
Toxic, radioactive, and disordered: a total scattering study of TlTcO4†
Bryce G.
Mullens
 a,
Frederick P.
Marlton
*b,
Matilde
Saura-Múzquiz
c,
Michelle
Everett
d,
Cheng
Li
d,
Alicia M.
Manjon-Sanz
d,
Matthew G.
Tucker
d,
Frederic
Poineau
e,
James
Louis-Jean
e,
Supratik
Mukherjee
f,
Subrata
Mondal
f,
Ganapathy
Vaitheeswaran
*g and
Brendan J.
Kennedy
a
a,
Frederick P.
Marlton
*b,
Matilde
Saura-Múzquiz
c,
Michelle
Everett
d,
Cheng
Li
d,
Alicia M.
Manjon-Sanz
d,
Matthew G.
Tucker
d,
Frederic
Poineau
e,
James
Louis-Jean
e,
Supratik
Mukherjee
f,
Subrata
Mondal
f,
Ganapathy
Vaitheeswaran
*g and
Brendan J.
Kennedy
a
aSchool of Chemistry, The University of Sydney, Sydney, New South Wales 2006, Australia. E-mail: brendan.kennedy@sydney.edu.au
bCentre for Clean Energy Technology, School of Mathematical and Physical Sciences, Faculty of Science, University of Technology Sydney, Sydney, New South Wales 2007, Australia. E-mail: frederick.marlton@uts.edu.au
cDepartamento de Física de Materiales, Facultad de Ciencias Físicas, Universidad Complutense de Madrid, 28040, Madrid, Spain
dNeutron Scattering Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
eDepartment of Chemistry, University of Nevada Las Vegas, Las Vegas, Nevada 89154, USA
fAdvanced Centre of Research in High Energy Materials (ACRHEM), University of Hyderabad, Prof. C. R. Rao Road, Gachibowli, 500046 Hyderabad, Telangana, India
gSchool of Physics, University of Hyderabad, Prof. C. R. Rao Road, Gachibowli, 500046 Hyderabad, Telangana, India. E-mail: vaithee@uohyd.ac.in
First published on 25th November 2024
Abstract
A detailed variable temperature neutron total scattering study of the potential nuclear waste matrix TlTcO4 was conducted. The long-range average structure of TlTcO4 undergoes an orthorhombic Pnma to tetragonal I41/amd phase transition below 600 K, consistent with previous synchrotron X-ray diffraction studies. However, several anomalies were observed in the Rietveld refinements to the neutron powder diffraction data, such as large atomic displacement parameters at low temperature and a shortening of the Tc–O bond distance upon heating. Modelling the short-range local structure of both the low- and high-temperature data required a lowering of symmetry to the monoclinic P21/c model due to the stereochemical activity of the Tl+ 6s2 lone pairs. Density functional theory calculations also verified this model to have a lower ground state energy than the corresponding long-range average structure. It is concluded that at low temperatures, the Tl+ 6s2 lone pairs are ‘frozen’ into the structure. Upon heating, the rigid TcO4 tetrahedra begin to rotate, as governed by the Γ3+ and M4+ modes. However, there is a disconnect between the two length scales, with the 6s2 lone pair electrons remaining stereochemically active on the local scale, as observed in the neutron pair distribution function fits. The orthorhombic Pnma to tetragonal I41/amd phase transition is seemingly the result of a change in the correlation length of the Tl+ 6s2 lone pairs, leading to a larger unit cell volume due to their uncorrelated displacements.
Introduction
The increasing concern of carbon emissions, in the context of climate change, has caused a global shift in the world's reliance on carbon-based fuels. This has led to a renewed interest in nuclear power which, according to the US Office of Nuclear Energy, provided almost half of the United States’ carbon-neutral energy in 2023. The safe storage of fission products from spent nuclear fuel remains a major challenge and is a constant impediment to the continued use of nuclear power. One of the most challenging fission products is 99Tc, with a cumulated yield of ∼6% from 235U fission. Technetium-99m (99mTc) is currently used as a medical radioisotope for selective imaging diagnosis due to its relatively short half-life, accounting for over half of all nuclear medicine procedures. However its decay product, 99Tc (upon decaying from 99mTc via isomeric transition), presents an environmental challenge due to its high mobility, β− emission, and half-life of around 211![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years.1
000 years.1
In developing technologies for the safe storage of Tc-containing waste, it is necessary to consider the chemistry of the commonly encountered oxidation states; namely Tc4+ and Tc7+. The Tc4+ oxidation state is only soluble in acidic conditions (below pH ∼3), meaning it often precipitates out as solid TcO2·nH2O under environmental conditions.2,3 Tc7+ forms the water-soluble and highly stable pertechnetate anion TcO4− which is much more mobile and can lead to the corrosion of Fe- and Zr-based storage containers.4–6 As a consequence, several materials have been investigated for storing Tc-containing waste to prevent its leaching into the environment. These storage mechanisms include metal oxides,7 glasses,8,9 and metal–organic frameworks (MOFs).10,11
Since Tc has no stable isotopes, it is common for studies to use Re as a nonradioactive surrogate.12–16 This is due to the predicted similarities in the chemical behaviour of Tc7+ and Re7+, which are attributed to their identical oxidation states and similar ionic radii (Tc7+ = 0.37 vs. Re7+ = 0.38 Å in tetrahedra geometry).17 However, a growing body of literature shows that Tc7+ and Re7+ do not always act identically. Lukens et al. reported that Tc7+ is significantly more volatile than Re7+, leading to a decrease in the amount stabilised in waste glasses.18 Gan et al. described the different behaviours of Tc and Re under various redox conditions, reflecting the lower standard reduction potential of Re7+ (ReO4−/ReO2 = 0.510 V) compared to Tc7+ (TcO4−/TcO2 = 0.738 V).19 These subtle differences are also evident in other oxides. For example, the perovskites CaTcO3 and SrTcO3 have exceptional magnetic properties, but their AReO3 analogues have yet to be synthesised.20–22 This suggests that differences in the electronic structures between the two cations may be important. Re has more radially-extended 5d valence orbitals than the 4d valence orbitals in Tc, potentially impacting its reactivity and redox stability.23 Similar differences have also been observed in other 4d and 5d metal oxides, where the more radially-extended 5d orbitals of Ta5+ lead to more covalent character in Ta–O bonds compared to Nb–O,24 and the lighter MoO4 tetrahedra undergo a greater amount of rotational disorder compared to the heavier WO4 analogues.25
Pertechnetate salts of the type ATcO4 (A+ = NH4, K, Cs) were among the first Tc oxides isolated and studied.26–28 Subsequently, several powder diffraction and single crystal studies have reported the structures and phase transitions in ATcO4 and AReO4 salts.26–31 Recent synchrotron X-ray diffraction (SXRD) studies of some ATcO4 and AReO4 oxides by Chay et al., Kennedy et al., and Weaver et al. showed that most are isostructural at room temperature.30,32,33 For both Tc and Re, the structures of the four salts (A+ = Na, K, Rb, Ag) were refined to the CaWO4, tetragonal scheelite, structure (space group I41/a, #88). In this structure, the AO8 dodecahedra contain two distinct A–O distances, whereas the BO4 tetrahedra possess a single B–O distance (see Fig. 1).34 The BO4 tetrahedra are isolated from each other and connected to the AO8 dodecahedra via bridging oxygen anions. Bastide showed that the structures of various ABO4 compounds can be predicted from the rA/rO and rB/rO ratios, where rX is the ionic radii of the A- and B-site cations and oxygen anion, respectively.35 This explains why the ATcO4 and AReO4 salts mostly exhibit the same room temperature structure (rTc/rO ∼ rRe/rO), as well as explaining why RbTcO4 and RbReO4 (rRb/rO ∼ 1.13) exhibit the tetragonal I41/a scheelite-type structure whilst the CsTcO4 and CsReO4 (rCs/rO ∼ 1.23) analogues exhibit an orthorhombic pseudo-scheelite structure (space group Pnma, #62. See Fig. 1 and Fig. S1, ESI†).28,30,32–34,36,37 Exceptions to these ionic radii rules have been observed, as reflected in the rich structural chemistry of the LnTaO4 (Ln3+ = lanthanoid La-Lu) oxides that is not replicated in the corresponding LnNbO4 oxides.38–43
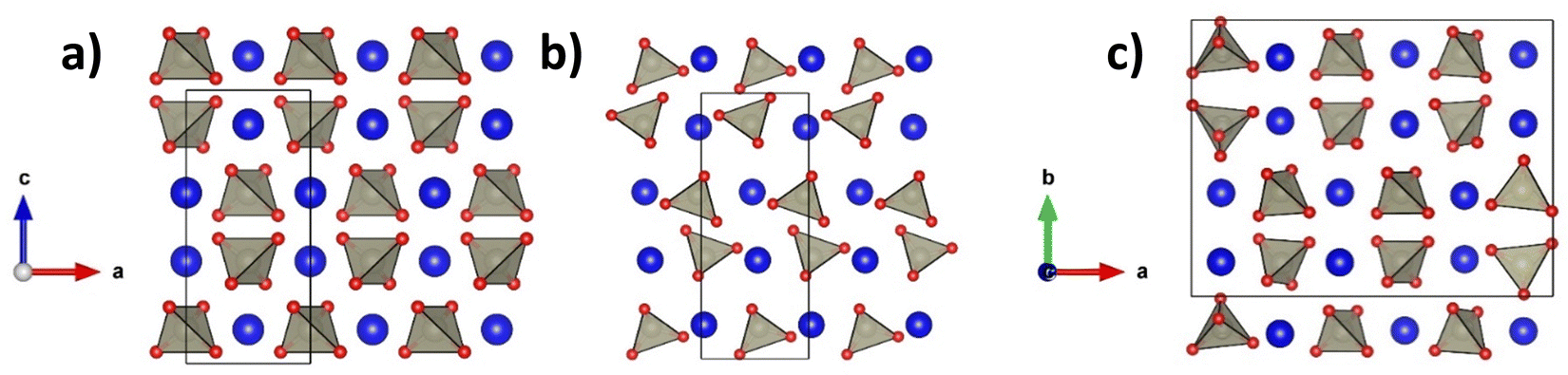 | ||
| Fig. 1 Crystal structures of the scheelite-type oxides for (a) tetragonal I41/a, (b) orthorhombic Pnma, and (c) monoclinic P21/c. Note that the monoclinic P21/c unit cell has been tripled along the a axis. | ||
Although Re7+ and Tc7+ are similar in size, previous studies have described differences between the ATcO4 and AReO4 salts. Reynolds et al. highlighted differences in the thermal behaviour between NH4TcO4 and NH4ReO4 attributed to subtle differences in the B–O (B7+ = Tc, Re) bonds within the BO4 tetrahedra.23 More dramatic differences are evident in the two thallium salts TlTcO4 and TlReO4. The former adopts the orthorhombic Pnma pseudo-scheelite structure at room temperature, whereas TlReO4 exhibits a monoclinic P21/c (space group #14) structure (Fig. 1).32,33,44 The temperature dependence of the two structures is also different, with TlReO4 exhibiting an unusual re-entrant phase transition.45–47 A neutron total scattering study by Saura-Múzquiz et al. revealed that the local structure of TlReO4 was always best fit to the monoclinic P21/c space group, irrespective of the long-range average structure, with a change in the correlation length of the Tl+ 6s2 lone pairs used to explain the re-entrant phase transition.48 This behaviour appears to be unique to TlReO4 and has not been reported in the closely related oxide TlTcO4 that undergoes an orthorhombic-to-tetragonal phase transition upon heating.33
In this work, the temperature dependence of the structure of TlTcO4 was investigated using neutron total scattering and pair distribution function (PDF) analysis. The thermal behaviour of TlTcO4 is analysed, and in-depth modelling of its long-range average and short-range local structure is presented. These results are then compared to TlReO4 and other 6s2-containing ABO4 materials to determine the origin of the differences between these materials.
Experimental methods
*** Caution! *** 99Tc is a β− emitter (Emax = 0.29 MeV). All operations relating to the synthesis of TlTcO4 were performed in a licensed radiochemical laboratory at the University of Las Vegas, Nevada. Appropriate shielding was employed during all manipulations and while loading the sample into a vanadium container for examination.*** Caution! *** Thallium salts are potentially fatal if swallowed. Appropriate personal protective equipment and safe work procedures were employed during sample preparation and sample manipulation.
Thallium pertechnetate was prepared by cation metathesis from KTcO4 and TlF in deionised water. A thallium(I) fluoride (1.223 g, 5.48 mmol) solution was added dropwise to a solution of KTcO4 (1.095 g, 5.42 mmol) and TlTcO4 precipitated immediately. The mixture was mechanically stirred at room temperature for 1 hour. The solid was allowed to settle, the supernatant was discarded, and the resulting white solid was washed with cold deionised water (3 × 5 mL), isopropanol (3 × 5 mL), and diethyl ether (3 × 5 mL) before being dried at 120 °C for 12 hours. After cooling, TlTcO4 was removed from the vial, weighed (1.477 g, 4.02 mmol, yield 74.2%) and finally sealed in a 6 mm vanadium can and shipped to Oak Ridge National Laboratory (ORNL) for neutron scattering measurements.
Neutron scattering data suitable for neutron PDF (NPDF) analysis were collected with a centre wavelength of 0.8 Å using the POWGEN diffractometer at the Spallation Neutron Source (SNS) located at ORNL.49 The data were corrected for instrument background, the incident neutron spectrum, absorption, and multiple scattering events before normalisation. The software PDFgetN3 was used to calculate the NPDF with a Qmax of 30.0 Å−1. A Savitzky–Golay filter was applied to the S(Q) for Q > 9.0 Å−1 to reduce the noise in the broad regions of the reciprocal space data. Additional details are provided in the ESI (Fig. S2–S13).†
Structural refinements using the Rietveld method were carried out using the program TOPAS 6.50 The background of each pattern was estimated using a 12th-order Chebyshev polynomial. The scale factor, lattice parameters, atomic positions, and atomic displacement parameters (ADPs) were refined simultaneously with the peak profile parameters. Partial NPDFs were calculated using RMCProfile,51 and the crystal structures were drawn using VESTA.52
The theoretical calculations for TlTcO4 were performed using the projector augmented wave (PAW) method,53 as implemented in the Vienna Ab initio Simulation Package (VASP) version 5.4.4.54 The Perdew–Burke–Ernzerhof (PBE) functional revised for solids (PBEsol) and the Armiento-Mattsson 2005 (AM05) exchange–correlation functionals were employed to study the ground state structure and electronic interactions.55–57 The basis sets considered for each atom were as follows: Tl: 6s26p1, Tc: 4d55s2, O: 2s22p4. The plane-wave cutoff energy was set to 700 eV. A k-point mesh of 11 × 11 × 7 was used in the irreducible Brillouin zone, utilising Monkhorst Pack (MP) grids.58 The force minimisation and convergence criteria were set to 10−9 eV for electronic convergence and 10−3 eV for ionic relaxation, respectively. To compute the electronic structure of TlTcO4, the Tran–Blaha modified Becke–Johnson (TB-mBJ) potential was employed in conjunction with the traditional generalised gradient approximation (GGA) method incorporated in VASP.59–62 The TB-mBJ potential is known to effectively address the band gap of materials, particularly for insulators and semiconductors. The impact of spin–orbit coupling (SOC) was also considered. Our results indicate that the inclusion of SOC has almost no impact on the partial density of states (PDOS) for the atoms. Isosurface maps illustrating bonding interactions through the electron localisation function (ELF) were generated for the structures of TlTcO4 determined from the NPDF fits at 50 K. The visualisation of the ELF was subsequently performed using VESTA.52
Results and discussion
Neutron powder diffraction and Rietveld analysis
Neutron powder diffraction (NPD) data were collected at 50, 300, 450, and 600 K (Table S1, ESI†). These temperatures were chosen based on the orthorhombic-to-tetragonal phase transition observed in the previous SXRD study.33 The 50 K data were measured to minimise thermal motion, thus allowing deconvolution of static and dynamic disorder. Although the fit to the 50 K data is not optimal, possibly due to disorder, the important structural features are evident. The orthorhombic Pnma pseudo-scheelite structure consists of a network of TlO10 polyhedra and TcO4 tetrahedra. Considering that the eight shortest Tl–O bond distances vary between 2.689(14)–3.052(6) Å, it appears that Tl+ is displaced towards the corner of an ideal cube by the Tl+ 6s2 lone pair electrons directed to the adjacent cube edge, resulting in two long Tl–O distances. There are two longer Tl–O contacts that become important in the high-temperature tetragonal structure (see below). Considering these, the bond distortion index of the resulting TlO12 polyhedra is 0.08205. Although the TcO4 tetrahedra are distorted, there is a narrow range of Tc–O bond lengths between 1.724(10)–1.767(16) Å with an average of 1.745(14) Å. The O–Tc–O bond angles in the tetrahedra range from 105.6(8)–111.9(5)°. The same orthorhombic Pnma model was also used to fit the data obtained at 300 and 450 K (Fig. 2 and Fig. S14, ESI†). | ||
| Fig. 2 Rietveld refinements to the neutron Bragg data of TlTcO4, collected on the POWGEN diffractometer at (a) 600 K refined to the tetragonal I41/amd space group, and (b) 50 K refined to the orthorhombic Pnma space group. The black circles represent the data, the red line represents the fit to the data, the green line represents the difference between the data and the fit, and the blue dashes represent the space group-allowed reflections. | ||
A previous SXRD study concluded that TlTcO4 underwent a phase transition from orthorhombic Pnma to tetragonal I41/amd at ∼500 K.33 This was based on a lack of evidence of the (114) reflection, which is allowed in I41/a but forbidden in I41/amd by the 2h + l = 4n reflection condition. The orthorhombic Pnma to tetragonal I41/amd phase transition is allowed to be continuous (by condensation of the M4+ mode, see Fig. S1, ESI†). However, other orthorhombic Pnma pseudo-scheelite structures (such as CsBO4, B7+ = Tc, Ru, Re, Os; and RbRuO4) display a first-order phase transition from orthorhombic Pnma to tetragonal I41/a.32,33,36,63 These two tetragonal structures differ by the rotation of the BO4 tetrahedra that is allowed in I41/a but not in I41/amd. Attempts to fit both models to the high temperature 600 K dataset returned similar qualities of fit (Rwp = 2.00% for I41/a vs. 2.02% for I41/amd; see Fig. S15, ESI†). Therefore, the tetragonal I41/amd model was adopted at 600 K in line with the suggestion of Kennedy et al.33 The quality of the fit to the 600 K data demonstrates the quality of the sample, further highlighting the anomalous nature of the 50 K dataset. The Tl+ cation is 12-coordinate in the tetragonal I41/amd structure with four Tl–O bonds at 2.942(7) and a further eight at 3.352(4). The bond distortion index of the TlO12 polyhedra is noticeably smaller than seen at 50 K (0.0567 vs. 0.08205).
The temperature dependence of the unit cell parameters and Tc–O distances is displayed in Fig. 3. Due to the sparsity of data points in the current NPD study, values from the previous SXRD study are also presented,33 and these are in good agreement with the current study. The lattice parameters display conventional thermal expansion, with the a and b lattice parameters becoming equal above 500 K, corresponding to the orthorhombic-to-tetragonal phase transition. The temperature dependence of the ADPs shows the values for the Tc7+ and O2− atoms increasing between 50 and 300 K before dropping at 450 K (Fig. S16, ESI†). This is thought to be a result of the rare isosymmetric orthorhombic Pnma to orthorhombic Pnma phase transition at ∼400 K.33
 | ||
| Fig. 3 Temperature dependence of the (a) and (b) lattice parameters, (c) unit cell volume, and (d) the Tc–O bond distances for TlTcO4 derived from the Rietveld refinements of the neutron powder diffraction data. Where not shown, the error bars are smaller than the symbols. The dashed lines in (a)–(c) are the results from the previous synchrotron X-ray diffraction study.33 The dotted line in (c) drawn to guide the eyes. | ||
As evident from Fig. 3, the unit cell volume of the tetragonal I41/amd phase is significantly larger than that of the extrapolated orthorhombic Pnma phase. Although only one temperature point is taken above the phase transition in the current NPD study, the previous SXRD refinements show the same trend.33 Similar behaviour was recently described in TlReO4, and Saura-Múzquiz et al. proposed that ordering of the Tl+ 6s2 lone pairs in the lower symmetry phase caused a more efficient packing of the structure.48 By analogy, we postulate that the 6s2 lone pairs in TlTcO4 are ordered in the orthorhombic phase and disordered in the tetragonal phase (Fig. S17, ESI†). This hypothesis is supported through a comparison of the monoclinic I2/b and tetragonal I41/a structures of NdNbO4 (Nd3+: 6s0) and BiVO4 (Bi3+: 6s2).24,64,65 The tetragonal I41/a unit cell volume in NdNbO4 is smaller than the unit cell volume extrapolated from the monoclinic I2/b phase. In contrast, it is larger in BiVO4 due to the disordering of the 6s2 lone pairs (Fig. S18, ESI†).
An additional advantage of NPD is that neutrons are scattered isotropically due to the short range of their interaction. This makes them more sensitive to the small displacements of atoms from their equilibrium positions, leading to greater precision in the refined ADPs compared to SXRD studies.66 Although oxygen is a relatively weak X-ray scatterer (Z = 8) compared to Tl+ (Z = 81) and Tc7+ (Z = 43), the coherent neutron scattering length of the three atoms are comparable (bO = 5.8, bTc = 6.8 fm, bTl = 8.8), leading to greater sensitivity to the Tc–O bond distances. As observed in Fig. 3, the Tc–O bond lengths decrease upon heating. This has been observed in several other ABO4 structures, including CaMoO4 and CsReO4,36,67 and has been correlated with the onset of rotational disorder in the rigid BO4 tetrahedra units. It is possible that the orthorhombic-to-tetragonal phase transition in TlTcO4 may be related to the onset of rotational disorder in the rigid TcO4 units. However, observation of the short-range local structure is required to verify this possibility.
Neutron total scattering and pair distribution function analysis
NPDF data of TlTcO4 at 50, 300, 450, and 600 K are plotted in Fig. 4. The first narrow peak at ∼1.72 Å corresponds to the first Tc–O pair within the TcO4 tetrahedra and shows neither splitting nor broadening at any of the measured temperatures. The first O–O distance, corresponding to the edge of the TcO4 tetrahedra, results in a consistent peak at ∼2.80 Å. Rietveld analysis of the NPD Bragg data shows the Tc–O bond lengths decrease upon heating, from an average of 1.731(14) Å at 50 K to 1.630(9) Å (a ∼6% decrease) at 600 K. However in the NPDF, the Tc–O bond length at ∼1.72 Å remains unchanged across all measured temperatures, suggesting the Tc–O bond length shortening observed in the NPD Bragg data is unphysical. That neither the first Tc–O nor O–O distances change significantly over the measured temperature range implies the presence of rigid TcO4 tetrahedra, consistent with other PDF studies of ABO4 scheelite-type oxides.34,36,48,67,68 | ||
| Fig. 4 Variable temperature neutron pair distribution function data of TlTcO4 at 50 (blue), 300 (green), 450 (orange), and 600 K (red). | ||
Stereochemically active ns2 lone pairs often cause cations to disorder through well-defined displacements from their centrosymmetric positions (giving a static contribution to the ADPs) as well as increased thermal motion (giving a dynamic contribution).69–71 Data were measured at 50 K to minimise the thermal disorder, resulting in sharper peaks at high r. Fits were conducted using the long-range average orthorhombic Pnma model over a range of 1.0–30 Å (Fig. 5). This model returned a poor fit especially at low r, with a large residual observed across the entire fitting range. The failure to accurately fit the high r region to the orthorhombic model suggests a subtle symmetry lowering. The feature at ∼2.84 Å was particularly poorly fitted, with partial PDFs revealing this region corresponds to the Tl–O distances within the TlO10 polyhedra and the O–O distances across the TlO10 polyhedra and TcO4 tetrahedra (Fig. S19, ESI†). Fits were attempted with both rigid (single Tc–O distance and O–Tc–O angles constrained to be 109.5°) and flexible TcO4 tetrahedra. However, neither approach was able to reproduce the sharp feature at ∼2.84 Å. Fits were also performed to the higher symmetry tetragonal I41/a and I41/amd scheelite-type structures, which also returned poor fits (Fig. S20, ESI†).
 | ||
| Fig. 5 Neutron pair distribution function data collected at 50 K on the POWGEN instrument. Fits to the data were conducted with (a) the long-range orthorhombic Pnma model, and (b) the long-range monoclinic P21/c model observed in TlReO4. The blue circles represent the data, the red line represents the fit to the data, and the green line represents the difference between the two. The dashed lines indicate the maximum and minimum in the difference curve across the plotted r ranges. | ||
Fits were then undertaken using the monoclinic P21/c structural model seen in other 6s2-containing ABO4 materials, such as the long-range average and short-range local structure of TlReO4 at room temperature and the short-range local structure of PbWO4.48,72 This monoclinic model involves more complex distortion of the TlO10 polyhedra associated with short-range disorder of the Tl+ 6s2 lone pair electrons and provides the best fit to the data over a range of 1.0–30 Å, as shown in Fig. 5. This implies that the local structures of TlTcO4 and TlReO4 are the same despite their different long-range average structures. A key feature of the monoclinic P21/c TlReO4 structure is the displacement of the Tl+ cations from the centre of the Tl+ polyhedra, resulting in more significant variability in the Tl–O bond lengths and rotation of the ReO4 tetrahedra. Evidently, similar displacements occur in TlTcO4, with the same features observed in the 100 K dataset taken on the NOMAD diffractometer (Fig. S21 and S22, ESI†), as well as the 300 and 450 K NPDF datasets (Fig. S23–S25, ESI†). As detailed below, density functional theory (DFT) calculations show the ground state energy of the monoclinic P21/c structure to be lower than that of the orthorhombic Pnma structure (Tables S2 and S3, ESI†).
Fits were also performed across different distance ‘windows’, with better fits achieved by using 10 Å windows across the data (Fig. S26, ESI†). This also resulted in a more distorted structure at low r and differences in the TcO4 volume. This suggests nanodomains may form at lower temperatures related to the ‘freezing in’ of Tl+ 6s2 lone pair disorder and the isosymmetric orthorhombic Pnma to orthorhombic Pnma phase transition that are difficult to model, also potentially explaining the suboptimal fitting to the NPD at 50 K. In fitting the NPDF in 10 Å windows, the less-local fits provide an ‘averaging’ of these more-local features to better fit the data. Further confirmation using additional techniques, such as transmission electron microscopy or solid-state nuclear magnetic resonance spectroscopy, would be required to better model these contributions.
There is an apparent disconnect between the long-range average and short-range local structures of TlTcO4. At 50 K, the long-range NPD Bragg data were best fit using the orthorhombic Pnma model consisting of TlO10 polyhedra with several different Tl–O bond lengths and distorted TcO4 tetrahedra. The short-range NPDF data revealed that the TcO4 tetrahedra are rigid and a greater distribution of Tl–O bond distances within the Tl+ polyhedra (Fig. S27, ESI†), where the long and flexible Tl–O bonds allow for the rigid TcO4 tetrahedra to rotate freely. This degree of rotational freedom in the ABO4 scheelite-derived structures is due to the A–O–B connectivity that acts as a ‘hinge’.67 Unlike in perovskite-derived structures where the BX6 octahedra are corner sharing, the TcO4 tetrahedra are not directly connected, relaxing the requirement of coherent tetrahedra rotations. It is this lack of connectivity that leads to incoherent TcO4 tetrahedra rotations and translations that Rietveld refinements may model as a combination of increased ADPs and Tc–O bond shortening, as also seen in RbReO4 and CsReO4.34,36 Furthermore, the monoclinic P21/c structural model can be derived from the tetragonal I41/amd aristotype by rotation of the TcO4 tetrahedra about the a-axis (similar to those observed in the orthorhombic Pnma structure) governed by a M4+ mode, and about the b-axis (analogous to that observed in the tetragonal I41/a structure) governed by a Γ3+ mode (Fig. S1, ESI†). It is postulated that locally both the M4+ and Γ3+ modes are present, and the lack of direct connectivity reduces the correlation of the rotations. This explains why the long-range average structure is best fitted to the orthorhombic Pnma (M4+) structure, as the TcO4 tetrahedra rotations governed by the Γ3+ mode are uncorrelated and separated by ∼18 Å.
The fitting process was repeated for the 600 K dataset (Fig. S28, ESI†), recalling that 600 K is above the orthorhombic-to-tetragonal phase transition.33 All four models (orthorhombic Pnma, tetragonal I41/a and I41/amd, and monoclinic P21/c) returned a similar goodness of fit, most likely due to the greater amount of thermal motion and broader NPDF peaks at higher temperatures. Despite these broadened features, the Tc–O peak at ∼1.72 Å remains relatively unchanged between the 50 and 600 K datasets, while the prominent peak at ∼2.84 Å shows significantly less thermal broadening compared to the other peaks within the NPDF datasets reflecting the rigid TcO4 tetrahedra (Fig. 4). Although the local features at ∼2.84 Å are best fit using the monoclinic P21/c model, the less local (10–30 Å) range is fit equally well between the four models (Fig. S29, ESI†). This suggests that the Tl+ 6s2 lone pairs are still sterically active at high temperatures, but with a short (>10 Å) correlation length.
Ground state energy calculations using density functional theory
Tables S2 and S3 report the ground state energies for the four possible structures of TlTcO4 from DFT calculations: tetragonal I41/a, tetragonal I41/amd, orthorhombic Pnma, and monoclinic P21/c calculated using the PBE, PBEsol, and AM05 functionals. For each structure, the calculations converged to a physically reasonable model and the calculated unit cell parameters are in acceptable agreement with the experimentally observed values.33 Irrespective of the functional employed, the tetragonal I41/a structure has the lowest energy and the tetragonal I41/amd the highest, with the ground state energy of the monoclinic P21/c structure invariably lower than that of the orthorhombic Pnma structure (Table S3, ESI†). That these functionals all predicted the tetragonal I41/a structure to be the ground state, in contrast to the experimental findings, reflects the challenges of investigating the structure and physical properties of materials with highly polarizable ions. The failure of these functionals to correctly predict the experimentally observed ground state mirrors the situation in BiVO4.73 Liu et al. reported that the relative energies of the experimentally observed tetragonal I41/a and monoclinic I2/b structures could only be reproduced using complex hybrid functionals as a consequence of the need to accurately describe the Bi3+ 6s2 lone pair electrons by including the polarizability of the Bi3+ cation.73 These authors did not consider the possibility of local scale distortions.Fig. 6 illustrates the band structure and PDOS for TlTcO4 in the monoclinic P21/c structure. Essentially identical results were obtained for calculations assuming the orthorhombic Pnma structure (Fig. S30, ESI†). This shows that TlTcO4 is a direct band gap semiconductor with a band gap of 2.53 eV, as estimated using the TB-mBJ potential. The valence band is predominantly compromised of the O p and Tl s states, while the conduction band is mainly derived from Tc d states. The Tl 6s states are present at the top of the valence band and show significant hybridisation with the O 2p states, with minimal involvement from the Tc states. This is a characteristic of stereochemical active 6s2 electrons.74,75 This is demonstrated through ELF calculations, illustrated for the 〈001〉 plane in Fig. 7, that indicate localisation of the Tl 6s states and presumed stereochemical activity. Conversely, the O 2p states exhibit moderate localisation, while the Tc states show very little.
 | ||
| Fig. 6 Electronic band structure and partial density of states (PDOS) of TlTcO4 in the monoclinic P21/c structure calculated using the Tran–Blaha modified Becke–Johnson (TB-mBJ) potential, showing a bandgap of 2.53 eV. | ||
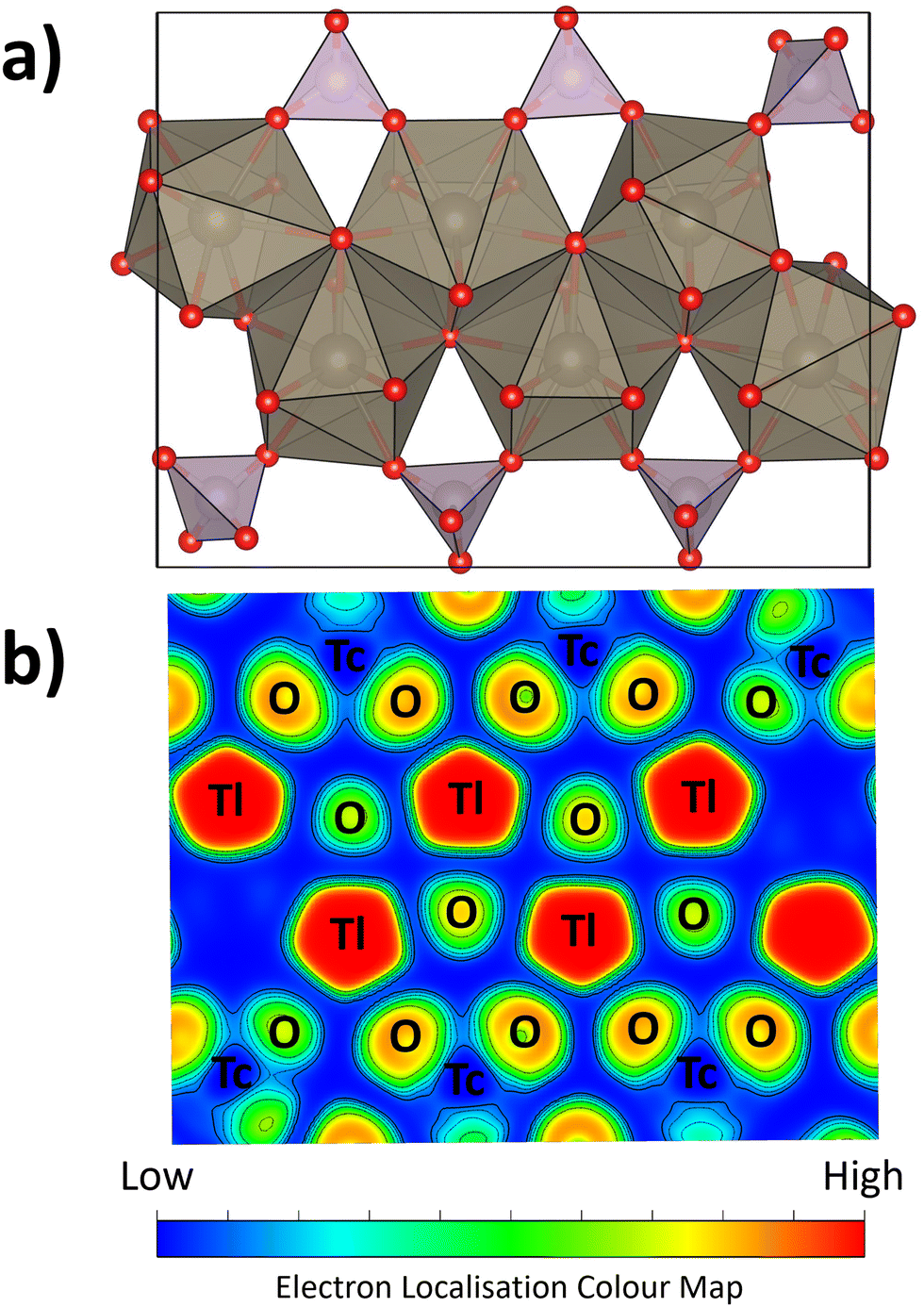 | ||
| Fig. 7 (a) Structural model of TlTcO4 in the monoclinic P21/c space group, fit from the 50 K POWGEN neutron pair distribution function data. (b) Isosurface maps illustrating bonding interactions through the electron localisation function (ELF) in the 〈001〉 plane generated for the geometry-optimised structure of TlTcO4 in the monoclinic P21/c space group. | ||
Comparison between ABO4 structures with 6s2 lone pair electrons
TlTcO4 can now be placed in the context of other ABO4 structures with 6s2 lone pair electrons. At 300 K, TlTcO4 (orthorhombic Pnma),33 TlReO4 (monoclinic P21/c),32 PbWO4 (tetragonal I41/a),72 and BiVO4 (monoclinic I2/b)76 each display different long-range average structures. The impact of the stereochemically active A-site 6s2 electron lone pairs on the local structure appears to be dependent on a combination of the size, oxidation state, and polarizability of the A- and B-site cations. These are shown in Fig. 8 and Table 1, and are discussed below. | ||
| Fig. 8 The A–O, B–O bond lengths, and O–B–O bond angles from (a)–(c) TlTcO4 from this work, (d)–(f) TlReO4 taken from Saura-Múzquiz et al.,48 (g)–(i) PbWO4 taken from Mullens et al.,72 and (j)–(l) BiVO4 taken from Sleight et al.76 All data is taken from the neutron pair distribution function fits at 300 K to the monoclinic P21/c model, except for BiVO4 (crossed histogram bars), which is the long-range average structure in the monoclinic I2/b space group. | ||
The A–O–B connectivity makes it reasonable to assume that distortions of the AO8 polyhedra and BO4 tetrahedra are related. Across the 5p block elements (A = Tl+, Pb2+, Bi3+), the oxidation state increases resulting in shorter A–O bonds and smaller AO8 polyhedra (Table 1). As seen in Fig. 8, the larger oxidation state A-site cations result in less flexible AO8 polyhedra and a smaller distribution of the A–O bond lengths. As the AO8 polyhedra get smaller, the flexibility of the BO4 tetrahedra becomes more important. While the TcO4 and ReO4 tetrahedra remain rigid, the WO4 tetrahedra distort through relaxation of the O–W–O bond angles and the VO4 tetrahedra distort through variations in both the O–V–O bond angles and the V–O bond lengths. Likewise, the choice of nd0 element on the B-site affects the degree of polarizability, with the VO4 tetrahedra in BiVO4 (V: 3d0) exhibiting a greater amount of distortion than the TcO4 tetrahedra in TlTcO4 (Tc: 4d0) and the ReO4 tetrahedra in TlReO4 (Re: 5d0).
The PDOS for TlTcO4 is very similar to that of TlReO4 and PbWO4 (Fig. S31, ESI†), despite these materials having different long-range average structures at 300 K.48,72 The PDOS is, however, significantly different from that reported for BiVO4 where the Bi 6s states are significantly further below the Fermi level (∼9 eV vs. 1 eV for TlTcO4).73 Presumably strong hybridisation of the 6s states of Pb and Tl with O p or metal d states pushes these levels closer to the conduction band edge, whereas the higher oxidation state of Bi reduces the hybridisation of the Bi 6s states with the oxygen states. Indeed, near the Fermi level BiVO4 features only O p states in the valence bands. The implication is that the 6s electrons do not participate significantly in bonding, rather they remain as lone pairs and exhibit an obvious stereochemical influence. It can be speculated that this difference in the PDOS may be correlated with the photocatalytic properties of these oxides.
The question remains as to why TlTcO4 and TlReO4 have the same monoclinic P21/c local structure but different long-range average structures. Previous studies have shown that all other ATcO4 and AReO4 salts (A+ = Na, K, Rb, Ag, Cs) are isostructural.30,32,33 Bastide's analysis of the rA/rO and rB/rO ratios in the ABO4 materials suggests that TlReO4 should adopt a tetragonal I41/a structure similar to RbReO4,35 as the two A-site cations are approximately the same size (Tl+ = 1.59 vs. Rb+ = 1.61 Å).17 TlReO4 exhibits the tetragonal I41/a structure at low (<150 K) and high temperatures (>400 K), with the room temperature monoclinic P21/c structure the result of long-range ordering of the Tl+ 6s2 lone pairs.48 Since Tc7+ is slightly smaller than Re7+ (0.37 vs. 0.38 Å),17 as evident from the sharp B–O peaks at 1.72 and 1.74 Å in the NPDF data of TlTcO4 and TlReO4 respectively, it is possible that this small change in the B-site cation ionic radii is sufficient to favour the formation of the long-range orthorhombic Pnma, rather than the tetragonal I41/a structure (rTc/rO ∼ 0.27 vs. rRe/rO ∼ 0.28).
The long-range average monoclinic P21/c structure of TlReO4 at room temperature is derived from the tetragonal I41/amd aristotype by rotations of the ReO4 tetrahedra associated with the irreps Γ3+ and M4+.32 In this structure, TlReO4 has two distinct ReO4 tetrahedra sites rotated about the b-axis (associated with the irrep Γ3+, analogous to the tetragonal I41/a to tetragonal I41/amd phase transition), and one distinct ReO4 tetrahedra site rotated about the a-axis (associated with the irrep M4+, analogous to the orthorhombic Pnma to tetragonal I41/amd phase transition). Although the same irreps are present in the local structure of TlTcO4, the opposite trend is observed. There are two distinct TcO4 tetrahedra sites rotated about the a-axis, and one rotated about the b-axis (Fig. S32, ESI†). Evidently, the ‘averaging’ of these leads to the observation of different long-range average structures.
A further difference between TlTcO4 and TlReO4 is the lack of a re-entrant phase transition observed in TlTcO4. Phase transitions can be driven by the ability of the 6s2 lone pairs to macroscopically align, leading to a lowering of symmetry and a decrease in unit cell volume due to the more compact structure. This is seen in TlReO4 where the room temperature structure is monoclinic P21/c due to the ability of the 6s2 to align macroscopically, before an increase in temperature leads to greater thermal motion and a breakdown of this long-range ordering.48 No such macroscopic ordering is observed in TlTcO4 due to the larger thermal motion of the TcO4 tetrahedra compared to ReO4 (Fig. S33, ESI†). This was similarly seen in work by Amarasinghe et al., where greater rotational disorder was observed in the lighter MoO4 tetrahedra of NaRE(BO4)2 (RE3+ = Ln, Y; B6+ = Mo, W) compared to the heavier WO4 tetrahedra (95 vs. 184 amu).25 As Tc7+ is almost half of the atomic mass of Re7+ (98 vs. 186 amu), a similar sensitivity to temperature and larger thermal displacements is expected. The greater susceptibility of the TcO4 tetrahedra to rotate may also explain the differences in the thermal expansion behaviour of the two ammonium salts NH4TcO4 and NH4ReO4 at different temperatures.23
Conclusions
A detailed variable temperature neutron total scattering study – combining both NPD and NPDF – to understand the long-range average and short-range local structure of TlTcO4 was conducted. In the long-range average structure, an orthorhombic Pnma to tetragonal I41/amd phase transition occurred before 600 K, consistent with previous SXRD studies.33 This is the first observation of such a phase transition, with other orthorhombic Pnma pseudo-scheelite structures undergoing a first order phase transition to the tetragonal I41/a scheelite-type structure.32,33,36,63 Modelling the short-range local structure required a lower symmetry monoclinic P21/c model due to the stereochemistry of the Tl+ 6s2 lone pairs. This is facilitated by the flexible Tl+ polyhedra and rigid TcO4 tetrahedra. The stereochemistry of the Tl+ 6s2 lone pair persisted above the long-range orthorhombic Pnma to tetragonal I41/amd phase transition, albeit on the local scale.Several anomalies were observed in the Rietveld refinements to the NPD data, such as large ADPs at low temperature and a shortening of the Tc–O bond distance upon heating. Using NPDF, these structural anomalies were resolved by devising a model with longer, more flexible Tl–O bonds that allow for the rotation of rigid TcO4 tetrahedra units. This model was then supported by DFT calculations. Considering both the long-range average and short-range local structures of TlTcO4, a more holistic structural model was developed. At low temperatures, the stereochemical activity of the Tl+ 6s2 lone pairs is ‘frozen’ into the structure, resulting in static rotations of the TcO4 tetrahedra. As the material is heated, the TcO4 tetrahedra begin to rotate as governed by the Γ3+ and M4+ modes. The Γ3+ rotations appear to be less correlated than the M4+ modes, resulting in the observation of the orthorhombic Pnma structure in the long range. At ∼500 K, the thermal motion is large enough such that the rotations shift from coherent to incoherent, resulting in an ‘averaging out’ of the rotations and the observation of the higher symmetry tetragonal I41/amd structure. However, the 6s2 lone pairs remain stereochemically active on the local scale, as observed in the NPDF fits. The orthorhombic Pnma to tetragonal I41/amd phase transition is seemingly the result of a change in the correlation length of the Tl+ 6s2 lone pairs, leading to a larger unit cell volume due to their uncorrelated displacements.
This study of TlTcO4 was put into the larger context of other ABO4 structures containing 6s2 lone pairs, including TlReO4, PbWO4, and BiVO4. Each of these oxides has a different long-range average structure at 300 K. However, common trends in the local scale distortions necessary to accommodate the stereochemically active 6s2 lone pair were identified. Although TlTcO4 and TlReO4 disorder in a similar way, a combination of the size of the A-site polyhedra and the choice of nd0 B-site cation leads to different disordering phenomena. These include large ranges of A–O bonds, flexibility in the O–B–O tetrahedra bond angles, and distortion of the B–O bond lengths. This gives a rich tapestry of mechanisms that can be further implemented when fine-tuning the local scale interactions of atoms for the next generation of functional energy materials. The role of polarizability of the B-site cations is worthy of further exploration, especially with the large distortions of the VO4 tetrahedra observed and the lack of NPDF data available for BiVO4. Unfortunately, no such BiBO4 analogous structures are currently known, with BiBO4 [B5+ = Nb (4d0), Sb (4d10), Ta (5d0)] crystallising in different structures consisting of BO6 octahedra.77–79
Optimisation of the structures using the commonly employed PBE, PBEsol, and AM05 exchange–correlation functionals within VASP indicated that the monoclinic P21/c structure was more stable than the orthorhombic Pnma model that describes the long-range average structure of TlTcO4 below 500 K. However, for each of these functionals the tetragonal I41/a structure was predicted to be the energetic ground state, demonstrating the challenges of accurately describing the 6s2 lone pair electrons and polarizability of the Tl+ cation. While it may be possible to identify a hybrid functional that correctly predicts the relative energy of these phases, it is evident that there is no unique universal exchange–correlation functional appropriate for these types of oxides, as seen with other ABO4 metal oxides.38,73
Finally, this work adds to the suite of studies demonstrating the necessity of considering both the long-range average and short-range local structure of ABO4 scheelite-related materials, which have found application as photocatalysts, selective oxidation catalysts, and potential nuclear waste forms.33,76,80 Relationships such as these are of great significance, and additional experimental studies of the local structures, partnered with DFT calculations, are required to identify additional ways of fine-tuning these local disordering features.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the support of the Australian Research Council for this work that was facilitated by access to Sydney Analytical, a core research facility at the University of Sydney. BGM thanks the Australian Institute for Nuclear Science and Engineering for a PGRA scholarship and the Australian Nuclear Science and Technology Organisation for a United Uranium Scholarship. MSM gratefully acknowledges the financial support from the Comunidad de Madrid, Spain, through an “Atracción de Talento Investigador” fellowship (2020-T2/IND-20581). A portion of this research used resources at the Spallation Neutron Source, a DOE Office of Science User Facility operated by the Oak Ridge National Laboratory [beamtime on NOMAD (IPTS #30368) and POWGEN (IPTS #26502) greatly acknowledged]. At UNLV, this material is based upon work performed under the auspices of the Consortium on Nuclear Security Technologies (CONNECT) supported by the Department of Energy/National Nuclear Security Administration under Award Number(s) DE-NA0003948. Supratik Mukherjee acknowledges DRDO, India, for the financial support provided through ACRHEM (DRDO/18/1801/2016/01038: ACRHEM-PHASE-III). Ganapathy Vaitheeswaran acknowledges the CMSD University of Hyderabad for providing the computational facilities, and expresses gratitude to the Institute of Eminence, University of Hyderabad (UOH-IOE-RC3-21-046), for their financial assistance.References
- W. Xie and M. Koyama, Theoretical design of a technetium-like alloy and its catalytic properties, Chem. Sci., 2019, 10(21), 5461–5469 RSC.
- F. N. Skomurski, K. M. Rosso, K. M. Krupka and B. P. McGrail, Technetium incorporation into hematite (α-Fe2O3), Environ. Sci. Technol., 2010, 44(15), 5855–5861 CrossRef CAS PubMed.
- R. Meyer, W. Arnold, F. Case and G. O'Kelley, Solubilities of Tc (IV) oxides, Radiochim. Acta, 1991, 55(1), 11–18 CrossRef CAS.
- K. Lieser and C. Bauscher, Technetium in the Hydrosphere and in the Geosphere, Radiochim. Acta, 1987, 42(4), 205–214 CrossRef CAS.
- C. D. Taylor, Surface segregation and adsorption effects of iron–technetium alloys from first-principles, J. Nucl. Mater., 2011, 408(2), 183–187 CrossRef CAS.
- D. Keiser Jr, D. Abraham and J. Richardson Jr, Influence of technetium on the microstructure of a stainless steel–zirconium alloy, J. Nucl. Mater., 2000, 277(2–3), 333–338 CrossRef.
- B. P. Burton-Pye, I. Radivojevic, D. McGregor, I. M. Mbomekalle, W. W. Lukens Jr and L. C. Francesconi, Photoreduction of 99Tc pertechnetate by nanometer-sized metal oxides: new strategies for formation and sequestration of low-valent technetium, J. Am. Chem. Soc., 2011, 133(46), 18802–18815 CrossRef CAS PubMed.
- I. L. Pegg, Behavior of technetium in nuclear waste vitrification processes, J. Radioanal. Nucl. Chem., 2015, 305, 287–292 CrossRef CAS PubMed.
- T. Jin, D. Kim, A. E. Tucker, M. J. Schweiger and A. A. Kruger, Reactions during melting of low-activity waste glasses and their effects on the retention of rhenium as a surrogate for technetium-99, J. Non-Cryst. Solids, 2015, 425, 28–45 CrossRef CAS.
- K. Kang, N. Shen, Y. Wang, L. Li, M. Zhang, X. Zhang, L. Lei, X. Miao, S. Wang and C. Xiao, Efficient sequestration of radioactive 99TcO4− by a rare 3-fold interlocking cationic metal-organic framework: A combined batch experiments, pair distribution function, and crystallographic investigation, Chem. Eng. J., 2022, 427, 130942 CrossRef CAS.
- Q.-H. Hu, Y.-Z. Shi, X. Gao, L. Zhang, R.-P. Liang and J.-D. Qiu, An alkali-resistant metal-organic framework as halogen bond donor for efficient and selective removing of ReO4−/TcO4−, Environ. Sci. Pollut. Res., 2022, 29(57), 86815–86824 CrossRef CAS PubMed.
- J. O. Dickson, J. B. Harsh, W. W. Lukens and E. M. Pierce, Perrhenate incorporation into binary mixed sodalites: The role of anion size and implications for technetium-99 sequestration, Chem. Geol., 2015, 395, 138–143 CrossRef CAS.
- A. Favre-Réguillon, M. Draye, G. Cote and K. R. Czerwinsky, Insights in uranium extraction from spent nuclear fuels using dicyclohexano-18-crown-6 – Fate of rhenium as technetium homolog, Sep. Purif. Technol., 2019, 209, 338–342 CrossRef.
- H. Fei, M. R. Bresler and S. R. Oliver, A new paradigm for anion trapping in high capacity and selectivity: crystal-to-crystal transformation of cationic materials, J. Am. Chem. Soc., 2011, 133(29), 11110–11113 CrossRef CAS PubMed.
- J. S. McCloy, B. J. Riley, A. Goel, M. Liezers, M. J. Schweiger, C. P. Rodriguez, P. Hrma, D.-S. Kim, W. W. Lukens and A. A. Kruger, Rhenium solubility in borosilicate nuclear waste glass: Implications for the processing and immobilization of technetium-99, Environ. Sci. Technol., 2012, 46(22), 12616–12622 CrossRef CAS PubMed.
- E. Strub, D. Grödler, D. Zaratti, C. Yong, L. Dünnebier, S. Bazhenova, M. Roca Jungfer, M. Breugst and M. Zegke, Pertechnetates – A structural study across the Periodic Table, Chem. - Eur. J., 2024, 30(26), e202400131 CrossRef CAS PubMed.
- R. D. Shannon, Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32(5), 751–767 CrossRef.
- W. W. Lukens, D. A. McKeown, A. C. Buechele, I. S. Muller, D. K. Shuh and I. L. Pegg, Dissimilar behavior of technetium and rhenium in borosilicate waste glass as determined by X-ray absorption spectroscopy, Chem. Mater., 2007, 19(3), 559–566 CrossRef CAS.
- H. Gan, D. A. McKeown, X. Xie and I. L. Pegg, Assessment of rhenium as a surrogate for technetium in Hanford low activity waste borosilicate glasses: Speciation, solubility, and redox effects, Int. J. Appl. Glass Sci., 2023, 14(1), 97–112 CrossRef CAS.
- M. Avdeev, G. J. Thorogood, M. L. Carter, B. J. Kennedy, J. Ting, D. J. Singh and K. S. Wallwork, Antiferromagnetism in a technetium oxide. Structure of CaTcO3, J. Am. Chem. Soc., 2011, 133(6), 1654–1657 CrossRef CAS PubMed.
- E. E. Rodriguez, F. Poineau, A. Llobet, B. J. Kennedy, M. Avdeev, G. J. Thorogood, M. L. Carter, R. Seshadri, D. J. Singh and A. K. Cheetham, High temperature magnetic ordering in the 4d perovskite SrTcO3, Phys. Rev. Lett., 2011, 106(6), 067201 CrossRef PubMed.
- G. J. Thorogood, M. Avdeev, M. L. Carter, B. J. Kennedy, J. Ting and K. S. Wallwork, Structural phase transitions and magnetic order in SrTcO3, Dalton Trans., 2011, 40(27), 7228–7233 RSC.
- E. M. Reynolds, M. Yu, G. J. Thorogood, H. E. Brand, F. Poineau and B. J. Kennedy, Thermal expansion of ammonium pertechnetate and ammonium perrhenate, J. Solid State Chem., 2019, 274, 64–68 CrossRef CAS.
- M. Saura-Múzquiz, B. G. Mullens, H. E. Maynard-Casely and B. J. Kennedy, Neutron diffraction study of the monoclinic-tetragonal phase transition in NdNbO4 and NdTaO4, Dalton Trans., 2021, 50, 11485–11497 RSC.
- D. K. Amarasinghe, S. S. Perera and F. A. Rabuffetti, Rotational disorder in scheelite-type NaRE(MO4)2 (RE = Rare-Earth, Y; M = Mo, W), Cryst. Growth Des., 2020, 20(5), 3442–3448 CrossRef CAS.
- G. Boyd, Technetium and promethium, J. Chem. Educ., 1959, 36, 1–14 CrossRef.
- B. Krebs and K.-D. Hasse, Refinements of the crystal structures of KTcO4, KReO4 and OsO4. The bond lengths in tetrahedral oxoanions and oxides of d0 transition metals, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32(5), 1334–1337 CrossRef.
- B. McDonald and G. Tyson, The crystal structure of caesium, ammonium and potassium pertechnetates, Acta Crystallogr., 1962, 15(1), 87 CrossRef CAS.
- R. Faggiani, R. Gillespie, C. Lock and J. Pocé, The structure of ammonium pertechnetate at 295, 208 and 141 K, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36(2), 231–233 CrossRef.
- J. Weaver, C. Z. Soderquist, N. M. Washton, A. S. Lipton, P. L. Gassman, W. W. Lukens, A. A. Kruger, N. A. Wall and J. S. McCloy, Chemical trends in solid alkali pertechnetates, Inorg. Chem., 2017, 56(5), 2533–2544 CrossRef CAS PubMed.
- B. Kanellakopulos, Zur kenntnis der hochtemperaturmodifikation einiger verbindungen des typs MeIXO4 (Me = Cs, Tl; X = Re, Tc, Cl), J. Inorg. Nucl. Chem., 1966, 28(3), 813–816 CrossRef CAS.
- C. Chay, M. Avdeev, H. E. Brand, S. Injac, T. A. Whittle and B. J. Kennedy, Crystal structures and phase transition behaviour in the 5d transition metal oxides AReO4 (A = Ag, Na, K, Rb, Cs and Tl), Dalton Trans., 2019, 48(47), 17524–17532 RSC.
- B. J. Kennedy, S. Injac, G. J. Thorogood, H. E. Brand and F. Poineau, Structures and phase transitions in pertechnetates, Inorg. Chem., 2019, 58(15), 10119–10128 CrossRef CAS PubMed.
- F. P. Marlton, B. G. Mullens, P. A. Chater and B. J. Kennedy, Tetrahedral displacive disorder in the scheelite-type oxide RbReO4, Inorg. Chem., 2022, 61(38), 15130–15137 CrossRef CAS PubMed.
- J. Bastide, Simplified systematics of the compounds ABX4 (X = O2−, F−) and possible evolution of their crystal-structures under pressure, J. Solid State Chem., 1987, 71(1), 115–120 CrossRef CAS.
- B. G. Mullens, F. P. Marlton, M. Saura-Múzquiz, P. A. Chater and B. J. Kennedy, Tetrahedra rotational and displacive disorder in the scheelite-type oxide CsReO4, Inorg. Chem., 2024, 63(22), 10386–10396 CrossRef CAS PubMed.
- P. B. Romero-Vázquez, S. López-Moreno and D. Errandonea, First-principles study of ATcO4 pertechnetates, J. Phys. Chem. Solids, 2022, 171, 110979 CrossRef.
- B. G. Mullens, M. Avdeev, H. E. A. Brand, S. Mondal, G. Vaitheeswaran and B. J. Kennedy, Insights into the structural variations in SmNb1-xTaxO4 and HoNb1-xTaxO4 combined experimental and computational studies, Dalton Trans., 2021, 50, 9103–9117 RSC.
- B. G. Mullens, M. Saura-Múzquiz, F. P. Marlton, M. Avdeev, H. E. Brand, S. Mondal, G. Vaitheeswaran and B. J. Kennedy, Beyond the ionic radii: A multifaceted approach to understand differences between the structures of LnNbO4 and LnTaO4 fergusonites, J. Alloys Compd., 2022, 930, 167399 CrossRef.
- M. Nyman, M. A. Rodriguez, L. E. Rohwer, J. E. Martin, M. Waller and F. E. Osterloh, Unique LaTaO4 polymorph for multiple energy applications, Chem. Mater., 2009, 21(19), 4731–4737 CrossRef CAS.
- I. Hartenbach, F. Lissner, T. Nikelski, S. F. Meier, H. Müller-Bunz and T. Schleid, Über oxotantalate der lanthanide des formeltyps MTaO4 (M = La–Nd, Sm–Lu), Z. Anorg. Allg. Chem., 2005, 631(12), 2377–2382 CrossRef CAS.
- K. P. Siqueira and A. Dias, Effect of the processing parameters on the crystalline structure of lanthanide orthotantalates, Mater. Res., 2014, 17, 167–173 CrossRef CAS.
- K. J. Cordrey, M. Stanczyk, C. A. Dixon, K. S. Knight, J. Gardner, F. D. Morrison and P. Lightfoot, Structural and dielectric studies of the phase behaviour of the topological ferroelectric La1−xNdxTaO4, Dalton Trans., 2015, 44(23), 10673–10680 RSC.
- P. Rögner and K. J. Range, The crystal structure of β-Thallium perrhenate, Z. Anorg. Allg. Chem., 1993, 619(6), 1017–1022 CrossRef.
- A. Jayaraman, G. Kourouklis, R. Fleming and L. Van Uitert, Temperature-induced phase transitions in TlReO4: A Raman spectroscopic and X-ray diffraction study, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37(1), 664 CrossRef CAS PubMed.
- D. de Waal and W. Kiefer, Raman Investigation of the Low Temperature Phase Transitions in Monoclinic TlReO4, Z. Anorg. Allg. Chem., 2004, 630(1), 127–130 CrossRef CAS.
- S. Mondal, G. Vaitheeswaran, B. J. Kennedy, C. Chay, S. Injac and D. Errandonea, Crystal structure and phase transition of TlReO4: a combined experimental and theoretical study, J. Phys.: Condens. Matter, 2020, 33(6), 065403 CrossRef PubMed.
- M. Saura-Múzquiz, F. P. Marlton, B. G. Mullens, A. M. Manjón-Sanz, J. C. Neuefeind, M. Everett, H. E. A. Brand, S. Mondal, G. Vaitheeswaran and B. J. Kennedy, Understanding the Re-entrant Phase Transition in a Non-magnetic Scheelite, J. Am. Chem. Soc., 2022, 144(34), 15612–15621 CrossRef PubMed.
- A. Huq, M. Kirkham, P. F. Peterson, J. P. Hodges, P. S. Whitfield, K. Page, T. Hűgle, E. B. Iverson, A. Parizzi and G. Rennich, POWGEN: rebuild of a third-generation powder diffractometer at the Spallation Neutron Source, J. Appl. Crystallogr., 2019, 52(5), 1189–1201 CrossRef CAS PubMed.
- A. A. Coelho, TOPAS and TOPAS-Academic: An optimization program integrating computer algebra and crystallographic objects written in C, J. Appl. Crystallogr., 2018, 51(1), 210–218 CrossRef CAS.
- M. G. Tucker, D. A. Keen, M. T. Dove, A. L. Goodwin and Q. Hui, RMCProfile: Reverse Monte Carlo for polycrystalline materials, J. Phys.: Condens. Matter, 2007, 19(33), 335218 CrossRef PubMed.
- K. Momma and F. Izumi, VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data, J. Appl. Crystallogr., 2011, 44(6), 1272–1276 CrossRef CAS.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186 CrossRef CAS PubMed.
- R. Armiento and A. E. Mattsson, Functional designed to include surface effects in self-consistent density functional theory, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72(8), 085108 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Restoring the density-gradient expansion for exchange in solids and surfaces, Phys. Rev. Lett., 2008, 100(13), 136406 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Special points for Brillouin-zone integrations, Phys. Rev. B: Solid State, 1976, 13(12), 5188–5192 CrossRef.
- D. Koller, F. Tran and P. Blaha, Merits and limits of the modified Becke-Johnson exchange potential, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83(19), 195134 CrossRef.
- D. Koller, F. Tran and P. Blaha, Improving the modified Becke-Johnson exchange potential, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85(15), 155109 CrossRef.
- D. J. Singh, Electronic structure calculations with the Tran-Blaha modified Becke-Johnson density functional, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82(20), 205102 CrossRef.
- F. Tran and P. Blaha, Accurate band gaps of semiconductors and insulators with a semilocal exchange-correlation potential, Phys. Rev. Lett., 2009, 102(22), 226401 CrossRef PubMed.
- S. Injac, A. K. Yuen, M. Avdeev, C.-H. Wang, P. Turner, H. E. Brand and B. J. Kennedy, Structural and magnetic studies of ABO4-type ruthenium and osmium oxides, Inorg. Chem., 2020, 59(5), 2791–2802 CrossRef CAS PubMed.
- B. G. Mullens, M. Saura-Múzquiz, G. Cordaro, F. P. Marlton, H. E. Maynard-Casely, Z. Zhang, G. Baldinozzi and B. J. Kennedy, Variable temperature in situ neutron powder diffraction and conductivity studies of undoped HoNbO4 and HoTaO4, Chem. Mater., 2024, 36(10), 5002–5016 CrossRef CAS.
- J. Sánchez-Martín, D. Errandonea, J. Pellicer-Porres, D. Vázquez-Socorro, D. Martínez-García, S. N. Achary and C. Popescu, Phase transitions of BiVO4 under high pressure and high temperature, J. Phys. Chem. C, 2022, 126(17), 7755–7763 CrossRef.
- M. Saura-Múzquiz, B. G. Mullens, M. Avdeev, P. K. Jharapla, G. Vaitheeswaran, M. Gupta, R. Mittal and B. J. Kennedy, Experimental and computational insights into the anomalous thermal expansion of (NH4)ReO4, J. Solid State Chem., 2022, 315, 123531 CrossRef.
- S. P. Culver and R. L. Brutchey, Thermally activated rotational disorder in CaMoO4 nanocrystals, CrystEngComm, 2016, 18(24), 4485–4488 RSC.
- F. A. Rabuffetti, S. P. Culver, L. Suescun and R. L. Brutchey, Structural disorder in AMoO4 (A = Ca, Sr, Ba) scheelite nanocrystals, Inorg. Chem., 2014, 53(2), 1056–1061 CrossRef CAS PubMed.
- E. S. Božin, C. D. Malliakas, P. Souvatzis, T. Proffen, N. A. Spaldin, M. G. Kanatzidis and S. J. Billinge, Entropically stabilized local dipole formation in lead chalcogenides, Science, 2010, 330(6011), 1660–1663 CrossRef PubMed.
- K. Knox, E. Bozin, C. Malliakas, M. Kanatzidis and S. Billinge, Local off-centering symmetry breaking in the high-temperature regime of SnTe, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89(1), 014102 CrossRef.
- G. Laurita and R. Seshadri, Chemistry, structure, and function of lone pairs in extended solids, Acc. Chem. Res., 2022, 55(7), 1004–1014 CrossRef CAS PubMed.
- B. G. Mullens, F. P. Marlton, M. K. Nicholas, A. J. Permana, M. Avdeev, S. Mukherjee, G. Vaitheeswaran, C. Li, J. Liu, P. A. Chater and B. J. Kennedy, Seeing the unseen: The structural influence of the lone pair electrons in PbWO4, Inorg. Chem., 2024, 63(24), 11176–11186 CrossRef CAS PubMed.
- T. Liu, X. Zhang, J. Guan, C. R. A. Catlow, A. Walsh, A. A. Sokol and J. Buckeridge, Insight into the fergusonite–scheelite phase transition of ABO4-type oxides by density functional theory: A case study of the subtleties of the ground state of BiVO4, Chem. Mater., 2022, 34(12), 5334–5343 CrossRef CAS.
- A. Walsh, D. J. Payne, R. G. Egdell and G. W. Watson, Stereochemistry of post-transition metal oxides: Revision of the classical lone pair model, Chem. Soc. Rev., 2011, 40(9), 4455–4463 RSC.
- A. Walsh, Y. Yan, M. N. Huda, M. M. Al-Jassim and S.-H. Wei, Band edge electronic structure of BiVO4: Elucidating the role of the Bi s and V d orbitals, Chem. Mater., 2009, 21(3), 547–551 CrossRef CAS.
- A. W. Sleight, H. Y. Chen, A. Ferretti and D. E. Cox, Crystal growth and structure of BiVO4, Mater. Res. Bull., 1979, 14(12), 1571–1581 CrossRef CAS.
- M. Subramanian and J. Calabrese, Crystal structure of the low temperature form of bismuth niobium oxide [α-BiNbO4], Mater. Res. Bull., 1993, 28(6), 523–529 CrossRef CAS.
- N. Zhuk, M. Krzhizhanovskaya, V. Belyy, N. Sekushin and A. Chichineva, The bismuth orthotantalate with high anisotropic thermal expansion, Scr. Mater., 2019, 173, 6–10 CrossRef CAS.
- D. Zhou, L.-X. Pang, H. Wang and X. Yao, Phase composition and phase transformation in Bi(Sb,Nb,Ta)O4 system, Solid State Sci., 2009, 11(11), 1894–1897 CrossRef CAS.
- M. Saura-Múzquiz, F. P. Marlton, B. G. Mullens, J. Liu, T. Vogt, H. E. Maynard-Casely, M. Avdeev, D. A. Blom and B. J. Kennedy, Cation and lone pair order–disorder in the polymorphic mixed metal bismuth scheelite Bi3FeMo2O12, Chem. Mater., 2023, 35(1), 123–135 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Variable temperature Rietveld refinements of TlTcO4, space group analysis at 600 K, atomic displacement parameters, depiction of structures, partial PDF fits, density functional theory calculations, and variable r range NPDF fits. See DOI: https://doi.org/10.1039/d4cp03707c |
| This journal is © the Owner Societies 2024 |