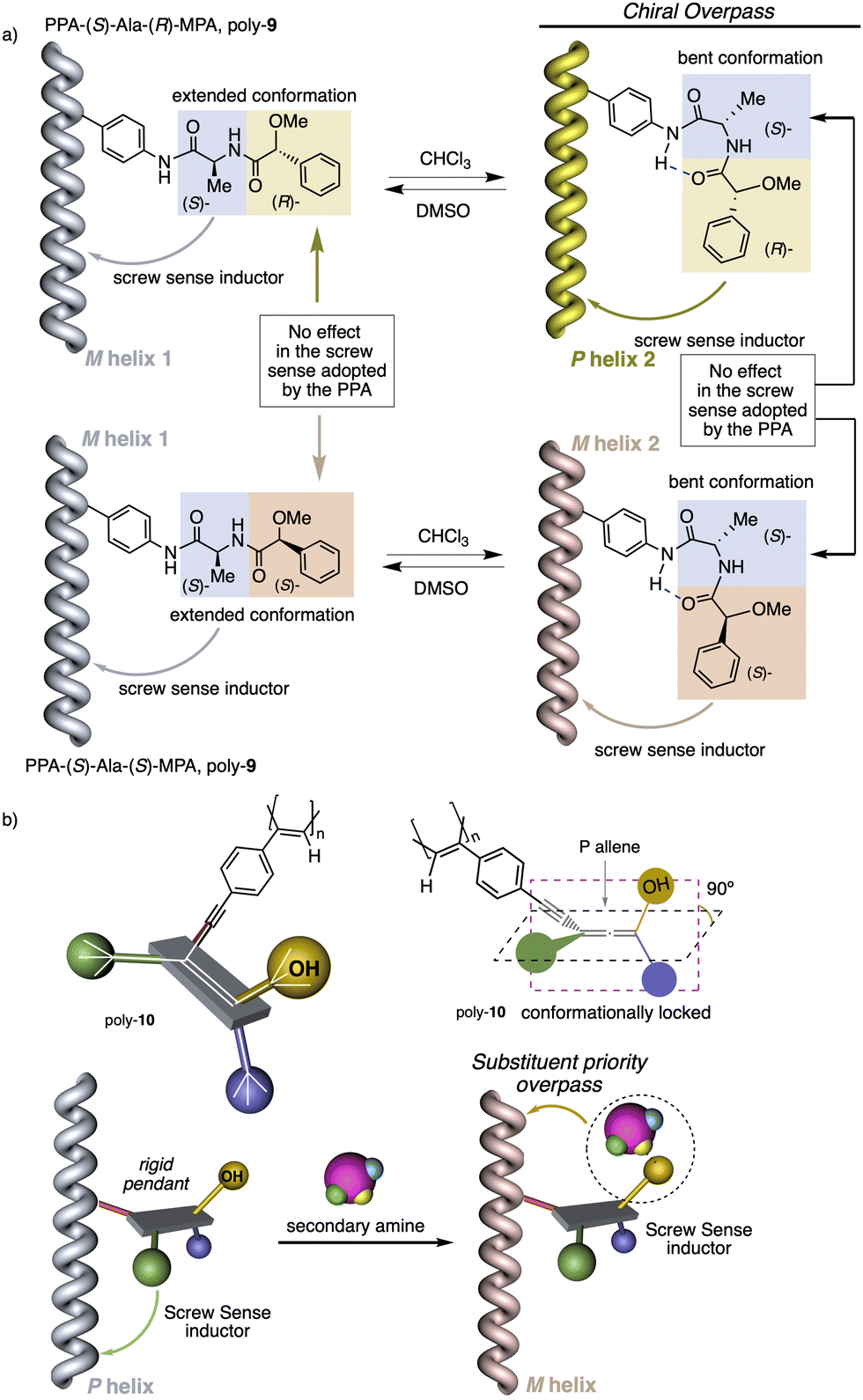 | ||
| Fig. 4 (a) Schematic illustration of the chiral overpass effect in poly(phenylacetylene)s. (b) Chiral amplification of a poly(phenylacetylene) bearing a chiral allene as pendant using chiral amines as external stimuli. | ||
However, if the (S)-Ala-(S)-MPA pendant is used, M helices are obtained in both extended and bent conformations (Fig. 4a).
1.2.1.3 Substituent priority overpass. This helix induction mechanism is found in dynamic helical polymers bearing conformationally locked chiral pendants such as chiral allenes.107 In these systems, the relative spatial orientation of the substituents is always the same, and it is not possible to modulate it by altering the conformational composition of the chiral moiety. In such cases, to invert the helical sense of the polymer it is necessary to modify the relative volume of one of the substituents.
As a result of this volume change, a new substituent located in a different position will be responsible for producing variations in the macromolecular structure and inducing the screw sense preference.
To show this effect, poly-10 was chosen as illustrative example.107 This polymer bears an allene pendant (Fig. 4b) in which its two consecutive double bonds place their four substituents in two perpendicular planes. If these substituents are different, the allene becomes chiral, as in poly-10, which shows P axial chirality. In poly-10, the screw sense is governed by the tert-butyl group located closest to the backbone (substituent a). However, if a secondary or tertiary amine are added to a solution containing poly-10, a supramolecular interaction is established between the alcohol group (substituent b) of the allene and the amine, increasing the size of this substituent. As a result, substituent b, placed in a different spatial orientation than substituent a, now controls the helical sense that poly-10 adopts, resulting in a helix inversion process.
1.2.2.1 Sergeants and Soldiers effect108–111 (Sergeant: chiral (R or S), minor component; Soldier: achiral, major component). The Sergeant orders the Soldier to adopt a conformation that promotes the same helical scaffold (sense and elongation) induced by the Sergeant.
1.2.2.2 Abnormal Sergeants and Soldiers effect94 (Sergeant: chiral, minor component (R or S); Soldier: achiral, major component). The Sergeant activates a conformation in the Soldier that promotes a different helical scaffold than the one adopted by the Sergeant (sense and/or elongation).
1.2.2.3 Chiral to chiral Sergeants and Soldiers effect112 (Sergeant: chiral, minor component (R or S); Soldier: chiral (R or S), major component). The Sergeant commands a specific conformation on the Soldier, which differs depending on the absolute configuration of the chiral Soldier (Fig. 5a and b). Thus, the two enantiomeric configurations of the Soldier can adapt to the scaffold induced by the Sergeant. For example, a PPA bearing the 4-anilide of (R or S)-methoxyphenylacetic acid (poly-11) behaves axially racemic (ECD = 0, mixture of P and M helices) even though the pendant is chiral.112 This fact is due to the presence of two conformers in equilibrium at the pendant, ap and sp, which place the substituents of the chiral moiety in different spatial locations, inducing opposite helical senses in the PPA (Fig. 5a). Interestingly, replacement of the phenyl group by an anthryl ring (poly-12) shifts the conformational composition of the pendant towards the ap conformer (Fig. 5a).111 When the monomers of poly-12 (Sergeant) and poly-11 (Soldier) are copolymerized to create poly[(S)-12(Sergeant)r-co-(S)-11Soldier(1−r)] and poly[(R)-12(Sergeant)r-co-(S)-11Soldier(1−r)] copolymer series (Fig. 5b), it was observed that both series adopt opposite helical senses, P and M, commanded by the (S)- or (R)-chirality of the Sergeant respectively. The (S)-Soldier fits into the P and M helical scaffolds adopting the ap or sp conformations respectively.112
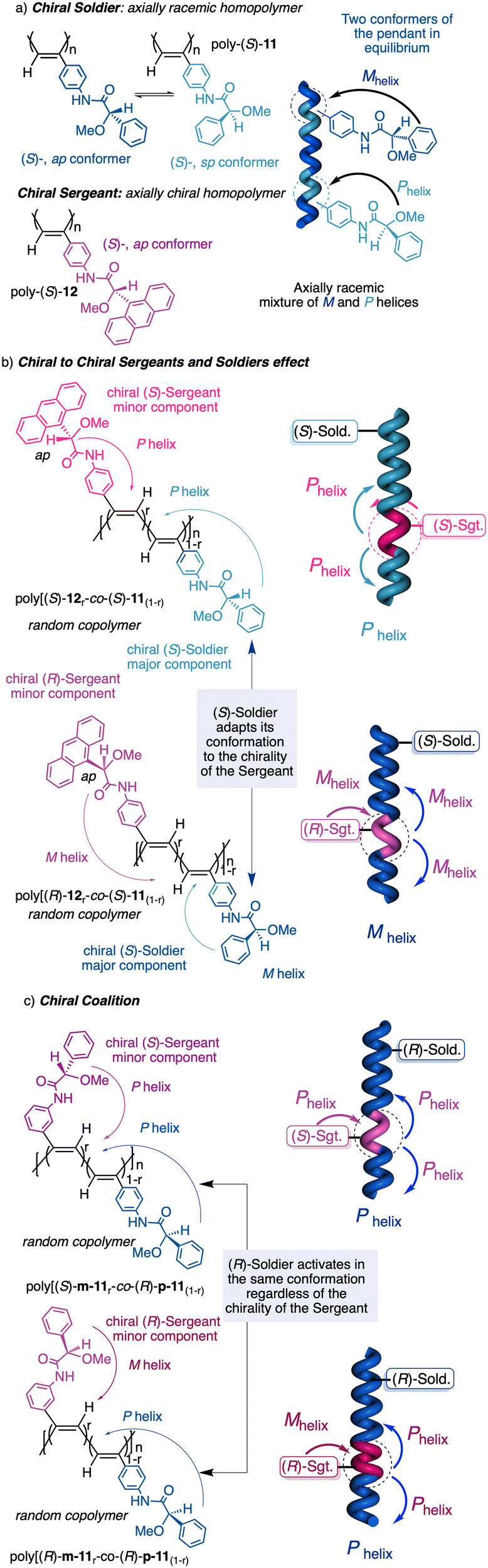 | ||
| Fig. 5 (a) Chiral Soldier. (b) Chiral to chiral Sergeants and Soldiers effect. (c) Chiral coalition. | ||
1.2.2.4 Chiral coalition or abnormal chiral to chiral Sergeants and Soldiers effect113 (Sergeant: chiral, minor component; Soldier: chiral, major component). In this case, the role of the Sergeant is to behave like a chiral dopant, triggering a specific conformation in the Soldier that is different depending on the chirality of the soldier. As a result, poly[(S)-Sergeantr-co-(S)-Soldier(1−r)] and poly[(R)-Sergeantr-co-(S)-Soldier(1−r)] adopt the same helical structure, which is commanded by the chirality of the Soldier, e.g., poly[(S)-5r-co-(S)-11(1−r)] and poly[(R)-5r-co-(S)-11(1−r)] (Fig. 5c).113 However, the Sergeant is necessary to trigger a specific conformation in the Soldier, otherwise, without its presence, the Soldier homopolymer behaves like an axially racemic helix.
1.2.2.5 Majority rules114 [Sergeant: chiral; Soldier: chiral (Sergeant's enantiomer)]. In this case, the screw sense preferences of copolymer series made by two enantiomeric monomers (identical chemical structure, mirror image relationship) are biased towards P or M helices, commanded by a slight enantiomeric excess of one of the enantiomers. For instance, in poly[(R)-Sergeant0.56-co-(S)-Soldier0.44], made by enantiomeric monomers of 2,6-dimethylheptyl isocyanate (56% (R)-enantiomer/44% (S)-enantiomer; ee 12%), the helix adopted by the copolymer is identical to that adopted by the (R)-homopolymer.114
1.2.2.6 Chiral accord and chiral conflict115–117. Other interesting phenomena can occur in chiral dynamic helical copolymers although there is not amplification of asymmetry due to the lack of communication between the two chiral components. Thus, if the two comonomers of the copolymer induce independently the same helical sense, a chiral accord116 emerges, whereas if both comonomers induce opposite helical senses, a chiral conflict116,117 is produced.
In short, the presence or absence of communication between co-monomers in dynamic helical polymers can produce interesting stimuli-responsive properties in the materials due to the dual or independent stimuli-responsiveness of the comonomers towards a specific stimulus.
1.3. Supramolecular helical polymers
In supramolecular helical polymers,5,18–38 helices are achieved by the presence of tilting degrees generated between building blocks during the supramolecular polymerizations. Positive or negative values of these tilting degrees are induced by the absolute configurations of the chiral groups introduced in the building blocks. Thus, depending on their absolute configurations, P or M supramolecular helices will be formed.The stimuli-responsive properties of those helices can produce the disassembly of the helical structures, helix inversion or the formation of other aggregates.
In this review, we will only focus on helical structural changes that result in a helix with opposite helical sense or the formation of another type of helical aggregate, such as J-type or H-type aggregates. To go from one supramolecular helical structure to another, two different mechanisms can be followed, a competitive pathway 118 or a consecutive one.119,120
In a competitive pathway, a kinetic supramolecular aggregate disassembles to provide the molecularly dissolved monomer that further aggregates (thermodynamic aggregate) into a different helical structure (sense and/or scaffold). The aggregation of an asymmetric oligo(phenyleneethynylene) (OPE) bearing the anilide of (S)-MPA [(S)-13] was chosen as illustrative example.118 In this case, the kinetic aggregate (AggI, M helix) was kinetically trapped in MCH/Tol/DCM solvent mixture at room temperature, while the thermodynamic aggregate (AggII, P helix) was obtained in MCH/DCM solvent mixture after thermal treatment (Fig. 6a).
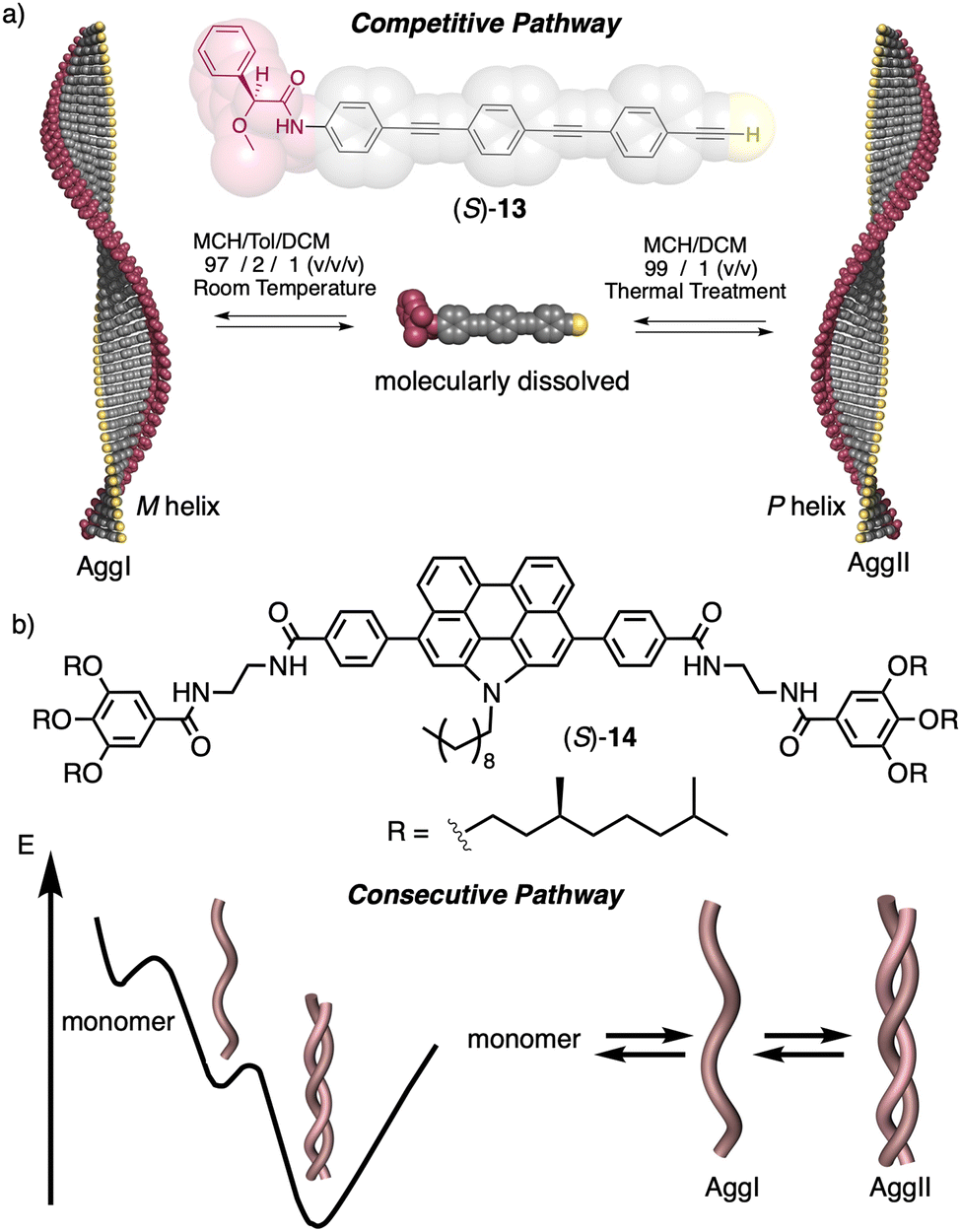 | ||
| Fig. 6 Supramolecular aggregation following a (a) competitive or a (b) consecutive pathway. | ||
In the consecutive pathway, one helical structure is transformed into a different one, without going through the monomeric state. For example, the N-annulated perylenetetracarboxamide (S)-14120 aggregates to form fibrillar structures that evolve to form thicker helical assemblies (Fig. 6b).
Supramolecular and helical polymers share many properties, such as the asymmetry amplifying effects found in their copolymers. Thus, Sergeants and Soldiers or Majority Rules can be found in both systems,121–123 although their information transmission mechanisms to induce screw sense excess are different.
2. Stimuli-responsive helical polymers
In this section we will use different examples found in the literature to show how helical structures adopted by foldamers, supramolecular and covalent helical polymers can be modified (helix inversion, amplification of asymmetry or variations in the helical scaffold) through interaction with different stimuli.124 We will focus our attention on those systems where the structural changes are clearly identified, although we also will go through other interesting examples where the structural changes associated to the presence of an external stimulus are not clear.2.1 External stimulus: temperature
Temperature is involved in the activation/deactivation of supramolecular forces and in the variation of the conformational composition of molecules. Hence, thermal control of screw sense in foldamers and polymers can be achieved by taking these parameters into account during macromolecule design.Inai designed a dynamic helical foldamer consisting of a nonapeptide made mostly by achiral residues such as Aib, β-Ala (β-alanine) and ΔZPh (Z-α,β-dehydrophenylalanine) and a single chiral residue,125L-Phe (L-phenylalanine), placed in an internal position [H-β-Ala-ΔZPh-Aib-(L)-Phe-(ΔZPh-Aib)2-OMe, 15] (Fig. 7a). The chiral residue is necessary to command a P helix in the foldamer, which is protected at the C-terminus with a methyl ester and has a free NH at the N-terminus, necessary to establish supramolecular interactions with chiral acids. Interestingly, when foldamer 15 interacts with Boc-D-amino acids such as Boc-D-Pro-OH or Boc-D-Leu-OH, a P to M helix inversion occurs in the foldamer. Activation/deactivation of the supramolecular amino/carboxylic acid interaction between the foldamer and the Boc-D-amino acids results in a reversible helix inversion process between a P helix (high temperature, supramolecular interaction OFF) and an M helix (low temperature, supramolecular interaction ON).125
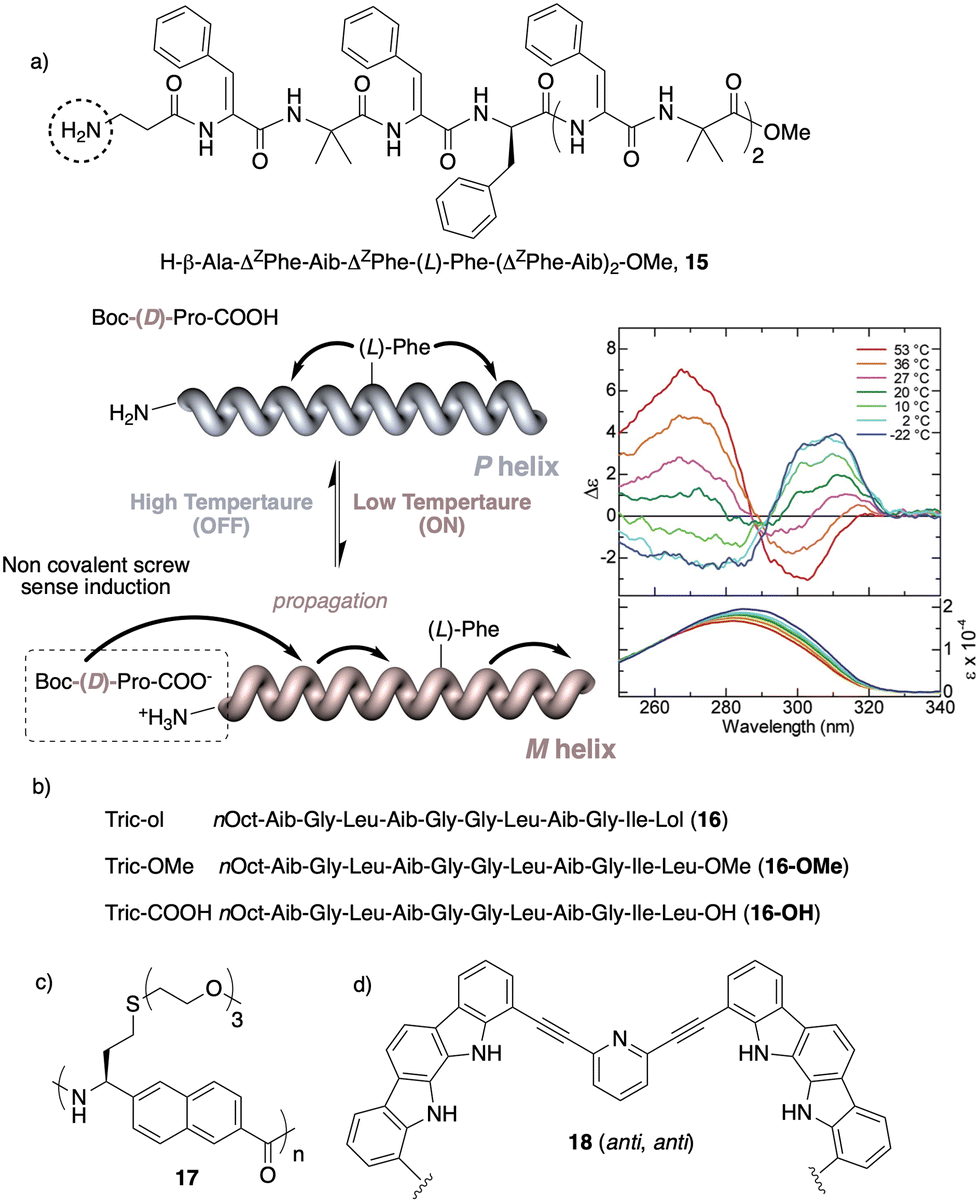 | ||
| Fig. 7 Temperature responsive foldamers: Helix inversion (a) 15 (reproduced from ref. 125 with permission from the American Chemical Society, copyright 2007) and (b) 16; elongation (c) 17 and (d) 18. | ||
In another work, Formaggio and De Zotti126 found that by replacing the amino alcohol leucenol (Lol) by leucine (Leu-OMe or Leu-OH) at the C-terminus of the natural occurring trichogin (a peptaibol, nOct-Aib-Gly-Leu-Aib-Gly-Gly-Leu-Aib-Gly-Ile-Lol, 16), the new peptides 16-OMe and 16-OH become thermo-responsive transitioning from a P to an M helix with increasing temperature (Fig. 7b). Again, these peptide sequences contain many flexible and achiral residues, such as Gly and Aib, that can be accommodated in the P and M helical senses.
In another example, Onitsuka and Kanbayashi127 used temperature to change the elongation of an arylopeptide that contains the axially unsymmetrical 2,6-napthylene as spacer between the amino and the acid group (17) (Fig. 7c). This amino acid has a long oligoether group in the side chain, a thermo-responsive substituent that exhibits lower critical solution temperature (LCST) behavior due to the transition in the hydration of the side chains. At 25 °C, foldamer 17 is soluble in water and adopts a 31-helix, while when heated this helical scaffold stretches and transforms into a 41helix. In the field of aromatic helical foldamers, Jeong128 prepared one that contains two indolocarbazole and two pyridine moieties (18) (Fig. 7d). The addition of Pd(CH3CN)2Cl2 generated a double-stranded helicate, which at low temperatures favors a more stretched helix (syn–anti orientation of the internal pyridine) and a more compact one at high temperatures (syn–syn orientation of the internal pyridine).
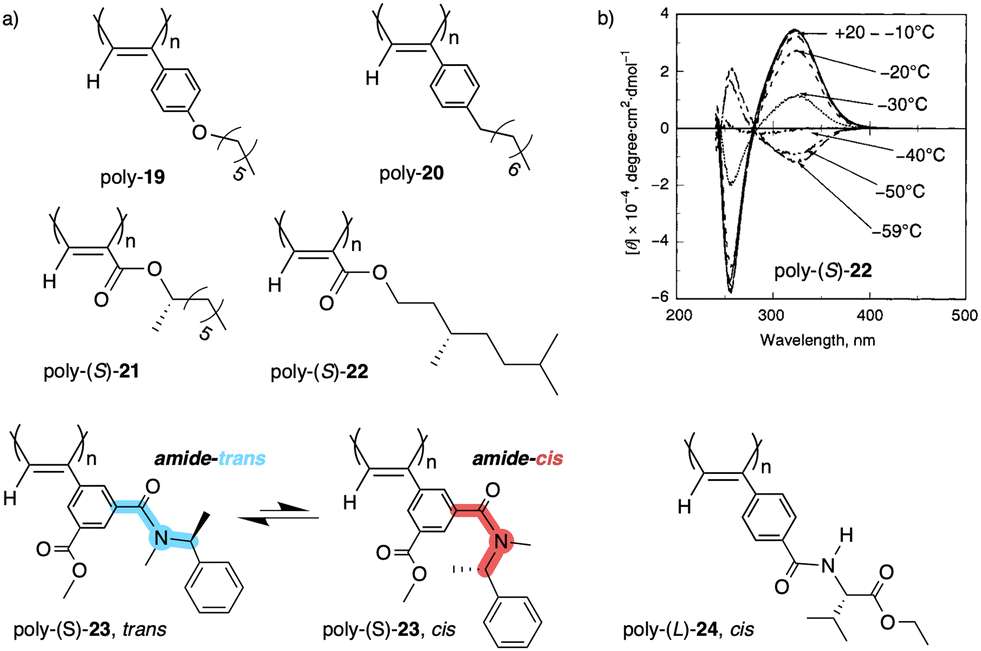 | ||
| Fig. 8 (a) Chemical structures of polymers poly-(19–24). (b) ECD spectra of poly-(S)-22 at different temperatures (ECD thermal switch) (reproduced from ref. 132 with permission from the American Chemical Society, copyright 2001). | ||
Another interesting example dealing with a temperature driven conformational change at the pendant was described by Wan133 for a 3,5-disubstitued PPA, poly{(−)-3-methoxycarbonyl-5-[N-methyl-N-(S)-(1-phenylethyl)carbamoyl]-phenylacetylene} (23) (Fig. 8a). In this case, the authors play with the cis/trans conformational composition of the tertiary amide using temperature.
Thus, while the trans isomer is favored at low temperatures, the cis ratio increases when the temperature is raised. As a result, a reversible helix inversion is produced generating a thermal helical switch. Zhang and Liu reported another PPA that works as thermal helical switch by using the benzamide of (L)-valine-OEt (24) as pendant (Fig. 8a).134 In this case, the macromolecular helical structure adopted by the polymer in chloroform at room temperature can be reversed by cooling the solution to 15 °C or lower. Following in the line of macromolecular helical changes induced by conformational changes driven by temperature in the pendant groups, an interesting example is the PPA designed by Bergueiro, Freire and Calderón,135 which includes elastin as pendant (poly-25) (Fig. 9a). Elastin is a thermoresponsive peptide, which undergoes conformational changes when the temperature of the solution varies.
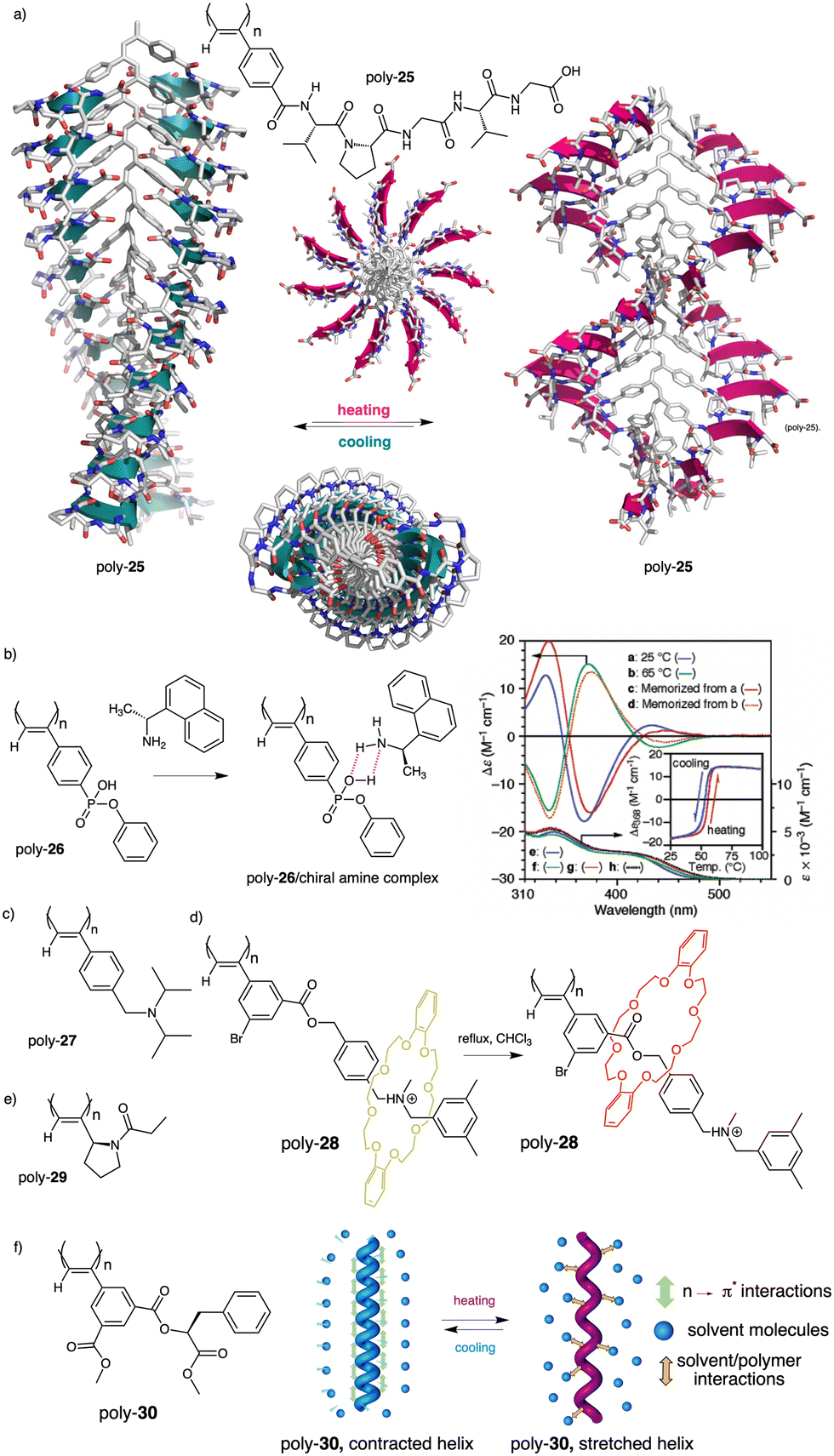 | ||
| Fig. 9 (a) Schematic representation of the thermal behaviour of a PPA bearing an elastin derivative as pendant group (poly-25). (b) Chemical structure of poly-26 and thermoresponsive behaviour of poly-26/(R)-1-(1-naphthyl)ethylamine complex (reproduced from ref. 136 with permission from the American Chemical Society, copyright 2005). Chemical structures of (c) poly-27, (d) poly-28 and (e) poly-(29). (f) Structure of poly-30 and its thermoresponsive behaviour. | ||
In this case, the thermal behavior of elastin with increasing temperature changes from a lower critical solution temperature (LSCT) to an upper critical solution temperature (UCST) once polymerized. Below the cloud point temperature, the elastin in the pendants adopts a hydrophobic state (β-spiral conformation), which results in the adoption of an elongated structure. Above the cloud point temperature, the pendant adopts a more hydrophilic state (extended conformation), which induces a more compressed helical scaffold. Yashima demonstrated, through a beautiful example, that a thermal switch can be created from a supramolecular PPA/chiral amine complex.136 Here, the PPA is achiral and bears a bulky phenyl phosphonate group on the pendant (26) (Fig. 9b).
The resulting polymer is achiral, and a mixture of P and M helical senses are obtained. However, by adding chiral amines such as (R)-1-(1-naphthyl)ethylamine, a chiral complex is formed in DMSO and a P screw sense excess is induced in poly-26 at 25 °C. In this complex, the helical sense is commanded by the phosphonate group, which became chiral after complexation with the chiral amine as inferred from VCD studies. Interestingly, when the DMSO solution of poly-26/(R)-1-(1-naphthyl)ethylamine complex heats up to 65 °C, a helix inversion from P to M is observed in the PPA, which is accompanied by a planarity of the phosphonate VCD signal. In this case, the authors indicate that phosphonate may racemize or exist as the achiral form due to the resonance effect of the P–OH and P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group, and therefore the helix induction comes from another moiety of the PPA/amino complex, i.e., the chiral amino group. Yashima created another thermal switch based on supramolecular complexes between an achiral PPA that bears a N,N-diisopropylaminomethyl group as pendant (27) (Fig. 9c) and chiral substrates such as chiral acids.137 In this example, no changes in the helical sense of the poly-27/chiral acid complex were observed by temperature changes. However, they found that a colorimetric thermal switch is obtained by variations in the polymer elongation. Thus, at low temperatures (0–20 °C), a compressed helical structure is induced in poly-27/chiral acid complex (yellowish solution) while at higher temperatures (25–40 °C), a stretched helix is obtained (orangish solution). In relation to the development of PPA thermal switches based on changes in polymer elongation, Takata reported a PPA that has a thermoresponsive rotaxane as pendant (poly-28) (Fig. 9d).138
O group, and therefore the helix induction comes from another moiety of the PPA/amino complex, i.e., the chiral amino group. Yashima created another thermal switch based on supramolecular complexes between an achiral PPA that bears a N,N-diisopropylaminomethyl group as pendant (27) (Fig. 9c) and chiral substrates such as chiral acids.137 In this example, no changes in the helical sense of the poly-27/chiral acid complex were observed by temperature changes. However, they found that a colorimetric thermal switch is obtained by variations in the polymer elongation. Thus, at low temperatures (0–20 °C), a compressed helical structure is induced in poly-27/chiral acid complex (yellowish solution) while at higher temperatures (25–40 °C), a stretched helix is obtained (orangish solution). In relation to the development of PPA thermal switches based on changes in polymer elongation, Takata reported a PPA that has a thermoresponsive rotaxane as pendant (poly-28) (Fig. 9d).138
Thus, depending on the position of the crown ether in the pendant, the polymer will adopt different degrees of stretching. At high temperatures, the ring of the rotaxane will sit close to the polyene backbone, resulting in a highly stretched helix. Conversely, at lower temperatures, the crown ether ring will be situated far from the backbone, adopting the PPA a more compressed helical structure. This structural change is accompanied by a color change of the solution, from red (high temperatures) to yellow (low temperatures) that also occurs in the solid state.
Wan reported a polyacetylene, poly[(S)-2-ethynyl-N-propionylpyrrolidine] (poly-29) (Fig. 9e),139 derived from proline, that shows a LCST behavior in water accompanied by elongation changes in the chiral polyacetylene helix. Thus, below 32 °C, the polymer adopts a compressed P helix that produces a clear solution, whereas at temperatures higher than 32 °C, a more stretched helix is obtained that generated a cloud solution due to the formation of aggregates. Zhang and Wan reported a 3,5-dibenzoate PPA (poly-30) (Fig. 9f)140 that also can vary its structure between compressed and stretched helical scaffolds in chloroform by deactivating, at high temperatures, the n–π* interactions between the oxygen of one carbonyl group and the carbon of another carbonyl in nearby upper or lower position within the helix. These n–π* interactions stabilize compressed cis–cisoidal polyene structures. At high temperatures, new interactions between solvent and pendants stabilize stretched cis–transoidal helices. Percec, Meijer and Zhang studied how temperature produces structural changes (elongation, helix sense) in dendronized PPAs. Thus, Meijer141 designed a PPA where the dendron grows directly from the phenylacetylene backbone. To do that, they did a 3,4,5-trisubstitution at the aryl ring with a chiral ether—poly[3,4,5-tris((S)-3,7-dimethyloctyloxy)phenylacetylene], poly-31 (Fig. 10a). The resulting polymer shows a helix inversion from M to P in a very narrow temperature range (M helix at 20 °C, P helix at 30 °C), which is accompanied by a stretching of the helical scaffold. Percec designed a dendronized PPA142,143 where the dendron and the PPA backbone are separated by a chiral ether group (poly-32) (Fig. 10b). This polymer shows a reversible cis–cisoidal to cis–transoidal (contraction/extension) thermoresponsive isomerization of the backbone that can act as a nanomechanical actuators. When extruded as fibers, they have been shown capable of work by displacing objects up to 250-times their mass (Fig. 10b). Li and Zhang144,145 designed water soluble PPAs bearing dendritic oligo(ethylene glycol) (OEG) as pendants using chiral alanine as spacer between the dendron and the PPA backbone. Different linkages were used to connect the amino acid to the PPA, such as anilide (poly-33) (Fig. 10c) or benzamide (poly-34) (Fig. 10d), which provide the polymer with different thermoresponsive properties such as reversible helix inversion (poly-33) or reversible contraction/extension (poly-34). This fact indicates that the amide linkage, benzamide/anilide, plays an important role in the thermal behavior of dendronized PPAs. Interestingly, the cloud point temperature (Tcp) of poly-33 can be tuned by salt-in and salt-out anions. Thus, while PF6− and SCN− (salt in anions) largely increased the Tcp, Cl− or SO42− (salt out anions) decreased the Tcp. In the case of adding salt in anions, the helix inversion occurs at the Tcp without structural changes in the PPA, whereas by adding salt out anions, temperature changes produce variation in the polymer elongation. This fact indicates that structural changes are produced by competitive interactions between OEG moieties, water, and anions (Fig. 10e).
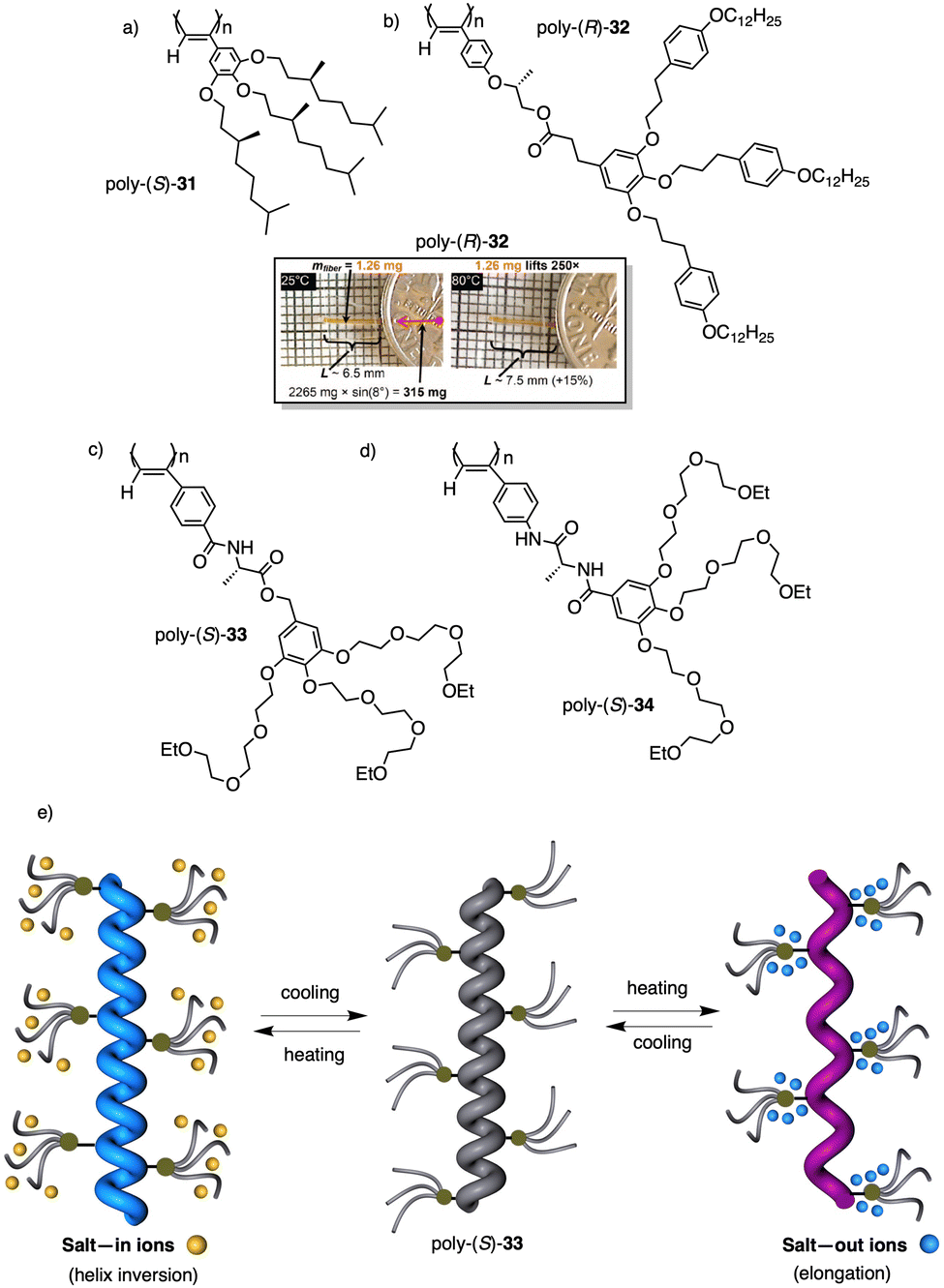 | ||
| Fig. 10 Structures of (a) poly-31, (b) poly-32 (reproduced from ref. 143 with permission from the American Chemical Society, copyright 2008), (c) poly-33 and (d) poly-34. Schematic representation of the thermal behaviour of poly-33. | ||
Apart from PPAs, there are other polyacetylene derivatives, such as poly(biphenylacetylene)s or PDPAs, that show thermoresponsiveness. In both systems, the large steric hindrance in the backbone results in a kinetically trapped helical structure with a certain helical sense excess that can evolve towards the thermodynamic helicity by temperature changes. For instance, Maeda146 designed a PPA that bears 4-biphenyl pendants with methoxymethyl (MOM) groups (poly-35) (Fig. 11a) at the 2,2′-position. This pendant is achiral, and therefore, the resulting polymer behaves as axially racemic.
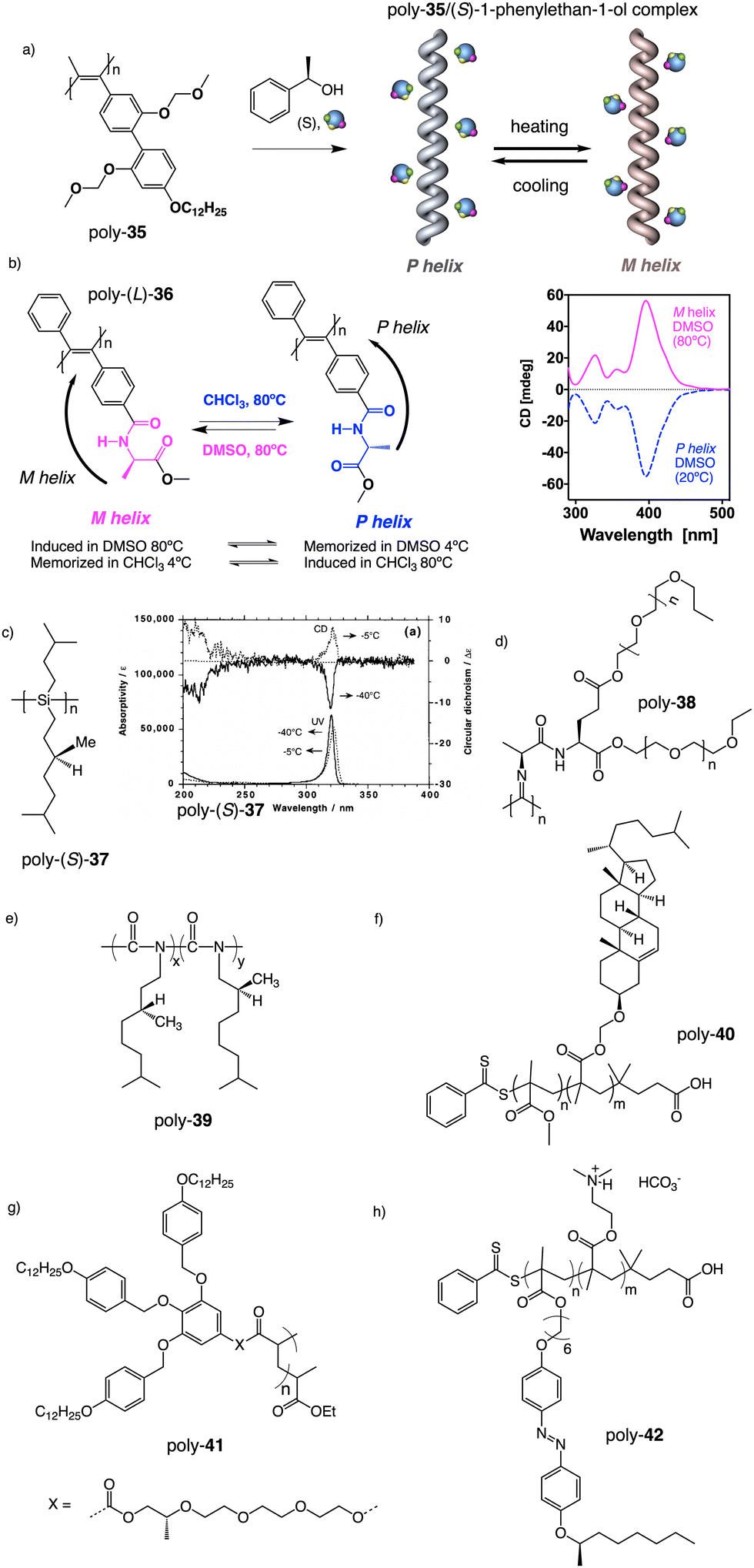 | ||
| Fig. 11 Thermal driven helix inversion of (a) PBPA poly-35, (b) PDPA poly-36 and (c) polysilylane, poly-37 (reproduced from ref. 149 with permission from the American Chemical Society, copyright 2000). Chemical structures of (d) polyisocyanide poly-38, (e) co-polyisocyanate poly-39, (f) PMMA poly-40, (g) polyacrylate poly-41, (h) PMMA poly-42. | ||
A screw sense preference can be induced in poly-35 (Fig. 11a) by adding a chiral alcohol as external stimulus. Interestingly, depending on the temperature used to form the complex, the crew sense induced in poly-35/chiral alcohol will be opposite, e.g., poly-35/(S)-1-phenylethan-1-ol complex shows a P helix at −10 °C and an M helix at 50°. In the field of PDPAs, Freire designed an asymmetric PDPA that bears the benzamide of (L)-alanine methyl ester as pendant (poly-36) (Fig. 11b).147 This polymer can adopt either P or M helical sense in different solvents at high temperatures (DMSO, M helix; CHCl3, P helix). This fact is due to a different conformational composition in the pendant. Interestingly, after solvent removal, the induced helical sense can be kinetically trapped after adding a solvent that induces the opposite helical sense. The evolution to the thermodynamic helix (opposite helical sense) can be controlled by temperature, allowing to create a long-lasting memory effect at low temperatures. Fujiki found that poly(silylene)s89,148–152 containing chiral side chains may exhibit temperature driven helical sense inversion, such as poly{(S)-3,7-dimethyloctyl-3-methylbutylsilylene} (poly-37) (Fig. 11c). Remarkably, in most of these polymers, the macromolecular helix inversion to form the diastereomeric helical structure is accompanied by a bathochromic or hypsochromic shift. Thus, when a depletion and shifting of the ECD signal occurs, in poly(silane)s partial helical inversion takes place. Li and Zhang,153 studied the thermal behavior of a polyisocyanide (poly-38) (Fig. 11d) that carries as pendants dipeptides derivatized with oligoethylene glycols (OEGs). In this polymer, the thermal behavior is provided by the OEG chains, while the secondary structure of the polymer remains unaffected. Thus, the OEG chains collapse at high temperatures on the polymer backbone, increasing the turbidity of the solution, while at low temperatures, those chains adopt an extended and solvated conformation.
Green and Selinger154 demonstrated in poly(isocyanate)s, e.g., poly-39 (Fig. 11e), how the absence of chiral-to-chiral communication in helical copolymers made by two chiral co-monomers can produce a material with interesting properties such as a thermal behavior. Thus, when a chiral conflict emerges, the two components of a copolymer induce opposite helical senses, and therefore, at a certain comonomer ratio, the copolymer is axially racemic. Interestingly, playing with the temperature, it is possible to modulate the conformational composition of the two components, and therefore the extension of the chiral conflict, which can result in the adoption of a P or M helical sense depending on the temperature used. Also, in the field of helical copolymers, Mehl, Zhang and Liu155 found that the thermal behavior of thin film block copolymers (BCPs), comprising poly(methylmethacrylate) (PMMA) and poly(cholesteryloxyhexylmethacrylate) (PChMA) blocks, depends on the relative lengths of the blocks (poly-40) (Fig. 11f).
Thus, when the PMMA block is short, a thermal helix inversion is produced in the BCP, while no structural changes are observed for those containing longer PMMA blocks.
They attribute this different behavior in the two-copolymer series to the number of methyl substituents in the backbone, which affect the copolymer packing. In polyacrylates and PMMAs, Percec156 and Zhang157 demonstrated through beautiful designs how, regardless of the tacticity or the helical structure adopted by the backbones, the pendants can self-assemble into P or M axial arrays by inserting into them a flexible chain used as a spacer that is connected to a planar and rigid fragment involved in π–π supramolecular interactions [aromatic dendrons, poly-41 (Percec) (Fig. 11g); azo derivatives, poly-42 (Zhang) (Fig. 11h)]. Thus, while Percec introduced the chiral center in the flexible spacer,156 Zhang introduced it as an external substituent of the azo group at the periphery of the helix.157
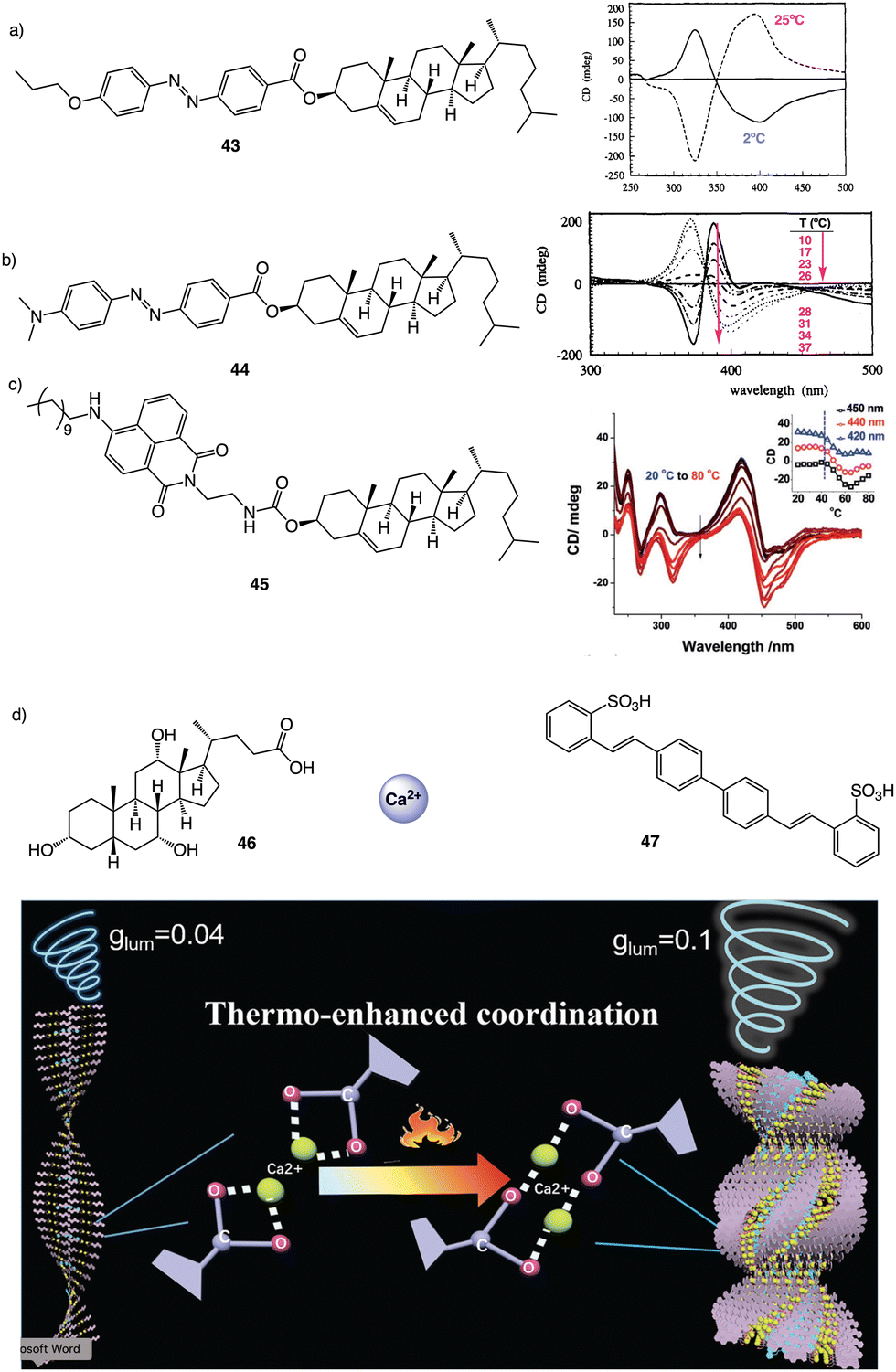 | ||
| Fig. 12 Thermal driven helix inversion of supramolecular aggregates using (a) and (b) azo (43–44) (reproduced from ref. 157 with permission from the American Chemical Society, copyright 1994) and (c) naphthalimide cholesterol derivatives (45) (reproduced from ref. 158 with permission from Wiley-VCH, copyright 2018) as building blocks. (d) CPL enhancement of 47 aligned in a thermoresponsive chiral metallosupramolecular polymer (46 + Ca2+) (reproduced from ref. 159 with permission from Wiley-VCH, copyright 2018). | ||
In the aggregate, the chirality of the cholesterol moiety was transferred to the naphthalimide group producing an M chiral aggregate, which progressively evolves towards an aggregate with opposite chirality by heating. This chiral inversion is attributed to the ability of the vesicles to entrap water, which can be released once the system is heated above 45 °C. Cholic acid (46) (Fig. 12d) was also used by Yan to create thermoresponsive metallo-supramolecular hydrogels.160 Cholic acid self-assembles in the presence of Ca2+ to form fibrillar structures that promote the formation of a chiral gel in water. The coordination mode between the carbonate groups of cholic acid and the Ca2+ ions released by Ca(NO3)2 changes from the metastable bidentate chelating mode to the thermally stable bridging mode with increasing temperature. As consequence, individual nanohelices bunch together to form large scale helical bundles. This effect was used to align a light emitting fluorescent dye within the fibers and create a circularly polarized light emitting system (CPL). The CPL intensity of the hydrogel doped with the fluorescent dye (47) (Fig. 12d) increases approximately 4-fold and the gem emission dissymmetry factor increases from 0.04 to 0.1 as the temperature goes from 25 to 50 °C.
Water can play an important role in the formation of supramolecular aggregates even when present as a trace in an organic solvent. Through elegant examples, Meijer showed that chiral supramolecular aggregates based on tetracarboxiamides can adopt different helical structures due to the incorporation of water as structural comonomer. This effect is found not only in homochiral aggregates (48) (Fig. 13a),161 but also in copolymer series that follow the Sergeants and Soldiers effect121–123 [48 (chiral, Sergeant) + 49 (achiral, Soldier)].162 Therefore, these systems depend on water and temperature variations led to different interactions between supramolecular aggregates and water molecules, producing different helical structures with diverse scaffolds and/or senses. This effect was also observed in the self-assembly of triarylamines (50, 51) (Fig. 13b) and chiral benzene tricarboxiamide (BTA) (52) (Fig. 13c).161,162 Choi and Jung developed trianilides with different D-alanine and/or glycine units (53, 54) (Fig. 13d).163 Interestingly, those containing one (53) or two (54) alanine segments can self-assemble into M or P supramolecular helical structures in toluene by playing with temperature. Thus, while an M supramolecular helix is obtained at low temperature (15 °C), an evolution to a supramolecular helix with opposite P chirality is observed by heating, reaching the maximum ECD spectra at ca. 45 °C. This helix inversion is attributed to conformational changes of the amide groups at the glycine residues attached to the trianilide core.
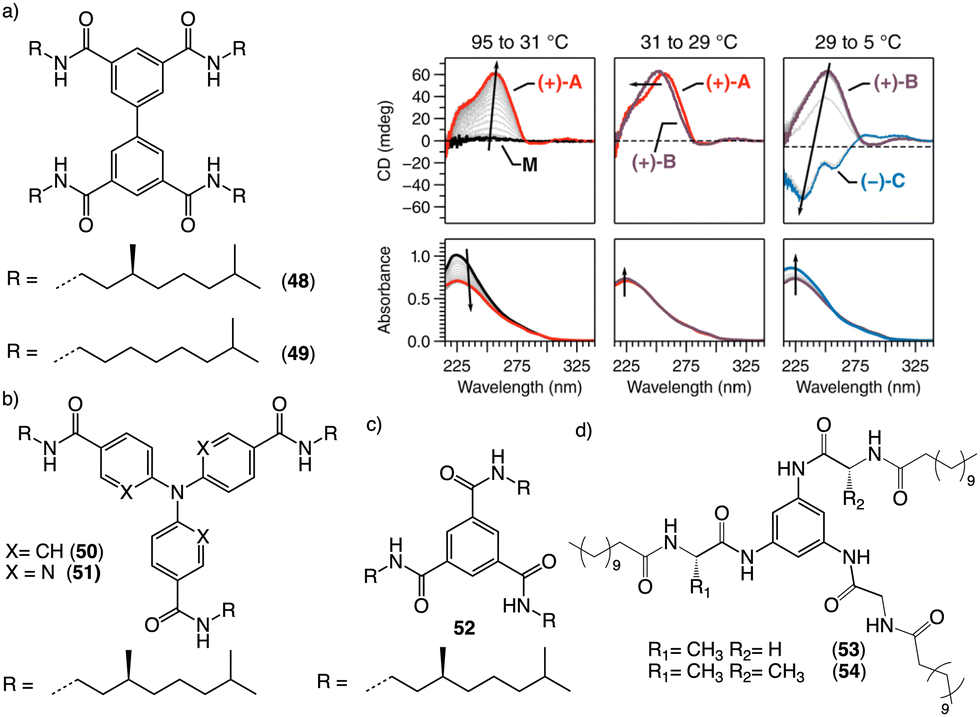 | ||
| Fig. 13 Water effects in the thermal driven helix inversion of supramolecular aggregates using (a) tetracarboxiamides derivatives (48, 49) (reproduced from ref. 162 with permission from the American Chemical Society, copyright 2020), (b) triarylamines (50, 51) and (c) chiral benzene tricarboxiamide (52). (d) Chemical structures of thermoresponsive chiral 1,3,5-trianilides (53, 54). | ||
Meijer and George reported another interesting example showing how the solvent–aggregate interaction affects the axial P/M chirality of the helical aggregate.164 They designed a coronene bisimide that has a 3,5-dialkoxy substitution in the imide phenyl groups (55) (Fig. 14a). This molecule self-assembles in methylcyclohexane (MCH), generating “molecular pockets” in the assembly. Solvent molecules can intercalate or form clathrates within the molecular pockets of the aggregate at low temperature (−10 °C), inducing an M helical structure. Interestingly, this weak solvent/aggregate interaction can be disrupted at 10 °C, resulting in M to P helix inversion.
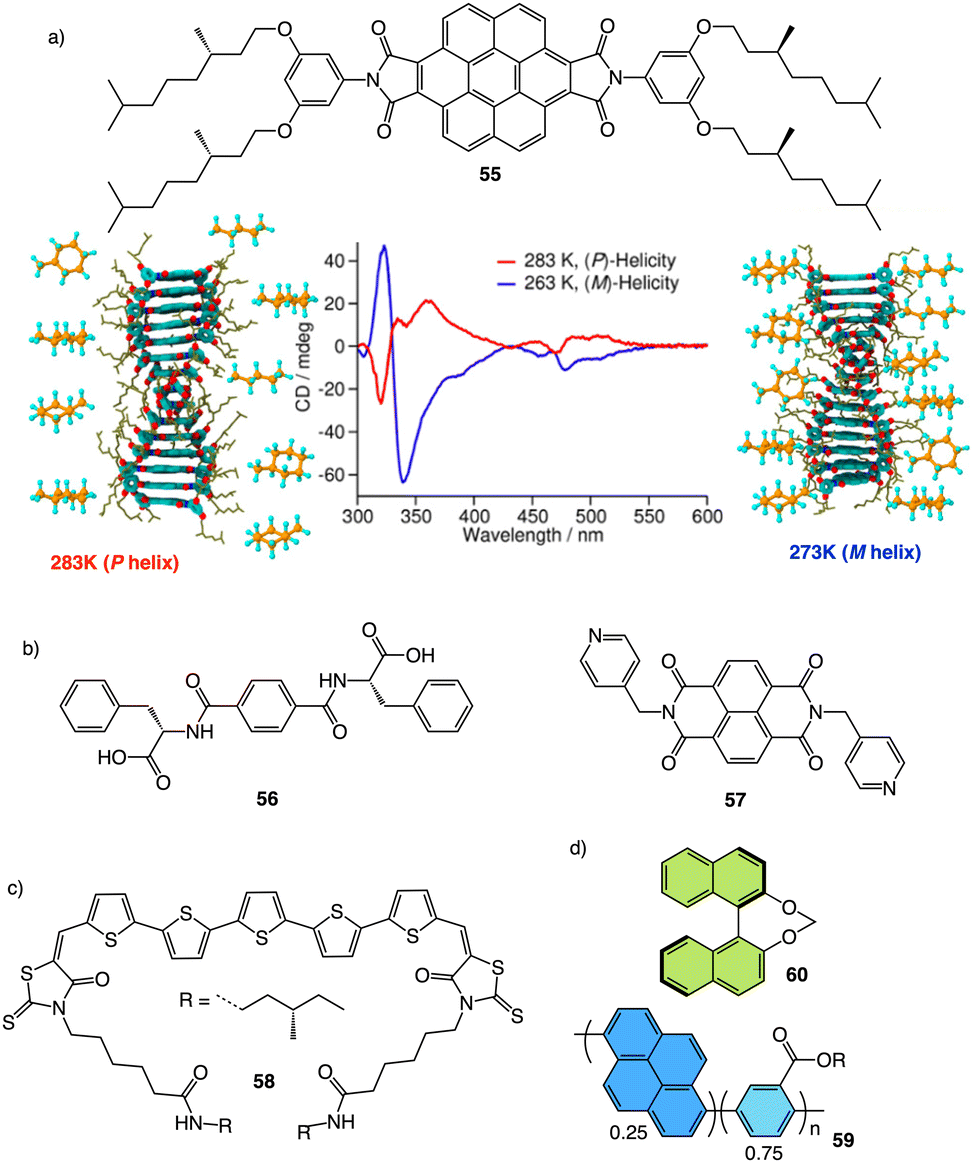 | ||
| Fig. 14 (a) “Clathrate effect” in the thermal behaviour of a coronene bisamide derivative (55) (reproduced from ref. 164 with permission from the American Chemical Society, copyright 2017). Chemical structures of (b) a two-component building block (56, 57) and (c) a quinquethiophene–rhodanine derivative (58) that generates chiral thermoresponsive aggregates. (d) Chiroptical thermal co-assembly based on achiral pyrenyl based polymer (59) and chiral binapthyl based inducers (60). | ||
Likewise, Percec showed that dendron-based C3-symmetric self-assembling dendrimers connected at their apex via trisesters and trisamides of 1,3,5-benzenetricarboxylic show thermal helical inversion due to the change of the lattice symmetry.165
Temperature can be used to discriminate between kinetic and thermodynamic aggregates that possess opposite chirality. Thus, Freire found that an asymmetric chiral OPE (13, Fig. 6a)118 can self-assemble into P or M supramolecular aggregates through a competitive aggregation pathway controlled by thermal treatment. Thus, while a kinetic P helix is obtained at room temperature in a DCM/MCH mixture, the thermodynamic M helix is formed at 0 °C after a heating/cooling cycle. Interestingly, the morphologies of these aggregates are different, thus while the kinetic P helix is formed by short oligomers that produced twisted sheets, the thermodynamic M helix forms columnar helical aggregates.
Dou and Feng found that a similar effect is obtained during the co-assembly of the two components of the building block.166 Thus, C2 phenylalanine-based aggregates (L-PF, 56) (Fig. 14b) can be transformed into kinetically trapped architectures with opposite helicity through a kinetic co-assembly pathway. In this case, a P helix was formed by heating/cooling or anti-solvent assembly of the L-phenylalanine derivative. Interestingly, the addition of an achiral pyridine derivative (57) (Fig. 14b), which can stablish supramolecular interactions with the L-PF derivative, generates achiral aggregates after thermal treatment. For its part, M chiral aggregates are obtained using the anti-solvent assembly protocol. The morphology of the aggregates depends also on the pathway followed by the self-assembly. In this sense, Carretero found that a quinquethiophene–rhodanine derivative (58) (Fig. 14c) self-assembles through hydrogen bonding interactions into opposite chiral helical structures before and after a heating/cooling cycle in pure toluene or in a 20/1 toluene/chloroform mixture.167
Supramolecular chemistry can also be used to induce chirality in an achiral polymer by interacting with a chiral molecule. Thus, Chen, Quan and Zheng created helical nanofibers by the co-assembly of achiral pyrenyl based polymer (59) and chiral binapthyl based inducers (60) (Fig. 14d) through π–π interactions. The co-assemblies show chiroptical properties due to the formation of helical fibers which can be tuned by thermal annealing towards P or M helical senses. The emissive properties of the pyrenil moiety allows to create a thermal CPL switch.168
2.2 External stimuli: anions
Negatively charged ions can be used as external stimuli to promote helix inversion of foldamers, dynamic covalent helical polymers and supramolecular polymers. Anions can establish supramolecular interactions with different functional groups, disrupting preexisting interactions or generating new ones that result in an inversion of the helical structure.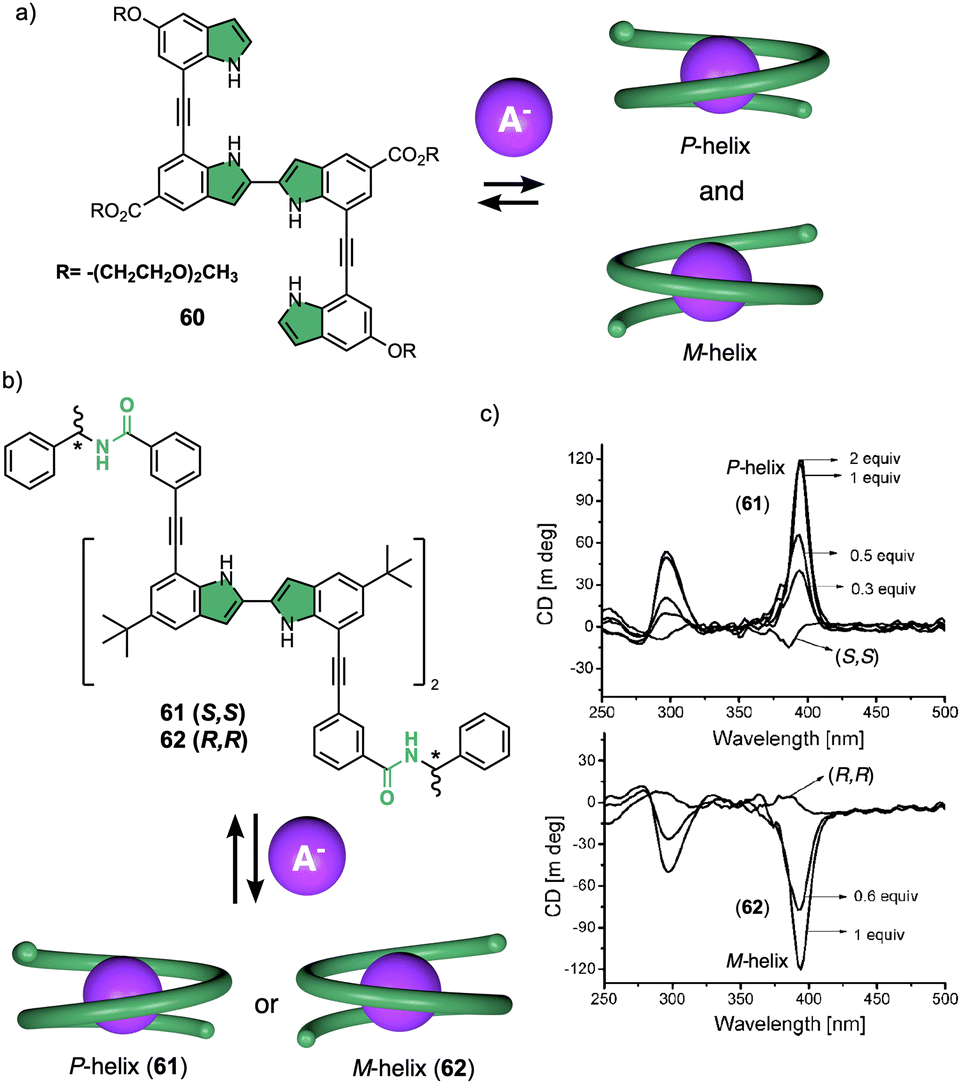 | ||
| Fig. 15 (a) Chemical structure of an oligoindole–ethynylene foldamer (60) and schematic illustration of its anion folding. (b) Chemical structure and (c) ECD spectra of an oligoindole based foldamer bearing (S)- (61) and (R)-phenylethylamido (62) groups as chiral termini (reproduced from ref. 171 with permission from the American Chemical Society, copyright 2008). | ||
However, in these works the helical scaffolds cannot be modulated from P to M or vice versa. So, the researchers took a step forward and reported the first example of a switchable anion-responsive foldamer based on a chiral indocarbazole dimer that folds into a helical conformation by intramolecular hydrogen bonding (63) (Fig. 16a).172 The helical sense of this dimer can be reversibly switched by binding it to a sulphate anion in CH2Cl2. Thus, the anion disrupts the intramolecular hydrogen bonds of the foldamer that stabilize the helical structure commanded by the chiral groups at the terminal ends. This leads to a different helical scaffold with opposite screw sense due to the formation of a new cavity with the NHs oriented towards the interior where the anion is located (Fig. 16b). In another example, they used a three-indolocarbazole-ethynylene trimer bearing a terminal amide-linked (S)-arylethylamido group at both ends (64).173 In this system, they found that a helix inversion can be achieved by using anions of appropriate size (Cl−, Br−, or CH3COO−) that fit in the foldamer cavity.
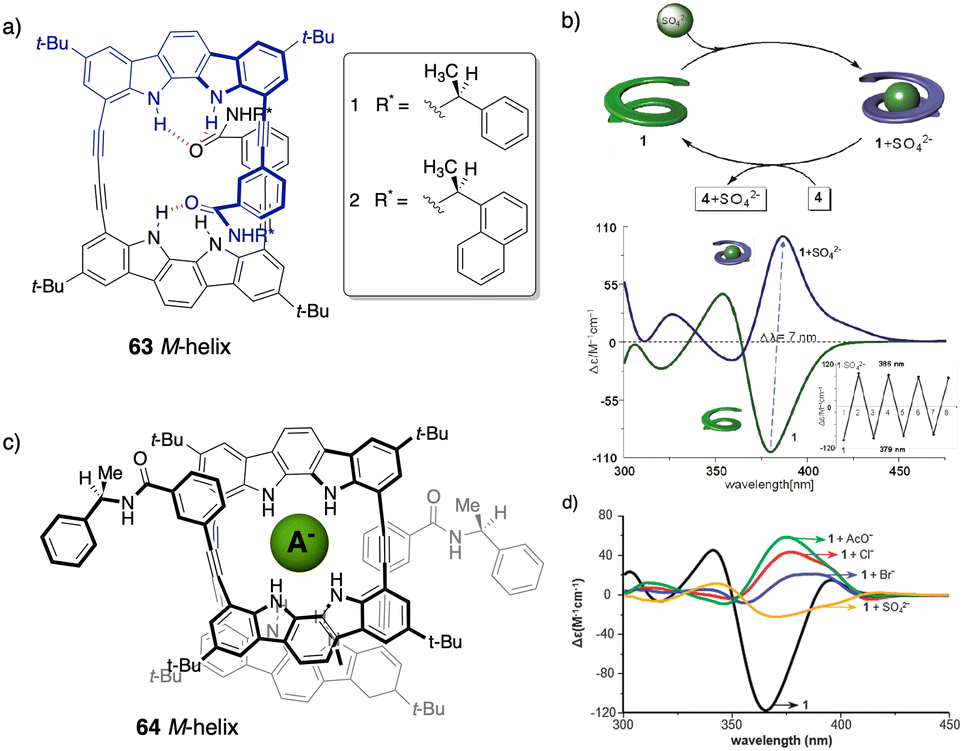 | ||
| Fig. 16 (a) Chemical structure of the foldamer (63). (b) Schematic representation and ECD spectra of the reversible helix inversion of 63 upon addition and removal of sulphate ions (reproduced from ref. 172 with permission from the American Chemical Society, copyright 2011). (c) Molecular structure of a complex between 64 and different anions. (d) ECD spectra of 64 in the absence (black) and in the presence of different tetrabutylammonium (TBA) anion salts (reproduced from ref. 173 with permission from the Royal Society of Chemistry, copyright 2011). | ||
Recently, Talukdar and coworkers reported a selective chloride anion responsive foldamer in which a double helical structure suffers a conformational change (65) (Fig. 17a).174 The foldamer-anion interaction causes the unwinding of the double helical structure to form an anion-coordinated supramolecular polymeric channel, and where the double helical structure can be recovered by adding Ag+ salts (Fig. 17b), demonstrating the reversibility of the process. Moreover, the formation of the anion-induced supramolecular ion channel results in efficient ion transport across lipid bilayer membranes with excellent chloride selectivity (Fig. 17c). This work demonstrates that anion–cation-assisted stimulus responsive unwinding and rewinding of artificial double-helix systems paves the way for developing smart materials with potential biomedical applications.
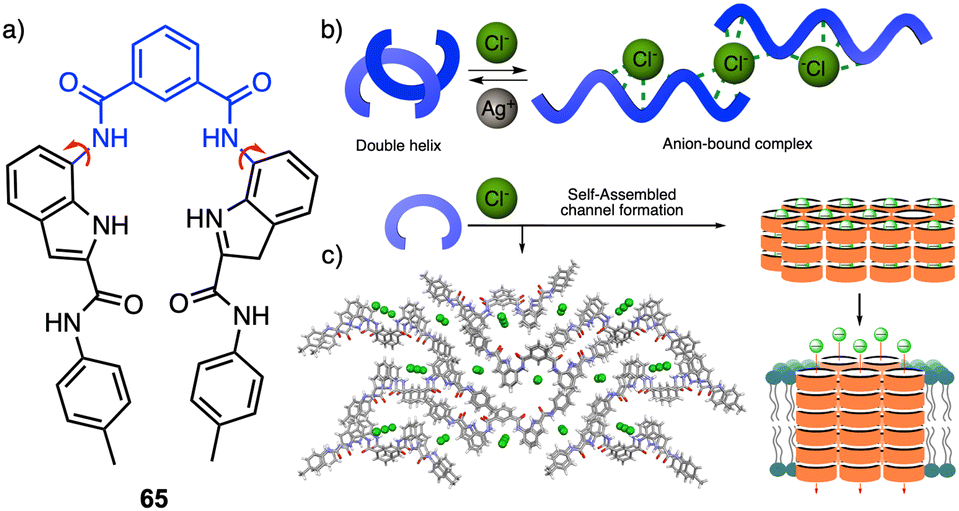 | ||
| Fig. 17 (a) Chemical structure of 65 that folds into a double helix. (b) Schematic illustration of conformational changes from double helix to anion-coordinated supramolecular assembly. (c) Anion-induced supramolecular channel (reproduced from ref. 174 with permission from Springer Nature, copyright 2022). | ||
In general, most of the reported anion-responsive foldamers are not selective for a single anion. They usually bind to different families of anions, e.g., halides (Cl−, Br−, and I−) and oxoanions (NO2−, H2PO4−, HSO4− and CH3COO−).175,176
However, selectivity is improving in recent years as the reversal mechanisms become clearer. In this regard, a selective oxoanion foldamer-switch was recently reported by Clayden,177 who developed axially racemic oligourea foldamers [66-(a–g)] (Fig. 18a) made from mesocyclohexane-1,2-diamine residues that can adopt a preferred screw-sense, differentiating their termini groups by the different characteristics of their hydrogen bonds.
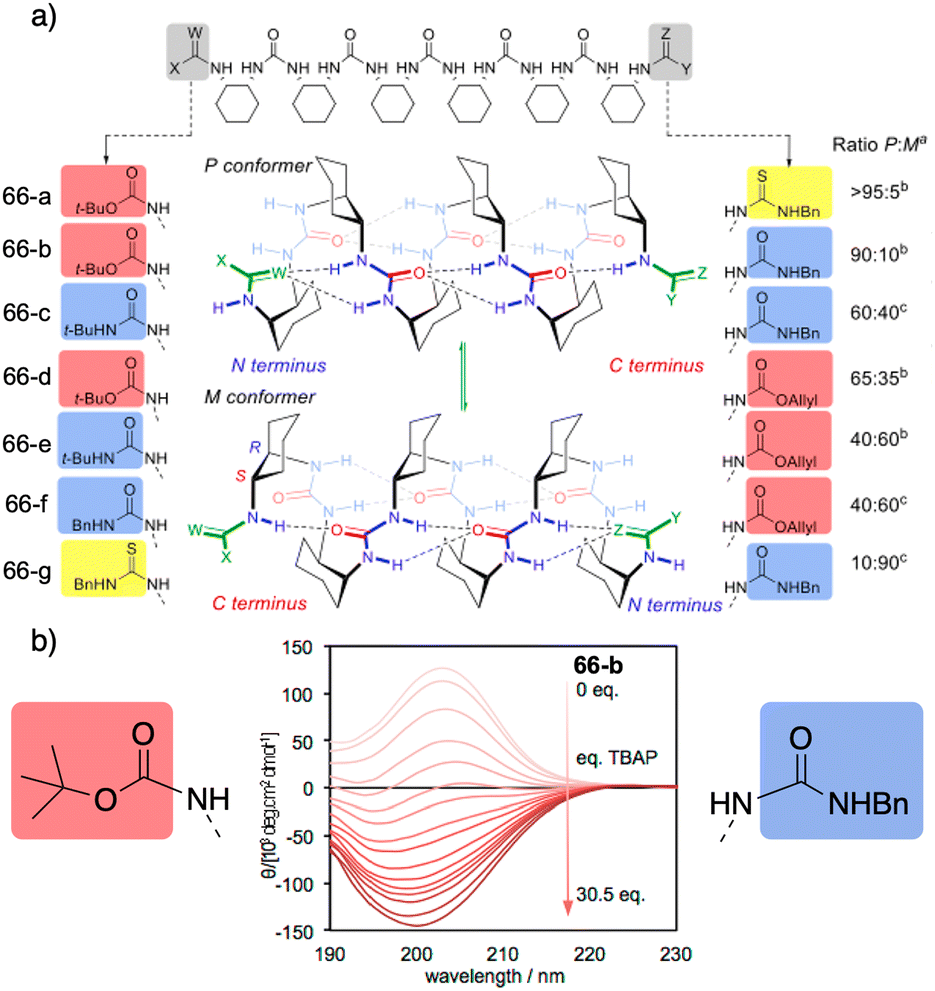 | ||
| Fig. 18 (a) 2.512/14-helical oligourea foldamers as torsion balances for hydrogen-bonding strength. (b) Screw-sense reversal and hydrogen-bond-directionality reversal upon addition of tetrabutylammonium phosphate (TBAP) (0.2 mM in MeCN). The end groups of the foldamer used in the titration are displayed on either side of the CD spectra. Reproduced from ref. 177 with permission from the American Chemical Society, copyright 2018. | ||
Thus, a dynamic equilibrium between two alternative screw sense conformers is set up whose relative population is determined by the competing hydrogen-bonding properties of the terminal groups, dictating the global hydrogen-bond directionality of the foldamer. In this case, these preferences are relatively insensitive to solvent effects and to neutral hydrogen-bond donors or acceptors. However, addition of a geometrically compatible anionic binding partner (acetate or phosphate) can induce a global refolding of the oligomer. This foldamer/anion interaction causes the exposure of previously embedded hydrogen-bond donors, resulting in the inversion of the conformational preference. For example, upon addition of achiral anion phosphate delivered from a tetrabutylammonium salt to a solution containing foldamer 66-b, an inversion of the CD spectra is observed. This suggests an inversion of the foldamer screw-sense (Fig. 18b) induced by anion binding to the thiourea-terminated foldamer that leads to a conformational change creating a switch in the hydrogen-bond directionality to favour the bind between the thiourea and the phosphate.
Helix inversion can also be achieved from metallo-foldamers when they interact with anions. Thus, Miyake created a peptide foldamer based on Inai's design82–85 that contains, in the middle of the sequence, a chiral N,N′-ethylenebis[N-methyl-(S)-alanine] connected to the N terminus of two H-(Aib-ΔPhe)2-Aib-OCH3 pentamers through a peptide bond (67).178 Addition of a hexacoordinated metal center [Co(II), Zn(II), or Ni(II), delivered as M(ClO4)2 salts] to a foldamer solution, forms a Λ–Δ complex with the N,N′-ethylenebis[N-methyl-(S)-alanine] of the foldamer that induces a P helix. Additional addition of NO3− anions (from TBANO3) produces a helix inversion from P to M due to a Λ- to Δ-form change in the metal complex, resulting in a chiral transfer from the metal center to the pentapeptide chains (Fig. 19a). Interestingly, analogous P to M helix inversion process was observed for Zn(II), Co(II) and Ni(II)-based complexes, although with a different time scale, which is based on the substitution lability of the metal center. Thus, the present metallo-peptide complex offers time-tunable peptide helix inversion that has a time scale from milliseconds to hours, as revealed by the stopped flow CD measurements (Fig. 19b).178
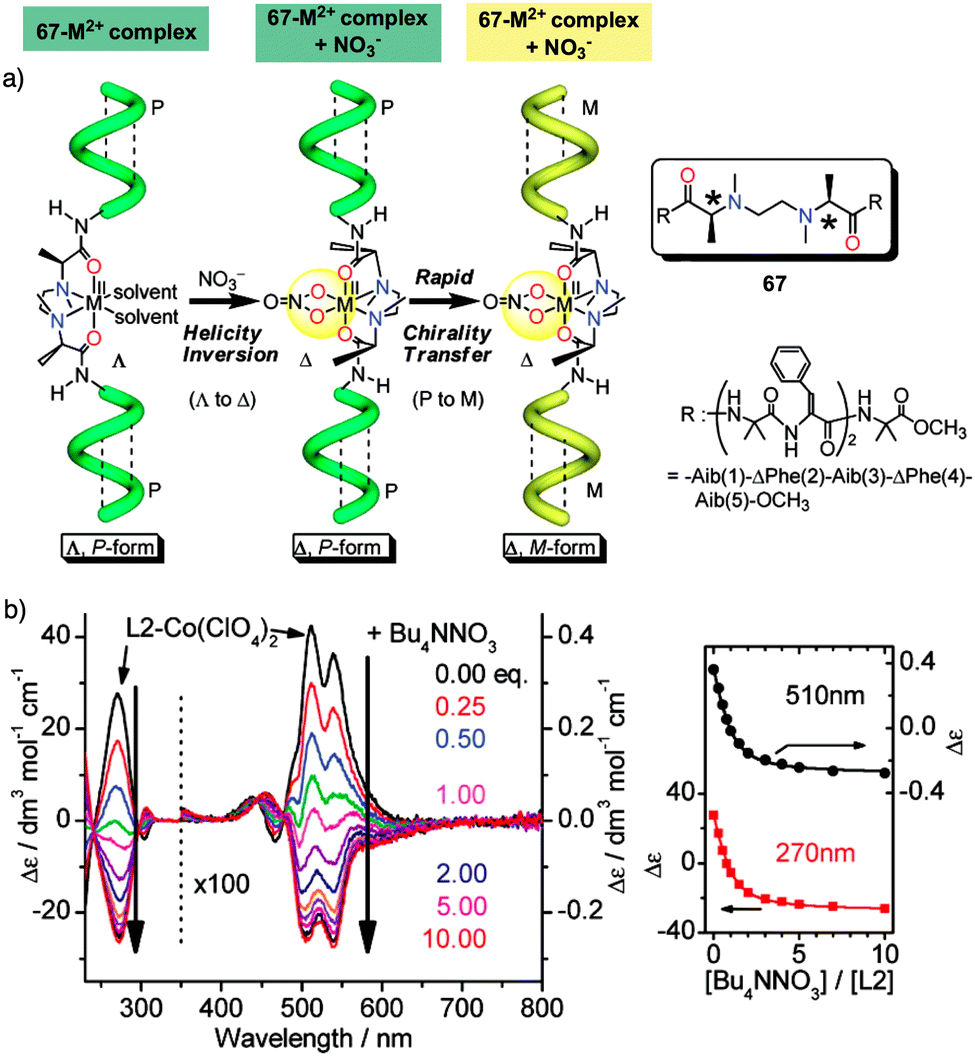 | ||
| Fig. 19 (a) Anion induced helix inversion of metallo-foldamer 67-M2+ (M2+ = Co, Zn, Ni). (b) CD spectra changes of the 67-Co(ClO4)2 complex upon addition of Bu4NNO3 and titration profiles of the CD wavelenght at 270 and 510 nm. Reproduced from ref. 178 with permission from the American Chemical Society, copyright 2008. | ||
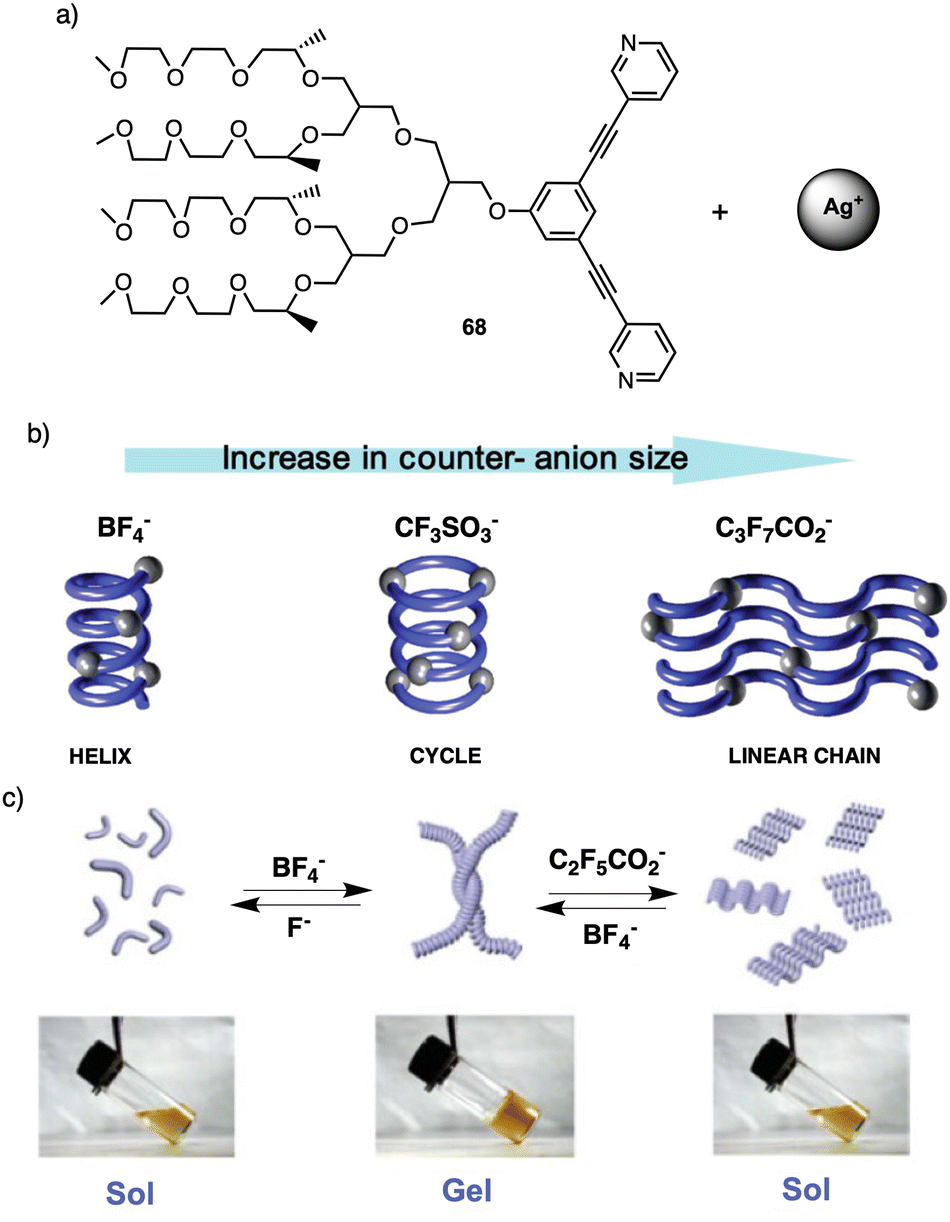 | ||
| Fig. 20 (a) Chemical structure of 68. (b) Schematic representation of the different morphologies adopted by 68-Ag+ coordination polymer induced by the anion size. Reproduced from ref. 179 with permission from the American Chemical Society, copyright 2004. (c) Schematic representation of the reversible conversion between folded and unfolded conformations of the 68-Ag+ coordination chain upon counteranion exchange Reproduced from ref. 180 with permission from John Wiley and Sons, copyright 2018. | ||
For instance, fibers obtained by combining 68 with silver nitrate (AgNO3) or silver tetrafluoroborate (AgBF4)—small anions—self-assemble into helical chains that are organized in a 2D hexagonal lattice (Fig. 20b). However, when a silver triflate salt is used [Ag(CF3SO3)], dimeric cycles are formed that are stacked on top of each other, giving rise to columns that are assembled laterally in a hexagonal fashion (Fig. 20b). This assembly behavior changes dramatically when silver salts containing large anions are used. For instance, metallo-supramolecular polymer chains based on silver salts as heptafluorobutyrate (CF3CF2CF2CO2) are organized in a lamellar structure (Fig. 20b). Therefore, these results demonstrate how a variation in the size of the counteranion can regulate the secondary structure of the coordination polymer chain, from helical, cyclic, to unfolded linear chain conformations in the solid state. This unique self-assembly behavior can be explained by considering the size of the counteranion and consequent chain conformation to maximize the electrostatic interactions.
This transition was attributed to the strong attraction between fluoride (F−) and silver ions leading to depolymerization of the helical polymer into individual molecules. In the case of C2F5CO2− Later, Lee found that anion exchange in the coordination polymer 68-Ag+ led to a reversible sol–gel transition in water.179 This sol–gel effect occurs due to a change in the secondary structure of the coordination polymer, from a folded helical conformation to an unfolded zigzag conformation. Thus, in the presence of BF4− anions, 68-Ag+ results in a gel that is liquified when fluoride (F−) or pentafluoropropionate (C2F5CO2−) are present (Fig. 20c), the gel liquefaction occurs due to the large size of this counterion compared to BF4− (Fig. 20c).180
More recently, Muranaka reported an anion-responsive π-conjugated supramolecular assembly based on the chiral anion receptor 69 (Fig. 21a).181 A tripropyl-substituted triazatriangulenium planar cation (TATA+) (Fig. 21a) forms an electrostatic interaction with anion receptor 69, which traps the Cl− anion delivered from the TATA-Cl salt. This electrostatic interaction results in a layer-by-layer chiral aggregate consisting of planar units with opposite charges (Fig. 21b). Aggregation studies of 69 in the absence and presence of TATA-Cl produce ECD and CPL spectra with opposite signs (Fig. 21c and d). Since the assembly modes depend on the ion-free and ion-pairing states of emissive π-conjugated molecules, the chiroptical properties change accordingly and thus result in readily tuneable CPL-active materials.
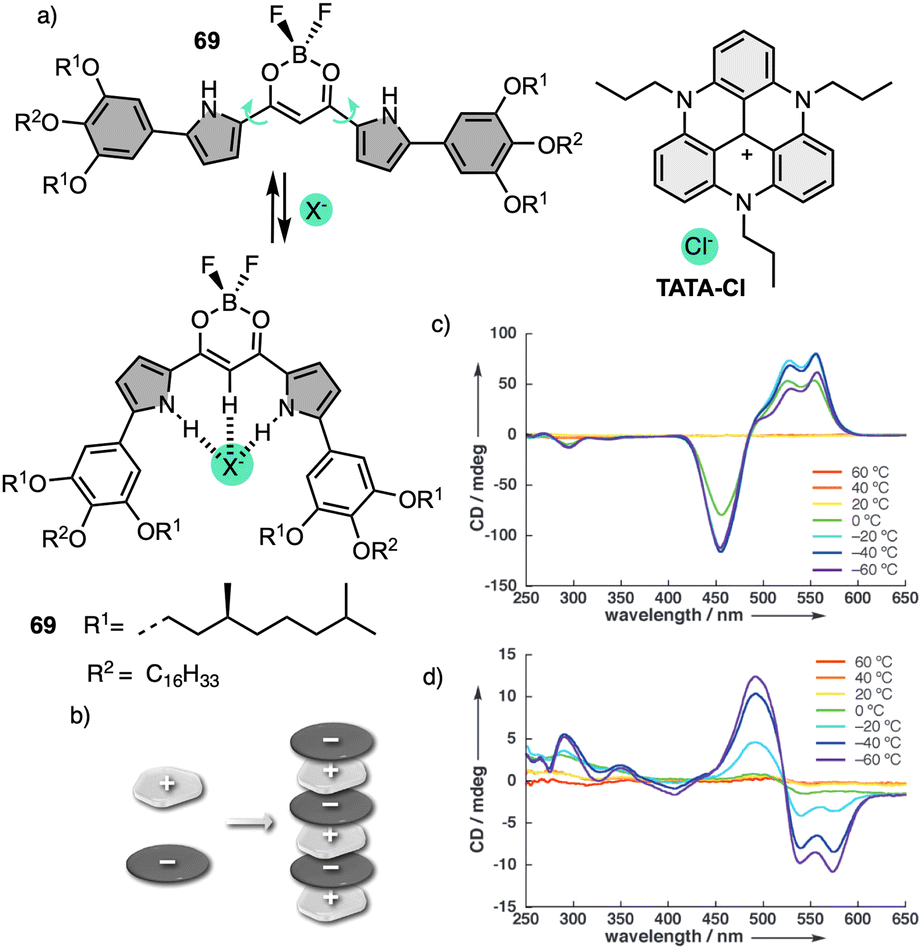 | ||
| Fig. 21 (a) Chemical structure and anion-binding modes of anion receptors 69. (b) Schematic model of charge-by-charge assembly. VT CD spectra of 69 in (c) absence Cl− and (d) presence of Cl− added as a TATA salt (1 equiv.). Reproduced from ref. 181 with permission from John Wiley and Sons, copyright 2013. | ||
In elongated PPA structures, the conjugation between alternating double bonds increases producing a red shift of the polyene band in the UV-vis spectra that is accompanied by a change in colour of the solution. Hua and Tian also reported another PPA that bears a naphthalimide as pendant connected to the PPA through an amide bond.186 This polymer works in a similar way to the previous ones, acting as a colorimetric and fluorescent chemosensor for the fluoride anion.
To create a selective anion detector based on a dynamic helical polymer, such as a PPA, the design of the monomer is crucial, as with many other stimuli. For instance, Kakuchi designed a chiral PPA that bears, as a pendant, an amino acid connected to the backbone through a urea group —L-leucine, L-glutamic acid, L-aspartic acid, L-phenylalanine, L-isoleucine, and L-alanine; PPA-Leu, PPA-Glu, PPA-Asp, PPA-Phe, PPA-Ile, and PPA-Ala, respectively.183,187 Thus, while the chiral amino acid is used to induce a specific helical sense in the PPA, the acidic urea groups are used to interact with anions. The anion/chiral pendant interaction causes a conformational change in the monomer repeating unit, which is harvested by the polyene backbone producing an elongation and/or a helical sense change. All these polymers showed modulable helicities by external stimuli. Interestingly, the addition of various anions as ammonium salts, such as tetra-n-butylammonium acetate (TBAA), benzoate (TBAB), nitrate (TBAN), azide (TBAN3), fluoride (TBAF), chloride (TBACl) and bromide (TBABr), to solutions containing the different polymers showed different ECD responses depending on polymer and anion (Fig. 22a for PPA-Leu). Therefore, the anionic signalling property of these polymers depends on both the size of the anion and the amino acid selected as the pendant group. For instance, while the addition of CH3COO− to a solution of PPA-Leu, PPA-Ala, and PPA-Ile induces a P helix in the polymer (ECD500 > 0), an M helix is induced in PPA-Phe, PPA-Asp, and PPA-Glu (ECD500 < 0) (Fig. 22b). Similar effects were observed with other anions, such as C6H5COO−, F−, Cl−, and Br−, indicating that these polymers can be used for distinguishing anionic hosts due to the resulting unique patterns. However, the mechanisms of helix modulation have not been fully described.
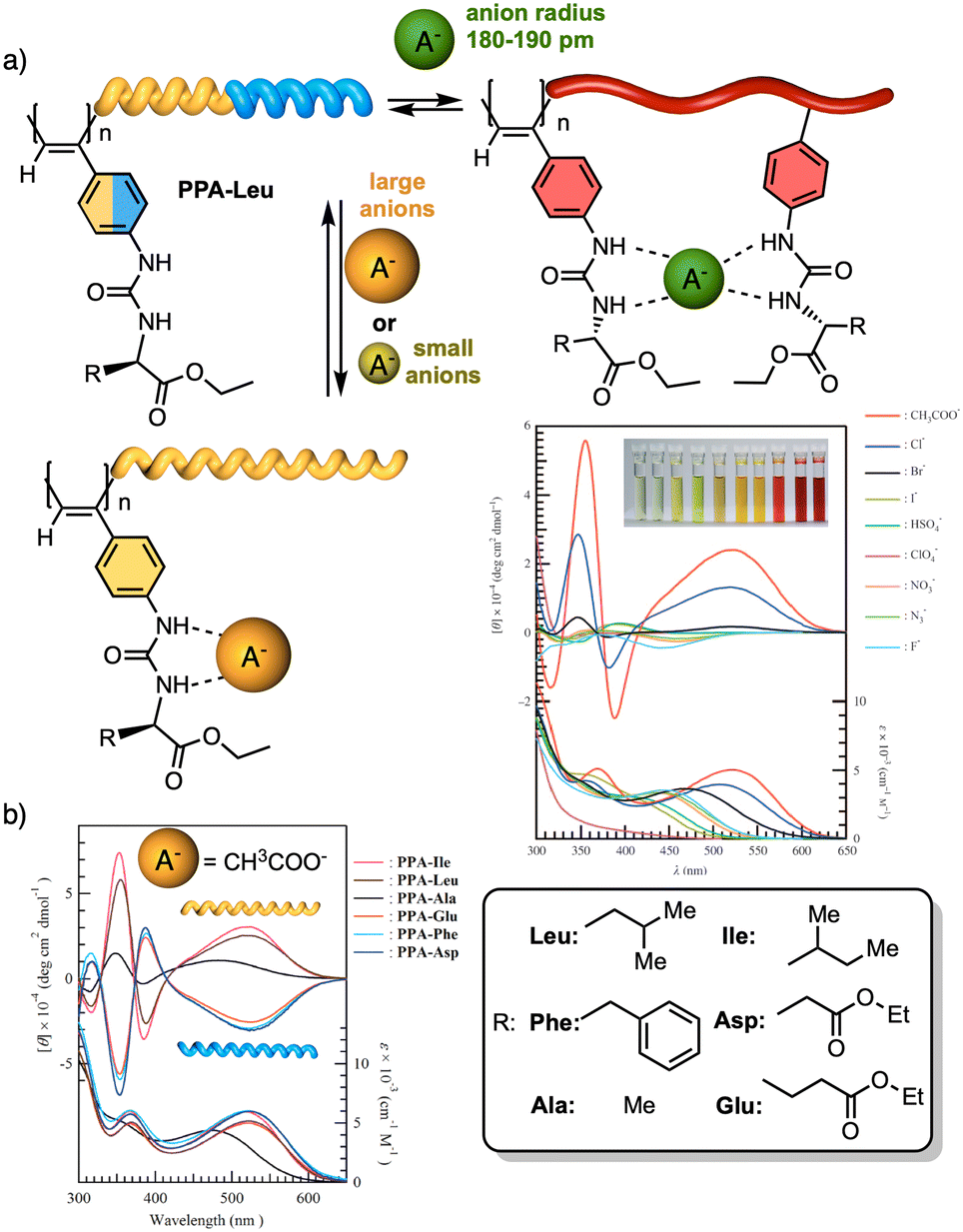 | ||
| Fig. 22 (a) Schematic representation of size-selective detection for anions based on a PPA. ECD (upper) and UV/Vis absorption (lower) spectra of poly-PPA-Leu upon addition of several anions (1 equiv. of anion to a 3.3 mmol L−1 THF solution of poly-PA-Leu) (reproduced from ref. 183 with permission from John Wiley and Sons, copyright 2008). (b) CD (upper) and UV-vis (lower) spectra of urea-functionalized polymers with CH3COO− Reproduced from ref. 187 with permission from the American Chemical Society, copyright 2004. | ||
Working in the same direction, Kakuchi subsequently reported analogous studies but using a different group for anionic binding, i.e., sulfonamide group.188 During the PPA/anion interaction studies, the behavior of sulfonamide–PPAs is coincident with that of urea–PPAs described above, confirming that the amino acid side chain plays a relevant role in the anion–PPA interaction.
On the other hand, PPAs can be used as chiroptical sensors to detect anions. Freire reported a PPA (poly-(R)-11) that bears an acidic anilide group as pendant (Fig. 23).189 This polymer behaves as axially racemic (mixture of P and M helices) due to the presence, in the pendant, of two conformers in equilibrium that have the carbonyl and methoxy groups [(O–C)–(CO)] in antiperiplanar (ap) or synperiplanar (sp) orientations. However, upon interaction with different TBAA salts, the equilibrium can shift toward the M or P helix due to a supramolecular interaction between the anilide group and the anion. Thus, while AcO−, BzO− and N3− stabilize the sp conformer in the pendant group of poly-(R)-11 and induce an M-helix, F− and CN− stabilize the ap conformer inducing a P-helix in the PPA (Fig. 23).
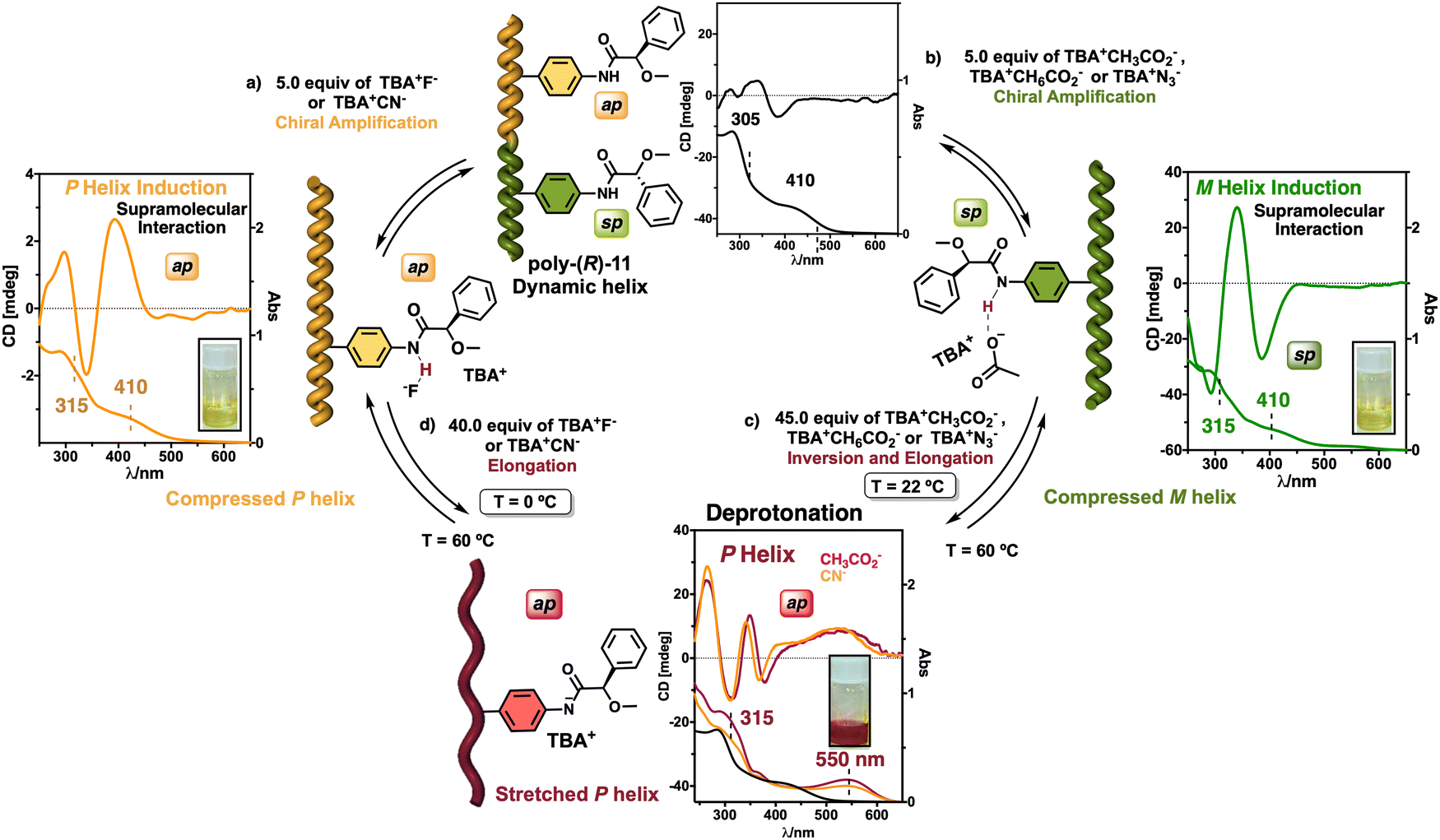 | ||
| Fig. 23 (a) P and (b) M helix induction in poly-(R)-11 by addition of 5.0 equiv. of TBAA (A = fluoride or cyanide) or (A = acetate, benzoate or azide) respectively. Elongation and P helix induction in poly-(R)-11 by addition of (c) 45.0 equiv. of TBAA (A = acetate, benzoate or azide) at rt or (d) 40.0 equiv. of TBAA (A = fluoride or cyanide) at 0 °C. Reproduced from ref. 189 with permission from Elsevier, copyright 2021. | ||
Interestingly, upon adding larger amounts of TBAA salts, a colorimetric shift from yellow to red is observed in the polymer solution (Fig. 23). This colour change is produced by the deprotonation of the anilide group, and the induced elongated P helix depends only on the accommodation of the deprotonated anilide(−)/TBA(+) ion pair within the helix, a phenomenon independent of the anion used to deprotonate the anilide group.
Recently, Gao designed an optically active helical polyacetylene carrying the amino acid (L)-alanine as pendant group, connected to the PA backbone through the C-terminus and functionalized at the N-terminus with a naphthalene diimide (NDIs) derivative (poly-70) (Fig. 24).190 In this polymer, the PA/anion interaction is used to regulate the stacking modes of π-conjugated NDIs building blocks. Thus, anions such as Cl− induce a dynamic reorganization of the molecular arrangement of NDIs within the helical scaffold, causing a transition from H-type to J-type aggregates, which activates the fluorescence emission at 515 nm and disrupts the induced helical structure of the polymer backbone.
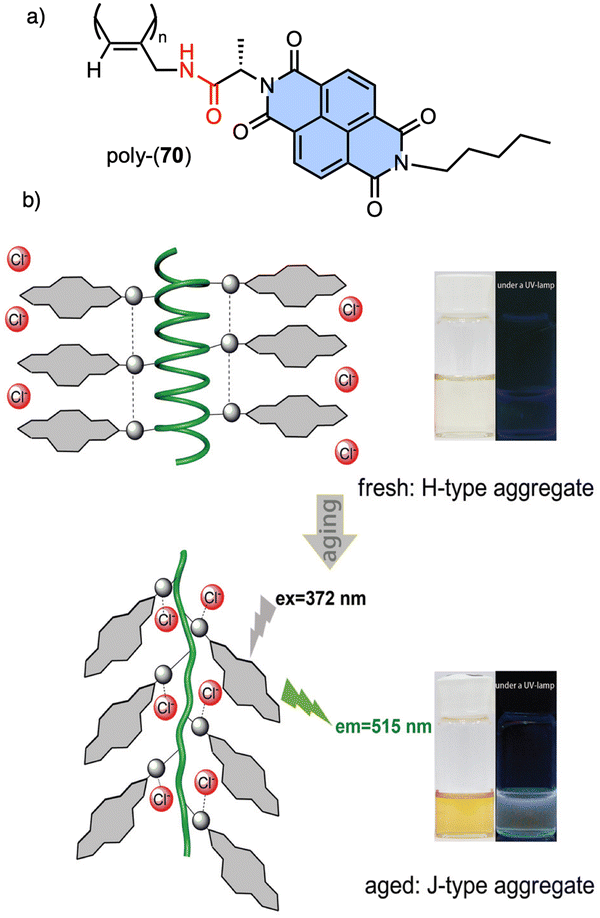 | ||
| Fig. 24 (a) Chemical structure of poly-70. (b) Schematic representation of time-dependent H to J structural changes in poly-70 by addition of Cl− and the corresponding fluorescence effects. Reproduced from ref. 190 with permission from the Royal Society of Chemistry, copyright 2021. | ||
Xie designed an asymmetrically 2,4-disubstituted PPA bearing a benzamide of (L)-alanine ethyl ester and a methoxycarbonyl group as chiral and achiral substituents respectively (poly-71) (Fig. 25). This polymer adopts a contracted helix stabilized by intramolecular hydrogen bonding networks among neighbouring pendants amides.191 This helical scaffold possesses small pockets suitable for accommodating the smallest anion, F−, which has a high basicity, excluding other anions. During the poly-71/F− interaction, the contracted cis–cisoidal backbone of poly-71 transforms into a stretched cis–transoidal exhibiting a nonpreferred-hand-sense helix, facilitating subsequent binding of F− anions. Such unzipping effect gives rise to an ultralow detection limit for F− (2.85 × 10−8 M or 0.54 ppb). Meanwhile, the colour of the solution undergoes a change from colourless to yellow due to the conjugated cis–transoidal conformation. As a result, the study achieves a remarkable naked-eye recognition of fluoride ions (F−) with high specificity and sensitivity.192
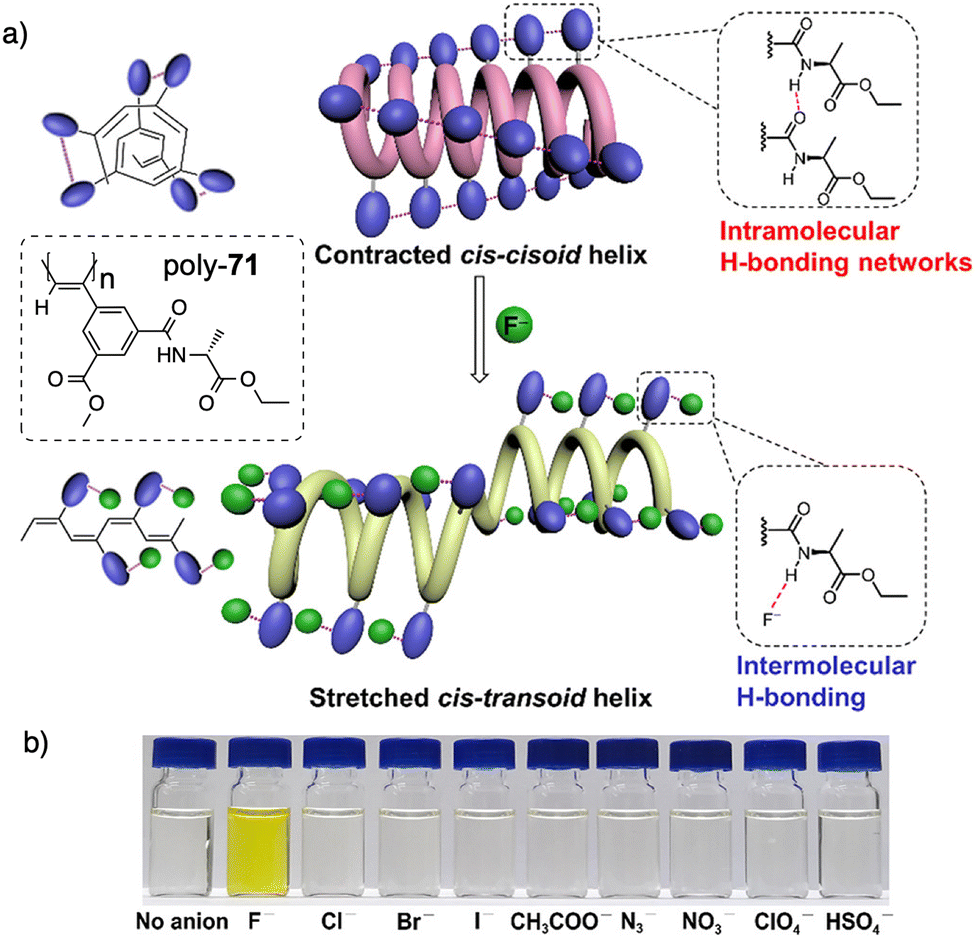 | ||
| Fig. 25 (a) Schematic mechanism of specific and (b) colorimetric fluoride ion recognition by poly-71. Reproduced from ref. 191 with permission from the American Chemical Society, copyright 2022. | ||
2.3 External stimuli: cations
Metal ions can coordinate to many functional groups inducing conformational changes, folding or aggregation of molecules or macromolecules. This ability, in combination with the toxicity properties of metals, makes helical polymers responsive to metal ion stimuli a very attractive field.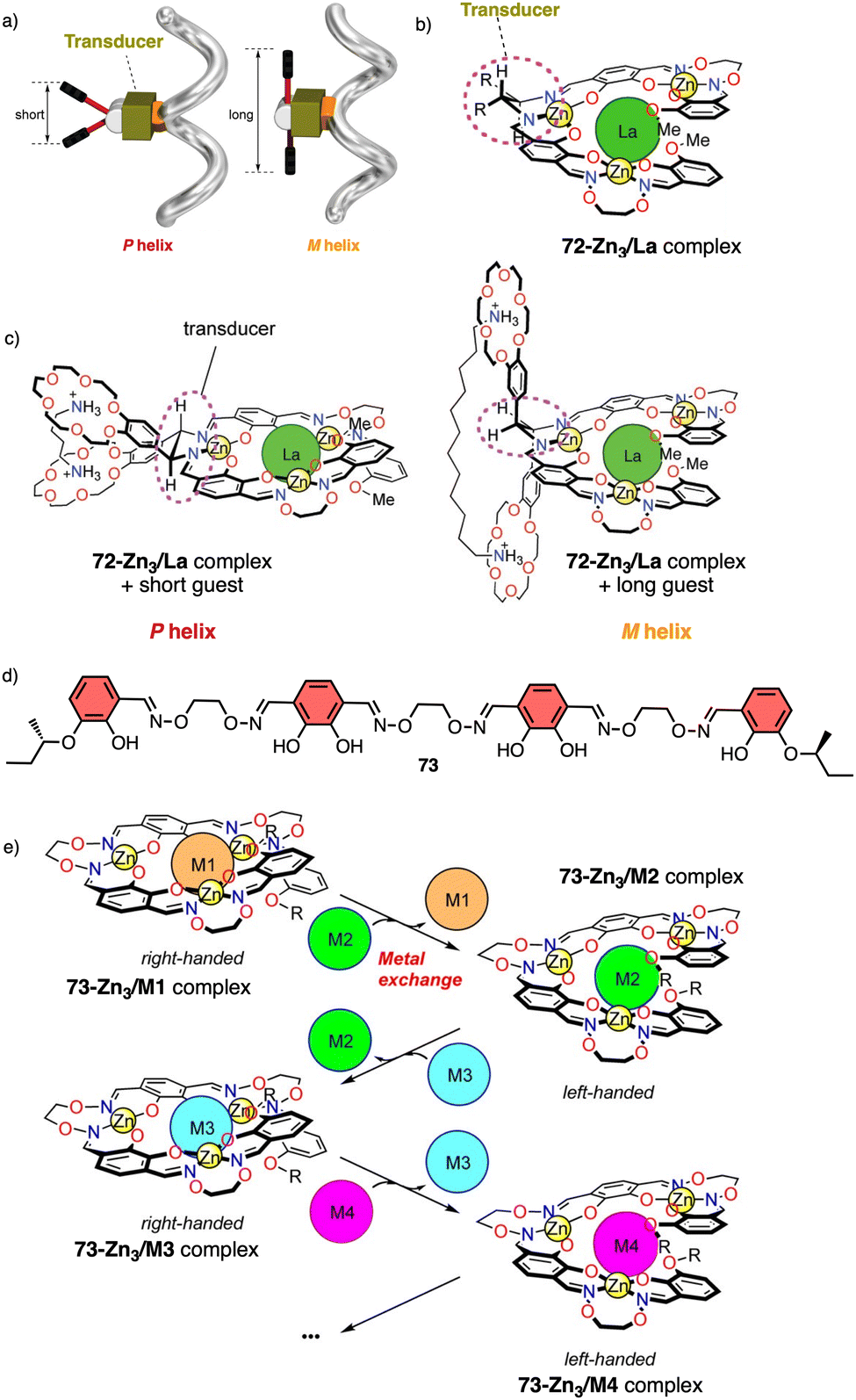 | ||
| Fig. 26 (a) Schematic representation of helix inversion based on a transducer mechanism. (b) chemical structure for 72-Zn3/La complex. (c) Helix induction of 72-Zn3/La complex by interaction with diamines (reproduced from ref. 197 with permission from the American Chemical Society, copyright 2011). (d) Chemical structure of ligand 73. (e) Schematic representation of helix inversion via metal exchange of helical complexes (reproduced from ref. 198 with permission from the American Chemical Society, copyright 2013). | ||
In another example, the authors use metal exchange of helical complexes to induce a helix inversion.198,199 In this case, the ligand H673 (Fig. 26d) underwent a four-step conversion (H673 → 73Zn3 → 73Zn5 → 73Zn3Ba → 73Zn3La) upon sequential metal addition (Zn2+, Ba2+, then La3+). Associated with the conversion, three-step helicity inversion took place (73Zn3, P helix → 73Zn5, M helix → 73Zn3Ba, P helix → 73Zn3La, M helix) (Fig. 26e).
Tanaka prepare a discrete chiral metallofoldamer composed of homochiral metallosalen complexes [(74RR-Ni)2Pd] (Fig. 27a).200 Connecting chiral Ni-salen moieties to the side chains with a transition metal center like Pd(II), at proper coordination bond angles, would lead to a helical organization of the columnar assembly. The metallosalen exhibited thermotropic columnar liquid crystalline properties and formed hexagonal columnar phases, which exhibited high thermal stability and showed orientation along the extrusion direction (Fig. 27b).
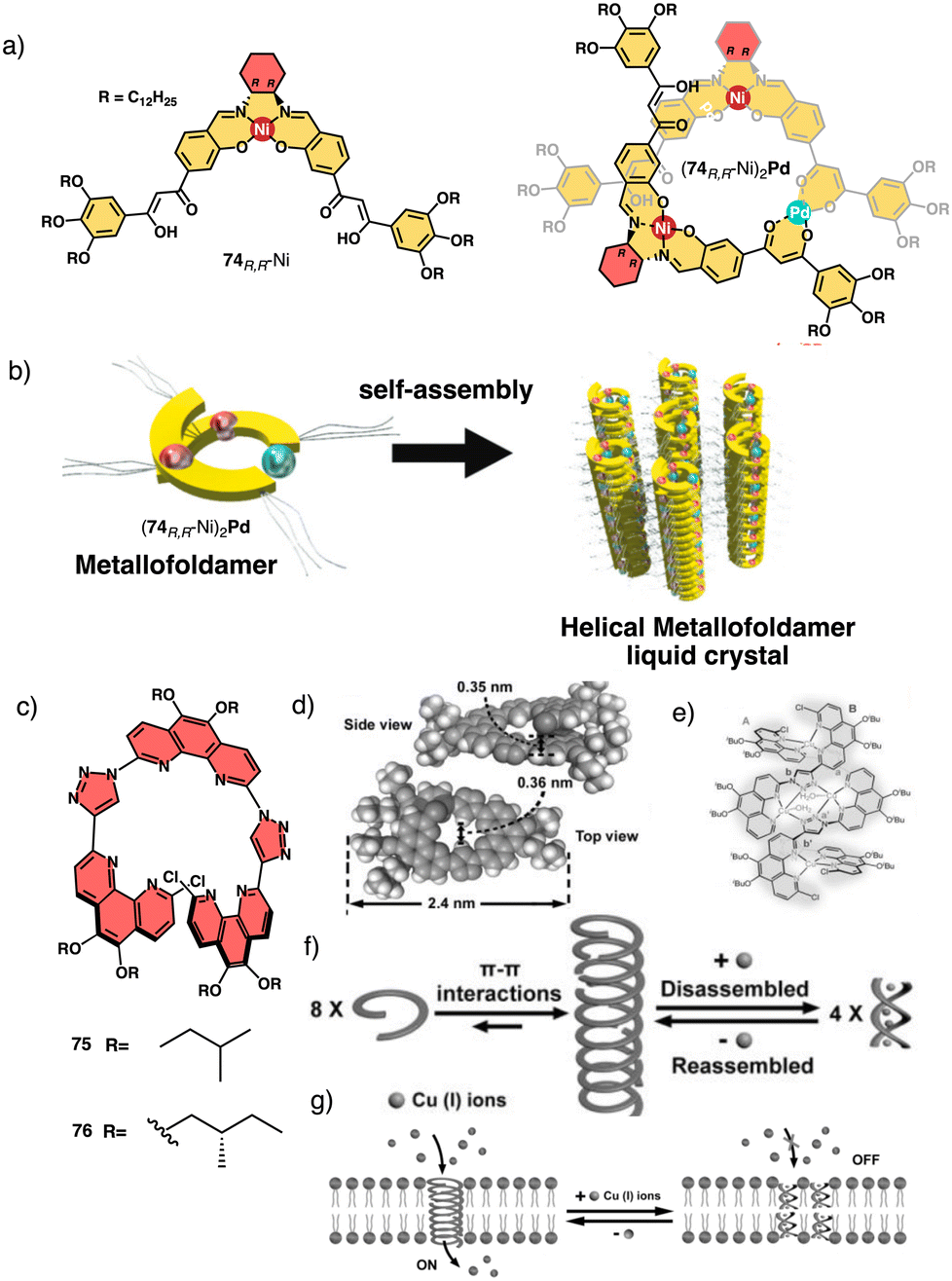 | ||
| Fig. 27 (a) Chemical structure for 74RR-Ni and (74RR-Ni)2Pd. (b) Helically assembled LC metallofoldamer (74RR-Ni)2Pd (reproduced from ref. 200 with permission from the Royal Society of Chemistry, copyright 2022). (c) Molecular structures of 75 and 76. (d) Crystal structure of 75. (e) Molecular structure of 752-[Cu(CH3CN)4PF6]4 (f) schematic representation of the tentative formation, disassembly and reassembly of the 1D hollow helical tubes from 75. (g) Schematic representation of the reversible ligand-gated ion channel formed by 75 triggered by Cu+ and NH3·H2O reproduced from ref. 201 with permission from John Wiley and Sons, copyright 2020. | ||
Another interesting example is the foldamer based on triazole-linked phenanthroline ligands (75–76) (Fig. 27c) designed by Zhu and Liu.201 In the extended solid-state, these ligands formed oligomeric channels through π–π interactions, that can trap small guest molecules (Fig. 27d). Upon complexation with copper ions (Fig. 27e), the foldamers intertwine to form a double-stranded helix consisting of two foldamers coordinated with four Cu+ ions, resulting in a metallofoldamers (Fig. 27e and f). This structural change is reversible, and it is possible to interconvert the hollow channels observed in the extended solid-state structure of the ligands into the “closed” structure of the copper complex (Fig. 27f) and vice versa (Cs+, Rb+, K+, Na+, Li+). By adding copper ions, ion transport is inhibited due to the structural change in the foldamer metal complex, which can be recovered once the Cu+ ions are removed (Fig. 27g).
Maayan demonstrated the positive allosteric cooperativity ability of a peptoid that incorporates four 8-hydroxyquinoline (HQ) ligands at fixed positions to create two distinct metal-binding sites (77) (Fig. 28).202 Peptoid 77 can bind a copper ion (Cu2+) in one site that leads a conformational change that in turn facilitates the coordination of a metal ion—Zn2+, Co2+—to the second metal binding site, demonstrating a positive allosteric cooperativity in peptidomimetics (Fig. 28). This finding was confirmed by competition experiments using a foldamer (78) that did not exhibit an increase in order upon complexation with Cu2+ (78Cu) (Fig. 28).
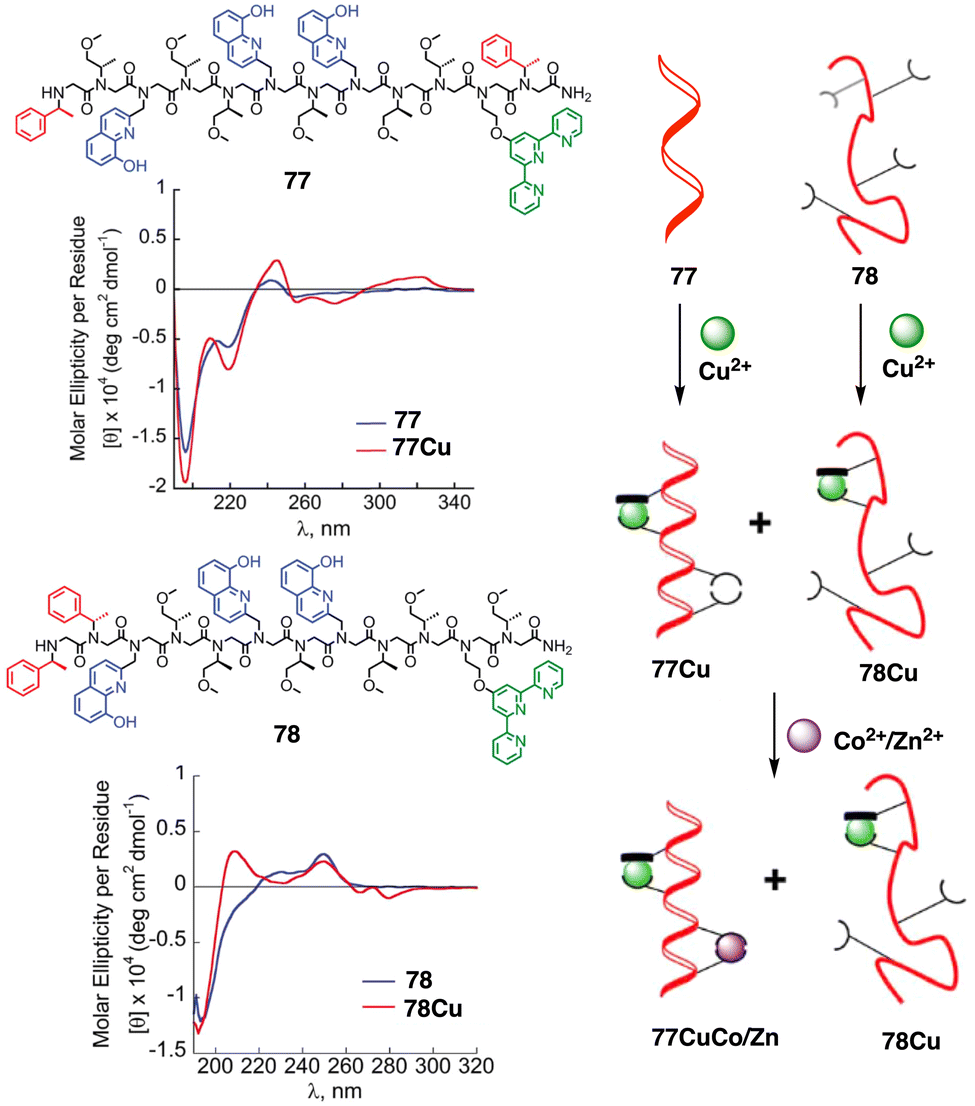 | ||
| Fig. 28 Chemical sequences of peptoid oligomers 77 and 78. CD spectra of peptoid 77 with 77Cu complex and 78 with 78Cu. A cartoon shows the positive allosteric cooperative binding of 77Cu and 78Cu. Reproduced from ref. 202 with permission from the Royal Society of Chemistry, copyright 2019. | ||
Jeong demonstrated the interplay between temperature and metal ions as stimuli in a double-stranded dinuclear helicate.128 Through self-assembly, the study combined an aromatic helical foldamer 18 with dichloropalladium(II) (Fig. 7), yielding a helicate that undergoes syn–anti and syn–syn conformational switching in response to temperature changes.128 In the field of aromatic oligoamides foldamers, Huc introduced, within a foldamer sequence, a central diacid residue known as pyridazine–pyridine–pyridazine (pyz–pyr–pyz) (Fig. 29a).203,204 The resulting aromatic helical foldamers 79 and 80 have a central residue that can effectively trap metal ions within their helix-shaped cavities (Fig. 29a). In the absence of metal ions, the central pyz–pyr–pyz residue is not folded. However, by addition of alkali or alkaline earth metal ions, the foldamer folds forming a helical structure. Interestingly, the metal ion coordination sphere remains accessible for guest recognition, where cooperative interactions with the helix host strengthen the binding of the guests (see Fig. 29b). Moreover, depending on the metal ion used, the metal-foldamer binding may involve first or second coordination spheres of the metal hydrates (Fig. 29c).
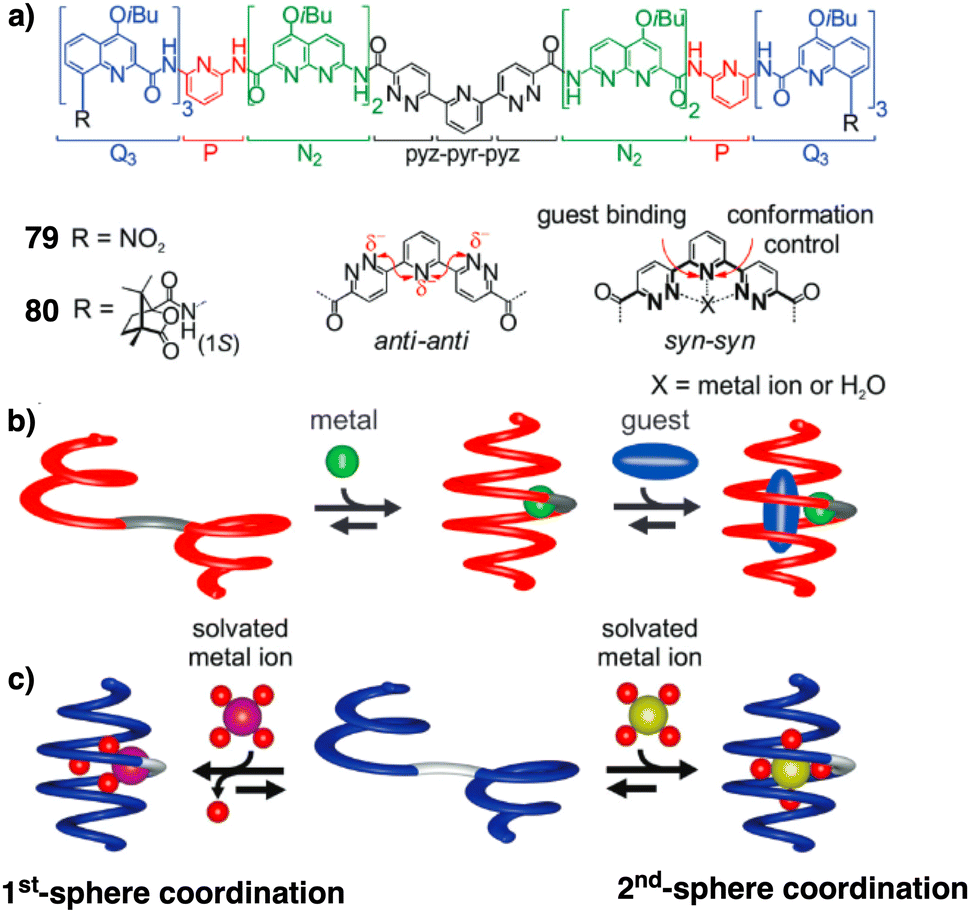 | ||
| Fig. 29 (a) Chemical of aromatic oligoamide sequences 79 and 80 containing the central diacid residue pyz–pyr–pyz and its preferred conformations in the absence and in the presence of a guest. (b) Depiction of helical-capsule folding upon metal coordination and guest binding in the capsule cavity assisted by the metal ion (reproduced from ref. 203 with permission from John Wiley and Sons, copyright 2017). (c) Schematic representation of metal-induced folding modes of a helical-capsule: first-sphere coordination (left) and second-sphere coordination (right). Reproduced from ref. 204 with permission from the Royal Society of Chemistry, copyright 2017. | ||
Contraction motions of double-stranded spiroborate helicates have been studied, in response to the binding and release of protons and/or metal ions, by Yashima.205,206 Thus, novel helicates composed of two tetraphenol strands bridged by two spiroborate groups (81, Fig. 30a) exhibited a unique unidirectional spring-like motion triggered by the presence of sodium cations. The contracted double-stranded helicate with embedded sodium ions undergoes a conformational change upon ion release with cryptand [2.2.1] that results in the extension of the helicate (Fig. 30b). However, when ortho-linked biphenol 82 or 6,6′-linked 2,2′-bipyridine (bpy) 83 are used as central residues (Fig. 30a), the extended form cannot be induced after addition of cryptand [2.2.1] due to their strong interaction with the sodium ion.
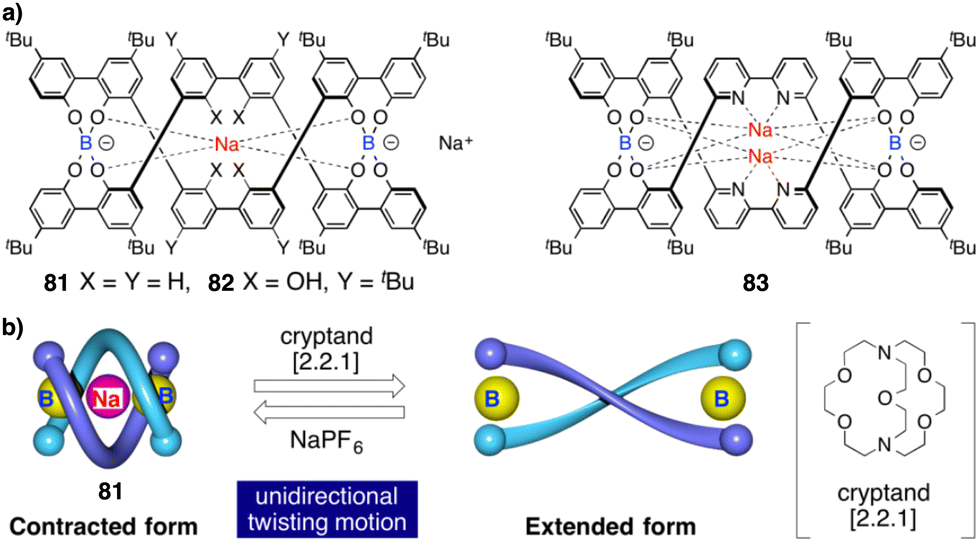 | ||
| Fig. 30 (a) Chemical structures of double-stranded spiroborate helicates 81, 82, and 83. (b) Schematic representation of the unidirectional spring-like motion upon Na+-ion release and binding in 81. Reproduced from ref. 206 with permission from the Royal Society of Chemistry, copyright 2019. | ||
In another example, Yam found that supramolecular helical metallofoldamers that conduct to a gel can be obtained by combining an oligomeric m-phenyleneethynylene backbone with Pt(II) ions. This metallofoldamer self-assembles through Pt–Pt and π–π interactions to create a gel.208 G. Feng reported also how, depending on the metal used, a ditopic ligand composed of terpyridine and acetylene segments can self-assemble into a helical (Cu2+) or non-helical (Zn2+) aggregate.209 C.-L. Feng reported the self-assembly of (L)-phenylalanine derivatives in presence of different metal ions that determine the helical sense induction of the assemblies.210
Continuing with metal–metal and π–π interactions, Akine contributed to the field with the study of metal complexes that exhibit stacking behaviour consisting of acyclic pyridine–phenol ligands and Ni2+ or Pd2+ ions.211 However, in these systems, the sense or morphology of the chiral aggregate cannot be controlled by the metal ion. In this line, Liu found during the coassembly of para-pyridine imine-linked cholesterol conjugate (pPMPCC, 84, Fig. 31a) with different metal salts that a precise control over the chirality of the resulting metal–organic supramolecular polymer (MOSP) aggregates can be obtained by choosing the correct metal ion (Fig. 31b).212 For instance, metallosupramolecular polymer obtained from 84 and Co2+, Cu2+ and Zn2+ produced chiral aggregates with opposite chirality to those obtained by combination of 84 and Ag+, Mn2+ and Bi3+. Interestingly, in the case of the 84/Bi3+ complex a transition between opposite chiral aggregates can be obtained by varying the 84/Bi3+ ratio (Fig. 31a).
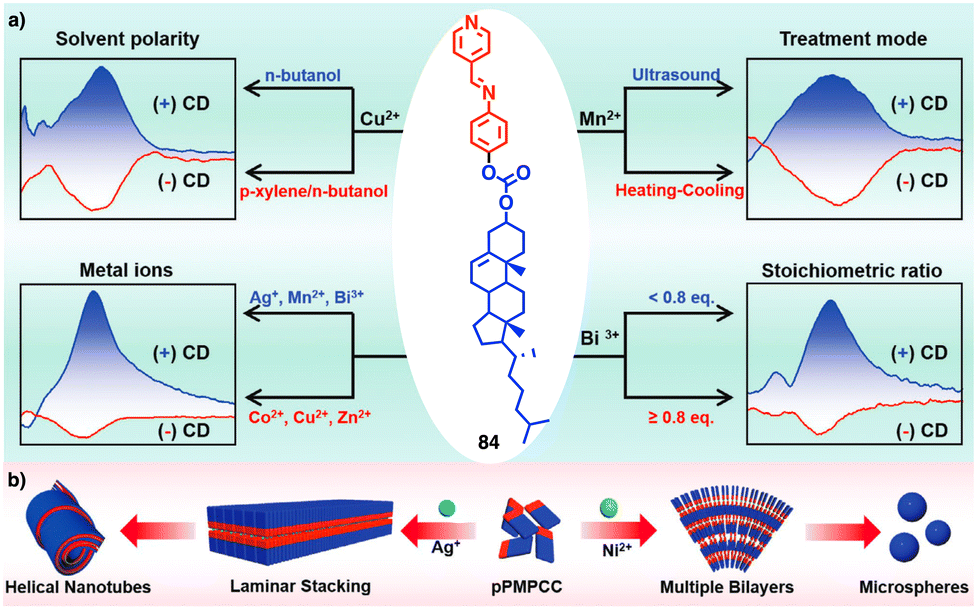 | ||
| Fig. 31 (a) Quadruple modulated dynamic SMCI of MOSP-based aggregation modulated by the stoichiometric ratio, type of metal ions, solvent polarity, and the treatment of heating–cooling and ultrasound. (b) Proposed self-assembly mechanism and aggregation evolution of 84 + AgNO3 and 84 + NiCl2 complex-based coassemblies. Reproduced from ref. 212 with permission from John Wiley and Sons, copyright 2023. | ||
Yashima and co-workers investigated helical foldamer 85, which has a flexible metal-coordination moiety of 2,6-pyridinebis(acylhydrazone) (Fig. 32a).213 This foldamer can adopt different helical structures in the presence or absence of silver metal ions. Moreover, foldamer and metallofoldamer self-assemble to form helical nanofibers with different shapes, which can undergo reversible transformations by adding or removing the silver ions. This macromolecular conformational change is driven by the metal ions and occurs in a cooperative and positive allosteric manner, meaning that the binding of one ion influences the binding of others. Thus, the metal-coordination-driven conformational change of 2,6-pyridinebis(acylhydrazone) linkers from a W-form to a U-form facilitates the formation of another helical foldamer composed of U-shaped linkers (Fig. 32b).
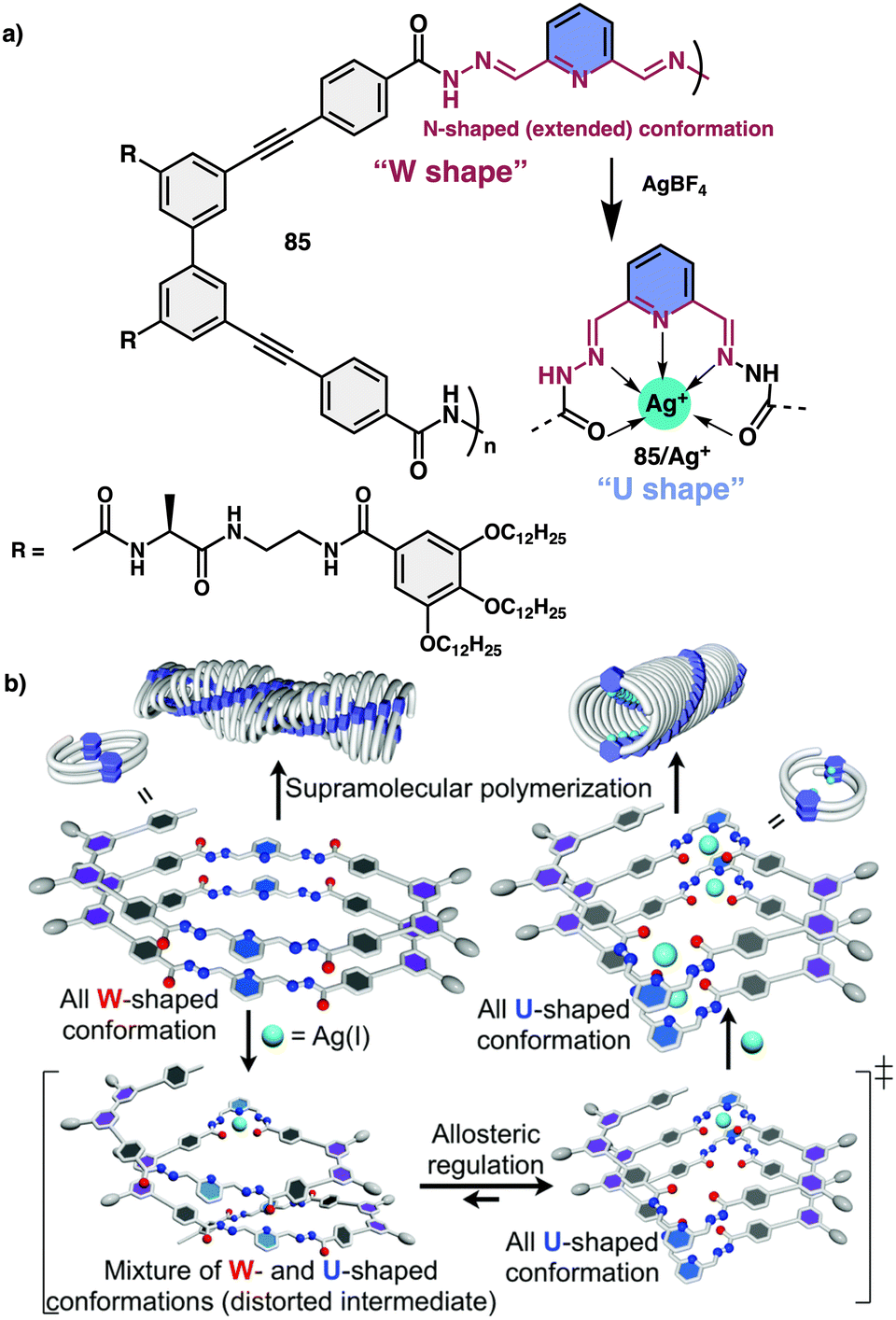 | ||
| Fig. 32 (a) Chemical structure of 85. (b) Schematic representation of Ag(I) ion-triggered W-to-U-shaped conformational changes of 2,6-pyridinebis(acylhydrazone) linker units. Reproduced from ref. 213 with permission from the Royal Society of Chemistry, copyright 2018. | ||
Lin studied the self-assembly and stimuli-responsive properties of a tripodal quinolinamido-based supramolecular organogel (86, TBT-gel) (Fig. 33).214 Through experimental and theoretical studies, it is demonstrated how the TBT-gel assembles into a one-dimensional helical supramolecular polymer through strong hydrogen bonds and π–π interactions. The addition of Fe3+ ions leads to the formation of a metallogel (86-Fe-gel) through coordination interactions. Interestingly, the TBT-gel selectively responds fluorescently to Fe3+ and F− ions via a competitive coordination mechanism, which is attributed to an intramolecular charge transfer process (Fig. 33).
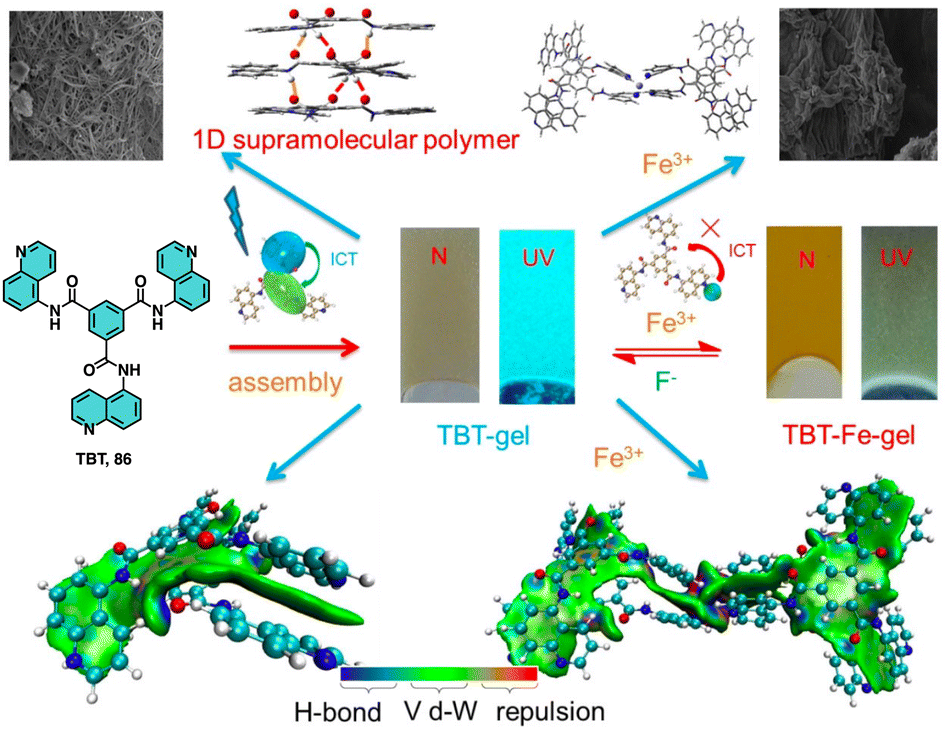 | ||
| Fig. 33 Structure of 86. Proposed assembly and ion response mechanisms of 86 with Fe3+ and F−. Reproduced from ref. 214 with permission from John Wiley and Sons, copyright 2021. | ||
Feng explored the control of circularly polarized luminescence (CPL) in supramolecular gels obtained by co-assembly of a chiral phenylalanine-derived hydrogelator (87) (Fig. 34) and achiral coumarin derivatives triggered by addition of metal ions.215 The study demonstrates that the handedness of CPL in these hydrogels can be efficiently inverted by the incorporation of coumarin derivatives through non-covalent interactions, and further tuned by the addition of metal ions. The key factors influencing CPL inversion are hydrogen bonds, coordination interactions, and steric hindrance.
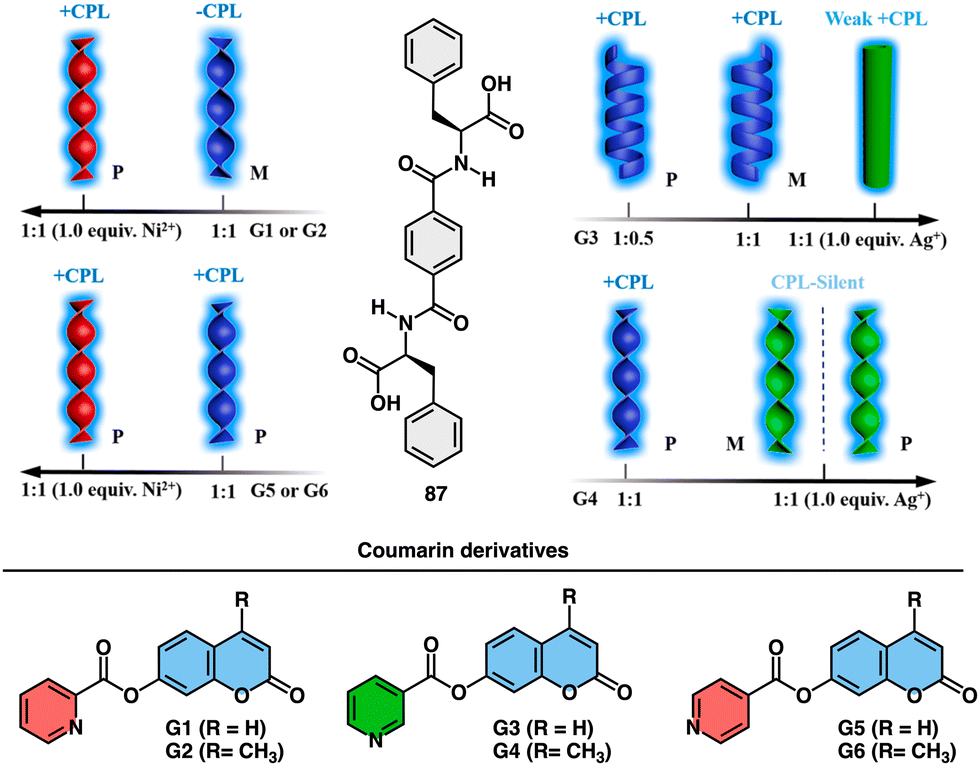 | ||
| Fig. 34 Schematic representation of the co-assembly of chiral LPF (87) and achiral fluorescent coumarin derivatives (G1–G6) and their response to metal ions (Ni2+ and Ag+). Reproduced from ref. 215 with permission from the American Chemical Society, copyright 2019. | ||
The co-assembled hydrogels exhibit chiral inversion and the formation of different nanostructures (nanotwists, nanohelices, and nanotubes) depending on the combination of components (Fig. 34).
Zhao investigated the self-assembly of a cholesterol-azopyridine conjugate known as PAzPCC (88) and the influence of light and metal ions in the supramolecular chirality of the aggregates (Fig. 35a).216 Thus, PAzPCC forms organogels that undergo a dimensional transition from 2D microbelts to 1D nanotubes and finally to 0D nanoparticles when exposed to UV light, which is driven by E/Z photoisomerization of the azopyridine unit. Furthermore, the self-assembled structures show helicity inversion mediated by metal ions through metal coordination. Interestingly, variations in the solvent while keeping the metal ion unaltered can lead to the formation of structures with opposite or racemic helical sense (Fig. 35b and c).
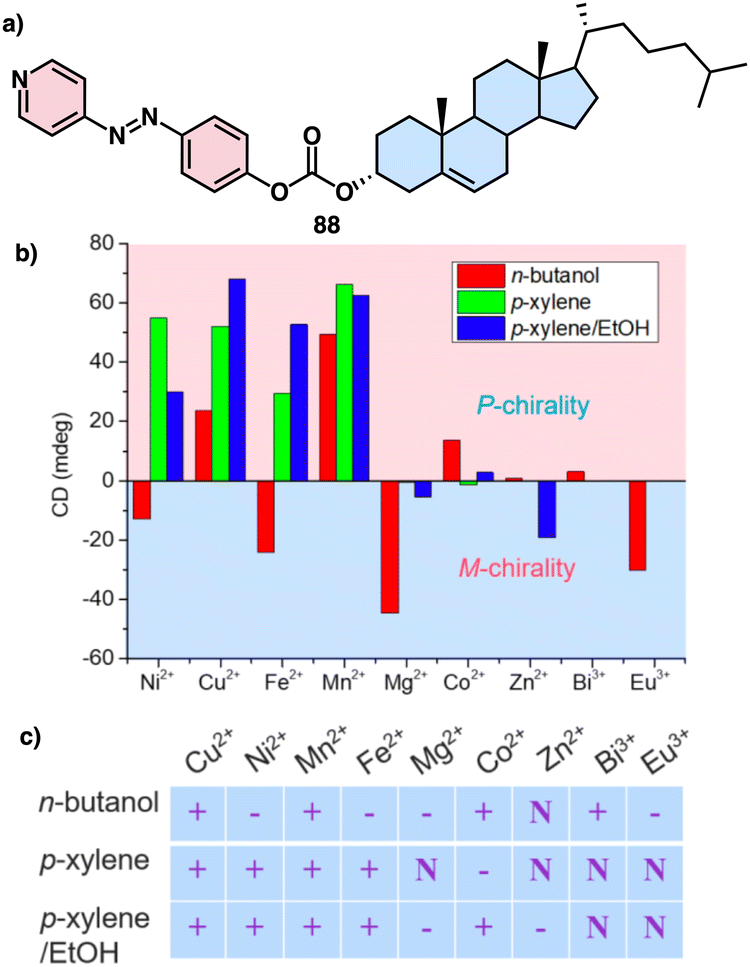 | ||
| Fig. 35 (a) Chemical structure of 88 (b) realtionship between the ECD absorption at 450 nm and corresponding chirality. (c) Supramolecular chirality of nanostructures based on PAzPCC and metal ions regulated by metal ion and solvent effects. “+” represents positive Cotton effect, “−” represents negative Cotton effect, and “N” stands for negligible supramolecular chirality. Reproduced from ref. 216 with permission from the American Chemical Society, copyright 2018. | ||
Jaworsky and Jung described a P/M reversible helical switch based on the formation of a chiral supramolecular gel using terpyridine-based ligands (89) and Co2+ ions at different metal/ligand ratios (Fig. 36a).217 Thus, at low Co2+ concentrations, octahedral complexes that promote the formation of M aggregates are formed, while above 0.6 equivalents, a co-existing square pyramidal complex that promotes the formation of P aggregates emerges (Fig. 36a). The helical inversion was fully reversible and controlled. Being enantiomeric ligands, 89 and 90 exhibited analogous Co2+ concentration-dependent switching mechanism (Fig. 36b).
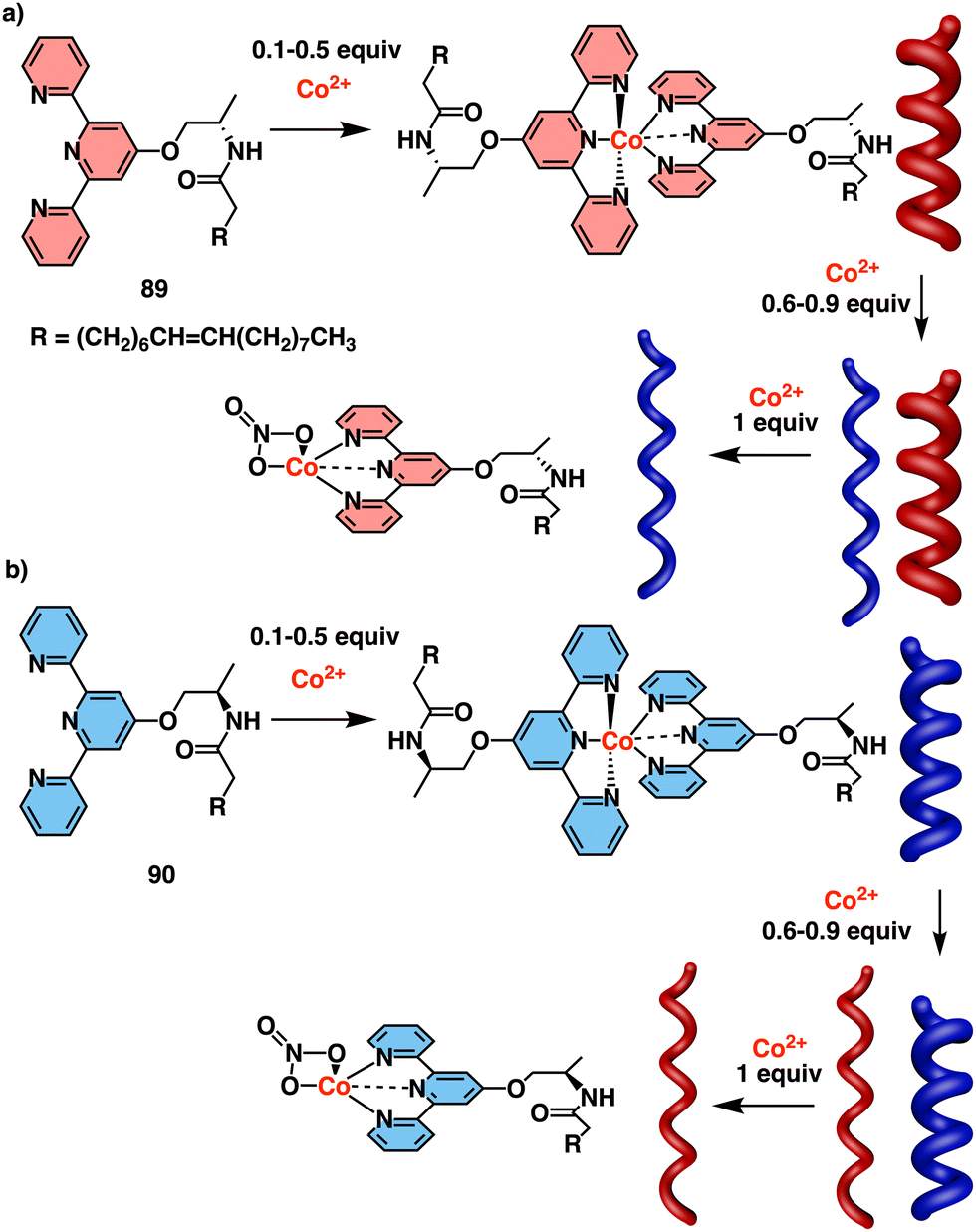 | ||
| Fig. 36 Structures of (a) 89 and (b) 90 and their coordination with different equivalents of Co2+. Reproduced from ref. 217 with permission from the Royal Society of Chemistry, copyright 2014. | ||
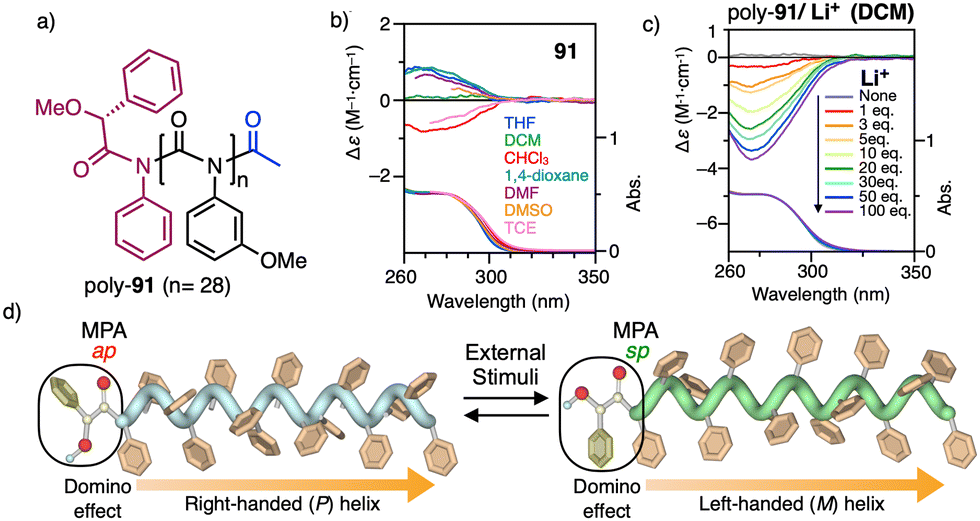 | ||
| Fig. 37 (a) Chemical structure for poly-91. (b) ECD spectra of poly-91 in different solvents and (c) in dichloromethane after addition of different amounts of LiClO4. (d) Schematic illustration of the switchable domino effect along a polymer chain. Reproduced from ref. 218 with permission from the Royal Society of Chemistry, copyright 2019. | ||
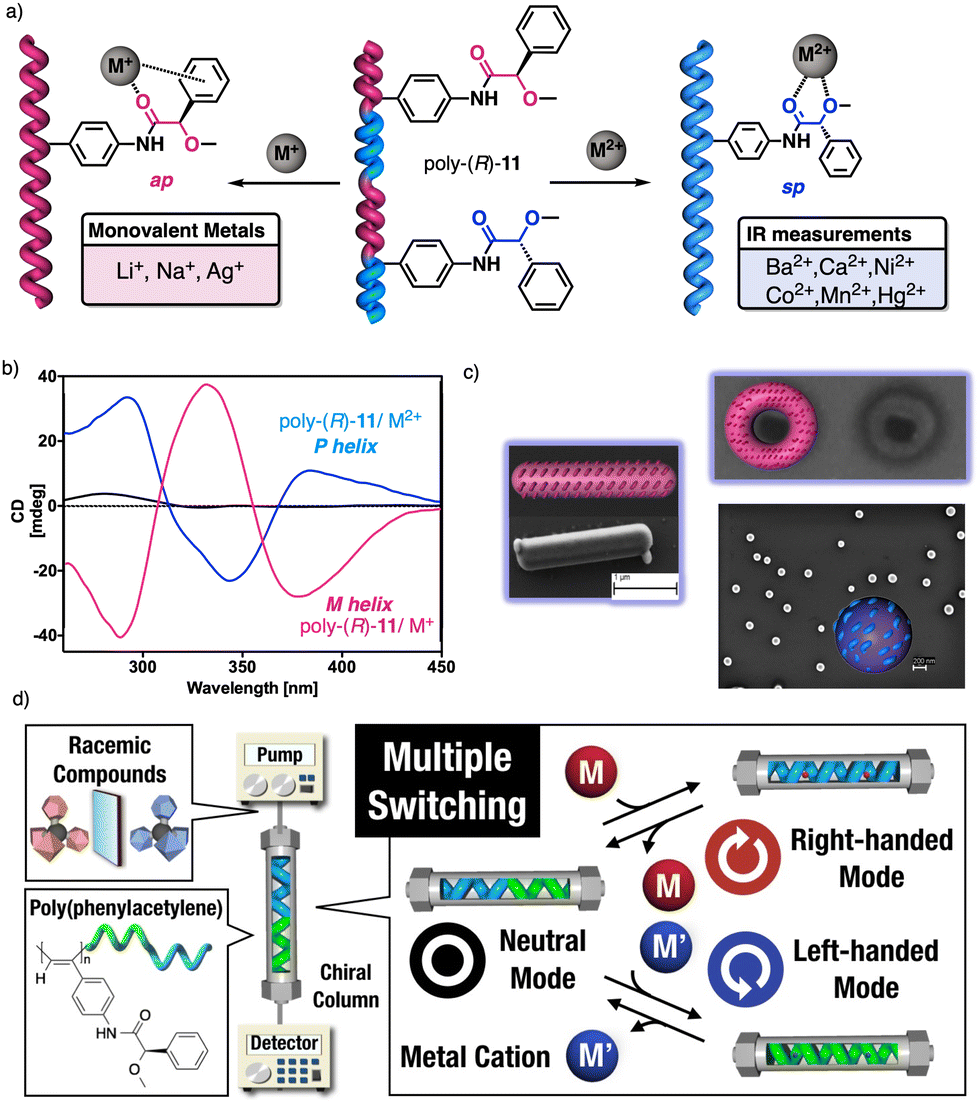 | ||
| Fig. 38 (a) Schematic illustration and (b) ECD spectra of the helical sense induction in poly-(R)-11 by monovalent and divalent metal ions. (c) Different nanostructures obtained for poly-(R)-11/Mn+. Reproduced from ref. 222 with permission from John Wiley and Sons, copyright 2014. (d) Application of poly-(R)-11/Mn+ as multiple-switch chiral stationary phase. Reproduced from ref. 225 with permission from the American Chemical Society, copyright 2019. | ||
The cross-linking ability of metal ions was employed to prepare nanospheres with tuneable size and chirality, which were achieved by controlling the metal-polymer ratio (Fig. 38c).220–223 Interestingly, in the case of poly-(R)-11/M+ helical polymer–metal complexes, a dual P or M helical sense can be induced by adding a cosolvent that switches the coordination mode between the monovalent ion and the pendant group, allowing tuning of the sp or ap conformer.224 The ability of poly-(R)-11 to adopt three different chiral states in the absence (axially racemic) and the presence of monovalent (M helix) and divalent (P helix) metal ions was used by Maeda and Freire to create a three-state switchable chiral stationary phase that allowed inverting the elution time of enantiomers by changing the helical sense of the polymer (Fig. 38d).225,226
The capacity of the MPA pendant group to interact selectively with monovalent and divalent metal ions was used by Freire and Riguera to explore the activation of the Sergeants and Soldiers effect in an axially racemic copolymer227 series—poly-11r-co-921−r—made by a dormant Sergeant (11) and an achiral Soldier, that bears the anilide of diphenyl acetic acid (92) (Fig. 39). By tuning the conformational composition of the chiral MPA pendant with monovalent and divalent metal ions in the poly-[11r-co-921−r] copolymer series, it was possible to selectively activate the Sergeants and Soldiers effect, adopting the copolymer a P or an M helical sense commanded by the Sergeant (Fig. 39). Additionally, it was also found that both P and M helical senses can be induced in poly-[11r-co-921−r] copolymer series by using monovalent metal ions and a cosolvent.228
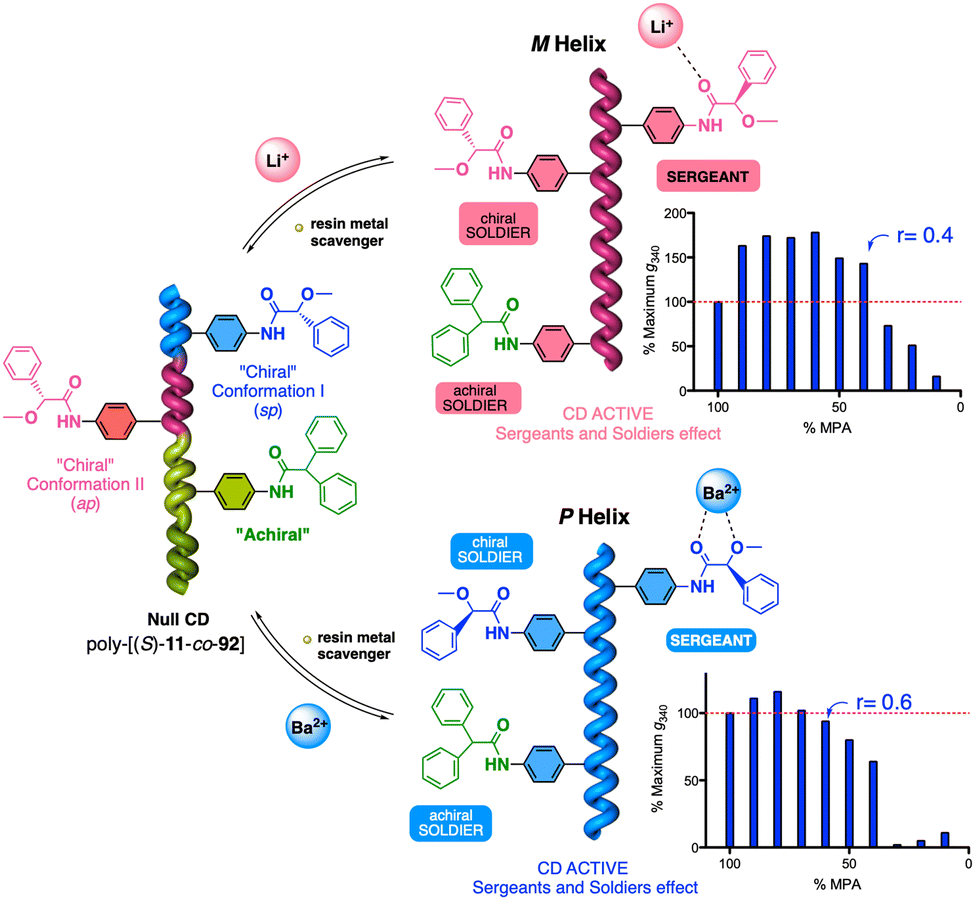 | ||
| Fig. 39 On/Off Sergeants and Soldiers effect in poly-[11r-co-921−r] copolymer series by metal ions (on) and resin metal scavengers (off). Reproduced from ref. 227 with permission from the Royal Society of Chemistry, copyright 2014. | ||
Working on the mechanisms of conformational communication along fully chiral copolymer chains, selective MPA/Mn+ (n = 1, 2) interactions were employed to describe the helix induction mechanisms in copolymer series that show chiral coalition,113,116 chiral to chiral Sergeants and Soldiers effect,112 chiral accord and chiral conflict.117
Riguera, Seco and Freire explored the coordination of metal ions with PPAs containing amino acids as pendant groups to change the conformational composition in the amino acids and induce a helical change (sense/elongation) in the PPAs. Thus, in seminal work, Riguera and Seco discovered that an anti–syn conformational change can triggered in a PPA bearing the benzamides of (L)-phenylglycine methyl ester (PGME, poly-5) as pendant when dissolved in low polar solvents.98 Therefore, if a metal ion is added to the two carbonyl groups of the pendant when they are oriented antiperiplanar (anti) in low polarity solvents, a chelate is produced between the metal ion and the amino acid that orients the two carbonyls in a synperiplanar (syn) manner.
This conformational change is accompanied by helical inversion of the PPA (Fig. 2b). Subsequently, Riguera and Freire demonstrated that PPAs containing aromatic amino acids as pendants, such as the benzamide of (L)-phenylalanine methyl ester (poly-93), or the benzamide of (L)-phenylglycine methyl ester (poly-5), can adopt up to four different helical scaffolds, in chloroform via dynamic coordination chemistry, when Li+ and a cosolvent as methanol are used as external stimuli (Fig. 40a).229 Thus, while the Li+ ion can create a tripodal coordination with the carbonyls and the aryl ring of the pendant, the cosolvent (MeOH) has the ability of change the coordination mode of the Li+ due to the selective disruption of Li+–OC and Li–π interactions (Fig. 40a).
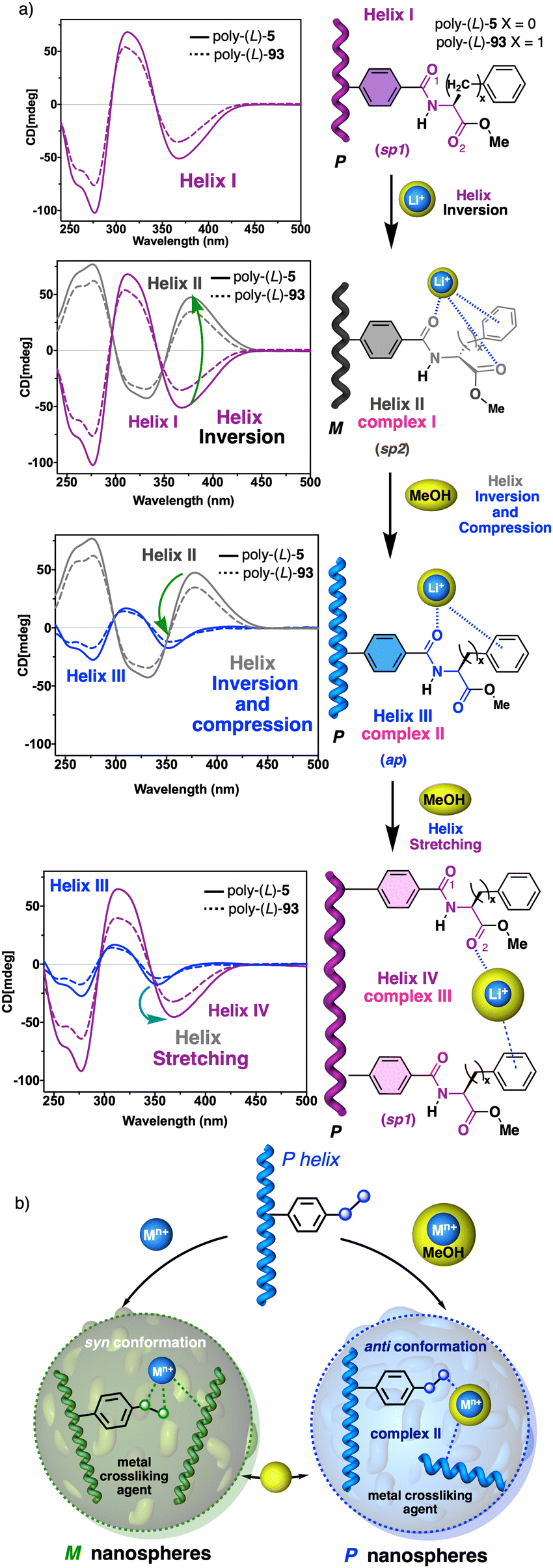 | ||
| Fig. 40 (a) Elongation and helical sense control via dynamic coordination chemistry in poly-(L)-5 and poly-(L)-93/Li+/MeOH complexes. (b) Illustration of nanostructuration via dynamic coordination chemistry. Reproduced from ref. 229 with permission from the Royal Society of Chemistry, copyright 2017. | ||
If other amino acids that lack an aromatic ring in the side chain are used as pendants, three different helices, instead of four, can be obtained from this dynamic metal-pendant coordination.
The crosslinking ability of the metal ions was used by Freire and Riguera to create P and M macroscopically chiral nanospheres from a single PPA-metal complex (Fig. 40b).230
2.4 External stimulus: redox
Metal complexes with a chiral response to redox stimuli231–234 have attracted the attention of the scientific community, as well as the chiral response of rotaxanes that exhibit redox responsiveness.235 Currently, there is growing interest in harnessing redox processes to control the properties and functions of larger macromolecules, such as polymers and foldamers (Fig. 41a), enabling the design of stimuli-responsive systems as well as the investigation of their properties.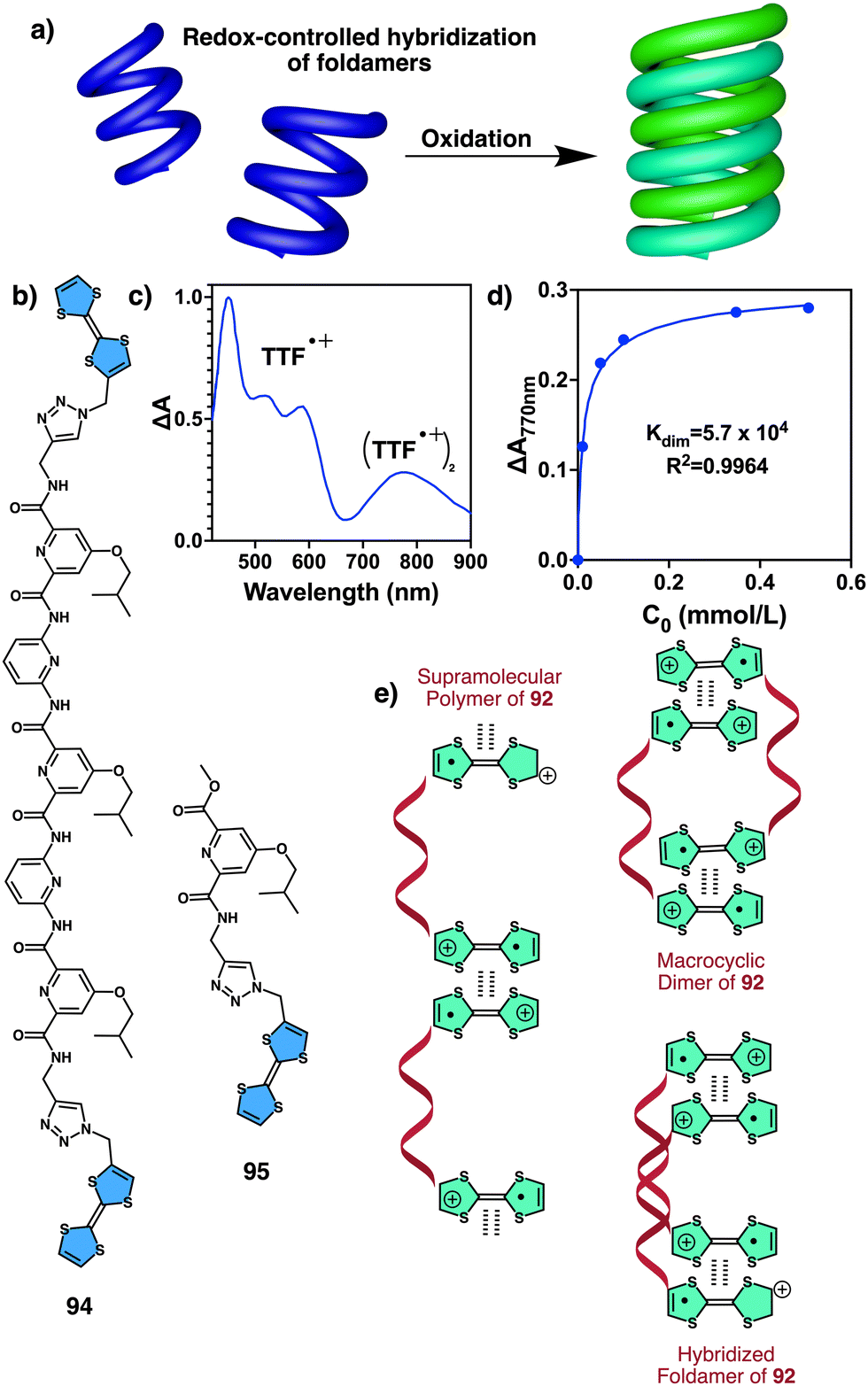 | ||
| Fig. 41 (a) Schematic representation of foldamer hybridization after oxidation. (b) Chemical structure of foldamer 94 and molecule 95. (c) Typical normalized spectrum obtained through spectroelectrochemical measurement upon oxidation of 94. (d) Evolution of ΔA770 upon oxidation of 94 (e) possible supramolecular arrangements accounting for intermolecular radical-cation dimerization of 94. Reproduced from ref. 236 with permission from the Royal Society of Chemistry, copyright 2019. | ||
The different spectroscopic data align with the dimerization of 94, while the possibility of supramolecular macrocycle formation is ruled out based on the high experimental value of Kdim (Fig. 41d and e), which cannot be achieved through π–dimer interactions alone, as it happens in the macrocycle structure.
They propose that the oligopyridine–dicarboxamide has stronger dimerization tendency in the oxidized foldamer compared to its neutral counterpart [Kdim(942(˙+)) = 102 × Kdim(94)]. Moreover, it was found that the length of the oligopyridine–dicarboxamide foldamer plays an important role in the supramolecular polymerization of 94. For instance, an oligomer containing the terminal TTF and a pyridine carboxiamide residue (95) (Fig. 41b) needs to increase the solution concentration up to 250-fold to force its aggregation. This result also indicates that the terminal TTF groups do not have propensity to form 1D polymers, and therefore excluding as aggregation products from 94, the formation of 1D polymers.
Stoddart studied the design, folding behaviour, and potential applications of redox-responsive foldamers constructed using oligobipyridinium chains.237 Varying the number of BIPY2+ units in oligoviologens 96-(2V4+), 96-(3V6+), 96-(4V8+), 96-(5V10+) and 96-(12V24+) (Fig. 42a), and utilizing short, flexible p-xylylene linkers, the researchers achieved extended conformations through Coulombic repulsion in the fully oxidized state. Upon reduction, the foldamers folded into stable conformations due to interactions between BIPY˙+ radical cations.
 | ||
| Fig. 42 (a) Structural formulas of 96-(2V4+), 96-(3V6+), 96-(4V8+), 96-(5V10+), and 96-(12V24+). PF6− counterions are omitted. (b) Schematic illustration of the switching process under redox stimulus. (c) Cyclic voltammograms of five oligoviologens. (d) Representations along the a-axis of the long-range packing order of the radical cationic species 3V3(˙+). Reproduced from ref. 237 with permission from the American Chemical Society, copyright 2015. | ||
This study shows how the folding behaviour of 96 was influenced by intra- and intermolecular radical–radical interactions (Fig. 42b), which depended on the chain length of the oligoviologens. Thus, longer oligoviologens, I.E., 96-(12V24+), showed greater radical stabilization, resulting in lower energy levels in their reduced states and the formation of intramolecular trisradical complexes. In contrast, the shorter oligoviologens, 96-(2-5Vn+), displayed reduced radical stabilization, rendering them unable to form triradical complexes (Fig. 42c). Consequently, foldamers with a reduced number of viologens units form radical–radical dimers and infinite packing due to intermolecular radical–radical interactions (Fig. 42d).
 | ||
| Fig. 43 (a) Structure of monomer 97. (b) Plot of CD intensity of LPFEG and LPFEG–GO hydrogels at around 270 nm versus UV irradiation time. (c) The representative transition stages from right-handed nanofibers to left-handed twists with increasing UV irradiation time. Reproduced from ref. 238 with permission from the American Chemical Society, copyright 2020. | ||
The redox phenomenon also plays a significant role in the sol–gel transition of various supramolecular systems containing cholesteric groups on their outer surface. A noteworthy study by Fang and co-workers showed the redox sensitivity of a chiral cholesteric ferrocene gel to different oxidants.239 Thus, the building block 98 (Fig. 44a) was designed introducing a redox sensitive ferrocene group derivatized, in each cyclopentadiene ring, with a glycine residue that has a cholesterol unit anchored at the C terminus. This molecule self-assembles to form a chiral gel, which can be oxidized using Ce4+ [(NH4)2Ce(NO3)6] and producing a colour change from yellow to green. This colour switch is attributed to the different oxidation states of the ferrocenyl residues within the gel, which are transformed into ferrocenium cations. The presence of positive charges at the core of the supramolecular aggregate disrupts the intermolecular hydrogen bonding network and the interaction of cholesteric residues resulting in a gel to sol process.
 | ||
| Fig. 44 (a) Self-assembly structure of 98 and gel–sol transition after oxidation with (NH4)2Ce(NO3)6. Reproduced from ref. 239 with permission from Elsevier, copyright 2015. (b) Structure of monomer 99 and the reversible gel–sol transition after oxidation and reduction. Reproduced from ref. 241 with permission from John Wiley and Sons, copyright 2005. (c) Structure of monomer 100. Formation of a gel made of J aggregates and gel–sol transition after oxidation with DTT. Reproduced from ref. 242 with permission from the Royal Society of Chemistry, copyright 2014. | ||
Conversely, although the addition of hydrazine for the reduction of ferrocenium cations to ferrocenyl produces the desired yellow colour, the gel state could not be recovered (Fig. 44a). Other examples of supramolecular assemblies involving ferrocene derivatized with peptide moieties have been used to demonstrated sol–gel transitions induced by a redox process, as observed in the work conducted by Kraatz and Adhikari on the supramolecular transformation of FcCO-Val-Phe-Phe-OMe from a nanofibrous gel to a spherical micelle.240 Shinkai241 designed an oligothiophene-based gelator bearing cholesteryl groups at both ends of the thiophene sequences (99) to study its redox properties. In this case, a reversible redox sol–gel transition was found by adding FeCl3 as an oxidant and ascorbic acid as a reductant (Fig. 44b). Ju described another interesting redox cholesteric gel using a disulfide bond as redox stimuli responsive group (100).242 Thus, compound 100 forms a gel through a combination of H-bonds and π–π interactions when the disulfide bond is present. However, addition of a reductant agent such as 1,4-dithiothreitol (DTT) destroys the fibres, obtaining a solution that can be monitored by SEM (Fig. 44c).
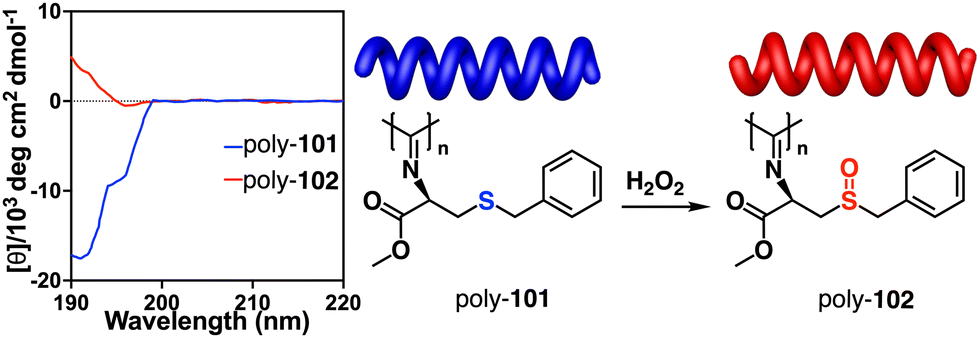 | ||
| Fig. 45 Chemical structures of poly-101 and poly-102. ECD spectra showing the helix inversion after oxidation of poly-101 to poly-102. Reproduced from ref. 243 with permission from De Gruyter, copyright 2019. | ||
In a different example, also using polyisocyanides as dynamic helical polymers, Wu studied the use of organoiodine-functionalized helical block copolymers as catalysts for asymmetric oxidations (poly-103) (Fig. 46).244 By immobilizing the organocatalyst on the helical polymers, the researchers not only facilitated catalyst recycling from homogeneous reactions, but also observed a significant boost in enantioselectivity. This immobilization strategy allowed the creation of an organoiodine-functionalized single left- and right-handed helical polyisocyanides block copolymerized with chiral monomers. These helical polyisocyanides served as catalysts for three asymmetric oxidations, delivering the desired products with high yields and excellent enantioselectivity (Fig. 46).
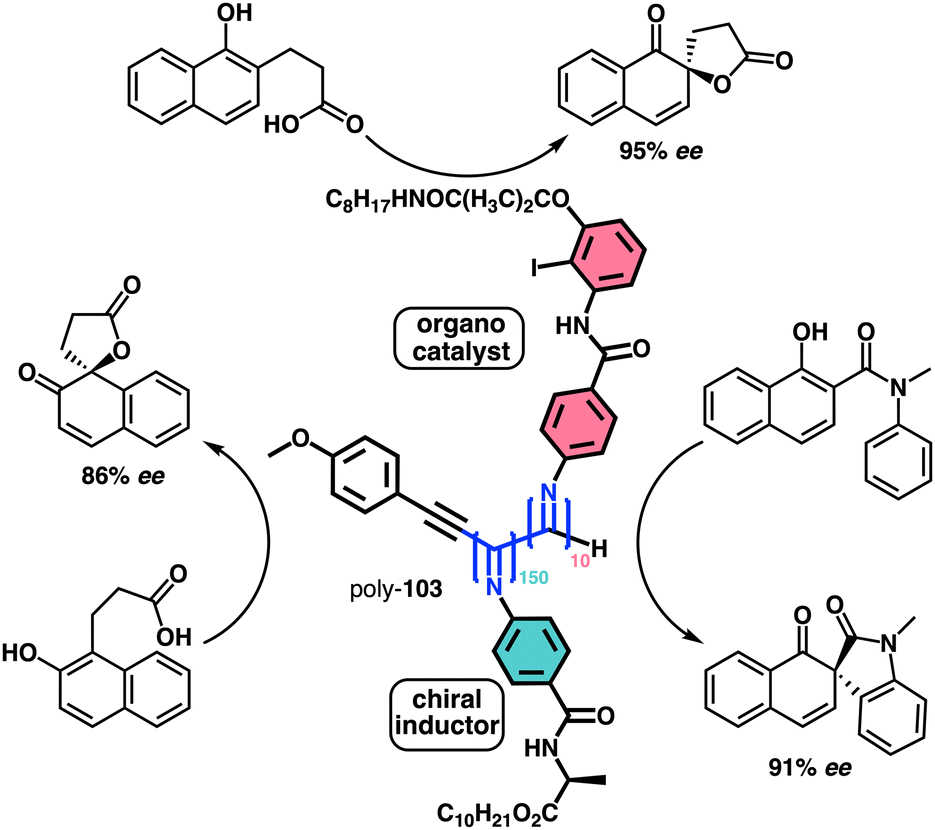 | ||
| Fig. 46 Structure of poly-103 and three asymmetric catalysed reactions. | ||
Amabilino explored the potential of electroactive organic polymers as chiral redox-polymer systems for applications in molecular electronics.245 Thus, a chiral polyisocyanide that incorporates a tetrathiafulvalene (TTF) derivatized with stereogenic centres (poly-104) was designed and prepared (Fig. 47). This polymer shows a reversible interconversion between three univalent and two mixed-valence redox states, each of which displays distinct chiroptical properties associated with the presence of helical structures, as deduced from ECD studies (Fig. 47).
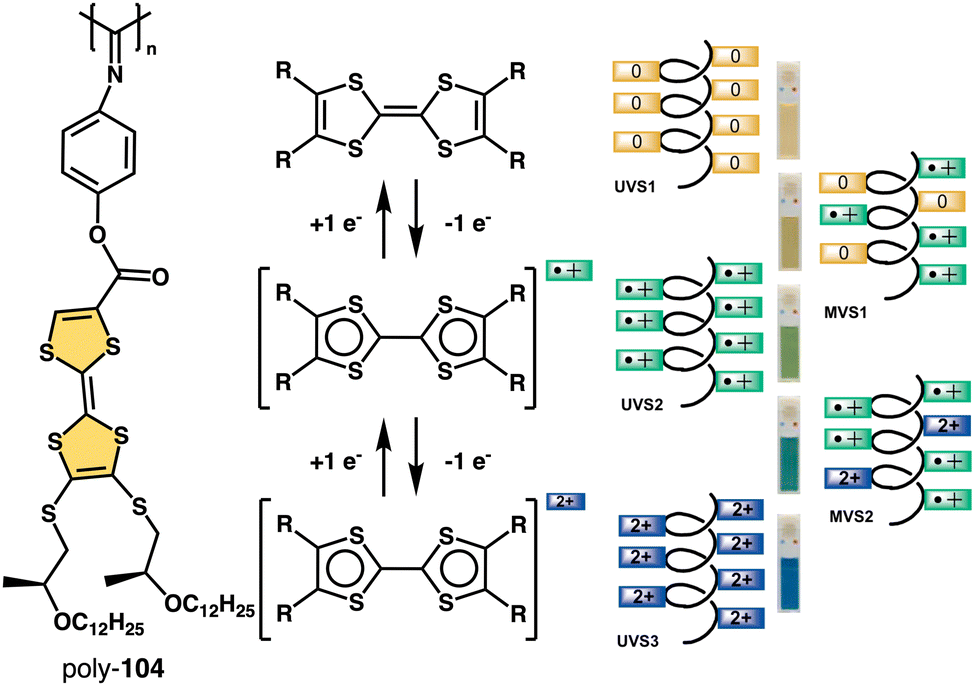 | ||
| Fig. 47 Cartoon representation of the redox states of poly-104 with TTF units in the side chains that can be interconverted through redox reactions and the colours shown by the solutions of these species. Reproduced from ref. 245 with permission from John Wiley and Sons, copyright 2005. | ||
Freire found that during the synthesis of hybrid materials from dynamic helical polymers and metal nanoparticles,246,247 adding HAuCl4 to an anilide-PPA [poly-(R)-11], oxidation of the polyene backbone occurs, which leads to the loss of the helical structure due to a delocalization of the radical along the polyene chain (Fig. 48a–c). To overcome this problem, TOAB (tetraoctylammonium bromide) was added to the polymer solution in order to trap the Au3+ cations and prevent the oxidation of the polyene (Fig. 48d).247
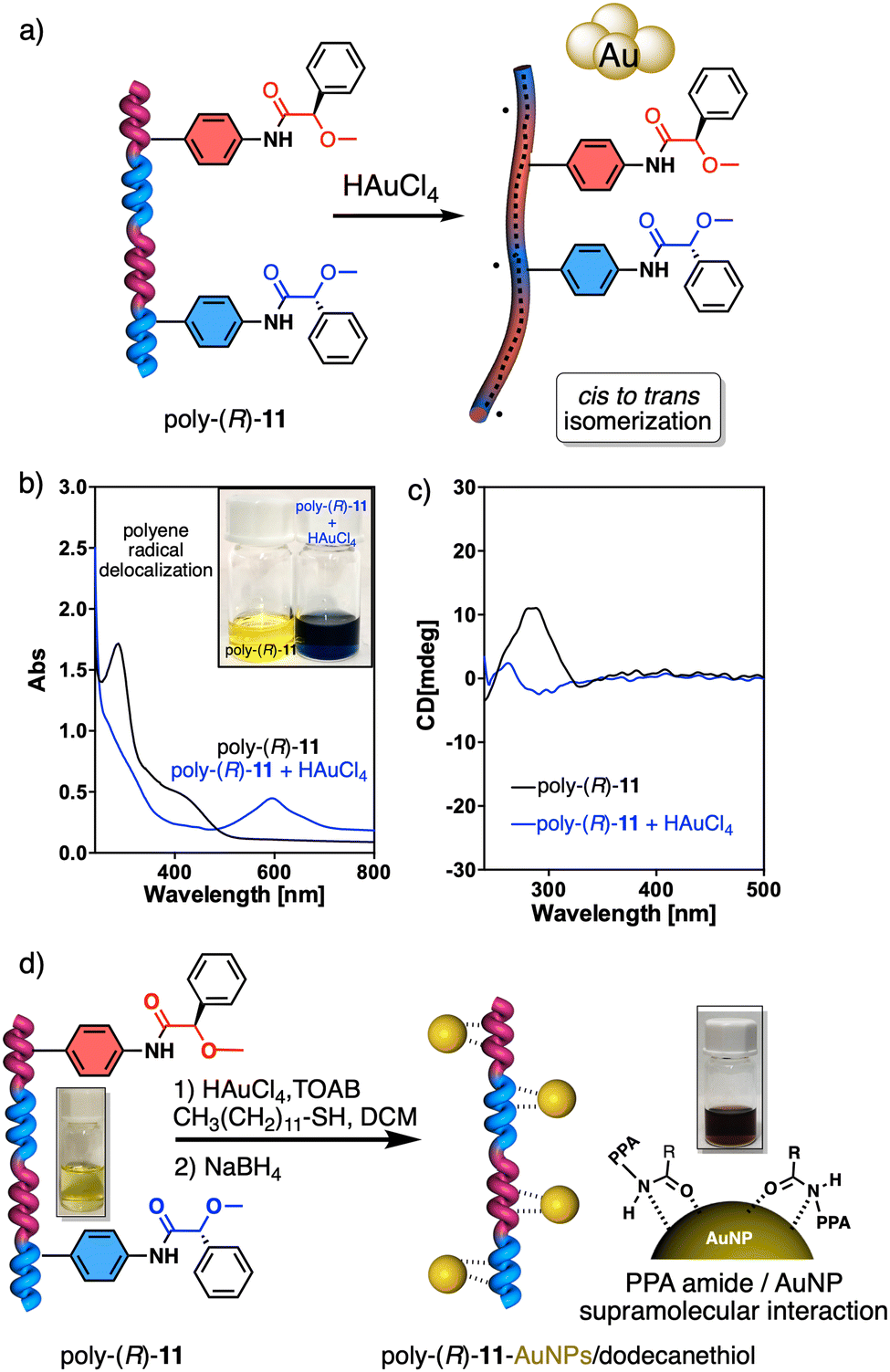 | ||
| Fig. 48 (a) Schematic representation for the formation of radicals in poly-(R)-1 in presence of HAuCl4. (b) UV-vis and (c) CD spectra of poly-(R)-11 and poly-(R)-11 in presence of HAuCl4. (d) Schematic representation for the formation of poly-(R)-11-AuNPs nanocomposites by a variation of Brust–Schiffrin method. Reproduced from ref. 247 with permission from John Wiley and Sons, copyright 2021. | ||
In this new protocol, gold interact with the polymer once it is reduced to Au0, which is obtained after the addition of NaBH4 as reducing agent. In this poly-11-AuNPs nanocomposite, the AuNPs are stabilized by supramolecular anilide–AuNPs interactions (Fig. 48d).
This protocol was also used to successfully prepare a poly-11-AgNPs nanocomposite.248 In a different example, Freire explored the helix inversion of a PPA during a redox metal translocation process, a crucial step during the synthesis of helical poly(phenylacetylene)-metal nanocomposites.249 A PPA copolymer made by two comonomers that contain the benzamides of methionine methyl ester [(L)-105] and (L)-alanine methyl ester [(L)-106] as pendants in 0.2 (L)-105/0.8 (L)-106 ratio—poly-[(L)-1050.2-co-(L)-1060.8]—was prepared. In a low-polarity solvent environment such as CHCl3, both pendant groups adopt an anti-conformation that induces an M helix into the copolymer (Fig. 49). However, upon complexation with gold (Fig. 49a–d) or silver (Fig. 49e–h) cations, the copolymer undergoes helix inversion from M to P due to the adoption of syn conformations in the pendants through chelation with metal ions. Further reduction of metal ions with NABH4 produces translocation of the metal towards the tioether group, which is accompanied by a conformational change from syn to anti in the pendants accompanied by a P to M helix inversion of the polymer (Fig. 49).
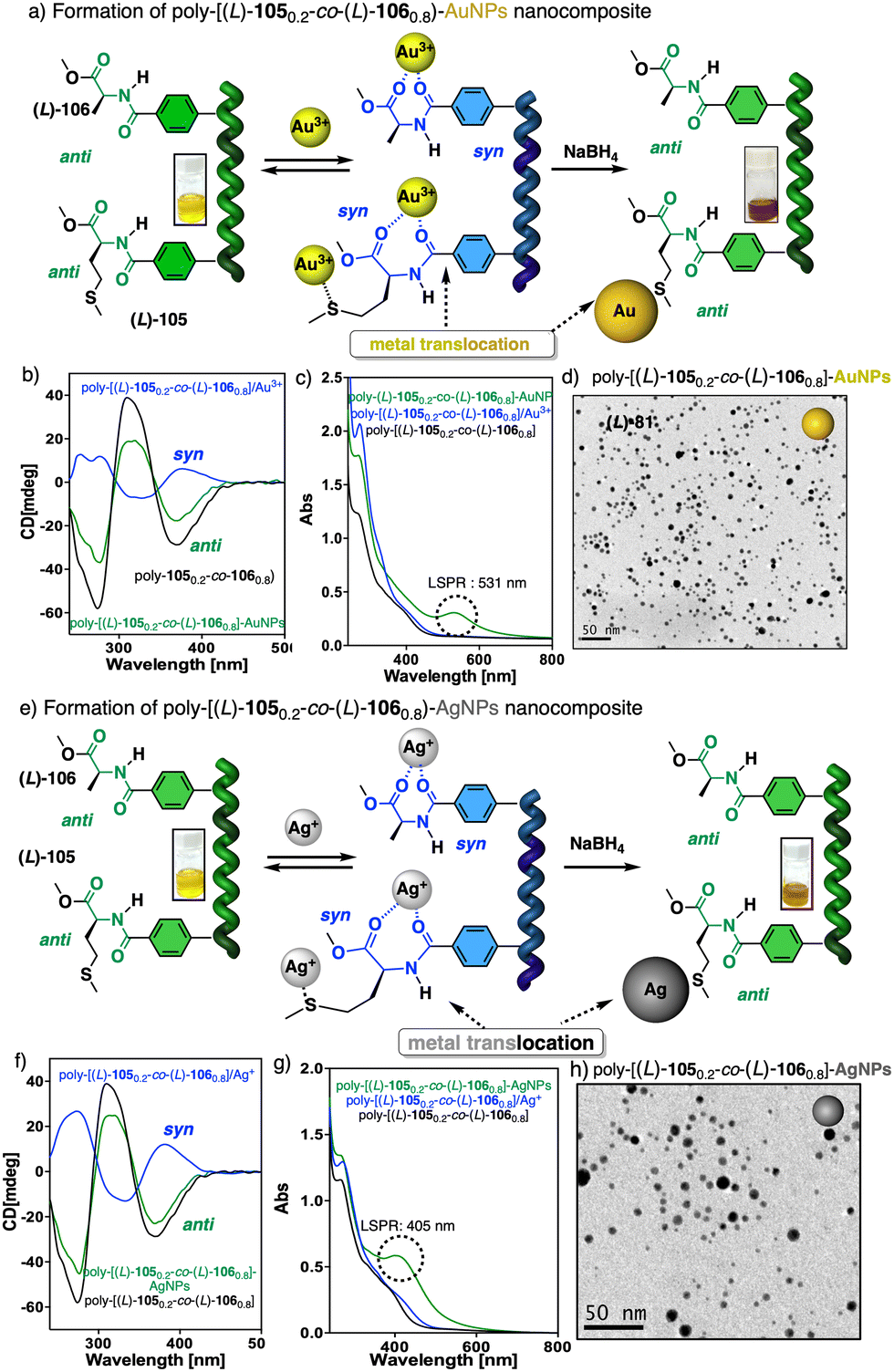 | ||
| Fig. 49 (a) Schematic illustration for the preparation of poly-[(L)-1050.2-co-(L)-1060.8]-AuNPs and its chiroptical responses associated to a redox driven translocation of a gold metal centre. (b) CD and (c) UV-vis spectra for poly-[(L)-1050.2-co-(L)-1060.8], poly-[(L)-1050.2-co-(L)-1060.8]/Au3+ and poly-[(L)-1050.2-co-(L)-1060.8]-AuNPs. (d) TEM image for poly-(1050.2-co-1060.8)-AuNPs nanocomposites. (e)–(h) Idem for silver. Reproduced from ref. 249 with permission from the Royal Society of Chemistry, copyright 2022. | ||
Yashima studied helical poly(phenylacetylene)s (PPAs) that bear riboflavin as pendants by cyclic voltammetry (poly-107) (Fig. 50a).250 The study aimed to understand the electron transfer capabilities of riboflavin pendants in these polymers. The values obtained were similar to those of the monomer, indicating that the redox behaviour was mainly governed by the riboflavin units. On the other hand, when using another connection point between riboflavin and PPA (poly-108) (Fig. 50a), a reduction curve was found at a more positive potential, indicating a relationship between the connector used to link riboflavin and PPA and its redox potential. These redox processes reversibly induced conformational changes in the flavin skeleton that are transmitted to the helical main chain (Fig. 50b and c).
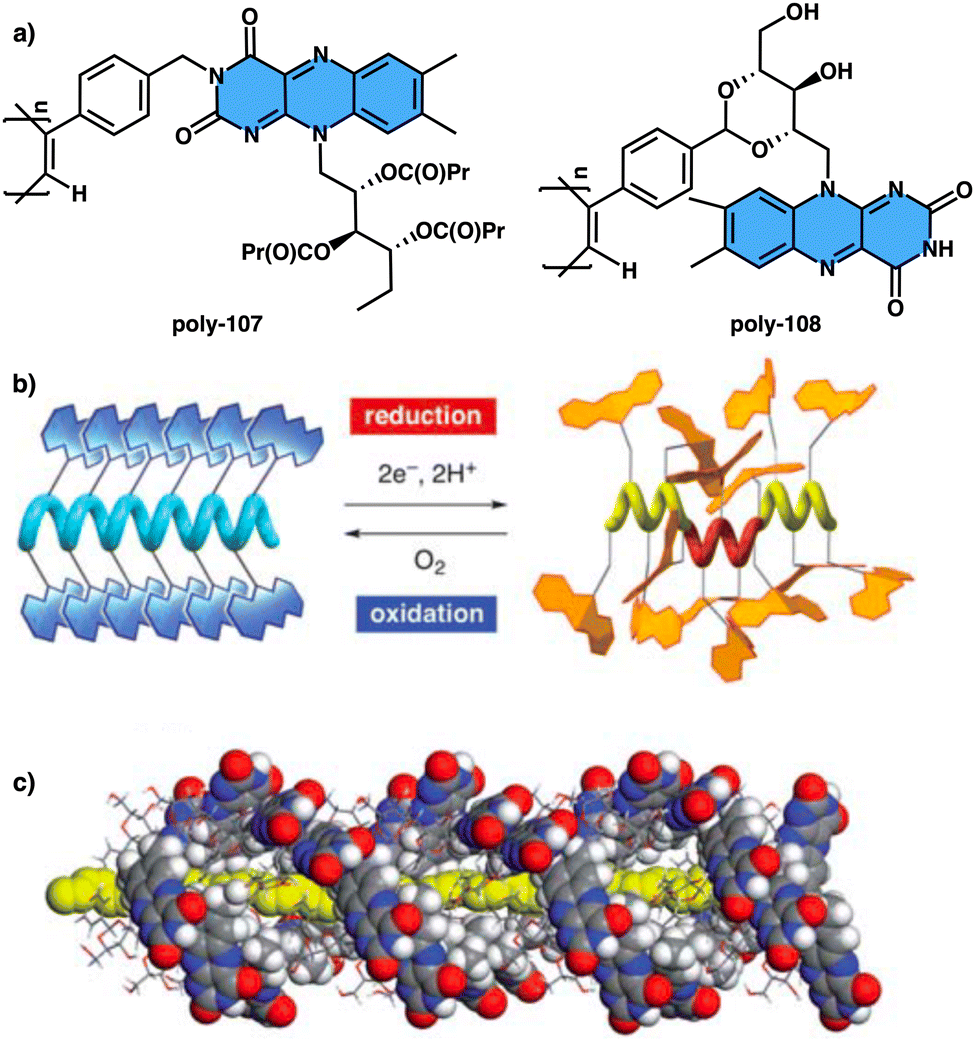 | ||
| Fig. 50 (a) Chemical structures of poly-107 and poly-108. (b) Schematic illustration of the redox-triggered switching of the helical structure of poly-108. (c) Possible helical structure of poly-108 (30-mer). Reproduced from ref. 250 with permission from the Royal Society of Chemistry, copyright 2010. | ||
Aida and Fukushima introduced an o-phenylene oligomer (109) (Fig. 51b) that exhibits dynamic motion triggered by redox reactions.251 Although the oligomer helices demonstrate rapid inversion in a solution environment, a remarkable phenomenon of chiral symmetry-breaking is observed during crystallization. Each crystal contains exclusively the right-handed or left-handed enantiomeric form, resulting in a mixture of non-racemic of crystals (Fig. 51a). Furthermore, the dynamic motion of the helical oligomer in solution is considerably suppressed upon one-electron oxidation (Fig. 51b). Using X-ray crystallography, analysis of the neutral and oxidized forms reveals the generation of a hole after oxidation, which is shared by the repeating o-phenylene units. This shared hole facilitates locking of the helix conformation, leading to sustained chiroptical memory. These findings offer valuable insights into the behaviour of redox-active helical polymers and their potential applications in various domains.
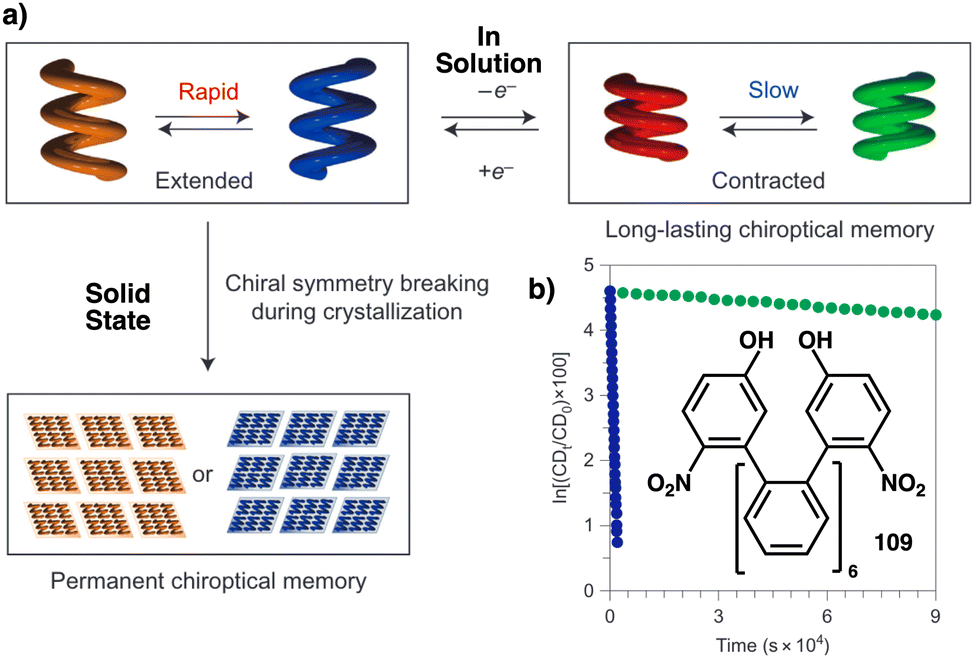 | ||
| Fig. 51 (a) Schematic representation of the helical inversion dynamics of oligomer 109 in solution and in solid state. (b) chemical structures of 109 and decay profiles of the CD intensities in spectra of the oligomer dissolved in MeCN at 265 nm and the oligomer dissolved in MeCN in the presence of the one-electron oxidant (4-BrC6H4)3N˙+ [SbCl6−] at 273 nm (green). Reproduced from ref. 251 with permission from Springer Nature, copyright 2010. | ||
The synthesis and properties of a series of π-conjugated polymers, formed by repeating units of tetrathiafulvalene vinylogue (TTFV) and phenylacetylene, were investigated as supramolecular hosts for non-covalent functionalization of single-walled carbon nanotubes (SWNTs). These polymers adopt a folding conformation in the neutral state, allowing them to interact effectively with individual SWNTs and form supramolecular complexes with good solubility in organic solvents. The polymers can be activated by redox stimuli to change their molecular shapes to linear structures in the oxidized state. It results in reversible dissociation of polymers from SWNTs and efficient release of pristine SWNTs from polymer dispersants.252,253 There are also examples of sol–gel transition compounds in which the disulphide bond can be cleaved with reducing agents such as dithiothreitol (DTT).254
2.5 External stimulus: light
The development of light-sensitive molecules has attracted the attention of numerous scientist due to the possibility of modulating the structures and functions of molecules after light irradiation. The introduction of these stimuli-responsive molecules or functional groups as components of macromolecules allows the generation of materials with unique light-responsive behavior. As a result, there are many examples in the literature of various types of well-established photo-responsive molecules, such as azobencene,255 diarylethenes,256–260 stilbenes,261,262 molecular motors,263–266 or acylhidrazones267,268 among others. In this section we will focus our attention on the photomodulation of the structure, properties, and functions of macromolecular helical structures.269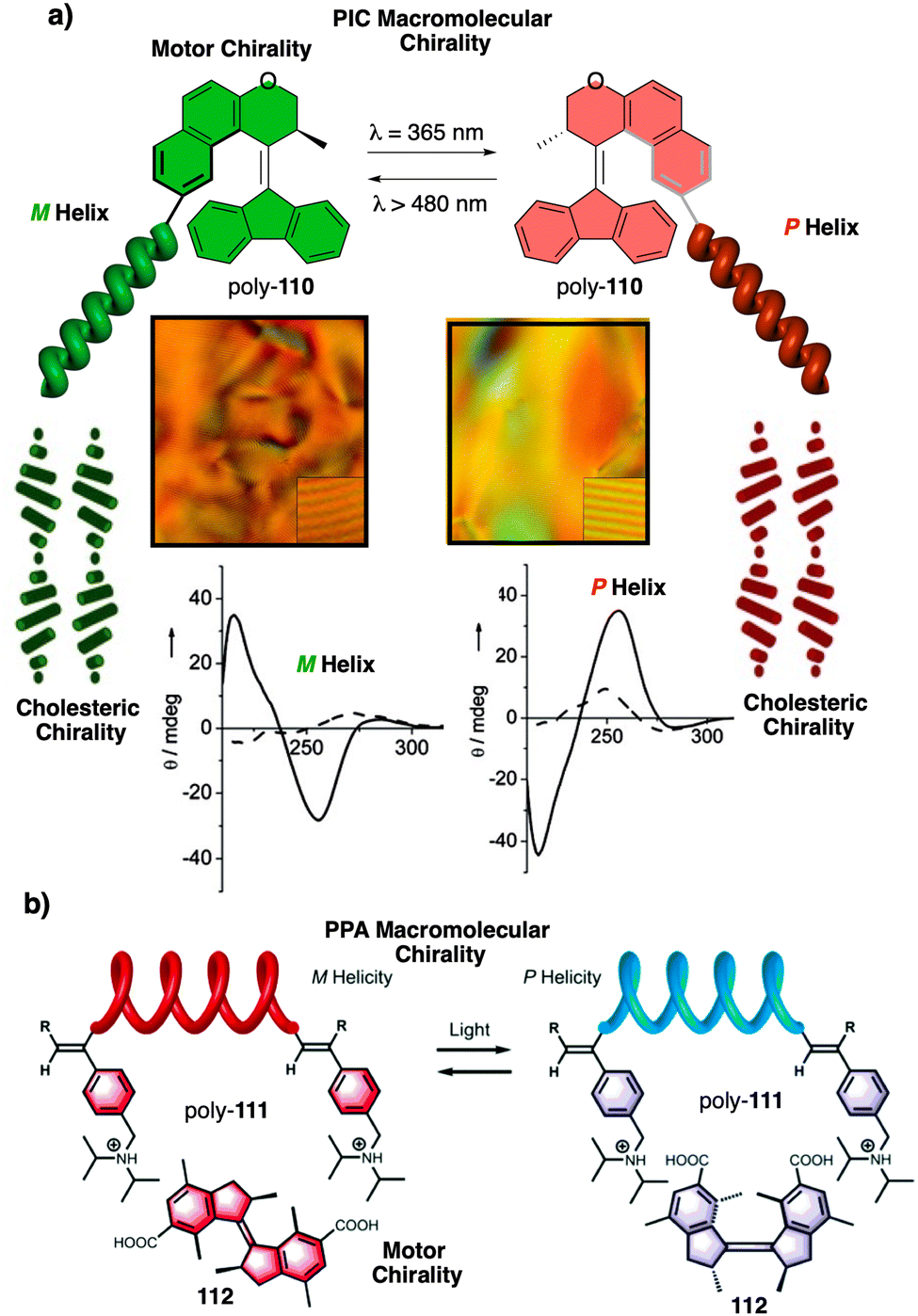 | ||
| Fig. 52 (a) Helix inversion process of poly-110 bearing a chiral photoactive molecular motor and subsequent hierarchical chiral transmission from molecular motor to helical PIC cholesteric phase. Reproduced from ref. 271 with permission from the American Chemical Society, copyright 2008. (b) Helix inversion process of PPA poly-111via salt-bridge formation with chiral molecular motor 112. Reproduced from ref. 272 with permission from the Royal Society of Chemistry, copyright 2017. | ||
The same group also reported the photoswitchable screw sense of an achiral PPA by stablishing a supramolecular interaction between the pendant and a chiral photoswitchable molecular motor. They prepared a PPA that bears a trialkyl amine as pendant (poly-111), which was combined with a chiral photoswitchable molecular motor functionalized with two carboxylic acids (Fig. 52b). From this system they observed that photomodulation of the motor chirality triggers the helical inversion of the PPA main chain, as inferred by ECD spectroscopy (Fig. 52b).272
Zentel reported several examples of light-driven helical inversion of PIC copolymers consisting of achiral monomers bearing an alkyl chain and chiral monomers bearing a photoswitchable azo group (poly-113 and poly-114) (Fig. 53).273–277 Depending on the chemical structure of the chiral azo monomer, the E/Z photoisomerization process produced either helical inversion (poly-113) or screw sense excess variation (poly-114) (Fig. 53a and b). Due to the good photostability and reversibility of the isomerization process, the PIC screw sense of poly-113 and poly-114 can be switched back and forth several times, opening venues for the preparation of chiroptical switches.
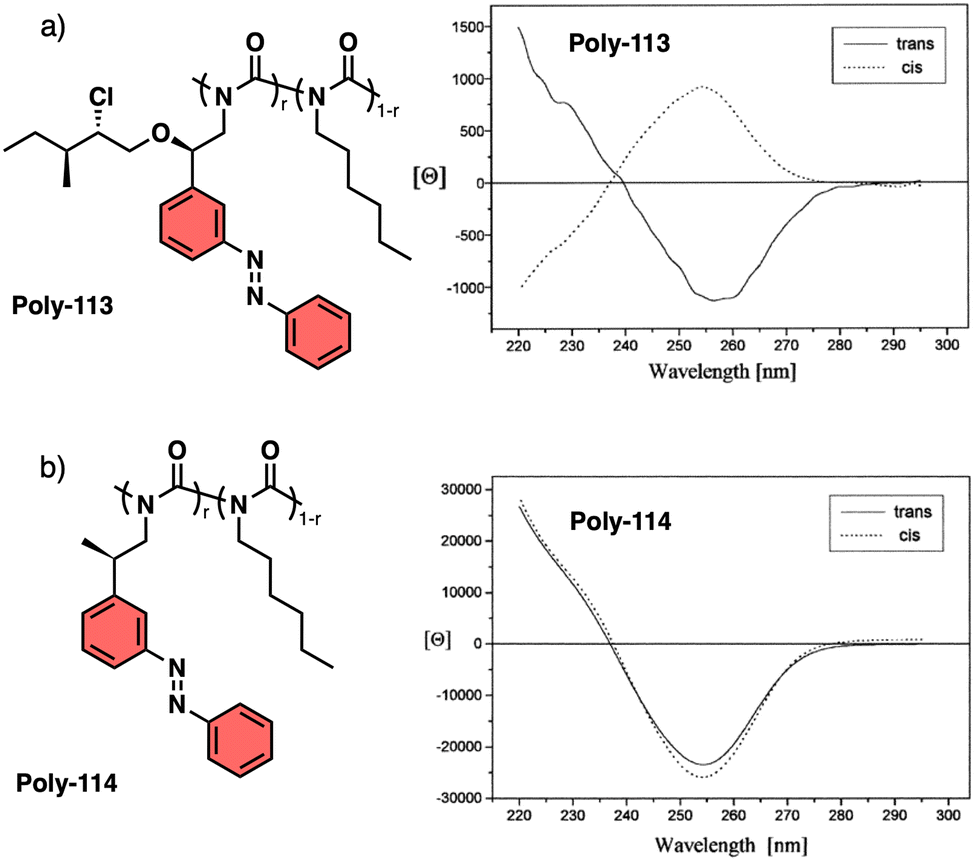 | ||
| Fig. 53 Structures of PICs (a) poly-113 and (b) poly-114 copolymers and CD monitoring of the helical variations in the E/Z photoisomerization processes. Reproduced from ref. 276 with permission from the American Chemical Society, copyright 1998. | ||
 | ||
| Fig. 54 (a) Structure of BTAS 115 and 116, bearing free and alkylated cysteine moieties respectively. (b) Schematic illustration of the photo-depolymerization process from one dimensional stack to homo/heterodimers as the alkylation of the cysteine moieties takes place. Reproduced from ref. 279 with permission from the Royal Society of Chemistry, copyright 2021. (c) Structure of BTAS 52 and 117. (d) Schematic illustration of the photo-triggered E/Z isomerization process of BTA 117 that triggers a change of function from intercalator to chain capper that is accompanied by a gel–sol state. Reproduced from ref. 280 with permission from the American Chemical Society, copyright 2020. | ||
This process modulates the polymer chain length and triggers a macroscopic sol–gel transition (Fig. 54c and d).280
Diarylethenes (DAEs) represent a family of photoswitches whose structure can be tuned from an open to a close conformation by light irradiation. This chemical transformation not only changes the optical properties of the molecule, but also its rigidity. This fact makes DAEs very interesting molecules to create photoactive functional supramolecular aggregates. For instance, Zhu reported the self-assembly of the DAE 118, which is functionalized with naphtylamide units carrying a phenylalanine-OH functional group (Fig. 55a). The supramolecular polymerization of the system is assisted by the formation of carboxylate bridges that generate macromolecular helical structures in CCl4. The handedness of the systems depends on the absolute configuration of the amino acid, while the morphology of the aggregate depends on the open/closed state of the DAE group. Photoisomerization of the DAE moiety triggers a reversible morphological transformation from helical structures to nanosheets related to the change in the DAE scaffold (Fig. 55a–d).281
 | ||
| Fig. 55 (a) Structures of DAE-naphtylamide-Phe derivative 118 in open and closed states. Morphological transformation from (b) helical structures to (c) nanosheets upon light irradiation. (d) Schematic illustration of carboxylate bridge-triggered self-assembly generating helical structures. Reproduced from ref. 281 with permission from the American Chemical Society, copyright 2016. (e) Structure of molecule 119. (f) Schematic representation of helix induction in open–closed sates of DAE-OPP derivatives. Reproduced from ref. 282 with permission from John Wiley and Sons, copyright 2014. | ||
Akagi reported the self-assembly of oligo(para-phenylenes) 119 functionalized with DAE bearing chiral esters. The macromolecular chirality of the system is reversible, showing inversion after irradiation with UV-vis light. This helical transformation is related to the open/closed form of the DAE moiety that promotes opposite handedness in the different states. Noteworthy, this process works both in solution and in solid state (Fig. 55e and f).282 Feringa introduced a photoswitchable molecular motor in a supramolecular helical polymer building block endowed with a urea side chain (120) for the construction of photo-responsive supramolecular chiral aggregates (Fig. 56a).283–287 In this system, the chirality of the molecular motor (P,P)-cis-120 is hierarchically transferred to supramolecular structures generating preferred-handed helical fibbers.
 | ||
| Fig. 56 (a) Structures of photoactive chiral molecular motor 120 and illustration of multistate rotation. Cryo-TEM image images of (b) (P,P)-cis-120 and (c) after irradiation with 312 nm light to reach (P,P)-trans-120. CD and UV-vis spectra of (d) (P,P)-cis-120 and (e) (P,P)-trans-120. Reproduced from ref. 290 with permission from the American Chemical Society, copyright 2022. | ||
Photoirradiation triggers rotation-induced morphological changes (e.g., chiral, and geometric) that dramatically influence the morphology and chirality of the supramolecular polymer (Fig. 56a–e). Through this process they achieved dynamic control of the supramolecular polymer in multiple self-assembled states. Therefore, a cascade of morphological transformations from initial helical fibers to micelles, worm-like micelles, to finally recovering helical fibers, is possible simply playing with light irradiation.288 Yagai designed a cone shape alkoxyazobenzene functionalized with chiral amides that self-assemble to generate uniform toroidal nanostructures that can hierarchically organize into nanotubes or axially chiral nanofibrils under the control by light, temperature, or concentration.289,290
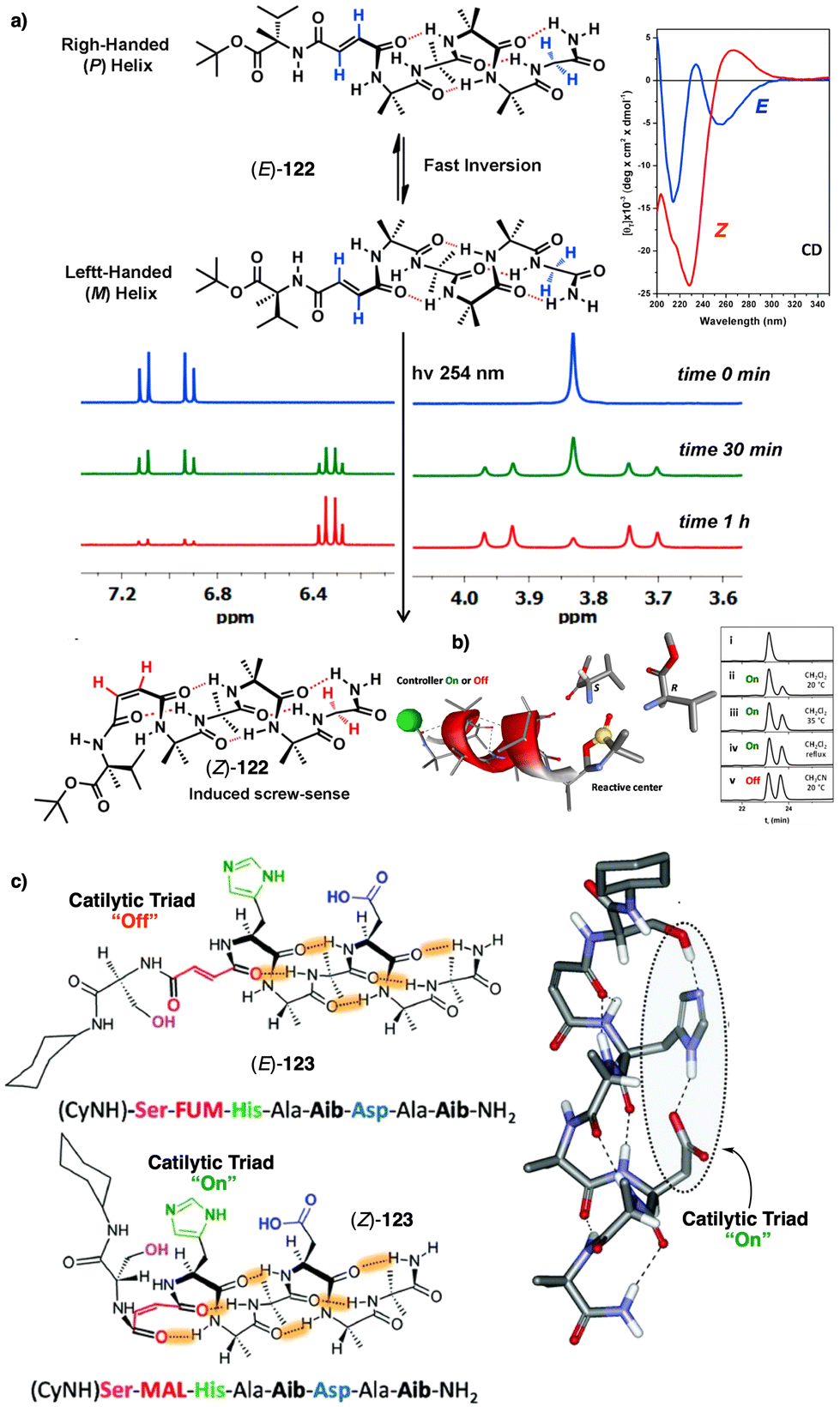 | ||
| Fig. 57 (a) Structure of molecular motors endowed with bipyridine units. (b) Optimized molecular structures of (P,P)-cis-121-Cu2 and (M,M)-cis-121-Cu2. (c) CD spectra showing the helical inversion process of these complexes. Reproduced from ref. 291 with permission from Springer Nature, copyright 2016. | ||
This different orientation of the coordinating units promotes the formation of polymeric or discrete helical structures (trans- and cis-conformations, respectively). In addition to modifying the cis-/trans-equilibrium of the molecular motor, the chirality of the system is also modulable with light, so the (P,P)-cis-121 and the (M,M)-cis-121 conformations induce opposite handedness in the discrete double helical structures (Fig. 57b and c).291
Moretto and Clayden reported a series of AIB-based foldamers containing photoswitchable fumaramide/maleamide linkers to tune the P/M screw sense excess of the foldamers by light. These moieties bearing carbonyl groups present different compatibility with the hydrogen-bonding pattern of the foldamers, with the Z maleamide being compatible with the HB network of the foldamer while the E fumaramide configuration is not. Photoswitching from the insulating fumaramide to the conducting maleamide allowed activation of the stereoselective reactivity of 122, as proven by chiral HPLC (Fig. 58a and b).292
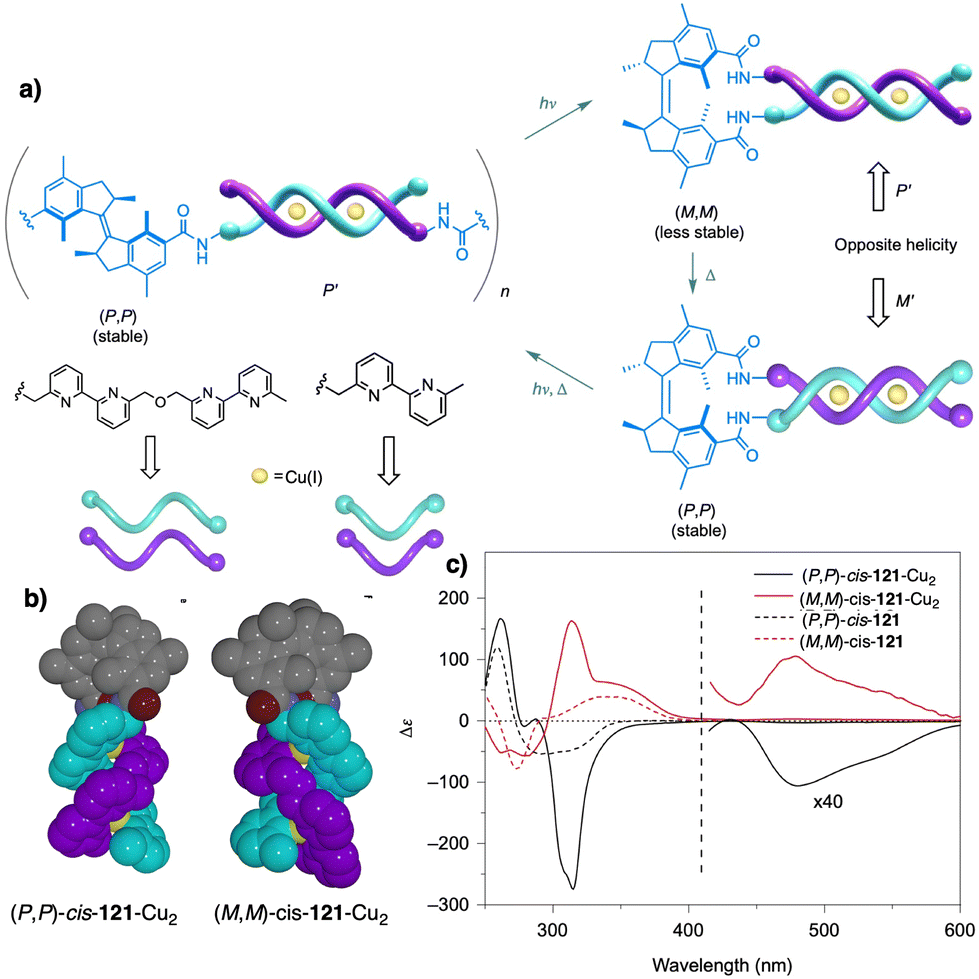 | ||
| Fig. 58 (a) Screw sense induction process by light irradiation in AIB foldamer 122 demonstrated by CD and NMR. (b) Schematic representation of the diastereoselective chainextension reactions followed by chiral HPLC. (c) Light-driven turn on/off catalytic activity of 123 bearing a catalytic triad esterase. Reproduced from ref. 292 with permission from the American Chemical Society, copyright 2016. (d) Switching the terminal azobenzene chromophore between E and Z configurations changes the population distribution between right- and left-handed conformations of foldamers 124. Reproduced from ref. 293 with permission from the Royal Society of Chemistry, copyright 2021. | ||
Following this work, they have also reported the photoactivation of a catalytic triad of reactive side chains – Ser, His and Asp – present in foldamer 123. Light modulation of the E/Z forms allows control of the reactivity of the system, as it only works in the maleamide form, where the reactive groups are placed in the proper orientation to catalyze the corresponding reaction (Fig. 58c).292 Clayden and coworkers also reported an azobenzene containing AIB foldamer 124 inserted into an artificial lipid membrane whose screw-sense was controlled by a configurational change in the azobencene unit. The E/Z conformers stabilize different hydrogen bond patterns within the AIB foldamers, biasing the rate of the helix inversion process (Fig. 58d).293 The introduction of fluorine atoms into the foldamer allowed direct monitoring of this process by 19F NMR experiments (Fig. 58).70
Huc reported an aromatic oligoamide (125) bearing a DAE central linker (S) (Fig. 59a and b). Binding of fructose results in the induction of opposite handedness depending on the open/close state of the DAE unit, as demonstrated by CD (Fig. 59c and d). This fact indicates that the DAE/fructose binding occurs in a different way. Interestingly, subsequent irradiation of the host–guest complex allows switching between P/M helical scaffolds (Fig. 59e).294 To date, many foldamer containing azobenzene groups has been reported, both as side chains and as part of the macromolecular main chain. The introduction and photomodulation of these azobenzene moieties allows control of the folding of the foldamer, and consequently, its properties.295 For instance, the E/Z structural variation of the azobenzene group in the side chains of the foldamer creates variations in the supramolecular interaction that governs the folding of the systems.296–301
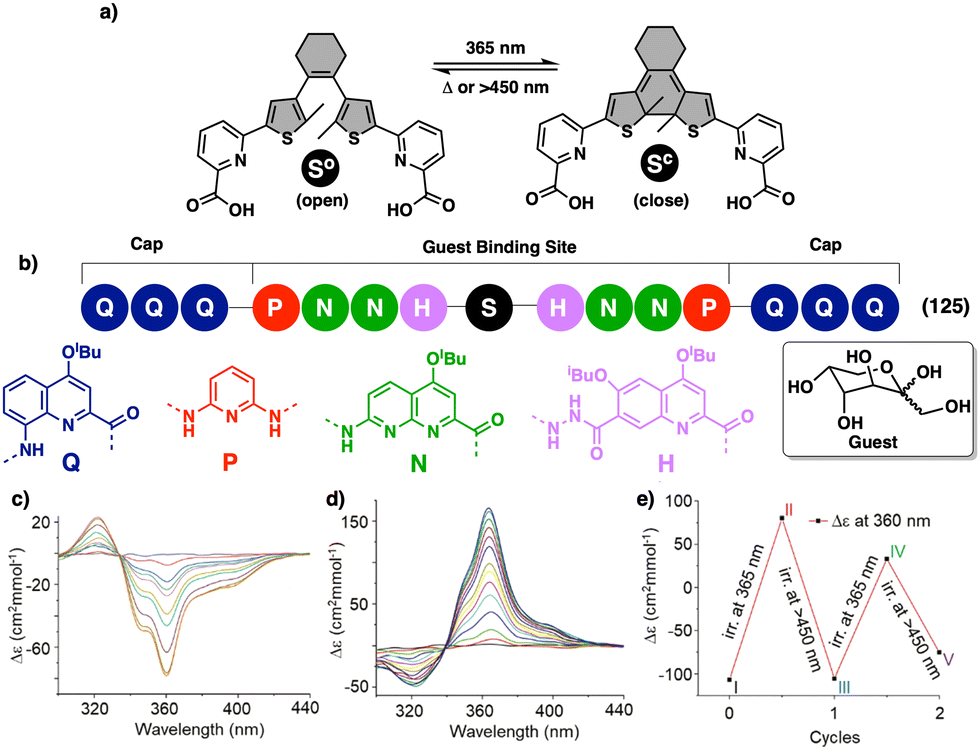 | ||
| Fig. 59 (a) Structure of DAE core and (b) oligoamide sequence of 125. CD spectra showing an increase of chiroptical activity after irradiation with different wavelengths: (c) λ = 365 and (d) λ = 450 nm. (e) CD switching upon subsequent irradiation cycles. Reproduced from ref. 294 with permission from the Royal Society of Chemistry, copyright 2021. | ||
Pieroni and Ciardelli296 reported examples on this topic in poly(glutamic) 126 derivatives and Ueno and Osa in poly(aspartic)300127 derivatives (Fig. 60a and b). This strategy was also employed by Ciardelli and coworkers using spiropyrans 128 as photoresponsive side chains instead of azo derivatives (Fig. 60c).302
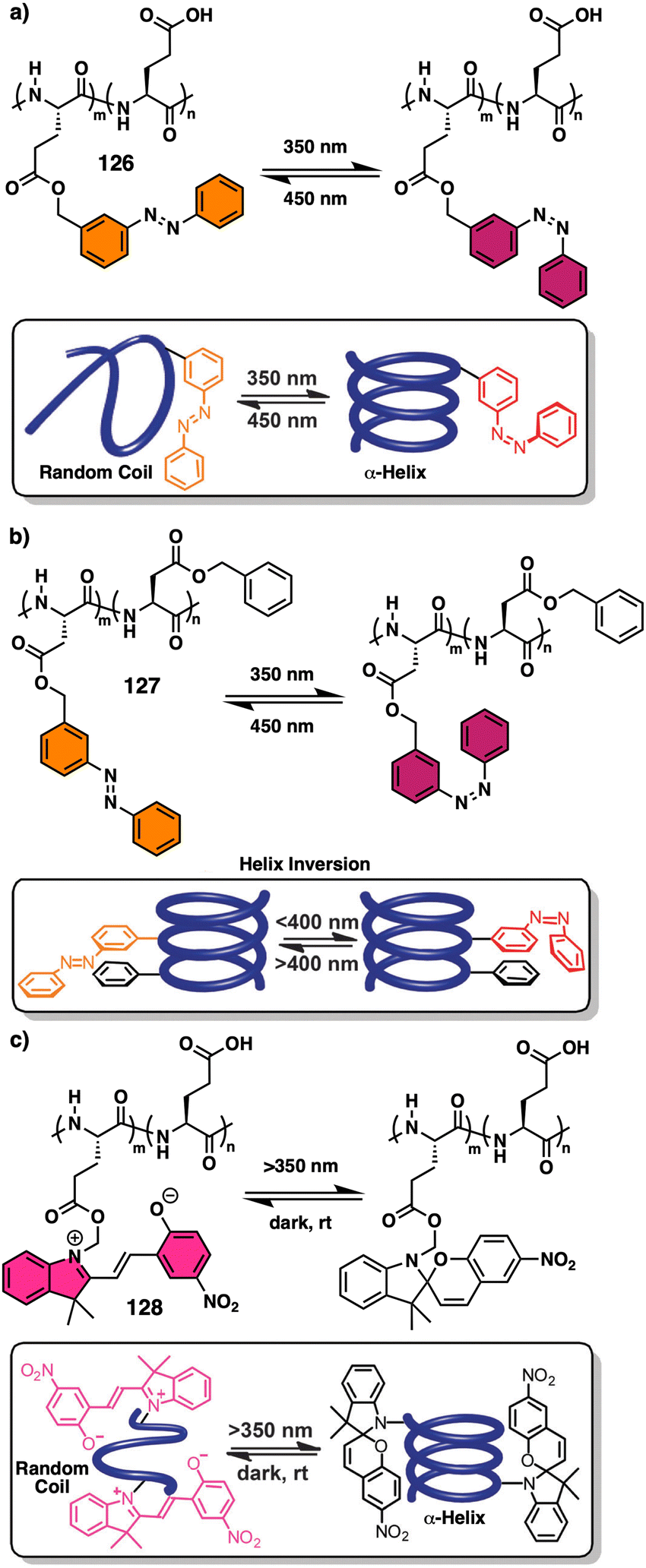 | ||
| Fig. 60 Structure and behavior with light of α-helical foldamers composed by (a) glutamic acid-azobenzene derivatives (126) and (b) aspartic acid-azobenzene derivatives (127). (c) Structure of glutamic acid-spiropyran derivative (128). A schematic illustration of the photoisomerization impact on the folding of the molecules is depicted below each section. Reproduced from ref. 295 with permission from the Royal Society of Chemistry, copyright 2016. | ||
Short helical foldamers containing photoresponsive units in the main chain or in the side chains can also be found in the literature.295 In these systems, the photoisomerization of the foldamer can trigger the unfolding of the system, a dimerization process, or affect their guest-binding ability.302–305 As an illustration, Jiang designed a small foldamer containing an azobenzene and two phenyl-1,2,3-triazole moieties (129) that, through photoisomerization of the foldamer backbone, changes the binding affinity for anions (Fig. 61a).306 Flood showed that foldamer 130, which contains a central benzene endowed with an amino acid (Leu) along with aryl triazoles-azobenzene units, can be used to bind or release small anions such as Cl−, depending on the E or Z configuration of the azo group.307
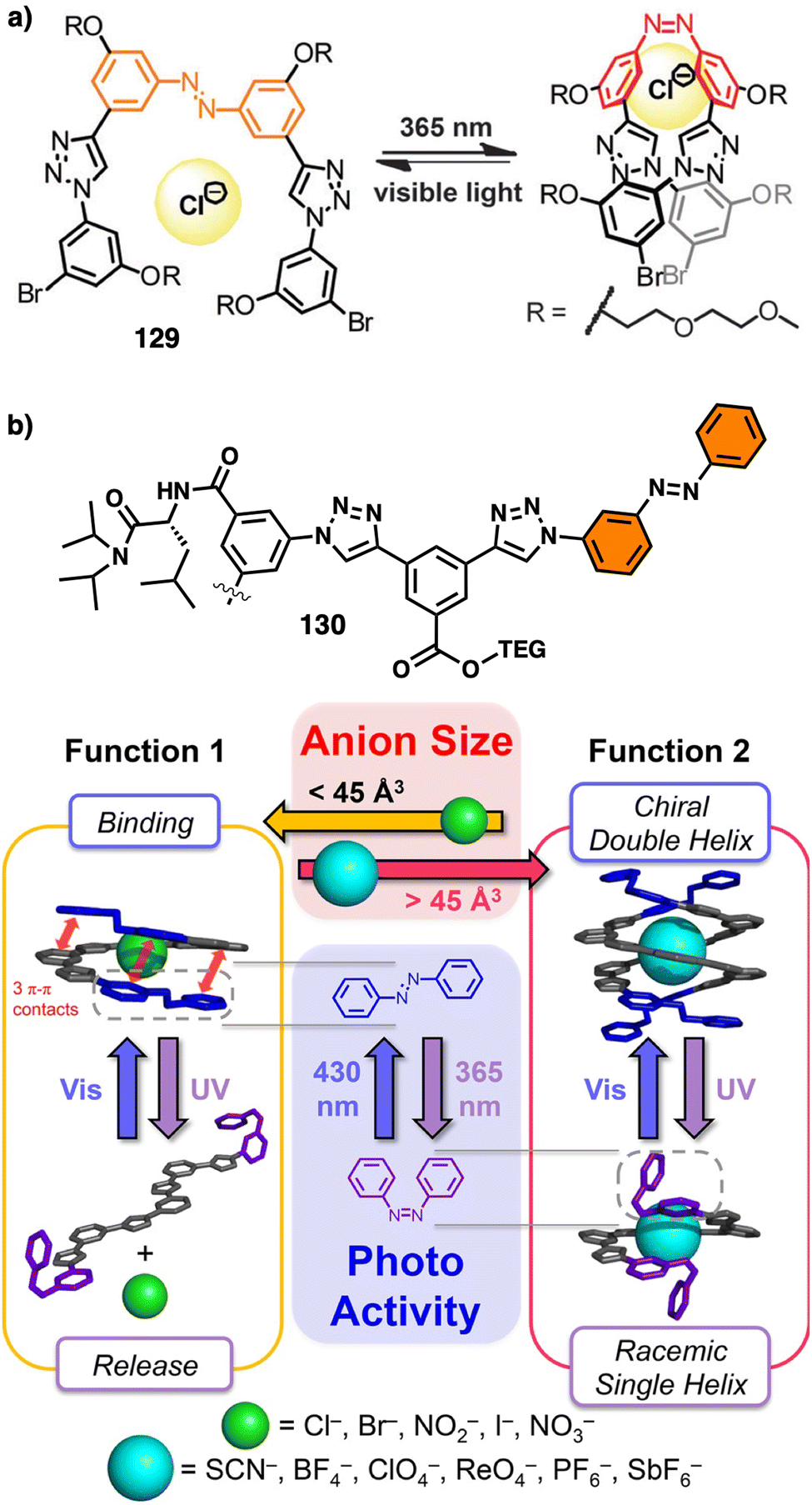 | ||
| Fig. 61 (a) Structure of foldamer 129, binding to Cl− and light-driven conformational change. Reproduced from ref. 303 with permission from the American Chemical Society, copyright 2010. (b) Structure of foldamer 130 and schematic illustration of the cascade of structural transformations mediated by photoirradiation and anion binding. Reproduced from ref. 307 with permission from the American Chemical Society, copyright 2018. | ||
In case of using large anions such as BF4− to interact with 130, the Z/E isomerization of the azo group leads to the formation of single or double helices.307
Foldamers based on oligo(meta-phenyleneethynylene)s (OmPEs) provided inspiration for the preparation of photoresponsive materials by including of photoresponsive azobenzene groups in the side chains or, mainly, within the sequence.308–314 Based on these designs, Hecht reported several examples of photoresponsive OmPEs foldamers bearing, for instance, only the central azo unit (131) (Fig. 62a),315–319 different alternating orientations of azobenzenes within the OmPEs-azo sequence (132) (Fig. 62b),313 or composed entirely of azobenzene units (133) (Fig. 62c).313,316 Most of the reported examples show variations in their folding degree upon photoirradiation. Generally, a decrease in the intensity of the CD signal is observed once the corresponding photostationary state is reached.295 Representative examples of the structures and their chiroptical properties are depicted in Fig. 62. Hecht also reported that slight variations in the connectivity of the units that make up the foldamers have a large impact on their chiroptical responses.
 | ||
| Fig. 62 Structures and CD spectra showing variations in the helical structures of foldamers containing (a) one central azo group (131) (reproduced from ref. 312 with permission from John Wiley and Sons, copyright 2006). (b) Alternating azo units (132) Reproduced from ref. 313 with permission from the Royal Society of Chemistry, copyright 2013 and (c) composed by azo units with alkynyl connectors of different lengths (133 and 134) Reproduced from ref. 314 with permission from John Wiley and Sons, copyright 2015. Otherwise, stated variations in the ECD spectra correlate with irradiation time. | ||
Replacing the alkynyl connector in 133 by a bisalkynyl unit resulted in self-assembly of foldamer 134, leading to a CD inversion. The assembly/disassembly process is easily controllable by simply irradiating the sample (Fig. 62c).314
2.6 External stimulus: circular polarized luminescence
Circular polarized luminescence (CPL) is an intriguing property of certain types of chiral luminophores that emit light with preferred handedness.317–322 In recent years, this type of materials at both the molecular,323–325 macromolecular and supramolecular levels326–329 have found applications in the development of CP-OLEDS,330–332 security encryption systems,333 tags for biology334 and asymmetric synthesis,335 in among many others. Although CPL emission is a well-established research topic whose importance is constantly growing year by year, the application of CP-light as an asymmetry inducer is still in its infancy. Therefore, a small number of examples on this topic can be found in the literature.336 Here we summarize some of the most relevant examples of asymmetry induction in covalent and supramolecular helical polymers. | ||
| Fig. 63 (a) Structure of the copolymers employed in this work 135, 136 and 137. (b) Schematic illustration of the photo-resolution of the prochiral moiety by irradiation with CP-light. (b) Structure of the copolymers employed in this work poly-135, poly-136 and poly-137. (c) ECD spectra of 136 showing the asymmetry induction after irradiation with (+) or (−)-CP-light. Reproduced from ref. 337 with permission from the American Chemical Society, copyright 2000. | ||
The ortho-, meta- or para-linkage of the photoactive moiety to the polymeric backbone is also a key factor in the transfer of asymmetry from the pendant to the main chain, losing effectiveness when we go from ortho, to meta and para. It is important to highlight that the helical sense of the polymer can be switched reversibly by alternating irradiation with (+)- or (−)-CP-light and return to the axially racemic state by irradiation with non-polarized light.21
Nakano and coworkers induced a preferred handedness in polyfluorene derivatives 138–140 by irradiation with either (+)- or (−)-CP-light (Fig. 64a).60 They found that the helix induction mechanism is based on a photoderacemization process in the photostationary state. This is conceivable through a chirality amplification process that takes place through the deactivation of a coplanar conformation in polyfluorene leading to right- and left-handed twisted dyads.
 | ||
| Fig. 64 (a) Schematic illustration of the asymmetry induction by CP-light irradiation and structures of polymers 138–140. (b) ECD monitoring of asymmetry induction after exposure to CP-light of poly(DOF) film (138). (c) CPL spectra of photo-induced films of poly(DOF) (138). Reproduced from ref. 50 with permission from the American Chemical Society, copyright 2018. | ||
Asymmetry induction could be achieved for polymers with longer alkyl chains [i.e., poly(DOF) 138, octyl chain, and poly(DDF) 139, dodecyl chain], while it was not feasible for the one carrying a shorter chain [hexyl chain, poly(DHF) 140], which required an auxiliary molecule to aid the induction of chirality. The chirality-induced samples in the film state remained stable for years if stored protected from light irradiation and at room temperature (Fig. 64b and c).60 Natansohn reported asymmetry induction in polymethylmethacrylate 141 endowed with photoresponsive azobencene units as side chains (Fig. 65a). The generated liquid crystalline phase takes advantage of the photoisomerization of all azobenzene groups in the film plane, inducing a preferred arrangement of the chromophores that changes from a random to a chiral environment (Fig. 65b). The chirality of the films can be exchanged after subsequent irradiation cycles with either (+) or (−)-CP-light, giving rise to interesting chiroptical switches in the condensed state (Fig. 65c and d).338 Similarly, Li, Yang and coworkers reported the impact of the linker between the azobencene unit that now holds a stereogenic center and the PMA backbone of 142(x) (x = 0–6) (Fig. 65e) in the light-driven asymmetry induction.
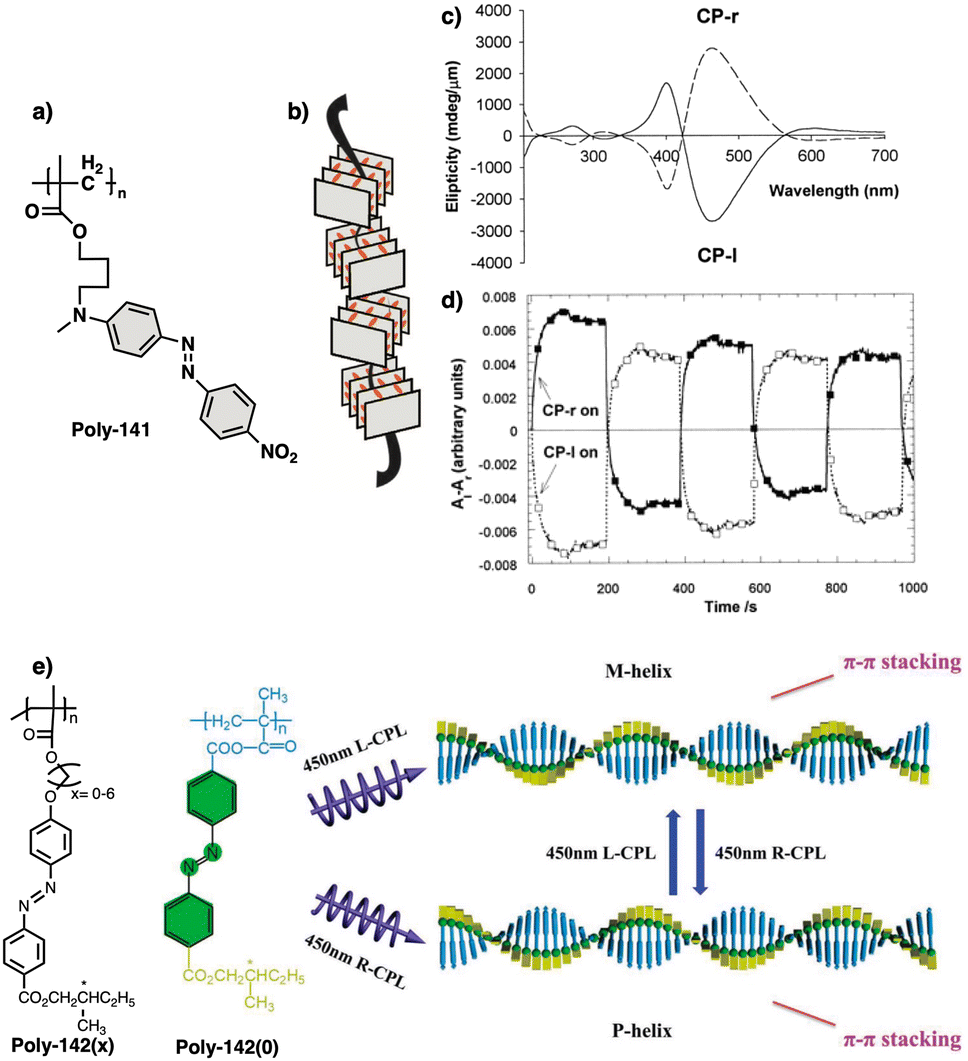 | ||
| Fig. 65 (a) Structure of poly-141. (b) Schematic illustration of the cholesteric phase generated upon CP irradiation. Reproduced from ref. 338 with permission from the American Chemical Society, copyright 2000. (c) ECD spectra of thin films showing the helical orientation of the molecules in the crystalline phase after irradiation with (L) or (r)-CP-light. (d) Several cycles of chirality switching in the crystalline phase by subsequent irradiation with CP-light of different chirality. (e) Structures of Azo polymers and schematic illustration of the supramolecular chiral structures of poly-142(0) induced by 450 nm CP-light. Reproduced from ref. 339 with permission from John Wiley and Sons, copyright 2022. | ||
They found that shorter linkers did not produce efficient chirality harvesting after thermal annealing, while the other derivatives, bearing longer linkers (from Azo-PMA-142(1) to Azo-PMA-142(6)), exhibited effective asymmetry transfer after thermal annealing. Remarkably, all derivatives show asymmetry induction after irradiation with (+) or (−)-CP-light, except 142(6) which can only be folded/unfolded by UV light irradiation.339 In the field of azobencene polymer derivatives, Meijer, Vantomme and Meskers reported alternating copolymers formed by azobenzene and fluorene units (poly-143) that form cholesteric phases with strong optical activity upon thermal annealing (Fig. 66a). Taking advantage of the trans/cis/trans isomerization of the azobencene units, the macromolecular helicity and organization of the polymer chains could be modulated by CP-light (Fig. 66b–e). Remarkably, due to the diastereomeric nature of the polymer chains and the light, there is asymmetry in the interconversion pathway.340 The same team also reported an azobencene–oligosiloxane alternating copolymer that shows a similar behavior, the formation of a liquid crystalline chirality that can be controlled by CP-light irradiation.341
 | ||
| Fig. 66 (a) Structure of alternating poly-143. (b) ECD spectra showing the helical orientation of the molecules in the crystalline phase after thermal annealing. (c) Schematic illustration of the cholesteric helical inversion after irradiation with (L)- or (R)-CP-light. (d) ECD spectra showing the helical inversion after irradiation with (L) or (R)-CP light. (e) Rewritable chirality of the crystalline phase by subsequent irradiation with CP-light of different chirality. Reproduced from ref. 340 with permission from John Wiley and Sons, copyright 2021. | ||
Kim, Seo and coworkers reported the asymmetry induction by CP-irradiation of triphenylamine342–346 derivative 144a bearing diacetylene units (Fig. 67a).
 | ||
| Fig. 67 (a) Structures of derivatives 144a-c and schematic illustration of the asymmetry induction and photopolymerization of 144a. ECD spectra showing the mirror images generated after (b) CP-irradiation and (c) photopolymerization. Reproduced from ref. 343 with permission from Springer Nature, copyright 2015. (d) Sergeants and Soldiers coassembly of 144a with (S)-144c as a molecular Sergeant: Schematic depiction of the copolymerization to M-helix and P-superhelix formation. (e) Amplifying and overriding molecular chiral information in the supramolecular state by CPL: Schematic depiction of 144a/(S)-144c copolymerization upon l- and r-CPL exposure. Reproduced from ref. 344 with permission from the American Chemical Society, copyright 2022. | ||
Irradiation with CP-light produced a chiral bias in the arrangement of the triphenylamine core that was subsequently amplified throughout the supramolecular polymerization process. Further irradiation with light triggered the photopolymerization of the diacetylene units, generating the corresponding covalent polymer with memorized chirality (Fig. 67a–c).343 They also showed that supramolecular chirality can emerge from a transiently chiral molecular building block by transmitting chiral information inherited from the CP-light irradiation.344 Consequently, a chiral radical cation was generated that aggregated into preferred-handed helical stacks (Fig. 67d). The nonlinear screw sense excess as a function of CPL irradiation time is reminiscent of the chiral induction expressed by the Sergeants-and-Soldier principle, where the co-assembly of a transiently chiral Soldier with a permanently chiral Sergeant takes place. Moreover, matching the handedness of the CPL to the Sergeant's direction produced an abnormally enhanced chiral response. However, in the mismatched scenario (CPL vs. mismatched Sergeant's direction) (Fig. 67e), a critical molar fraction of the Sergeant was found to bifurcate the bias: above a critical point, the helicity determined by the Sergeant persisted. Below the critical point, the CPL can override the molecular chirality with increasing irradiation time. Therefore, the authors demonstrated that supramolecular chirality can be developed following the handedness of the external stimulus and overturn—in some extent—the one imposed via molecular constraints (Fig. 67e).344 Recently, Raynal and coworkers demonstrated how the presence of radical species in triarylamine trisamides affects to its self-assembly.345 Zou reported a similar CP-light asymmetry induction in porphyrins 145 endowed with diacetylene moieties (Fig. 68a).
 | ||
| Fig. 68 (a) Structure of the porphyrin 145 used in this work. (b) Schematic illustration of the photoinduced asymmetry induction. (c) ECD showing the induction of mirror image helical structures upon irradiation with (+)- or (−)-CPL. (d) TEM image of the supramolecular fibbers generated upon self-assembly of 145. Reproduced from ref. 346 with permission from the Royal Society of Chemistry, copyright 2019. | ||
Irradiation of these molecules with (+) or (−)-CP-light generated photo-induced protonation of the basic internal rim of the macrocycle, producing P- or M-helical stacks, respectively (Fig. 68b–d). Further irradiation with non-polarized light triggered the polymerization of the diacetylene moieties yielding the corresponding covalent polymer with memorized handedness.346 Sánchez reported asymmetry induction in the supramolecular polymerization of achiral N-annulated perylene derivative 146 (Fig. 69a). Irradiation with (+) or (−)-CP-light during the elongation process results in the adoption of preferred P or M helical assemblies as determined by ECD (Fig. 69b and c). Additionally, after removing CP irradiation, these systems exhibit a “memory” that shows, after 2 hours, an ECD trace virtually identical to that initially prepared.120
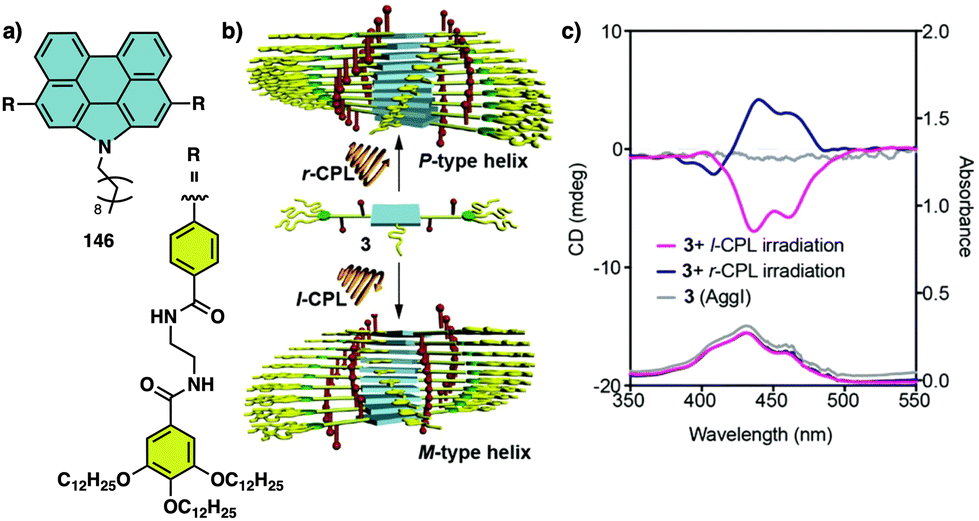 | ||
| Fig. 69 (a) Structure of 146. (b) Schematic illustration of the asymmetry induction by CP-light irradiation. (c) ECD Spectra of the opposite handed helical structures generated by irradiation with CP-light. Reproduced from ref. 120 with permission from the Royal Society of Chemistry, copyright 2020. | ||
Lee and co-workers reported the co-assembly of amidoamine 147 with monoacid 148 bearing diacetylene units that self-assemble to generate supramolecular ribbons in both gel and film states (Fig. 70). After irradiation with (+) or (−) CP-light, these ribbons fold into preferred left- or right-handed structures, as determined by ECD and SEM visualization.347
 | ||
| Fig. 70 (a) Structures 147 and 148 and schematic illustration of the asymmetry induction by CP-light irradiation of 147-148 co-assembly. (b) ECD spectra showing opposite chirality after irradiation with L/R CP-light. (c) SEM images of the gels with different handedness. Reproduced from ref. 347 with permission from the American Chemical Society, copyright 2022. | ||
2.7 External stimuli: solvents
The main endoergic contribution to solute–solvent interactions is the formation of a cavity in the solvent to accommodate the solute. This is a chemical property of the solvent, which depends on the association of its molecules in a liquid state.348 The interactions between solvents and solutes are of paramount significance in the field of organic chemistry. They play a central role in governing various aspects, such as solubility, reactivity, and structural characteristics of the compounds involved.An important parameter that affects the folding of the foldamers is the nature of the solvent, which greatly affects their secondary structure. For instance, oligophenylethynylenes are also called “solvophobic foldamers” due to their sensitivity to solvents.350–352 On the other hand, the aromatic amide foldamers developed by Huc and co-workers showed remarkable solvent folding stability in a wide range of solvents,353 which is quite unique compared to other foldamers. However, this apparent and atypical lack of solvent dependence led them to carry out more in-depth studies on the subject. Thus, a helical aromatic oligoamide foldamer (149) bearing triethyleneglycol (Tg) side chains (Fig. 71a) was prepared to study its folding in a wide range of solvents.354
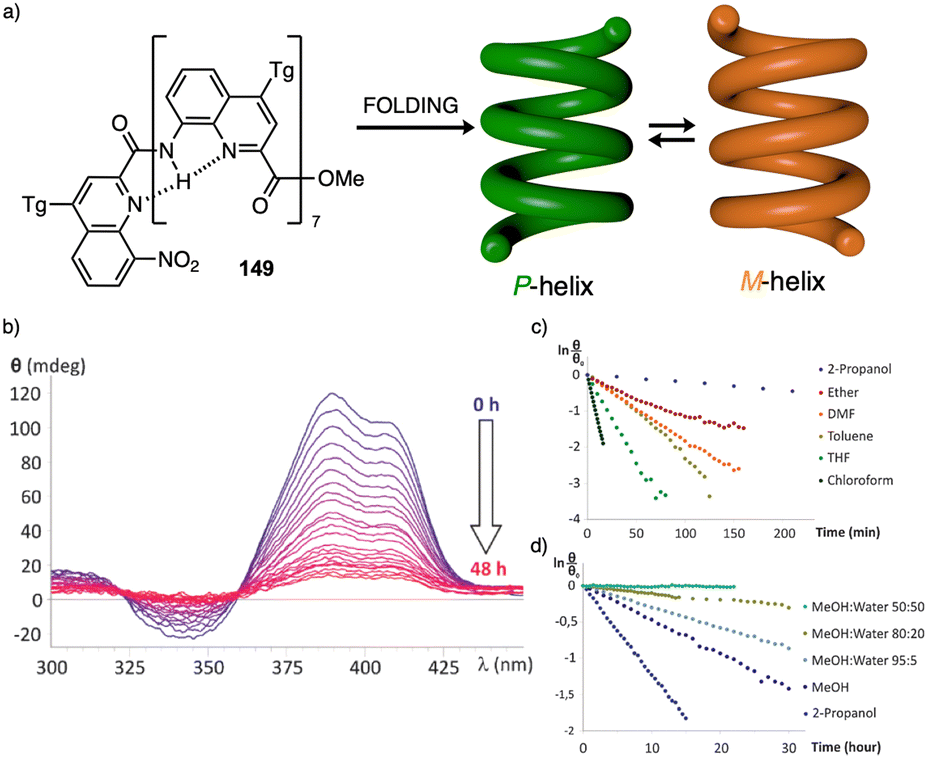 | ||
| Fig. 71 (a) Chemical structure of foldamer 149 and its folding into a mixture of P and M helices. Time dependent ECD racemization studies for P-149 as a function of solvent in (b) MeOH, (c) different solvents and (d) MeOH:Water mixtures. Reproduced from ref. 354 with permission from the Royal Society of Chemistry, copyright 2012. | ||
Although the polymer is achiral, the two P and M helical structures of 149 were isolated by chiral HPLC when folded into an axially racemic mixture, allowing their exceptional stability to be determined (Fig. 71a). The helices did not racemize during the chromatography studies. By performing time dependent ECD studies in different solvents (Fig. 71b–d), they found that in chloroform, which was believed to provide highly stable conformations, turned out to be one of the least favourable solvents for these foldamers (Fig. 71c).
On the other hand, protic solvents,355 such as methanol–water mixtures, demonstrated a remarkable ability to stabilize helical conformations (Fig. 71d).
Another example of solvent-dependent aromatic foldamers was reported by Li who prepared hybrid oligomers containing one aromatic amide trimer or pentamer and one or two aromatic 1,2,3-triazole tetramers (150) (Fig. 72a).356 In this case, the researchers explored a conformational communication mechanism to induce a helical sense in the 1,2,3-triazole tetramer from the aromatic amide region. It was found that while in polar solvents the conformational communication from the aromatic amide to the triazole tetramer stops, in low polar solvents the conformational communication flows between the two fragments (Fig. 72b).
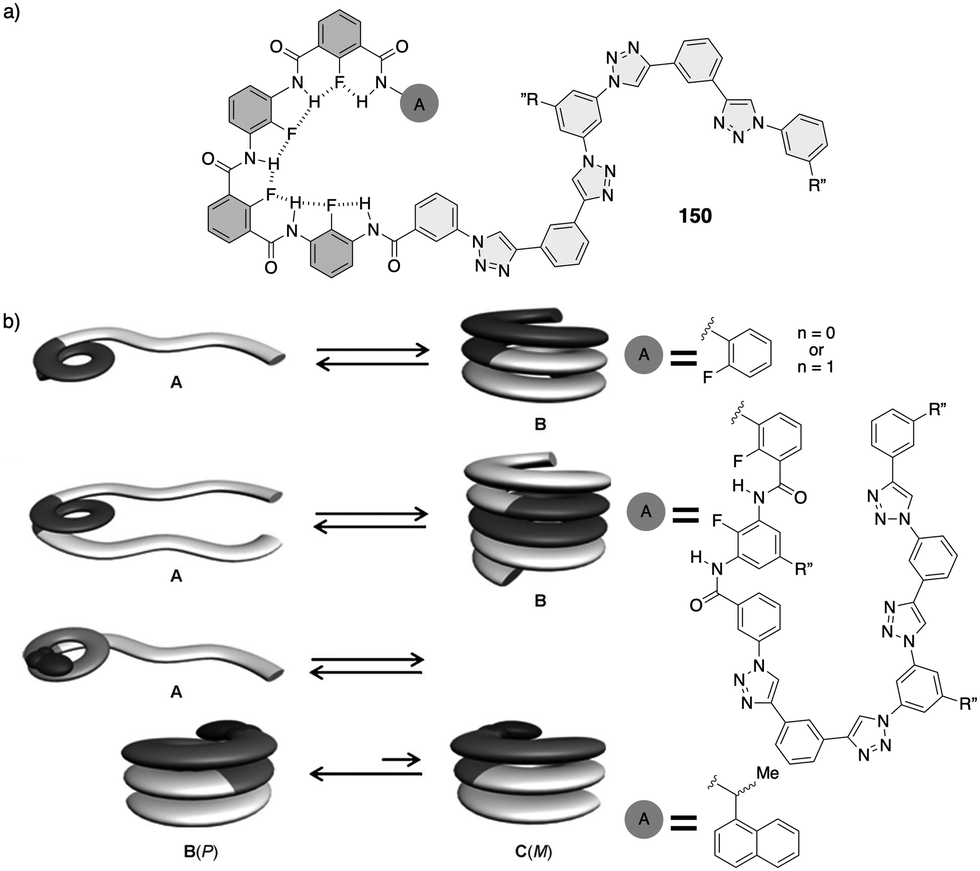 | ||
| Fig. 72 (a) General foldamer structures 150 used in this work. (b) Schematic representations of the solvent-dependent (A polar; B low-polar) folding-induced processes for the different 150 derivatives. Reproduced from ref. 356 with permission from John Wiley and Sons, copyright 2014. | ||
Flood and Liu described a more complex system where two stimuli, solvent, and anions, were combined to produce a structural change from single to double helices.357 Thus, they studied a series of C2-symmetric aryl-triazole foldamers (151) (Fig. 73a) in which the interconversion between chloride-stabilized single and double helices can be understood and controlled by sequence modification strategies. Interestingly, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 single to 2:2 duplex (Fig. 73c) helical switch could be obtained at room temperature by increasing the solvent polarity (Fig. 73b and d), indicating that solvophobically driven π-stacking plays a critical role in stabilization of the 2:2 double helix compared to the single helix. For instance, there was a notable increase in the population of the double helix as the proportion of acetonitrile in chloroform or DCE increased (Fig. 73b). This indicates that the enhanced stability of double helices in polar solvents is associated with greater burial of π surfaces within the double helix structure.
:1 single to 2:2 duplex (Fig. 73c) helical switch could be obtained at room temperature by increasing the solvent polarity (Fig. 73b and d), indicating that solvophobically driven π-stacking plays a critical role in stabilization of the 2:2 double helix compared to the single helix. For instance, there was a notable increase in the population of the double helix as the proportion of acetonitrile in chloroform or DCE increased (Fig. 73b). This indicates that the enhanced stability of double helices in polar solvents is associated with greater burial of π surfaces within the double helix structure.
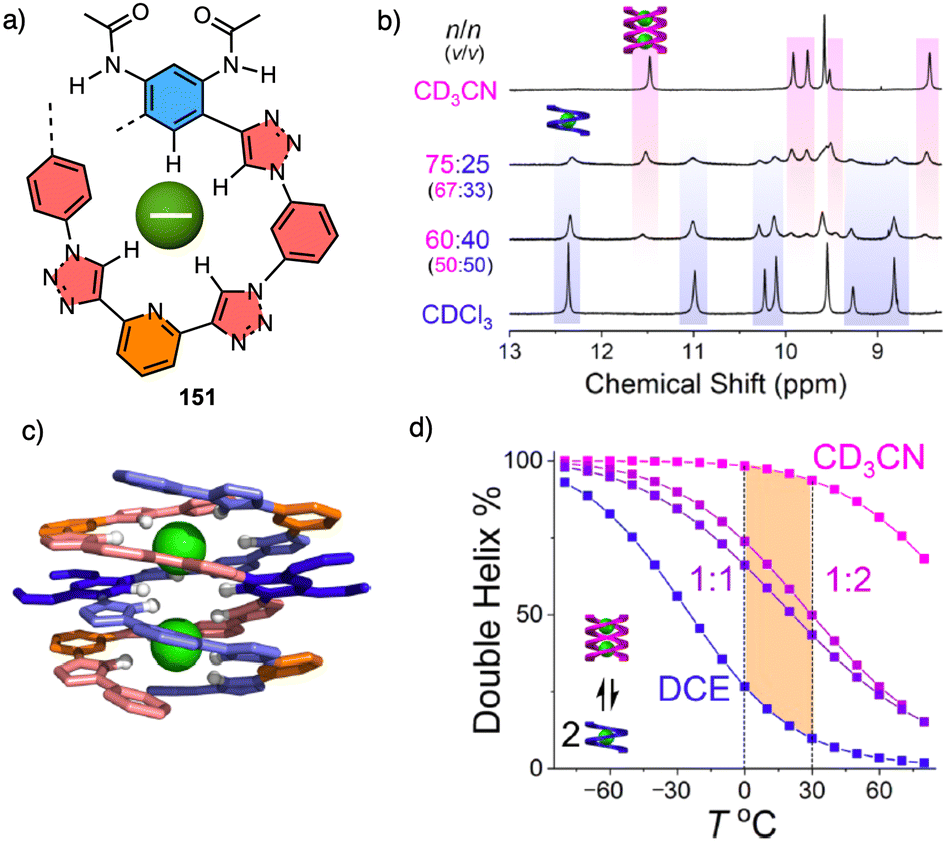 | ||
| Fig. 73 (a) Chemical structure of anion foldamer host 151. (b) Double helix formation of 151-Cl− in response to solvent polarity (5 mM, 15 equiv. of TEACl). (c) DFT-optimized geometry of the 2:2 double helix formed by 151-Cl. (d) Two factors combined showing maximal solvent-induced swings in double helix population around ambient temperatures (orange shade). Reproduced from ref. 357 with permission from the American Chemical Society, copyright 2018. | ||
Another family of foldamers whose helical structure is also solvent dependent are the AIB containing foldamers described by Clayden.1 These foldamers tend to form a 310-helix that shows a solvent dependent decay of screw-sense excess induced by chiral residues introduced in both ends (152) or at one end (153) of the foldamer chain.358,359 The observed signal decay was attributed to the occurrence of ‘tendril perversions’, which are characterized by random breaks in hydrogen bonds along the helix (Fig. 74a). These breaks cause a helical reversal in the specific location where they occur, producing the observed decay in the signal. After exhaustive study using a variety of analytical and computational techniques, the intrusion of ‘tendril perversions’ was shown to have an effect analogous to a γ-turn, a less prevalent turn that involves the formation of a hydrogen bond between residues i and i + 2 (Fig. 74b). Furthermore, γ-turn conformations were also found to be more frequent in polar solvents, as tendril perversion increases from 0.5% per residue in THF to 6% per residue in methanol (Fig. 74c).
 | ||
| Fig. 74 (a) Tendril perversions and (b) γ-turn in AIB foldamer 152. (c) Chemical shift differences (Δδ) measured in a series of three ‘frame-shifted’ oligomers, each containing three pairs of abundance-labelled (100%, 50% or 25%) 13C labels. Screw-sense preference exhibited by Aib oligomer 153 decays as a terminal chiral influence becomes more distant. Reproduced from ref. 358 with permission from the Royal Society of Chemistry, copyright 2016. | ||
In supramolecular helical polymers, monomeric repeating units are held together by non-covalent interactions. The dynamic nature of these interactions makes that the nature of the solvent can affect supramolecular polymerization pathways.360 Consequently, investigating interactions between supramolecular polymers and solvents remains challenging as solvent molecules will participate in the molecular stacking arrays of various ways. So, in recent years detailed solvent effects on supramolecular polymers can be found in several excellent works, focusing on stability, kinetics, pathway selection, and structure.28,361–364 This review will focus mainly on supramolecular polymers whose supramolecular helix evolves into a different one due to the action of solvents.
 | ||
| Fig. 75 Chemical structure of amphiphilic BiPy-BTA (154) and its hierarchical self-assembly from supramolecular fibre to triple helical bundle depending on the water content of water–isopropanol mixtures. Reproduced from ref. 365 with permission from the American Chemical Society, copyright 2014. | ||
Meijer and George reported a coronene bisimide bearing a 3,5-dialkoxy substitution on the imide phenyl groups (55) (Fig. 14) that shows solvent-dependent aggregation behavior.164 Thus, helix inversion is observed when cyclic solvents are used to form the chiral aggregate, e.g., cis/trans-decalin mixture or cyclohexane. Zhao reported another example of aggregation solvent dependence using, as building block, a molecule that contains naphthalimide and cholesteryl groups (155) (Fig. 76a).366,367 This molecule self-assembles in low-polarity solvents (THF/hexane) into right-handed supramolecular helices (ECD > 0) leading to M-oriented fibres observed in transmission electron microscopy studies (TEM) (Fig. 76b and c). In solvents with increased polarity (THF/water), this molecule self-assembles into left-handed supramolecular helices (ECD < 0), that promote the formation of toroidal structures formed by P fibres (Fig. 76b and d).
 | ||
| Fig. 76 (a) Chemical structure of 155. (b) Normalized CD spectra of a vesicle sample (mixture of THF/water) and a gel sample (mixture of THF/hexane). TEM images of (c) M- and (d) P-type twist nanofibers observed in hexane and water, respectively Reproduced from ref. 366 with permission from the American Chemical Society, copyright 2016. | ||
Ajayaghosh reported a chiral OPE (156) that assembles, in n-decane, into a helical supramolecular polymer that in turn, with increasing building block concentration, assembles into superhelices of opposite handedness (Fig. 77a). Interestingly, the addition of chloroform recoils the superhelices to get the initial supramolecular helices.33
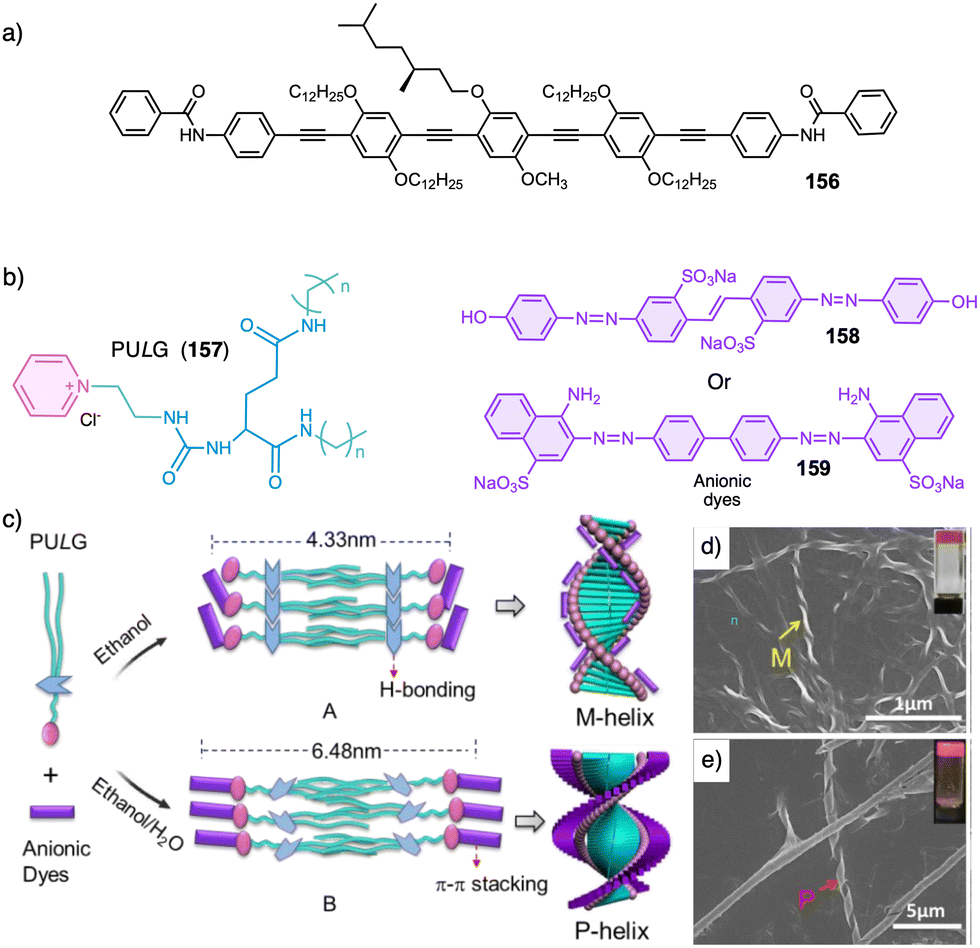 | ||
| Fig. 77 (a) Chemical structure of OPE 156. (b) Chemical structures of L-glutamide derivatives (157) and achiral dyes 158 and 159. (c) Schematic representation of different helical co-assemblies of 157 with 158 or 159 induced by solvent conditions. (e) SEM images of aggregates showing an M helix obtained from ethanol. (d) SEM images of aggregates showing a P helix obtained from ethanol/water. Reproduced from ref. 368 with permission from the Royal Society of Chemistry, copyright 2019. | ||
Liu reported another example in which the solvent dominates the helical structure over the rest of the stimuli.368 The co-assembly of L-glutamide derivatives (157 = PULG) with achiral anionic dyes (158, 159) (Fig. 77b) promotes the formation of P or M helical structures in presence or absence of water. Thus, while co-assembly of monomers in ethanol results in the formation of M-oriented nanoribbon-like structures (Fig. 77c and d), addition of water to the ethanol solution led to a helical inversion into a P-helix (Fig. 77c and e). The amplification of asymmetry was mainly driven by electrostatic interaction between the gelator (157) and the achiral dyes (158, 159). Remarkably, this interaction caused the transformation of the nanofibrous structures of the gelator into uniform left-handed helices stabilized by hydrogen bonds of 157, while addition of water produced the formation of right-hand helices stabilized by π–π stacking of dyes 158 or 159. Ortí and Sanchez designed a chiral N-annulated perylene bisimide that self-assembles into supramoplecular helical polymers with different handedness depending on the solvent (methylclyclohexane or toluene).369
Therefore, PPAs carrying chiral amino acid derivatives as pendants usually show helix inversion in different solvents due to conformational changes in the pendant group (Fig. 2b),98 which places the substituents of the chiral pendant in different spatial orientations. In this way, a mechanism is produced that allows opposite helical senses to be induced.
For instance, Tang and Yashima described PPAs bearing L-leucine370 and D-/L-alanine371 derivatives as pendants that showed solvent-induced helix inversion. Moreover, Yashima, using a PPA with a chiral alanine as pendant (poly-160), was able to visualize, through high-resolution AFM images, the two helical senses adopted by poly-160 in solvents with different polarity, e.g., THF and benzene (Fig. 78).372,373 Helix inversion also occurs in the solid state by placing a 2D crystal in vapors of a solvent that induces the opposite chirality (Fig. 78).
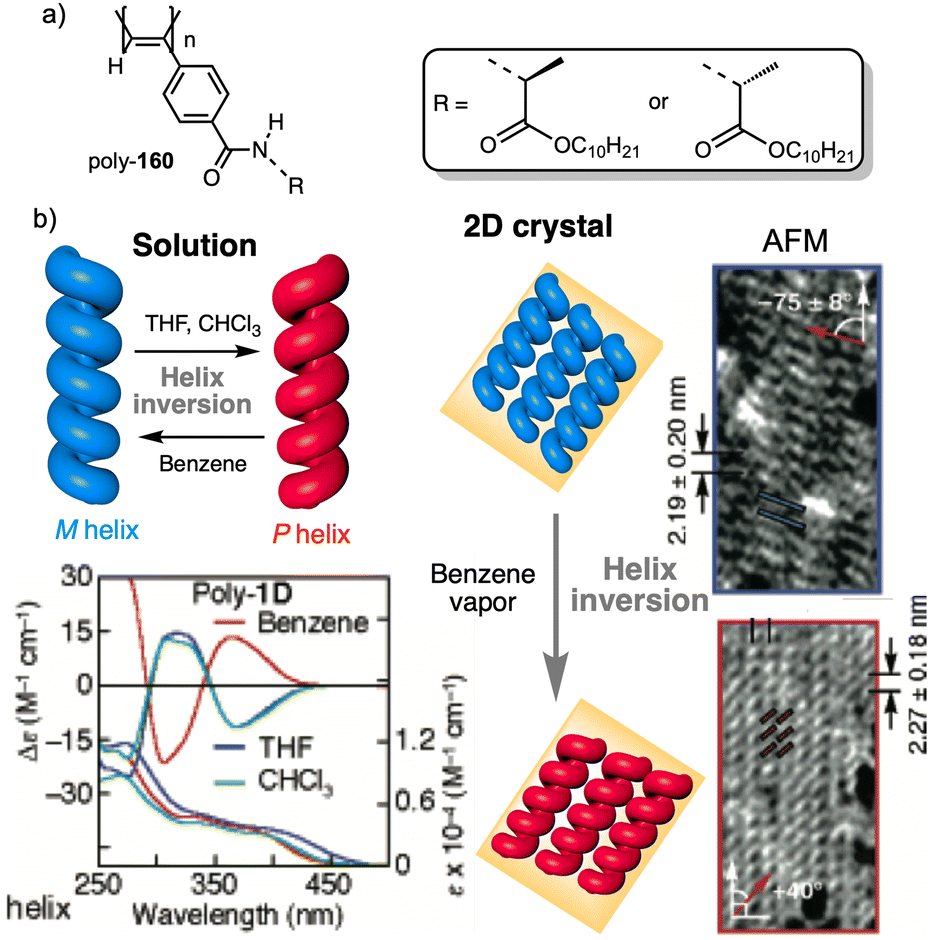 | ||
| Fig. 78 (a) Chemical structure of poly-L/D-160. (b) Schematic representation of poly-D-158 and its macromolecular helicity inversion in solution and in 2D crystal state Reproduced from ref. 372 with permission from the American Chemical Society, copyright 2006. | ||
This dynamic behavior of PPAs in the solid state was also employed by Tsuchihara to produce a colour change in a PPA film under solvent exposure.374 Thus, a chiral para-substituted ether-PPA bearing an alcohol group and a long alkyl chain (poly-161) (Fig. 79a) shows a colour change in the film state prepared in diethylamine upon exposure of the film to solvent vapors such as acetone, methanol, THF, and CHCl3. The yellow to red colour change of the film is accompanied by a bathochromic UV shift from 400 to 500 nm, and by a new ECD pattern with a positive Cotton band at 550 nm, which indicates the presence of a highly stretched helix (Fig. 79a).
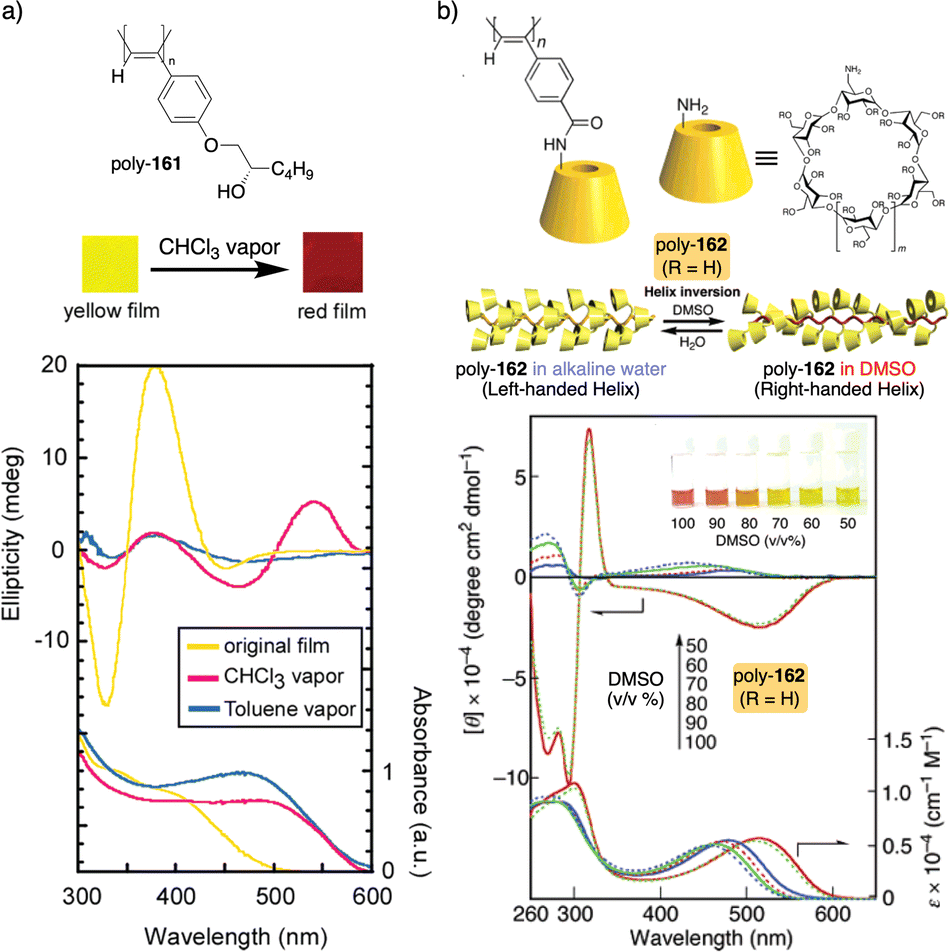 | ||
| Fig. 79 (a) Chemical structure of poly-161 (top), colour change of its film under vapor exposure (middle), and ECD and UV spectra of its films in different solvents (bottom). (b) Chemical structure of poly-162 (top), schematic representation of its helical inversion (middle) and CD and UV-vis spectra in DMSO/water at varied ratios. Reproduced from ref. 374 and 383 with permission from the American Chemical Society, copyright 2006. | ||
Another interesting examples of helix inversion under different solvent vapor exposures can be found in literature of PPAs375,376 and PDPAs.377 In some cases, solvent can produce changes in both the elongation and the helical sense of dynamic helical polymers. Yashima described PPA poly-162 containing optically active cyclodextrin (CyD) as pendant (Fig. 79b),378 whose elongation and screw sense depend on the solvent used to dissolve the polymer. Thus, while poly-162 adopts a more compressed helix in alkaline water, when the DMSO content increases in the solvent mixture, a bathochromic shift is observed in the polyene band, which is accompanied by a colour change of the solution from yellow to red and a helix inversion, as inferred from ECD studies (Fig. 79b). These structural changes are due to a different array of the CyD units along the polymer chains in both solvents.
Over the last few years, several examples have been reported involving solvent-induced helical structural changes in dynamic helical polymers (sense and/or elongation) induced by solvents that affect intra-pendant interactions within a helix.378–383
Recently, Wan384 reported a PPA bearing achiral alkoxycarbonyl and chiral alkylamide substituents at meta-positions of its phenyl ring (poly-163) (Fig. 80a). In this case, a variation in the elongation of the polymer between a cis–transoid (stretched) and cis–cisoid (compressed) can be produced by playing with the polarity of the solvent (Fig. 80b–d).
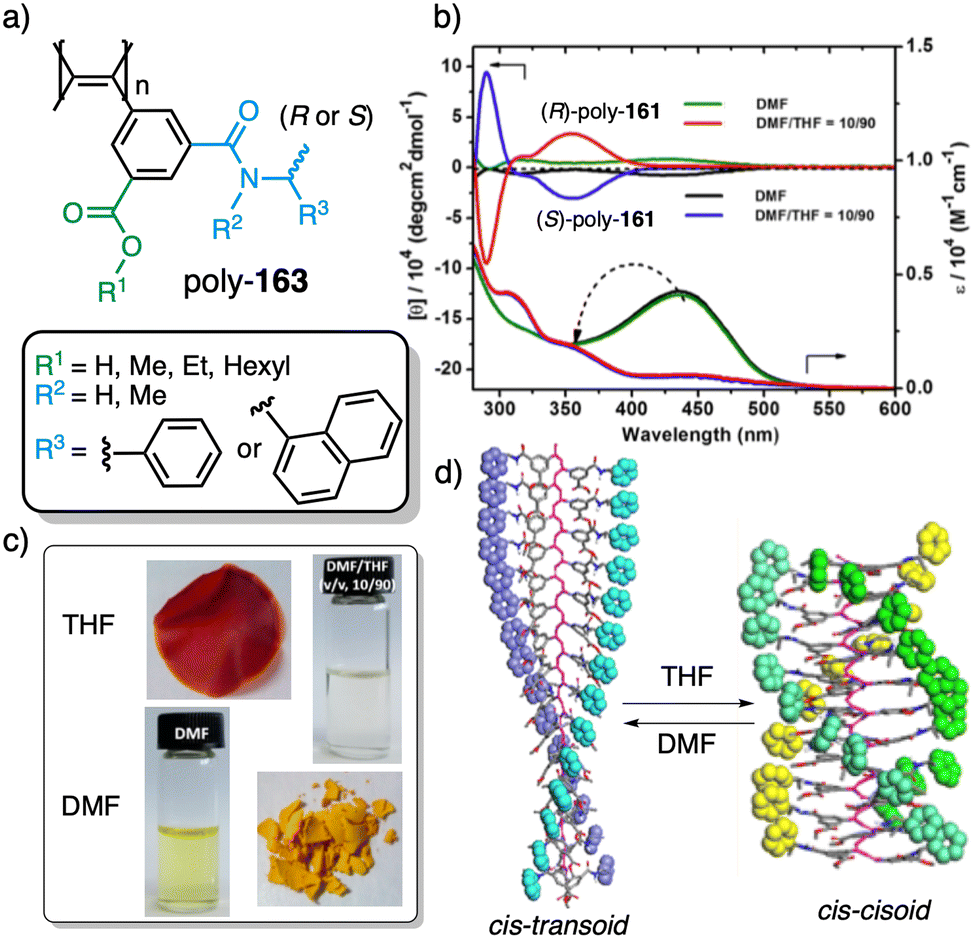 | ||
| Fig. 80 (a) Chemical structure of poly-163. (b) CD and UV-Vis spectra of (R)/(S)-poly-163 in DMF and DMF/THF mixture (v/v, 10/90). (c) Photographs of poly-163 in solid and solution states in THF and in DMF Reproduced from ref. 384 with permission from the American Chemical Society, copyright 2017. | ||
Another family of dynamic helical polymers, whose helical scaffold shows solvent dependence, are polyquinoxalines (PQXs) developed by Suginome and coworkers. In a first example, they reported a poly(quinoxaline-2,3-diyl)s that bears chiral (R)-2-butoxy groups as side chains (poly-164).385 This polymer adopts a P helix in CHCl3, whereas an M helix is adopted in 1,1,2-trichloroethane (1,1,2-TCE) (Fig. 81a), a helix inversion triggered by a conformational change in the pendant. In a different example, a polyquinoxanoline based phosphine (PQXphos) copolymer, consisting of a chiral comonomer carrying (R)-2-butoxymethyl groups and an achiral comonomer with a bulky phosphorous group in a 950/50 ratio, also shows a helix inversion in chloroform and in 1,1,2-trichloroethane/toluene (2/1) mixture.386
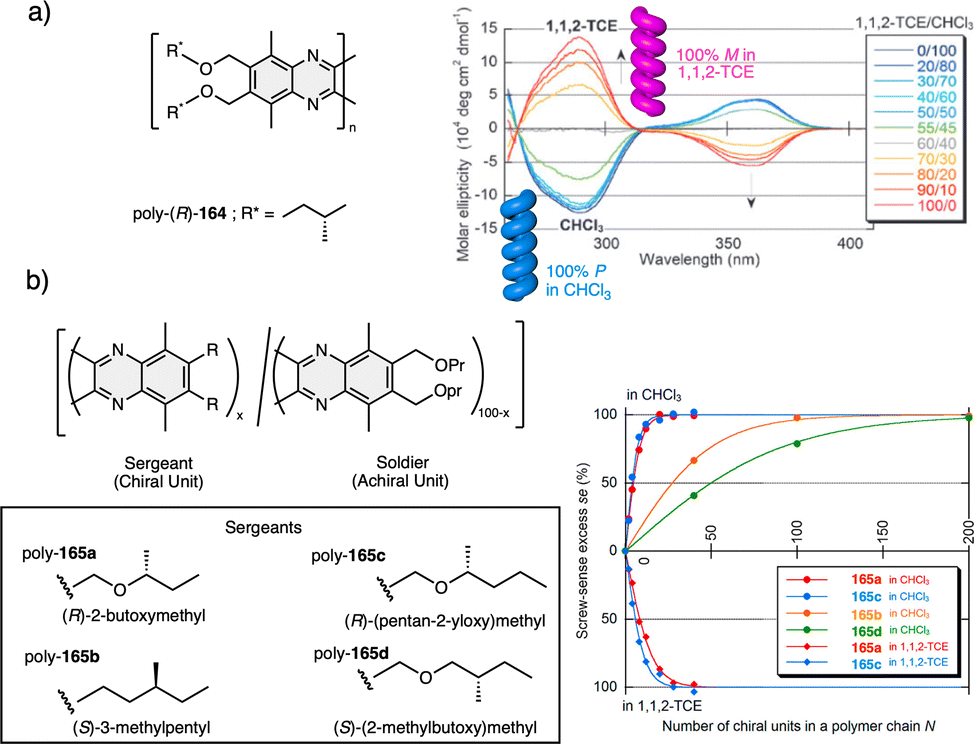 | ||
| Fig. 81 (a) Chemical structure of poly-(R)-164 and its CD spectra in 1,1,2-TCE/CHCl3 with varied ratios. Reproduced from ref. 385 with permission from the Royal Society of Chemistry, copyright 2010. (b) General chemical structures of copolymers series 165a–d and their screw sense excess in different solvents Reproduced from ref. 387 with permission from the American Chemical Society, copyright 2013. | ||
Next, the same group explored how solvents can affect the Sergeants and Soldiers effect in poly(quinoxaline-2,3-diyl) copolymers. Thus, different copolymer series were prepared using, as chiral Sergeants, the monomeric repeating units carrying (R)-2-butoxymethyl (165a), (S)-3-methylpentyl (165b), (R)-(pentan-2-yloxy)methyl (165c) and (S)-(2-methylbutoxy)methyl (165d) side chains at positions 6 and 7 of the quinoxaline rings. Each Sergeant was copolymerized with an achiral Soldier that bears an achiral propoxymethyl chain also at positions 6 and 7 of the quinoxaline rings (Fig. 81b).387 In all cases, an effective P or M solvent-dependent Sergeants and Soldier effect was observed. The helical sense adopted by the copolymer is that dictated by the chiral comonomer (Fig. 81b). The ability of these polymers to adopt a single-handed helix, even when copolymerized with an achiral quinoxanoline, was employed by the same group to prepare a PQXphos acting as a catalysts, whose helical structure can be transferred to an enantiospecific hydrosilylation reaction.388 These PQXs were also used to create a solvent-induced CPL switch.389 Thus, after annealing in CHCl3, a PQX film displayed reflection of right-handed CPL, whereas chiral reversion of reflected CPL was observed after annealing in 1,2-DCE vapor.
Although most examples deal with the polarity of the solvents, there is another relevant parameter that solvents possess and that must be considered, the donor character, which is the ability to solvate cations and Lewis's acids and is related to the presence of atoms with free electron pairs. Freire and Riguera developed a PPA that was able to differentiate solvents based on their polarity and donor number, due to the presence of two independent tuneable bonds in the pendant. To do that, a PPA that bears (R)-α-methoxy-α-(trifluoromethyl)phenyl acetic acid (MTPA) as pendant [poly-(R)-166] was prepared (Fig. 82).390 In this polymer, the amide bond (H–)N–C(O) is sensitive to the donor ability of the solvent—trans, non-donor solvents (CHCl3); cis, donor solvents (THF), while the (O)C–C(–O) bond responds to the polarity of the solvent—sp, polar solvents (DMSO); ap, low-polar solvents (DCM). The sp/ap conformational change (O)C–C(–O) produces a helical inversion in the polymer due to a different spatial location of the substituents, while the cis/trans conformational change in the amide (H–)N–C(O) produces changes in the elongation of the polymer. As a result, four different helical scaffolds are obtained for poly-(R)-166 taken into account the donor character (compressed/stretched) and the polarity (P/M) of the solvent (Fig. 82a). Moreover, this solvent behaviour for poly-(R)-167 is observed in both solution and film state. The authors used the ability of this polymer to form cis amide bonds in THF to create a stereocomplex, where poly-(R)-166 interacts with its enantiomeric and complementary helix to form fibre-like aggregates that evolve into a gel at high concentrations.391
 | ||
| Fig. 82 (a) Correspondence between the four different structural states of poly-(R)-166 and the polar/low-polar and donor/non-donor properties of the solvents. (b) Structure of poly-(R)-166 and PPAs with ortho-, meta- and para-aromatic substitution and anilide and benzamide connectors. Reproduced from ref. 391 with permission from the Royal Society of Chemistry, copyright 2015. | ||
Freire performed also solvent-dependent studies on PDPAs functionalized with chiral amino acids as pendants,147,392,393 obtaining syn/anti conformational equilibria similar to those obtained for PPAs, although the activation energy to promote helix inversion is very high providing the polymers a memory effect. Also, the same group studied how the aromatic substitution in chiral PPAs (i.e., ortho, meta, para) and/or the connector used to link pendant and backbone (i.e., anilide, benzamide) affects the dynamic behaviour of PPAs and their solvent response (Fig. 82b).394,395
The conformational control of the pendants by solvents was also employed by Freire and Riguera to study conformational communication mechanisms between chiral comonomers along the polymer chain. Thus, they were able to prepare several series of fully chiral copolymers in which the different conformations adopted by the chiral Sergeants in various solvents are transmitted to the chiral Soldiers through Sergeants and Soldiers112,115 or Chiral Coalition effects.113
Most of the solvent response behavior of dynamic covalent polymers found in the literature is based on a teleinduction phenomenon, where the chiral information flows through the different spatial orientation of substituents at the chiral centre towards the polyene backbone. Lately, Freire's group reported two different chiral harvesting mechanisms to induce a helical sense in the polymer (Fig. 3),102,103,396,397 where the conformational information in the chiral pendant placed at a remote position is transferred to an achiral spacer and then harvested by the polyene backbone. Another helix induction mechanism that can be activated/deactivated by solvents is the Chiral Overpass effect105,106 (Fig. 4a), where different chiral residues of a multichiral pendant group can control the helical sense of the helical polymer depending on the conformation adopted by the pendant.
2.8 External stimuli: chiral molecules
Chiral molecules have been widely used as a stimulus to induce the transfer of asymmetry from the molecular to the macromolecular level. Different strategies have been employed, from simple chiral solvation to the development of sophisticated receptors for specific targets. In this section we will show different examples on the topic. | ||
| Fig. 83 (a) Structures of polymers poly-6 and poly-35. Schematic illustration of asymmetry induction and memory effect after (b) replacing the chiral molecules by achiral ones (first generation, dynamic memory) and (c) removal of the chiral molecules (second generation, static memory). Reproduced from ref. 422 with permission from the Chemical Society of Japan, copyright 2021. | ||
This protocol has been successfully applied by Yashima's group working not only with PPAs with the aforementioned receptors but also with other types of polymers such as poly(isocyanide)s (PICs),416,417 poly(phospazanes)418,419 or PDPAs420 among others, giving rise to different types of memory effects. Similar effects occur in double-helical polymers stabilized by carboxylate bridges through the intercalation of chiral amines.421 A detailed explanation of each receptor and the memory effects related to the nature of the polymer backbone can be found in specific reviews.422
Yashima also reported efficient asymmetry transfer during the formation of inclusion complexes in the helical cavity of PMMAs. Syndiotactic PMA (st-PPMA, poly-167) is known to encapsulate fullerenes within its helical cavity.423 Therefore, they prepared a fullerene functionalized with a P or an M helical 310 peptide (168) (Fig. 84a). Upon encapsulation of the corresponding fullerene, poly-167 folds into a preferred helical conformation, as demonstrated by VCD. Similarly, they found that encapsulation of fullerene bearing P or M polylactic acid also resulted in the folding of poly-167 into a preferred handedness (Fig. 84b).424
 | ||
| Fig. 84 (a) Schematic illustration of the asymmetry induction of PMMA poly-167 upon encapsulation of fullerenes derivatives functionalized with P or M 310-helical peptide (168) or with (b) P or M polylactic acid. Reproduced from ref. 424 with permission from the American Chemical Society, copyright 2020. | ||
Apart from directed supramolecular interactions between a designed receptor and a target chiral molecule, in the literature we can find a large number of examples of efficient asymmetry transfer from solvent to solute (i.e., helical macromolecule). Therefore, the use of chiral solvents as a source of chirality has been applied for the helical induction in different macromolecules. For instance, Kwak reported the asymmetry induction of a series of PDPAs containing trialkylsilyl moieties as pendant groups (poly-169) by thermal annealing in (R)- or (S)-limonene (Fig. 85a). During the annealing process, the macromolecule concomitantly adopts a preferred helical sense that depends on the absolute configuration of the solvent, as demonstrated by ECD and CPL spectroscopies (Fig. 85b).425
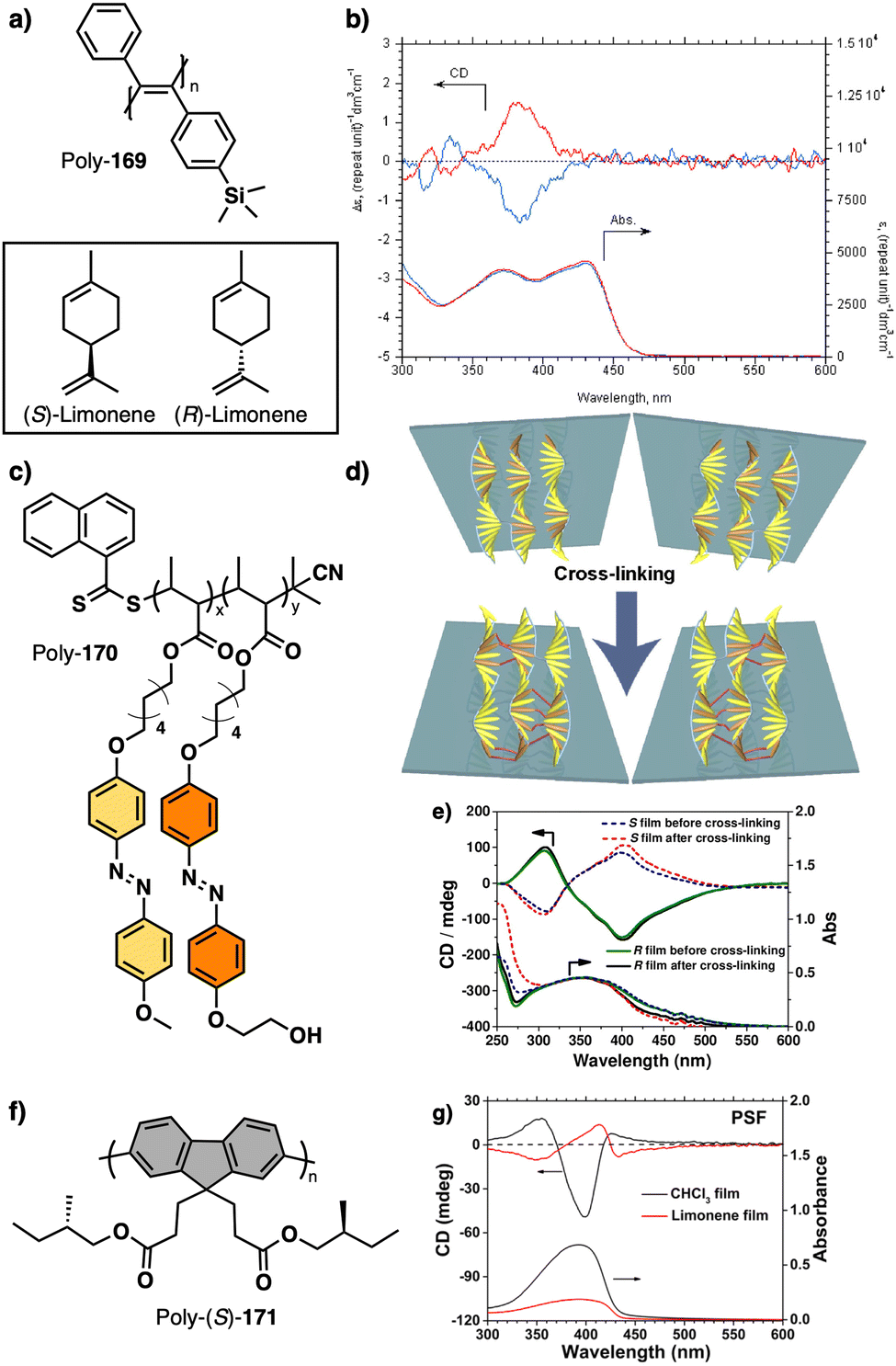 | ||
| Fig. 85 (a) Structure of poly-169 and (b) ECD and UV spectra in (S)- and (R)-limonene (red and blue respectively). (c) Structure of poly-170, (d) Schematic illustration of the solvent drive symmetry breaking and further crosslinking upon self-assembly. (e) CD and UV spectra of chiral poly-170 films before and after cross-linking. Reproduced from ref. 425 with permission from the American Chemical Society, copyright 2012. (f) Structure of poly-(S)-171. (g) CD an UV-Vis spectra of poly-(S)-171 showing the helix inversion of the films from chloroform solution and from assemblies in limonene. Reproduced from ref. 428 with permission from John Wiley and Sons, copyright 2017. | ||
In another example, Zhang employed the chirality of limonene as an efficient asymmetry inductor in achiral PMMA-azo (poly-170) (Fig. 85c and d)426 or crosslinked films of achiral poly(fluorene) derivatives427 with chiral storage and self-recovering properties. Thus, the chiroptical activity of poly-170 after induction with limonene can be easily analyzed by CD spectroscopy due to the presence of a strong bisignate band in the azo region (Fig. 85e).426
Interestingly, in the case of chiral polyfluorene derivatives [poly-(S)-171, Fig. 85f], Zhang induced opposite handedness in films generated from CHCl3 or limonene solutions, because the chirality came from a combination of the polymer side chains and the solvents (Fig. 85g).428 Following this protocol, this group has also prepared efficient multicolor CPL sources.429
Suginome reported different examples of PQXs endowed with solubilizing achiral lateral chains that adopt a preferred-handed helical structure upon dissolving in chiral solvents, such as poly-172 (Fig. 86a) that adopts a P helix in the presence of (R)-limonene (Fig. 86b). This induction of asymmetry later occurred in asymmetric catalysts for Suzuki, asymmetric silaborative C–C cleavage and hydrosilylation reactions (Fig. 86c).430
 | ||
| Fig. 86 (a) Structure of poly-172. (b) CD spectra of poly-172 upon helical induction with (R)-limonene. (c) Enantioselective reaction catalysed by poly-172 and induced by (R)-limonene. Reproduced from ref. 430 with permission from the American Chemical Society, copyright 2019. | ||
Nitschke and coworkers reported chiral induction, assisted by chiral solvents, in double-helical metallopolymers 173 prepared via subcomponent self-assembly (Fig. 87a). During these studies, it was found that the metallopolymer/chiral solvent interaction (i.e., (S)-ethyl lactate) induces the adoption of P double helices with a degree of asymmetry similar to the use of a chiral initiator (Fig. 87b). Remarkably, asymmetry induction works only during the self-assembly process, since the addition of the chiral solvent once the polymer was generated did not trigger any screw sense excess, as demonstrated by CD spectroscopy (Fig. 87b).431
 | ||
| Fig. 87 (a) Subcomponent self-assembly polymerization of double helical metallopolymer 173 in the presence of a chiral solvent. (b) CD spectra showing the formation of enantiomeric helical structures as function of the enantiomeric form of the chiral solvent. Reproduced from ref. 431 with permission from the American Chemical Society, copyright 2018. | ||
 | ||
| Fig. 88 (a) Structure of BTA-(S)-174 and CD spectra showing the impact of chiral solvent on the screw sense excess of the supramolecular polymer. Reproduced from ref. 121 with permission from John Wiley and Sons, copyright 1997. (b) Structures of TTAs 175 and 176. Structures of TTAs (S)-177 and (R)-177 and CD spectra showing their temperature dependent stereomutation in (S)-CldMeOct. Reproduced from ref. 433 with permission from Springer Nature, copyright 2021. (c) Structures of the OPVs 178–180 and CD spectra showing the adoption of opposite handed helices depending on absolute configuration of the chiral solvent. (d) Schematic illustration of the solvent drive symmetry breaking during self-assembly. Reproduced from ref. 435 with permission from the Royal Society of Chemistry, copyright 2011. | ||
This group also reported the impact of chiral solvents on the self-assembly of TTAs containing chiral side chains [(S)-177 and (R)-177]. They discovered that depending on the temperature, the degree of solvation of the supramolecular polymer changes and, consequently, the point chirality of the TTAS side chains or the chirality of the solvents govern the screw sense of the supramolecular polymer. Therefore, at low temperatures, when the polymer is more solvated, chiral solvents govern the helicity of the systems, while at high temperatures, when the polymer is less solvated, point chirality is what controls the handedness of the polymers (Fig. 88b).433 Moving from discotic to linear molecules, the same group reported the self-assembly of three different achiral oligo(p-phenylenevinylene)s (OPV)s with diaminotriazine (A-OPVTs) (178) or ureidotriazine (A-OPVUTs) (179, 180) units devoid of chiral side chains (Fig. 88c). As expected, aggregation in non-polar solvents gave rise to a racemic mixture of helical aggregates, while the inclusion of a small percentage of a chiral alcohol [i.e., (R)- or (S)-citronellol] produced a chiral bias towards M- or P-helical stacks, respectively, as inferred by ECD spectroscopy (Fig. 88d). Intriguingly, they found that chiral alkanes were not able to efficiently transfer asymmetry to the aggregate, thus indicating the presence of precise supramolecular forces between solvent and solute (e.g., hydrogen bond between the alcohol and the triazine unit) as a key factor in biasing the handedness of the aggregates.435 Bouteiller and coworkers reported on the asymmetry induction in the formation of supramolecular nanotubes composed by benzene bisureas. In this system, self-assembly of the achiral derivatives in the presence of (R)- or (S)-limonene provided 33% of the maximum net helicity achieved for analogous building blocks containing chiral side chains.436
The chiral solvent-mediated asymmetry induction found in supramolecular polymers in the solution state also operates at the solid–liquid interface. Thus, De Feyter reported the multicomponent assembly of coronene, isophthalic acid and dehydrobenzo[12]annulene at the solid–liquid interface, where the chirality of the system depended solely on the chiral solvent used.437 Another strategy to study the transfer of asymmetry from a chiral molecule to supramolecular polymers is based on the inclusion of a receptor at the periphery of the assembling molecule. Following this idea, George and coworkers reported on the formation of supramolecular polymers based on naphthalene diimide that carried zinc-complexes as substituents (NDPA, 181). Opposite handedness is induced in the supramolecular polymer that binds adenophosphates to these receptors as a function of the number on phosphates anchored to the chiral adenosine (i.e., ATP, P helix; AMP and ADP, M helix). Additionally, the enzymatic transformation from ATP to ADP mediated by alkaline phosphatase produces helix inversion (Fig. 89).438,439
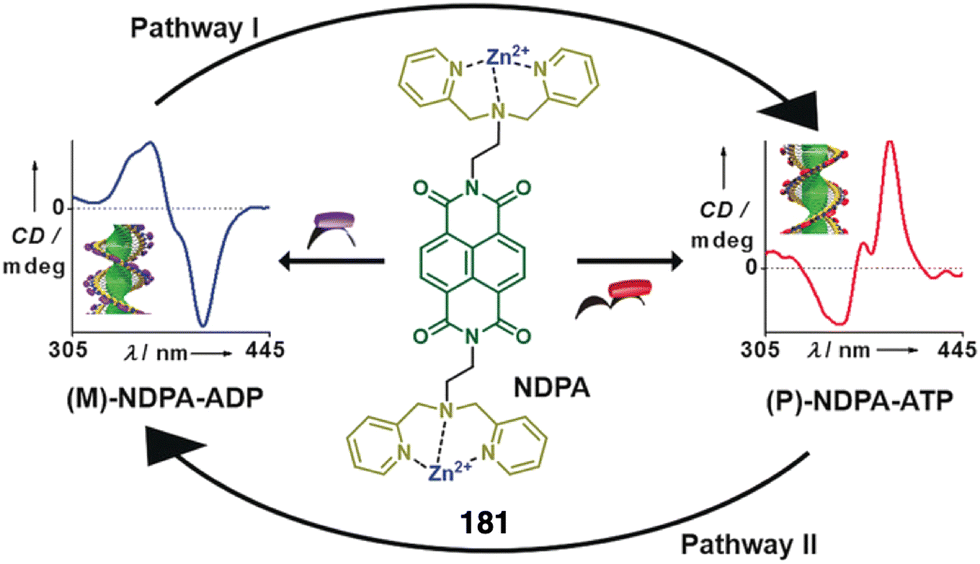 | ||
| Fig. 89 Chemical structure of naphthalene diimide 181, bearing as substituents zinc-complexes, that promotes P or M helical coassemblies by interaction with ATP or ADP respectively. Reproduced from ref. 438 with permission from John Wiley and Sons, copyright 2017. | ||
This asymmetry induction mechanism was exploited by Clayden and coworkers to develop stimuli-responsive foldamers based on 310-helical Aib peptides (Fig. 1)358 and oligoureas 66 (Fig. 18).177 Different receptors such as amino groups, ureas, boronic esters, pyridines, or metal complexes that upon interaction with the corresponding guests induced changes in the helical scaffold were introduced into the N or C terminus of the foldamer. These interactions produced variations in the spectroscopic data from NMR, CD or photoluminescence among other techniques employed.60,70,72–74,78–81,442–444 As an example, the authors reported the preparation of an Aib foldamer carrying a 2-(aminomethyl)phenylboronic acid derivative (182)—known to interact with geminal diols—placed at the N-terminal position (Fig. 90a). Coordination of the diol 183 with the boron atom results in the adoption of a preferred screw sense in the Aib foldamer. The helical bias is monitored by the presence of evident chemical shifts and diastereotopic NMR signals in 11B and 1H experiments, respectively (Fig. 90b).445 A very interesting asymmetry induction found in foldamers is based on the encapsulation of achiral/chiral guests within the helical cavity.13,445–449 Huc and Ferrand reported different oligoamide-based foldamers (e.g., 184, Fig. 91a) and metallofoldamers that encapsulate chiral guests such as tartaric acid450 or different saccharides.13 Encapsulation of the corresponding guest induced the adoption of a preferred-handed helical structures (Fig. 91b and c). Remarkably, modifications in certain regions of the foldamer with light291 (Fig. 59) or addition of metals induced additional helix inversion processes (Fig. 29b and c).203,204 Li studied other examples of foldamers that encapsulate saccharides, and designed oligohydrazide-type foldamers capable of encapsulating glucose through hydrogen bonds. Changes in foldamer length allowed the binding of different saccharides to be modified, while the inclusion of chiral handles, such as proline residues, allowed the binding events to be monitored using CD spectroscopy.451–453
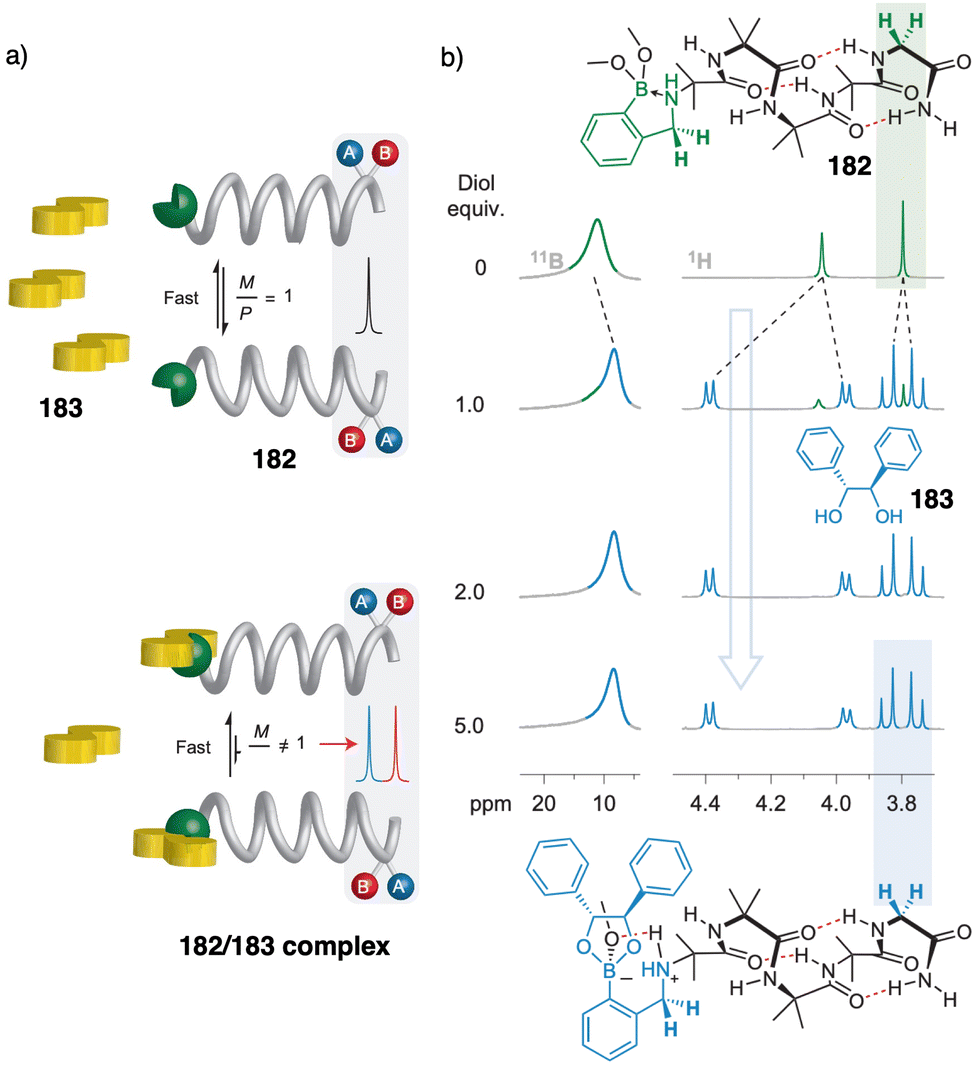 | ||
| Fig. 90 (a) Schematic illustration of the helical bias upon binding of a substrate in the receptor of the Aib foldamer 182 and the corresponding NMR spectroscopic output upon binding event. (b) Structure of the Aib based foldamer 182 containing the boronate receptor for diols (top) and 11B and 1H NMR experiments showing the diastereotopic signals of 182 upon adoption of preferred screw-sense excess when interacts with the chiral diol 183. Reproduced from ref. 445 with permission from Springer Nature, copyright 2013. | ||
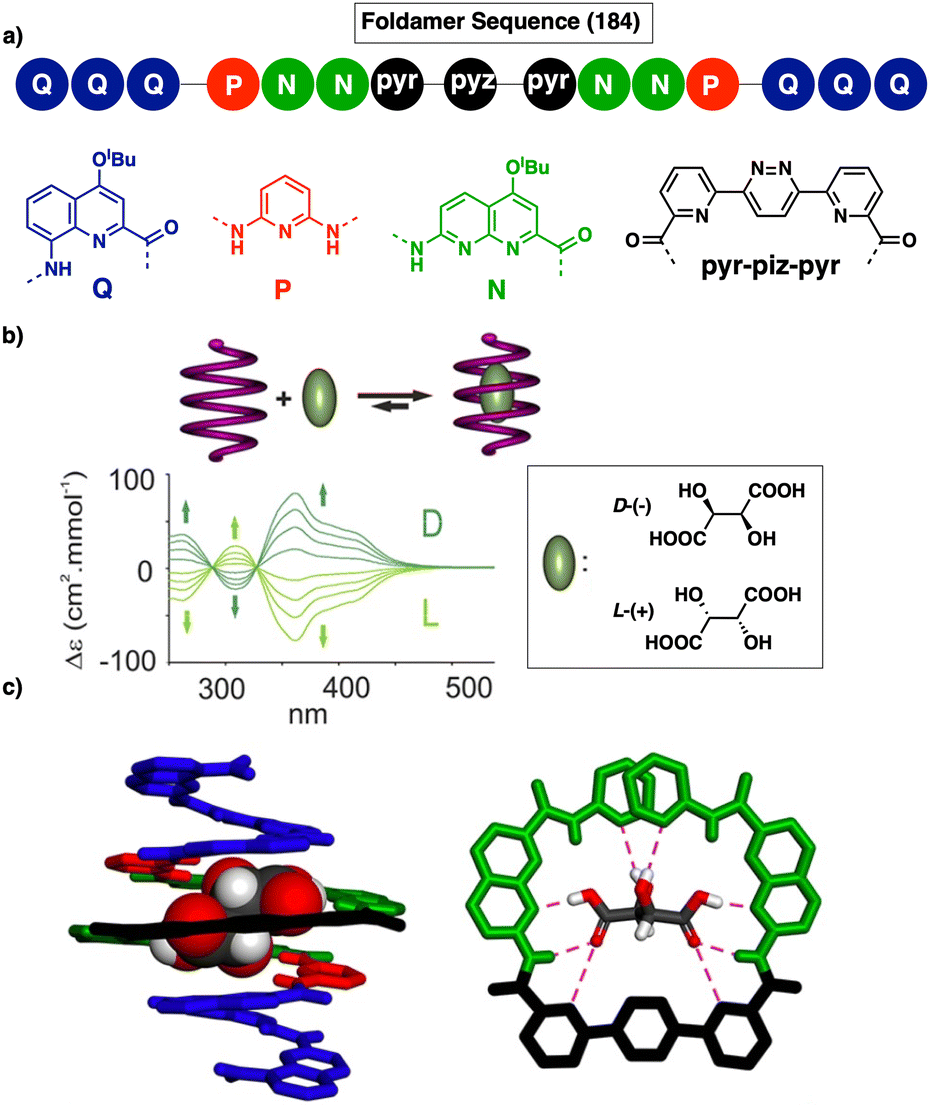 | ||
| Fig. 91 (a) Structure of foldamer 184. (b) Schematic illustration of the host–guest formation monitored by CD and structures of the guests. (c) Host–guest complex formed by foldamer 184 and D-tartaric acid. Reproduced from ref. 450 with permission from the American Chemical Society, copyright 2010. | ||
meta-Phenylene ethynylenes (mPEs) represent another interesting family of foldamers that adopt helical structures induced by solvophobic effects.454–458 For instance, Moore reported that mPE 185 (Fig. 92a), with a triethyleneglycol side chain, folds into a helical structure with no screw sense preference in MeCN/H2O mixtures. Upon addition of chiral terpenes, such as pinene, a CD signal arises for both compounds indicating the adoption of preferred handed helices. The asymmetry induction comes from the formation of inclusion complexes, where pinene is encapsulated within the helical cavity of the mPE foldamer. Moore also prepared longer derivatives of up to 28 mPE units and also mPEs with different core linkers, which were able to form inclusion complexes with chiral aromatic pillars such as 186, 187 (Fig. 92b–d).454–458
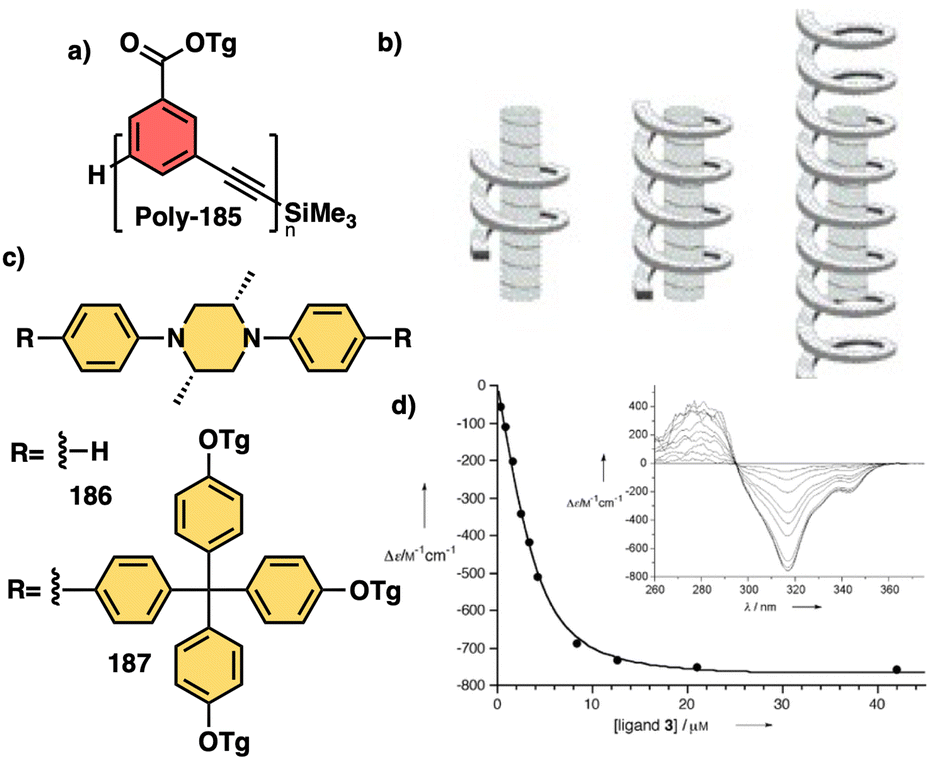 | ||
| Fig. 92 (a) Structure of foldamer 185. (b) Schematic illustration of the host–guest formation with (c) chiral pillars 186 and 187 (reproduced from ref. 457 with permission from the American Chemical Society, copyright 2001). ECD spectra upon the addition of a chiral molecule to foldamer 185. Reproduced from ref. 458 with permission from John Wiley and Sons, copyright 2002. | ||
Inoue developed an m-pyridine ethynylene foldamer that, upon binding to β-glucosides, folds into a helical structure with preferred handedness in MeOH/H2O mixtures. Protonation of the pyridine units also resulted in direct folding into helical structures. Curiously, in this state, the chirality of the foldamer (i.e., P and M helical conformations) depends on the mutarotation of glucose. Additional modifications to the sequence of the foldamer allowed the preparation of systems that self-assemble into double-helical structures, which can be disassembled upon binding the corresponding guests.459,460
2.9 External stimulus: pH
The influence of pH on stimuli-responsive helical polymers has attracted significant attention from scientists working in this field due to the possibility of controlling the helical sense through the presence of positive or negative charges in the chiral material.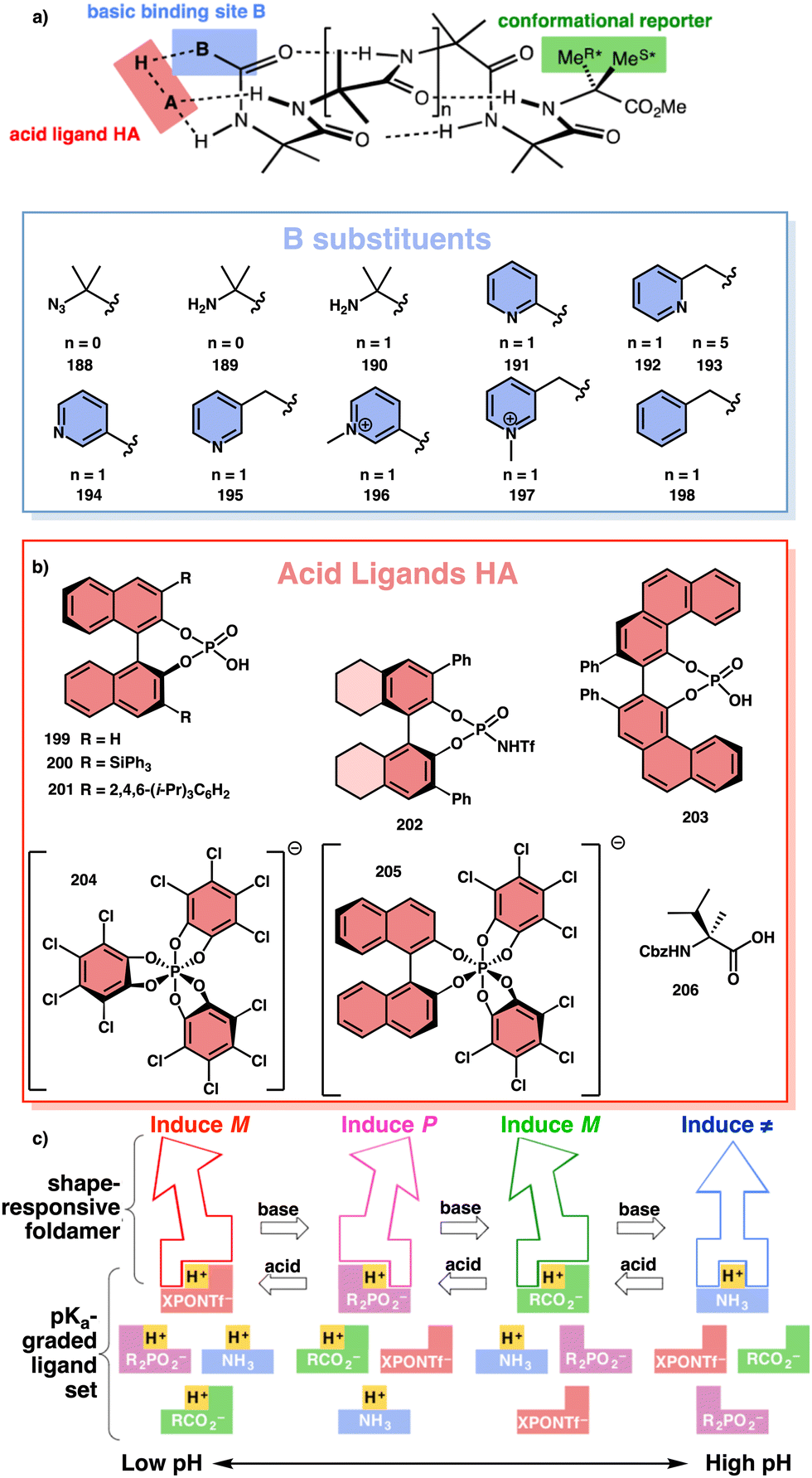 | ||
| Fig. 93 (a) Achiral foldamers (188–198) and their binding sites. (b) Chiral acids (199–203, 206) and anion ligands (204, 205). (c) Schematic representation of the helical sense of the foldamer at different pH after the coordination with the ligands. Reproduced from ref. 444 with permission from the American Chemical Society, copyright 2015. | ||
In a project led by Huc, a helix-rod was designed with host–guest functionality.461 This innovative design involved the folding, binding, and random sliding motions of helical foldamers (specifically, aromatic oligoamides such as 207) along rod-like guests (Fig. 94a). To facilitate this process, two distinct binding stations were created along the rod-like guest 208: one with a heptyl segment and the other with a diethylamine segment.
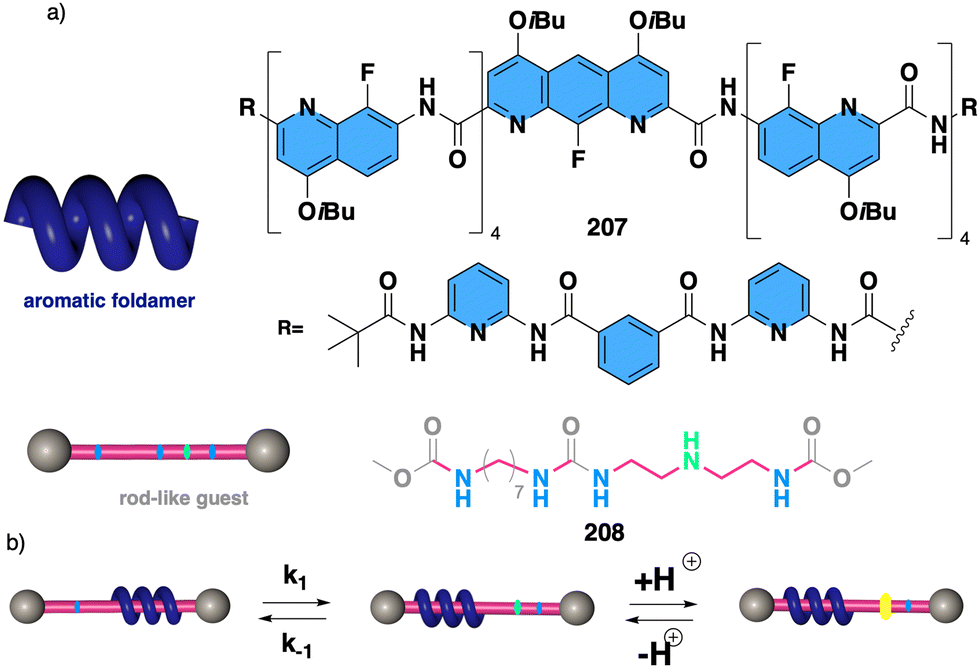 | ||
| Fig. 94 (a) Molecular structure of aromatic oligoamide 207 and the rod like guest 208. (b) Scheme of helix sliding along a nondegenerate guest possessing a station that can be blocked or unblocked upon protonation or deprotonation, respectively. The green dot symbolizes the amine function, which can be included in the helical cavity. The yellow dot is the corresponding ammonium, which is not included in the helical cavity. | ||
The host–guest complex demonstrated negligible bias between the two binding sites. However, during the experimental work, an interesting observation was made. By adding an acid, the researchers were able to selectively trap the host–guest complex at a single binding site. Conversely, the addition of a base, which served to deprotonate, successfully reactivated the dynamic motion of the system (Fig. 94b).
Similarly, in another system investigated by Bhosale and coworkers using NDI-L-Glu monomers (209), chiral supramolecular helices were formed (Fig. 95a).463 In this case, the pH response was as observed in the previous example. At acidic pH, the monomers interacted through hydrogen bonding between the amides and carboxylic acids, while at high pH the carboxylic acids were deprotonated, and electrostatic repulsion stabilized the opposite helical sense (Fig. 95b). Additionally, in this system, it was found that the P/M helical sense of the supramolecular aggregate could be switched and interconverted after several pH cycles (Fig. 95c).
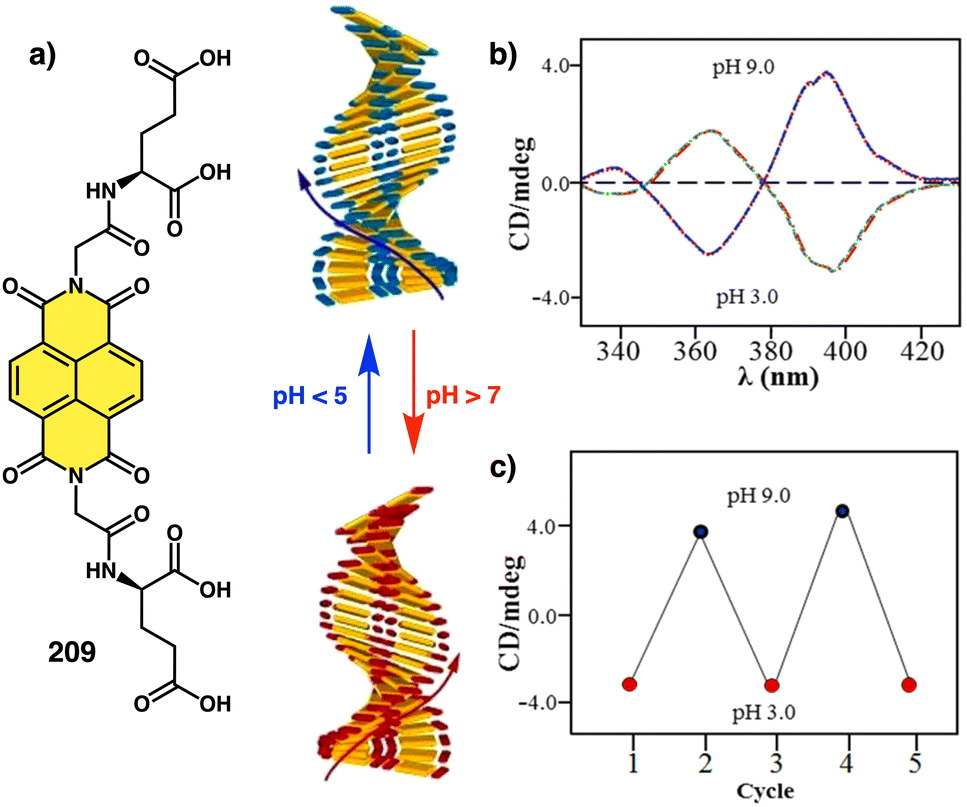 | ||
| Fig. 95 (a) Molecular structure of 209 and graphical illustration of P or M helical senses of the supramolecular aggregate tuneable by the action of pH. (b) CD spectra of 209 at pH 3 and 9. (c) pH chiroptical switch of a supramolecular aggregate of 209 after different cycles at pH = 3 and pH = 9. Reproduced from ref. 463 with permission from Springer Nature, copyright 2018. | ||
The pH-responsive self-assembly of a histidine-functionalized perylene diimide (HPH, 210) was studied by Govindaraju (Fig. 96a). This molecule exhibited a pH reversible chiroptical supramolecular aggregate switch with tunable 1D nanostructures between thin or thick fibrils and belts.464 The pH-induced self-assembly of 210 was confirmed by CD measurements (Fig. 96b and c).
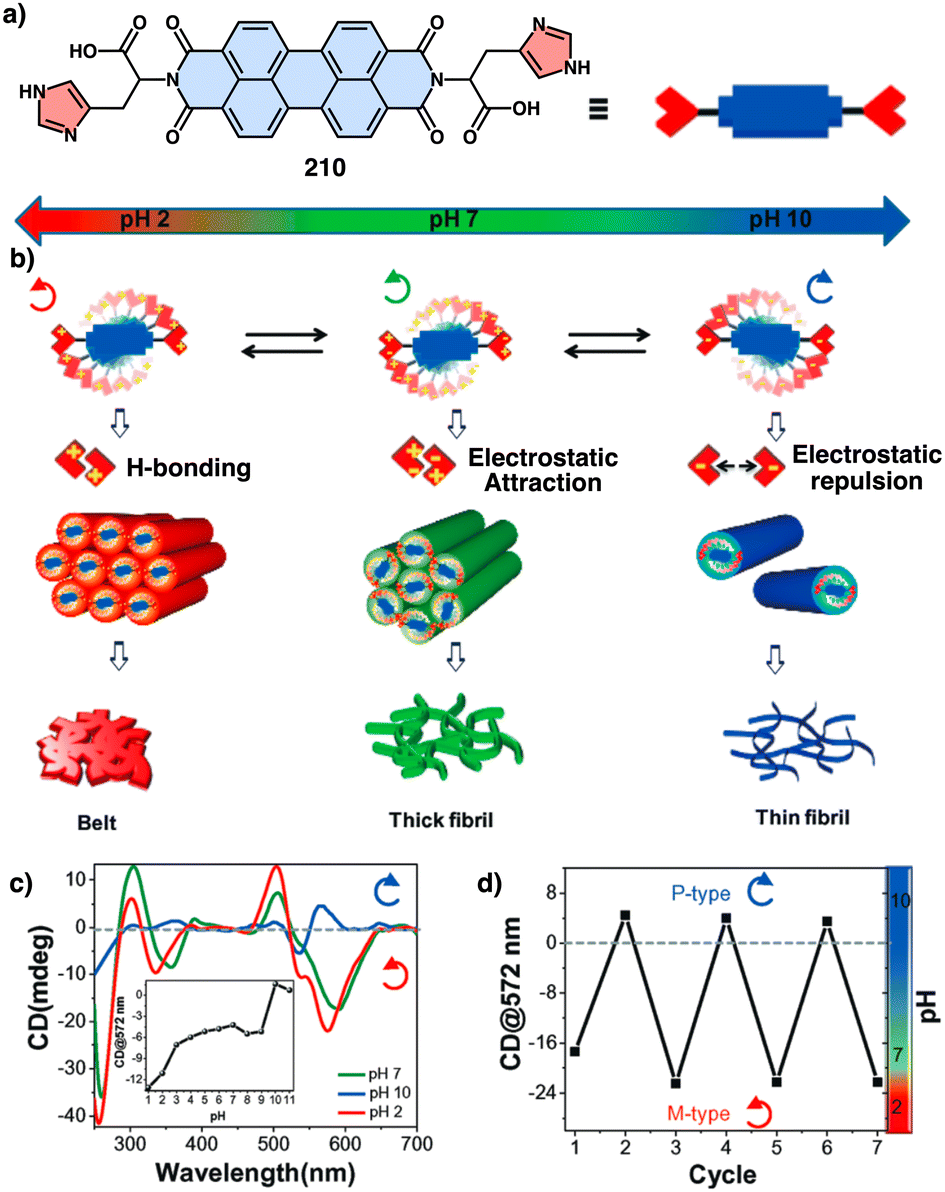 | ||
| Fig. 96 (a) Molecular structure and schematic representation of 210. (b) Schematic illustration of the pH-responsive reversible chiroptical aggregate switch and its tuneable 1D morphology induced by protonation–deprotonation. Curly arrows indicate the handedness of the molecular assembly of 210 (c) CD spectra of 210 at different pH. Inset: Plot of CD intensity at 572 nm versus pH. (d) Reversible chiroptical switching at 572 nm as a function of pH cycles. Curly arrows indicate the handedness of the molecular assembly of 210. Reproduced from ref. 464 with permission from the Royal Society of Chemistry, copyright 2016. | ||
Besenius also explored pH-responsive supramolecular aggregates using oppositely charged phenylalanine-based dendritic peptide amphiphiles (211 and 212, Fig. 97a), which enabled selective homopolymerization by pH modulation and charge switching into comonomers.465 The strategy involves attractive Coulomb interactions in oppositely charged dendritic peptide amphiphiles that reinforce weak non-covalent interactions, resulting in the formation of alternating supramolecular copolymers. By altering the pH and switching the peptidic monomers, attractive electrostatic interactions are disrupted, leading to selective polymerization and the formation of homopolymers 211 of the basic comonomer at high pH and homopolymers 212 of the acidic comonomer at low pH. The transition between copolymers and homopolymers is reversible (Fig. 97b).
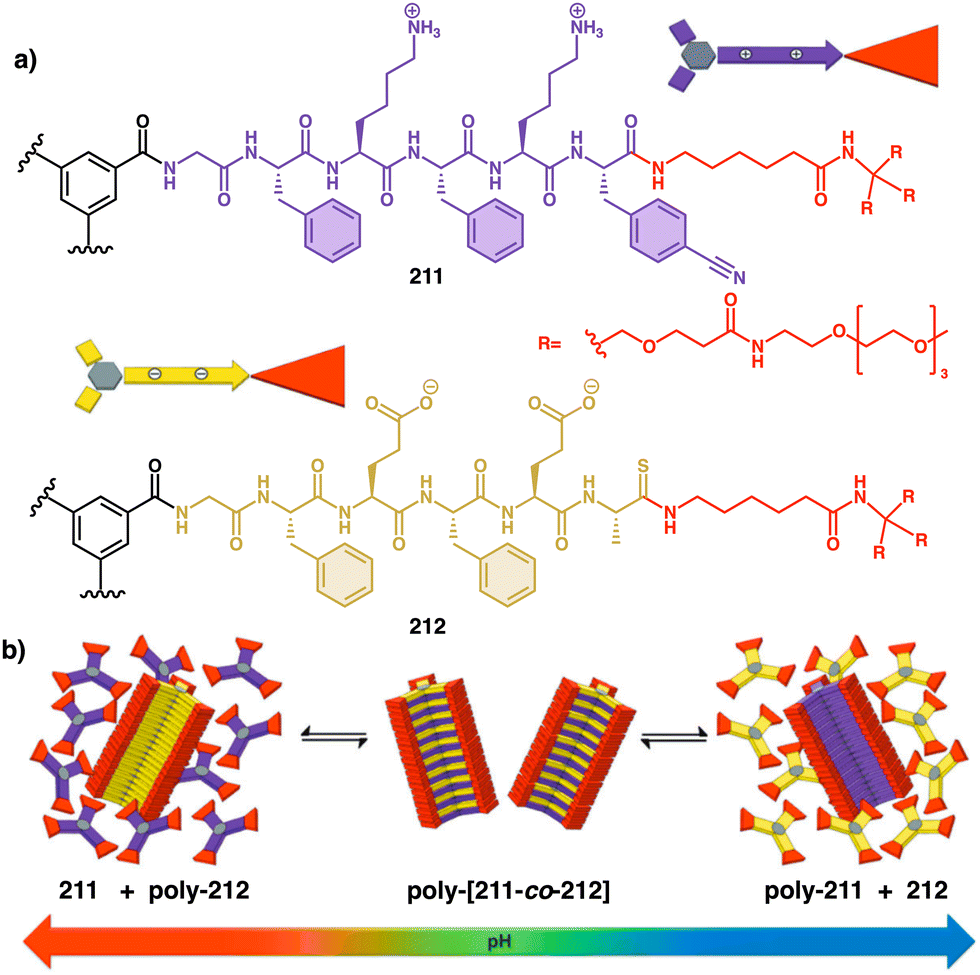 | ||
| Fig. 97 (a) Chemical structures of the C3-symmetric dendritic peptide comonomers 211 and 212 and (b) their pH-regulated supramolecular polymerization into homopolymers of 211 and 212, at high and low pH respectively, and 211–212 copolymers at neutral pH. Reproduced from ref. 465 with permission from John Wiley and Sons, copyright 2015. | ||
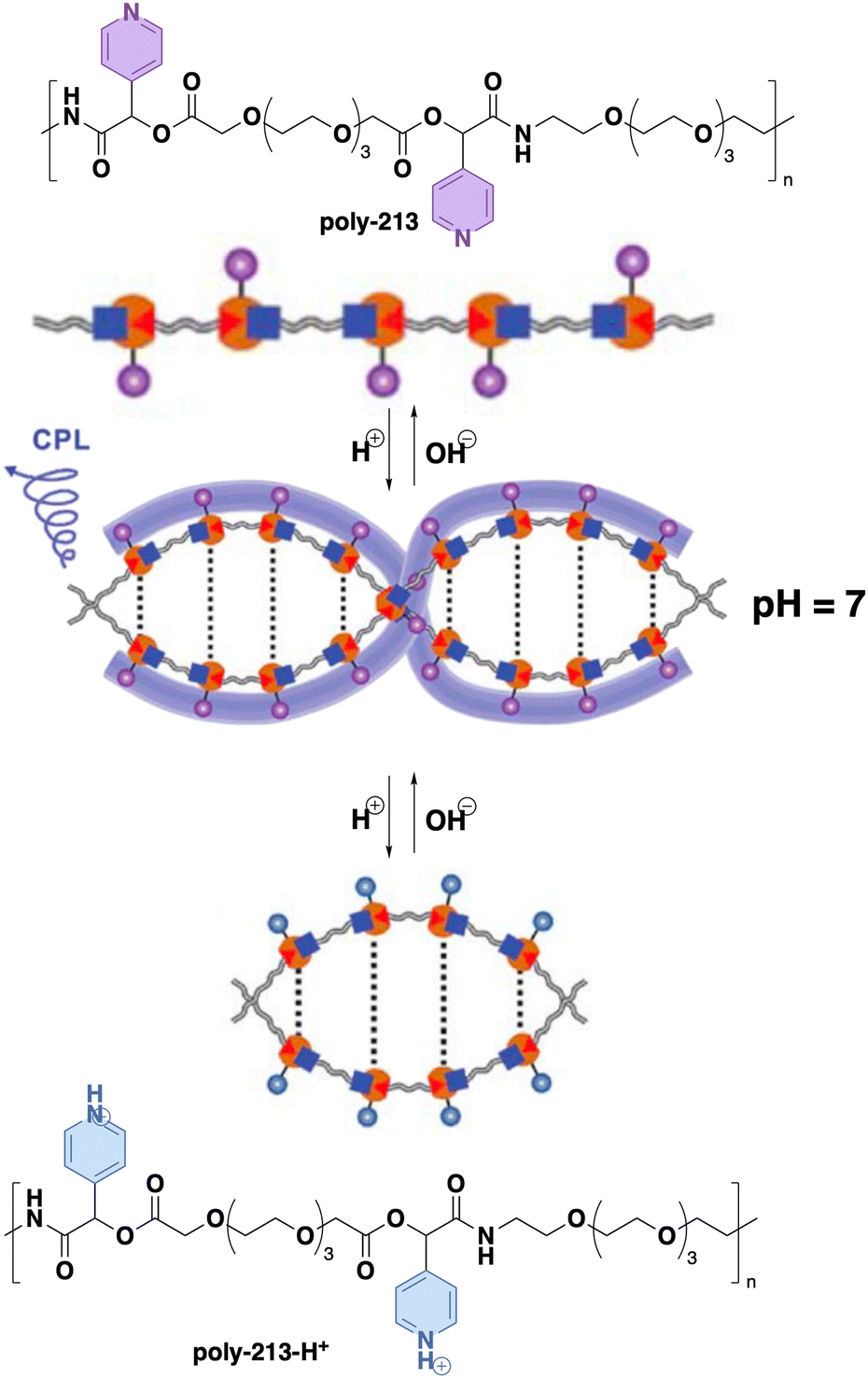 | ||
| Fig. 98 Schematic illustration of double-helical AIE-active polymers 213 with multiple responsiveness via the PMPIA. Reproduced from ref. 466 with permission from the Royal Society of Chemistry, copyright 2020. | ||
Freire successfully synthesized water-soluble helical amine-PPAs with pH-responsive behaviour, such as poly-214.467 Importantly, the location of the amino group within the polymer determines the mechanism and direction of the helical response. At acidic pH, the preferred helical conformation is stabilized by a hydrogen bond between the ammonium and carbonyl groups, while at higher pH values, when the amino group is deprotonated, an antiperiplanar orientation is preferred between both groups, placing the substituents of the chiral centre in different locations and therefore, promoting opposite helical senses. Helix inversion could be observed in MeOH, but not in water, due to solubility problems of the neutral form of poly-214 (Fig. 99).
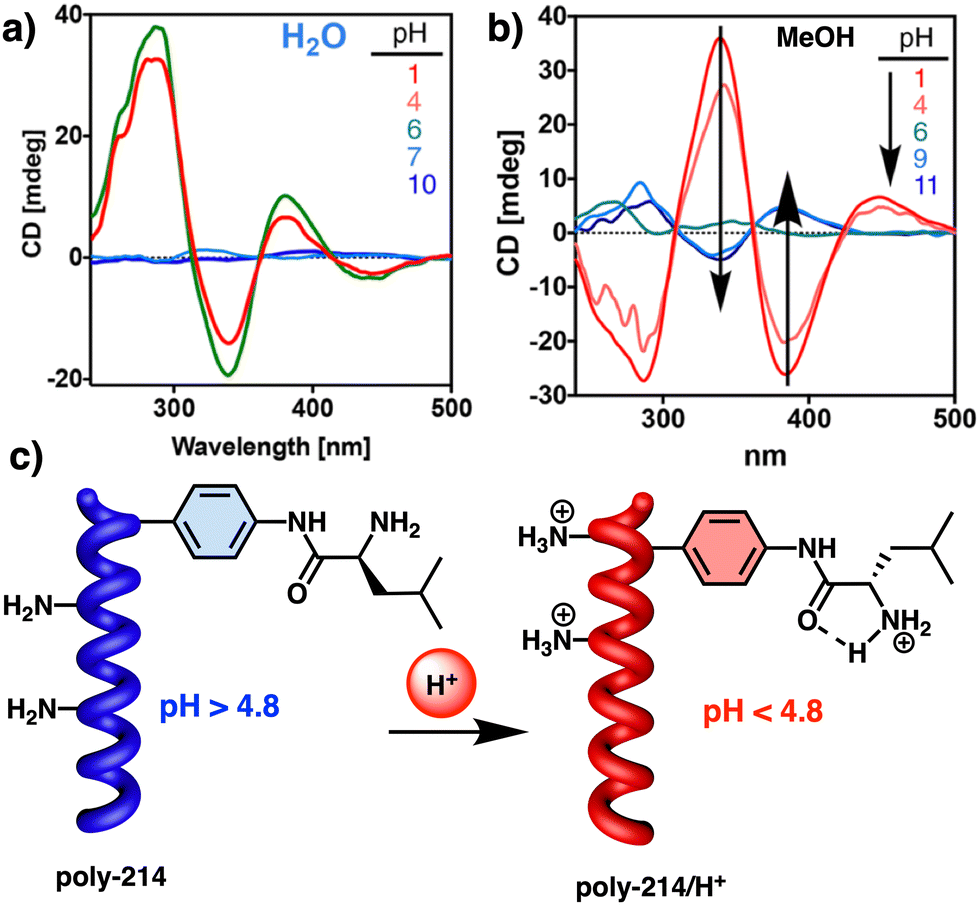 | ||
| Fig. 99 CD spectra for poly-214 at different pH in (a) water and (b) MeOH. (c) Conformational composition of amino-PPAs in basic and acidic media. [amino-PPA] = 0.5 mg mL−1. Reproduced from ref. 467 with permission from the American Chemical Society, copyright 2018. | ||
Masuda reported poly(phenyleneethynylene-pyromellitic diimide)s,468 which exhibited stable helical conformations that could be disrupted by the addition of NaOH and subsequently recovered by the addition of HCl. Zheng and Bao studied the self-assembly of 1D gadolinium(III) phosphonate coordination polymers (CPs) using different metal sources and ligands. They achieved the formation of helical superstructures with controllable twist directions by manipulating interchain interactions and pH. The specific counter-anions present in the system and the pH determined the chemical composition and morphology of the CPs.469 Other examples dealing with a helix to random coil transition triggered by pH are found in the literature for unnatural polypeptides470 and graft copolymers,471 however, the main goal of this review is to describe systems that form helical structures whose scaffolds can be altered by the presence of external stimuli.
3 Conclusions and outlook
Dynamic helical polymers (covalent and supramolecular) and foldamers share the helix as structural motif, which can be altered (sense and elongation) by the presence of external stimuli. In this review, we highlight the most important helix induction and conformational communication mechanisms triggered by the presence of different external stimuli, which provoke conformational or structural changes in the monomer repeating units or building blocks and which are shared by these different materials.Thus, although helical polymers (covalent and supramolecular) and foldamers belong to different research areas, the knowledge and advances achieved in the different materials can also be applied to others. Therefore, through a rational design of the components of helical polymers, it is possible to create dynamic helices with stimuli-responsive properties that can be used in different applications such as sensing, molecular recognition, asymmetric synthesis, signalling, information storage, and son on.
In summary, this review shows the main stimuli-responsive properties of dynamic chiral helical polymers, and the mechanisms involved in the structural changes induced in macromolecular and supramolecular helices. However, although the helices are different and each one has its own intrinsic properties, the stimuli-response capacity of the building blocks or monomer repeating units, and the mechanisms involved in the structural changes of the helices are similar, making it possible to extract information from a specific system and employ it in another different material. Thus, by combining information from different systems, it is possible to search for novel helical polymers with new or improved functionalities.
Author contributions
All authors have contributed to the preparation of the manuscript and agreed with the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from AEI (PID2022-136848NB-I00), Xunta de Galicia (ED431C 2022/21), Centro Singular de Investigación de Galicia acreditación 2019–2022, ED431G 2019/03, and the European Regional Development Fund (ERDF) and is gratefully acknowledged. R. R. and M. F.-M. thanks MICINN for a Juan de la Cierva Incorporación and FPI contracts. M. L. thanks Xunta de Galicia for a predoctoral contract.Notes and references
- D. T. J. Morris and J. Clayden, Chem. Soc. Rev., 2023, 52, 2480–2496 RSC.
- T. Seedorf, A. Kirschning and D. Solga, Chem. – Eur. J., 2021, 27, 7321–7339 CrossRef PubMed.
- R. Aksakal, C. Mertens, M. Soete, N. Badi and F. Du Prez, Adv. Sci., 2021, 8, 20004038 Search PubMed.
- H.-W. Schmidt and F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 8766–8775 CrossRef.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef PubMed.
- A. Das and S. Ghosh, Chem. Commun., 2016, 52, 6860–6872 RSC.
- M. Shigeno, Y. Kushida and M. Yamaguchi, Chem. Commun., 2016, 52, 4955–4970 RSC.
- D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef PubMed.
- Z. C. Girvin and S. H. Gellman, J. Am. Chem. Soc., 2020, 142(41), 17211–17223 CrossRef.
- P. Rivera-Fuentes and F. Diederich, Angew. Chem., Int. Ed., 2012, 51, 2818–2828 CrossRef PubMed.
- G. Maayan, Eur. J. Org. Chem., 2009, 5699–5710 CrossRef.
- A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2038–2054 CrossRef PubMed.
- Y. Ferrand and I. Huc, Acc. Chem. Res., 2018, 51, 970–977 CrossRef PubMed.
- I. Huc and S. Hecht, Foldamers: Structure, Properties, and Applications, John Wiley & Sons, Weinheim, 2007 Search PubMed.
- E. A. John, C. J. Massena and O. B. Berryman, Chem. Rev., 2020, 120, 2759–2782 CrossRef PubMed.
- D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4012 CrossRef PubMed.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef.
- O. J. G. M. Goor, S. I. S. Hendrikse, P. Y. W. Dankers and E. W. Meijer, Chem. Soc. Rev., 2017, 46, 6621–6637 RSC.
- M. H. Bakker, C. C. Lee, E. W. Meijer, P. Y. W. Dankers and L. Albertazzi, ACS Nano, 2016, 10, 1845–1852 CrossRef CAS PubMed.
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef.
- S. Biswas, K. Kinbara, N. Oya, N. Ishii, H. Taguchi and T. Aida, J. Am. Chem. Soc., 2009, 131, 7556–7557 CrossRef PubMed.
- O. Dumele, J. Chen, J. V. Passarelli and S. I. Stupp, Adv. Mater., 2020, 1907247 CrossRef.
- Q. Zhang, R. Toyoda, L. Pfeifer and B. L. Feringa, J. Am. Chem. Soc., 2023, 145, 6976–6985 CrossRef PubMed.
- Y. Yamaguchi, Y. Ochi, S. Miyamura, T. Tanaka, S. Kobayashi, T. Wakamiya, Y. Matsubara and Z. Yoshida, J. Am. Chem. Soc., 2006, 128, 4504–4505 CrossRef PubMed.
- A. P. H. J. Schenning and E. W. Meijer, Chem. Commun., 2005, 3245–3258 RSC.
- M. Raynal, F. Portier, P. W. N. M. van Leeuwen and L. Bouteiller, J. Am. Chem. Soc., 2013, 135, 17687–17690 CrossRef.
- C. Kulkarni, E. W. Meijer and A. R. A. Palmans, Acc. Chem. Res., 2017, 50, 1928–1936 CrossRef PubMed.
- T. F. A. de Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754 CrossRef.
- Y. Dorca, J. Matern, G. Fernández and L. Sánchez, Isr. J. Chem., 2019, 59, 869–880 CrossRef CAS.
- A. Sorrenti, J. Leira-Iglesias, A. J. Markvoort, T. F. A. de Greef and T. M. Hermans, Chem. Soc. Rev., 2017, 46, 5476–5490 RSC.
- E. E. Greciano, J. Calbo, E. Ortí and L. Sánchez, Angew. Chem., Int. Ed., 2020, 132, 17670–17677 CrossRef.
- F. Würthner, C. R. S. Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef.
- M. Hifsudheen, R. K. Mishra, B. Vedhanarayanan, V. K. Praveen and A. Ajayaghosh, Angew. Chem., Int. Ed., 2017, 56, 12634–12638 CrossRef.
- Y. Dorca, E. E. Greciano, J. S. Valera, R. Gómez and L. Sánchez, Chem. – Eur. J., 2019, 25, 5848–5864 CrossRef PubMed.
- Y. Yang, Y. Zhang and Z. Wei, Adv. Mater., 2013, 42, 6039–6049 CrossRef PubMed.
- J.-M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 11, 1304–1319 CrossRef.
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS.
- F. García, R. Gómez and L. Sánchez, Chem. Soc. Rev., 2023, 52, 7524–7548 RSC.
- E. Yashima, K. Maeda and Y. Furusho, Acc. Chem. Res., 2008, 41, 1166–1180 CrossRef CAS.
- E. Yashima, K. Maeda, H. Iida and Y. Furusho, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS.
- J. Liu, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2009, 109, 5799–5867 CrossRef CAS PubMed.
- T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013–4038 CrossRef CAS PubMed.
- R. Sakai, E. B. Barasa, N. Sakai, S. I. Sato, T. Satoh and T. Kakuchi, Macromolecules, 2012, 45, 8221–8227 CrossRef.
- E. Yashima and K. Maeda, Macromolecules, 2008, 41, 3–12 CrossRef.
- K. Maeda and E. Yashima, Top. Curr. Chem., 2006, 265, 47–88 CrossRef.
- C. Zhang, Y. Qiu, S. Bo, F. Wang, Y. Wang, L. Liu, Y. Zhou, H. Niu, H. Dong and T. Satoh, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1024–1031 CrossRef.
- J. Shen and Y. Okamoto, Chem. Rev., 2016, 116, 1094–1138 CrossRef.
- C. I. Simionescu and V. Percec, Prog. Polym. Sci., 1982, 8, 133–214 CrossRef.
- M. Milton, R. Deng, A. Mann, C. Wang, D. Tang and M. Weck, Acc. Chem. Res., 2018, 54, 2397–2408 CrossRef.
- J. G. Rudick and V. Percec, Acc. Chem. Res., 2008, 41, 1641–1652 CrossRef CAS PubMed.
- N. Liu, L. Zhou and Z. Q. Wu, Acc. Chem. Res., 2021, 54, 3953–3967 CrossRef CAS PubMed.
- R. P. Megens and G. Roelfes, Chem. – Eur. J., 2011, 17, 8514–8523 CrossRef CAS.
- Y. Nagata, R. Takeda and M. Suginome, ACS Cent. Sci., 2019, 5, 1235–1240 CrossRef CAS PubMed.
- K. Maeda, D. Hirose, M. Nozaki, Y. Shimizu, T. Mori, K. Yamanaka, K. Ogino, T. Nishimura, T. Taniguchi, M. Moro and E. Yashima, Sci. Adv., 2021, 7, eabg5381 CrossRef PubMed.
- B. A. San Jose, S. Matsushita and K. Akagi, J. Am. Chem. Soc., 2012, 134, 19795–19807 CrossRef CAS PubMed.
- S. Wang, D. Hu, X. Guan, S. Cai, G. Shi, Z. Shuai, J. Zhang, Q. Peng and X. Wan, Angew. Chem., Int. Ed., 2021, 60, 21918–21926 CrossRef.
- F. Freire, E. Quiñoá and R. Riguera, Chem. Rev., 2016, 116, 1242–1271 CrossRef.
- J. J. L. M. Cornelissen, A. E. Rowan, R. J. M. Nolte and N. A. J. M. Sommerdijk, Chem. Rev., 2001, 101, 4039–4070 CrossRef PubMed.
- E. Schwartz, M. Koepf, H. J. Kito, R. J. M. Nolte and A. E. Rowan, Polym. Chem., 2011, 2, 33–47 RSC.
- Y. Wang, T. Harada, L. Q. Phuong, Y. Kanemitsu and T. Nakano, Macromolecules, 2018, 51, 6865–6877 CrossRef.
- E. Kervio, A. Hochgesand, U. E. Steiner and C. Richert, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12074–12079 CrossRef PubMed.
- A. Brewer and A. P. Davis, Nat. Chem., 2014, 6, 569–574 CrossRef CAS PubMed.
- D. M. Lovinger, Alcohol Res. Health, 2008, 31, 196–214 Search PubMed.
- B. Liu, C. G. Pappas, J. Ottelé, G. Schaeffer, C. Jurissek, P. F. Pieters, M. Altay, I. Maric, M. C. A. Stuart and S. Otto, J. Am. Chem. Soc., 2020, 142, 4184–4192 CrossRef CAS.
- T. Kosikova, N. I. Hassan, D. B. Cordes, A. M. Z. Slawin and D. Philp, J. Am. Chem. Soc., 2015, 137, 16074–16083 CrossRef CAS PubMed.
- G. Clixby and L. Twyman, Org. Biomol. Chem., 2016, 14, 4170–4184 RSC.
- G. De, Bo, M. A. Y. Gall, S. Kuschel, J. DeWinter, P. Gerbaux and D. A. Leigh, Nat. Nanotechnol., 2018, 13, 381–385 CrossRef.
- R. K. O’Reilly, A. J. Turberfield and T. R. Wilks, Acc. Chem. Res., 2017, 50, 2496–2509 CrossRef.
- F. G. A. Lister, B. A. F. Le Bailly, S. J. Webb and J. Clayden, Nat. Chem., 2017, 9, 420–425 CrossRef.
- M. de Poli, W. Zawodry, O. Quinonero, M. Lorch, S. J. Webb and J. Clayden, Science, 2016, 352, 575–580 CrossRef PubMed.
- J. Zhang, D. Luo, C. Ma and Q. Gan, Nat. Commun., 2021, 12, 2659 CrossRef PubMed.
- S. M. Wales, D. T. J. Morris and J. Clayden, J. Am. Chem. Soc., 2022, 144, 2841–2846 CrossRef.
- D. T. J. Morris, S. M. Wales, D. Tilly, E. H. E. Farrar, M. N. Grayson, J. W. Ward and J. Clayden, Chem, 2021, 7, 2460–2472 Search PubMed.
- J. E. Jones, V. Diemer, C. Adam, J. Raftery, R. E. Ruscoe, J. Sengel, M. I. Wallace, A. Bader, S. L. Cockroft, J. Clayden and S. J. Webb, J. Am. Chem. Soc., 2016, 132, 688–695 CrossRef.
- Y. Zhong, T. A. Sobiech, B. Kauffmann, B. Song, X. Li, Y. Ferrand, I. Huc and B. Gong, Chem. Sci., 2023, 14, 4759–4768 RSC.
- J. Wang, B. Wicher, A. Méndez-Ardoy, X. Li, G. Pecastaings, T. Buffeteau, D. M. Bassani, V. Maurizot and I. Huc, Angew. Chem., Int. Ed., 2021, 60, 18461–18466 CrossRef.
- M. De Zotti and J. Clayden, Org. Lett., 2019, 21, 2209–2212 CrossRef PubMed.
- D. P. Tilly, J. P. Heeb, S. J. Webb and J. Clayden, Nat. Commun., 2023, 14, 2647–2653 CrossRef PubMed.
- F. G. A. Lister, N. Eccles, S. J. Pike, R. A. Brown, G. F. S. Whitehead, J. Raftery, S. J. Webb and J. Clayden, Chem. Sci., 2018, 9, 6860–6870 RSC.
- J. Solá, S. P. Fletcher, A. Castellanos and J. Clayden, Angew. Chem., Int. Ed., 2010, 49, 6836–6839 CrossRef PubMed.
- J. Solá, M. Helliwell and J. Clayden, J. Am. Chem. Soc., 2010, 132, 4548–4549 CrossRef.
- Y. Inai, N. Ousaka and T. Okabe, J. Am. Chem. Soc., 2003, 125, 8151–8162 CrossRef PubMed.
- Y. Inai, K. Tagawa, A. Takasu, T. Hirabayashi, T. Oshikawa and M. Yamashita, J. Am. Chem. Soc., 2000, 122, 11731–11732 CrossRef.
- Y. Inai, Y. Ishida, K. Tagawa, A. Takasu and T. Hirabayashi, J. Am. Chem. Soc., 2002, 124(11), 2466–2473 CrossRef.
- Y. Inai and H. Komori, Biomacromolecules, 2004, 5, 1231–1240 CrossRef PubMed.
- N. Ousaka, Y. Takeyama, H. Iida and E. Yashima, Nat. Chem., 2011, 3, 856–861 CrossRef.
- G. Lautrette, B. Wicher, B. Kauffmann, Y. Ferrand and I. Huc, J. Am. Chem. Soc., 2016, 138, 10314–10322 CrossRef PubMed.
- M. Fijiki, J. Am. Chem. Soc., 1994, 116, 6017–6018 CrossRef.
- A. Ohira, M. Kunitake, M. Fujiki, M. Naioto and A. Saxena, Chem. Mater., 2004, 16, 3919–3923 CrossRef.
- M. Fujiki, Chem. Rec., 2009, 9, 271–298 CrossRef PubMed.
- M. Suginome, Molecular Technology, John Wiley & Sons, Weinheim, 2018 Search PubMed.
- Y. Nagata, T. Nishikawa, M. Suginome, S. Sato, M. Sugiyama, L. Porcar, A. Martel, R. Inoue and N. Sato, J. Am. Chem. Soc., 2018, 140, 2722–2726 CrossRef PubMed.
- Y. Nagata, T. Kuroda, K. Takagi and M. Suginome, Chem. Sci., 2014, 5, 4953–4956 RSC.
- Y. Nagata, T. Nishikawa and M. Suginome, Chem. Commun., 2018, 54, 6867–6870 RSC.
- Y. Nagata, T. Nishikawa, K. Terao, H. Hasegawa and M. Suginome, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 260–263 CrossRef.
- T. Nishikawa, Y. Nagata and M. Suginome, ACS Macro Lett., 2017, 6, 431–435 CrossRef PubMed.
- T. Yamamoto, R. Murakami, S. Komatsu and M. Suginome, J. Am. Chem. Soc., 2018, 140, 3867–3870 CrossRef PubMed.
- I. Louzao, J. M. Seco, E. Quiñoá and R. Riguera, Angew. Chem., Int. Ed., 2010, 49, 1430–1433 CrossRef PubMed.
- K. Maeda, K. Morino, Y. Okamoto, T. Sato and E. Yashima, J. Am. Chem. Soc., 2004, 126(13), 4329–4342 CrossRef PubMed.
- E. Yashima, K. Maeda and Y. Okamoto, Nature, 1999, 399, 449–451 CrossRef CAS.
- Y.-Z. Ke, Y. Nagata, T. Yamada and M. Suginome, Angew. Chem., Int. Ed., 2015, 54, 9333–9337 CrossRef CAS.
- R. Rodríguez, E. Suárez-Picado, E. Quiñoá, R. Riguera and F. Freire, Angew. Chem., Int. Ed., 2020, 59, 8616–8622 CrossRef.
- Z. Fernández, B. Fernández, E. Quiñoá, R. Riguera and F. Freire, Chem. Sci., 2020, 11, 7182–7187 RSC.
- J. J. L. M. Cornelissen, J. J. J. M. Donners, R. De Gelder, W. S. Graswinckel, G. A. Metselaar, A. E. Rowan, N. A. J. M. Sommerdijk and R. J. M. Nolte, Science, 2001, 293, 676–680 CrossRef PubMed.
- E. Suárez-Picado, E. Quiñoá, R. Riguera and F. Freire, Angew. Chem., Int. Ed., 2020, 59, 4537–4543 CrossRef.
- R. Rodríguez, E. Rivadulla-Cendal, M. Fernández-Míguez, B. Fernández, E. Quiñoá and F. Freire, Angew. Chem., Int. Ed., 2022, 61, e202209953 CrossRef PubMed.
- M. Lago-Silva, M. M. Cid, E. Quiñoá and F. Freire, Angew. Chem., Int. Ed., 2023, 62, e202303329 CrossRef PubMed.
- M. M. Green, M. P. Reidy, R. D. Johnson, G. Darling, D. J. O’Leary and G. Willson, J. Am. Chem. Soc., 1989, 111, 6452–6454 CrossRef.
- M. M. Green, N. C. Peterson, T. Sato, A. Teramoto, R. Cook and S. Lifson, Science, 1995, 268, 1860–1866 CrossRef.
- H. Gu, Y. Nakamura, T. Sato, A. Teramoto, M. M. Green, S. K. Jha, C. Andreola and M. P. Reidy, Macromolecules, 1998, 31, 6362–6368 CrossRef.
- M. M. Green, J. W. Park, T. Sato, A. Teramoto, S. Lifson, R. L. Selinger and J. V. Selinger, Angew. Chem., Int. Ed., 1999, 38, 3138–3154 CrossRef PubMed.
- K. Cobos, E. Quiñoá, R. Riguera and F. Freire, J. Am. Chem. Soc., 2018, 140, 12239–12246 CrossRef PubMed.
- S. Arias, R. Rodríguez, E. Quiñoá, R. Riguera and F. Freire, J. Am. Chem. Soc., 2018, 140, 667–674 CrossRef PubMed.
- M. M. Green, B. A. Garetz, B. Munoz, H. Chang, S. Hoke and R. G. Cooks, J. Am. Chem. Soc., 1995, 117(14), 4181–4182 CrossRef.
- K. Cobos, R. Rodríguez, E. Quiñoá, R. Riguera and F. Freire, Angew. Chem., Int. Ed., 2020, 59, 23724–23730 CrossRef PubMed.
- V. Jain, K.-S. Cheon, K. Tang, S. Jha and M. M. Green, Isr. J. Chem., 2011, 51, 1067–1074 CrossRef.
- M. Alzubi, S. Arias, R. Rodríguez, E. Quiñoá, R. Riguera and F. Freire, Angew. Chem., Int. Ed., 2019, 58, 13365–13369 CrossRef.
- Z. Fernández, B. Fernández, E. Quiñoá and F. Freire, Angew. Chem., Int. Ed., 2021, 60, 9919–9924 CrossRef.
- M. A. Martínez, E. E. Greciano and L. Sánchez, Chem. – Eur. J., 2019, 25, 16012–16016 CrossRef.
- E. E. Greciano, R. Rodríguez, K. Maeda and L. Sánchez, Chem. Commun., 2020, 56, 2244–2247 RSC.
- A. R. A. Palmans and E. W. Meijer, Angew. Chem., Int. Ed., 2007, 46, 8948–8968 CrossRef.
- A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1997, 36, 2648–2651 CrossRef.
- M. M. J. Smulders, P. J. M. Stals, T. Mes, T. F. E. Paffen, A. P. H. J. Schenning, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2010, 132(2), 620–626 CrossRef PubMed.
- G. Liu, M. G. Humphrey, C. Zhang and Y. Zhao, Chem. Soc. Rev., 2023, 52, 4443–4487 RSC.
- H. Komori and Y. Inai, J. Org. Chem., 2007, 72, 4012–4022 CrossRef PubMed.
- M. De Zotti, V. N. Syryamina, R. Hussain, E. Longo, G. Siligardi, S. A. Dzuba, L. Stella and F. Formaggio, ChemBioChem, 2019, 20, 2125–2132 CrossRef.
- Y. Ishido, N. Kanbayashi, T. Okamura and K. Onitsuka, Macromol. Rapid Commun., 2021, 42, 2100250 CrossRef.
- H. G. Jeon, H. K. Lee, S. Lee and K. S. Jeong, Chem. Commun., 2018, 54, 5740–5743 RSC.
- A. Motoshige, Y. Mawatari, Y. Yoshida, R. Motoshige and M. Tabata, Poly. Chem., 2014, 5, 971–978 RSC.
- A. Motoshige, Y. Mawatari, Y. Yoshida, C. Seki, H. Matsuyama and M. Tabata, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3008–3015 CrossRef.
- Y. Yoshida, Y. Matawari, A. Motoshige, R. Motoshige, T. Hiraoki, M. Wagner, K. Mullen and M. Tabata, J. Am. Chem. Soc., 2013, 135, 4110–4116 CrossRef PubMed.
- H. Nakako, R. Nomura and T. Masuda, Macromolecules, 2001, 34, 1496–1502 CrossRef.
- X. Guan, S. Wang, G. Shi, J. Zhang and X. Wan, Macromolecules, 2021, 54, 4592–4600 CrossRef.
- Y. Zhou, C. Zhang, Y. Qiu, L. Liu, T. Yang, H. Dong, T. Satoh and Y. Okamoto, Molecules, 2016, 21, 1583 CrossRef PubMed.
- S. Arias, F. Freire, M. Calderón and J. Bergueiro, Angew. Chem., Int. Ed., 2017, 56, 11420–11425 CrossRef.
- T. Miyagawa, A. Furuko, K. Maeda, H. Katagiri, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2005, 127, 5018–5019 CrossRef CAS.
- K. Nagai, K. Maeda, Y. Takeyama, T. Sato and E. Yashima, Chem. – Asian J., 2007, 2, 1314–1321 CrossRef CAS PubMed.
- N. Zhu, K. Nakazono and T. Takata, Chem. Commun., 2016, 52, 3647–3649 RSC.
- G. Shi, S. Wang, X. Guan, J. Zhang and X. Wan, Chem. Commun., 2018, 54, 12081–12084 RSC.
- S. Wang, S. Xie, H. Du, H. Zeng, J. Zhang and X. Wan, Sci. China: Chem., 2023, 66, 887–895 CAS.
- A. P. H. J. Schenning, M. Fransen and E. W. Meijer, Macromol. Rapid Commun., 2002, 23, 265–270 CrossRef CAS.
- V. Percec, M. Peterca, J. G. Rudick, E. Aqad, M. R. Imam and P. A. Heiney, Chem. – Eur. J., 2007, 13, 9572–9581 CrossRef.
- V. Percec, J. G. Rudick, M. Peterca and P. A. Heinei, J. Am. Chem. Soc., 2008, 130, 7503–7508 CrossRef CAS.
- F. Wang, C. Zhou, K. Liu, J. Yan, W. Li, T. Masuda and A. Zhang, Macromolecules, 2019, 52, 8631–8642 CrossRef CAS.
- Y. Cao, L. Ren, Y. Zhang, X. Lu, X. Zhan, J. Yan, W. Li, T. Masuda and A. Zhang, Macromolecules, 2021, 54, 7621–7631 CrossRef CAS.
- K. Maeda, K. Shimomura, T. Ikai, S. Kanoh and E. Yashima, Macromolecules, 2017, 50, 7801–7806 CrossRef CAS.
- J. J. Tarrío, R. Rodríguez, J. Crassous, E. Quiñoá and F. Freire, Angew. Chem., Int. Ed., 2023, 62, e202307059 CrossRef PubMed.
- J. K. Koe, M. Fujiki, M. Motonaga and H. Nakashima, Chem. Commun., 2000, 389–390 RSC.
- M. Fujiki, J. Am. Chem. Soc., 2000, 122, 3336–3343 CrossRef CAS.
- M. Fujiki, J. R. Koe, M. Motonaga, H. Nakashima, K. Terao and A. Teramoto, J. Am. Chem. Soc., 2001, 123, 6253–6261 CrossRef CAS.
- M. Fujiki, Macromol. Rapid Commun., 2001, 22, 539–563 CrossRef CAS.
- H. Nakashima, M. Fujiki, J. R. Koe and M. Motonaga, J. Am. Chem. Soc., 2001, 123, 1963–1969 CrossRef CAS.
- G. Hu, W. Li, Y. Hu, A. Xu, J. Yan, L. Liu, X. Zhang, K. Liu and A. Zhang, Macromolecules, 2013, 46, 1124–1132 CrossRef CAS.
- K. Tang, M. M. Green, K. S. Cheon, J. V. Selinger and B. A. Garetz, J. Am. Chem. Soc., 2003, 125, 7313–7323 CrossRef CAS.
- V. Krishnasamy, W. Qu, C. Chen, H. Huo, K. Ramanagul, V. Gothandapani, G. H. Mehl, Q. Zhang and F. Liu, Macromolecules, 2020, 53, 4193–4203 CrossRef CAS.
- K. A. Andreopoulou, M. Peterca, D. A. Wilson, B. E. Partridge, P. A. Heiney and V. Percec, Macromolecules, 2017, 50, 5271–5284 CrossRef CAS.
- Y.-J. Sun, X.-X. Cheng, T.-F. Miao, H.-T. Ma, W. Zhang and X.-L. Zhu, Chin. J. Polym. Sci., 2022, 40, 56–66 CrossRef CAS.
- K. Murata, M. Aoki, T. Suzuki, T. Harada, H. Kawabata, T. Komori, F. Ohseto, K. Ueda and S. Shinkai, J. Am. Chem. Soc., 1994, 116, 6664–6676 CrossRef CAS.
- P. Xing, Y. Li, Y. Wang, P.-Z. Li, H. Chen, S. Z. F. Phua and Y. Zhao, Angew. Chem., Int. Ed., 2018, 57, 7774–7779 CrossRef CAS.
- W. Qi, C. Ma, P. Liao, H. Li, T. Gu, T. Wu, J. Huang and Y. Yan, Adv. Optical Mater., 2023, 11, 2201229 CrossRef CAS.
- N. J. V. Zee, B. Adelizzi, M. F. J. Mabesoone, X. Meng, A. Aloi, H. Zha, M. Lutz, I. A. W. Filot, A. R. A. Palmans and E. W. Meijer, Nature, 2018, 558, 100–103 CrossRef PubMed.
- N. J. V. Zee, M. F. J. Mabesoone, B. Adelizzi, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 20191–20200 CrossRef.
- M. Go, H. Choi, K. Y. Kim, C. J. Moon, Y. Choi, H. Miyake, S. S. Lee, S. H. Jung, M. Y. Choi and J. H. Jung, Org. Chem. Front., 2019, 6, 1100–1108 RSC.
- C. Kulkarny, P. A. Korevaar, K. K. Bejagam, A. R. A. Palmans, E. W. Meijer and S. J. George, J. Am. Chem. Soc., 2017, 139, 13867–13875 CrossRef.
- M. Peterca, M. R. Iman, C.-H. Ahn, V. S. Balagurusamy, D. A. Wilson, B. M. Rosen and V. Percec, J. Am. Chem. Soc., 2011, 133, 2311–2328 CrossRef CAS.
- L. Gao, C. Xing, X. Dou, Y. Zou, C. Zhao and C. Feng, Angew. Chem., Int. Ed., 2022, 61, e202211812 CrossRef CAS PubMed.
- A. M. García and A. Ruiz-Carretero, Chem. Commun., 2022, 58, 529–532 RSC.
- Y. Zhang, H. Li, Z. Geng, W.-H. Zheng, Y. Quan and Y. Cheng, ACS Nano, 2022, 16, 3173–3181 CrossRef CAS PubMed.
- H. Juwarker and K. S. Jeong, Chem. Soc. Rev., 2010, 39, 3664–3674 RSC.
- K. J. Chang, B. N. Kang, M. H. Lee and K.-S. Jeong, J. Am. Chem. Soc., 2005, 127, 12214–12215 CrossRef CAS.
- M. C. Kim, J. Suk, H. J. Kim, E. Sim, M. Lee, K.-S. Jeong and V. R. Naidu, Org. Lett., 2008, 10, 5373–5376 CrossRef.
- J. M. Suk, V. R. Naidu, X. Liu, M. S. Lah and K. S. Jeong, J. Am. Chem. Soc., 2011, 133, 13938–13941 CrossRef CAS PubMed.
- D. A. Kim, P. Kang, M. G. Choi and K. S. Jeong, Chem. Commun., 2013, 49, 9743–9745 RSC.
- D. Mondal, M. Ahmad, B. Dey, A. Mondal and P. Talukdar, Nat. Commun., 2022, 13, 6507 CrossRef CAS.
- Q. Li, F. Huang, J. Shang, Y. Che, Y. Wang, W. Zhao and H. Jiang, Tetrahedron Lett., 2016, 57, 1691–1694 CrossRef.
- J. V. Gavette, C. J. Evoniuk, L. N. Zakharov, M. E. Carnes, M. M. Haley and D. W. Johnson, Chem. Sci., 2014, 5, 2899–2905 RSC.
- R. Wechsel, M. Žabka, J. W. Ward and J. Clayden, J. Am. Chem. Soc., 2018, 140, 3528–3531 CrossRef CAS.
- H. Miyake, H. Kamon, I. Miyahara, H. Sugimoto and H. Tsukube, J. Am. Chem. Soc., 2008, 130, 792–793 CrossRef CAS PubMed.
- H.-J. Kim, W.-C. Zin and M. Lee, J. Am. Chem. Soc., 2004, 126, 7009–7014 CrossRef CAS PubMed.
- H.-J. Kim, J.-H. Lee and M. Lee, Angew. Chem., Int. Ed., 2005, 44, 5810–5814 CrossRef CAS PubMed.
- H. Maeda, W. Hane, Y. Bando, Y. Terashima, Y. Haketa, H. Shibaguchi, T. Kawai, M. Naito, K. Takaishi, M. Uchiyama and A. Muranaka, Chem. – Eur. J., 2013, 19, 16263–16271 CrossRef CAS PubMed.
- R. Kakuchi, S. Nagata, Y. Tago, R. Sakai, I. Otsuka, T. Satoh and T. Kakuchi, Macromolecules, 2009, 42, 1476–1481 CrossRef CAS.
- R. Kakuchi, S. Nagata, R. Sakai, I. Otsuka, H. Nakade, T. Satoh and T. Kakuchi, Chem. – Eur. J., 2008, 14, 10259–10266 CrossRef CAS.
- R. Sakai, S. Okade, E. B. Barasa, R. Kakuchi, M. Ziabka, S. Umeda, K. Tsuda, T. Satoh and T. Kakuchi, Macromolecules, 2010, 43, 7406–7411 CrossRef CAS.
- R. Kakuchi, T. Kodama, R. Shimada, Y. Tago, R. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2009, 42, 3892–3897 CrossRef CAS.
- Y. Qu, J. Hua, Y. Jiang and H. Tian, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1544–1552 CrossRef CAS.
- R. Kakuchi, Y. Tago, R. Sakai, T. Satoh and T. Kakuchi, Macromolecules, 2009, 42, 4430–4435 CrossRef CAS.
- R. Kakuchi, R. Shimada, Y. Tago, R. Sakai, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1683–1689 CrossRef CAS.
- S. Leiras, E. Suárez-Picado, E. Quiñoá, R. Riguera and F. Freire, Giant, 2021, 7, 100068 CrossRef CAS.
- Y. Gao, T. Gao, L. Wang, X. Ma, R. Jin, C. Kang and L. Gao, New J. Chem., 2021, 45, 5093 RSC.
- Y. Qiu, Y. Zhang, Q. Jiang, H. Wang, Y. Liao, H. Zhou and X. Xie, Macromolecules, 2022, 55, 9057–9065 CrossRef CAS.
- M. H.-Y. Chan, S. Y.-L. Leung and V. W.-W. Yam, J. Am. Chem. Soc., 2019, 141, 12312–12321 CrossRef CAS.
- J. Liu, C. Sun, W. Ma, Y.-J. Lu, L. Yu, K. Zhang and H. Zeng, RSC Adv., 2014, 4, 54469–54473 RSC.
- Y.-C. Lin and C.-T. Chen, Chem. – Eur. J., 2012, 19, 2531–2538 CrossRef PubMed.
- S. Y.-L. Leung, A. Y.-Y. Tam, C.-H. Tao, H. S. Chow and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 134, 1047–1056 CrossRef.
- S. Akine, T. Matsumoto and T. Nabeshima, Chem. Commun., 2008, 4604–4606 RSC.
- S. Akine, S. Hotate and T. Nabeshima, J. Am. Chem. Soc., 2011, 133, 13868–13871 CrossRef CAS PubMed.
- S. Akine, S. Sairenji, T. Taniguchi and T. Nabeshima, J. Am. Chem. Soc., 2013, 135, 12948–12951 CrossRef CAS PubMed.
- S. Sairenji, S. Akine and T. Nabeshima, Dalton Trans., 2016, 45, 14902–14906 RSC.
- S. Kawano, K. Narita, Y. Ikemoto, A. Sasaki and K. Tanaka, Chem. Commun., 2022, 58, 3274–3277 RSC.
- D. Bai, T. Yan, S. Wang, Y. Wang, J. Fu, X. Fang, J. Zhu and J. Liu, Angew. Chem., Int. Ed., 2020, 59, 13602–13607 CrossRef CAS.
- M. Baskin, H. Zhu, Z.-W. Qu, J. H. Chill, S. Grimme and G. Maayan, Chem. Sci., 2019, 10, 620–632 RSC.
- M. Horeau, G. Lautrette, B. Wicher, V. Blot, J. Lebreton, M. Pipelier, D. Dubreuil, Y. Ferrand and I. Huc, Angew. Chem., Int. Ed., 2017, 56, 6823–6827 CrossRef CAS.
- P. Mateus, B. Wicher, Y. Ferrand and I. Huc, Chem. Commun., 2017, 53, 9300–9303 RSC.
- K. Miwa, Y. Furusho and E. Yashima, Nat. Chem., 2010, 2, 444–449 CrossRef CAS PubMed.
- D. Taura, K. Shimizu, C. Yokota, R. Ikeda, Y. Suzuki, H. Iida, N. Ousaka and E. Yashima, Chem. Commun., 2019, 55, 12084–12087 RSC.
- B. Maiti, S. Bhattacharjee and S. Bhattacharya, Nanoscale, 2019, 11, 2223–2230 RSC.
- M. H.-Y. Chan, M. Ng, S. Y.-L. Leung, W. H. Lam and V. W.-W. Yam, J. Am. Chem. Soc., 2017, 139, 8639–8645 CrossRef CAS.
- X. Yu, Z. Wang, Y. Li, L. Geng, J. Ren and G. Feng, Inorg. Chem., 2017, 56, 7512–7518 CrossRef CAS.
- F. Wang and C.-L. Feng, Angew. Chem., Int. Ed., 2018, 57, 5655–5659 CrossRef CAS.
- S. Akine, H. Nagumo and T. Nabeshima, Inorg. Chem., 2012, 51, 5506–5508 CrossRef CAS.
- C. Liu, K. Fu, S. Zheng, L. Yao, W. Zhou and G. Liu, Adv. Opt. Mater., 2023, 11, 2300019 CrossRef CAS.
- S. Kawabata, N. Ousaka and E. Yashima, Chem. Commun., 2018, 54, 2417–2420 RSC.
- Z. Li, H. Yang, K. M. Adam, H. Yao, T. Wei, Y. Zhang and Q. Lin, ChemPlusChem, 2021, 86, 146–154 CrossRef CAS.
- F. Wang, W. Ji, P. Yang and C.-L. Feng, ACS Nano, 2019, 13, 7281–7290 CrossRef CAS.
- G. Liu, J. Sheng, W. L. Teo, G. Yang, H. Wu, Y. Li and Y. Zhao, J. Am. Chem. Soc., 2018, 140, 16275–16283 CrossRef CAS.
- S. H. Park, S. H. Jung, J. Ahn, J. H. Lee, K.-Y. Kwon, J. Jeon, H. Kim, J. Jaworski and J. H. Jung, Chem. Commun., 2014, 50, 13495–13498 RSC.
- M. Fukuda, R. Rodríguez, Z. Fernández, T. Nishimura, D. Hirose, G. Watanabe, E. Quiñoá, F. Freire and K. Maeda, Chem. Commun., 2019, 55, 7906–7909 RSC.
- F. Freire, J. M. Seco, E. Quiñoá and R. Riguera, Hierarchical Macromolecular Structures: 60 Years after the Staudinger Nobel Prize II, 2013, pp. 123–140 Search PubMed.
- F. Freire, J. M. Seco, E. Quiñoá and R. Riguera, Angew. Chem., Int. Ed., 2011, 50, 11692–11696 CrossRef CAS PubMed.
- F. Freire, J. M. Seco, E. Quiñoá and R. Riguera, J. Am. Chem. Soc., 2012, 134, 19374–19383 CrossRef CAS.
- S. Arias, F. Freire, E. Quiñoá and R. Riguera, Angew. Chem., Int. Ed., 2014, 53, 13720–13724 CrossRef CAS PubMed.
- R. Rodríguez, S. Arias, E. Quiñoá, R. Riguera and F. Freire, Nanoscale, 2017, 9, 17752–17757 RSC.
- S. Arias, F. Freire, E. Quiñoá and R. Riguera, Polym. Chem., 2015, 6, 4725–4733 RSC.
- D. Hirose, A. Isobe, E. Quiñoá, F. Freire and K. Maeda, J. Am. Chem. Soc., 2019, 141, 8592–8598 CrossRef CAS.
- M. Fukuda, T. Nishimura, D. Hirose and K. Maeda, Chem. Lett., 2023, 52, 136–139 CrossRef CAS.
- J. Bergueiro, F. Freire, E. P. Wendler, J. M. Seco, E. Quiñoá and R. Riguera, Chem. Sci., 2014, 5, 2170–2176 RSC.
- S. Arias, J. Bergueiro, F. Freire, E. Quiñoá and R. Riguera, Small, 2015, 12, 238–244 CrossRef PubMed.
- M. Alzubi, S. Arias, I. Louzao, E. Quiñoá, R. Riguera and F. Freire, Chem. Commun., 2017, 53, 8573–8576 RSC.
- S. Arias, M. Núñez-Martínez, E. Quiñoá, R. Riguera and F. Freire, Polym. Chem., 2017, 8, 3740–3745 RSC.
- Y.-H. Chiu, O. dos Santos and J. W. Canary, Tetrahedron, 1999, 55, 12069–12078 CrossRef CAS.
- S. Zahn and J. W. Canary, Science, 2000, 288, 1404–1407 CrossRef CAS PubMed.
- J. Liang and J. W. Canary, Chirality, 2010, 23, 24–33 CrossRef PubMed.
- F. Wendt, C. Näther and F. Tuczek, J. Biol. Inorg. Chem., 2016, 21, 777–792 CrossRef CAS PubMed.
- M. Gaedke, F. Witte, J. Anhäuser, H. Hupatz, H. V. Schröder, A. Valkonen, K. Rissanen, A. Lützen, B. Paulus and C. A. Schalley, Chem. Sci., 2019, 10, 10003–10009 RSC.
- L. Faour, C. Adam, C. Gautier, S. Goeb, M. Allain, E. Levillain, D. Canevet and M. Sallé, Chem. Commun., 2019, 55, 5743–5746 RSC.
- Y. Wang, M. Frasconi, W.-G. Liu, Z. Liu, A. A. Sarjeant, M. S. Nassar, Y. Y. Botros, W. A. Goddard III and J. F. Stoddart, J. Am. Chem. Soc., 2015, 137, 876–885 CrossRef CAS.
- Y. Zhang, M. Qin, C. Xing, C. Zhao, X. Dou and C. Feng, ACS Nano, 2020, 14, 17151–17162 CrossRef CAS.
- J. Yan, J. Liu, H. Lei, Y. Kang, C. Zhao and Y. Fang, J. Colloid Interface Sci., 2015, 448, 374–379 CrossRef CAS.
- B. Adhikari and H. B. Kraatz, Chem. Commun., 2014, 50, 5551–5553 RSC.
- S. Kawano, N. Fujita and S. Shinkai, Chem. – Eur. J., 2005, 11, 4735–4742 CrossRef CAS.
- Y. Gao, J. Lu, J. Wu, J. Hu and Y. Ju, RSC Adv., 2014, 4, 63539–63543 RSC.
- L.-Y. Guo, H. Zhang, S.-T. Huang, M. Di, G. Yao and S. Wu, e-Polymers, 2019, 19, 377–384 CAS.
- Z.-Q. Wu, X. Song, Y.-X. Li, L. Zhou, Y.-Y. Zhu, Z. Chen and N. Liu, Nat. Commun., 2023, 14, 566–577 CrossRef CAS PubMed.
- E. Gomar-Nadal, J. Veciana, C. Rovira and D. B. Amabilino, Adv. Mater., 2005, 17, 2095–2098 CrossRef CAS.
- J. Bergueiro, M. Núñez-Martínez, S. Arias, E. Quiñoá, R. Riguera and F. Freire, Nanoscale Horiz., 2020, 5, 495–500 RSC.
- M. Núñez-Martínez, S. Arias, J. Bergueiro, E. Quiñoá, R. Riguera and F. Freire, Macromol. Rapid Commun., 2022, 43, 2100616 CrossRef PubMed.
- M. Núñez-Martínez, S. Arias, E. Quiñoá, R. Riguera and F. Freire, Chem. Mater., 2021, 33, 4805–4812 CrossRef.
- M. Núñez-Martínez, E. Quiñoá and F. Freire, Nanoscale, 2022, 14, 13066–13072 RSC.
- H. Iida, T. Mizoguchi, S.-D. Oh and E. Yashima, Polym. Chem., 2010, 1, 841–848 RSC.
- E. Ohta, H. Sato, S. Ando, A. Kosaka, T. Fukushima, D. Hashizume, M. Yamasaki, K. Hasegawa, A. Muraoka, H. Ushiyama, K. Yamashita and T. Aida, Nat. Chem., 2010, 3, 68–73 CrossRef PubMed.
- S. Liang, G. Chen and Y. Zhao, J. Mater. Chem. C, 2013, 1, 5477–5490 RSC.
- S. Liang, G. Chen, J. Peddle and Y. Zhao, Chem. Commun., 2012, 48, 3100–3102 RSC.
- S. Anantharaj and M. Jayakannan, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 2864–2875 CrossRef CAS.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- M. Ire, Chem. Rev., 2000, 100, 1685–1716 CrossRef.
- M. Ire, T. Fukaminato, K. Matsuda and S. Kobakate, Chem. Rev., 2014, 114, 12174–12277 CrossRef.
- K. Matsuda and M. Irie, J. Photochem. Photobiol., C, 2004, 5, 169–182 CrossRef CAS.
- M. Li and W. H. Zhu, Acc. Chem. Res., 2022, 55, 3136–3149 CrossRef CAS.
- G. Likhtenshtein, Stilbenes: Applications in Chemistry, Life Sciences and Materials Science, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2009 Search PubMed.
- T. Dünnebacke, K. K. Kartha, J. M. Wiest, R. Q. Albuquerque and G. Fernández, Chem. Sci., 2020, 11, 10405–10413 RSC.
- R. Costil, M. Holsheimer, S. Crespi, N. A. Simeth and B. L. Feringa, Chem. Rev., 2021, 121, 13213–13237 CrossRef CAS.
- J. C. M. Kistemaker, A. S. Lubbe and B. L. Feringa, Mater. Chem. Front., 2021, 5, 2900–2906 RSC.
- D. R. S. Pooler, A. S. Lubbe, S. Crespi and B. L. Feringa, Chem. Sci., 2021, 12, 14964–14986 RSC.
- B. L. Feringa, J. Org. Chem., 2007, 72, 6635–6652 CrossRef CAS.
- S. Kassem, T. Van Leeuwen, A. S. Lubbe, M. R. Wilson and B. L. Feringa, Nat. Rev. Chem., 2017, 1, 0096 CrossRef.
- X. Su and I. Aprahamian, Org. Lett., 2011, 13, 30–33 CrossRef CAS.
- B. Shao and I. Aprahamian, Chem, 2020, 6, 2162–2173 CAS.
- F. Xu and B. L. Feringa, Adv. Mater., 2023, 35, 2204413 CrossRef CAS.
- D. Pijper and B. L. Feringa, Angew. Chem., Int. Ed., 2007, 46, 3693–3696 CrossRef CAS PubMed.
- D. Pijper, M. G. M. Jongejam, A. Meetsma and B. L. Feringa, J. Am. Chem. Soc., 2008, 130, 4541–4552 CrossRef CAS.
- T. Van Leeuwen, G. H. Heideman, D. Zhao, S. J. Wezenberg and B. L. Feringa, Chem. Commun., 2017, 53, 6393–6396 RSC.
- M. Muellerand and R. Zentel, Macromolecules, 1994, 27, 4404–4406 CrossRef.
- G. Maxeinand and R. Zentel, Macromolecules, 1995, 28, 8438–8440 CrossRef.
- M. Müllerand and R. Zentel, Macromolecules, 1996, 29, 1609–1617 CrossRef.
- S. Mayer, G. Maxeinand and R. Zentel, Macromolecules, 1998, 31, 8522–8525 CrossRef CAS.
- S. Mayerand and R. Zentel, Macromol. Chem. Phys., 1998, 199, 1675–1682 CrossRef.
- X. Yao, T. Li, J. Wang, X. Ma and H. Tian, Adv. Opt. Mater., 2016, 4, 1322–1349 CrossRef CAS.
- K. M. Vonk, E. W. Meijer and G. Vantomme, Chem. Sci., 2021, 12, 13572–13579 RSC.
- E. Weyandt, G. M. Ter Huurne, G. Vantomme, A. J. Markvoort, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 6295–6303 CrossRef CAS PubMed.
- Y. Cai, Z. Guo, J. Chen, W. Li, L. Zhong, Y. Gao, L. Jiang, L. Chi, H. Tian and W. H. Zhu, J. Am. Chem. Soc., 2016, 138, 2219–2224 CrossRef CAS PubMed.
- B. A. San Jose, T. Ashibe, N. Tada, S. Yorozuka and K. Akagi, Adv. Funct. Mater., 2014, 24, 6166–6171 CrossRef CAS.
- F. K. C. Leung, T. Van Den Enk, T. Kajitani, J. Chen, M. C. A. Stuart, J. Kuipers, T. Fukushima and B. L. Feringa, J. Am. Chem. Soc., 2018, 140, 17724–17733 CrossRef CAS.
- S. Chen, L. Yang, F. K. C. Leung, T. Kajitani, M. C. A. Stuart, T. Fukushima, P. Van Rijn and B. L. Feringa, J. Am. Chem. Soc., 2022, 144, 3543–3553 CrossRef CAS.
- F. Xu, L. Pfeifer, S. Crespi, F. K. C. Leung, M. C. A. Stuart, S. J. Wezenberg and B. L. Feringa, J. Am. Chem. Soc., 2021, 143, 5990–5997 CrossRef CAS PubMed.
- T. S. C. MacDonald, B. L. Feringa, W. S. Price, S. J. Wezenberg and J. E. Beves, J. Am. Chem. Soc., 2020, 142, 20014–20020 CrossRef CAS PubMed.
- S. Chen, S. Chen, F. K. C. Leung, M. C. A. Stuart, C. Wang and B. L. Feringa, J. Am. Chem. Soc., 2020, 142, 10163–10172 CrossRef CAS.
- F. Xu, S. Crespi, G. Pacella, Y. Fu, M. C. A. Stuart, Q. Zhang, G. Portale and B. L. Feringa, J. Am. Chem. Soc., 2022, 144, 6019–6027 CrossRef CAS.
- S. Yagai, M. Yamauchi, A. Kobayashi, T. Karatsu, A. Kitamura, T. Ohba and Y. Kikkawa, J. Am. Chem. Soc., 2012, 134, 18205–18208 CrossRef CAS.
- A. Suzuki, K. Aratsu, S. Datta, N. Shimizu, H. Takagi, R. Haruki, S.-I. Adachi, M. Hollambi, F. Silly and S. Yagai, J. Am. Chem. Soc., 2010, 141, 13196–13202 CrossRef.
- D. Zhao, T. Van Leewen, J. Cheng and B. L. Feringa, Nat. Chem., 2017, 9, 250–256 CrossRef CAS.
- D. Mazzier, M. Crisma, M. De Poli, G. Marafon, C. Peggion, J. Clayden and A. Moretto, J. Am. Chem. Soc., 2016, 138, 8007–8018 CrossRef CAS.
- M. Pollastrani, G. Marafon, J. Clayden and A. Moretto, Chem. Commun., 2021, 57, 2269–2272 RSC.
- S. Pramanik, B. Kauff, S. Hecht, Y. Ferrand and I. Huc, Chem. Commun., 2021, 57, 93–96 RSC.
- Z. Yu and S. Hecht, Chem. Commun., 2016, 52, 6639–6653 RSC.
- O. Pieroni, J. L. Houben, A. Fissi, P. Costantino and F. Ciardelli, J. Am. Chem. Soc., 1980, 102, 5913–5915 CrossRef CAS.
- J. L. Houben, A. Fissi, D. Bacciola, N. Rosato, O. Pieroni and F. Ciardelli, Int. J. Biol. Macromol., 1983, 5, 94–100 CrossRef CAS.
- O. Pieront, D. Fabbri, A. Fissi and F. Ciardelli, Macromol. Rapid Commun., 1988, 9, 637–640 CrossRef.
- A. Ueno, J.-i Anzai, T. Osa and Y. Kadoma, Bull. Chem. Soc. Jpn., 1979, 52, 549–554 CrossRef CAS.
- A. Ueno, K. Takahashi, J. Anzai and T. Osa, J. Am. Chem. Soc., 1981, 103, 6410–6415 CrossRef CAS.
- A. Ueno, K. Adachi, J. Nakamura and T. Osa, J. Polym. Sci., Part A: Polym. Chem., 1990, 28, 1161–1170 CrossRef CAS.
- F. Ciardelli, D. Fabbri, O. Pieroni and A. Fissi, J. Am. Chem. Soc., 1989, 111, 3470–3472 CrossRef CAS.
- E. D. King, P. Tao, T. T. Sanan, C. M. Hadad and J. R. Parquette, Org. Lett., 2008, 10, 1671–1674 CrossRef CAS.
- C. J. Gabriel and J. R. Parquette, J. Am. Chem. Soc., 2006, 128, 13708–13709 CrossRef CAS.
- Y. Hua and A. H. Flood, J. Am. Chem. Soc., 2010, 132, 12838–12840 CrossRef CAS.
- Y. Wang, F. Bie and H. Jiang, Org. Lett., 2010, 12, 3630–3633 CrossRef CAS.
- F. C. Parks, Y. Liu, S. Debnath, R. Stutsman, K. Raghavachari and A. H. Flood, J. Am. Chem. Soc., 2018, 140, 17711–17723 CrossRef CAS PubMed.
- A. Khan, C. Kaiser and S. Hecht, Angew. Chem., Int. Ed., 2006, 45, 1878–1881 CrossRef CAS.
- Z. Yu and S. Hecht, Angew. Chem., Int. Ed., 2011, 50, 1640–1643 CrossRef CAS PubMed.
- J. Moore and C. Ray, Adv. Polym. Sci., 2005, 177, 91–149 CrossRef.
- S. Hecht and A. Khan, Angew. Chem., Int. Ed., 2003, 115, 6203–6206 CrossRef.
- A. Khan and S. Hecht, Chem. – Eur. J., 2006, 12, 4764–4774 CrossRef CAS PubMed.
- Z. Yu, S. Weidner, T. Risse and S. Hecht, Chem. Sci., 2013, 4, 4156–4167 RSC.
- Z. Yu and S. Hecht, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 313–318 CrossRef CAS.
- R. B. Prince, J. G. Saven, P. G. Wolynes and J. S. Moore, J. Am. Chem. Soc., 1999, 121, 3114–3121 CrossRef CAS.
- S. Hecht and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2001, 40, 74–91 CrossRef CAS.
- P. D. Kiser, M. Golczakak and K. Palczewski, Chem. Rev., 2014, 114, 194–232 CrossRef CAS PubMed.
- J. P. Riehl and F. S. Richardson, Chem. Rev., 1986, 86(1), 1–16 CrossRef CAS.
- J. P. Riehl, Chiroptical spectroscopy, emission theory, Elsevier Ltd., 3rd edn, 2016 Search PubMed.
- Y. Zhang, S. Yu, B. Han, Y. Zhou, X. Zhang, X. Gao and Z. Tang, Matter, 2022, 5, 837–875 CrossRef CAS.
- G. Longhi, E. Castiglioni, J. Koshoubu, G. Mazzeo and S. Abatte, Chirality, 2016, 28, 696–707 CrossRef CAS PubMed.
- S. Yang, S. Zhang, F. Hu, J. Han and F. Li, Coord. Chem. Rev., 2023, 485, 215116 CrossRef CAS.
- E. M. Sánchez-Carretero, A. R. Agarrabeitia, F. Moreno, B. L. Maroto, G. Muller, M. J. Ortiz and S. de la Moya, Chem. – Eur. J., 2015, 21, 13488–13500 CrossRef.
- L. Arrico, L. di Bari and F. Zinna, Chem. – Eur. J., 2021, 27, 2920–2934 CrossRef CAS PubMed.
- S. Y. Li, L. Xu, R. T. Gao, Z. Chen, N. Liu and Z. Q. Wu, J. Mater. Chem. C, 2022, 11, 1242–1250 RSC.
- K. Watanabe and K. Akagi, Sci. Technol. Adv. Mater., 2014, 15, 044203 CrossRef CAS.
- X. Liu and R. H. Jin, Chem. Synth., 2022, 1–29 Search PubMed.
- Y. Sang, J. Han, T. Zhao, P. Duan and M. Liu, Adv. Mater., 2020, 32, 1900110 CrossRef CAS.
- J. Kumar, T. Nakashima and T. Kawai, J. Phys. Chem. Lett., 2015, 6, 3445–3452 CrossRef CAS PubMed.
- K. Dhbaibi, L. Abella, S. Meunier-Della-Gatta, T. Roisnel, N. Vanthuyne, B. Jamoussi, G. Pieters, B. Racine, E. Quesnel, J. Autschbach, J. Crassous and L. Favereau, Chem. Sci., 2021, 12, 5522–5533 RSC.
- J. R. Brandt, X. Wang, Y. Yang, A. J. Campbell and M. J. Füchter, J. Am. Chem. Soc., 2016, 138, 9743–9746 CrossRef CAS.
- Y. Yang, R. Correa da Costa, D. M. Smilgies, A. J. Campbell and M. J. Fuchter, Adv. Mater., 2013, 25, 2624–2628 CrossRef CAS.
- L. E. MacKenzie and R. Pal, Nat. Rev. Chem., 2021, 5, 109–124 CrossRef CAS.
- P. Stachelek, L. MacKenzie, D. Parker and R. Pal, Nat. Commun., 2022, 13, 1–8 Search PubMed.
- C. He and Y. Li, Chin. Chem. Lett., 2022, 108077 Search PubMed.
- J. S. Kang, N. Kim, T. Kim, M. Seo and B. S. Kim, Macromol. Rapid Commun., 2022, 43, 1–21 Search PubMed.
- J. Li, G. B. Schuster, K. S. Cheon, M. M. Green and J. V. Selinger, J. Am. Chem. Soc., 2000, 122, 2603–2612 CrossRef CAS.
- G. Iftime, F. L. Labarthet, A. Natansohn and P. Rochon, J. Am. Chem. Soc., 2000, 122, 12646–12650 CrossRef CAS.
- G. Li, M. Xu, S. Zhang, G. Yang and W. Li, Macromol. Rapid Commun., 2022, 43, 2100904 CrossRef CAS.
- C. Kulkarni, R. H. N. Curvers, G. Vantomme, D. J. Broer, A. R. A. Palmans, S. C. J. Meskers and E. W. Meijer, Adv. Mater., 2021, 33, 1–7 CrossRef.
- H. Sakaino, B. A. G. Lamers, S. C. J. Meskers, E. W. Meijer and G. Vantomme, J. Polym. Sci., 2021, 59, 1131–1141 CrossRef CAS.
- E. Moulin, J. J. Armao IV and N. Giuseppone, Acc. Chem. Res., 2019, 52, 975–983 CrossRef CAS PubMed.
- J. Kim, J. Lee, W. Y. Kim, H. Kim, S. Lee, H. C. Lee, Y. S. Lee, M. Seo and S. Y. Kim, Nat. Commun., 2015, 6, 6959 CrossRef CAS.
- J. S. Kang, S. Kang, J. M. Suh, S. M. Park, D. K. Yoon, M. H. Lim, W. Y. Kim and M. Seo, J. Am. Chem. Soc., 2022, 144, 2657–2666 CrossRef CAS PubMed.
- Q. Sallembien, P. Aoun, S. Blanchard, L. Bouteiller and M. Raynal, Chem. – Eur. J., 2023, 29, e202203199 CrossRef CAS.
- J. Hu, Y. Xie, H. Zhang, C. He, Q. Zhang and G. Zou, Chem. Commun., 2019, 55, 4953–4956 RSC.
- H. Wang, Y. Liu, L. Wang, B. Raktani, T. Yang, K. M. Tran, Y. Luo, J. Yu, H. Lee and H. Lee, ACS Mater. Lett., 2022, 4, 1954–1961 CrossRef CAS.
- Y. Marcus, Chem. Soc. Rev., 1993, 22, 409–416 RSC.
- G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933 RSC.
- D. J. Hilland and J. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5053 CrossRef.
- J.-L. Hou, M.-X. Jia, X.-K. Jiang, Z.-T. Liand and G.-J. Chen, J. Org. Chem., 2004, 69, 6228 CrossRef CAS.
- A. J. Zychand and B. L. Iverson, J. Am. Chem. Soc., 2000, 122, 8898 CrossRef.
- I. Huc, Eur. J. Org. Chem., 2004, 17–29 CrossRef CAS.
- T. Qi, V. Maurizot, H. Noguchi, T. Charoenraks, B. Kauffmann, M. Takafuji, H. Iharacg and I. Huc, Chem. Commun., 2012, 48, 6337–6339 RSC.
- M. Kudo, V. Maurizot, B. Kauffmann, A. Tanatani and I. Huc, J. Am. Chem. Soc., 2013, 135, 9628–9631 CrossRef CAS.
- C.-F. Wu, Z.-M. Li, X.-N. Xu, Z.-X. Zhao, X. Zhao, R.-X. Wang and Z.-T. Li, Chem. – Eur. J., 2014, 20, 1418–1426 CrossRef CAS PubMed.
- Y. Liu, F. C. Parks, W. Zhao and A. H. Flood, J. Am. Chem. Soc., 2018, 140, 15477–15486 CrossRef CAS PubMed.
- B. A. F. Le Bailly and J. Clayden, Chem. Commun., 2016, 52, 4852–4863 RSC.
- M. Tomsett, I. Maffucci, B. A. F. Le Bailly, L. Byrne, S. M. Bijvoets, M. G. Lizio, J. Raftery, C. P. Butts, S. J. Webb, A. Contini and J. Clayden, Chem. Sci., 2017, 8, 3007–3018 RSC.
- D. Zwaag, P. A. Pieters, P. A. Korevaar, A. J. Markvoort, A. J. H. Spiering, T. F. A. de Greef and E. W. Meijer, J. Am. Chem. Soc., 2015, 137, 12677–12688 CrossRef PubMed.
- L. Brunsveld, B. J. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4098 CrossRef CAS.
- S. I. Stupp and L. C. Palmer, Chem. Mater., 2014, 26, 507–518 CrossRef CAS.
- J. Matern, Y. Dorca and L. Sánchez, Angew. Chem., Int. Ed., 2019, 58, 16730–16740 CrossRef CAS.
- M. F. J. Mabesoone, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2020, 142, 19781–19798 CrossRef CAS PubMed.
- M. A. J. Gillissen, M. M. E. Koenigs, J. J. H. Spiering, J. A. J. M. Vekmemans, A. R. A. Palmans, I. K. Voets and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 336–343 CrossRef CAS.
- P. Xing, H. Chen, L. Bai, A. Hao and Y. Zhao, ACS Nano, 2016, 10, 2716–2727 CrossRef CAS.
- P. Xing and Y. Zhao, Acc. Chem. Res., 2018, 51, 2324–2334 CrossRef CAS.
- C. Liu, D. Yang, L. Zhang and M. Liu, Soft Matter, 2019, 15, 6557–6563 RSC.
- M. A. Martínez, A. Doncel-Giménez, J. Cerdá, J. Calbo, R. Rodríguez, J. Aragó, J. Crassous, E. Ortí and L. Sánchez, J. Am. Chem. Soc., 2021, 143, 13281–13291 CrossRef.
- K. K. L. Cheuk, J. W. Y. Lam, J. Chen, L. M. Lai and B. Z. Tang, Macromolecules, 2003, 36, 5947–5959 CrossRef CAS.
- K. Okoshi, S. Sakurai, S. Ohsawa, J. Kumaki and E. Yashima, Angew. Chem., Int. Ed., 2006, 45, 8173–8176 CrossRef CAS PubMed.
- S. Sakurai, K. Okoshi, J. Kumaki and E. Yahima, J. Am. Chem. Soc., 2006, 128, 5650–5651 CrossRef CAS.
- S. Sakurai, K. Okoshi, J. Kumaki and E. Yashima, Angew. Chem., 2006, 118, 1267–1270 CrossRef.
- T. Fukushima, K. Takachi and K. Tsuchihara, Macromolecules, 2006, 39, 3103–3105 CrossRef CAS.
- T. Fukushima, H. Kimura and K. Tsuchihara, Macromolecules, 2009, 42, 8619–8626 CrossRef CAS.
- T. Fukushima and K. Tsuchihara, Macromolecules, 2009, 42, 5453–5460 CrossRef CAS.
- K. K. L. Cheuk, J. W. Y. Lam, B. S. Li, Y. Xie and B. Z. Tang, Macromolecules, 2007, 40, 2633–2642 CrossRef CAS.
- K. Maeda, H. Mochizuki, M. Watanabe and E. Yashima, J. Am. Chem. Soc., 2006, 128, 7639–7650 CrossRef CAS PubMed.
- C. Zhang, F. Liu, Q. Geng, S. Zhang, X. Shen, R. Kakuchi, H. Misaka, T. Kakuchi, T. Satoh and R. Sakai, Eur. Polym. J., 2011, 47, 1923–1930 CrossRef CAS.
- H. Zhao, F. Sanda and T. Masuda, Macromol. Chem. Phys., 2005, 206, 1653–1658 CrossRef CAS.
- H. Zhao, F. Sanda, T. J. Masuda, H. Zhao, F. Sanda and T. J. Masuda, Polym. Sci., Part A: Polym. Chem., 2005, 43, 5168–5176 CrossRef CAS.
- F. Sanda, H. Araki and T. Masuda, Macromolecules, 2005, 38, 10605–10608 CrossRef CAS.
- A. Ikeda, K. Terada, M. Shiotsuki and F. Sanda, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3783–3796 CrossRef CAS.
- S. Wang, X. Feng, J. Zhang, P. Yu, Z. Guo, Z. Li and X. Wan, Macromolecules, 2017, 50, 3489–3499 CrossRef CAS.
- T. Yamada, Y. Nagata and M. Suginome, Chem. Commun., 2010, 46, 4914–4916 RSC.
- T. Yamamoto, T. Yamada, Y. Nagata and M. Suginome, J. Am. Chem. Soc., 2010, 132, 7899–7901 CrossRef CAS.
- Y. Nagata, T. Yamada, T. Adachi, Y. Akai, T. Yamamoto and M. Suginome, J. Am. Chem. Soc., 2013, 135, 10104–10113 CrossRef CAS.
- Y. Nagata, T. Nishikawa and M. Suginome, J. Am. Chem. Soc., 2015, 137, 4070–4073 CrossRef CAS PubMed.
- Y. Nagata, K. Takagi and M. Suginome, J. Am. Chem. Soc., 2014, 136, 9858–9861 CrossRef CAS.
- S. Leiras, F. Freire, J. M. Seco, E. Quiñoá and R. Riguera, Chem. Sci., 2013, 4, 2735–2743 RSC.
- S. Leiras, F. Freire, E. Quiñoá and R. Riguera, Chem. Sci., 2015, 6, 246–253 RSC.
- J. J. Tarrío, R. Rodríguez, B. Fernández, E. Quiñoá and F. Freire, Angew. Chem., Int. Ed., 2022, 61, e20211507 Search PubMed.
- J. J. Tarrío, B. Fernández, E. Quiñoá and F. Freire, J. Mater. Chem. C, 2023, 11, 8378–8382 RSC.
- R. Rodríguez, E. Quiñoá, R. Riguera and F. Freire, J. Am. Chem. Soc., 2016, 138, 9620–9628 CrossRef.
- K. Cobos, R. Rodríguez, O. Domarco, B. Fernández, E. Quiñoá, R. Riguera and F. Freire, Macromolecules, 2020, 53, 3182–3193 CrossRef CAS.
- R. Rodríguez, E. Quiñoá, R. Riguera and F. Freire, Small, 2019, 15, 1970070 CrossRef.
- Z. Fernández, B. Fernández, E. Quiñoá and F. Freire, J. Am. Chem. Soc., 2021, 143, 20962–20969 CrossRef PubMed.
- E. Yashima, T. Matsushima and Y. Okamoto, J. Am. Chem. Soc., 1995, 117, 11596–11597 CrossRef CAS.
- K. Morino, N. Watase, K. Maeda and E. Yashima, Chem. – Eur. J., 2004, 10, 4703–4707 CrossRef CAS PubMed.
- K. Morino, K. Maeda and E. Yashima, Macromolecules, 2003, 36, 1480–1486 CrossRef CAS.
- E. Yashima, T. Matsushima and Y. Okamoto, J. Am. Chem. Soc., 1997, 119, 6345–6359 CrossRef CAS.
- R. Nonokawa and E. Yashima, J. Am. Chem. Soc., 2003, 125, 1278–1283 CrossRef CAS PubMed.
- R. Nonokawa, M. Oobo and E. Yashima, Macromolecules, 2003, 36, 6599–6606 CrossRef CAS.
- K. Morino, M. Oobo and E. Yashima, Macromolecules, 2005, 38, 3461–3468 CrossRef CAS.
- R. Sakai, I. Otsuka, T. Satoh, R. Kakuchi, H. Kaga and T. Kakuchi, Macromolecules, 2006, 39, 4032–4037 CrossRef CAS.
- E. Yashima, T. Nimura, T. Matsushima and Y. Okamoto, J. Am. Chem. Soc., 1996, 118, 9800–9801 CrossRef CAS.
- T. Nishimura, K. Tsuchiya, S. Ohsawa, K. Maeda, E. Yashima, Y. Nakamura and J. Nishimura, J. Am. Chem. Soc., 2004, 126, 11711–11717 CrossRef CAS.
- H. Onouchi, T. Miyagawa, A. Furuko, K. Maeda and E. Yashima, J. Am. Chem. Soc., 2005, 127, 2960–2965 CrossRef CAS.
- Y. Nishikawa, D. Hirose, S. Sona and K. Maeda, Chem. Commun., 2023, 59, 8226–8229 RSC.
- H. Onouchi, T. Hasegawa, D. Kashiwagi, H. Ishiguro, K. Maeda and E. Yashima, Macromolecules, 2005, 38, 8625–8633 CrossRef CAS.
- K. Maeda, Y. Takeyama, K. Sakajiri and E. Yashima, J. Am. Chem. Soc., 2004, 126, 16284–16285 CrossRef CAS.
- K. Nagai, K. Maeda, Y. Takeyama, K. Sakajiri and E. Yashima, Macromolecules, 2005, 38, 5444–5451 CrossRef CAS.
- K. Maeda, H. Tsukui, Y. Matsushita and E. Yashima, Macromolecules, 2007, 40, 7721–7726 CrossRef CAS.
- K. Maeda, H. Mochizuki, K. Osato and E. Yashima, Macromolecules, 2011, 44, 3217–3226 CrossRef CAS.
- T. Ikai, M. Ando, M. Ito, R. Ishidate, N. Suzuki, K. Maeda and E. Yashima, J. Am. Chem. Soc., 2021, 143, 12725–12735 CrossRef CAS.
- Y. Hase, M. Ishikawa, R. Muraki, K. Maeda and E. Yashima, Macromolecules, 2006, 39, 6003–6008 CrossRef CAS.
- Y. Hase, K. Nagai, H. Iida, K. Maeda, N. Ochi, K. Sawabe, K. Sakajiri, K. Okoshi and E. Yashima, J. Am. Chem. Soc., 2009, 131, 10719–10732 CrossRef CAS.
- E. Yashima, K. Maeda and T. Yamanaka, J. Am. Chem. Soc., 2000, 122, 7813–7814 CrossRef CAS.
- K. Maeda, K. Kuroyanagi, S. I. Sakurai, T. Yamanaka and E. Yashima, Macromolecules, 2011, 44, 2457–2464 CrossRef CAS.
- K. Maeda, M. Nozaki, K. Hashimoto, K. Shimomura, D. Hirose, T. Nishimura, G. Watanabe and E. Yashima, J. Am. Chem. Soc., 2020, 142, 7668–7682 CrossRef CAS PubMed.
- W. Makiguchi, S. Kobayashi, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2013, 52, 5275–5279 CrossRef CAS PubMed.
- K. Maeda and E. Yashima, Bull. Chem. Soc. Jpn., 2021, 94, 2637–2661 CrossRef.
- N. Ousaka, F. Mamiya, Y. Iwata, K. Nishimura and E. Yashima, Angew. Chem., Int. Ed., 2017, 56, 791–795 CrossRef CAS PubMed.
- T. Ikai, S. Kawabata, F. Mamiya, D. Taura, N. Ousaka and E. Yashima, J. Am. Chem. Soc., 2020, 142, 21913–21925 CrossRef CAS.
- D. Lee, Y. J. Jin, H. Kim, N. Suzuki, M. Fujiki, T. Sakaguchi, S. K. Kim, W. E. Lee and G. Kwak, Macromolecules, 2012, 45, 5379–5386 CrossRef CAS.
- T. Miao, X. Cheng, H. Ma, Z. He, Z. Zhang, N. Zhou, W. Zhang and X. Zhu, Angew. Chem., Int. Ed., 2021, 60, 18566–18571 CrossRef CAS PubMed.
- J. Liu, Y. Zhao, H. Chen, Z. Zhang, W. Zhang and X. Zhu, React. Funct. Polym., 2017, 121, 76–81 CrossRef CAS.
- Y. Zhao, L. Yin, J. Liu, H. Chen and W. Zhang, Chirality, 2017, 29, 107–114 CrossRef CAS.
- G. Zhang, Y. Bao, M. Pan, N. Wang, X. Cheng and W. Zhang, Sci. China: Chem., 2023, 66, 1169–1178 CrossRef CAS.
- Y. Nagata, R. Takeda and M. Suginome, ACS Cent. Sci., 2019, 5, 1235–1240 CrossRef CAS PubMed.
- J. L. Greenfield, E. W. Evans, D. Di Nuzzo, M. Di Antonio, R. H. Friend and J. R. Nitschke, J. Am. Chem. Soc., 2018, 140, 10344–10353 CrossRef.
- S. Xue, P. Xing, J. Zhang, Y. Zeng and Y. Zhao, Chem. – Eur. J., 2019, 25, 7426–7437 CrossRef CAS PubMed.
- M. L. Ślęczkowski, M. F. J. Mabesoone, P. Ślęczkowski, A. R. A. Palmans and E. W. Meijer, Nat. Chem., 2021, 13, 200–207 CrossRef.
- A. K. Mondal, M. D. Preuss, M. L. Ślȩczkowski, T. K. Das, G. Vantomme, E. W. Meijer and R. Naaman, J. Am. Chem. Soc., 2021, 143, 7189–7195 CrossRef CAS PubMed.
- S. J. George, Ž. Tomović, A. P. H. J. Schenning and E. W. Meijer, Chem. Commun., 2011, 47, 3451–3453 RSC.
- B. Isare, M. Linares, L. Zargarian, S. Fermandjian, M. Miura, S. Motohashi, N. Vanthuyne, R. Lazzaroni and L. Bouteiller, Chem. – Eur. J., 2010, 16, 173–177 CrossRef CAS.
- I. Destoop, E. Ghijsens, K. Katayama, K. Tahara, K. S. Mali, Y. Tobe and S. De Feyter, J. Am. Chem. Soc., 2012, 134, 19568–19571 CrossRef CAS PubMed.
- S. Dhiman, A. Jain and S. J. George, Angew. Chem., Int. Ed., 2017, 56, 1329–1333 CrossRef CAS PubMed.
- S. Dhiman, A. Jain, M. Kuma and S. J. George, J. Am. Chem. Soc., 2017, 139, 16568–16575 CrossRef CAS PubMed.
- N. Ousaka, Y. Inai and R. Kuroda, J. Am. Chem. Soc., 2008, 130, 12266–12267 CrossRef CAS.
- N. Ousaka and Y. Inai, J. Am. Chem. Soc., 2006, 128, 14736–14737 CrossRef CAS.
- T. Sanji, K. Takase and H. Sukaria, J. Am. Chem. Soc., 2001, 123, 12690 CrossRef CAS PubMed.
- B. A. F. Le Bailly, L. Byrne and J. Clayden, Angew. Chem., Int. Ed., 2016, 55, 2132–2136 CrossRef CAS.
- J. Brioche, S. J. Pike, S. Tshepelevitsh, I. Leito, G. A. Morris, S. J. Webb and J. Clayden, J. Am. Chem. Soc., 2015, 137, 6680–6691 CrossRef CAS.
- R. A. Brown, V. Diemer, S. J. Webb and J. Clayden, Nat. Chem., 2013, 5, 853–860 CrossRef CAS.
- S. Saha, B. Kauffmann, Y. Ferrand and I. Huc, Angew. Chem., Int. Ed., 2018, 57, 13542–13546 CrossRef CAS.
- P. Mateus, B. Wicher, Y. Ferrand and I. Huc, Chem. Commun., 2018, 54, 5078–5081 RSC.
- M. Vallade, P. Sai Reddy, L. Fischer and I. Huc, Eur. J. Org. Chem., 2018, 5489–5498 CrossRef CAS.
- P. Mateus, N. Chandramouli, C. D. Mackereth, B. Kauffmann, Y. Ferrand and I. Huc, Angew. Chem., Int. Ed., 2020, 59, 5797–5805 CrossRef CAS.
- Y. Ferrand, A. M. Kendhale, B. Kauffmann, A. Grélard, C. Marie, V. Blot, M. Pipelier, D. Dubreuil and I. Huc, J. Am. Chem. Soc., 2010, 132, 7858–7859 CrossRef CAS.
- J. L. Hou, X.-B. Shao, G. J. Chen, Y. X. Zhou, X. K. Jiang and Z. T. Li, J. Am. Chem. Soc., 2004, 126, 12386 CrossRef CAS.
- C. Li, G. T. Wang, H. P. Yi, X. K. Jiang, Z. T. Li and R. X. Wang, Org. Lett., 2007, 9, 1797 CrossRef CAS.
- M. Inouye, M. Waki and H. Abe, J. Am. Chem. Soc., 2004, 126, 2022–2027 CrossRef CAS PubMed.
- J. C. Nelson, J. G. Saven, J. S. Moore and P. G. Wolynes, Science, 1997, 277, 1793–1796 CrossRef CAS PubMed.
- R. B. Prince, S. A. Barnes and J. S. Moore, J. Am. Chem. Soc., 2000, 122, 2758–2762 CrossRef CAS.
- M. T. Stone and J. S. Moore, Org. Lett., 2004, 6, 469–472 CrossRef CAS PubMed.
- A. Tanatani, M. J. Mio and J. S. Moore, J. Am. Chem. Soc., 2001, 123, 1792–1793 CrossRef CAS PubMed.
- A. Tanatani, T. S. Hughes and J. S. Moore, Angew. Chem., Int. Ed., 2002, 41, 325–328 CrossRef CAS.
- H. Abe, N. Masuda, M. Waki and M. Inouye, J. Am. Chem. Soc., 2005, 127, 16189–16196 CrossRef CAS PubMed.
- H. Abe, H. Machiguchi, S. Matsumoto and M. Inouye, J. Org. Chem., 2008, 73, 4650–4661 CrossRef CAS PubMed.
- Q. Gan, Y. Ferrand, C. Bao, B. Kauffmann, A. Grélard, H. Jiang and I. Huc, Science, 2011, 331, 1172–1175 CrossRef CAS PubMed.
- A. Kousar, J. Liu, N. Mehwish, F. Wang, A. Y. Dang-i and C. Feng, Mater. Today Chem., 2019, 11, 217–224 CrossRef CAS.
- S. Goskulwad, D. D. La, M. A. Kobaisi, S. V. Bhosale, V. Bansal, A. Vinu, K. Ariga and S. V. Bhosale, Sci. Rep., 2018, 8, 11220 CrossRef PubMed.
- M. Pandeeswar and T. Govindaraju, Mol. Syst. Des. Eng., 2016, 1, 202–207 RSC.
- H. Frisch, Y. Nie, S. Raunser and P. Besenius, Chem. – Eur. J., 2015, 21, 3304–3309 CrossRef CAS.
- J. Liu, Z. Luo, L. Yu, P. Zhang, H. Wei and Y. Yu, Chem. Sci., 2020, 11, 8224–8230 RSC.
- E. Suárez-Picado, E. Quiñoá, R. Riguera and F. Freire, Chem. Mater., 2018, 30, 6908–6914 CrossRef.
- R. Liu, F. Sanda and T. Masuda, Macromol. Chem. Phys., 2009, 210, 331–339 CrossRef CAS.
- L. Wu, Y. Xu, T. Hou, J. Jia, X. Huang, G. Weng, S. Bao and L. Zheng, Chem. – Eur. J., 2021, 27, 16722–16734 CrossRef CAS.
- J. Yuan, Y. Zhang, Y. Sun, Z. Cai, L. Yang and H. Lu, Biomacromolecules, 2018, 19, 2089–2097 CrossRef CAS PubMed.
- R. Barman, P. Dey, T. Mondal and S. Ghosh, Chem. – Asian J., 2019, 14, 4741–4747 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |