
Heterometallic cages: synthesis and applications
Lana K.
Moree
 ab,
Logan A. V.
Faulkner
ab and
James D.
Crowley
*ab
ab,
Logan A. V.
Faulkner
ab and
James D.
Crowley
*ab
aDepartment of Chemistry, University of Otago, PO Box 56, Dunedin 9054, New Zealand. E-mail: jcrowley@chemistry.otago.ac.nz
bMacDiarmid Institute for Advanced Materials and Nanotechnology, Wellington 6140, New Zealand
First published on 1st December 2023
Abstract
High symmetry metallosupramolecular architectures (MSAs) have been exploited for a range of applications including molecular recognition, catalysis and drug delivery. Recently there have been increasing efforts to enhance those applications by generating reduced symmetry MSAs. While there are several emerging methods for generating lower symmetry MSAs, this tutorial review examines the general methods used for synthesizing heterometallic MSAs with a particular focus on heterometallic cages. Additionally, the intrinsic properties of the cages and their potential emerging applications as host–guest systems and reaction catalysts are described.
 Lana K. Moree | Lana is completing a Master's degree at the University of Otago under the supervision of James Crowley, synthesising Pd(II)-based cages for catalysis and drug delivery. She is interested in developing new catalysts for the Suzuki–Miyaura cross coupling reaction. |
 Logan A. V. Faulkner | Logan is completing a Master's degree at the University of Otago under the supervision of James Crowley, working on synthesizing catenanes and rotaxanes for antibacterial testing. He is interested in self-assembly and the development of supramolecular machines. |
 James D. Crowley | James Crowley obtained his BSc(Hons) and MSc from Victoria University of Wellington and completed his PhD at the University of Chicago under the direction of Prof. Brice Bosnich. In 2005 he moved to Prof. David Leigh's group at the University of Edinburgh, where he was awarded a British Ramsay Memorial Trust Fellowship (2006–2008), to carry out research on molecular machines. He started his independent career at the University of Otago in 2008 and has moved through the ranks to Professor (2019). His major research interests are in catalysis, the self-assembly of metallocages, molecular recognition, and the development of switchable molecular materials. |
Key learning points(1) The subcomponent self-assembly, symmetry interaction and directional bonding approaches may be utilised in the formation of heterometallic supramolecular cages.(2) The unique properties imbued on these heterometallic cages via the metal centres enable many potential applications such as spin crossover (SCO) and light-harvesting. (3) Guest binding within the cavities of heterometallic cages offers further applications in molecular recognition and catalysis. |
Introduction
Nature exploits self-assembly to generate a wide variety of sophisticated functional (supra)molecular architectures. Natural systems exploit non-covalent/supramolecular interactions such as hydrogen bonding, π–π interactions, dispersion/van der Waals forces and solvophobic effects in order to generate these large complex architectures efficiently. Inspired by the natural systems, chemists have used these same non-covalent forces to synthesise large complex supramolecular architectures.1 In addition to this fully biomimetic approach, coordination chemists have been using labile metal ions and polytopic linker ligands to construct large (metallo)supramolecular assemblies under mostly thermodynamic control for almost thirty years. These metal–ligand assemblies have been called metal–organic cages (MOCs),2–8 metal–organic polygons or polyhedra (MOPs),9–11 coordination cages (CCs),5 supramolecular coordination complexes (SCC)12,13 amongst others. Herein, we will refer to these types of assemblies as metallosupramolecular architectures (MSAs)14–18 as this older name not only includes cages but also metallomacrocycles, helicates and grid structures assembled from appropriately designed linker ligands and metal ions. There are three general methods used for the formation of these MSAs: (1) the directional bonding (or ligand directed or molecular library)19,20 approach (subsets of this method include the molecular panelling21 and molecular clip22,23 methods), (2) the symmetry interaction24 method and (3) the weak-link approach.20 Overarching these three general approaches is the sub-component self-assembly (SCSA) method,25 where the linker ligands are also self-assembled from simple building blocks using dynamic covalent chemistry. The SCSA method can potentially be applied to all of the aforementioned procedures. These versatile general methods coupled to the broad range of metal ions and linker ligands have enabled an expansive set of MSAs to be synthesised and studied. These systems have been examined for their new and emerging biological,26–28 electronic,29–31 redox32,33 and photophysical34–36 properties. Additionally, the majority of these architectures inherently contain molecular cavities, which have been exploited for their molecular recognition properties. Cavity containing MSAs have been used to encapsulate environmental pollutants37,38 and reactive species39–41 while others have been leveraged for catalytic,42–46 photochemical and electrochemical sensing, storage and drug delivery47–50 applications.While these efforts have been extremely impressive, they have for the most part been achieved with high symmetry MSAs. Nature certainly generates and uses high symmetry self-assembled constructs (for example the iron storage protein Ferritin or virus capsids). However, many of the sophisticated biological architectures are lower symmetry and this is key to their function and selectivity. This is particularly true of enzyme active sites that many of the applications of MSAs are attempting to mimic. As such, in the past ten years there have been increasing efforts to develop synthetic approaches that afford lower symmetry MSAs. One approach exploits low/reduced symmetry linker ligands to generate homometallic MSAs. While this method can lead to a mixture of isomers it is widely applied and has been generally successful.51–53 A second method to lower symmetry metallo-constructs is the heterometallic approach.
In this tutorial review we give an overview of the different approaches to heterometallic MSAs with a particular focus on heterometallic cages (i.e. systems that feature a cavity). In addition, we examine some of the properties and applications of these heterometallic assemblies. The area has been examined before by others54–57 and as such the current tutorial review makes no attempt to be comprehensive, rather we illustrate the general principles of the field and highlight more recent advances and applications.
Generic design principles for the synthesis of heterometallic metallosupramolecular architectures and cages
The general design strategies used for the synthesis of heterometallic MSAs are essentially the same as those used for homometallic systems, but they need to include methods for inserting two (or more) different metal ions into the assemblies (Fig. 1). The simplest approach to heterometallic MSAs uses lower symmetry linker ligands with two (or more) different metal binding sites that can selectively interact with different metal ions (Fig. 1a) to generate the assembly under thermodynamic control. This can be commonly achieved in two ways: (1) design a ligand that features both hard and soft donor sites that can be used to selectively interact with hard and soft metal ions,58,59 and (2) generate a low symmetry ligand that contains metal binding sites of different denticity and exploit the coordination geometry preferences of certain metal ions to drive the site specific complexation.60–62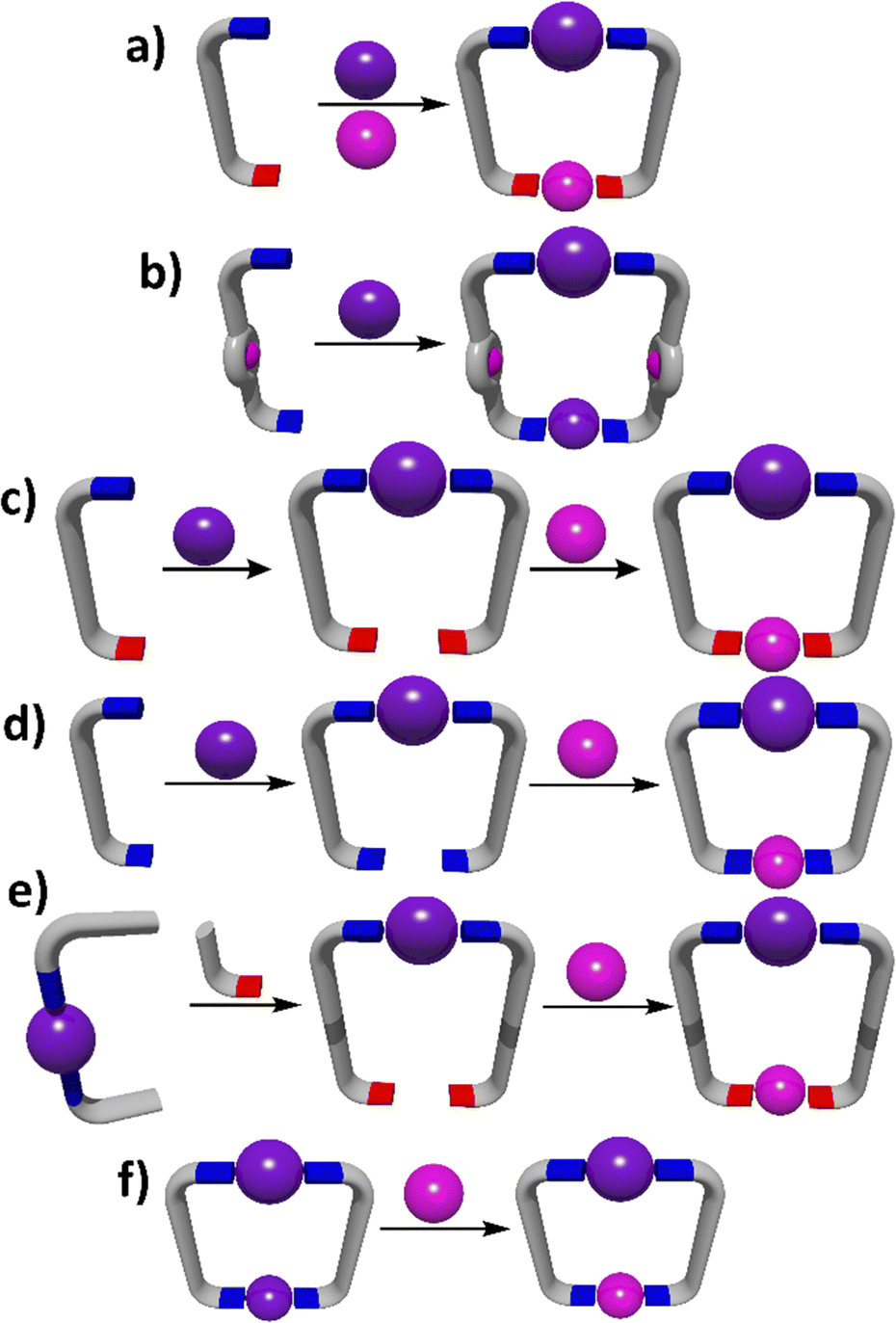 | ||
| Fig. 1 Cartoon diagrams showing the generic synthetic strategies to the assembly of heterometallic MSAs. The examples shown are all metallo-macrocycles. However, these same strategies can be applied to generate a wide range of MSAs including heterometallic cages. | ||
While the fully thermodynamic self-assembly approaches mentioned above have been used it is far more common to exploit step-wise metallo-ligand strategies to synthesise heterometallic MSAs (Fig. 1b–f). These approaches generally insert a kinetically inert metal ion into the linker ligand architecture in an initial step and then a more labile metal ion is used to cyclise/complete the MSAs. Most early work exploited symmetric ligands that featured macrocyclic (porphyrins) or organometallic (ferro/metallocenes) metal binding pockets (Fig. 1b). More recently, metallo-ligands have been synthesised by mono-metalating either symmetric or lower symmetry ligands with a relatively inert metal ion and then further reacting the resulting metallo-ligands with a second more labile metal ion to obtain heterometallic MSAs (Fig. 1c and d).63–67
Finally, heterometallic systems can also be generated using post-assembly modification (Fig. 1e and f). In one approach a pre-formed metal complex can be modified by the addition of a new metal binding pocket and then a labile metal ion added to complete the structure (Fig. 1e).68,69 In an alternative post-assembly modification method a homometallic MSA can be transmetalated to generate a heterometallic system. While this approach is commonly used with metal–organic frameworks (MOFs),70,71 it remains rare in discrete MSAs (Fig. 1f). These general design principles for heterometallic architectures, as illustrated in Fig. 1, can be applied across the four assembly strategies (directional bonding, symmetry interaction, weak link and SCSA approaches) that will be described in the next sections. We will examine specific examples of heterometallic cage systems generated using these methods with a focus on materials that have been shown to have other useful properties.
Subcomponent self-assembly approaches to heterometallic cages
SCSA is a powerful method which has been utilised for the formation of MSAs (Fig. 2).25 Subcomponent self-assembly often exploits the formation of dynamic-covalent (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and coordinative (ligand → metal) bonds during a ‘one pot’ synthesis.57,72 The metal ions used therefore are also selected based on a variety of factors, such as coordination geometry and kinetic lability, which drive the selectivity of the assembly process.24 When these smaller, simple building blocks come together in one pot, they generate much larger and increasingly complex structures.73 Traditionally, SCSA has been used to generate a variety of symmetrical and homometallic architectures and cages.56 However, due to increasing interest in more complex, lower symmetry heterometallic architectures, a variety of SCSA methods have been explored for the formation of new heterometallic cages.72
C) and coordinative (ligand → metal) bonds during a ‘one pot’ synthesis.57,72 The metal ions used therefore are also selected based on a variety of factors, such as coordination geometry and kinetic lability, which drive the selectivity of the assembly process.24 When these smaller, simple building blocks come together in one pot, they generate much larger and increasingly complex structures.73 Traditionally, SCSA has been used to generate a variety of symmetrical and homometallic architectures and cages.56 However, due to increasing interest in more complex, lower symmetry heterometallic architectures, a variety of SCSA methods have been explored for the formation of new heterometallic cages.72
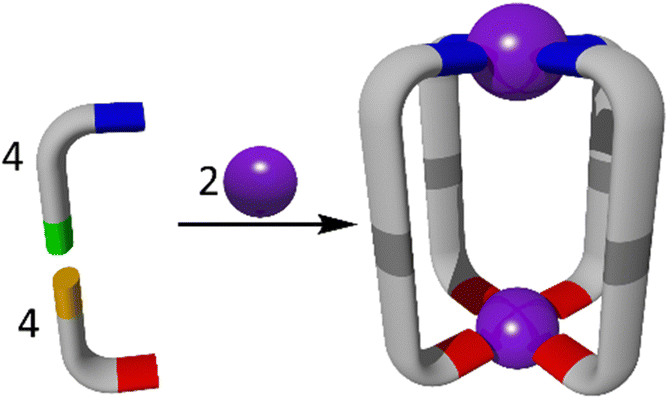 | ||
| Fig. 2 A cartoon representation of the sub-component self-assembly (SCSA) method. The example shows the assembly of an M2L4 cage system from two metal ions and two different ligand sub-components. The ligands (L) are assembled in situ via dynamic-covalent reactions. While the example provided is a homometallic M2L4 cage, similar strategies can be applied to generate a wide range of MSAs. | ||
By combining the heterometallic methods illustrated in Fig. 1 with SCSA (Fig. 2) a range of functional heterometallic cages have been generated. Homometallic [Pt2L4]4+ and [Pd2L4]4+ systems have been extensively studied.74–76 In 2020, work by Crowley and co-workers synthesized the first [PdPtL4]4+ heterometallic cage.77 Platinum(II) is a relatively inert metal ion, so it was first heated in the presence of 3-pyridylcarboxyaldehyde (Pyald) and AgBF4 to generate an inert [Pt(Pyald)4]2+ building block.78 This was then combined with [Pd(CH3CN)4](BF4)2 and an amine containing half-ligand (L1) which also featured a pyridine site able to coordinate to Pd(II) ions (Scheme 1). Under SCSA conditions the [Pt(Pyald)4]2+ complex underwent four imine condensation reactions with L1, and Pd(II) coordinated to the pyridyl units of L1′ resulting in the self-assembly of the [PdPtL1′4]4+ cage. The authors also showed the heterometallic cage was switchable and could be reversibly opened and closed, even in the presence of a guest (Scheme 1) when exposed to a chemical stimulus.
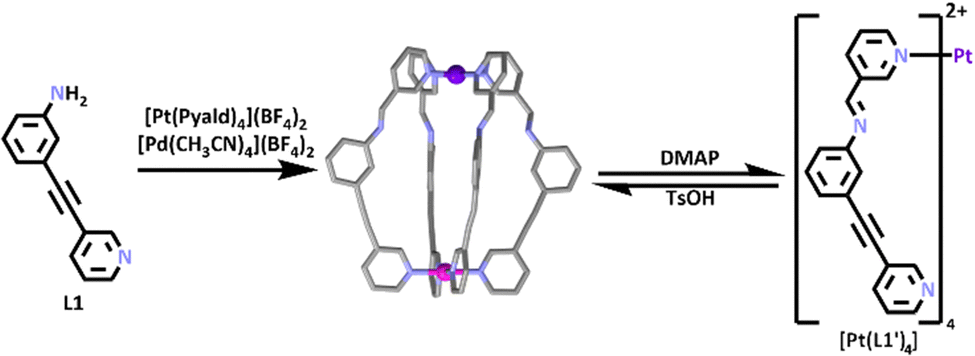 | ||
| Scheme 1 Subcomponent self-assembly of the [PdPtL1'4]4+ cage, which could open and close using DMAP and TsOH respectively. Element colours: carbon (black in chemdraws, grey in crystal structure), nitrogen (lavender), palladium (magenta), platinum (purple). Solvent molecules, counter anions and hydrogen atoms removed for clarity. | ||
Host–guest (HG) chemistry is an important potential application for cage structures,79 and it is extensively used in biology, with enzymes playing a key role in reversibly binding substrates in order to catalyse biological processes.80 The guest binding of this heterometallic [PdPtL1′4]4+ cage was examined and Crowley and co-workers showed that the cage was able to interact with benzoquinone (BQ) and also 2,6-diaminoanthraquinone (DAQ). Fitting the titration data to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding model using Bindfit,81,82 gave a binding constant K of 23 ± 0.8 M−1 for the [PdPtL1′4⊂BQ]4+ interaction and 2900 ± 300 M−1 for [PdPtL1′4⊂DAQ]4+. The 1:1 stoichiometry of the HG complex was confirmed by electrospray ionization mass spectrometry (ESI-MS) and X-ray crystallography (Fig. 3). While this approach provided the first example of a [PdPtL4]4+ heterometallic cage, the use of imine chemistry limited the functionality of the cage because it was not stable in water or aqueous mixtures. The same authors have gone on to show that a second-generation system featuring a hydrazone linker is more robust in aqueous mixtures. Additionally, they examined the in vitro antiproliferative activity of both heterometallic cages against a series of cancer cell lines. While the cages were active (IC50 = 25–35 μM) they were less effective than the well-known metallodrugs cisplatin and oxaliplatin.83
:1 binding model using Bindfit,81,82 gave a binding constant K of 23 ± 0.8 M−1 for the [PdPtL1′4⊂BQ]4+ interaction and 2900 ± 300 M−1 for [PdPtL1′4⊂DAQ]4+. The 1:1 stoichiometry of the HG complex was confirmed by electrospray ionization mass spectrometry (ESI-MS) and X-ray crystallography (Fig. 3). While this approach provided the first example of a [PdPtL4]4+ heterometallic cage, the use of imine chemistry limited the functionality of the cage because it was not stable in water or aqueous mixtures. The same authors have gone on to show that a second-generation system featuring a hydrazone linker is more robust in aqueous mixtures. Additionally, they examined the in vitro antiproliferative activity of both heterometallic cages against a series of cancer cell lines. While the cages were active (IC50 = 25–35 μM) they were less effective than the well-known metallodrugs cisplatin and oxaliplatin.83
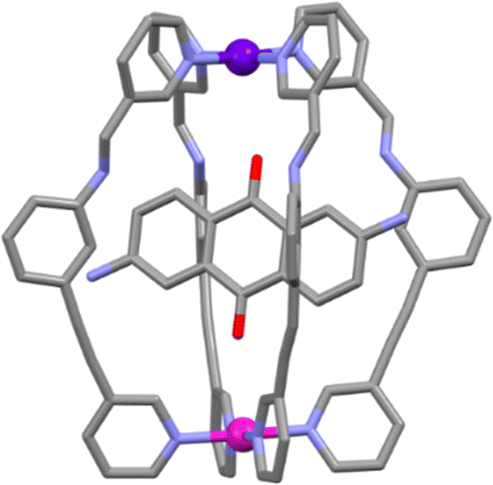 | ||
| Fig. 3 Molecular structure of the [PdPtL1'4⊂DAQ]4+ host–guest adduct. Colours: carbon (grey), nitrogen (lavender), oxygen (red), palladium (magenta), platinum (purple). Solvent molecules, counter anions and hydrogen atoms removed for clarity. | ||
More recently, this approach has been extended to generate larger, more complex heterometallic cages. While homometallic Pd(II) containing cages with multiple discrete cavities have been generated previously,48,84–88 Crowley and co-workers synthesised the first heterometallic [PdPt2L4]6+ and [Pd2PtL4]6+ double cavity systems.89 These cages were generated using a SCSA approach similar to the method that provided the parent [PdPtL1′4]4+ cage. A preformed ligand (L2) which contained a pyridine binding site and two free terminal amines, was reacted with [Pt(Pyald)4]2+ and [Pd(CH3CN)4](BF4)2 in a one pot synthesis, resulting in the formation of the [Pd2PtL2′4]6+ double cavity structure. A second ligand (L3) which consisted of two pyridine groups and one terminal amine was reacted with [Pt(Pyald)4]2+ and [Pd(CH3CN)4](BF4)2, forming the cage [PdPt2L3′4]6+ (Scheme 2).
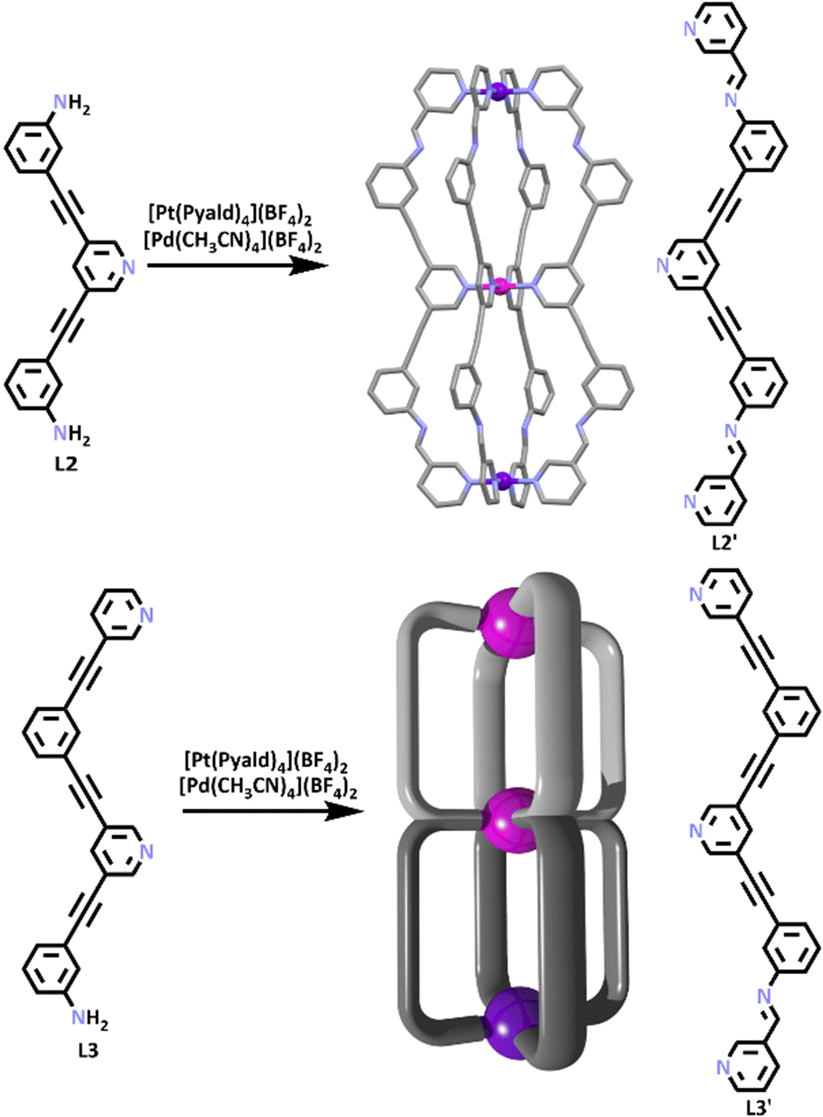 | ||
| Scheme 2 Molecular structure and cartoon model of the [PdPt2L2′4]6+ and [Pd2PtL3′4]6+ double cavity cages. Colours: carbon (black in the chemical structures, grey in the molecular structure), nitrogen (lavender), palladium (magenta), platinum (purple). Solvent molecules, counter ions and hydrogen atoms removed for clarity. | ||
Given the previous success that Crowley and co-workers had binding DAQ within the single cavity cage, DAQ was then used again in guest binding studies with the two double cavity architectures. The 1H nuclear magnetic resonance (NMR) spectra showed broad signals but also demonstrated multiple complexation-induced shifts, which indicated guest binding between DAQ and [PdPt2L2′4]6+ and [Pd2PtL3′4]6+ respectively. ESI-MS data obtained from the host–guest mixtures suggested the formation of a 1:2 host–guest adduct (peaks consistent with both 1:1 and 1:2 host–guest species were observed). The molecular structure of the HG adduct was determined using X-ray crystallography data (Fig. 4) showing the presence of one DAQ molecule in each cavity. The authors then examined the ability of the cages to bind other guests. [PdPt2L2′4]6+ was shown to bind two molecules of 5-fluorouracil (5-FU) in a statistical (non-cooperative) manner. In contrast, [Pd2PtL3′4]6 was found to interact with two different guests, the anti-cancer drugs 5-FU and cisplatin, selectively. The cage was shown to bind two molecules of cisplatin in the Pd–Pd cavity, and one molecule of 5-FU in the Pd–Pt cavity.
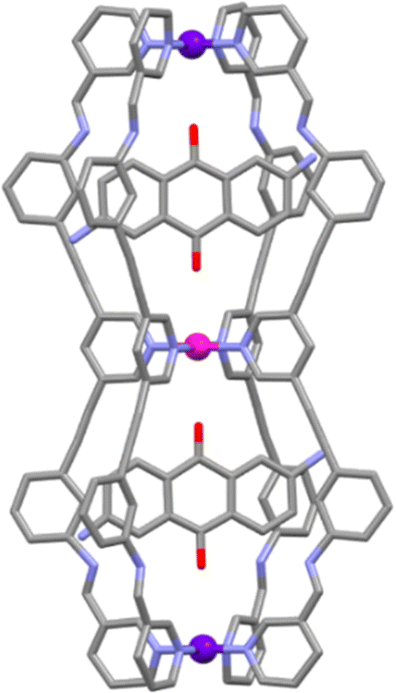 | ||
| Fig. 4 Molecular structure of the [PdPt2L2'4⊂(DAQ)2]6+ host–guest adduct. Colours: carbon (grey), nitrogen (lavender), palladium (magenta), platinum (purple). Solvent molecules, counter anions and hydrogen atoms removed for clarity. | ||
Recent work from the Nitschke group has also utilised the SCSA approach to build two new heterometallic [Cd4Cu4L4]12+ cages (Scheme 3).90 These cages were synthesized overnight by combining [Cd(CH3CN)4](ClO4)2, Cu(ClO4), 3-(diphenylphospino)benzaldehyde (L4) and the tri-amine ligand (L5). These components were combined in a one pot synthesis, in which an imine condensation occurred between the aldehyde of L4 and each of the amines of L5, generating a larger ligand (L4–L5) which contained both nitrogen and phosphorus donor atoms. This ligand then assembled with Cd(II) and Cu(I) ions. This can be explained by the hard and soft acids and bases theory (HSAB). HSAB states that soft acids will react faster and form stronger bonds with soft bases, while hard acids will react faster and form stronger bonds with hard bases.91 In this case, the soft Cu(I) ions bound to the soft phosphorus donors, while the hard Cd(II) ions interacted with the imine and pyridine nitrogen donors.
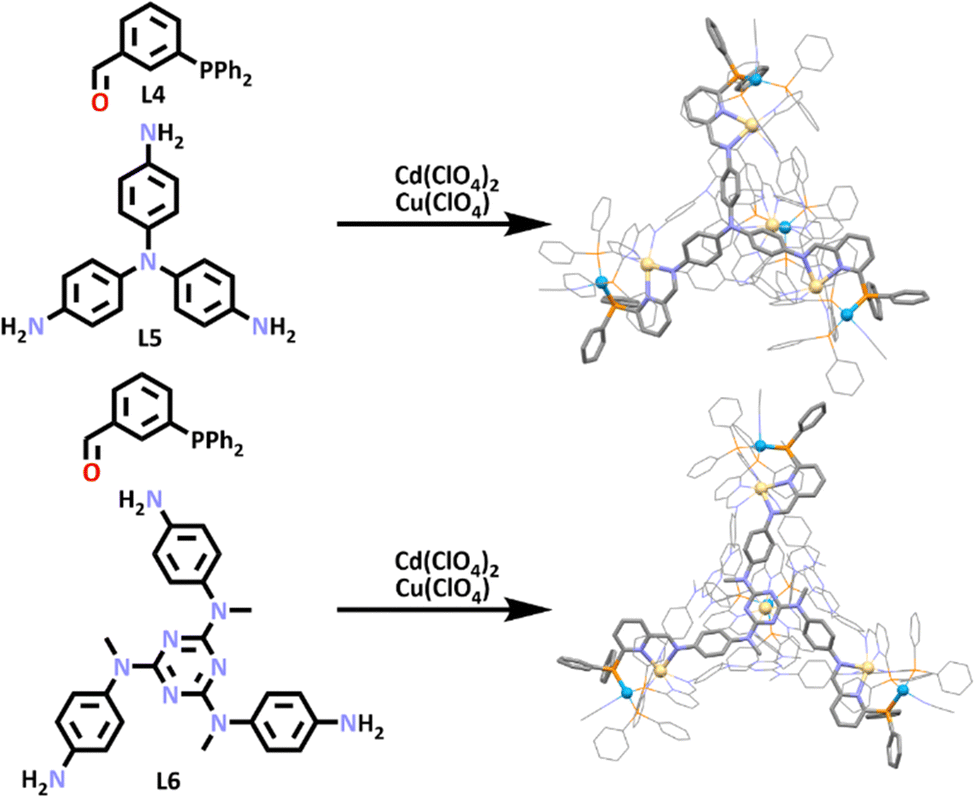 | ||
| Scheme 3 SCSA of two new Cd4Cu4 cages. Colours: carbon (grey), nitrogen (lavender), phosphorus (orange), cadmium (beige), copper (blue). Solvent molecules, counter anions and hydrogen atoms removed for clarity. | ||
The authors then used the slightly more complex triamine subcomponent (L6), which had been shown to form large [M4L4]8+ tetrahedra.92,93 Exploitation of this larger subcomponent enabled the generation of a larger heterometallic cage, formed in the same way as the first cage. Molecular structures of both cages were obtained using X-ray crystallography (Scheme 3), which confirmed the pyramid-like cage structure of these heterometallic architectures.
In other recent work on heterometallic cages from the Nitschke group, SCSA was utilised in the formation of another tetrahedron shaped heterometallic cage.94 In this system a lanthanum ion acted as a template, which 3 equivalents of the ligand (L7) coordinated around. Due to their close proximity to each other, these ligands then underwent a series of imine condensations, resulting in the reversible formation of a macrocycle with three pyridyl triazole arms which coordinated to the zinc(II) ions, generating the tetrahedron. The structure of the [La4Zn4L712]20+ tetrahedron was confirmed by 1H NMR spectroscopy, which showed a single imine peak present, which is consistent with T point symmetry. Diffusion ordered spectroscopy (DOSY) NMR spectroscopy showed a single diffusion constant (3.2 × 10−10 m2 s−1), which was consistent with related structures of similar sizes. ESI-MS data displayed peaks consistent with cage formulation and the molecular structure was also obtained (via X-ray crystallography), confirming the formation of the tetrahedral heterometallic cage (Scheme 4).
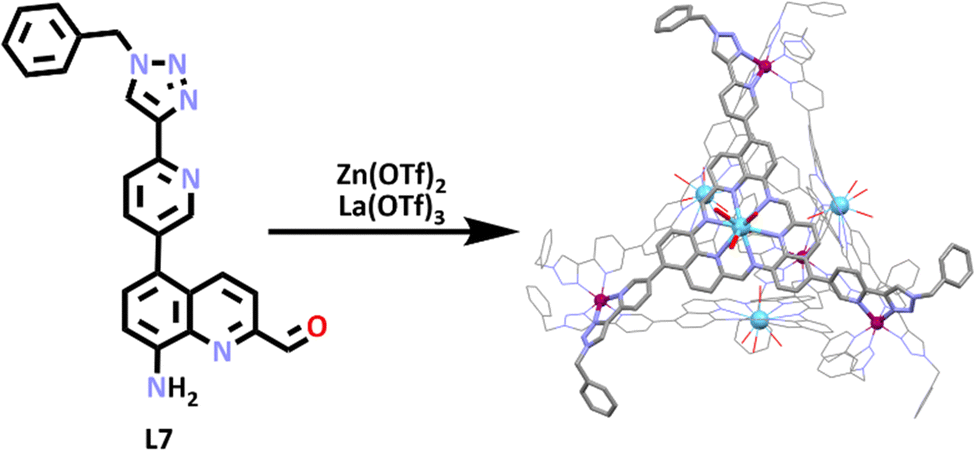 | ||
| Scheme 4 SCSA of a lanthanum–zinc heterometallic cage. Colours: carbon (grey), nitrogen (lavender), oxygen (red), zinc (mulberry), lanthanum (light blue). Solvent molecules, counter anions and hydrogen atoms removed for clarity. | ||
Host guest titrations were monitored by 1H NMR spectroscopy, and were conducted by adding [ReO4]−, [ClO4]− and [OTf]− as tetrabutylammonium (TBA) salts into solutions of [La4Zn4L712]20+ respectively. [ReO4]− was found to have the strongest interaction with the cage. The binding constants (K) were calculated using the Hill equation.95 The binding constant for [La4Zn4L712⊂ReO4]19+ was found to be (8.88 ± 0.01) × 103 M−1. The fluorescence emission of the cages was also examined. The ligand L7 exhibited fluorescent emission centred at 622 nm. When the cage formed, the intensity of the peak was enhanced and had a blue shift of 68 nm. Upon the titration of [ReO4]− into a solution of the cage, the fluorescence intensity was progressively diminished, reaching a plateau after 16 equivalents of [ReO4]− were added.
Lützen and co-workers exploited a Pt(II) symmetric metalloligand previously designed by the Nitschke group96 to generate a series of heterometallic cubes.96,97 Four equivalents of 4-(4′-pyridyl)aniline were reacted with PtCl2 in the presence of AgBF4, resulting in the formation of the square planar Pt(II) metallo-ligand (L8).96,97 This ligand was separately reacted with a series of heterocyclic aldehydes and either Fe(II) or Zn(II) providing a family of three new [Pd6Fe8L86]28+ and three [Pd6Zn8L86]28+ cages.98 The structures of these cube complexes were confirmed by 1H NMR and DOSY spectroscopy and ESI-MS data. X-ray crystallography was used to further characterise all three [Pt6Fe8L8]28+ cages and one [Pt6Zn8L8]28+ cage (Scheme 5), and confirmed the heterometallic cube shape of these MSAs.
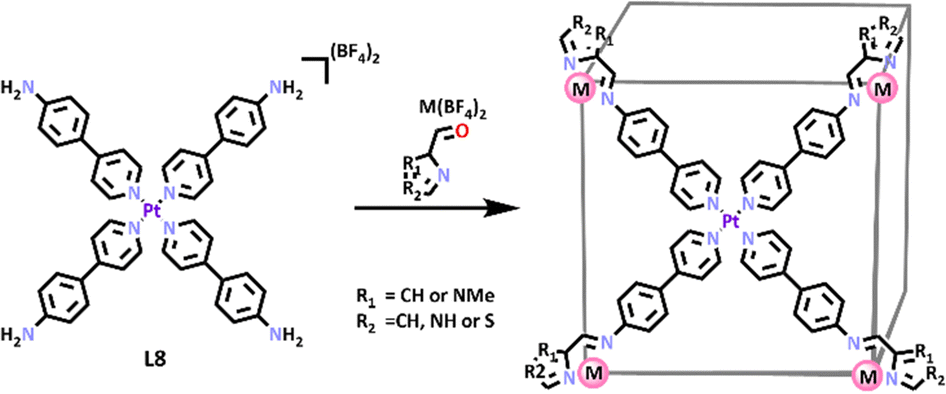 | ||
| Scheme 5 SCSA of heterometallic cuboid cages. Colours: carbon (black), nitrogen (lavender), oxygen (red), phosphorus (orange), zinc (mulberry), platinum (purple), iron (rust). | ||
These cuboid cages have interesting potential applications as molecular switches based on the spin crossover (SCO) phenomenon. Spin crossover is the transition of 3d metal ions (d4–d7) between the low spin (LS, maximum number of paired electrons) and high spin (HS, maximum number of unpaired electrons) states.99 Most 3d metal complexes exist in one state or the other, only when the ligand field strength is ‘just right’ can SCO occur.100 The most common way to induce SCO is by altering the temperature.101 Spin crossover materials are of particular interest because they have potential applications in information storage, sensors, electronic switches, and display devices.102 The [Pt6Fe8L8]28+ cubes showed spin transition temperatures T1/2 of 215 for R1 = NH, R2 = CH, 281 K for R1 = CH, R2 = NMe, and 324 K for R1 = S, R2 = CH. There are also possible future applications investigating the host–guest potential of these heterometallic cubes.
Heterometallic cages generated using the symmetry interaction approach
The symmetry interaction approach is commonly used in the synthesis of supramolecular cage structures (Fig. 5).103 This method, defined by Raymond and co-workers, describes how naked metal ions with preferred coordination numbers and geometries will form complexes with pre-designed ligands to predictably form supramolecular structures based on the symmetries of the components.24 Often for heterometallic cages, this first involves the formation of a relatively inert metallo-ligand complex before addition of the second more labile metal to complete the assembly of the cage structure.59,104–114 Use of metallo-ligands in place of organic ligands potentially provides the resulting structures advantages such as new functionality, flexible geometric control and the ability to assemble many components into a discrete entity.114 Exploiting the symmetry interaction approach outlined in Fig. 5 in conjunction with the heterometallic methods illustrated in Fig. 1 has enabled the synthesis of a variety of functional heterometallic cages.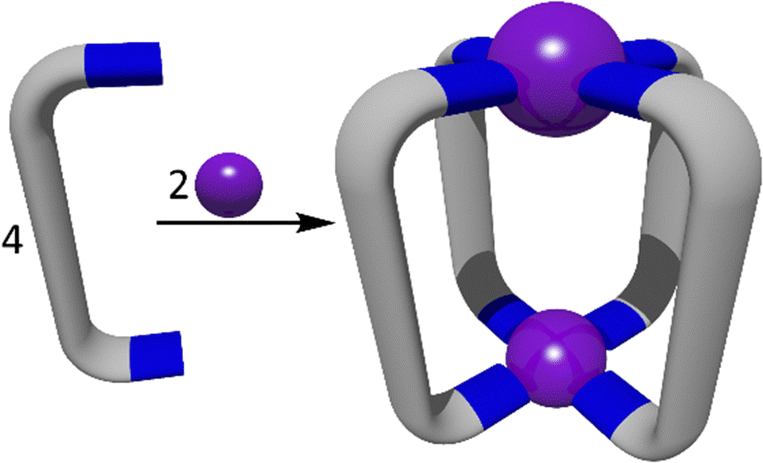 | ||
| Fig. 5 A cartoon representation of the symmetry interaction approach to a generic M2L4 cage. This method uses “naked” or very weakly ligated metal ions and bridging ligands to generate MSAs based on the symmetries of the metal ions and ligands. | ||
An early example of this method includes the synthesis of a capped octahedron [Pd6(AlL3)8](NO3)12 (C9, Scheme 6) by Wu and Wang using hard soft acid base principles.114,115 In general, species with a small atomic radius, low polarizability and high effective nuclear charge are ‘hard’ species, while those with the opposite characteristics are described as being ‘soft’. These principles have been used often in the design and synthesis of supramolecular architectures. With this concept in mind, Wu and Wang started their synthesis with the complexation of aluminium in the form of Al(NO3)3 with a rigid ditopic ligand (L9) containing β-diketone and pyridine binding sites.59 Due to aluminium being a six-coordinate metal, and the diketone donors having an affinity for hard metals, the metal ion preferentially bound at these sites, forming AlL93. To form the cubic cage, the aluminium metallo-ligand was subjected to a square-planar ligand containing a four-coordinate palladium metal. The coordination requirements of the palladium(II) metal ion resulted in it selectively complexing to the pyridine nitrogen donor over the β-diketone, forming the final heterometallic aluminium palladium cubic cage, C9.
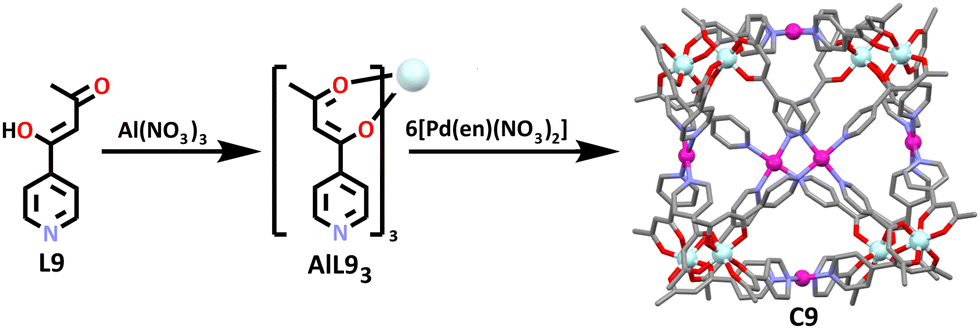 | ||
| Scheme 6 The synthesis of the aluminium metallo-ligand (AlL93) from the rigid ditopic precursor ligand (L9), and the subsequent formation of heterometallic C9 upon addition of palladium to the metallo-ligand. Element colours: carbon (grey), nitrogen (lavender), oxygen (red), palladium (magenta), aluminium (pale blue). Hydrogen atoms, counter anions and solvent molecules have been omitted for clarity. | ||
More recent work undertaken by Brechin and co-workers demonstrates how the metals used in this hard soft acid base approach can imbue unique properties on the resulting cage structure.110,116,117 Since 2015, a wide range of heterometallic cubes, similar in structure to those made by Wu and Wang, have been synthesised by this group, beginning with the synthesis of two [Cr8M6L1024]12+ cages, where M = Cu(II) or Co(II) (Fig. 6).117 In these syntheses, a metallo-ligand containing chromium(III) was first generated, followed by addition of either Cu(NO3)2·3H2O or Co(ClO4)·6H2O salts. The naked metal ions in the salts interacted with the terminal binding sites of the metallo-ligands in a way which resulted in a square planar arrangement of the pyridyl ligands around the metal ions (Cu or Co), with the remaining two coordination sites being occupied by an oxygen donor from either water or nitrate molecules. The final arrangement around each metal were regular octahedral arrangements for chromium(III) and cobalt(II), and a Jahn–Teller distorted geometry for copper(II). These structures were both shown to exhibit weak ferromagnetic exchange interactions.
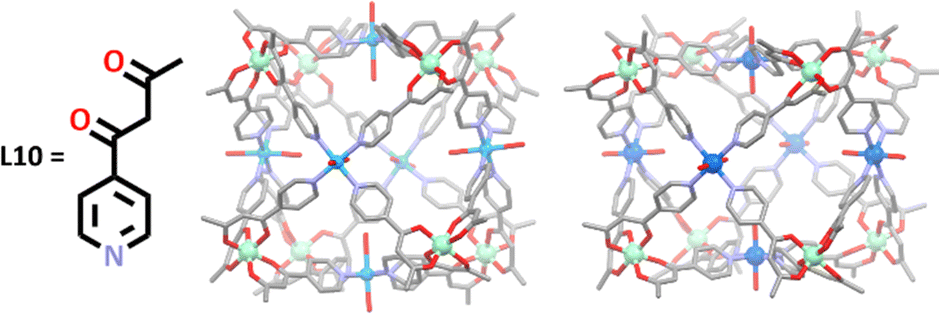 | ||
| Fig. 6 Structural diagram of the ligand, L10, and the molecular structures of the [Cr8Cu6L1024]12+ (left) and [Cr8Co6L1024]12+ (right) cubic cages. Hydrogen atoms, counter anions and solvent molecules have been omitted for clarity. Element colours: carbon (black in the chemical structure, grey in the molecular structures), oxygen (red), nitrogen (lavender), chromium (mint green), copper (blue), cobalt (deep blue). | ||
Further work by the same group demonstrated the scope of this method, using the same ligand, L10 (Fig. 6), to construct a small family of five new paramagnetic coordination cubes of the formula [Fe8M6L1024]n+, where M = Co(II), Ni(II), Pd(II) or Cu(II).110 Similar to their previous work, the corner Fe(III) metals displayed a regular octahedral geometry, while the face capping metals displayed either square planar, square pyramidal or octahedral geometries. In each cage, the metallo-ligands were arranged in a square plane around the face capping metals, with anions present in the starting materials occupying the apical sites. Magnetic susceptibility measurements revealed cages [Fe8Cu6L1024Br4]8+, [Fe8Ni6L1024(SCN)11Cl] and [Fe8Co6L1024(SCN)10]2+ exhibited weak antiferromagnetic exchange between the paramagnetic metal cations.
The family was again extended in 2021, with the group demonstrating the ease of this synthetic method through the construction of three new heterometallic cubic cages. The previous chromium metallo-ligand was combined with various nickel(II) salts giving cages [Cr8Ni6L1024(H2O)12](NO3)12, [Cr8Ni6L1024(MeCN)7(H2O)5](ClO4)12, and [Cr8Ni6L1024Cl12] (Fig. 7). Interestingly, these cages exhibited the presence of weak ferromagnetic exchange between the Cr(III) and Ni(II) ions. This work highlighted the diverse heterometallic structures that can be accessed through hard soft acid base chemistry, concluding that potentially any M(II) ion may be used to construct the initial metallo-ligand, and the face capping ions can potentially be any M(II) ion that can adopt the necessary square planar, square pyramidal or octahedral geometry. These robust design principles should enable the development of magnetically tuneable cage materials.
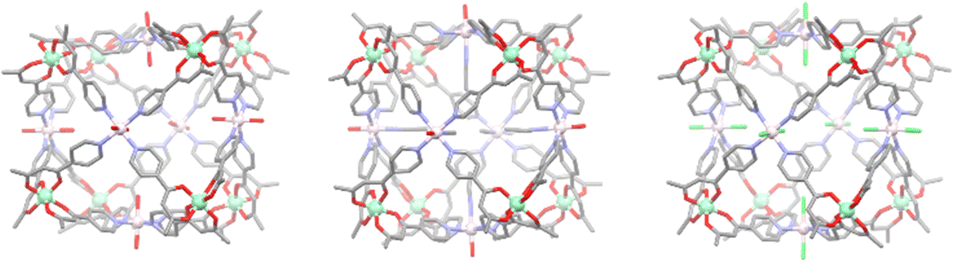 | ||
| Fig. 7 Molecular structures of cages [Cr8Ni6L1024(H2O)12](NO3)12, [Cr8Ni6L1024(MeCN)7(H2O)5](ClO4)12, and [Cr8Ni6L1024Cl12] (from left to right). All hydrogen atoms, counter anions and solvent molecules have been omitted for clarity. Element colours: carbon (grey), oxygen (red), nitrogen (lavender), chromium (mint green), nickel (pale pink), chlorine (green). | ||
Similar to the preceding examples, Wang and co-workers synthesised a series of chiral cubic heterometallic Zn(II)/Pd(II) cages (Scheme 7).112 Through the reaction of L11 with either S-1-phenylethylamine or R-1-phenylethylamine, chiral heterotopic ligands, L11a and L11b, were synthesised in a manner where absolute configuration was able to be controlled. This approach enabled the authors to generate the first homochiral cubic structures.114,118–121 In contrast to the previous family of heterometallic cubes, hard–soft acid–base principles were not used in this work, instead the coordination geometry preference of the different metal ions was exploited in order to obtain selectivity. Similar to the aluminium metallo-ligand employed by Wu and Wang, reaction of six coordinate zinc(II) with either L11a or L11b gave octahedral chiral (Δ or Λ) metallo-ligands where the zinc(II) preferentially bound to the bidentate amine and pyridine donor motif at the top end of each ligand. Subsequent addition of square planar palladium(II) completed cage structures providing C11a-all Δ and C11b-all Λ, with the four-coordinate metal ion preferring the monodentate pyridine donor at the bottom end of each ligand. The group used this approach to synthesise a small series of larger heterometallic homochiral cubes, where the backbone of the ligands was extended by an alkyne group. Again, the stereochemical control afforded by the phenylethylamine precursor enabled the cubes to be synthesised diastereoselectively. Substitution of the chiral phenylethylamine portion of the ligand with achiral 4-methylaniline gave a racemic mixture of cubic cages.
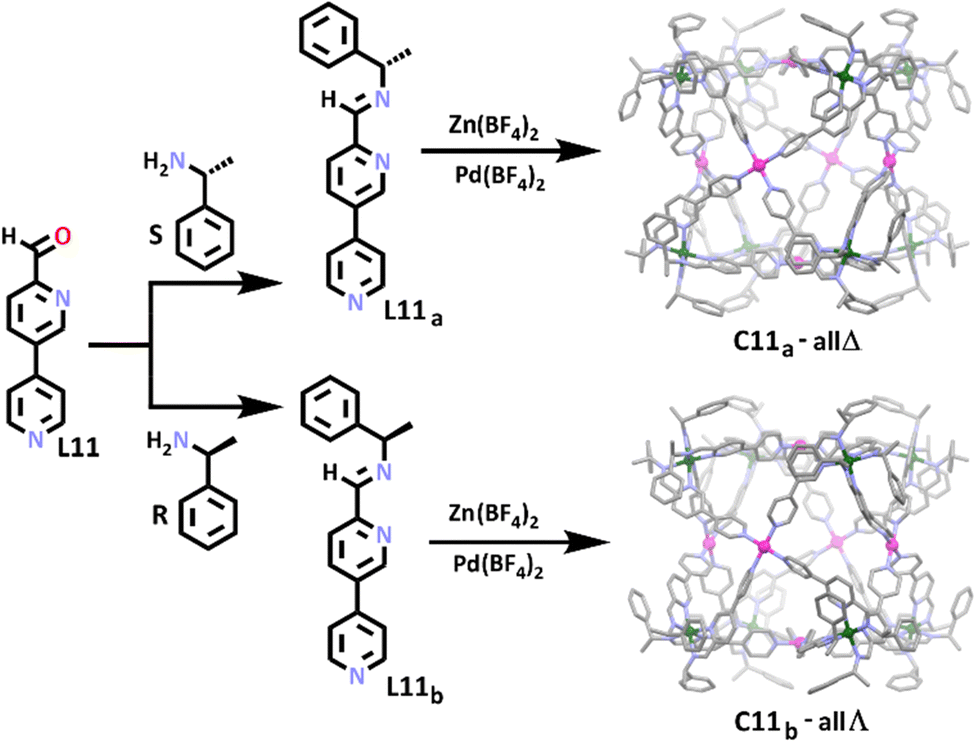 | ||
| Scheme 7 The synthesis of ligands L11a and L11b, and for the resulting homochiral cages C11a and C11b. Cages C11a and C11b shown as molecular structures. Element colours: carbon (black in the chemical structures, grey in the molecular structures), nitrogen (lavender), palladium (magenta), zinc (dark green). All hydrogen atoms, counter anions and solvent molecules have been omitted for clarity. | ||
The same group expanded on this work in 2018, synthesising a further four new enantiomerically pure heterometallic cubic cages using L11a and L11b.109 In the new cages, the initial octahedral metallo-ligand was constructed using copper(II), similar to the zinc(II) metallo-ligand used previously. To complete the cages, face capping metal cations nickel(II) or cobalt(II) were added. Addition of the copper(II) ions to the cages resulted in structures that all demonstrated antiferromagnetic interactions. This work thus offers a platform for further study of host–guest chemistry of potentially chiral magnetic hosts.
A commonly utilised approach to the synthesis of heterometallic cubes via symmetry interaction method involves the use of tripodal metallo-ligands, complexed with square planar metal ions to face cap the resulting cube.54,122,123 A recent example of this is the iron(II) and palladium(II) based cage by Lützen and co-workers (Scheme 8).107 Similar to above, bidentate pyridylimine nitrogen donors were used to complex to the iron(II) ions while palladium(II) bound to monodentate pyridyl units. Interestingly, it was found that steric strain generated by the methyl group in the first metallo-ligand stabilised the iron(II) in its paramagnetic high spin state. Furthermore, when 100 equivalents of the de-methylated ligand was added (L12), the group found the cube still formed but with the iron(II) in the low spin state. Thus, the group was able to tune the ligand field, selectively altering the spin state of the metal centre via variations in the sterics of the ligand, a phenomenon also observed for the iron(II) metallo-ligand on its own. This work was a rare example of a heterometallic cage exhibiting spin cross over, with majority of iron containing cages stabilising the iron in its low spin state only.56,102
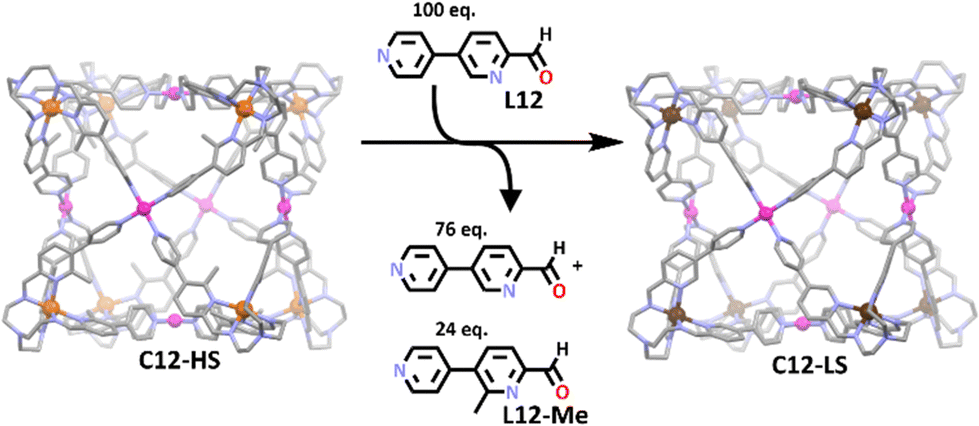 | ||
| Scheme 8 The conversion of the iron(II) atoms from high spin state to low spin state upon addition of competing ligand L12. Element colours: carbon (grey), nitrogen (lavender), oxygen (red), palladium (magenta), iron (rust for high spin, chocolate brown for low spin). Hydrogen atoms, counter anions and solvent molecules have been omitted for clarity. | ||
Interestingly, the heterometallic Fe(II)/Pd(II) cage also proved to be stimuli responsive. When 6 equivalents of 1,3-bis(diphenyl-phosphine)propane were added to the cage a rearrangement occurred generating the smaller heterometallic trigonal bipyramidal structure, C12′, along with 4 equivalents of an [FeL12-Me]2+ complex. The structure of this new heterometallic cage was supported by ESI-MS data and 1H NMR spectroscopy. The molecular structures of the C12’ trigonal bipyramid and the [FeL12-Me]2+ complex were determined using X-ray crystallography, confirming the shape of the new cage (Scheme 9). Like the cubic cage, the new trigonal bipyramidal cage was shown to possess spin crossover capabilities. When C12′ was exposed to 12 equivalents of L12, the Fe(II) centres changed from the high spin state into the low spin state. The low spin trigonal bipyramidal structure could also be obtained by reacting the Fe(II) low spin state cage with 1,3-bis(diphenyl-phosphine)propane.
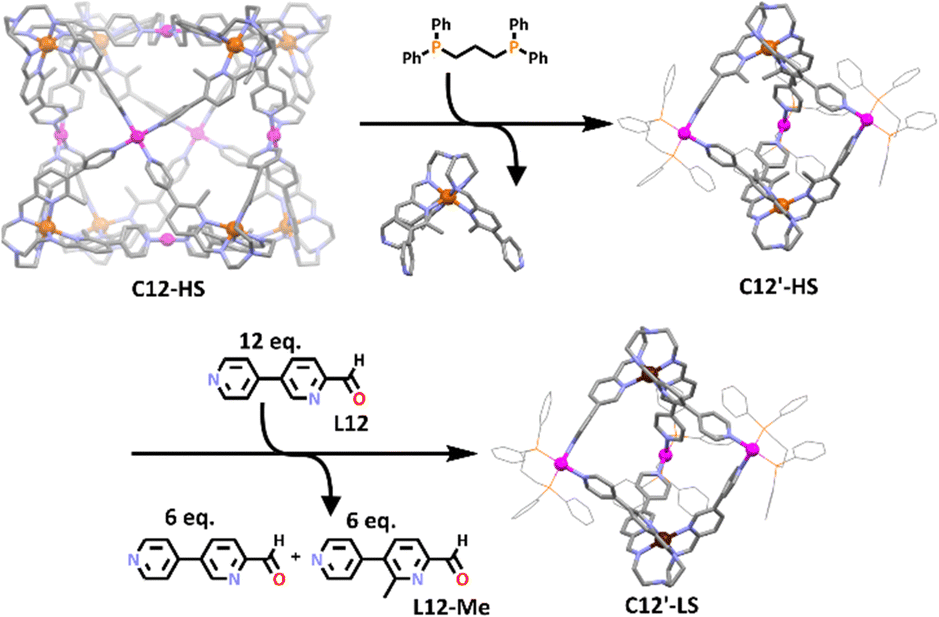 | ||
| Scheme 9 Structural change from the 3D cube to a trigonal bipyramidal structure, which could be switched between its Fe(II) high and low spin states. Colours: carbon (grey), nitrogen (lavender), oxygen (red) palladium (magenta), iron high spin (rust), iron low spin (chocolate brown). Solvent atoms, counter anions and hydrogen atoms removed for clarity. | ||
Li and co-workers demonstrated the generality of this method, constructing a series of new heterobimetallic iron(II)/palladium(II) cages employing the use of both pyridine and imidazole donors (Scheme 10).105 In this approach, the group took the well-studied tris(imidazolimine) ligand and altered its ligand field through addition of three 4-pyridyl units.124–126 When complexed with Fe(II) this gave a tripodal tris-imidazolimine metallo-ligand (FeL13) where the iron was high spin. Further complexation with palladium(II) cations generated the corresponding high spin heterometallic cage (C13). Building on those result the same group investigated the SCO abilities of the related cages where the Pd(II) ions where replaced with Ni(II), giving C13a.127 Solid state magnetic susceptibility measurements of the Fe(II)/Ni(II) cage showed three of the eight Fe(II) metal centres exhibited a gradual, incomplete spin transition between 50 K at 24.19 cm3 K mol−1 to 400 K at 34.64 cm3 K mol−1. Notably, a LIESST (light-induced excited spin-state trapping) effect was also seen, corresponding to approximately two Fe(II) centres being excited by both red and green light. The authors suggested there were multiple structural differences between the Pd(II) and Ni(II) based cages which allowed for SCO in C13a, but not C13. For example, the Ni(II) centre in C13a exhibited a distorted octahedral coordination environment, permitting the reorganisation of the fairly rigid metallo-ligands required for SCO, compared to the square planar arrangement of the Pd(II) metal centre in C13. Furthermore, C13a was observed to pack in a more offset manner compared to C13, evidenced by a move from 33.4 to 28.7% in the packing coefficients of C13 and C13a, respectively. This was attributed to the presence of axially coordinated acetonitrile ligands around the Ni(II) ions, enabling further conformational freedom. This study highlighted the role different metal centres may play in the overall conformation of a structure, allowing for an architecture to undergo SCO or not.
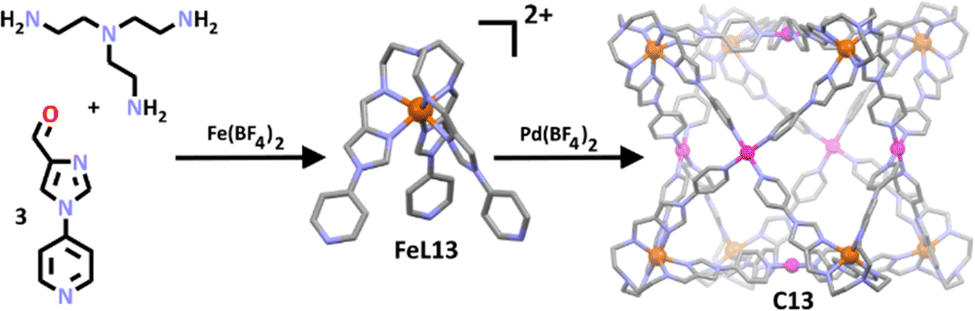 | ||
| Scheme 10 The formation of the tripodal iron-based metallo-ligand (FeL13) and the final cubic cage structure (C13) upon addition of Pd2+ ions. The metallo-ligand and cage are depicted as molecular structures, with hydrogen atoms, counter anions and solvent molecules omitted for clarity. Element colours: carbon (black in the chemical structures, grey in the molecular structures), oxygen (red), nitrogen (lavender), iron (rust), palladium (magenta). | ||
In a rather unique synthesis, Englert and co-workers utilised the HSAB theory to construct a new cube-like heterometallic architecture in 2021.106 Inspired by early work from Rauchfuss and Wrobleski, their unique ligand design featured three possible donor sites: a β-diketone, nitrogen and phosphorus group (Scheme 11, L14).128,129 The hard diketone O, O donors showed preferential complexation with hard metal cations such as iron(III) or aluminium(III), as has been demonstrated many times in literature, giving metallo-ligand ML143.59,110,112,114,116,117 The two alternate phosphorus and nitrogen soft donor sites may have in principle competed for any soft metal cation they were subjected to, however being a stronger donor than nitrogen, the phosphorus donor showed selectivity in reactions with soft metals in model studies. Thus, when the soft metal cation, copper(I), was added to the aluminium metallo-ligand (AlL143), the metal complexed with the phosphorus donor over the nitrogen donor. The shape of resulting cage, C14, was rather uncommon. Unlike previous heterometallic cubes, the cuboid here sported all eight corners capped with a metal, rather than the usual arrangement where the corners are capped by the hard octahedral metallo–ligand and the faces capped with the square planar, square pyramidal or octahedral soft metal.
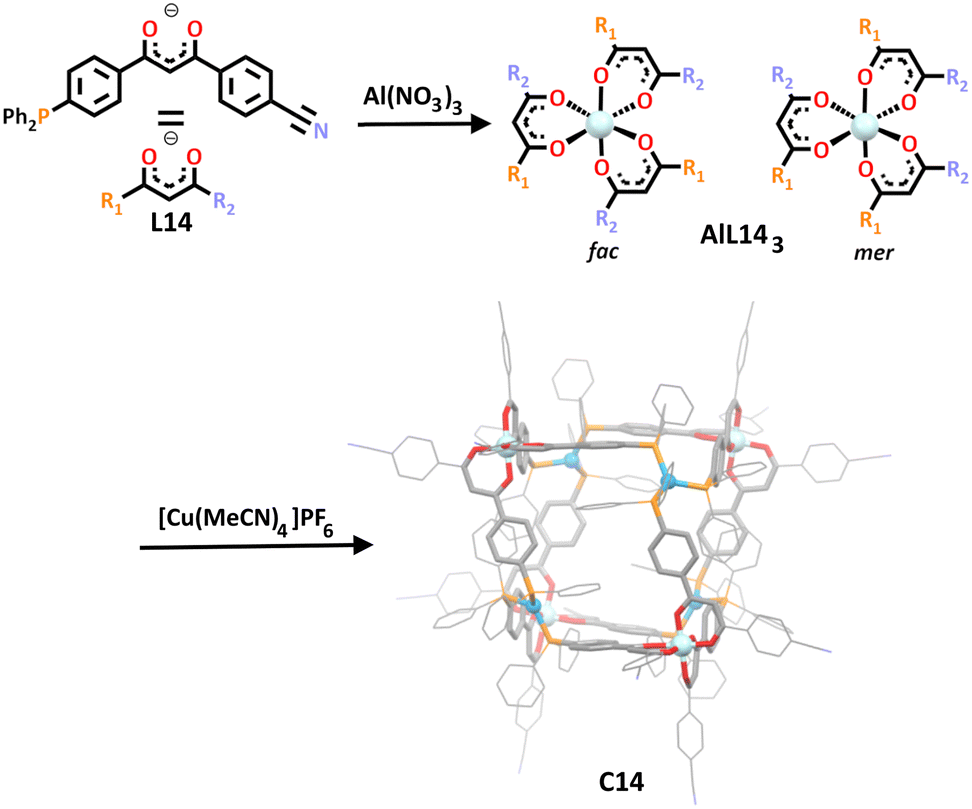 | ||
| Scheme 11 The synthesis of the C14 from the aluminium metallo–ligand (AlL143). The final cage is shown as its molecular structure. Element colours: carbon (black in the chemical structures, grey in the molecular structure), nitrogen (lavender), phosphorus (orange), aluminium (pale blue), copper (blue). Hydrogen atoms, counter anions and solvent molecules have been omitted for clarity. | ||
It is clear to see that heterometallic cubic cages are abundant in the literature. Less common are the “donut” shaped cages. However, interest in these MSAs is increasing.104,111 In a recent example, 1,3-propanediamine was combined with a pyridyl-imidazole based carboxaldehyde to form a ditopic ligand (L15) (Scheme 12).104 Addition of copper(II) ions to the mixture resulted in the subcomponent self-assembly of a copper(II)-based metallo–ligand (CuL15). Complexation with palladium(II) cations gave the final tubular “donut” shaped structure (C15), which was found to be an efficient heterogenous catalyst for the epoxidation of styrene and styrene derivatives (Scheme 12). When styrene was reacted with C15 and TBHP under optimised conditions for eight hours, conversion reached ∼99% with epoxide selectivity reaching 90%.
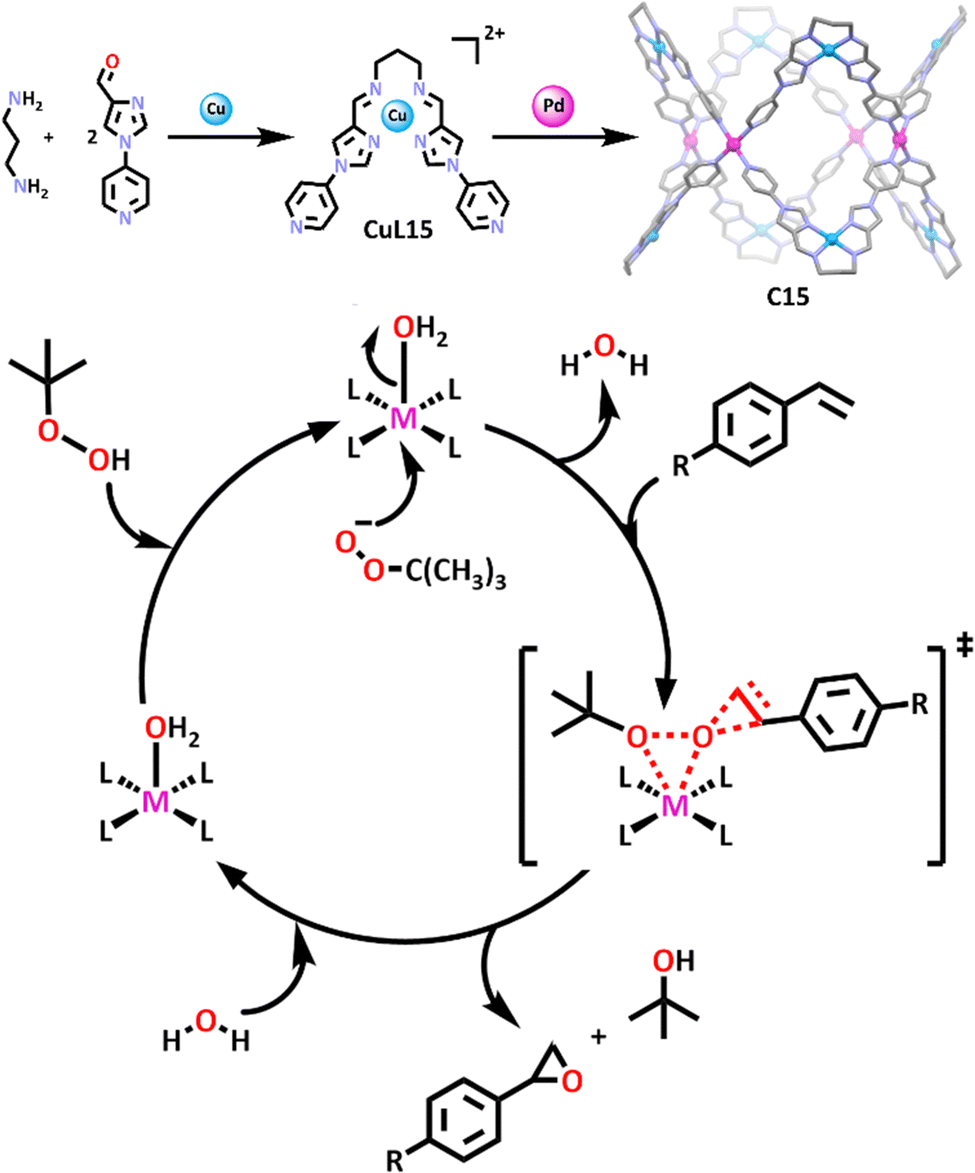 | ||
| Scheme 12 Reaction scheme (top) depicting the formation of the copper-based dipodal metallo–ligand (CuL15) and the final copper/palladium donut cage (C15). The molecular structure of the cage has been depicted with hydrogen atoms, counter anions and solvent molecules omitted for clarity. Element colours: carbon (black in the chemical structures, grey in the molecular structure), oxygen (red), nitrogen (lavender), copper (blue), palladium (magenta). The catalytic cycle (bottom) leading to epoxide formation proposed by the authors (R = H, F, Cl, Br, CH3, CH3COO, or CF3). | ||
Under identical conditions, CuL15, Cu(NO3)2, Pd(NO3)2 and a mixture of Cu(NO3)2 and Pd(NO3)2 were also tested for their catalytic activity. The conversion and epoxide selectivity were highest with the cage. In addition to this, the group also tested previously reported heterogenous catalysts in the epoxidation of styrene, again concluding that C15 performed better. This was attributed to the confined space of the cage enhancing both the conversion and selectivity of epoxidation of styrene. The authors did not confirm whether the copper or palladium ions acted as the catalytic centre, however based on these results, it is likely both were involved in the catalysis.
In a related example, a heterometallic platinum(II)–palladium(II) donut shaped cage was synthesised by the Crowley group.111 Heterometallic platinum(II)–palladium(II) MSAs are becoming more common but for the most part the interest has been purely structural.72,130,131 This Pd–Pt donut was shown to be catalytically active. By taking a ligand containing a 2-pyridyl-1,2,3-triazole component (L16), a bidentate binding pocket was formed allowing for complexation of one platinum(II) cation with two ligands in a head to tail fashion around the square planar metal (Scheme 13). The remaining free monodentate nitrogen binding site on the pyridine ring was then able to complex with palladium(II) cations, generating the final cage structure. The three planar, cationic, panel-containing clefts in the cage structure allowed for planar aromatic rings such as anthracene to bind within the cavity. This prompted the group to explore the applications of the cage, concluding that the structure had potential in being used as a molecular flask for anthracenyl substrates and also in photocatalysis applications, allowing for anthracenyl endoperoxides to be formed in a catalytic manner (Scheme 13).132 Endoperoxides often appear as intermediates in organic transformations and have been shown to exhibit bioactive properties.133
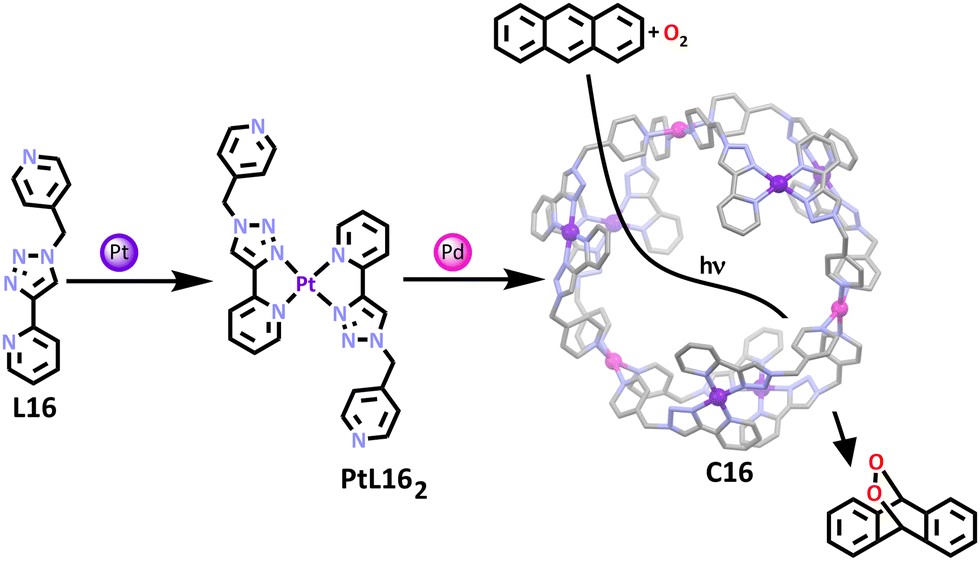 | ||
| Scheme 13 Formation of the platinum(II) metallo–ligand (PtL162) and the final platinum(II)/palladium(II) containing donut shaped cage (C16). The molecular structure is depicted with all hydrogen atoms, counter anions and solvent molecules omitted for clarity. The oxidation of anthracene catalysed by C16 is also depicted. Element colours: carbon (black in the chemical structures, grey in the molecular structure), oxygen (red), nitrogen (lavender), platinum (purple), palladium (magenta). | ||
To investigate the effect of the cage on this cycloaddition process, three aerobic solutions with anthracene (10 mM) and [D3]acetonitrile were left to stand in direct sunlight for 10 hours. The solution containing only free anthracene displayed 35% conversion of anthracene to endoperoxide, while the solution containing PtL162 complex gave a conversion of 42%. One common limitation to using metallo-supramolecular architectures as catalysts is the failure of the structure to effectively release product once it has been formed, preventing catalytic turnover.134,135 Fortunately, for C16, it was shown via1H NMR and DOSY spectroscopy that the resulting endoperoxide did not interact with the cage, suggesting C16 shows promise in the catalytic turnover of substrate.
The use of lower symmetry ligands that feature a monodentate and bi/tridentate metal bind sites is a common approach to heterometallic cages. While a wide range of metal ion combinations have been exploited to synthesise these types of heterometallic MSAs, ruthenium(II) and palladium(II) containing systems66,136–139 are a particularly well studied sub-class of compound for two reasons (1) the inert nature of the Ru(II) metalloligands means that they can be readily synthesised and purified and (2) they often display interesting photophysical/chemical properties.140–142 One elegant example of this is the stepwise synthesis of a Ru(II)/Pd(II) heterometallic cage displaying a rare rhombododechahedral shape from the Su group.113 As illustrated in Scheme 14, the synthesis began with the construction of a bulky triangular Ru(II) metallo–ligand, which when exposed to naked Pd(II) metal cations formed the thermodynamically favoured Pd6(RuL173)8 cage. The cage showcased the 6 vertices of the truncated octahedron occupied by Pd(II) ions, while the 8 faces were capped with RuL173 ligands, forming a large hydrophobic cavity with relatively narrow rhombic windows. The group first explored guest binding behaviour of the cage using aromatic molecules such as phenanthrene (Phen), pyrene, anthracene and perylene. 1H NMR data showed significant upfield shifts of guest protons upon Phen encapsulation, with integration of proton peaks and NMR/DOSY titrations suggesting approximately 18 Phen molecules were trapped within the cage cavity. Molecular dynamic simulations (MDS) revealed 7 Phen molecules can reside within the cavity, while a further 17 can potentially reside in the rhombic windows via π–π interactions. While pyrene or anthracene molecules were also shown to guest bind, the larger perylenes were not.
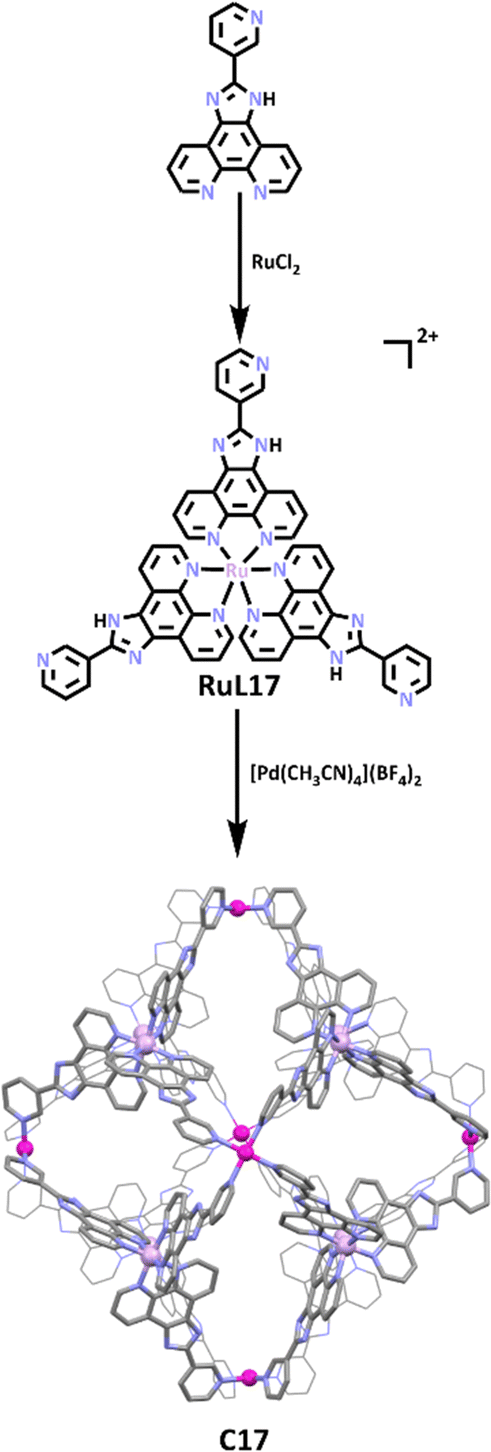 | ||
| Scheme 14 Formation of C17 from metallo–ligand RuL173. The molecular structure of C17 has been shown with all hydrogen atoms, counter anions and solvent molecules omitted for clarity. Element colours: carbon (black in the chemical structures, grey in the molecular structure), nitrogen (lavender), palladium (magenta), ruthenium (light purple). | ||
Encouraged by the above results, the group moved on to analyse the ability of C17 to trap and stabilize photosensitive molecules. The three molecules tested for this purpose, 2,2-dimethoxy-2-phenylacetophenone (DMPA), 1-hydroxycyclohexyl phenyl ketone (HCPK), and 2hydroxy-2-methylpropiophenone (HMPP), are all widely used in ink and paint as light-curing agents. Similar to previous guest binding studies, large upfield shifts were observed in 1H NMR spectra for guest molecules upon inclusion, suggesting a host–guest interaction. UV radiation studies suggested the cage had a protective effect on the photoinitiators. When exposed to 365 nm UV light for 12 hours, the free guests showed photolysis, while encapsulated guests did not. Control studies showed this protective effect was greater for the cage than for the RuL173 metallo–ligand, with C17 suppressing photolysis for up to 120 hours compared to RuL173 suppressing photolysis for only 24 hours. These studies show C17 has potential application in photocuring materials, as well as in drug delivery of photosensitisers and photodynamic therapy, as has been achieved by previously reported Ru cages.143,144 The authors have gone on to show that the inherently redox- and photo-active C17 has been shown to have applications in many areas. These include photochemical hydrogen production,145,146 enantioseparation of atropisomeric molecules,147 regio- and enantioselective photodimerization of naphols and its derivatives,148 use as a redox-guest-induced multimode photoluminescence switch for sequential logic gates,149 photocatalysis of asymmetric [2+2] cycloadditions of an ACE derivative150 and α,β-unsaturated carbonyl compounds,151 light imaging and tracking of the living cell membrane,152 a multirole and multi-way catalytic platform for H/D exchange and unconventional Glaser coupling of terminal alkynes153 and as an enzyme-mimicking catalytic system for the green visible light driven photoinduced Meerwein arylation.154
Liu and co-workers have synthesized a series of lanthanide-containing heterometallic cages and examined their use as catalysts for the conversion of CO2 under mild conditions.155 An application of increasing interest in recent years is the conversion of CO2 into less harmful or useful substrates, such as cyclic carbonates.156–160 Liu and co-workers designed their first cage using previous knowledge that Lewis-basic groups such as amides and having multiple Lewis-acid sites has been shown to be an effective way of enhancing catalytic reactivity and selectivity.161 These heterometallic cages were used as catalysts, along with tetrabutylammonium bromide (TBAB) as the cocatalyst, for the coupling of CO2 and styrene oxide (SO). They found that the catalytic architecture could be recycled. After undergoing CO2 conversion, the cage could be recovered, and still possessed the same structural integrity and lanthanide luminescence. Further testing demonstrated that it still had the same level of catalytic activity.
Liu and co-workers then designed two new heterometallic [Zn4Ln8L186]32+ cages.162 The ligand (L18) was synthesized by refluxing 2,2′-oxide(acetohydrazide) and 2-hydroxy3-methoxybenzaldehyde in ethanol overnight. ZnCl2 was then added, with the zinc ion occupying the amide pocket of the ligand. TEA and Ln(NO3)3 (where Ln = Eu(III) or Tb(III)) were then added to the mixture, allowing the ligand to coordinate around the lanthanoid ions, which occupied the end of the ligand pockets, resulting in the formation of two new heterometallic, lanthanoid containing cages. Single-crystals were obtained, and showed that the cages were isostructural. The lanthanoid ions were captured at the end of the ligand pockets and were found to be nine-coordinate, with seven Ln–O bonds from the ligands, and two solvent molecules attached. The Zn(II) ions inhabited the central amide pockets and were further coordinated with two Cl− ligands (Scheme 15). These lanthanide-containing cages are particularly interesting because they contain both 3d (Zn) and 4f (Ln) metal centres. These metal centres tend to show distinct binding and catalytic ability.
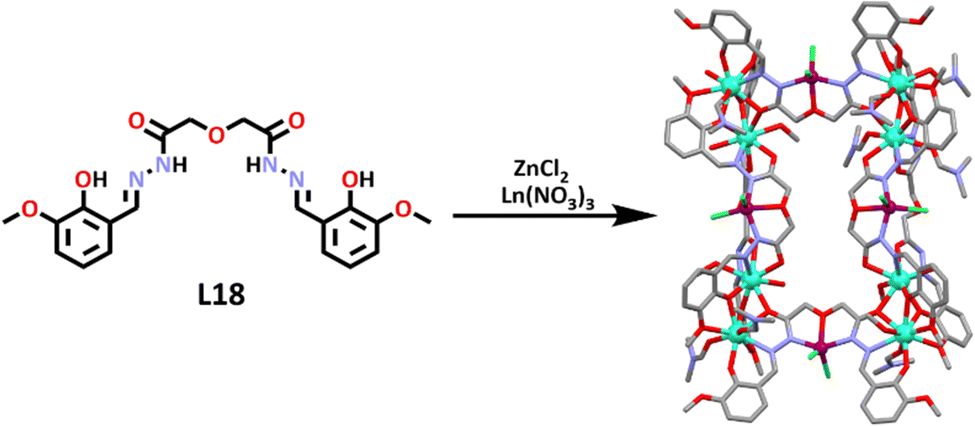 | ||
| Scheme 15 One pot self-assembly of lanthanoid-zinc heterometallic cages for use as CO2 converters. Colours: carbon (grey), nitrogen (lavender), oxygen (red), zinc (mulberry), chlorine (green), europium (aqua marine), terbium (turquoise). Solvent molecules, counter anions and hydrogen atoms removed for clarity. | ||
The authors tested the catalytic ability of these new cages. They found that these systems had high CO2 conversion rates of 93% for [Eu8Zn4L186]32+ and 92% for [Tb8Zn4L186]32+ (Scheme 16). [Eu8Zn4L186]32+ was found to be very stable, and once again maintained its structural integrity and properties after undergoing catalysis. Control tests conducted showed that neither the cage nor the TBAB could catalyse the CO2 conversion on their own, so they were both essential for this process.
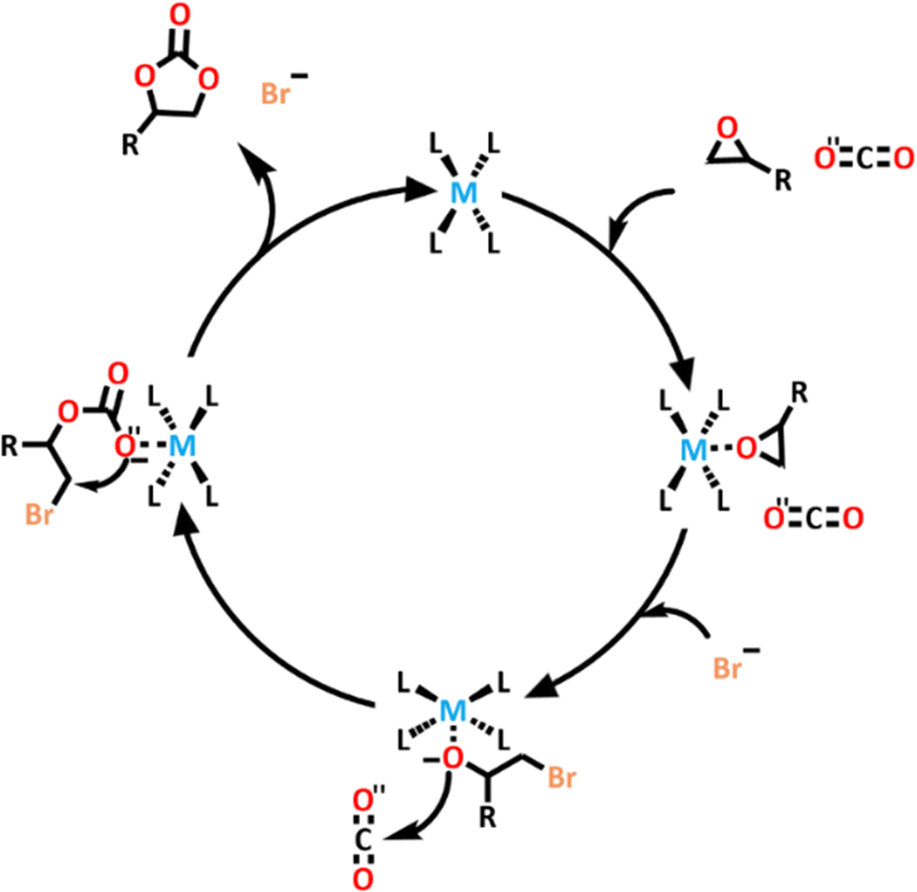 | ||
| Scheme 16 Proposed mechanism for the catalytic process of the [Tb8Zn4L186]32+ and [Eu8Zn4L186]32+ cages. | ||
Related lanthanide containing heterometallic cages have been generated by Sun and co-workers.163 The authors synthesised four new chiral MSAs incorporating Eu(III), Yb(III), Nd(III) and Lu(III). Two featured the Ln2Pt3L196 formulation, while two others were a larger Ln4Pt6L1912 organisation. The two cage sizes were achieved by altering the bending angle in the initial metallo–ligands (Fig. 8). By using a 90° angle in L19a, the smaller architecture prevailed, while using a bending angle of 180° in L19b gave an architecture with a larger cavity.
 | ||
| Fig. 8 Chemical structures of the metallo–ligands used in the formation heterometallic lanathanide-based cages. On the left is a ligand containing an 90° bending angle (L19a), used to form Ln2Pt3L196 cages, while on the right is a ligand with a 180° bending angle (L19b) used to form the larger Ln4Pt6L1912 architectures. | ||
The structures of each cage were confirmed by 1H NMR spectroscopy, 1H NMR DOSY spectroscopy and ESI-TOF mass spectrometry. Unfortunately, no crystal structures were obtained of the chiral cages, however slow vapor diffusion of isopropyl ether into an acetonitrile solution of a related achiral cage Yb4(L3)6(OTF)12 (Fig. 9) gave high quality single crystals suitable for X-ray crystallography. The resulting X-ray diffraction experiment unambiguously confirmed the tetrahedral geometry of the cage, proving the feasibility of these syntheses. Discrete d/f heterometallic cages and metallocycles show potential in applications such as electro-optic and magnetic materials. In fact, two of the cages produced in this work show promise as potential luminescent temperature sensors, suggested by linear temperature-dependant red and NIR (near infra-red) emissions observed for these complexes.
 | ||
| Fig. 9 Molecular structure of Yb4(L3)6(OTf)12 determined by X-ray crystallography. Element colours: carbon (grey), oxygen (red), nitrogen (lavender), phosphorus (orange), platinum (purple), ytterbium (forest green). All hydrogen atoms, counter anions and solvent molecules have been omitted for clarity. | ||
Very recently Crowley and co-workers synthesized a second generation [PdPtL4]4+ cage.164 Unlike the first-generation cage which was synthesised using a subcomponent self-assembly pathway, this new cage was generated using the stepwise metallo–ligand symmetry interaction approach. A low symmetry ligand (L20) which featured both imidazole and pyridine units was developed. The differing donor strengths of the two coordinating units enabled selective complexation of the Pt(II) ion at the imidazole unit to provide the [Pt(L20)4]4+ metallo–ligand (Scheme 17). Subsequent addition of palladium(II) ion completed the [PdPtL204]4+ cage formation. 1H NMR spectra confirmed the selective metalation of the ligand and formation of the cage. 1H DOSY spectroscopy provided further evidence and ESI-MS showed sequential loss of three BF4− counter anions, consistent with formation of C20. The formation of the heterometallic architecture was confirmed using X-ray crystallography (Scheme 17). Similar to the first-generation cage the new system was stimuli responsive and could be opened upon addition of competing ligand DMAP and subsequently closed using TsOH or MsOH. This new [PdPtL4]4+ cage displayed somewhat limited HG chemistry, none of the neutral guest examined were found to bind. The system could bind anions (for example, methanesulfonate, p-toluenesulfonate, tolyltriflouroborate, and dimethylphosphate) and most interestingly due to the low symmetry of the cage, the anions were found to bind selectively at the palladium end of the cage.
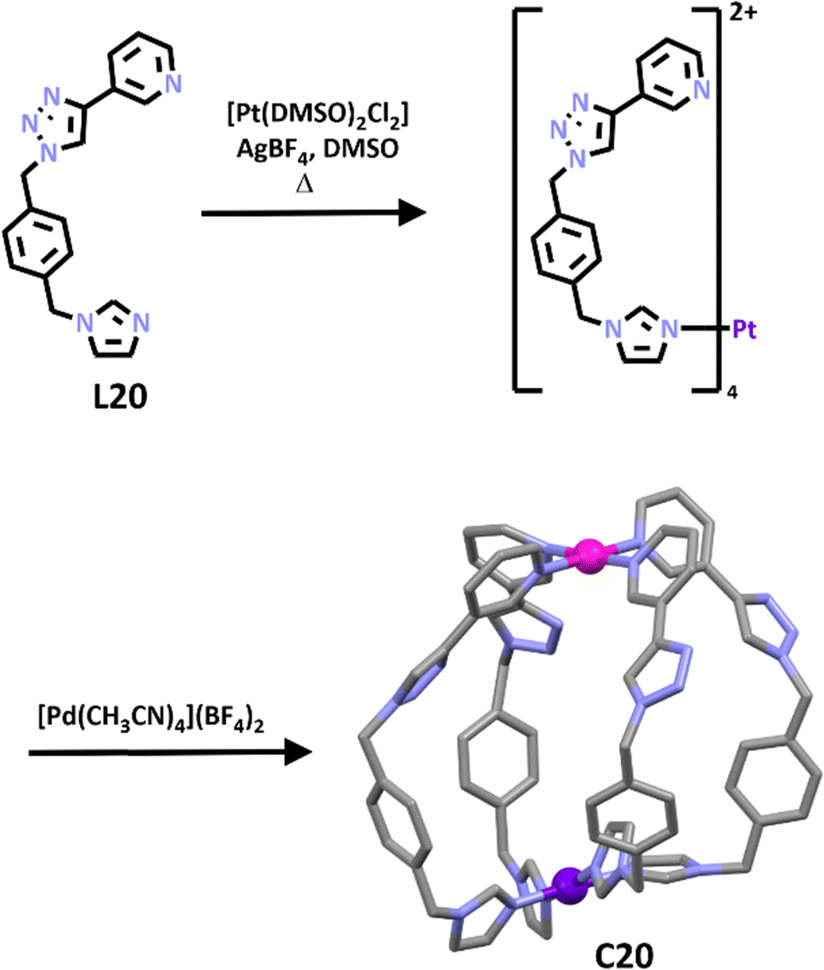 | ||
| Scheme 17 Formation of heterometallic platinum–palladium cage C20. The molecular structure of the final cage has been depicted with hydrogen atoms, counter anions and solvent molecules omitted for clarity. Element colours: carbon (grey), nitrogen (lavender), palladium (magenta), platinum (purple). | ||
Directional bonding approach to heterometallic cages
The directional bonding approach (Fig. 10) to the construction of heterometallic three dimensional architectures has been widely used in recent years.107,163,165–167 Unlike the symmetry interaction approach, the directional bonding method utilises capped metal ions, whereby the kinetically inert capping ligands direct the approach of the remaining bridging ligands or metallo–ligands to predictably form the resulting architecture. Commonly shaped structures include prisms,165–167 and lanterns.107,114,163,168 A wide range of functional heterometallic cages have been generated by exploiting the directional bonding approach and combining it with the heterometallic methods outlined in Fig. 1.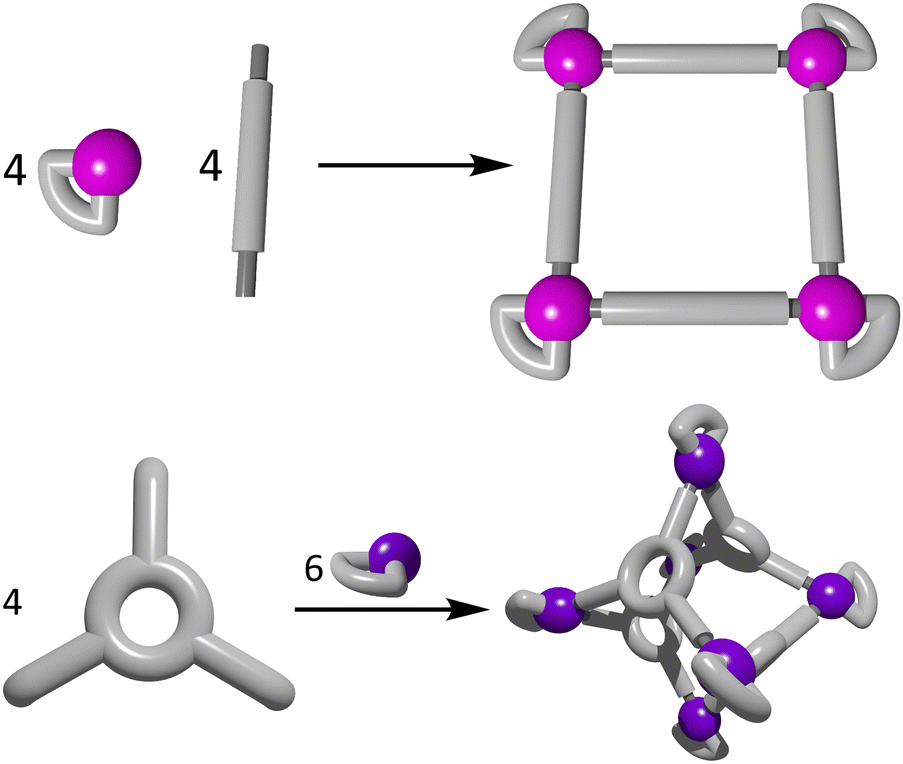 | ||
| Fig. 10 Cartoon representations of the directional bonding approach showing the formation of a 2D molecular square (top) and a 3D octahedral cage (bottom). | ||
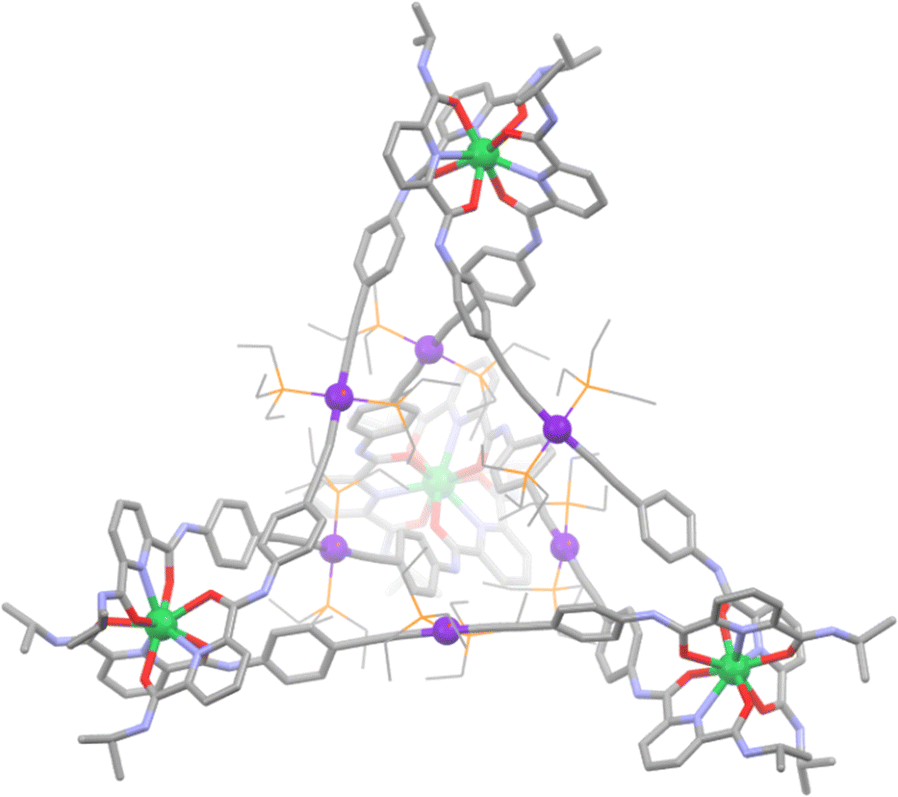
One such prism design was achieved by Mukherjee and Adeyemo in 2018.165 In this work, a triplatinum organometallic ligand (L21) was first synthesised in a 4 step procedure, where a Sonogashira-like coupling reaction was used to give the final product. This is a different method than is typically used to synthesise metallo–ligands, where often the metal ion is incorporated into the ligand via nitrogen, oxygen or even phosphorus donor groups. The terminal pyridyl donors of this ligand remained free to complex with the appropriate dinuclear arene-ruthenium(II) clips (1a or 1b). The final trigonal prismatic cages were isolated as nitrate complexes, and their structures confirmed with multinuclear NMR and ESI-MS studies (Scheme 18). Unfortunately, no crystal structures were able to be obtained to unambiguously confirm their structures.
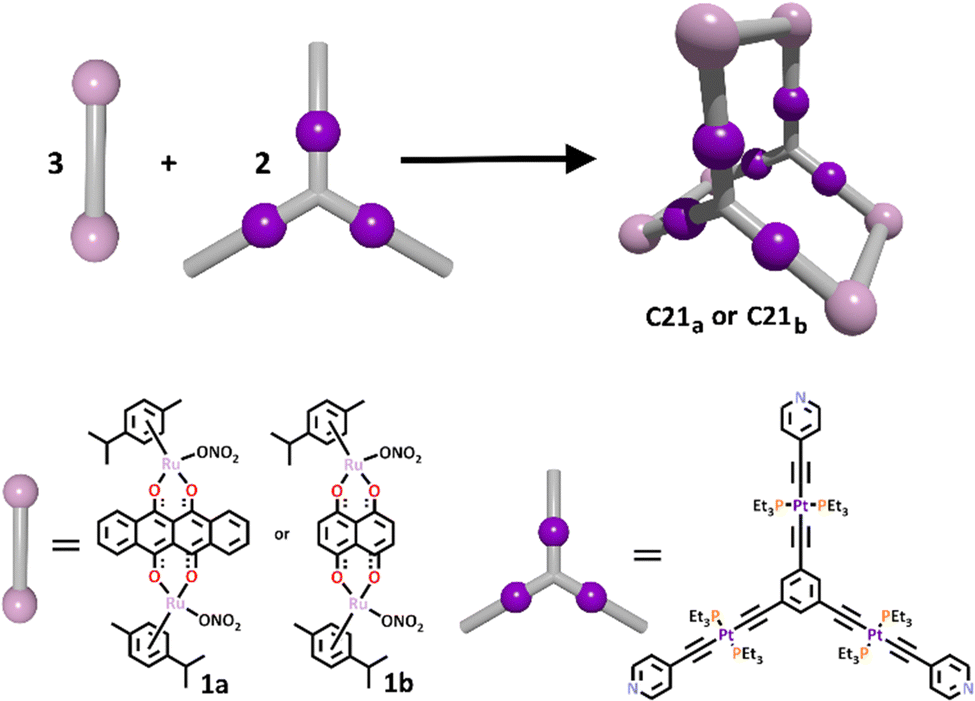 | ||
| Scheme 18 The formation of heterometallic Pt(II)/Ru(II) prismatic cage C21 from the corresponding dinuclear arene-ruthenium(II) clips (1a or 1b) and tri-platinum(II) metallo–ligand. | ||
In a different approach, Lin and Liu constructed a series of rectangular prism shaped cages using a heterometallic metallo–ligand.166 The group first synthesised a di-rhodium complex, before complexing this with either zinc, nickel or cobalt to form the corresponding heterometallic metallo–ligand, L22. The final cage structures were achieved by treating this metallo–ligand with a rigid linker ligand (either La, Lb or Lc) containing a nitrogen donor at each end in the form of either a pyridine or imidazole ring (Scheme 19). It was shown that the linker ligands could be altered in a variety of ways to lengthen the chain, and as long as the linker remained fairly rigid, a cage was able to form. Molecular structures were obtained for three rhodium(III)/zinc(II) heterometallic cages, showcasing the linkers complexing to the rhenium metals, forming the four edges of the prism (C22a and C22b). In one case, the group also observed the linker ligand complexing to the central zinc metal, forming a chain through the centre of the prism between the two metal sites (C22c). This approach was uncommon, with most heterometallic cages synthesised using homometallic ligands instead.
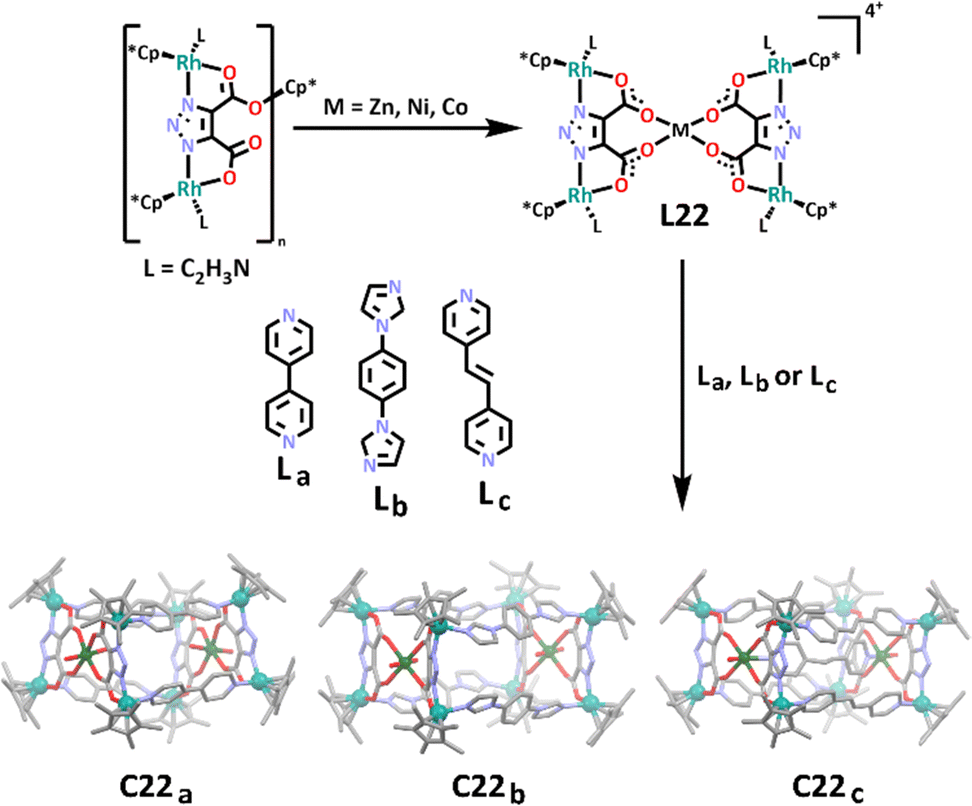 | ||
| Scheme 19 The synthesis of heterometallic, prismatic Rh(III)/M(II) cages C22a, C22b and C22c from their corresponding heterometallic metallo–ligand and rigid linker ligands (either La, Lb or Lc). The cages have been shown as molecular structures, with all solvent molecules, counter anions and hydrogen atoms omitted for clarity. Element colours: carbon (black in the chemical structures, grey in the molecular stuctures), oxygen (red), nitrogen (lavendar), rhodium (teal), zinc (dark green). | ||
Similar to the above example, the Ma group have also recently used a heterometallic ligand to synthesise a prismatic rhodium/copper heterometallic cage (Scheme 20).167 In this work, heterometallic ligand L23a and organic ligand L23b were combined in a one pot synthesis to afford C23. Two of the heterometallic ligands coordinated with the organic ligands via a rhodium-pyridyl bond forming a macrocyclic shape. The remaining two heterometallic ligands completed the cage structure by coordinating to one side each of the macrocycle via a rhodium-imidazole bond. The host–guest chemistry of the cage was briefly explored by the group, it was found that C23 was a good host for two isopropyl ether molecules.
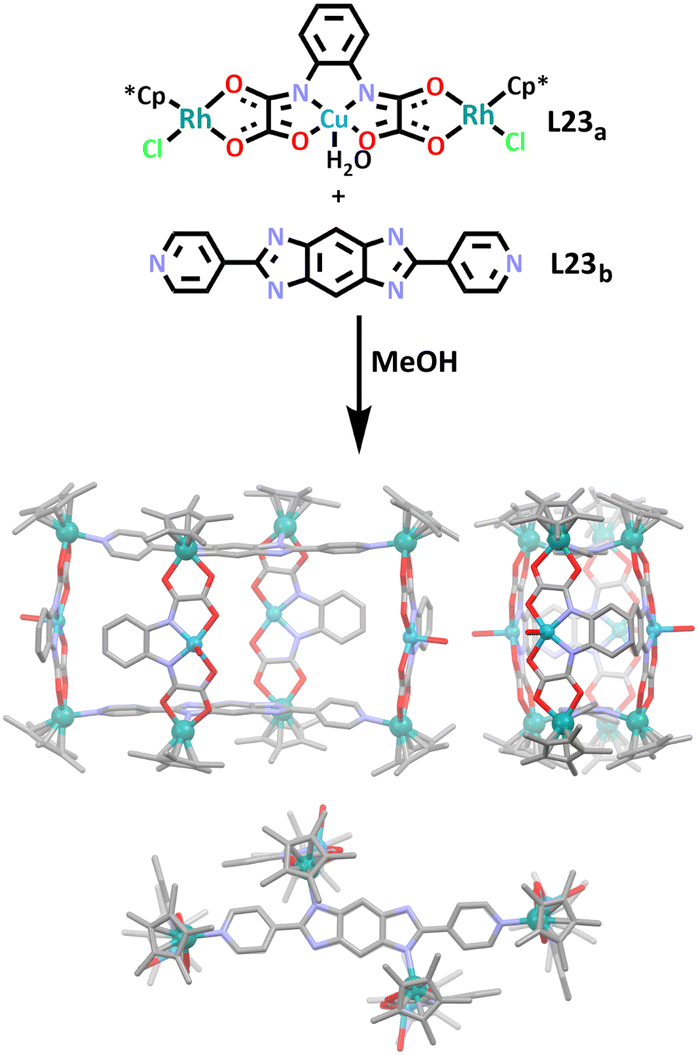 | ||
| Scheme 20 Synthesis of C23 from heterometallic ligand L23a and organic ligand L23b. The molecular structure is displayed in three orientations (front, side and top views) (from left to right). Hydrogen atoms, counter anions and solvent molecules have been omitted for clarity. Element colours: carbon (black in the chemical structures, grey in the molecular structures), oxygen (red), nitrogen (lavender), copper (blue), rhodium (teal). | ||
Similar to what is often seen in the synthesis of heterometallic cages using the symmetry interaction approach, the HSAB theory can also be applied to the synthesis of architectures using a directional bonding method. Ye and co-workers exploited these principles in the construction of two neutral heterometallic M2Pd3 lantern shaped cages, [Al2Pd3L246Cl6] (C24a) and [Fe2Pd3L246Cl6] (C24b) (Scheme 21).168 Similar to previous work, a β-diketone aluminium or iron metallo–ligand was first synthesised via a Claisen condensation, before addition of a four-coordinate square planar palladium(II) ion to the soft imidazole nitrogen donor to complete the cage. The chlorine atoms coordinated to the palladium(II) metal directed the approach of the metallo–ligand in a trans fashion.
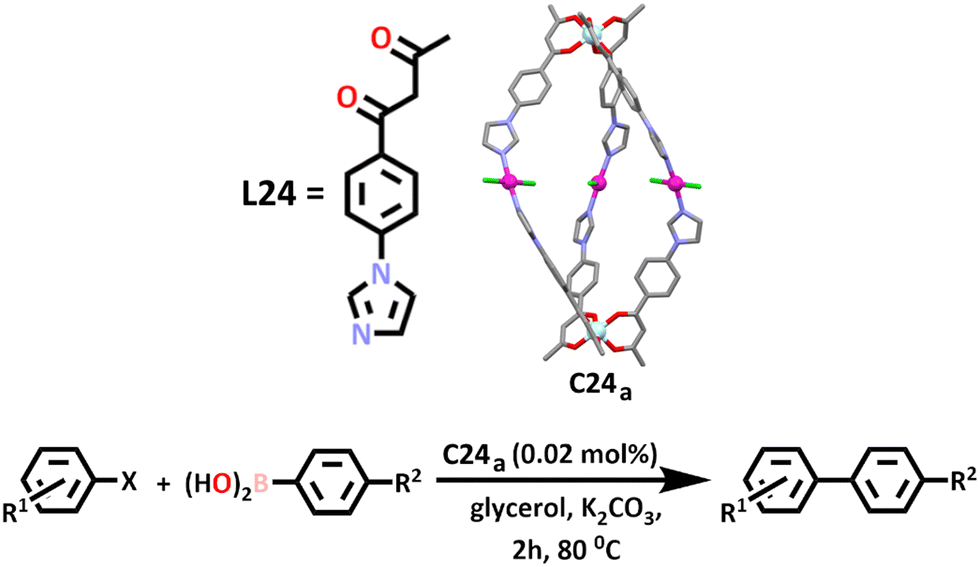 | ||
| Scheme 21 Chemical structure of the ligand, L24, and molecular structure of heterometallic [Al2Pd3L246Cl6] supramolecular cage (C24a). Colours: aluminium (pale blue), palladium (magenta), oxygen (red), carbon (black in the chemical structures, grey in the molecular structure), nitrogen (lavender), chlorine (green). Hydrogen atoms and solvents atoms have been omitted for clarity. The Suzuki–Miyaura coupling reaction catalysed by C24a and C24b has also been depicted, where R1 = COCH3, COOH, CN, NO2, CH3, H, OCH3 or Br, R2 = H, F, CH3, CHO, COCH3 or OCH3 and X = Br or Cl. | ||
A slightly larger cavity was afforded by addition of a benzene linker in the metallo–ligand, allowing for separation of the potential catalytic metal centres. It was found that both cages were able to catalyse the Suzuki–Miyaura coupling reaction, whereby an alkynyl, aryl or alkynyl organoborane is coupled with a halide or triflate in the presence of a base. In this work, the catalytic activity of both cages was investigate using a series of aryl halides and aryl boronic acids. A range of eco-friendly and bio renewable solvents were tested, with glycerol chosen over ethanol and water as the Pd(II) ion was observed to leach in these solvents, reducing the reusability of the catalyst. Under an ambient atmosphere and at 80–90 °C, the cross-coupled product reached yields of 99% in most cases when C24a was used as a catalyst. Similar results were obtained upon using C24b as a catalyst in the same reactions under the same conditions.
Mukherjee and co-workers have also synthesised a series of lantern shaped heterometallic cages incorporating the less often used zirconium(IV) metal ions along with commonly used Pd(II) cations.169 As is common when incorporating Zr(IV) into supramolecular architectures,2 two trinuclear zirconium metallo–ligands were first synthesised through the hydrolysis of Cp2ZrCl2 in the presence of a carboxylic acid (Scheme 22). The tripodal ligands this synthesis afforded showcased the hard Zr(IV) cation coordinated with the hard oxygen donors of the carboxylic acid. The nitrogen donors incorporated in the functional groups of the carboxylic acids provided the soft donors necessary for the subsequent coordination of ethylenediamine (en)-capped Pd(II) metal cations. This gave the final cages, C25a and C25b, where the zirconium-based ligands were directed by the en capping ligands in a cis fashion around the square planar palladium cation. Using cis-[(TMEDA)Pd(NO3)2] as opposed to cis-[(en)Pd(NO3)2] in combination with L25a or L25b also afforded two new Zr(IV)/Pd(II) cages.
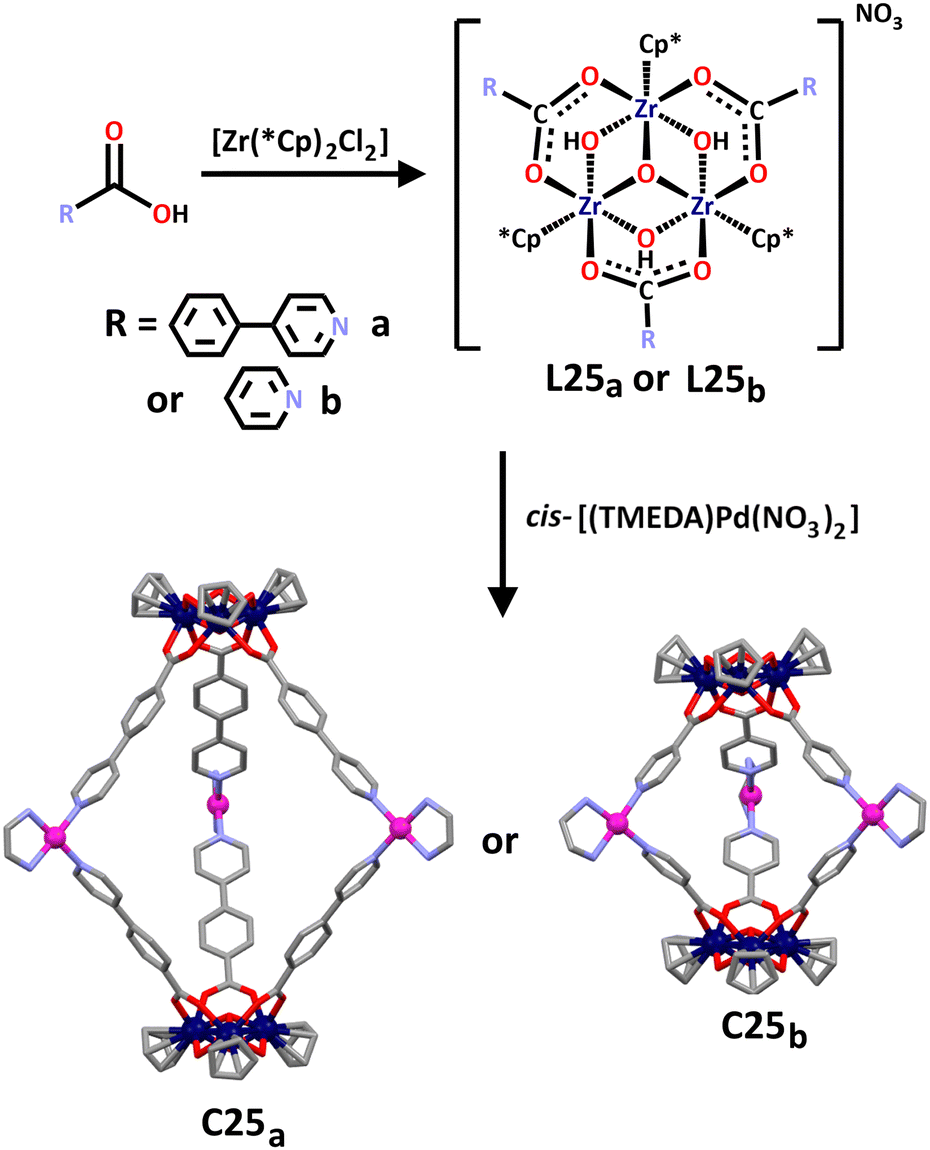 | ||
| Scheme 22 Synthesis of trinuclear Zr(IV) tripodal ligands L25a and L25b, and formation of resulting cages C25a and C25b. Cages C25a and C25b have been shown as molecular structures, with solvent molecules, counter anions and hydrogen atoms omitted for clarity. Element colours: carbon (black in the chemical structures, grey in the molecular structures), oxygen (red), nitrogen (lavender), palladium (magenta), zirconium (dark blue). | ||
Intriguingly, the group found C25b had guest binding abilities. Due to the water-soluble nature of this cage, the hydrophobic effect could be exploited allowing for organic fluorophores such as naphthalene and 2-naphthaldehyde to be bound within the cage interior. This was supported by 1H NMR data showing large upfield shifts of the α-pyridyl and aromatic protons in naphthalene when stirred in D2O with C25b. Additional support for this host–guest interaction was provided by NOE interactions between α and β protons of C25b with naphthalene protons observed in 2D NOESY NMR studies. Similar 1H NMR and 2D NOESY NMR results also supported the observed encapsulation of 2-naphthaldehyde within C25b. A host–guest stoichiometry of 1:2 was found in both cases, with binding constants between C25b and naphthalene calculated to be 1120 ± 41 and 96 ± 12 M−1, and binding constants between C25b and 2-naphthladehyde calculated to be 2488 ± 27 and 310 ± 32 M−1.
Weak link and post-assembly modification approaches
The weak link approach (Fig. 11) initially generates macrocyclic MSAs featuring hemilabile ligands that are bonded to the metal ions in a bidentate fashion in such a way that one of the metal–ligand bonds is stronger than the other. The addition of a second, usually small, co-ligand (or cofactor) breaks the weak link (weaker metal ligand interaction) generating the final cavity containing macrocycle or cage.170–172 This method has been extensively used, by the Mirkin group, to construct homometallic MSAs. They have also developed several heterometallic MSAs173 and used some of them as allosteric catalysts.174–176 However, most recent examples of MSAs generated via the weak-link approach have been homometallic177,178 systems.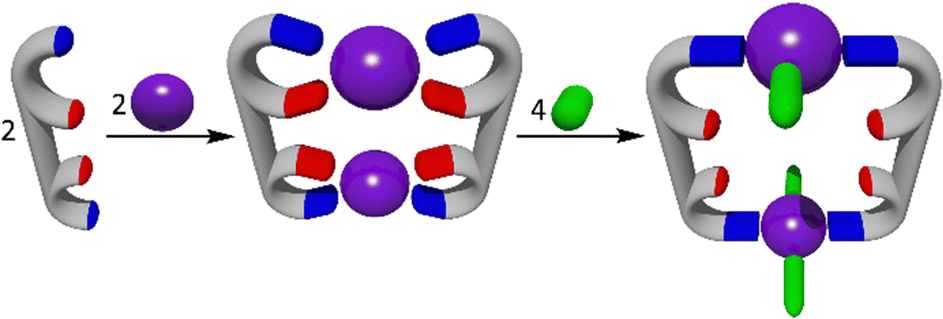 | ||
| Fig. 11 A carton representation of the weak link approach to MSAs. Initially a metallomacrocycle featuring hemilabile ligands that are bonded to the metal ions through both strong (blue) and weak (red) metal–ligand interactions is synthesised. Then a co-ligand (green) which breaks the weak link is added to generate the final cavity containing MSAs. | ||
Post-assembly modification is when an already synthesized structure is further modified. Post-assembly modification of supramolecular cages is limited to structures which are stable enough to withstand the modification conditions.179 There are potentially many types of post-assembly modifications, metals can be exchanged,180 additional groups such as proteins or alkyl chains can be added,181 or reversible bonds such as imines are reduced.7 The goals of post-assembly modification can be a wide range of things, from increasing the host–guest interactions,182 to making more stable/soluble complexes,181 or altering the structures’ spin crossover properties.183 Pre-assembly functionalisation is often impractical as it may be too difficult or might result in the formation of other, less desired structures, so the supramolecular architectures are formed and then altered after they have assembled.184 In order for post assembly modification to be a viable option, the modification must be highly efficient (high yields) and ideally should occur under mild reaction conditions.185
Law and co-workers synthesized a variety of homometallic helicates by reacting two equivalents of Ln(OTf)3 (where Ln = La(III), Sm(III), Eu(III), Gd(III) and Lu(III)) with three equivalents of the previously synthesized ligand (L26).186 This resulted in the formation of five homometallic helicates. When two species of helicates were crystallised together, they underwent a transformation/post-assembly modification in which the helicates rearranged into a range of heterometallic tetrahedra. Combining the helicates to generate mixtures of [Eu2L263]6+ and [Ln2L263]6+ (Ln = Gd, Tb and Dy) resulted in the formation of a mixture of heterometallic [EunLn4−nL266] (n = 0–4) cages.187 The tetrahedral shape of the heterometallic cages was confirmed by single crystal X-ray crystallography, showing the assemblies contained six L26 and 4 lanthanoid ions. ESI-MS was also used to confirm the formulation of these heterometallic tetrahedra. For all cages, MS deconvolution showed 5 sets of peak series which corresponded to the different possible combinations of Ln and L26 (Scheme 23). The most populous species was found to be [Eu2Ln2L266]8+. Achieving this selectivity from a one pot self-assembly was not possible because L26 showed no selectivity towards lanthanide ions. These heterometallic tetrahedra have potential applications to act as chiroptical sensors and materials.
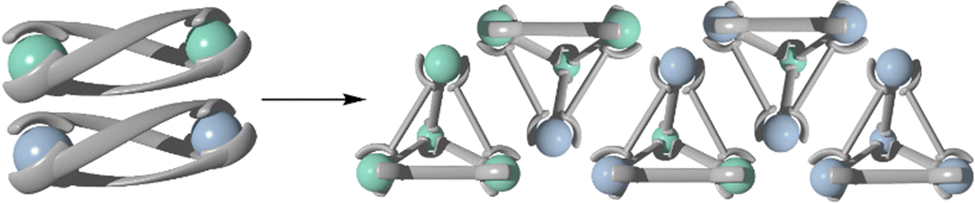 | ||
| Scheme 23 Cartoon representation of the rearrangement of two homometallic helicates into a range of homo and heterometallic tetrahedral cages. Colours: europium (aqua marine), gadolinium (pastel blue). | ||
Shionoya and co-workers synthesized a heterometallic cage containing a zinc-panel ligand and cobalt(II) ions (Scheme 24). The ligand was synthesized by refluxing the previously synthesized 5-methyl-2,2′-bipyridine188 with pyrrole and 4-pyridinecarboxaldehyde in propionic acid, which was then treated with MeI and AgOTf, resulting in the charged Zn(II)-porphyrin-based tris-bidentate ligand. The ligand was then treated with 1.33 equivalents of Co(OTf)2 in CD3CN/D2O = 19:1 at 70 °C. This resulted in the formation of a [Co4L273]8+ complex. Shionoya and co-workers were then able to selectively oxidise a single cobalt centre using one equivalent of cerium(II) ammonium nitrate at room temperature. The resulting cage was treated with excess Ni(OTf)2 and heated at 70 °C for 6 days. This led to the transmetalation of the three Co(II) ions with Ni(II), as the bpy motif/unit has a higher affinity for Ni(II) than Co(II). The structure of the supramolecular cage was confirmed using NMR spectroscopy and ESI-MS spectrometry. The molecular structure was determined by X-ray crystallography. Within the heterometallic structure, the Co(III) ions were found to inhabit the binding sites formed by three bidentate bpy units. The Co(III) could then be reduced back to Co(II) giving an overall [CoNi3L273]8+ structure.
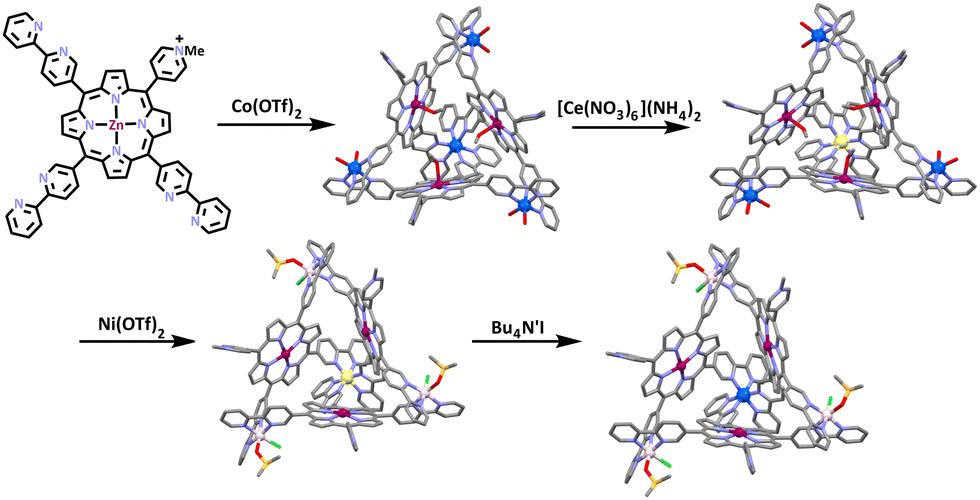 | ||
| Scheme 24 Synthetic route to the formation of the heterometallic panel cage achieved by selective oxidation and reduction of a single metal centre. Solvent molecules, counter anions and hydrogen atoms omitted for clarity. Colours: carbon (grey), nitrogen (lavender), oxygen (red), zinc (mulberry), chlorine (green), cobalt (deep blue), cobalt(III) (pale yellow), nickel (pale pink). | ||
The [CoNi3L273]8+ cage showed no sign of decomposition upon extended heating at 70 °C and even in the presence of DMSO-d6 acting as a strongly coordinating solvent, although it was thermodynamically unstable due to the higher affinity of the bpy units for Ni(II) than Co(II). The metal-selective reduction of the Co(III) site formed the [CoNi3L273]8+ cage, which could not be synthesized by conventional approaches, direct synthesis from the subcomponents, or a simple transmetalation. Thus, the reduction/oxidation of the Co(II) was necessary as a post assembly modification to enable the synthesis of this heterometallic cage.
Bloch and co-workers have very recently synthesized a new heterometallic paddle-wheel cage using the post assembly modification approach.189 The ligand (L28) was synthesized by a Suzuki coupling between bis(4-iodo-3,5-dimethyl-1H-pyrazol-1-yl)methane and 3-carboxyphenylboronic acid. L28 was then reacted with one equivalent of Cu2(OAc)4 in DMF, which resulted in the formation of the homometallic Cu paddlewheel cage (Scheme 25). This cage was also shown to be water stable.
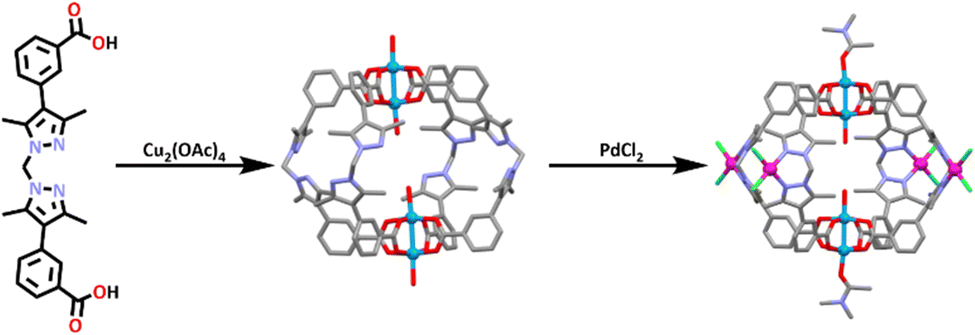 | ||
| Scheme 25 Synthesis of homometallic Cu(II) cage and post assembly modification forming the heterometallic [Cu2(PdCl2)4L284]. Molecular structures of both cages are shown. Solvent molecules and hydrogen atoms omitted for clarity. Colours: carbon (grey), nitrogen (lavender), oxygen (red), copper (blue), palladium, (magenta), chlorine (green). | ||
Reacting L28 with an excess of Cu(OAc)2 resulted in the immediate precipitation of an amorphous solid. This suggested that the reaction was under stoichiometric control and the assembly of the Cu paddle wheel complex did not appear to be in competition with the formation of other coordination networks, which has often been the case for other Cu2 paddle wheel containing cages.190
The homometallic copper cage was then combined with four equivalents of [Pd(CH3CN)2]Cl2 at room temperature to undergo a post-assembly metal insertion, resulting in the formation of two different isomers of a heterometallic [Cu4(L28(PdCl2)4)] cage. The structure of this cage was confirmed by 1H NMR spectroscopy which showed changes in the methylene (CH2) and methyl (CH3) resonances of the ligand (L28) which shifted downfield by 0.8 and 0.5 ppm respectively. The downfield shift of these protons was consistent with Pd(II) coordination to the pyrazole nitrogen donors in solution. CO2 gas adsorption measurements were carried out with the heterometallic CuPd systems able to take more CO2 than the homometallic copper cage.
Conclusions
The interest in (metallo)-supramolecular structures has increased exponentially in recent years. Focus has moved away from simple, symmetrical structures, towards more complex low symmetry systems and this has led to the synthesis of a wide variety of heterometallic architectures. This tutorial review has examined the different approaches (subcomponent self-assembly, symmetry interaction and directional bonding) to the synthesis of novel heterometallic metallosupramolecular cages. Similar to the related homometallic systems there are now numerous robust methods of the construction of heterometallic cages. These generic methods have mostly been developed for the generation of heterometallic cage systems. However, they could equally be applied to the synthesis of other types of heterometallic MSAs (macrocycles, helicates, and grids) which would further enhance the structural diversity of those systems. With a range of facile methods available for their synthesis there has been increasing interest in the properties of heterometallic MSAs. The lower symmetry and presence of two (or more) different metal ions in these MSAs has led to new properties. For example, some heterometallic MSAs have displayed host–guest chemistry that is distinct to what has been observed with symmetric single cavity homometallic hosts. In addition, systems that selectivity sequester different guests within discrete segregated compartments contained within single metallosupramolecular structures have been created. These host–guest properties could be potentially useful in numerous applications, including dual guest delivery or sequestration, energy transfer processes and enzyme-like multi-component reactions and catalysis. Similarly, the presence of two (or more) different metal ions within the MSAs has led to new electronic and photophysical behaviour due to synergistic/cooperative interactions between the metal ions. These types of systems may lead to new emergent behaviour that is not observed in the simpler homometallic MSAs. As with the electronic and photophysical properties the presence of multiple different metal ions within the MSAs could lead to new and interesting catalytic behaviour. Indeed, there are many heterometallic MSAs continuing to emerge that feature superior catalytic properties when compared to homometallic complexes.191–194 The coupling of photo- or redox-active metal ions within a single MSA catalyst is an excellent strategy for generating either new or better catalytic activity. These types of catalytic heterometallic MSA systems could play a key role in providing future energy solutions or offer alternative ways to mitigate climate change. The possibilities with these heterometallic architectures are almost endless and limited only by the imagination of the designer(s).However, there are limitations that also need to be overcome. The vast majority of homometallic MSAs are synthesised from labile metal ions to enable the formation of the thermodynamic product. For some biological and catalytic applications systems with labile metal ions can be decomposed/rearranged by the presence of biological nucleophiles and/or nucleophilic substrates. Therefore, to enhance these types of applications there is a need to develop more kinetically robust MSAs.195–198 The majority of heterometallic MSAs developed to date feature both inert and liable metal ions. Thus, it would be expected that those systems would be more robust than the related homometallic systems. However, so far there are very few studies that have confirmed this77,83 and this needs to be examined more in the future. Additionally, methods that enable the synthesis of heterometallic cages that feature only inert metal ions are desirable as these more robust systems may further enhance biological and catalytic applications.
Moreover, the host–guest chemistry of heterometallic MSAs needs to be examined in more detail as the lower symmetry cavity environments that these systems offer should lead to greater guest selectivity, which in turn could result in better, more enzyme-like, catalysis.199
Conflicts of interest
There are no conflicts to declare.Acknowledgements
LKM thanks the University of Otago for a R & E Seelye Trust Master's Scholarship. JDC thanks the MacDiarmid Institute for funding. JDC is also grateful to the Marsden Fund (UOA1726) for support.Notes and references
- J. W. Steed and J. L. Atwood, Supramolecular chemistry, Wiley, Chichester, 2000 Search PubMed.
- E.-S. M. El-Sayed, Y. D. Yuan, D. Zhao and D. Yuan, Acc. Chem. Res., 2022, 55, 1546–1560 CrossRef CAS PubMed.
- D. Zhang, T. K. Ronson, Y.-Q. Zou and J. R. Nitschke, Nat. Rev. Chem., 2021, 5, 168–182 CrossRef CAS PubMed.
- J. Liu, Z. Wang, P. Cheng, M. J. Zaworotko, Y. Chen and Z. Zhang, Nat. Rev. Chem., 2022, 6, 339–356 CrossRef PubMed.
- T. Tateishi, M. Yoshimura, S. Tokuda, F. Matsuda, D. Fujita and S. Furukawa, Coord. Chem. Rev., 2022, 467, 214612 CrossRef CAS.
- E.-S. M. El-Sayed and D. Yuan, Chem. Lett., 2020, 49, 28–53 CrossRef CAS.
- B. S. Pilgrim and N. R. Champness, ChemPlusChem, 2020, 85, 1842–1856 CrossRef CAS PubMed.
- E. Benchimol, B.-N. T. Nguyen, T. K. Ronson and J. R. Nitschke, Chem. Soc. Rev., 2022, 51, 5101–5135 RSC.
- S. Mollick, S. Fajal, S. Mukherjee and S. K. Ghosh, Chem. – Asian J., 2019, 14, 3096–3108 CrossRef CAS PubMed.
- S. Lee, H. Jeong, D. Nam, M. S. Lah and W. Choe, Chem. Soc. Rev., 2021, 50, 528–555 RSC.
- H. Vardhan, M. Yusubov and F. Verpoort, Coord. Chem. Rev., 2016, 306, 171–194 CrossRef CAS.
- N. Dey and C. J. E. Haynes, ChemPlusChem, 2021, 86, 418–433 CrossRef CAS PubMed.
- G. Moreno-Alcántar and A. Casini, FEBS Lett., 2023, 597, 191–202 CrossRef PubMed.
- M. J. Hannon, C. L. Painting and N. W. Alcock, Chem. Commun., 1999, 2023–2024, 10.1039/A905795A.
- E. C. Constable and A. M. W. C. Thompson, J. Chem. Soc., Dalton Trans., 1994, 1409–1418 RSC.
- E. C. Constable, Pure Appl. Chem., 1996, 68, 253–260 CrossRef CAS.
- C. M. Hartshorn and P. J. Steel, Chem. Commun., 1997, 541–542 RSC.
- V. G. Machado, P. N. W. Baxter and J.-M. Lehn, J. Braz. Chem. Soc., 2001, 12(4), 431–462 CrossRef CAS.
- S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853–908 CrossRef CAS PubMed.
- B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022–2043 CrossRef CAS PubMed.
- M. Fujita, K. Umemoto, M. Yoshizawa, N. Fujita, T. Kusukawa and K. Biradha, Chem. Commun., 2001, 509–518 RSC.
- S. R. Seidel and P. J. Stang, Acc. Chem. Res., 2002, 35, 972–983 CrossRef CAS PubMed.
- C. J. Kuehl, S. D. Huang and P. J. Stang, J. Am. Chem. Soc., 2001, 123, 9634–9641 CrossRef CAS PubMed.
- D. L. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS.
- J. R. Nitschke, Acc. Chem. Res., 2007, 40, 103–112 CrossRef CAS PubMed.
- B. Therrien, CrystEngComm, 2015, 17, 484–491 RSC.
- R. A. Kaner and P. Scott, Future Med. Chem., 2015, 7, 1–4 CrossRef CAS PubMed.
- R. A. S. Vasdev, D. Preston, S. Ø. Scottwell, H. J. L. Brooks, J. D. Crowley and M. P. Schramm, Molecules, 2016, 21, 1548 CrossRef PubMed.
- R. A. Bilbeisi, S. Zarra, H. L. C. Feltham, G. N. L. Jameson, J. K. Clegg, S. Brooker and J. R. Nitschke, Chem. – Eur. J., 2013, 19, 8058–8062 CrossRef CAS PubMed.
- A. Ferguson, M. A. Squire, D. Siretanu, D. Mitcov, C. Mathonière, R. Clérac and P. E. Kruger, Chem. Commun., 2013, 49, 1597–1599 RSC.
- R. J. Archer, C. S. Hawes, G. N. L. Jameson, V. McKee, B. Moubaraki, N. F. Chilton, K. S. Murray, W. Schmitt and P. E. Kruger, Dalton Trans., 2011, 40, 12368–12373 RSC.
- M. Frank, J. Hey, I. Balcioglu, Y.-S. Chen, D. Stalke, T. Suenobu, S. Fukuzumi, H. Frauendorf and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 10102–10106 CrossRef CAS PubMed.
- V. Croué, S. Goeb and M. Sallé, Chem. Commun., 2015, 51, 7275–7289 RSC.
- L. Xu, Y.-X. Wang and H.-B. Yang, Dalton Trans., 2015, 44, 867–890 RSC.
- O. Chepelin, J. Ujma, X. Wu, A. M. Z. Slawin, M. B. Pitak, S. J. Coles, J. Michel, A. C. Jones, P. E. Barran and P. J. Lusby, J. Am. Chem. Soc., 2012, 134, 19334–19337 CrossRef CAS PubMed.
- M. Han, R. Michel, B. He, Y.-S. Chen, D. Stalke, M. John and G. H. Clever, Angew. Chem., Int. Ed., 2013, 52, 1319–1323 CrossRef CAS PubMed.
- V. Blanco, M. D. García, A. Terenzi, E. Pía, A. Fernández-Mato, C. Peinador and J. M. Quintela, Chem. – Eur. J., 2010, 16, 12373–12380 CrossRef CAS PubMed.
- C. Peinador, E. Pía, V. Blanco, M. D. García and J. M. Quintela, Org. Lett., 2010, 12, 1380–1383 CrossRef CAS PubMed.
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS PubMed.
- M. Yamashina, Y. Sei, M. Akita and M. Yoshizawa, Nat. Commun., 2014, 5, 4662 CrossRef CAS PubMed.
- I. A. Riddell, M. M. J. Smulders, J. K. Clegg and J. R. Nitschke, Chem. Commun., 2011, 47, 457–459 RSC.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012–3035 CrossRef CAS PubMed.
- S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC.
- A. M. Lifschitz, M. S. Rosen, C. M. McGuirk and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 7252–7261 CrossRef CAS PubMed.
- M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS PubMed.
- R. J. Hooley, Synlett, 2020, 1448–1463 CrossRef CAS.
- J. E. M. Lewis, E. L. Gavey, S. A. Cameron and J. D. Crowley, Chem. Sci., 2012, 3, 778–784 RSC.
- D. Preston, J. E. M. Lewis and J. D. Crowley, J. Am. Chem. Soc., 2017, 139, 2379–2386 CrossRef CAS PubMed.
- B. Therrien, G. Süss-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, Angew. Chem., Int. Ed., 2008, 47, 3773–3776 CrossRef CAS PubMed.
- Y.-R. Zheng, K. Suntharalingam, T. C. Johnstone and S. J. Lippard, Chem. Sci., 2015, 6, 1189–1193 RSC.
- J. E. M. Lewis and J. D. Crowley, ChemPlusChem, 2020, 85, 815–827 CrossRef CAS PubMed.
- S. Khanra, M. Helliwell, F. Tuna, E. J. L. McInnes and R. E. P. Winpenny, Dalton Trans., 2009, 6166–6174 RSC.
- A. Tarzia, J. E. M. Lewis and K. E. Jelfs, Angew. Chem., Int. Ed., 2021, 60, 20879–20887 CrossRef CAS PubMed.
- F. Li and L. F. Lindoy, Aust. J. Chem., 2019, 72, 731–741 CrossRef CAS.
- H. Li, Z.-J. Yao, D. Liu and G.-X. Jin, Coord. Chem. Rev., 2015, 293–294, 139–157 CrossRef CAS.
- M. Hardy and A. Lützen, Chem. – Eur. J., 2020, 26, 13332–13346 CrossRef CAS PubMed.
- Y.-Y. Zhang, W.-X. Gao, L. Lin and G.-X. Jin, Coord. Chem. Rev., 2017, 344, 323–344 CrossRef CAS.
- Y. Sakata, S. Hiraoka and M. Shionoya, Chemistry, 2010, 16, 3318–3325 CrossRef CAS PubMed.
- X. Sun, D. W. Johnson, D. L. Caulder, K. N. Raymond and E. H. Wong, J. Am. Chem. Soc., 2001, 123, 2752–2763 CrossRef CAS PubMed.
- M. L. Saha, S. Neogi and M. Schmittel, Dalton Trans., 2014, 43, 3815–3834 RSC.
- M. L. Saha, J. W. Bats and M. Schmittel, Org. Biomol. Chem., 2013, 11, 5592–5595 RSC.
- M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2013, 135, 17743–17746 CrossRef CAS PubMed.
- A. J. Metherell and M. D. Ward, Chem. Sci., 2016, 7, 910–915 RSC.
- Q. Yang and J. Tang, Dalton Trans., 2019, 48, 769–778 RSC.
- S. Chakraborty and G. R. Newkome, Chem. Soc. Rev., 2018, 47, 3991–4016 RSC.
- A. K. Pal, B. Laramée-Milette and G. S. Hanan, RSC Adv., 2014, 4, 21262–21266 RSC.
- A. R. Stefankiewicz, J. Harrowfield, A. Madalan, K. Rissanen, A. N. Sobolev and J.-M. Lehn, Dalton Trans., 2011, 40, 12320–12332 RSC.
- M. Lalonde, W. Bury, O. Karagiaridi, Z. Brown, J. T. Hupp and O. K. Farha, J. Mater. Chem. A, 2013, 1, 5453–5468 RSC.
- N. C. Fletcher, R. T. Brown and A. P. Doherty, Inorg. Chem., 2006, 45, 6132–6134 CrossRef CAS PubMed.
- M. Viciano-Chumillas, X. Liu, A. Leyva-Pérez, D. Armentano, J. Ferrando-Soria and E. Pardo, Coord. Chem. Rev., 2022, 451, 214273 CrossRef CAS.
- N. M. Padial, B. Lerma-Berlanga, N. Almora-Barrios, J. Castells-Gil, I. da Silva, M. A. de la Mata, S. I. Molina, J. Hernández-Saz, A. E. Platero-Prats, S. Tatay and C. Marti-Gastaldo, J. Am. Chem. Soc., 2020, 142, 6638–6648 CrossRef CAS PubMed.
- H.-Y. Lin, Y.-T. Wang, D. Zhang and L. Xu, in Supramolecular Coordination Complexes, ed. S. Shanmugaraju, Elsevier, 2023, ch. 4, pp. 69–100 DOI:10.1016/B978-0-323-90582-4.00013-0.
- D. Zhang, T. K. Ronson and J. R. Nitschke, Acc. Chem. Res., 2018, 51, 2423–2436 CrossRef CAS PubMed.
- T. Tateishi, S. Takahashi, A. Okazawa, V. Martí-Centelles, J. Wang, T. Kojima, P. J. Lusby, H. Sato and S. Hiraoka, J. Am. Chem. Soc., 2019, 141, 19669–19676 CrossRef CAS PubMed.
- S. M. Jansze, M. D. Wise, A. V. Vologzhanina, R. Scopelliti and K. Severin, Chem. Sci., 2017, 8, 1901–1908 RSC.
- G. H. Clever and P. Punt, Acc. Chem. Res., 2017, 50, 2233–2243 CrossRef CAS PubMed.
- L. S. Lisboa, J. A. Findlay, L. J. Wright, C. G. Hartinger and J. D. Crowley, Angew. Chem., Int. Ed., 2020, 59, 11101–11107 CrossRef CAS PubMed.
- S. Bandi and D. K. Chand, Chem. – Eur. J., 2016, 22, 10330–10335 CrossRef CAS PubMed.
- A. Schmidt, A. Casini and F. E. Kuehn, Coord. Chem. Rev., 2014, 275, 19–36 CrossRef CAS.
- M. Pahwa, M. Sharma, S. K. Meethal and S. S. Agasti, Bull. Mater. Sci., 2020, 43, 1–7 CrossRef.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- L. S. Lisboa, M. Riisom, H. J. Dunne, D. Preston, S. M. F. Jamieson, L. J. Wright, C. G. Hartinger and J. D. Crowley, Dalton Trans., 2022, 51, 18438–18445 RSC.
- M. D. Johnstone, E. K. Schwarze, G. H. Clever and F. M. Pfeffer, Chem. – Eur. J., 2015, 21, 3948–3955 CrossRef CAS PubMed.
- S. Bandi, S. Samantray, R. D. Chakravarthy, A. K. Pal, G. S. Hanan and D. K. Chand, Eur. J. Inorg. Chem., 2016, 2816–2827 CrossRef CAS.
- S. Bandi, A. K. Pal, G. S. Hanan and D. K. Chand, Chem. – Eur. J., 2014, 20, 13122–13126 CrossRef CAS PubMed.
- R. Zhu, I. Regeni, J. J. Holstein, B. Dittrich, M. Simon, S. Prévost, M. Gradzielski and G. H. Clever, Angew. Chem., Int. Ed., 2018, 57, 13652–13656 CrossRef CAS PubMed.
- K. Wu, B. Zhang, C. Drechsler, J. J. Holstein and G. H. Clever, Angew. Chem., Int. Ed., 2021, 60, 6403–6407 CrossRef CAS PubMed.
- L. S. Lisboa, D. Preston, C. J. McAdam, L. J. Wright, C. G. Hartinger and J. D. Crowley, Angew. Chem., Int. Ed., 2022, 61, e202201700 CrossRef CAS PubMed.
- J. P. Carpenter, T. K. Ronson, F. J. Rizzuto, T. O. Heliot, P. Grice and J. R. Nitschke, J. Am. Chem. Soc., 2022, 144, 8467–8473 CrossRef CAS PubMed.
- P. K. Chattaraj, H. Lee and R. G. Parr, J. Am. Chem. Soc., 1991, 113, 1855–1856 CrossRef CAS.
- R. A. Bilbeisi, J. K. Clegg, N. Elgrishi, X. de Hatten, M. Devillard, B. Breiner, P. Mal and J. R. Nitschke, J. Am. Chem. Soc., 2012, 134, 5110–5119 CrossRef CAS PubMed.
- M. Kieffer, R. A. Bilbeisi, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2020, 59, 11369–11373 CrossRef CAS PubMed.
- D. Yang, J. L. Greenfield, T. K. Ronson, L. K. S. von Krbek, L. Yu and J. R. Nitschke, J. Am. Chem. Soc., 2020, 142, 19856–19861 CrossRef CAS PubMed.
- H. Takezawa, T. Murase, G. Resnati, P. Metrangolo and M. Fujita, J. Am. Chem. Soc., 2014, 136, 1786–1788 CrossRef CAS PubMed.
- M. M. J. Smulders, A. Jiménez and J. R. Nitschke, Angew. Chem., Int. Ed., 2012, 51, 6681–6685 CrossRef CAS PubMed.
- R. Li, M. P. Martin, Y. Liu, B. Wang, R. A. Patel, J.-Y. Zhu, N. Sun, R. Pireddu, N. J. Lawrence, J. Li, E. B. Haura, S.-S. Sung, W. C. Guida, E. Schonbrunn and S. M. Sebti, J. Med. Chem., 2012, 55, 2474–2478 CrossRef CAS PubMed.
- M. Hardy, J. Tessarolo, J. J. Holstein, N. Struch, N. Wagner, R. Weisbarth, M. Engeser, J. Beck, S. Horiuchi, G. H. Clever and A. Lützen, Angew. Chem., Int. Ed., 2021, 60, 22562–22569 CrossRef CAS PubMed.
- P. Gütlich and H. A. Goodwin, Spin Crossover in Transition Metal Compounds I, 2004, pp. 1–47 Search PubMed.
- S. Brooker, Chem. Soc. Rev., 2015, 44, 2880–2892 RSC.
- O. Kahn, Chem. Br., 1999, 35, 24–27 CAS.
- A. J. McConnell, Supramol. Chem., 2018, 30, 858–868 CrossRef CAS.
- A. J. McConnell, Chem. Soc. Rev., 2022, 51, 2957–2971 RSC.
- X.-C. Zhou, L.-X. Wu, X.-Z. Wang, Y.-L. Lai, Y.-Y. Ge, J. Su, X.-P. Zhou and D. Li, Inorg. Chem., 2022, 61, 5196–5200 CrossRef CAS PubMed.
- H. Min, A. R. Craze, T. Taira, M. J. Wallis, M. M. Bhadbhade, R. Tian, D. J. Fanna, R. Wuhrer, S. Hayami, J. K. Clegg, C. E. Marjo, L. F. Lindoy and F. Li, Chemistry, 2022, 4, 535–547 CrossRef CAS.
- H. Gildenast, F. Busse and U. Englert, Cryst. Growth Des., 2021, 21, 5807–5817 CrossRef CAS.
- M. Hardy, N. Struch, J. J. Holstein, G. Schnakenburg, N. Wagner, M. Engeser, J. Beck, G. H. Clever and A. Lützen, Angew. Chem., Int. Ed., 2020, 59, 3195–3200 CrossRef CAS PubMed.
- R. A. S. Vasdev, J. A. Findlay, A. L. Garden and J. D. Crowley, Chem. Commun., 2019, 55, 7506–7509 RSC.
- Y. Yang, Y. Wu, J.-H. Jia, X.-Y. Zheng, Q. Zhang, K.-C. Xiong, Z.-M. Zhang and Q.-M. Wang, Cryst. Growth Des., 2018, 18, 4555–4561 CrossRef CAS.
- S. Sanz, H. M. O’Connor, P. Comar, A. Baldansuren, M. B. Pitak, S. J. Coles, H. Weihe, N. F. Chilton, E. J. L. McInnes, P. J. Lusby, S. Piligkos and E. K. Brechin, Inorg. Chem., 2018, 57, 3500–3506 CrossRef CAS PubMed.
- D. Preston, J. J. Sutton, K. C. Gordon and J. D. Crowley, Angew. Chem., Int. Ed., 2018, 57, 8659–8663 CrossRef CAS PubMed.
- Y. Yang, J.-H. Jia, X.-L. Pei, H. Zheng, Z.-A. Nan and Q.-M. Wang, Chem. Commun., 2015, 51, 3804–3807 RSC.
- K. Li, L.-Y. Zhang, C. Yan, S.-C. Wei, M. Pan, L. Zhang and C.-Y. Su, J. Am. Chem. Soc., 2014, 136, 4456–4459 CrossRef CAS PubMed.
- H.-B. Wu and Q.-M. Wang, Angew. Chem., Int. Ed., 2009, 48, 7343–7345 CrossRef CAS PubMed.
- T.-L. Ho, Chem. Rev., 1975, 75, 1–20 CrossRef CAS.
- H. M. O’Connor, S. Sanz, A. J. Scott, M. B. Pitak, W. T. Klooster, S. J. Coles, N. F. Chilton, E. J. L. McInnes, P. J. Lusby, H. Weihe, S. Piligkos and E. K. Brechin, Molecules, 2021, 26, 757 CrossRef PubMed.
- S. Sanz, H. M. O'Connor, E. M. Pineda, K. S. Pedersen, G. S. Nichol, O. Mønsted, H. Weihe, S. Piligkos, E. J. L. McInnes, P. J. Lusby and E. K. Brechin, Angew. Chem., Int. Ed., 2015, 54, 6761–6764 CrossRef CAS PubMed.
- W. J. Ramsay, T. K. Ronson, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 13439–13443 CrossRef CAS PubMed.
- W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2011, 50, 3479–3483 CrossRef CAS PubMed.
- F. Li, N. F. Sciortino, J. K. Clegg, S. M. Neville and C. J. Kepert, Aust. J. Chem., 2014, 67, 1625 CrossRef CAS.
- F. Reichel, J. K. Clegg, K. Gloe, K. Gloe, J. J. Weigand, J. K. Reynolds, C.-G. Li, J. R. Aldrich-Wright, C. J. Kepert, L. F. Lindoy, H.-C. Yao and F. Li, Inorg. Chem., 2014, 53, 688–690 CrossRef CAS PubMed.
- M. B. Duriska, S. M. Neville, J. Lu, S. S. Iremonger, J. F. Boas, C. J. Kepert and S. R. Batten, Angew. Chem., Int. Ed., 2009, 48, 8919–8922 CrossRef CAS PubMed.
- M. B. Duriska, S. M. Neville, B. Moubaraki, J. D. Cashion, G. J. Halder, K. W. Chapman, C. Balde, J.-F. Létard, K. S. Murray, C. J. Kepert and S. R. Batten, Angew. Chem., Int. Ed., 2009, 48, 2549–2552 CrossRef CAS PubMed.
- A. Craze, N. Sciortino, M. Badbhade, C. Kepert, C. Marjo and F. Li, Inorganics, 2017, 5, 62 CrossRef.
- A. Craze, M. Bhadbhade, C. Kepert, L. Lindoy, C. Marjo and F. Li, Crystals, 2018, 8, 376 CrossRef.
- A. R. Craze, M. M. Bhadbhade, Y. Komatsumaru, C. E. Marjo, S. Hayami and F. Li, Inorg. Chem., 2020, 59, 1274–1283 CrossRef CAS PubMed.
- H. Min, A. R. Craze, M. J. Wallis, R. Tokunaga, T. Taira, Y. Hirai, M. M. Bhadbhade, D. J. Fanna, C. E. Marjo, S. Hayami, L. F. Lindoy and F. Li, Chem. – Eur. J., 2023, 29, e202203742 CrossRef CAS PubMed.
- T. B. Rauchfuss, S. R. Wilson and D. A. Wrobleski, J. Am. Chem. Soc., 1981, 103, 6769–6770 CrossRef CAS.
- D. A. Wrobleski and T. B. Rauchfuss, J. Am. Chem. Soc., 1982, 104, 2314–2316 CrossRef CAS.
- N. R. Lagesse, K. Y. L. Tan, J. D. Crowley and J. A. Findlay, Chem. – Asian J., 2020, 15, 1567–1573 CrossRef CAS PubMed.
- D. Preston, R. A. J. Tucker, A. L. Garden and J. D. Crowley, Inorg. Chem., 2016, 55, 8928–8934 CrossRef CAS PubMed.
- C. Colomban, C. Fuertes-Espinosa, S. Goeb, M. Sallé, M. Costas, L. Blancafort and X. Ribas, Chem. – Eur. J., 2018, 24, 4371–4381 CrossRef CAS PubMed.
- M. Balci, Chem. Rev., 1981, 81, 91–108 CrossRef CAS.
- M. Yoshizawa, Y. Takeyama, T. Okano and M. Fujita, J. Am. Chem. Soc., 2003, 125, 3243–3247 CrossRef CAS PubMed.
- S. Karthikeyan and V. Ramamurthy, Tetrahedron Lett., 2005, 46, 4495–4498 CrossRef CAS.
- D. Rota Martir, D. B. Cordes, A. M. Z. Slawin, D. Escudero, D. Jacquemin, S. L. Warriner and E. Zysman-Colman, Chem. Commun., 2018, 54, 6016–6019 RSC.
- D. Rota Martir and E. Zysman-Colman, Chem. Commun., 2019, 55, 139–158 RSC.
- C. Shen, A. D. W. Kennedy, W. A. Donald, A. M. Torres, W. S. Price and J. E. Beves, Inorg. Chim. Acta, 2017, 458, 122–128 CrossRef CAS.
- J. Yang, M. Bhadbhade, W. A. Donald, H. Iranmanesh, E. G. Moore, H. Yan and J. E. Beves, Chem. Commun., 2015, 51, 4465–4468 RSC.
- Y.-L. Li, A.-J. Li, S.-L. Huang, J. J. Vittal and G.-Y. Yang, Chem. Soc. Rev., 2023, 52, 4725–4754 RSC.
- E. T. Luis, H. Iranmanesh and J. E. Beves, Polyhedron, 2019, 160, 1–9 CrossRef CAS.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- B. Therrien, Chem. – Eur. J., 2013, 19, 8378–8386 CrossRef CAS PubMed.
- F. Schmitt, J. Freudenreich, N. P. E. Barry, L. Juillerat-Jeanneret, G. Süss-Fink and B. Therrien, J. Am. Chem. Soc., 2012, 134, 754–757 CrossRef CAS PubMed.
- S. Chen, K. Li, F. Zhao, L. Zhang, M. Pan, Y.-Z. Fan, J. Guo, J. Shi and C.-Y. Su, Nat. Commun., 2016, 7, 13169 CrossRef CAS PubMed.
- K. Wu, K. Li, S. Chen, Y. J. Hou, Y. L. Lu, J. S. Wang, M. J. Wei, M. Pan and C. Y. Su, Angew. Chem., Int. Ed., 2020, 59, 2639–2643 CrossRef CAS PubMed.
- K. Wu, K. Li, Y.-J. Hou, M. Pan, L.-Y. Zhang, L. Chen and C.-Y. Su, Nat. Commun., 2016, 7, 10487 CrossRef CAS PubMed.
- J. Guo, Y.-W. Xu, K. Li, L.-M. Xiao, S. Chen, K. Wu, X.-D. Chen, Y.-Z. Fan, J.-M. Liu and C.-Y. Su, Angew. Chem., 2017, 129, 3910–3914 CrossRef.
- K. Wu, Y. J. Hou, Y. L. Lu, Y. Z. Fan, Y. N. Fan, H. J. Yu, K. Li, M. Pan and C. Y. Su, Chem. – Eur. J., 2019, 25, 11903–11909 CrossRef CAS PubMed.
- J. Guo, Y. Z. Fan, Y. L. Lu, S. P. Zheng and C. Y. Su, Angew. Chem., Int. Ed., 2020, 59, 8661–8669 CrossRef CAS PubMed.
- J.-S. Wang, K. Wu, C. Yin, K. Li, Y. Huang, J. Ruan, X. Feng, P. Hu and C.-Y. Su, Nat. Commun., 2020, 11, 4675 CrossRef CAS PubMed.
- Y.-P. Wang, K. Wu, M. Pan, K. Li, J.-T. Mo, X.-H. Duan, H.-Z. He, J. Shen and C.-Y. Su, ACS Appl. Mater. Interfaces, 2020, 12, 35873–35881 CrossRef CAS PubMed.
- K. Li, K. Wu, Y. L. Lu, J. Guo, P. Hu and C. Y. Su, Angew. Chem., Int. Ed., 2022, 61, e202114070 CrossRef CAS PubMed.
- Y. Wang, J. Chen, J. Yang, Z. Jiao and C. Y. Su, Angew. Chem., Int. Ed., 2023, 62, e202303288 CrossRef CAS PubMed.
- L. Wang, R. Zhang, Q. Han, C. Xu, W. Chen, H. Yang, G. Gao, W. Qin and W. Liu, Green Chem., 2018, 20, 5311–5317 RSC.
- R.-P. Ye, J. Ding, W. Gong, M. D. Argyle, Q. Zhong, Y. Wang, C. K. Russell, Z. Xu, A. G. Russell, Q. Li, M. Fan and Y.-G. Yao, Nat. Commun., 2019, 10, 5698 CrossRef CAS PubMed.
- M. Alves, B. Grignard, R. Mereau, C. Jerome, T. Tassaing and C. Detrembleur, Catal. Sci. Technol., 2017, 7, 2651–2684 RSC.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- G. A. Bhat and D. J. Darensbourg, Green Chem., 2022, 24, 5007–5034 RSC.
- L. Guo, K. J. Lamb and M. North, Green Chem., 2021, 23, 77–118 RSC.
- L. Wang, C. Xu, Q. Han, X. Tang, P. Zhou, R. Zhang, G. Gao, B. Xu, W. Qin and W. Liu, Chem. Commun., 2018, 54, 2212–2215 RSC.
- H. Yang, Y. Xie, W. Chen, X. Tang, M. Hu, Y. Shu, L. Wang and W. Liu, J. CO2 Util., 2021, 54, 101780 CrossRef CAS.
- Q.-Y. Zhu, L.-P. Zhou, L.-X. Cai, S.-J. Hu, X.-Z. Li and Q.-F. Sun, Inorg. Chem., 2022, 61, 16814–16821 CrossRef CAS PubMed.
- A. C. Pearcy, L. S. Lisboa, D. Preston, N. B. Page, T. Lawrence, L. J. Wright, C. G. Hartinger and J. D. Crowley, Chem. Sci., 2023, 14, 8615–8623 RSC.
- A. A. Adeyemo and P. S. Mukherjee, Beilstein J. Org. Chem., 2018, 14, 2242–2249 CrossRef CAS PubMed.
- X.-C. Liu and L. Lin, Inorg. Chem. Commun., 2021, 129, 108660 CrossRef CAS.
- L.-L. Dang, T. Chen, T.-T. Zhang, T.-T. Li, J.-L. Song, K.-J. Zhang and L.-F. Ma, Molecules, 2022, 27, 3756 CrossRef CAS PubMed.
- X.-R. Wu, S.-Y. Yao, L.-Q. Wei, L.-P. Li and B.-H. Ye, Inorg. Chim. Acta, 2018, 482, 605–611 CrossRef CAS.
- M. Maity, P. Howlader and P. S. Mukherjee, Cryst. Growth Des., 2018, 18, 6956–6964 CrossRef CAS.
- N. C. Gianneschi, M. S. Masar and C. A. Mirkin, Acc. Chem. Res., 2005, 38, 825–837 CrossRef CAS PubMed.
- A. H. Eisenberg, F. M. Dixon, C. A. Mirkin, C. L. Stern, C. D. Incarvito and A. L. Rheingold, Organometallics, 2001, 20, 2052–2058 CrossRef CAS.
- J. R. Farrell, C. A. Mirkin, I. A. Guzei, L. M. Liable-Sands and A. L. Rheingold, Angew. Chem., Int. Ed., 1998, 37, 465–467 CrossRef CAS PubMed.
- A. H. Eisenberg, M. V. Ovchinnikov and C. A. Mirkin, J. Am. Chem. Soc., 2003, 125, 2836–2837 CrossRef CAS PubMed.
- N. C. Gianneschi, P. A. Bertin, S. T. Nguyen, C. A. Mirkin, L. N. Zakharov and A. L. Rheingold, J. Am. Chem. Soc., 2003, 125, 10508–10509 CrossRef CAS PubMed.
- C. G. Oliveri, J. Heo, S. T. Nguyen, C. A. Mirkin and Z. Wawrzak, Inorg. Chem., 2007, 46, 7716–7718 CrossRef CAS PubMed.
- H. J. Yoon, J. Heo and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 14182–14183 CrossRef CAS PubMed.
- A. I. d’Aquino, H. F. Cheng, J. Barroso-Flores, Z. S. Kean, J. Mendez-Arroyo, C. M. McGuirk and C. A. Mirkin, Inorg. Chem., 2018, 57, 3568–3578 CrossRef PubMed.
- J. Mendez-Arroyo, A. I. d’Aquino, A. B. Chinen, Y. D. Manraj and C. A. Mirkin, J. Am. Chem. Soc., 2017, 139, 1368–1371 CrossRef CAS PubMed.
- R. J. Severinsen, G. J. Rowlands and P. G. Plieger, J. Inclusion Phenom. Macrocyclic Chem., 2020, 96, 29–42 CrossRef CAS.
- M. E. Carnes, M. S. Collins and D. W. Johnson, Chem. Soc. Rev., 2014, 43, 1825–1834 RSC.
- A. Carné-Sánchez, J. Albalad, T. Grancha, I. Imaz, J. Juanhuix, P. Larpent, S. Furukawa and D. Maspoch, J. Am. Chem. Soc., 2019, 141, 4094–4102 CrossRef PubMed.
- C. T. McTernan, T. K. Ronson and J. R. Nitschke, J. Am. Chem. Soc., 2019, 141, 6837–6842 CrossRef CAS PubMed.
- A. Enríquez-Cabrera, L. Routaboul, L. Salmon and A. Bousseksou, Dalton Trans., 2019, 48, 16853–16856 RSC.
- H. Zeng, L. Stewart-Yates, L. M. Casey, N. Bampos and D. A. Roberts, ChemPlusChem, 2020, 85, 1249–1269 CrossRef CAS PubMed.
- D. A. Roberts, B. S. Pilgrim and J. R. Nitschke, Chem. Soc. Rev., 2018, 47, 626–644 RSC.
- K.-H. Yim, C.-T. Yeung, H.-Y. Wong and G.-L. Law, Inorg. Chem. Front., 2021, 8, 2952–2964 RSC.
- K.-H. Yim, C.-T. Yeung, M. Y.-M. Wong, M. R. Probert and G.-L. Law, Chem. – Eur. J., 2022, 28, e202201655 CrossRef CAS PubMed.
- T. Nakamura, H. Ube, M. Shiro and M. Shionoya, Angew. Chem., Int. Ed., 2013, 52, 720–723 CrossRef CAS PubMed.
- M. T. Yong, O. M. Linder-Patton and W. M. Bloch, Inorg. Chem., 2022, 61, 12863–12869 CrossRef CAS PubMed.
- O. Barreda, G. Bannwart, G. P. Yap and E. D. Bloch, ACS Appl. Mater. Interfaces, 2018, 10, 11420–11424 CrossRef CAS PubMed.
- R. Ham, C. J. Nielsen, S. Pullen and J. N. H. Reek, Chem. Rev., 2023, 123, 5225–5261 CrossRef CAS PubMed.
- L. J. Jongkind, X. Caumes, A. P. T. Hartendorp and J. N. H. Reek, Acc. Chem. Res., 2018, 51, 2115–2128 CrossRef CAS PubMed.
- E. O. Bobylev, J. Ruijter, D. A. Poole III, S. Mathew, B. de Bruin and J. N. H. Reek, Angew. Chem., Int. Ed., 2023, 62, e202218162 CrossRef CAS PubMed.
- E. O. Bobylev, Y. Zeng, K. Weijgertse, E. Koelman, E. M. Meijer, B. de Bruin, A. Kros and J. N. H. Reek, Chem, 2023, 9, 1578–1593 CAS.
- E. O. Bobylev, D. A. Poole III, B. de Bruin and J. N. H. Reek, Chem. – Eur. J., 2021, 27, 12667–12674 CrossRef CAS PubMed.
- E. O. Bobylev, R. A. Knol, S. Mathew, D. A. Poole, I. Kotsogianni, N. I. Martin, B. de Bruin, A. Kros and J. N. H. Reek, Chem. Sci., 2023, 14, 6943–6952 RSC.
- B. P. Burke, W. Grantham, M. J. Burke, G. S. Nichol, D. Roberts, I. Renard, R. Hargreaves, C. Cawthorne, S. J. Archibald and P. J. Lusby, J. Am. Chem. Soc., 2018, 140, 16877–16881 CrossRef CAS PubMed.
- P. Su, W. Zhang, C. Guo, H. Liu, C. Xiong, R. Tang, C. He, Z. Chen, X. Yu, H. Wang and X. Li, J. Am. Chem. Soc., 2023, 145, 18607–18622 CrossRef CAS PubMed.
- T. K. Piskorz, V. Martí-Centelles, R. L. Spicer, F. Duarte and P. J. Lusby, Chem. Sci., 2023, 14, 11300–11331 RSC.
| This journal is © The Royal Society of Chemistry 2024 |